Stochastic vs. Triggered: Decoding the Dual Mechanisms of Bacterial Persister Cell Formation
This article provides a comprehensive analysis of the two primary paradigms in bacterial persister cell formation: stochastic, internal fluctuations and triggered, external stress-response pathways.
Stochastic vs. Triggered: Decoding the Dual Mechanisms of Bacterial Persister Cell Formation
Abstract
This article provides a comprehensive analysis of the two primary paradigms in bacterial persister cell formation: stochastic, internal fluctuations and triggered, external stress-response pathways. Tailored for researchers and drug development professionals, it synthesizes foundational concepts, current methodological approaches for studying these dormant cells, the significant challenges in eradicating them, and a comparative evaluation of the mechanisms. By integrating the latest research on (p)ppGpp signaling, toxin-antitoxin modules, and metabolic dormancy, this review aims to bridge mechanistic understanding with the development of novel therapeutic strategies to combat chronic and relapsing infections.
Defining the Dormant State: Core Principles of Stochastic and Triggered Persistence
Historical Context and Definition
Bacterial persisters are a subpopulation of genetically drug-susceptible, quiescent bacteria that survive exposure to high concentrations of antibiotics and other environmental stresses, only to resume growth once the stress is removed [1] [2]. The phenomenon was first identified in 1942 by Gladys Hobby, who observed that penicillin killed approximately 99% of bacteria, leaving 1% surviving [1]. In 1944, Joseph Bigger named these surviving cells "persisters" and suggested pulsed antibiotic therapy as a potential treatment strategy [1].
The critical distinction between persisters and other survival mechanisms lies in their non-heritable, phenotypic nature. The table below clarifies the key differences between bacterial persistence, antibiotic resistance, and tolerance.
Table 1: Key Characteristics of Bacterial Survival Strategies
| Feature | Persister Cells | Antibiotic-Resistant Cells | Tolerant Cells |
|---|---|---|---|
| Genetic Basis | Non-heritable, phenotypic variant | Heritable (mutations or acquired genes) | Can be non-heritable or genotypic |
| Minimum Inhibitory Concentration (MIC) | Unchanged | Increased | Unchanged |
| Mechanism | Dormancy (non-growing or slow-growing) | Prevents drug from binding to target | Population-wide slowing of death rate |
| Reversibility | Reversible upon antibiotic removal | Not reversible | Reversible |
| Population Heterogeneity | Small subpopulation | Entire population | Can be population-wide or a subpopulation |
Mechanisms of Persister Formation
The formation of persister cells is a complex process governed by multiple mechanisms, which can be broadly categorized into stochastic (random) and triggered (induced) pathways.
Stochastic Formation
Stochastic formation implies that persister formation occurs randomly within a bacterial population, even in the absence of external stress. This is thought to be due to natural fluctuations in the expression of key regulatory genes and proteins [3]. Models suggest that low ATP levels can induce protein aggregation, favoring the stochastic formation of non- or slow-growing persister cells [3].
Triggered Formation
Triggered formation occurs in response to specific environmental stresses. Key inducers include:
- Antibiotic exposure: The primary trigger studied in clinical contexts [1].
- Host immune factors: Reactive oxygen and nitrogen species (ROS/RNS) produced by macrophages during infection are major inducers of metabolic dormancy and antibiotic tolerance in intracellular pathogens like Staphylococcus aureus and Mycobacterium tuberculosis [4].
- Nutrient starvation and acidic pH: Common stressors encountered during infection [1].
- DNA damage: Can activate the SOS stress response, upregulating DNA repair functions and facilitating persister survival [2].
The following diagram illustrates the core signaling pathways and environmental triggers involved in persister cell formation.
Detection and Isolation Methodologies
Accurately detecting and isolating persister cells is crucial for research. The following workflow outlines a standard protocol for generating and quantifying persisters in vitro.
Table 2: Key Experimental Protocol for Persister Isolation and Time-Kill Assay
| Step | Protocol Detail | Purpose | Key Considerations |
|---|---|---|---|
| 1. Culture Preparation | Grow bacterial culture to mid-log or stationary phase. | To obtain a population with a baseline level of persisters. | Persister frequency is typically higher in stationary phase [2]. |
| 2. Antibiotic Challenge | Expose culture to a high concentration of a bactericidal antibiotic (e.g., 100x MIC) for a defined period (e.g., 3-5 hours). | To kill the majority of growing, susceptible cells. | Use concentrations significantly above MIC to ensure complete killing of non-persisters. |
| 3. Drug Removal | Wash cells via centrifugation and resuspension in fresh, antibiotic-free medium. | To remove the antibiotic and allow for resuscitation. | Incomplete removal can inhibit subsequent outgrowth. |
| 4. Viability Quantification | Serially dilute and spot-plate on antibiotic-free agar plates. Count Colony Forming Units (CFUs). | To quantify the number of surviving persister cells. | Survivors are defined as persisters if they retain genetic susceptibility upon regrowth [1]. |
For studying intracellular persisters, more complex models are required. A 2025 study used a high-throughput screen with a bioluminescent MRSA strain (JE2-lux) to probe the metabolic activity of bacteria inside macrophages, identifying host-directed compounds that could sensitize intracellular persisters to antibiotics [4].
Clinical Significance and Therapeutic Strategies
Persister cells are a major culprit in the recalcitrance of chronic and biofilm-associated infections. They are clinically significant because they underlie treatment failure, infection relapse, and may serve as a reservoir for the development of full-blown antibiotic resistance [1] [2].
Table 3: Anti-Persister Therapeutic Strategies and Compounds
| Therapeutic Strategy | Mechanism of Action | Example Compounds/Agents | Development Status |
|---|---|---|---|
| Direct Killing | Targets growth-independent structures like cell membranes or causes uncontrolled protein degradation. | XF-73, SA-558, Pyrazinamide (PZA), ADEP4 | Preclinical; PZA is in clinical use for TB [5]. |
| Preventing Formation | Inhibits pathways leading to dormancy, such as alarmone synthesis or quorum sensing. | CSE inhibitors, H2S scavengers, Quorum Sensing inhibitors (e.g., brominated furanones) | Mostly preclinical research [5]. |
| Resuscitation & Sensitization | Awakens persisters, making them susceptible to conventional antibiotics. | Host-directed adjuvant KL1, Membrane permeabilizers (e.g., SPR741) | KL1 identified in 2025 high-throughput screen [4] [5]. |
| Rational Drug Discovery | Uses tailored chemoinformatic models to find compounds with ideal properties for penetrating persisters. | Eravacycline, Minocycline derivatives | Proof-of-concept studies identifying new leads [6]. |
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Research Reagents for Studying Bacterial Persisters
| Reagent / Tool | Function in Research | Example Application |
|---|---|---|
| High-Persistence (hip) Mutant Strains | Models with genetically elevated persister formation for consistent study. | E. coli HM22 (hipA7 allele) used in screening anti-persister compounds [6]. |
| Bioluminescent Reporter Strains | Probes real-time metabolic activity of bacteria, especially inside host cells. | MRSA JE2-lux used to screen for metabolic potentiators in macrophages [4]. |
| Fluorescent Reporter Strains | Visualizes and isolates dormant subpopulations via microscopy or flow cytometry. | Inducible GFP reporters used to track S. aureus survival in mouse kidney cells [4]. |
| Membrane-Active Compounds | Increases permeability of persister cell envelopes to facilitate antibiotic uptake. | Polymyxin B nonapeptide (PMBN), synthetic retinoids used in combination therapies [5]. |
| XMD8-87 | XMD8-87, MF:C24H27N7O2, MW:445.5 g/mol | Chemical Reagent |
| LY 3000328 | LY 3000328, CAS:1373215-15-6, MF:C25H29FN4O5, MW:484.5 g/mol | Chemical Reagent |
Bacterial persisters, born from a delicate interplay between stochastic intracellular events and environmentally triggered responses, represent a profound challenge in managing chronic infections. Their ability to survive antibiotic assault without genetic resistance directly contributes to relapsing and biofilm-associated diseases. Understanding the dual nature of their formation is fundamental to devising effective countermeasures. While significant progress has been made in detecting persisters and identifying potential therapeutic targets, the translation of anti-persister strategies into clinical practice remains a critical frontier. Future research leveraging high-throughput screening, host-directed therapies, and rational drug design holds the promise of finally overcoming the resilience of these dormant cells.
Stochastic phenotype switching describes the ability of an isogenic cell population to generate multiple distinct phenotypes in the absence of genetic differences. This phenomenon represents a fundamental bet-hedging strategy that enhances population survival in unpredictably fluctuating environments [7] [8]. In essence, by randomly distributing different phenotypic states across a population, organisms ensure that at least a subpopulation will survive a sudden environmental stress, such as antibiotic exposure, that would eliminate a uniformly specialized population [9].
This review examines the mechanisms and implications of stochastic switching models, with particular focus on their role in generating non-genetic heterogeneity in bacterial persister cells and therapy-tolerant cancer cells. We frame this discussion within the ongoing scientific exploration of stochastic versus triggered persister cell formation mechanisms. While cells can certainly switch phenotypes in direct response to environmental cues (triggered switching), evidence increasingly supports the existence of intrinsically stochastic switching that occurs prior to environmental challenges, providing a survival advantage when changes are sudden and lethal [7] [8] [10]. This distinction has profound implications for understanding treatment failure in infectious diseases and cancer, and for developing more effective therapeutic strategies.
Theoretical Foundations: From Evolutionary Concepts to Mathematical Frameworks
Bet-Hedging as an Evolutionary Strategy
Bet-hedging represents an evolutionary optimal strategy for populations facing unpredictable environmental fluctuations. The concept derives from economic theory, where it describes diversifying investments to reduce risk, but in biological systems, it manifests as phenotypic heterogeneity within clonal populations [8] [9]. This heterogeneity ensures that even if the majority phenotype perishes under sudden stress, a minor subpopulation possessing a different phenotype might survive and eventually repopulate.
Theoretical models indicate that the effectiveness of bet-hedging depends critically on the frequency and predictability of environmental changes [8]. When environmental fluctuations occur rapidly, a generalist phenotype adapted to average conditions typically prevails. Conversely, when environmental changes happen on an intermediate timescale, a bet-hedging strategy with bimorphic phenotype distribution becomes optimal. For very slow environmental fluctuations, specialized monomorphic phenotypes adapted to each specific environment tend to dominate [8].
Mathematical Modeling of Stochastic Switching
The dynamics of stochastic phenotype switching can be captured mathematically through several modeling approaches:
Stochastic Differential Equations (SDEs) provide a powerful framework for describing systems evolving under inherent randomness. The canonical SDE form is:
[ dXt = f(Xt,t)dt + G(Xt,t)dWt ]
where (Xt) represents the state vector, (f(Xt,t)) defines the deterministic drift, (G(Xt,t)) is the diffusion coefficient matrix, and (Wt) is a Wiener process representing stochastic noise [11]. In biological contexts, SDEs can model how protein concentrations or other intracellular components fluctuate stochastically, potentially driving phenotype transitions.
Markov chain models offer another approach, treating phenotype switching as stochastic transitions between discrete states. These models are particularly useful for describing the random emergence of bacterial persisters or drug-tolerant cancer cells [12]. Recent advances have integrated non-genetic inheritance and cellular memory into these models, recognizing that daughter cells often inherit parental phenotypic states with probabilities that decay over time [12].
Table 1: Key Mathematical Frameworks for Stochastic Switching
| Model Type | Key Features | Biological Applications |
|---|---|---|
| Stochastic Differential Equations | Continuous-time, noise-driven dynamics | Protein concentration fluctuations, gene expression noise |
| Markov Chain Models | Discrete states with transition probabilities | Bacterial persistence, cancer cell phenotype switching |
| Structured Population Models | PDEs tracking population distributions | Age-structured phenotype inheritance, tumor heterogeneity |
| Adaptive Dynamics | Evolutionary optimization of switching rates | Bet-hedging strategy evolution in fluctuating environments |
Experimental Evidence Across Biological Systems
Bacterial Persistence: A Paradigm of Stochastic Phenotype Switching
Bacterial persisters represent a non-growing or slow-growing subpopulation that survives antibiotic treatment despite genetic susceptibility to these drugs [1]. These cells were first identified by Gladys Hobby in 1942 and named by Joseph Bigger in 1944, who observed that penicillin killed most staphylococci but left a small subset surviving [1].
Seminal work by Balaban et al. established that E. coli populations contain multiple persister types [9]. Type I persisters emerge during stationary phase and exhibit complete growth arrest, while Type II persisters grow slowly but steadily during normal growth conditions and maintain this slow growth rate under antibiotic stress [9]. This phenotypic heterogeneity occurs without genetic differences, strongly supporting a stochastic switching mechanism.
Recent research has identified energy management as a key factor in persister formation. Studies demonstrate that bacterial cells with low ATP levels show increased survival under antibiotic treatment [13]. Single-cell analysis using an ATP reporter (iATPSnFr1.0) revealed that a subpopulation with reduced ATP before antibiotic exposure exhibits enhanced tolerance, suggesting that stochastic fluctuations in energy-generating enzymes drive persistence formation [13].
Table 2: Bacterial Persister Types and Characteristics
| Persister Type | Formation Trigger | Growth State | Metabolic Characteristics | Molecular Mechanisms |
|---|---|---|---|---|
| Type I | Stationary phase transition | Non-growing | Metabolic dormancy | toxin-antitoxin modules (HipA), stringent response |
| Type II | Stochastic during growth | Slow-growing | Reduced ATP, impaired Krebs cycle | Stochastic fluctuations in energy-generating enzymes |
| Type III (VBNC) | Severe stress | Dormant | Very low metabolic activity | Protein degradation, RNA stability |
Stochastic Switching in Cancer: Drug-Tolerant Persisters
Cancer biology has revealed striking parallels to bacterial persistence in the form of drug-tolerant persisters (DTPs). These cells exhibit reversible resistance to targeted therapies and chemotherapeutic agents without possessing genetic resistance mutations [7].
In non-small cell lung cancer (NSCLC), treatment with EGFR inhibitors at concentrations 100-fold higher than the IC₅₀ value led to the isolation of DTPs that regained drug sensitivity when propagated in drug-free media [7]. Similarly, melanoma tumors resistant to BRAF inhibition upregulated EGFR in a reversible manner, with drug holidays restoring treatment sensitivity [7]. Single-cell transcriptomics has revealed that these transitions often involve shifts from drug-naïve "melanocytic" states to drug-resistant "mesenchymal-like" states driven by underlying signaling network dynamics rather than selection of pre-existing genetic mutants [7].
The cancer stem cell (CSC) model further illustrates stochastic phenotype switching, with dynamic interconversion between CSCs and non-CSCs maintaining phenotypic equilibrium within tumors [7]. This plasticity enables tumors to withstand therapeutic assaults and represents a significant clinical challenge for achieving durable remissions.
Methodologies: Experimental Protocols and Analytical Approaches
Isolation and Characterization of Bacterial Persisters
Protocol 1: Persister Isolation via Antibiotic Killing Curves
- Culture Preparation: Grow bacterial culture to mid-exponential phase (OD₆₀₀ ≈ 0.5) in appropriate liquid medium.
- Antibiotic Exposure: Add ciprofloxacin (or other bactericidal antibiotic) at 10-100× MIC concentration.
- Incubation and Sampling: Incubate culture at 37°C with shaking. Remove samples at predetermined time points (e.g., 0, 2, 4, 8, 24 hours).
- Viability Assessment: Serially dilute samples and plate on drug-free agar plates. Count colony-forming units (CFUs) after 24-48 hours incubation.
- Data Analysis: Plot log CFU/mL versus time to generate biphasic killing curves. The initial steep decline represents killing of normal cells, while the subsequent plateau indicates persister survival [1].
Protocol 2: Single-Cell ATP Analysis Using Microfluidics
- Strain Engineering: Chromosomally integrate ratiometric ATP reporter iATPSnFr1.0 under constitutive promoter.
- Microfluidics Setup: Load stationary-phase culture into mother machine microfluidic device.
- Media Flow: Flow fresh medium for 1 hour to revive cells, then switch to medium containing ampicillin.
- Time-Lapse Microscopy: Image cells every 15-30 minutes for 5+ hours using dual excitation (405 nm and 488 nm).
- ATP Quantification: Calculate 488â‚‘â‚“/405â‚‘â‚“ emission ratio for each cell. Correlate pre-treatment ATP levels with survival outcomes [13].
Monitoring Phenotype Switching in Cancer Cells
Protocol 3: Drug-Tolerant Persister Isolation in Cancer Models
- Cell Culture: Maintain NSCLC cell lines (e.g., PC9) expressing fluorescent viability reporters.
- Drug Treatment: Expose to high concentrations of EGFR inhibitors (e.g., erlotinib at 1-10 µM) for 72 hours.
- Persister Enrichment: Wash away dead cells and continue culture in drug-free medium for recovery.
- Flow Cytometry Analysis: Sort viable cells based on fluorescence markers. Confirm DTP phenotype by re-challenging with original drug.
- Single-Cell RNA Sequencing: Profile transcriptomes of parental cells, DTPs, and recovered populations to identify transitional states [7].
The Scientist's Toolkit: Essential Research Reagents and Methods
Table 3: Key Research Reagents and Methods for Studying Stochastic Switching
| Reagent/Method | Function/Application | Key Features | Example Uses |
|---|---|---|---|
| iATPSnFr1.0 Reporter | Ratiometric ATP sensing | Dual excitation (405/488 nm), self-normalizing | Single-cell ATP monitoring in microfluidics [13] |
| Mother Machine Microfluidics | Single-cell culture and imaging | High-temporal resolution, controlled environment | Long-term tracking of phenotype switching [13] |
| Fluorescence-Activated Cell Sorting (FACS) | Population separation based on protein levels | High-throughput, multi-parameter analysis | Isolation of dim/bright subpopulations [13] |
| Toxin-Antitoxin Mutants (hipA7) | Persistence gene manipulation | Gain-of-function mutations | Study of Type I persister mechanisms [9] |
| Krebs Cycle Enzyme Reporters (GltA, Icd, SucA) | Monitoring metabolic heterogeneity | mVenus translational fusions | Correlation of metabolic state with persistence [13] |
| MDI-2268 | MDI-2268|PAI-1 Inhibitor|Research Compound | MDI-2268 is a potent PAI-1 inhibitor with antithrombotic properties. This product is for Research Use Only and not for human or veterinary diagnosis or therapy. | Bench Chemicals |
| BET bromodomain inhibitor | BET Bromodomain Inhibitor for Epigenetic Research | Explore high-purity BET bromodomain inhibitors for cancer, inflammation, and epigenetic research. This product is For Research Use Only (RUO). Not for personal use. | Bench Chemicals |
Signaling Networks and Molecular Mechanisms
The molecular basis of stochastic phenotype switching involves complex regulatory networks that exhibit bistability and stochastic noise. In bacteria, toxin-antitoxin (TA) modules have been strongly implicated in persistence formation. The HipA toxin, part of the HipBA TA system, phosphorylates glutamyl-tRNA synthetase, inhibiting translation and inducing dormancy [1]. Stochastic fluctuations in HipA levels can trigger transition to the persistent state.
Metabolic regulation represents another key mechanism. Studies demonstrate that isocitrate dehydrogenase (Icd) mutants in E. coli produce more persisters and have lower ATP levels, directly linking Krebs cycle activity to persistence formation [13]. Cells with diminished expression of Krebs cycle enzymes show enriched survival under antibiotic challenge.
In cancer cells, phenotype switching between drug-sensitive and drug-tolerant states often involves chromatin remodeling and epigenetic regulation. For example, in NSCLC, chromatin modifications that alter gene expression patterns can create transiently resistant subpopulations without genetic mutations [7]. These epigenetic states can be stable through cell divisions but remain reversible.
Therapeutic Implications and Future Directions
Understanding stochastic switching mechanisms has profound implications for combating chronic infections and therapy-resistant cancers. Traditional antibiotic and anticancer therapies often fail because they primarily target rapidly dividing cells, leaving persister populations intact and enabling disease recurrence [7] [1].
Novel therapeutic approaches aim to target persister cells specifically or prevent their formation. For bacterial infections, this includes developing compounds that disrupt TA modules, metabolic pathways essential for persistence, or membrane integrity of dormant cells [1]. In cancer, strategies focus on combination therapies that simultaneously target both proliferating cells and drug-tolerant persisters, or epigenetic modulators that lock cells in drug-sensitive states [7] [12].
Evolutionarily-informed treatment schedules represent another promising approach. Mathematical modeling suggests that adaptive therapy—modulating drug pressure based on tumor burden—can suppress the expansion of drug-tolerant subpopulations by maintaining competitive suppression from drug-sensitive cells [12]. This approach has shown promise in both experimental cancer models and clinical trials.
Future research directions include:
- Single-cell multi-omics to comprehensively characterize the molecular features of persister cells across different biological systems.
- High-throughput screening for novel anti-persister compounds with specific activity against dormant cell populations.
- Clinical translation of evolutionary principles into treatment schedules that manage rather than eliminate persistent cell populations.
- Quantitative modeling integrating stochastic switching dynamics with pharmacokinetic/pharmacodynamic parameters to optimize treatment timing and dosing.
The continued investigation of stochastic switching models and their role in bet-hedging strategies remains essential for addressing some of the most challenging problems in modern medicine, particularly the crisis of therapeutic resistance in infectious diseases and cancer.
Bacterial persistence represents a significant challenge in clinical settings, underlying chronic infections, treatment failures, and relapsing diseases. Unlike antibiotic resistance, which involves genetic mutations that permit growth in the presence of drugs, persistence describes a phenotypically dormant state in which susceptible bacterial cells survive antibiotic exposure without genetic alteration [1] [5]. These dormant persister cells can resuscitate after antibiotic removal, leading to recurrent infections. The formation of persister cells occurs through two primary mechanisms: stochastic persistence, which arises spontaneously in a subset of cells through non-deterministic processes, and triggered persistence, induced by environmental stressors [14] [15]. This whitepaper examines the principal environmental triggers of bacterial persistence—antibiotic stress, nutrient starvation, and host immune factors—within the broader context of stochastic versus triggered formation mechanisms. Understanding these pathways is critical for developing novel therapeutic strategies against persistent infections.
Distinguishing Bacterial Persistence: Definitions and Key Concepts
Table 1: Key Definitions and Distinctions in Bacterial Survival Strategies
| Term | Definition | Key Characteristics | Reference |
|---|---|---|---|
| Antibiotic Persistence | A phenomenon where a susceptible bacterial subpopulation survives antibiotic exposure by entering a transient, dormant state. | Reversible, non-genetic, phenotypic heterogeneity within clonal population; biphasic killing curve. | [14] [1] |
| Antibiotic Resistance | The inherited ability of bacteria to grow at high antibiotic concentrations, typically measured by elevated MIC. | Genetic basis (mutations or gene acquisition); stable inheritance; allows growth under antibiotic pressure. | [16] |
| Antibiotic Tolerance | The ability of the entire bacterial population to survive antibiotic treatment for extended periods, often due to slow growth. | Population-wide survival; delayed killing; often induced by environmental conditions like starvation. | [1] [17] |
| Stochastic Persistence | Persister formation that occurs spontaneously at low frequency in homogeneous, unstressed cultures. | Probabilistic single-cell switching; pre-existing heterogeneity; not induced by external triggers. | [14] [15] |
| Triggered Persistence | Persister formation induced by environmental stressors such as antibiotics, nutrient limitation, or immune factors. | Response to external signals; often higher frequency than stochastic persistence; potentially preventable. | [14] [4] |
The distinction between persistence, resistance, and tolerance has important clinical implications. While resistance mechanisms are readily identifiable through standard microbiological assays like minimum inhibitory concentration (MIC) testing, persistence is difficult to measure and often missed in clinical diagnostics, potentially leading to unexplained treatment failures [16]. Persister cells exhibit metabolic heterogeneity, ranging from complete growth arrest to slow growth, and exist along a continuum from "shallow" to "deep" persistence states [1]. The relative contribution of stochastic versus triggered mechanisms to persister formation varies depending on bacterial species, growth conditions, and the specific environmental stressors encountered.
Molecular Mechanisms of Persister Formation
Core Signaling Pathways and Networks
The molecular machinery governing persister formation involves interconnected networks that sense environmental stress and modulate bacterial metabolism toward dormancy. Key systems include toxin-antitoxin (TA) modules, stringent response pathways, and secondary messenger systems.
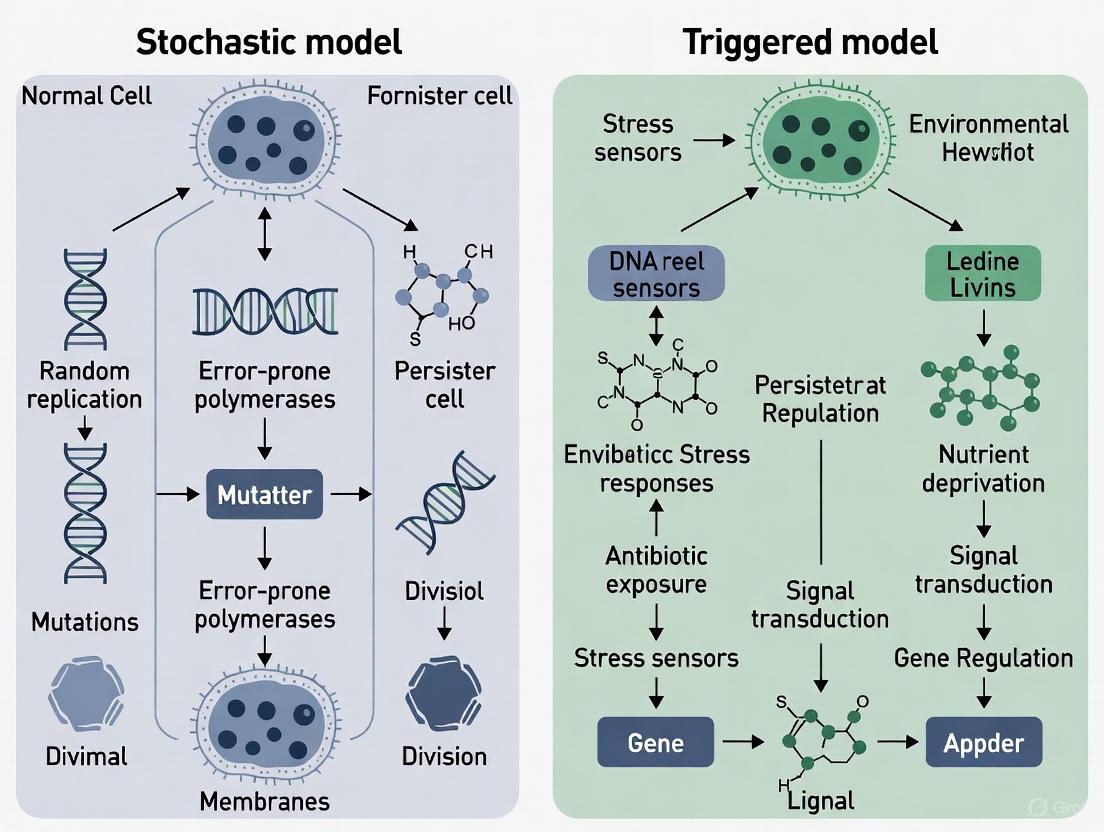
Figure 1: Molecular Pathways of Triggered Persister Formation. Environmental triggers activate cellular sensors that initiate signaling cascades leading to growth arrest and antibiotic tolerance. Key pathways include HipA-mediated stringent response, (p)ppGpp-polyphosphate signaling, and toxin-antitoxin system activation. PPX = exopolyphosphatase; ROS/RNS = reactive oxygen/nitrogen species.
The stringent response serves as a master regulator connecting various environmental stresses to persistence. Through the signaling nucleotides (p)ppGpp, bacteria can modulate cellular physiology in response to nutrient limitation and other stressors [18]. The HipA toxin exemplifies this integration: when activated by antibiotic stress, HipA phosphorylates glutamyl-tRNA synthetase (GltX), leading to accumulation of uncharged tRNAGlu that activates RelA and (p)ppGpp synthesis [18]. Elevated (p)ppGpp inhibits exopolyphosphatase (PPX), causing polyphosphate accumulation that activates Lon protease to degrade antitoxins, thereby freeing TA-encoded toxins such as mRNA-degrading nucleases that inhibit translation and induce dormancy [18].
Stochastic Switching Mechanisms
Stochastic persister formation occurs through probabilistic switching in individual cells without requiring external triggers. Single-cell studies of E. coli have revealed that in exponentially growing populations, most persisters against ampicillin and ciprofloxacin were actually growing before antibiotic treatment, displaying heterogeneous survival dynamics including continuous growth with L-form-like morphologies, responsive growth arrest, or post-exposure filamentation [15]. This demonstrates that growth arrest is not mandatory for persistence against all antibiotics. The stochastic variation in (p)ppGpp levels represents a key mechanism driving spontaneous persistence, as fluctuations in this global regulator can trigger TA system activation through the polyphosphate-Lon protease pathway [18].
Major Environmental Triggers of Persistence
Antibiotic Stress
Antibiotics themselves can induce persistence by activating stress response pathways. For example, sublethal antibiotic exposure triggers the SOS response and TA system activation through pathways involving (p)ppGpp [14] [18]. In Acinetobacter baumannii, exposure to imipenem or ciprofloxacin induces persistence through specific TA systems including AbkA/AbkB and HigBA [14]. Antibiotic-induced persistence demonstrates a paradoxical feedback loop wherein the therapeutic agent intended to eliminate bacteria instead promotes the formation of treatment-refractory subpopulations.
Nutrient Starvation
Nutrient limitation represents a potent trigger for persistence by activating the stringent response and shifting metabolism toward dormancy. However, the protective effect of nutrient deprivation exhibits important distinctions between phenotypic tolerance and genuine persistence. Stationary-phase cultures and nutrient-starved E. coli display complete survival when treated with antibiotics including ciprofloxacin, ampicillin, kanamycin, and mitomycin C—behavior characteristic of tolerance rather than persistence [17]. This protection is readily reversible; adding nutrients to stationary-phase cultures before ciprofloxacin treatment restores antibiotic killing to 0.001% survival [17]. In contrast, high-persistence mutants (e.g., hipA7 and metG2) maintain high survival (∼10%) even after nutrient restoration, indicating a genetically programmed persistent state distinct from phenotypic tolerance [17].
Table 2: Comparative Analysis of Nutrient Deprivation Effects on Bacterial Survival
| Condition | Survival After Antibiotic Treatment | Response to Nutrient Restoration | Classification | Reference |
|---|---|---|---|---|
| Exponential Phase (nutrient replete) | 0.0001% ( extensive killing) | Not applicable | Sensitive population | [17] |
| Stationary Phase (nutrient depleted) | 10-100% ( complete survival) | Extensive killing (0.001% survival) | Phenotypic tolerance | [17] |
| hipA7 mutant (stationary phase) | 10% ( high survival) | Maintains high survival (∼10%) | Genetic persistence | [17] |
| Starvation in saline | High survival | Killing upon nutrient addition | Phenotypic tolerance | [17] |
Host Immune Factors
The host intracellular environment represents a significant niche for antibiotic-tolerant persisters. Professional phagocytes like macrophages provide physical protection and induce metabolic dormancy through multiple mechanisms. Host-produced reactive oxygen and nitrogen species (ROS/RNS) have been strongly implicated in inducing antibiotic tolerance in pathogens including S. aureus, M. tuberculosis, Yersinia pseudotuberculosis, and Salmonella enterica Typhimurium [4]. Additional intracellular stressors such as phagosome acidification and nutrient deprivation further contribute to tolerance induction [4]. This host-induced tolerance has dominant effects; clinical S. aureus isolates showing 200-fold differences in persister frequency in vitro produced similarly high tolerance levels (26-51% survival) after macrophage internalization [4].
A novel screening approach identified KL1, a host-directed compound that increases intracellular bacterial metabolic activity and sensitizes persisters to antibiotics without causing cytotoxicity or bacterial outgrowth [4]. KL1 modulates host immune response genes and suppresses ROS production in macrophages, alleviating a key inducer of antibiotic tolerance [4]. This approach demonstrates that targeting host pathways controlling bacterial dormancy represents a promising strategy against persistent infections.
Experimental Approaches and Methodologies
Persister Detection and Quantification Methods
Table 3: Methodologies for Studying Persister Cells
| Method Category | Specific Techniques | Key Applications | Advantages/Limitations |
|---|---|---|---|
| Population Level Assays | Time-kill curves, Replica Plating Tolerance Isolation System (REPTIS), Tolerance Disk (TD) assay | Quantification of persister frequencies; antibiotic susceptibility profiling | Population averages; misses single-cell heterogeneity [16] [1] |
| Single-Cell Analysis | Microfluidics with membrane-covered microchamber arrays (MCMA), growth reporters, ScanLag | Tracking individual cell histories before/during/after antibiotic exposure; heterogeneity analysis | Reveals rare events; technically challenging; low-throughput [14] [15] |
| Genetic/ Molecular Screens | Mutant libraries, -omics techniques (transcriptomics, proteomics), mathematical modeling | Identifying genes and pathways involved in persister formation | Comprehensive mechanism identification; complex data interpretation [14] [1] |
| Metabolic Activity Probes | Lux-based bioluminescent reporters, ATP assays, ROS detection | Monitoring bacterial metabolic state and energy status | Functional readout of dormancy; may not correlate directly with viability [4] [17] |
Detailed Experimental Protocols
Protocol 1: Microfluidic Single-Cell Analysis of Persister Dynamics [15]
- Device Fabrication: Create a microfluidic device with a membrane-covered microchamber array (MCMA) featuring 0.8-µm deep microchambers etched on a glass coverslip.
- Cell Loading: Enclose E. coli cells in microchambers by covering with a cellulose semipermeable membrane via biotin-streptavidin bonding.
- Medium Control: Establish controlled medium flow above the membrane to flexibly manipulate environmental conditions around cells.
- Antibiotic Treatment: Introduce antibiotics at lethal concentrations (e.g., 200 µg/mL ampicillin [12.5×MIC] or 1 µg/mL ciprofloxacin [32×MIC]) through the medium flow.
- Time-Lapse Imaging: Monitor individual cell growth, division, and survival dynamics before, during, and after antibiotic exposure using phase-contrast and fluorescence microscopy.
- Data Analysis: Track cell lineages and classify survival behaviors based on pre-exposure growth status and post-exposure responses.
Protocol 2: High-Throughput Screening for Intracellular Persister Sensitizers [4]
- Reporter Strain Preparation: Utilize bioluminescent MRSA strain JE2-lux, where lux activity correlates with metabolic activity (ATP levels, NAD(P)H, FMNH2).
- Macrophage Infection: Infect bone marrow-derived macrophages (BMDMs) with JE2-lux and eliminate extracellular bacteria with gentamicin-containing media.
- Compound Screening: Dispense infected macrophages into 384-well plates containing test compounds from a drug-like library (>4,700 compounds).
- Dual-Parameter Detection: Measure bacterial bioluminescence (metabolic activity) and host viability after 4-hour incubation.
- Hit Validation: Identify compounds that increase bioluminescence >1.5-fold without cytotoxicity, then test their ability to enhance antibiotic killing of intracellular persisters.
- Mechanistic Studies: Employ transcriptomic analysis and ROS detection assays to elucidate compound mechanisms of action.
Protocol 3: Distinguishing Persistence from Phenotypic Tolerance via Nutrient Restoration [17]
- Culture Preparation: Grow wild-type and high-persistence mutant (e.g., hipA7) E. coli to stationary phase in rich medium.
- Antibiotic Challenge: Treat stationary-phase cultures with ciprofloxacin at 20×MIC for 3-5 hours.
- Nutrient Restoration: Dilute cultures 20-fold into fresh, pre-warmed medium containing the same antibiotic concentration.
- Viability Assessment: Sample cultures at intervals during antibiotic exposure, wash to remove antibiotics, and plate on drug-free agar to quantify surviving colony-forming units (CFUs).
- Data Interpretation: Classify based on survival patterns: persistent populations maintain high survival after nutrient addition, while phenotypically tolerant populations show extensive killing.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Persistence Research
| Reagent/Category | Specific Examples | Function/Application | Research Context |
|---|---|---|---|
| Bacterial Strains | E. coli MG1655 (wild-type), E. coli hipA7 (high-persister mutant), MRSA JE2-lux (bioreporter) | Model organisms for persistence mechanisms; lux reporter enables metabolic activity monitoring | Fundamental persistence studies [18] [4] [15] |
| Microfluidic Systems | Membrane-covered microchamber arrays (MCMA) | Single-cell analysis of persister dynamics under controlled environmental conditions | Tracking individual cell histories [15] |
| Metabolic Reporters | Lux-based bioluminescence, ATP assays, ROS-sensitive dyes (e.g., H2DCFDA) | Probing bacterial metabolic state and oxidative stress responses | Assessing dormancy depth and resuscitation [4] [17] |
| TA System Modulators | Inducible toxin expression vectors, antitoxin overexpression plasmids | Dissecting specific TA module contributions to persistence | Mechanistic studies of persistence pathways [14] [18] |
| Host Cell Systems | Bone marrow-derived macrophages (BMDMs), human primary neutrophils | Modeling intracellular persistence in host environments | Studying host-induced tolerance [4] |
| Stringent Response Agents | RelA mutants, (p)ppGpp analogs, PPX inhibitors | Manipulating stringent response pathways to probe persistence connections | Investigating stress signaling to dormancy [18] |
| Pkm2-IN-1 | Pkm2-IN-1, MF:C18H19NO2S2, MW:345.5 g/mol | Chemical Reagent | Bench Chemicals |
| Hdac8-IN-1 | Hdac8-IN-1, MF:C22H19NO3, MW:345.4 g/mol | Chemical Reagent | Bench Chemicals |
Therapeutic Implications and Future Directions
The distinction between stochastic and triggered persistence has profound implications for antimicrobial therapy development. While stochastic persistence might be addressed through combination therapies that increase the killing cascade effectiveness, triggered persistence offers potential intervention points to prevent persister formation by disrupting environmental sensing or response pathways.
Table 5: Anti-Persister Therapeutic Strategies
| Strategy | Mechanism of Action | Examples | Current Status |
|---|---|---|---|
| Membrane-Targeting Agents | Disrupt bacterial membranes independently of metabolic state | XF-73, SA-558, thymol triphenylphosphine conjugates | Preclinical development [5] |
| Metabolic Resuscitation | Reactivate dormant persisters to sensitize them to conventional antibiotics | KL1 (host-directed adjuvant) | Preclinical validation in animal models [4] |
| TA System Disruption | Interfere with toxin-antitoxin modules that maintain persistence | Peptide inhibitors of TA interaction | Early research stage [14] |
| Stringent Response Modulation | Inhibit (p)ppGpp synthesis or signaling | RelA inhibitors, alarmone analogs | Target identification and validation [18] |
| Combination Therapies | Target multiple vulnerabilities simultaneously | Aminoglycoside-polymyxin combinations against Gram-negative persisters | In vitro validation [17] |
| Host-Directed Therapies | Modulate host pathways that induce bacterial dormancy | ROS suppression agents, immune modulators | Early experimental stage [4] |
Current research highlights several promising approaches. The combination of aminoglycosides with polymyxins effectively kills E. coli persisters by synergistic membrane disruption, working through ROS-independent mechanisms that bypass the antioxidant defenses of dormant cells [17]. Host-directed adjuvants like KL1 represent another innovative approach, modulating the host environment to reduce ROS production and resuscitate intracellular bacteria for enhanced antibiotic killing [4]. Additionally, membrane-active compounds such as XF-73 and SA-558 directly target the structural integrity of persister cells, overcoming the limitations of conventional antibiotics that require metabolic activity [5].
Environmental triggers—antibiotic stress, nutrient starvation, and host immune factors—play pivotal roles in bacterial persistence by activating specific molecular pathways that induce dormancy and antibiotic tolerance. The interplay between stochastic and triggered persistence mechanisms underscores the complexity of this phenomenon and the challenges it poses for therapeutic intervention. While stochastic persistence may represent an inevitable bet-hedging strategy in bacterial populations, triggered persistence offers potential intervention points for preventing persister formation by modulating environmental signals or blocking their transduction. Future research should focus on elucidating the precise molecular connections between environmental sensing and persistence effectors, developing standardized methodologies for distinguishing persistence from tolerance in clinical isolates, and translating mechanistic insights into novel therapeutic strategies that target both stochastic and triggered persistence pathways. The integration of single-cell analysis, host-pathogen models, and innovative screening approaches will be essential for advancing our understanding and combating this clinically significant bacterial survival strategy.
This technical guide provides an in-depth examination of the key molecular players in bacterial persistence, focusing on the stringent response and toxin-antitoxin (TA) systems. Within the context of the ongoing debate between stochastic and triggered persistence mechanisms, we synthesize current research on how (p)ppGpp signaling and TA system activation contribute to bacterial survival under stress. The review integrates quantitative data on transcriptional regulation, detailed experimental methodologies for studying persistence, and visual representations of core signaling pathways. Additionally, we provide a comprehensive toolkit of research reagents to facilitate further investigation into these fundamental bacterial survival mechanisms, aiming to support therapeutic development against persistent bacterial infections.
Bacterial persisters represent a non-growing or slow-growing subpopulation of genetically susceptible cells that survive antibiotic treatment and other environmental stresses, contributing significantly to chronic and relapsing infections [1]. The formation of these persister cells is explained by two non-mutually exclusive conceptual frameworks: stochastic formation and triggered induction [13] [1]. The stochastic model posits that persisters arise randomly in bacterial populations through inherent noise in gene expression and biochemical processes, resulting in phenotypic heterogeneity even in uniform environments. In contrast, the triggered induction model suggests that specific environmental stresses actively signal and promote the transition to a persistent state through dedicated molecular pathways.
At the heart of this scientific discourse lie two key molecular systems: the stringent response with its central alarmone (p)ppGpp, and diverse toxin-antitoxin (TA) systems. These systems represent fascinating intersections of stochastic and triggered mechanisms, as they can be activated by specific environmental cues yet also exhibit stochastic activation patterns. This review comprehensively examines the molecular mechanisms, interactions, and experimental evidence surrounding these systems, providing researchers with the technical foundation needed to advance our understanding of bacterial persistence and develop novel therapeutic interventions.
The Stringent Response: (p)ppGpp as Master Regulator
Molecular Mechanisms and Signaling Pathways
The stringent response is a universal bacterial stress adaptation system mediated by the alarmones guanosine tetraphosphate (ppGpp) and guanosine pentaphosphate (pppGpp), collectively known as (p)ppGpp [19] [20]. These nucleotides function as master regulators that extensively rewire cellular physiology in response to nutrient deprivation and other stresses, modulating the expression of up to one-third of all bacterial genes [21] [20]. The synthesis and hydrolysis of (p)ppGpp are primarily governed by enzymes of the RelA/SpoT Homologue (RSH) superfamily [19].
In Escherichia coli and other Gammaproteobacteria, RelA is a ribosome-associated (p)ppGpp synthetase that becomes activated by direct binding to ribosomes harboring deacylated tRNA in the A-site – a clear signal of amino acid starvation [19]. This activation mechanism represents a classic triggered response pathway. Structural studies using cryo-EM have revealed that RelA adopts an open conformation on the ribosome that facilitates (p)ppGpp synthesis [19]. In contrast, SpoT is a bifunctional enzyme capable of both synthesizing and hydrolyzing (p)ppGpp, responding to a broader range of stresses including fatty acid limitation, iron restriction, and other environmental challenges [19] [20].
Table 1: Enzymes Involved in (p)ppGpp Metabolism and Their Regulation
| Enzyme | Primary Function | Activation Signals | Cellular Targets |
|---|---|---|---|
| RelA | (p)ppGpp synthesis | Deacylated tRNA in ribosomal A-site (amino acid starvation) | RNA polymerase, transcriptional machinery |
| SpoT | (p)ppGpp synthesis AND hydrolysis | Fatty acid limitation, iron restriction, multiple stresses | Multiple targets including RNA polymerase |
| Small Alarmone Synthetases (SAS) | (p)ppGpp synthesis | Various stresses in species-specific manners | Species-specific targets |
Once synthesized, (p)ppGpp exerts profound effects on cellular physiology through multiple mechanisms. Its primary target in Gammaproteobacteria is RNA polymerase, where it acts in concert with the cofactor DksA to dramatically alter transcriptional patterns [21]. This interaction promotes the expression of biosynthetic genes while repressing transcription of ribosomal RNA and transfer RNA genes, effectively redirecting cellular resources from growth to maintenance and survival [19] [20]. Additionally, (p)ppGpp directly binds to and modulates the activity of numerous metabolic enzymes involved in nucleotide synthesis, ribosome biogenesis, and lipid metabolism [19].
Figure 1: The Stringent Response Signaling Pathway. Environmental stresses activate RelA and SpoT enzymes, leading to (p)ppGpp accumulation that binds RNA polymerase with DksA to drive transcriptional reprogramming and cellular adaptation toward a persistent state.
Quantitative Aspects of (p)ppGpp Signaling
Recent research has revealed that the stringent response operates not as a simple binary switch but as a graded system where (p)ppGpp accumulation is proportional to stress severity. A 2025 study on Pseudomonas aeruginosa demonstrated that treatment with increasing concentrations of serine hydroxamate (SHX) – a serine analog that induces amino acid starvation – resulted in dose-dependent (p)ppGpp accumulation and corresponding growth inhibition [21]. The IC₅₀ for SHX-mediated growth inhibition was determined to be 128 ± 24 µM, with (p)ppGpp levels increasing 1.33-fold, 1.39-fold, and 1.48-fold under mild, intermediate, and acute stringent response conditions, respectively [21].
This graded (p)ppGpp response produces correspondingly graded transcriptional changes. Transcriptomic analysis revealed that mild stringent response conditions affected just 4% of the genome (227 differentially expressed genes), while intermediate and acute conditions affected 20% (1,197 genes) and 25% (1,508 genes) of the genome, respectively [21]. The regulatory pattern follows a "layer-by-layer" principle, where increasing stress severity engages progressively more cellular pathways in the transcriptional response.
Table 2: Graded Transcriptional Response to (p)ppGpp Levels in P. aeruginosa
| Stringent Response Level | SHX Concentration | (p)ppGpp Increase (fold) | Differentially Expressed Genes | Key Affected Pathways |
|---|---|---|---|---|
| Mild | 100 µM | 1.33× | 227 (4% of genome) | Initial metabolic adjustments, motility suppression |
| Intermediate | 500 µM | 1.39× | 1,197 (20% of genome) | Growth and metabolism reduction, biofilm promotion |
| Acute | 1000 µM | 1.48× | 1,508 (25% of genome) | Virgene repression, ribosome biogenesis downregulation, strong biofilm induction |
The functional consequences of this graded response include progressive impairment of motility, promotion of biofilm formation, and development of antimicrobial tolerance [21]. At higher (p)ppGpp levels, biofilm-related genes are upregulated at the expense of virulence factors, promoting the formation of condensed biofilms with enhanced tolerance properties [21].
Toxin-Antitoxin Systems: Multifaceted Genetic Elements
Classification and Molecular Mechanisms
Toxin-antitoxin (TA) systems are genetic modules composed of a stable toxin protein that inhibits bacterial growth and a labile antitoxin that neutralizes the toxin [22] [23]. These systems are classified into six types (I-VI) based on the nature of the antitoxin and its mechanism of action [22] [23].
Type I systems feature a protein toxin whose translation is inhibited by an antisense RNA antitoxin that binds complementarily to the toxin mRNA. The toxins are typically small, hydrophobic proteins that form pores in bacterial membranes, disrupting membrane potential and ATP synthesis [22] [23]. Well-characterized examples include hok/sok and tisB/istR, with the latter being induced during the SOS response to DNA damage [22].
Type II systems represent the most extensively studied class, comprising a protein toxin neutralized by a protein antitoxin that forms a stable complex with the toxin [22] [23]. The toxins employ diverse mechanisms including mRNA cleavage (MazF, RelE), DNA gyrase inhibition (CcdB), translation inhibition through distinct mechanisms (VapC, HipA), and cell wall synthesis disruption (PezT) [22] [23]. These systems typically autoregulate their own transcription through TA complex binding to promoter regions.
Type III systems consist of a protein toxin neutralized directly by an RNA antitoxin that binds and inhibits the toxin protein [22]. The toxIN system from Erwinia carotovora was the first identified example of this type.
Types IV-VI represent more recently discovered mechanisms. Type IV systems feature antitoxins that prevent toxin binding to cellular targets rather than directly binding the toxin itself. Type V systems contain antitoxins that are RNAases specifically cleaving toxin mRNAs. Type VI systems have antitoxins that promote degradation of the toxin protein [22].
Figure 2: Toxin-Antitoxin System Regulation. Environmental stress triggers antitoxin degradation and increased TA transcription. However, recent evidence suggests antitoxin in complex with toxin is protected from degradation, challenging the traditional view of stress-induced toxin activation.
Biological Functions and Controversies
TA systems were originally discovered as plasmid stabilization elements through "post-segregational killing" - a process where plasmid-free daughter cells are eliminated due to persistent toxin activity after antitoxin degradation [22] [23]. Chromosomal TA systems have been proposed to serve various functions including stress management, phage defense through abortive infection, and persister formation [22].
However, the functional roles of chromosomal TA systems remain controversial and actively debated. While early studies suggested that TA systems were key players in stress response and persister formation, recent research has challenged these paradigms [24]. A comprehensive 2020 study demonstrated that although diverse stresses (amino acid starvation, translation inhibition, oxidative stress, heat shock) induce substantial transcriptional activation of TA systems (often exceeding 10-50 fold increases in mRNA levels), this transcriptional induction does not necessarily lead to toxin liberation or activity [24].
The mechanistic insight underlying this discrepancy involves differential stability of antitoxin pools. Free antitoxin is rapidly degraded by proteases like Lon and ClpP during stress, but antitoxin in complex with toxin is protected from proteolysis, preventing significant toxin release [24]. This protection mechanism maintains toxin inhibition despite dramatic increases in TA transcription. Supporting this model, a strain of E. coli lacking 10 chromosomal TA systems (Δ10TA) showed no growth advantage following exposure to multiple stresses compared to wild-type strains [24].
Integration and Interplay in Persister Formation
Molecular Convergence on Persistence
The stringent response and TA systems represent complementary and potentially interconnected mechanisms in bacterial persistence. While early models proposed a hierarchical relationship with (p)ppGpp activating TA systems through stimulation of Lon protease-mediated antitoxin degradation [19], recent evidence challenges this linear pathway [24]. Instead, both systems appear to converge on similar cellular outcomes through parallel mechanisms.
Both systems implement a growth-to-survival transition by inhibiting energy-intensive processes. The stringent response achieves this through transcriptional reprogramming that downregulates ribosome biogenesis, DNA replication, and flagellar assembly while upregulating stress response and amino acid biosynthesis genes [21]. TA systems directly target central cellular processes including translation (MazF, RelE, VapC), DNA replication (CcdB), and cell wall synthesis (PezT) [22] [23].
A key point of integration involves metabolic regulation. The stringent response directly modulates metabolic enzymes and pathways, while recent research has demonstrated that stochastic fluctuations in energy-generating enzymes themselves can drive persister formation [13]. Cells with lower levels of Krebs cycle enzymes (isocitrate dehydrogenase, citrate synthase, α-ketoglutarate dehydrogenase) show enhanced survival against ciprofloxacin, and direct measurement of ATP levels using ratiometric sensors revealed that subpopulations with low ATP are enriched in persisters [13].
Resolving Stochastic versus Triggered Mechanisms
The interplay between the stringent response and TA systems provides a framework for reconciling stochastic and triggered persistence mechanisms. The graded nature of the (p)ppGpp response [21] allows for proportional cellular adaptation to stress severity, representing a triggered mechanism. However, heterogeneity in (p)ppGpp accumulation or response thresholds within isogenic populations could introduce stochastic elements.
For TA systems, the balance appears shifted toward stochasticity. While TA transcription is strongly induced by stress [24], the protection of TA complexes from degradation [24] means that stochastic fluctuations in TA expression or complex formation in subpopulations may be more significant for persistence than stress-induced activation. This model is consistent with the "low energy" hypothesis of persister formation, where stochastic heterogeneity in energy-generating components creates subpopulations with low ATP that are intrinsically tolerant to antibiotics [13].
Table 3: Comparative Analysis of Stringent Response and TA Systems in Persistence
| Feature | Stringent Response | Toxin-Antitoxin Systems |
|---|---|---|
| Primary signaling molecule | (p)ppGpp | Protein toxins (various types) |
| Activation mechanism | Triggered by nutrient limitation and other stresses | Both stochastic expression and stress-induced transcription |
| Major molecular targets | RNA polymerase, metabolic enzymes, replication initiation | mRNA (type II RNases), DNA gyrase, cell wall synthesis, translation |
| Temporal response | Rapid (minutes) | Varies by system |
| Evidence for persistence role | Strong and consistent across studies | Controversial, with recent challenges |
| Therapeutic targeting potential | High (central regulator) | Uncertain due to functional redundancy |
Experimental Approaches and Methodologies
Key Protocols for Investigating Persistence Mechanisms
Inducing and Measuring the Stringent Response:
The stringent response can be experimentally induced using serine hydroxamate (SHX), a serine analog that inhibits seryl-tRNA synthetase and creates amino acid starvation conditions [21]. Standard protocol involves adding SHX at concentrations ranging from 10-1000 µM to exponentially growing cultures, with higher concentrations producing more severe stringent response activation [21]. The (p)ppGpp levels can be quantified using thin-layer chromatography or high-performance liquid chromatography to separate and measure nucleotide pools [21]. For transcriptional analysis, RNA sequencing reveals the comprehensive transcriptomic changes associated with different levels of stringent response activation.
Single-Cell ATP Measurement:
To investigate the relationship between energy status and persistence, the iATPSnFr1.0 ratiometric ATP sensor can be employed [13]. This reporter incorporates a circularly permuted superfolder GFP and an ATP-binding subunit from Bacillus PS3 F0F1 ATP synthase. It is excited at two wavelengths (405 nm and 488 nm) with emission at 515 nm, and the ratio between signals (488ex/405ex) provides a self-normalized measure of ATP concentration that is independent of reporter expression levels [13]. This sensor can be expressed from the chromosome under a constitutive promoter for continuous monitoring.
Microfluidics and Time-Lapse Microscopy:
Mother machine microfluidic devices enable tracking of individual bacterial lineages under controlled conditions [13]. These devices contain growth trenches orthogonal to a media channel, with bacteria at the dead ends of trenches ("mother cells") serving as progenitors for linearly growing populations. This setup allows for continuous observation of growth, division, and survival of thousands of individual cells before, during, and after antibiotic exposure when combined with time-lapse microscopy [13].
TA System Transcriptional Analysis:
Quantitative reverse transcription PCR (qRT-PCR) can monitor TA system transcription under various stress conditions [24]. Stresses including amino acid starvation (SHX), translation inhibition (chloramphenicol), DNA synthesis inhibition (trimethoprim), oxidative stress (hydrogen peroxide), and heat shock effectively induce TA transcription. For population heterogeneity assessment, promoter-yfp fusions can determine whether transcriptional responses occur uniformly or in subpopulations [24].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Investigating Persistence Mechanisms
| Reagent/Tool | Function/Application | Key Features/Considerations |
|---|---|---|
| Serine Hydroxamate (SHX) | Induces amino acid starvation and stringent response | Dose-dependent effects (10-1000 µM); IC₅₀ ~128 µM in P. aeruginosa |
| iATPSnFr1.0 ATP sensor | Ratiometric measurement of ATP at single-cell level | Dual excitation (405/488 nm), emission at 515 nm; self-normalizing via ratio |
| Mother machine microfluidic devices | Single-cell tracking under controlled conditions | Enables observation of thousands of individual lineages; compatible with time-lapse microscopy |
| Δ10TA E. coli strain | Testing TA system functions in stress response | Lacks 10 chromosomal type II TA systems; must verify absence of ϕ80 prophage contaminants |
| QUEEN series ATP sensors | Alternative ATP measurement tools | Lower fluorescence in some bacterial systems; potential toxicity at high expression |
| Promoter-yfp fusions | Monitoring transcriptional dynamics in single cells | Enables assessment of population heterogeneity in stress responses |
| K-Ras(G12C) inhibitor 12 | K-Ras(G12C) inhibitor 12, MF:C15H17ClIN3O3, MW:449.67 g/mol | Chemical Reagent |
| ALK inhibitor 2 | ALK inhibitor 2, CAS:761438-38-4, MF:C23H28ClN7O3S, MW:518.0 g/mol | Chemical Reagent |
The intricate interplay between the stringent response and TA systems represents a complex regulatory network that influences bacterial persistence through both stochastic and triggered mechanisms. The graded nature of the (p)ppGpp response enables precise adaptation to stress severity, while the stochastic activation of TA systems and heterogeneity in energy metabolism contribute to phenotypic variation in isogenic populations.
Future research should focus on quantifying the relative contributions of these systems to persistence across different bacterial species and stress conditions. The development of improved dynamic reporters for both (p)ppGpp levels and TA system activity in single cells will be crucial for understanding the temporal dynamics of persistence formation. Additionally, exploring the potential crosstalk between these systems and other regulatory networks, such as those controlling metabolism and epigenetic modifications, may reveal novel layers of regulation.
From a therapeutic perspective, targeting the stringent response represents a promising approach for combating persistent infections, given its central role in coordinating stress adaptation. However, the conservation of (p)ppGpp signaling across bacteria and its connection to essential physiological processes present challenges for specific inhibition. TA systems offer more targeted opportunities but face challenges due to functional redundancy and questions about their precise roles in persistence. As our understanding of these key molecular players continues to evolve, so too will our ability to develop innovative strategies against persistent bacterial infections.
Drug tolerance, a reversible non-genetic capacity of bacterial cells to survive antibiotic treatment, represents a significant challenge in managing persistent infections. Unlike genetic resistance, tolerance is a phenotypic state characterized by reduced metabolic activity and growth arrest, allowing pathogens to withstand lethal antibiotic concentrations. This whitepaper examines the central role of ATP depletion and metabolic dormancy as fundamental hallmarks of the drug-tolerant persister phenotype. Within the context of ongoing scientific debate, we analyze evidence supporting both stochastic and triggered formation mechanisms of persister cells. Through synthesis of current research findings, experimental protocols, and emerging therapeutic strategies, this review provides a technical framework for researchers and drug development professionals seeking to overcome the limitations of conventional antibiotics against dormant bacterial populations.
Bacterial persisters represent a growth-arrested subpopulation of cells that exhibit extraordinary tolerance to antibiotics without undergoing genetic mutations [5] [25]. These phenotypic variants can survive antibiotic concentrations that kill the majority of their genetically identical counterparts and possess the capacity to resume growth once antibiotic pressure is removed, potentially leading to relapsing infections [26] [27]. This phenomenon differs fundamentally from antibiotic resistance, which is heritable and involves genetic changes that increase the minimum inhibitory concentration (MIC) [28]. In contrast, drug tolerance does not alter the MIC but extends the time required to kill a bacterial population, characterized by an increased minimum duration for killing (MDK99) [28].
The clinical importance of persister cells is profound, as they contribute significantly to recalcitrant infections that are difficult to eradicate. Persisters play established roles in chronic lung infections in cystic fibrosis patients, medical device-associated infections, and Lyme disease [5] [25]. Their presence in bacterial populations provides a reservoir from which antibiotic-resistant mutants may emerge over time, compounding the global antimicrobial resistance crisis [25]. Understanding the metabolic underpinnings of drug tolerance, particularly the roles of ATP depletion and growth arrest, is therefore critical for developing novel therapeutic interventions against persistent infections.
Core Metabolic Mechanisms of Drug Tolerance
Energy Depletion and ATP Reduction
A fundamental metabolic hallmark of persister cells is a significantly reduced ATP pool, which creates a state of metabolic quiescence incompatible with antibiotic-mediated killing. Direct measurements using ratiometric ATP sensors such as iATPSnFr1.0 have demonstrated that bacterial subpopulations with low intracellular ATP levels exhibit enhanced survival under antibiotic pressure [13]. This correlation was established through microfluidics time-lapse microscopy experiments tracking single-cell ATP levels and survival outcomes, providing robust evidence for the low-energy mechanism of persister formation.
The connection between impaired energy metabolism and persistence is further supported by genetic studies. Escherichia coli mutants lacking isocitrate dehydrogenase (icd), which catalyzes the rate-limiting step of the Krebs cycle, displayed lower ATP levels and increased persistence to ciprofloxacin compared to wild-type strains [13]. Similarly, mutants in sucB (α-ketoglutarate dehydrogenase) and ubiF (involved in ubiquinone biosynthesis) demonstrated deficient energy production and altered persister formation [26] [27]. These findings collectively indicate that disruptions in core energy-generating pathways promote a drug-tolerant state through ATP depletion.
Metabolic Slowdown and Growth Arrest
Antibiotics typically target active cellular processes such as cell wall synthesis, DNA replication, and protein translation, making rapidly dividing cells particularly vulnerable [28] [5]. Persister cells evade these mechanisms through coordinated metabolic slowdown, reducing or suspending the processes targeted by conventional antibiotics. This programmed dormancy represents a successful survival strategy against antimicrobial agents whose efficacy depends on metabolic activity [28].
This metabolic reprogramming involves reduced tricarboxylic acid (TCA) cycle activity and a shift toward lipid anabolism in Mycobacterium tuberculosis [28]. The subsequent thickening of the mycobacterial cell wall further reduces drug penetration, creating a physical barrier that complements metabolic dormancy. Similar observations of metabolic downshifting have been reported in Staphylococcus aureus persisters, which maintain active but reconfigured metabolic pathways, including glycolysis, TCA cycle, and pentose phosphate pathway, albeit at reduced levels [26].
Table 1: Key Metabolic Features of Drug-Tolerant Persister Cells
| Metabolic Feature | Functional Consequence | Experimental Evidence |
|---|---|---|
| Reduced ATP levels | Decreased energy availability for cellular processes | Single-cell ATP measurements using iATPSnFr1.0 reporter [13] |
| Impaired Krebs cycle activity | Limited ATP production through oxidative phosphorylation | icd mutants with increased persistence [13] |
| Metabolic slowdown | Reduced activity of antibiotic targets | Transcriptional downregulation of metabolic genes [26] [27] |
| Metabolic shifting | Alternative pathway utilization for homeostasis | Shift from TCA cycle to lipid anabolism in M. tuberculosis [28] |
| Proton motive force disruption | Collapse of membrane energetics | Thioridazine sensitivity studies [29] |
Regulatory Systems Controlling Metabolic Dormancy
The transition to a metabolically dormant state is orchestrated by sophisticated regulatory networks that sense environmental stress and implement survival programs. The stringent response, mediated by the alarmone ppGpp, serves as a master regulator of persistence under nutrient limitation [26] [27]. ppGpp accumulation triggers comprehensive transcriptional reprogramming that redirects cellular resources from growth to maintenance, promoting antibiotic tolerance across multiple bacterial species.
Toxin-antitoxin (TA) systems represent another key regulatory module in persister formation. These genetic elements encode a stable toxin that disrupts essential cellular processes and a labile antitoxin that neutralizes the toxin [28] [26]. Under stress conditions, activation of TA systems through antitoxin degradation or impaired synthesis leads to toxins such as HipA phosphorylating glutamyl-tRNA synthetase, which inhibits translation and mimics nutrient starvation, thereby inducing persistence [26] [13]. Additional toxins including TisB and HokB form membrane channels that dissipate the proton motive force, further reducing ATP levels and promoting dormancy [13].
In Mycobacterium tuberculosis, WhiB transcription factors and sigma factors function as critical stress regulators that coordinate the response to antibiotic pressure [28]. WhiB7 activation upon drug exposure upregulates drug efflux pumps and readjusts cellular processes to compensate for metabolic disruptions, while sigma factors SigB, SigE, and SigF modulate transcription to favor survival under stress.
Stochastic Versus Triggered Persister Formation Mechanisms
The origin of persister cells within isogenic bacterial populations remains a subject of intense investigation, with evidence supporting both stochastic and environmentally triggered formation mechanisms.
Stochastic Formation Model
The stochastic formation model posits that persisters arise spontaneously in growing populations due to random fluctuations in gene expression or protein abundance, without requiring external triggers. Support for this model comes from single-cell studies demonstrating that fluctuations in the expression of energy-generating enzymes precede persister formation [13]. When populations were sorted based on expression levels of Krebs cycle enzymes (GltA, Icd, SucA), subpopulations with low abundance of these proteins showed significantly higher survival rates under antibiotic treatment.
This model is further supported by the observed heterogeneity in ATP levels within bacterial populations, where cells with naturally low ATP before antibiotic exposure are more likely to survive treatment [13]. The stochastic variation in metabolic components creates a pre-existing reservoir of potential persister cells that become selected under antibiotic pressure, rather than being induced by the stress itself.
Triggered Formation Model
In contrast, the triggered formation model suggests that persisters form in direct response to environmental cues or stresses. Numerous studies have demonstrated that various stressors can induce persistence as an adaptive response. These inducing factors include:
- Nutrient limitation: Starvation for amino acids, carbon sources, or other nutrients dramatically increases persister frequency [26] [27]
- Antibiotic exposure: Sublethal antibiotic concentrations can activate stress responses that promote persistence to subsequent lethal doses [28]
- Other environmental stresses: pH change, oxidative stress, and immune system components can trigger persister formation [5]
These triggers often activate the stringent response and TA systems, initiating a programmed transition to dormancy [26]. The identification of specific signaling molecules that modulate persistence, including quorum sensing peptides and metabolic byproducts, provides additional support for regulated, inducible formation mechanisms [5] [25].
Integrated Perspective
The stochastic and triggered models are not mutually exclusive, and current evidence suggests both mechanisms likely operate simultaneously or sequentially in bacterial populations. Random fluctuations in metabolic proteins may establish a baseline persister level, while environmental stressors can amplify this subpopulation through induced dormancy programs. This integrated perspective acknowledges the complex interplay between intrinsic heterogeneity and responsive regulation in bacterial survival strategies.
Table 2: Comparative Analysis of Stochastic vs. Triggered Persister Formation
| Aspect | Stochastic Model | Triggered Model |
|---|---|---|
| Primary driver | Random fluctuations in gene expression and protein abundance | Environmental stressors and signaling molecules |
| Key evidence | Single-cell heterogeneity in Krebs cycle enzymes and ATP before antibiotic exposure [13] | Increased persister levels following nutrient limitation or sublethal antibiotic exposure [28] [27] |
| Timeframe | Pre-existing before antibiotic challenge | Induced during or before antibiotic challenge |
| Regulatory systems | Natural variation in metabolic components | Stringent response, TA systems, sigma factors [28] [26] |
| Therapeutic implications | Targeting variable subpopulations; preventing formation | Intercepting stress signaling; blocking induction |
Experimental Approaches and Methodologies
Single-CATP Level Monitoring
Cutting-edge approaches for monitoring persister metabolism employ ratiometric ATP biosensors such as iATPSnFr1.0, which enable real-time tracking of ATP dynamics in individual cells [13]. This reporter system utilizes a circularly permuted superfolder GFP coupled to an ATP-binding domain from Bacillus PS3 F0F1 ATP synthase, producing distinct fluorescence signals at two excitation wavelengths (405 nm and 488 nm) with emission at 515 nm.
Protocol for single-cell ATP monitoring:
- Clone iATPSnFr1.0 under a constitutive promoter into the chromosome of target bacteria
- Grow cultures to desired growth phase and load into microfluidics devices (e.g., mother machine)
- Acquire time-lapse images using dual-excitation microscopy (405 nm and 488 nm)
- Calculate 488ex/405ex ratio for individual cells to determine ATP concentrations
- Track cell survival and division events following antibiotic exposure
- Correlate pre-treatment ATP levels with survival outcomes
This methodology enabled the demonstration that E. coli cells with low ATP before ampicillin treatment had significantly higher survival rates, providing direct evidence for the energy depletion mechanism of persistence [13].
Metabolic Flux Analysis
Isotopolog profiling using 13C-labeled substrates represents another powerful technique for investigating persister metabolism [26]. This approach enables mapping of relative metabolic pathway activities by tracking the incorporation of labeled carbon atoms into metabolic intermediates.
Protocol for 13C-isotopolog profiling of persisters:
- Grow bacterial cultures to stationary phase to enrich for persisters
- Challenge with bactericidal antibiotics (e.g., daptomycin) at appropriate concentrations
- Feed 13C-labeled carbohydrates (e.g., glucose, pyruvate) to surviving persisters
- Extract intracellular metabolites at specific time points
- Analyze labeling patterns in amino acids and other metabolites via GC-MS or LC-MS
- Reconstruct relative metabolic flux through glycolysis, TCA cycle, and pentose phosphate pathway based on isotopolog distributions
Application of this technique to S. aureus persisters revealed active amino acid anabolism and continued operation of central metabolic pathways, albeit at altered levels compared to normal cells [26].
Genetic Approaches
Genetic screens have identified numerous metabolic genes that influence persister formation when disrupted [27]. These include:
- Transposon mutagenesis screens for persistence mutants
- Construction of chromosomal fluorescent protein fusions to monitor enzyme expression
- Targeted gene knockouts in metabolic pathways (e.g., icd, sucB, glpD)
- Reporter systems for stress response pathways (ppGpp, TA systems)
Visualization of Metabolic Pathways in Persister Formation
The following diagrams illustrate key signaling pathways and metabolic transitions involved in persister formation and drug tolerance.
Metabolic Regulation of Bacterial Persistence
Experimental Workflow for Persister Metabolism Analysis
Therapeutic Implications and Research Directions
Anti-Persister Therapeutic Strategies
The unique metabolic features of persister cells present opportunities for targeted therapeutic interventions aimed at eradicating these treatment-recalcitrant subpopulations.
Direct killing approaches focus on targets that remain vulnerable in dormant cells, particularly the bacterial membrane. Membrane-active agents such as XF-70, XF-73, and SA-558 disrupt membrane integrity independently of metabolic activity, effectively killing persisters through physical lysis [5] [25]. Similarly, pyrazinamide, a key anti-tuberculosis drug, targets membrane energetics in M. tuberculosis persisters by disrupting proton motive force and binding to PanD, thereby triggering its degradation [25].
Metabolic potentiation strategies seek to reverse or exploit persister dormancy to enhance conventional antibiotic efficacy. Thioridazine, a phenothiazine derivative, disrupts proton motive force and cellular energy metabolism, demonstrating strong synergy with ofloxacin particularly in pre-treatment and co-treatment conditions [29]. This approach effectively eradicates antibiotic-tolerant cells by targeting their energy metabolism, highlighting the potential of metabolic inhibitors as anti-persistence adjuvants.
Combination therapies represent another promising avenue, where membrane-permeabilizing compounds are paired with conventional antibiotics. MB6, CD437, and CD1530 disrupt MRSA membrane integrity, increasing uptake of gentamicin and resulting in potent anti-persister activity [25]. Similarly, H2S scavengers and cystathionine γ-lyase (CSE) inhibitors sensitize persisters to aminoglycosides by disrupting bacterial defense mechanisms against oxidative stress [25].
Research Reagent Solutions
Table 3: Essential Research Tools for Persister Metabolism Studies
| Reagent/Tool | Function/Application | Key Features |
|---|---|---|
| iATPSnFr1.0 ATP reporter | Single-cell ATP monitoring | Ratiometric measurement; 488ex/405ex excitation ratio correlates with ATP levels [13] |
| 13C-labeled substrates (glucose, pyruvate) | Metabolic flux analysis | Enables tracking of carbon fate through metabolic pathways [26] |
| Microfluidics devices (mother machine) | Single-cell culture and monitoring | Enables long-term tracking of individual cells under controlled conditions [13] |
| Chromosomal fluorescent protein fusions (mVenus) | Monitoring enzyme expression levels | Reports native expression patterns of metabolic enzymes [13] |
| Thioridazine and other phenothiazines | PMF disruption and metabolic inhibition | Anti-persister adjuvant that targets cellular energy metabolism [29] |
ATP depletion and growth arrest represent fundamental metabolic hallmarks of drug-tolerant persister cells across bacterial species. The integrated evidence from single-cell studies, genetic screens, and metabolic profiling supports a model wherein energy limitation creates a state of metabolic quiescence that protects bacteria from antibiotic-mediated killing. While stochastic fluctuations in metabolic components generate pre-existing low-energy subpopulations, environmental triggers can actively induce dormancy programs through conserved regulatory systems including the stringent response and toxin-antitoxin modules.
The ongoing delineation of stochastic versus triggered persister formation mechanisms continues to shape our fundamental understanding of bacterial survival strategies and informs therapeutic development. Future research directions should focus on elucidating the precise molecular connections between metabolic status and antibiotic tolerance, developing more sophisticated tools for monitoring persister physiology, and translating these insights into novel treatment paradigms that effectively target metabolically dormant populations. As our technical capabilities for studying these rare and transient phenotypic variants advance, so too will our capacity to overcome the clinical challenges posed by persistent infections.
Tools and Techniques: Profiling and Targeting Persister Cell Subpopulations
Bacterial persistence is a phenomenon in which a small subpopulation of genetically susceptible cells survives lethal doses of antibiotics, only to resume growth once the treatment ceases. These bacterial persisters are increasingly recognized as a critical factor in recurrent infections and treatment failure. The central debate in persistence research revolves around their origin: are they primarily stochastic persisters that arise spontaneously from random fluctuations in gene expression, or triggered persisters induced by external environmental signals? Single-cell technologies are pivotal for resolving this debate, as they can track the behavior of individual cells before, during, and after antibiotic challenge, revealing heterogeneity that bulk population studies inevitably mask [1] [30].
This technical guide details how the combined use of microfluidics and fluorescent reporters creates a powerful experimental framework for directly observing persister cell dynamics. By enabling long-term, high-resolution imaging of individual cells under controlled conditions, these technologies provide quantitative data on the growth, gene expression, and ultimate fate of persisters, offering unprecedented insights into the mechanisms underlying this phenotype [31] [15].
Core Methodologies: Integrating Microfluidics and Fluorescence
Microfluidic Platforms for Single-Cell Cultivation and Imaging
Microfluidic devices are engineered to create environments where individual bacterial cells can be trapped, cultured, and exposed to dynamically changing conditions while being imaged over time. Two primary designs are prominent in persister research.
The membrane-covered microchamber array (MCMA) operates by enclosing bacterial cells within shallow (e.g., 0.8 µm deep) microchambers etched on a glass coverslip. A semipermeable membrane covers the array, allowing for rapid medium exchange from the flow channel above while maintaining cells in a monolayer for clear microscopy. This setup is ideal for observing two-dimensional microcolony growth and tracking cell lineages [15].
An alternative multi-condition microfluidic platform significantly increases experimental throughput. One such design incorporates 32 pairs of microchambers fed by independent reservoirs. This allows for simultaneous testing of multiple compounds or conditions on a single device without cross-contamination, making it suitable for screening applications [32].
Table 1: Key Microfluidic Device Configurations for Persister Research
| Device Type | Key Features | Typical Applications | Throughput | References |
|---|---|---|---|---|
| Membrane-Covered Microchamber (MCMA) | Monolayer cell growth, rapid medium exchange, lineage tracking | Long-term imaging of persister formation and resuscitation | Up to thousands of cells per device | [15] |
| Multi-Condition Platform | Multiple independent microchambers, parallel compound testing | High-content screening of pheno-tuning compounds | 32+ conditions per device | [32] |
| Droplet-Based Microfluidics | Encapsulation of single cells in picoliter droplets | High-throughput single-cell sequencing, microbial interactions | 1,000 - 10,000 cells per run | [33] |
Fluorescent Reporter Systems for Monitoring Cellular States
Fluorescent reporters are the molecular tools that illuminate cellular processes, transforming physiological states into quantifiable signals.
- Transcriptional Reporters: Fusions of a promoter of interest to a gene encoding a fluorescent protein (e.g., GFP, mCherry) allow real-time monitoring of gene expression. In persistence research, the SOS response pathway is a frequent target. A
psulA::gfpreporter is widely used as a sensitive indicator of SOS induction, reporting on DNA damage after fluoroquinolone exposure [31]. - Protein Localization and Expression Reporters: A gene can be fused directly to a fluorescent protein gene to create a protein-FP fusion. This reveals the protein's abundance, localization, and dynamics. For example, an HU-GFP fusion can be used to visualize nucleoids and quantify DNA content within single cells [31].
- Physiological Biosensors: A newer class of tools includes biosensors for key metabolites. The QUEEN biosensor, for instance, is a modified ATP synthase subunit that changes fluorescence properties upon ATP binding, enabling direct measurement of single-cell energetics, a crucial parameter in dormancy [30].
Experimental Workflows and Key Insights
A standard integrated experiment for tracking persister dynamics typically follows a multi-stage workflow, as exemplified by several seminal studies.
A Standard Protocol for Tracking Persister Histories
The following protocol, adapted from Goormaghtigh and Van Melderen (2019) and Elowitz et al. (2024), outlines a core workflow for single-cell persister tracking [31] [15]:
- Strain Preparation: Engineer or select an E. coli strain (e.g., MG1655) harboring the desired fluorescent reporters (e.g.,
psulA::gfpfor SOS response and/or HU-GFP for nucleoid visualization). - Culture Growth & Device Inoculation:
- Grow a culture to stationary phase.
- Dilute into fresh medium and grow to mid-log phase (OD600 ~0.3) to ensure a population of actively dividing cells.
- Inoculate the cell suspension into the microfluidic device.
- Pre-Treatment Growth Phase (5-7 hours): Perfuse the device with fresh, antibiotic-free medium (e.g., MOPS-glucose). Acquire time-lapse images (e.g., every 15 minutes) to establish single-cell growth rates and baseline fluorescence before stress.
- Antibiotic Treatment Phase (5-7 hours): Switch the perfusion medium to one containing a lethal concentration of antibiotic (e.g., 5 µg/mL ofloxacin, ~60x MIC). Continue time-lapse imaging to monitor cell division arrest, filamentation, and induction of stress responses (e.g., SOS).
- Post-Treatment Recovery Phase (≥24 hours): Re-perfuse the device with antibiotic-free medium. Continue imaging to identify persister cells—those that resume division and give rise to a new population—and monitor their recovery dynamics (e.g., filament division).
- Data Analysis: Use automated or manual cell tracking software to analyze the collected images. For each cell, quantify parameters such as generation time, cell area, and fluorescence intensity over the entire time course.
Quantitative Findings on Persister Origins and Behavior
Single-cell studies have yielded critical quantitative data challenging traditional views, summarized in the table below.
Table 2: Key Single-Cell Findings on Persister Cell Dynamics
| Experimental Condition | Key Finding | Quantitative Result | Interpretation & Implication |
|---|---|---|---|
| E. coli + Ofloxacin (Exponential Phase) | Most persisters were actively growing before treatment. | Persister frequency: ~10â»Â² to 10â»â¶ within a clonal population. | Challenges the dogma that persisters originate solely from a pre-existing, non-growing dormant subpopulation (Type II persisters). Supports a stochastic element. |
| E. coli + Ampicillin (From Exponential Phase) | Growing persisters showed heterogeneous survival dynamics. | Dynamics included continuous growth as L-forms, responsive arrest, and post-exposure filamentation. | Reveals multiple pathways to survival even from active growth, highlighting profound phenotypic heterogeneity among persisters. |
| E. coli + Ciprofloxacin (From Post-Stationary Phase) | All identified persisters were growing before antibiotic treatment. | 100% of tracked persisters (n=all observed) were in a growing state pre-treatment. | Suggests that for some antibiotic classes, the growing cell fraction is the primary source of persisters, even after stationary phase. |
| E. coli + Ampicillin (From Stationary Phase) | Most persisters originated from non-growing cells. | Incubation under stationary phase conditions increased the frequency and survival probability of non-growing cells. | Supports the classical "Type I" persistence model where pre-existing dormant cells from stationary phase survive treatment. |
The following diagram illustrates the core experimental workflow and the divergent survival paths observed in persister cells.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of these experiments requires a suite of specialized reagents and tools.
Table 3: Research Reagent Solutions for Single-Cell Persister Studies
| Item | Function/Description | Example Use Case |
|---|---|---|
| Fluorescent Reporter Plasmids | Engineered genetic constructs for monitoring gene expression or protein localization. | psulA::gfp plasmid to report induction of the SOS response via GFP fluorescence [31]. |
| HU-GFP Fusion Strain | Chromosomally encoded fusion of the nucleoid-associated protein HU to GFP. | Visualizing nucleoid morphology and quantifying DNA content in individual cells during antibiotic stress [31]. |
| Microfluidic Device (MCMA) | A chip with microchambers for single-cell trapping and culturing. | Long-term imaging of E. coli cell lineages under controlled perfusion of antibiotics and nutrients [15]. |
| Time-Lapse Fluorescence Microscope | Microscope system with environmental control for automated, multi-position imaging. | Capturing phase-contrast and fluorescence images of microcolonies inside the device at set intervals (e.g., every 15 min) [31] [15]. |
| RpoS-mCherry Reporter | Fluorescent reporter for the general stress response sigma factor RpoS. | Investigating correlation between stress response activation and persister formation (note: functional defects of fusions must be verified) [15]. |
| Cellular ATP Biosensor (e.g., QUEEN) | A protein-based sensor that changes fluorescence upon ATP binding. | Quantifying single-cell metabolic activity and energy status to define dormancy depth [30]. |
| MELK-8a hydrochloride | MELK-8a hydrochloride, MF:C25H33ClN6O, MW:469.0 g/mol | Chemical Reagent |
| Melk-IN-1 | Melk-IN-1, MF:C31H33N5O4, MW:539.6 g/mol | Chemical Reagent |
Signaling Pathways in Persister Formation and Survival
The molecular mechanisms of persistence are complex, but single-cell studies have clarified the role of specific pathways. A key finding is that survival is not due to the simple absence of damage, but rather the execution of specific survival programs.
The SOS response, a conserved bacterial DNA repair pathway, is strongly implicated in persistence to fluoroquinolones like ofloxacin and ciprofloxacin. These antibiotics inhibit DNA gyrase and topoisomerase IV, causing DNA double-strand breaks. The resulting DNA damage leads to RecA activation, which facilitates the auto-cleavage of the LexA repressor. LexA cleavage derepresses the SOS regulon, including genes for DNA repair (recA, uvrA), cell division inhibition (sulA), and in some cases, the toxin-antitoxin module tisB/istR. The TisB toxin can depolarize the membrane and reduce ATP levels, potentially inducing a dormant state [31] [1].
Crucially, single-cell analysis reveals that both persister and non-persister cells endure DNA damage and induce the SOS response during antibiotic treatment [31]. The critical difference often manifests during recovery: persister cells show a prolonged SOS response, reaching a peak hours after antibiotic removal, and undergo extensive filamentation before resuming division. This suggests that the efficiency of DNA repair and recovery management, rather than the avoidance of damage, may be a defining feature of a persister cell.
The integration of microfluidics and fluorescent reporters has fundamentally shifted our understanding of bacterial persistence from a monolithic concept of "dormancy" to a complex spectrum of survival states with diverse origins. The evidence from single-cell studies strongly argues against a purely stochastic or purely triggered model. Instead, it demonstrates that both mechanisms operate, with their relative importance depending critically on the antibiotic class, bacterial growth phase, and environmental history [31] [15].
This refined understanding opens new avenues for therapeutic development. Future research will leverage these single-cell technologies to screen for pheno-tuning compounds (PTCs) that manipulate phenotypic variation to make bacterial populations more homogeneously susceptible to antibiotics [32]. Furthermore, the growing ability to study microbial interactions and resuscitation dynamics at the single-cell level within complex communities will provide a more ecologically relevant perspective on persistence during actual infections [3]. As these tools continue to evolve, they will undoubtedly uncover further layers of complexity, guiding the development of novel strategies to eradicate persistent infections.
Bacterial persisters, a subpopulation of cells capable of surviving antibiotic treatment without genetic resistance, pose a significant challenge in treating chronic and recurrent infections. A key mechanism underlying this tolerance is a metabolically dormant, low-energy state. This whitepaper provides an in-depth technical guide to genetically encoded reporter systems, such as iATPSnFr1.0, that are critical for quantifying ATP levels and metabolic activity in single bacterial cells. Framed within the broader debate on stochastic versus triggered persister formation, this document details experimental protocols, presents quantitative data for easy comparison, and visualizes core concepts and workflows. It is designed to equip researchers and drug development professionals with the tools to investigate and target the energetic basis of bacterial persistence.
Bacterial persistence is a phenomenon of phenotypic heterogeneity where a small, genetically susceptible subpopulation survives exposure to lethal antibiotics [1]. A paradigm shift in the field has established a low-energy state as a core mechanism of this tolerance. Persisters exhibit reduced ATP levels and diminished metabolic activity, which decreases the efficacy of bactericidal antibiotics that corrupt active cellular processes [13] [34] [35].
The formation of these low-energy persisters can be understood through two non-exclusive frameworks:
- Stochastic Formation: Random fluctuations in the expression of core metabolic components, such as tricarboxylic acid (TCA) cycle enzymes, naturally generate a subpopulation of cells with low ATP in a growing culture [13] [34].
- Triggered Formation: Specific stress responses, such as the SOS response or toxin-antitoxin (TA) system activation (e.g., TisB, HokB), can be induced by environmental cues, leading to a deliberate downregulation of energy generation [13] [1] [35].
Quantifying ATP and metabolic activity at the single-cell level is therefore essential for dissecting these mechanisms. Genetically encoded fluorescent reporters provide the necessary resolution to observe this heterogeneity and directly correlate a cell's energetic state with its outcome following antibiotic challenge.
Reporter Systems for ATP Quantification
Genetically encoded biosensors allow for real-time, non-destructive monitoring of intracellular ATP dynamics. The table below summarizes key ATP reporters used in microbial research.
Table 1: Genetically Encoded ATP Reporters
| Reporter Name | Basis of Operation | Spectral Properties | Key Features and Applications | Affinity/Notes |
|---|---|---|---|---|
| iATPSnFr1.0 [13] [36] | Circularly permuted superfolder GFP fused to the ATP-binding ε subunit of Bacillus PS3 F0F1 ATP synthase. | Ratiometric excitation: 405 nm (low ATP) vs 488 nm (high ATP). Emission: 515 nm. | Self-normalizing for expression level variation. Fast response time (<10 ms). Ideal for single-cell microscopy (e.g., microfluidics) and flow cytometry. | Validated in E. coli and Pseudomonas putida. Strong correlation with luciferase assay data [36]. |
| QUEEN [34] [37] | GFP fused to an ATP-binding subunit of Bacillus PS3 F0F1 ATP synthase. | Ratiometric excitation: 405 nm & 488 nm. Emission: 513 nm. Ratio reports ATP concentration. | Earlier generation sensor. Signal can be weak and temperature-sensitive. Requires codon optimization for use in some species like S. aureus [34]. | |
| Perceval [38] | Circularly permuted Venus fluorescent protein fused to the bacterial regulatory protein GlnK1 from Methanococcus jannaschii. | Ratiometric excitation: 405 nm (low ratio) vs 490 nm (high ratio). | Reports the ATP/ADP ratio, not absolute ATP concentration. Behavior is sensitive to the ratio due to competition between ATP and ADP for the same binding site. | Extremely high affinity for Mg-ATP (Kd ~0.04 μM). KR (half-maximal ratio) is ~0.2 [38]. |
| rrnB P1-GFP [37] | Synthetic promoter (rrnB P1) whose transcription initiation rate is sensitive to high ATP concentrations, fused to a fast-folding, fast-degrading GFP. | Monochromatic GFP signal. | Indirect reporter of ATP. Correlates well with bulk ATP levels across growth phases. Simple and effective for population-level studies and flow cytometry. | Functions as an "ATP integrator." Response is also influenced by ppGpp levels [37]. |
The following diagram illustrates the core operating principles of the rationetric ATP biosensors iATPSnFr1.0 and Perceval.
Quantitative Data on ATP and Persistence
Research using these biosensors has generated robust quantitative evidence linking low ATP to antibiotic tolerance.
Table 2: Key Experimental Findings Linking Low ATP to Persister Formation
| Experimental Approach | Organism | Key Quantitative Finding | Implication for Persistence |
|---|---|---|---|
| Microfluidics + iATPSnFr1.0 [13] | E. coli | A subpopulation of cells with low ATP survived ampicillin treatment, while cells with high ATP died. | Direct observational evidence that low ATP cells are persisters. |
| FACS Sorting of Low-Enzyme Cells [13] | E. coli | Cells with "Dim" levels of Krebs cycle enzymes (GltA, Icd, SucA) had significantly higher survival after ciprofloxacin treatment than "Bright" cells. | Stochastic fluctuation in energy-generating enzymes is a mechanism for persister formation. |
| TCA Cycle Mutants [34] | S. aureus | Mutants in TCA cycle genes (e.g., sucA, fumC) had lower ATP levels and persister frequencies of nearly 10% upon antibiotic challenge, vs. much lower in WT. | Targeted disruption of energy generation increases tolerance. |
| FACS Sorting of Low-TCA Reporters [34] | S. aureus | Cells with low expression of TCA promoters (PsucA, PfumC) showed a nearly 100-fold enrichment in persisters compared to cells with high expression. | Native stochastic variation in metabolic gene expression creates a low-energy, persistent subpopulation. |
| Carbon Source & ATP Dynamics [36] | E. coli, P. putida | A transient ATP surplus was observed during the transition from exponential to stationary phase. Carbon sources like acetate (in E. coli) elevated steady-state ATP. | ATP dynamics are growth-phase and nutrient-dependent, influencing bioproduction and potentially persistence. |
Detailed Experimental Protocols
Objective: To calibrate the iATPSnFr1.0 biosensor and measure ATP heterogeneity in a bacterial population at the single-cell level.
Materials:
- Bacterial strain expressing chromosomally integrated or plasmid-borne iATPSnFr1.0 under a constitutive promoter.
- Appropriate growth medium.
- Arsenate solution (100-400 mM): Used as a positive control for ATP depletion.
- Microfluidic device (e.g., mother machine) or flow cytometer.
- Fluorescence microscope with capabilities for ratiometric imaging or a flow cytometer with multiple laser lines.
- Luciferase-based ATP assay kit for bulk validation.
Method:
- Culture Growth: Grow the sensor strain to the desired growth phase (e.g., mid-exponential phase).
- Biosensor Calibration:
- Divide the culture into aliquots.
- Treat aliquots with increasing concentrations of sodium arsenate (e.g., 0, 1, 2, 4 mM) for a defined period (e.g., 30 minutes) to titrate ATP levels [13].
- For each aliquot, measure the fluorescence intensity at two excitation wavelengths (405 nm and 488 nm) with emission at ~515 nm.
- Calculate the ratiometric value (488ex/405ex) for each cell. A decrease in this ratio indicates ATP depletion.
- In parallel, measure the bulk ATP concentration of the same aliquots using a commercial luciferase assay to confirm the correlation between the sensor ratio and absolute ATP [13] [36].
- Single-Cell ATP Measurement:
- For time-lapse microscopy: Load the untreated sensor strain into a microfluidic device (e.g., mother machine). Flow fresh medium to allow growth and division, then switch to medium containing a lethal antibiotic (e.g., ampicillin). Image the trapped cells over several hours to track the ATP ratio (488ex/405ex) and cell survival (lysis or lack of elongation) simultaneously [13].
- For snapshot population analysis: Use flow cytometry to analyze the untreated sensor strain. Measure the 405ex and 488ex fluorescence for thousands of cells to generate a population distribution of the ATP ratio, revealing the natural heterogeneity in ATP levels [36].
Objective: To isolate and study persister cells by sorting a population based on the expression level of a metabolic gene reporter.
Materials:
- Bacterial strain with a chromosomal fusion of a TCA cycle promoter (e.g., sucA or fumC promoter) to a stable GFP.
- Appropriate antibiotics for challenge (e.g., ciprofloxacin).
- Fluorescence-Activated Cell Sorter (FACS).
Method:
- Culture and Challenge: Grow the reporter strain to late exponential phase. Challenge the culture with a lethal dose of antibiotic (e.g., 10x MIC of ciprofloxacin) for a period sufficient to kill the majority of the population (e.g., 3-5 hours) [34].
- FACS Analysis and Sorting: After antibiotic treatment, analyze the culture by FACS.
- Gate the population based on GFP intensity into "Dim," "Middle," and "Bright" subpopulations. The "Dim" population represents cells that had low expression of the metabolic gene at the time of antibiotic addition.
- Sort these gated populations directly onto nutrient agar plates.
- Viability Assessment: Count the colony-forming units (CFUs) that grow on the plates after incubation. The survival rate (CFUs after treatment / CFUs before treatment) is calculated for each sorted subpopulation.
- Expected Result: The "Dim" subpopulation will show a significantly higher survival rate (enrichment of persisters) compared to the "Bright" subpopulation, often by orders of magnitude [34].
The workflow for this FACS-based protocol is summarized below.
The Scientist's Toolkit: Essential Research Reagents
The following table lists critical reagents and tools for investigating the role of ATP in bacterial persistence.
Table 3: Essential Research Reagents and Materials
| Item | Function/Description | Example Use Case |
|---|---|---|
| iATPSnFr1.0 Plasmid/Strain [13] [36] | Genetically encoded, ratiometric ATP biosensor. | Real-time monitoring of ATP dynamics in single cells under antibiotic stress. |
| QUEEN Sensor (Codon-Optimized) [34] | Ratiometric ATP biosensor, requires optimization for non-model organisms. | Measuring ATP heterogeneity in pathogens like S. aureus [34]. |
| Microfluidic Devices (Mother Machine) [13] | Microfabricated device for long-term single-cell imaging and perturbation. | Tracking ATP levels and cell fate of individual lineages before, during, and after antibiotic exposure [13]. |
| Fluorescence-Activated Cell Sorter (FACS) [13] [34] | Instrument for analyzing and sorting cells based on fluorescence. | Isolating subpopulations with high or low metabolic activity (GFP reporters) or ATP levels for downstream persistence assays. |
| Luciferase ATP Assay Kit [36] [37] | Biochemical kit for quantifying ATP concentration in cell lysates. | Validating and calibrating the readings from genetically encoded ATP biosensors in bulk populations. |
| TCA Cycle Mutant Libraries [34] | Collection of defined gene knockouts in tricarboxylic acid cycle enzymes. | Studying the direct impact of impaired energy generation on persister frequency across antibiotic classes. |
| Sodium Arsenate [13] | Chemical that induces a futile cycle, depleting cellular ATP. | Used as a positive control to experimentally lower ATP and validate sensor response and its link to tolerance. |
| Methacycline Hydrochloride | Methacycline Hydrochloride, CAS:3963-95-9, MF:C22H23ClN2O8, MW:478.9 g/mol | Chemical Reagent |
| Methylstat | Methylstat, CAS:1310877-95-2, MF:C28H31N3O6, MW:505.6 g/mol | Chemical Reagent |
The development and application of genetically encoded reporters like iATPSnFr1.0 have transformed our ability to quantify the metabolic state of individual bacterial cells. The data generated by these tools provide compelling evidence that stochastic fluctuations in core metabolism represent a fundamental mechanism for the formation of low-energy persister cells. Furthermore, they allow researchers to dissect the contribution of triggered mechanisms, such as toxin-antitoxin systems, that converge on the same low-energy phenotype.
Moving forward, the integration of these reporters with advanced techniques like microfluidics and high-throughput screening will be crucial for:
- Systematically identifying genetic and environmental factors that influence metabolic heterogeneity.
- Screening for compounds that effectively kill low-energy persisters or disrupt their formation.
- Precisely mapping the complex signaling networks that bridge stochastic and triggered persistence pathways.
This detailed understanding of the "quantifiable persistence" mediated by ATP dynamics will open new avenues for developing therapeutic strategies aimed at eradicating chronic and recurrent bacterial infections.
Persister cells represent a transient, non-genetically heritable subpopulation of cells that exhibit remarkable tolerance to lethal stressors, such as antibiotics in bacteria or chemotherapeutic agents in cancer. Unlike resistant cells, which possess stable genetic mutations conferring survival, persisters evade treatment through reversible phenotypic adaptations, often entering a dormant or slow-cycling state. This phenomenon was first described in bacterial populations by Bigger and has since been identified as a critical clinical challenge in chronic infections and cancer relapse [39]. The core debate in persister biology centers on their formation mechanisms: whether they arise stochastically within a naive population, independent of environmental cues, or are triggered in response to external stressors, such as drug treatment. Elucidating the genetic basis of these formation pathways is paramount for developing strategies to eradicate these resilient cells. Modern functional genomics, particularly CRISPR-based genetic screens, coupled with multi-omics technologies, provide powerful, systematic tools to dissect these mechanisms, identify key persister genes, and reveal novel therapeutic vulnerabilities.
Core Mechanisms: Stochastic versus Triggered Persister Formation
The formation of persister cells is understood through two primary, non-mutually exclusive models: stochastic and triggered. The table below summarizes the key characteristics of, and supporting evidence for, these mechanisms.
Table 1: Comparative Analysis of Stochastic and Triggered Persister Formation Mechanisms
| Feature | Stochastic Formation | Triggered Formation |
|---|---|---|
| Definition | Spontaneous, pre-existing phenotypic variation in an isogenic population [13] | Induction of the persister state in response to external environmental stress [39] |
| Dependency on Stressor | Formation is independent of the drug/stressor; occurs prior to exposure [13] | Formation is directly dependent on the application of the stressor [39] |
| Proposed Molecular Basis | Fluctuations in core cellular processes like energy generation (e.g., low ATP levels) [13] | Activation of specific survival programs (e.g., epigenetic reprogramming, signaling pathways) [40] [39] |
| Key Supporting Evidence | Microfluidics shows subpopulation with low ATP survives ampicillin [13]; Sorted E. coli with low Krebs cycle enzyme levels have higher survival [13] | In EGFR-mutant NSCLC, osimertinib exposure engages Hippo/YAP-TEAD signaling [40]; Cancer DTPs exhibit therapy-induced epigenetic and metabolic shifts [39] |
| Heterogeneity & Plasticity | Population is pre-heterogeneous due to random fluctuations | Treatment pressure can induce multiple, distinct DTP phenotypic states (e.g., mesenchymal-like, luminal-like) [39] |
Methodological Toolkit: Genetic Screens and Omics Technologies
CRISPR-Cas Functional Genomic Screens
CRISPR-Cas technology has revolutionized the systematic identification of genes involved in persister formation. It enables targeted perturbation of genes across the genome, whose effects on persister frequency can be quantified.
Table 2: Key CRISPR-Cas Screening Modalities for Persister Gene Discovery
| Screening Modality | Mechanism of Action | Application in Persister Research |
|---|---|---|
| CRISPR Knockout (KO) | Utilizes Cas9 nuclease to create double-strand breaks, leading to frameshift mutations and gene knockout [41]. | Identifies genes whose loss increases (negative selection) or decreases (positive selection) persister frequency, revealing inhibitory and essential pathways [40] [41]. |
| CRISPR Activation (CRISPRa) | Uses nuclease-inactive dCas9 fused to transcriptional activation domains to overexpress target genes [41]. | Uncovers genes whose overexpression confers a survival advantage or induces the persister state. |
| CRISPR Interference (CRISPRi) | Uses dCas9 fused to transcriptional repressors to inhibit gene expression [41]. | Determines genes required for persistence; less toxic than KO, allowing for finer dissection of essential genes. |
| In Vivo CRISPR Screens | CRISPR-mutagenized cells are implanted into animal models (e.g., mice) [41]. | Identifies persister genes and dependencies within a clinically relevant tumor microenvironment. |
| Chemogenetic Screens | CRISPR perturbation is combined with drug treatment [41]. | Directly identifies genetic modifiers of drug tolerance and synthetic lethal interactions with therapies. |
| Mevociclib | Mevociclib (SY-1365)|Selective Covalent CDK7 Inhibitor | Mevociclib is a potent, selective covalent CDK7 inhibitor for cancer research (e.g., AML, breast). It shows anti-tumor activity. For Research Use Only. Not for human use. |
| MI 14 | MI 14, MF:C19H23ClN6O3S, MW:450.9 g/mol | Chemical Reagent |
Protocol: A Typical Pooled CRISPR-KO Screen for Persister Genes
- Library Design & Transduction: A pooled library of lentiviral vectors, each encoding a specific sgRNA and a unique barcode, is transduced into a target cell population (e.g., cancer cells, bacteria) at a low Multiplicity of Infection (MOI) to ensure most cells receive only one sgRNA.
- Selection & Population Expansion: Cells are selected with an antibiotic (e.g., puromycin) to eliminate untransduced cells, and expanded to create a representative "T0" population.
- Treatment & Persister Selection: The population is split. One arm is treated with a lethal dose of the drug of interest (e.g., osimertinib for cancer, ampicillin for bacteria), while the other is an untreated control. After a defined period, the surviving persister cells are recovered.
- DNA Extraction & NGS: Genomic DNA is extracted from both the pre-treatment (T0), untreated control, and persister populations. The sgRNA barcode regions are amplified via PCR and subjected to Next-Generation Sequencing (NGS) to quantify the abundance of each sgRNA.
- Computational Analysis: Bioinformatic tools (e.g., MAGeCK, BAGEL) compare sgRNA depletion or enrichment in the persister population versus the control to identify genes that significantly modulate persister formation [41].
Diagram 1: CRISPR Screen Workflow
Computational Analysis of CRISPR Screen Data
The raw NGS data from CRISPR screens requires sophisticated computational analysis to account for various biases and identify true hits.
- Key Algorithms: Commonly used tools include MAGeCK, which uses a negative binomial model to prioritize sgRNAs, genes, and pathways, and BAGEL, a Bayesian method for identifying essential genes [41].
- Bias Correction: A major challenge is correcting for technical biases, notably copy number (CN) bias (false essentiality calls in amplified genomic regions) and proximity bias (correlated fitness effects for proximal genes, potentially due to chromosomal truncations) [42]. Methods like CRISPRcleanR (unsupervised) and AC-Chronos (supervised, requires CN data) are benchmarked to effectively correct these biases and improve data quality [42].
Multi-Omics Integration
Omics approaches provide a multi-dimensional view of the molecular state of persister cells, complementing genetic screens.
- Genomics/Resistome Profiling: Whole-genome sequencing (WGS) of clinical persister isolates (e.g., Pseudomonas aeruginosa) reveals a broad collection of antibiotic-resistance genes, efflux pumps, and a distinct phylogenetic clade, linking persistent infections to MDR [43].
- Transcriptomics: Single-cell RNA-seq (scRNA-seq) reveals dynamic gene expression patterns and the coexistence of multiple transcriptional states (e.g., mesenchymal-like, luminal-like) within DTP populations, highlighting their heterogeneity and plasticity [39].
- Proteomics & Metabolomics: These technologies offer insights into the protein expression and metabolic pathways (e.g., Krebs cycle fluctuations, ATP levels) crucial for detoxification and survival. In bacteria, a drop in ATP is a key mechanism for stochastic persister formation [13] [44] [45].
Integrating data from these different omics platforms (genomics, transcriptomics, proteomics, metabolomics) holds immense potential for constructing a comprehensive map of the molecular basis of persistence, identifying regulatory mechanisms, and revealing novel therapeutic targets [44] [45] [46].
Key Experimental Findings and Pathways
Bacterial Systems: The Low-Energy Persister
In E. coli, stochastic heterogeneity in energy-generating processes is a key mechanism. Fluorescence-activated cell sorting (FACS) of cells with low levels of Krebs cycle enzymes (GltA, Icd, SucA) showed significant enrichment for persisters tolerant to ciprofloxacin, unlike cells with low levels of the glyoxylate shunt enzyme AceA [13]. Using a ratiometric ATP sensor (iATPSnFr1.0) in microfluidics time-lapse microscopy directly demonstrated that a subpopulation of cells with low ATP levels before treatment was better able to survive ampicillin killing [13]. This points to a general "low energy" mechanism for stochastic persister formation.
Table 3: Key Research Reagents for Bacterial Persister Studies
| Research Reagent / Tool | Function/Description | Application in Evidence Generation |
|---|---|---|
| Chromosomal mVenus Fusions (e.g., Icd-mVenus) | Translational fusions that report on the abundance of specific metabolic enzymes (e.g., Krebs cycle) in single cells. | Used in FACS to sort cells based on enzyme levels and demonstrate that "Dim" cells (low enzyme levels) have higher survival rates [13]. |
| iATPSnFr1.0 ATP Reporter | A genetically encoded, ratiometric fluorescent biosensor for measuring intracellular ATP levels at single-cell resolution. | Enabled real-time tracking of ATP in microfluidics, directly linking pre-existing low-ATP states to antibiotic survival [13]. |
| Mother Machine Microfluidics | A high-throughput microfluidic device for tracking thousands of individual bacterial lineages over time. | Allowed for the identification and observation of rare, low-ATP persister cells before, during, and after antibiotic exposure [13]. |
Cancer Systems: The Drug-Tolerant Persister (DTP)
In EGFR-mutant Non-Small Cell Lung Cancer (NSCLC), genome-wide CRISPR knockout and activation screens identified the Hippo signaling pathway and YAP/TAZ-TEAD transcriptional axis as a critical mediator of the DTP state triggered by EGFR inhibitors like osimertinib [40]. Persister cells survive initial treatment by engaging this non-genetic, transcriptional adaptation. Co-inhibition of EGFR and the Hippo/YAP-TEAD axis synergistically reduced cell viability in both cell lines and patient-derived organoids, proposing a promising combinatorial therapeutic strategy [40].
Cancer DTPs exhibit significant plasticity, engaging diverse adaptive programs. They share features with other resilient cell states like cancer stem cells (CSCs) and senescent cells but are uniquely defined by their induction by standard-of-care therapy [39]. For example, in colorectal cancer, chemotherapy can induce DTPs that resemble slow-cycling CSCs and undergo oncofetal reprogramming, a state maintained by YAP/AP-1 signaling [39].
Diagram 2: Signaling Pathways in Persistence
The Evolutionary Bridge from Tolerance to Resistance
Persister cells act as a reservoir for the evolution of stable genetic resistance. In P. aeruginosa, persister cells surviving meropenem treatment were serially passaged under antibiotic pressure. This experimental evolution led to the emergence of low-level resistant mutants with various mutations, followed by the sequential acquisition of mutations in the oprD porin gene and the mexR repressor (derepressing the MexAB-OprM efflux pump), resulting in high-level, multi-drug resistance [47]. This study highlights the dynamic interplay where non-genetic persistence facilitates the acquisition of genetic resistance.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Persister Gene Discovery
| Reagent / Material | Function in Persister Research |
|---|---|
| Genome-wide sgRNA Libraries | Pooled libraries of guide RNAs for targeting every gene in the genome in CRISPR knockout, activation, or interference screens [41]. |
| Patient-Derived Organoids (PDOs) | 3D ex vivo cultures derived from patient tumors that better recapitulate in vivo physiology and tumor heterogeneity, used for validating hits from cell line screens [40] [39]. |
| Single-Cell RNA-Seq Kits | Reagents for preparing next-generation sequencing libraries from individual cells, enabling deconvolution of transcriptional heterogeneity within persister populations [39]. |
| Ratiometric Fluorescent Biosensors (e.g., iATPSnFr) | Genetically encoded tools for quantifying dynamic changes in metabolites (e.g., ATP) or signaling activities in live cells at high resolution [13]. |
| Microfluidic Devices (e.g., Mother Machine) | Platforms for long-term, high-throughput imaging and tracking of single-cell behaviors under controlled environmental conditions, ideal for studying rare persister events [13]. |
| MI-192 | MI-192, MF:C24H21N3O2, MW:383.4 g/mol |
| MI-463 | MI-463, MF:C24H23F3N6S, MW:484.5 g/mol |
The integration of high-throughput genetic screens, such as CRISPR-Cas9, with multi-omics technologies is powerfully elucidating the complex pathways underlying persister cell formation. Evidence robustly supports the coexistence of both stochastic mechanisms, driven by fluctuations in core cellular processes like energy metabolism, and triggered mechanisms, induced by therapy-driven activation of specific survival signaling axes like YAP/TEAD. The plasticity and heterogeneity of persisters, revealed by single-cell omics, underscore that these cells can occupy multiple, reversible biological states. Furthermore, the role of persisters as a nidus for genetic resistance mutations highlights the clinical urgency of targeting them. Future research must leverage integrated computational and experimental approaches to bridge the gap between bench-side discoveries and bedside therapies, ultimately developing strategies to eradicate the persistent reservoir of cells that drive relapse in chronic infections and cancer.
Bacterial persisters are a transiently tolerant, dormant subpopulation capable of surviving high-dose antibiotic treatment without undergoing genetic mutation. Within structured biofilm communities, these cells are significantly enriched and are a major cause of recurrent and chronic infections. Understanding their formation and behavior is critical for developing effective antimicrobial strategies. Current research primarily investigates two formation mechanisms: stochastic formation, which occurs randomly in a portion of the population as a "bet-hedging" strategy, and triggered formation, which is induced in response to specific environmental stresses such as antibiotic exposure, nutrient starvation, or host immune factors [9] [48]. This technical guide provides researchers with established methodologies for quantifying, analyzing, and characterizing persister cells within these complex structured communities, with a focus on differentiating between these mechanistic pathways.
Quantitative Profiling of Persister Subpopulations
A critical first step in persister research is accurately quantifying the different subpopulations within a biofilm. Population analysis profiling reveals that biofilms contain a heterogeneous mix of cells with varying antibiotic tolerance levels, moving beyond the simple binary of normal and persister cells.
Population Analysis and Profiling
Research on Staphylococcus epidermidis biofilms has identified at least three distinct subpopulations. The table below summarizes their key characteristics.
Table 1: Bacterial Subpopulations within Biofilms
| Subpopulation | Definition | Antibiotic Response | Approximate Proportion in Biofilms |
|---|---|---|---|
| Normal Cells | The majority, antibiotic-susceptible population. | Killed by antibiotic concentrations at or near the planktonic Minimum Bactericidal Concentration (MBC). | >99.99% [49] |
| Tolerant-but-Killable (TBK) Cells | A persister subpopulation that survives high, but not maximal, antibiotic concentrations. | Killed by prolonged exposure or concentrations above 8x MIC, but below the biofilm MBC (MBCbiofilm). | ~0.01% of total cells [49] |
| Dormant Cells | A highly tolerant persister subpopulation in a deep dormant state. | Survive exposure to MBCbiofilm levels of antibiotics for extended periods (24-48 hours). | ~1 in 10^6 cells [49] |
The following workflow outlines the key steps for isolating and quantifying these subpopulations from a biofilm.
This methodology isolates single cells from the biofilm matrix, which is crucial for obtaining an accurate measure of persister prevalence without the confounding protective effects of the extracellular polymeric substance (EPS) [49].
Metabolic Activity Assessment
Metabolic activity is a key differentiator between persister subpopulations. A standard protocol for this uses differential staining to simultaneously identify all cells and those with active respiration.
Table 2: Key Reagents for Metabolic Staining of Biofilm Persisters
| Research Reagent | Function/Staining Target | Key Application in Persister Research |
|---|---|---|
| 5-Cyano-2,3-ditolyl Tetrazolium Chloride (CTC) | RedoxSensor dye; fluoresces red (CTF) when reduced by metabolically active cells. | Stains respiring cells. Persisters often show little to no CTC signal, indicating dormancy [50]. |
| SYTO 40 | Nucleic acid stain; fluoresces blue. | Stains all cells, providing a baseline count for the total population [50]. |
| Ciprofloxacin | Fluoroquinolone antibiotic. | Used at high concentrations (e.g., 150x MIC) to eliminate normal and TBK cells, enriching for and isolating the persister population for study [50]. |
| Flow Cell Reactor | Continuous culture system for biofilm growth. | Allows for standardized, in-situ development of biofilms on surfaces like coverslips under controlled shear stress and nutrient conditions [50]. |
The typical workflow involves growing a biofilm in a flow cell reactor for several days, treating it with a high concentration of ciprofloxacin to eliminate non-persisters, and then staining with CTC and SYTO 40. Analysis via confocal laser scanning microscopy (CLSM) follows, where the ratio of red (CTC-positive, active) to blue (total) cells reveals the fraction of metabolically active survivors [50]. Software like Intensity Luminance V1 or COMSTAT (as a plugin for ImageJ) can be used to quantify the fluorescence from the resulting image stacks [50].
Advanced Imaging and Image Analysis for Biofilm Persisters
Advanced imaging techniques are indispensable for characterizing the spatial distribution and morphology of persister cells within the 3D structure of a biofilm.
Scanning Electron Microscopy (SEM) and Machine Learning Segmentation
SEM provides high-resolution images that can show individual bacteria and the biofilm structure. To move from qualitative observation to quantitative data, machine learning-based image segmentation is required. The following workflow is used to quantify biofilm coverage from SEM images.
This method uses the Trainable Weka Segmentation plugin within the open-source Fiji/ImageJ platform. The researcher manually trains the algorithm by marking example regions of the biofilm and the background surface. The algorithm then learns to distinguish based on features like texture and edges, not just pixel intensity, making it effective for complex, rough surfaces like sandblasted, acid-etched (SLA) titanium. This automated process provides an objective and quantifiable measure of biofilm area before and after an intervention, with studies achieving a mean segmentation sensitivity (true positive rate) of 0.74–0.80 [51].
3D Image Cytometry with BiofilmQ
For a more comprehensive analysis of the biofilm's 3D internal architecture, BiofilmQ software is used. This tool acts as a "flow cytometer for biofilms," enabling the quantification of fluorescent reporters and structural features with spatial resolution [52]. If the image resolution is insufficient for single-cell segmentation, BiofilmQ can dissect the biofilm biovolume into a grid of cubes. For each cube, it calculates dozens of properties, including local biomass density, fluorescence intensity, and distance to the biofilm surface [52]. This allows researchers to correlate the location of persister cells (identified via fluorescent reporter systems) with microenvironments inside the biofilm, such as nutrient gradients, to investigate triggered formation mechanisms.
Experimental Protocols for Key Persister Assays
Protocol: Population Analysis of Biofilm Persister Cells
This protocol is adapted from methods used to characterize S. epidermidis subpopulations [49].
- Biofilm Cultivation: Grow biofilms in a suitable model system (e.g., flow cell reactor, Calgary Biofilm Device, or on coupons in a CDC biofilm reactor) for a defined period.
- Harvesting and Homogenization: Gently scrape or sonicate the biofilm to dislodge it from the surface. Resuspend the biomass in a neutral buffer such as phosphate-buffered saline (PBS).
- Single-Cell Isolation: Pass the resulting suspension through a 1.2 μm syringe filter to remove cell clumps and obtain a single-cell suspension. Validate the representativeness of the filtered population by comparing viable counts or flow cytometry profiles (e.g., using a GFP-expressing strain) before and after filtration.
- Antibiotic Challenge for TBK Cells:
- Expose the single-cell suspension to an antibiotic at a concentration of 8x the MIC for 3 hours.
- After exposure, pellet the cells by centrifugation, wash to remove the antibiotic, and serially dilute the suspension.
- Plate the dilutions on agar plates and incubate to determine the number of surviving colony-forming units (CFUs). These survivors represent the TBK persister subpopulation.
- Antibiotic Challenge for Dormant Cells:
- Take the remaining single-cell suspension and challenge it with the antibiotic at the predetermined MBCbiofilm concentration.
- Incubate for an extended period (24-48 hours).
- Pellet, wash, serially dilute, and plate the cells. The surviving CFUs represent the highly tolerant dormant persister subpopulation.
Protocol: Assessing Respiratory Activity in Biofilm Persisters
This protocol outlines the use of CTC staining to measure the metabolic activity of cells within a biofilm, particularly after antibiotic treatment [50].
- Biofilm Development: Grow biofilms in a flow cell system for a set duration (e.g., 6 days at room temperature).
- Antibiotic Treatment (for Persister Enrichment): Expose the mature biofilms to a high concentration of an antibiotic like ciprofloxacin (e.g., 150x MIC) or a control solution (saline) for 18 hours.
- Metabolic Staining:
- Prepare a staining solution in saline containing 5 μM SYTO 40 and 5 mM CTC.
- Perfuse this solution through the flow cell and incubate for 60 minutes in the dark.
- Confocal Microscopy:
- Acquire Z-stack images of the biofilm using a confocal laser scanning microscope with the appropriate settings:
- SYTO 40 (all cells): excitation 405 nm, emission 450-455 nm.
- CTC (active cells): excitation 476 nm, emission 650-680 nm.
- Acquire Z-stack images of the biofilm using a confocal laser scanning microscope with the appropriate settings:
- Image Quantification:
- Use image analysis software (e.g., Intensity Luminance V1, COMSTAT, or BiofilmQ) to quantify the fluorescence channels.
- The percentage of respiratory-active cells is calculated as:
(Biovolume of CTC signal / Biovolume of SYTO 40 signal) * 100.
The Scientist's Toolkit: Essential Reagents and Software
Table 3: Essential Tools for Studying Biofilm Persisters
| Category/Tool | Specific Example | Primary Function in Research |
|---|---|---|
| Biofilm Growth Models | Flow Cell Reactor [50], CDC Biofilm Reactor | Provide standardized, in-situ environments for growing mature, structured biofilms under controlled conditions. |
| Antibiotics for Selection | Ciprofloxacin [50], Vancomycin, Oxacillin [49] | Used at specific concentrations (8x MIC, MBCbiofilm) to selectively eliminate non-persister cells and isolate the tolerant subpopulations. |
| Viability Stains | SYTO 40 [50], SYTO 9/Propidium Iodide (Live/Dead) | Distinguish between total cells, live cells, and dead cells via fluorescence microscopy or flow cytometry. |
| Metabolic Activity Probes | CTC RedoxSensor [50] | Identify the fraction of cells within a population that are actively respiring, highlighting metabolically dormant persisters. |
| Image Analysis Software | BiofilmQ [52], COMSTAT [50], ImageJ/Fiji [51] | Quantify 3D biofilm architecture, biomass, thickness, and fluorescence signals from confocal or other micrographs. |
| Segmentation Software | Trainable WEKA Segmentation (in Fiji) [51] | Use machine learning to accurately segment and quantify biofilm coverage from complex SEM or other images. |
| Statistical Analysis | R, PRISM, Python | Perform significance testing (e.g., ANOVA with Tukey's comparison) to validate results from experimental replicates [50]. |
Bacterial persisters are a non-growing or slow-growing subpopulation of cells that exhibit transient, phenotypic tolerance to high doses of conventional antibiotics without acquiring genetic resistance [53] [5]. These dormant cells are a major culprit behind chronic and relapsing infections, including tuberculosis, recurrent urinary tract infections, and biofilm-associated infections on medical devices [53] [54]. Their recalcitrance stems from the fact that most conventional antibiotics target active cellular processes like cell wall synthesis, DNA replication, and protein synthesis—functions that are largely suspended in metabolically dormant persisters [5] [55]. The critical barrier to eradicating persistent infections is the lack of anti-persister therapeutics, a gap that originates from an incomplete understanding of the molecular pathways driving persister formation [53] [56].
A fundamental framework for understanding persistence centers on two primary formation mechanisms: stochastic versus triggered formation. Stochastic persister formation occurs spontaneously in a small subset of cells within an isogenic population due to random, non-genetic fluctuations in key molecular circuits [53] [13]. In contrast, triggered persistence is a bet-hedging strategy induced in response to external environmental stresses, such as antibiotic exposure, nutrient starvation, or host immune factors, which mimic conditions within an infection site [53] [56]. This dichotomy is not merely academic; it dictates divergent strategies for drug discovery. Screens targeting stochastic persistence must identify compounds that exploit inherent vulnerabilities like low energy states, while strategies against triggered persistence can focus on disrupting specific stress-response pathways activated by the host environment [56] [54]. This guide details how to leverage these distinct molecular insights into the design of robust discovery campaigns for the next generation of anti-persister therapeutics.
Molecular Mechanisms: Informing Target Selection
The Stochastic "Low-Energy" Model
Emerging research posits that stochastic fluctuations in central energy metabolism are a primary driver of persister formation. A landmark study demonstrated that a subpopulation of E. coli cells with low ATP levels is enriched for survival upon antibiotic challenge [13]. This heterogeneity arises from random variations in the expression of energy-generating enzymes.
- Key Evidence: An E. coli mutant for isocitrate dehydrogenase (
icd), which catalyzes a rate-limiting step in the Krebs cycle, exhibited both lower intracellular ATP and a higher persister frequency against ciprofloxacin [13]. - Single-Cell Analysis: Using a ratiometric ATP sensor (
iATPSnFr1.0) in a microfluidics time-lapse microscopy setup, researchers directly observed that individual cells with low ATP before antibiotic treatment were the ones that survived ampicillin exposure [13]. - Implication: The dormant, low-energy state itself is the primary tolerance mechanism, making cellular energetics a compelling target area.
Triggered Persistence via Stress-Response Pathways
In response to host and therapeutic pressures, bacteria activate defined genetic programs that induce a protective dormant state. Key pathways include:
- Toxin-Antitoxin (TA) Systems: Under stress, labile antitoxins are degraded, freeing toxins to inhibit essential processes. For example, the HipA toxin phosphorylates glutamyl-tRNA synthetase, inhibiting translation, while the TisB toxin forms membrane pores that reduce proton motive force and ATP [13].
- Stringent Response: Nutrient starvation triggers the RelA enzyme to synthesize the alarmone (p)ppGpp, which dramatically reprograms cellular metabolism away from growth and toward maintenance [53] [56].
- Transcriptional and Epigenetic Reprogramming: Stressors can induce regulators like RpoS or lead to epigenetic modifications that silence metabolic genes, locking the cell into a non-growing state [53].
The following table summarizes the core characteristics of these two formation mechanisms.
Table 1: Core Mechanisms of Persister Cell Formation
| Feature | Stochastic Persistence | Triggered Persistence |
|---|---|---|
| Induction | Spontaneous, random fluctuations | Directed response to external stress (e.g., antibiotics, starvation, acidic pH) |
| Predictability | Non-deterministic, heterogeneous within a population | More deterministic, can be synchronized in vitro |
| Key Molecular Drivers | Fluctuations in Krebs cycle enzymes & ATP levels [13] | TA system activation (e.g., HipBA, TisB/IstR) [13], Stringent Response [53] |
| Primary Model | "Low-Energy" hypothesis | "Active Stress Response" hypothesis |
| In Vitro Modeling | Tracking single-cell ATP heterogeneity in microfluidics [13] | In vitro stress models (e.g., Wayne hypoxia, nutrient starvation) [56] |
Diagram 1: Stochastic vs. Triggered Persister Formation Pathways
From Mechanisms to Screening Campaigns
The choice of screening strategy is paramount and must be guided by the targeted persister formation mechanism. Campaigns can be designed to either eradicate existing persisters or prevent their formation.
Screening for Eradication of Stochastic Persisters
The "low-energy" model suggests that stochastic persisters, while dormant, possess unique physiological vulnerabilities.
- Direct Killing Strategies: These approaches target growth-independent cellular structures.
- Membrane-Targeting Agents: Compounds like XF-73 and synthetic cation transporters (e.g., SA-558) disrupt membrane integrity, causing cell lysis independent of metabolic activity [5].
- Proteolytic Activation: ADEP4 activates the ClpP protease, forcing it to degrade proteins indiscriminately without the need for ATP, which effectively "digests" the cell from within [5].
- Screening Protocol: ATP-Level-Based Persister Enrichment
- Culture Preparation: Grow the target bacterium (e.g., E. coli or S. aureus) to mid-exponential phase.
- Persister Enrichment: Treat the culture with a high concentration of a bactericidal antibiotic (e.g., 10x MIC of ciprofloxacin or ampicillin) for 3-4 hours to kill all actively growing cells.
- Washing: Pellet the cells and wash twice with fresh, pre-warmed medium to remove the antibiotic.
- Resuspension: Resuspend the pellet in fresh medium. The surviving cells are highly enriched for persisters.
- Compound Screening: Dispense the persister-enriched suspension into a 96-well or 384-well plate containing the compound library. Incubate for 16-24 hours.
- Viability Readout: Use ATP-based luminescence (e.g., BacTiter-Glo) or resazurin reduction assays to quantify surviving cells. A significant reduction in signal compared to a DMSO control indicates anti-persister activity [13] [54].
Screening for Prevention of Triggered Persisters
This strategy aims to interfere with the specific molecular pathways that induce the persistent state, sensitizing the population to conventional antibiotics.
- Inhibition of Persister Formation:
- Quorum Sensing Inhibitors: Compounds with a benzamide-benzimidazole backbone can inhibit the MvfR regulon in P. aeruginosa, reducing persister formation without affecting growth [5].
- H2S Biogenesis Inhibitors: Inhibiting bacterial cystathionine γ-lyase (bCSE) reduces H2S-mediated protection, decreasing biofilm formation and persister numbers [5].
- Screening Protocol: Co-administration with Triggering Stressor
- Culture Preparation: Grow the target bacterium to mid-exponential phase.
- Co-treatment: In a microtiter plate, simultaneously expose the bacterial culture to: a) the environmental stressor that triggers persistence (e.g., sub-lethal antibiotic, low pH medium, nutrient limitation), and b) the compound from the library.
- Incubation: Incubate the plate for a defined period (e.g., 5-6 hours) to allow persister formation in untreated controls.
- Lethal Challenge: Add a high dose of a bactericidal antibiotic to all wells to kill all non-persister cells.
- Washing and Outgrowth: Wash the cells to remove antibiotics and resuspend in fresh medium. Monitor regrowth over 24-48 hours.
- Hit Identification: A hit compound is one where no regrowth occurs, indicating that it successfully prevented cells from entering the tolerant persister state, leaving them susceptible to the lethal challenge [54].
Table 2: Quantitative In Vitro Stress Models for Studying Triggered Persistence
| Stress Model | Inducing Signal | Key Molecular Readouts | Pathogen Example |
|---|---|---|---|
| Nutrient Starvation | Incubation in phosphate-buffered saline [56] | Upregulation of 279 genes; downregulation of energy metabolism & lipid biosynthesis genes [56] | M. tuberculosis [56] |
| Hypoxic Wayne Model | Gradual oxygen depletion [56] | Induction of ~50 genes in Non-Replicating Persistence (NRP) state; high expression of Rv0251c, Rv1874 [56] | M. tuberculosis [56] |
| Drug Persister Model | Antibiotic stress (e.g., D-cycloserine) [56] | Transcriptome analysis reveals specific tolerance pathways [56] | M. tuberculosis [56] |
| Reactive Oxygen/Nitrogen Species | Exposure to H2O2 or NO donors [5] | Induction of antioxidant enzymes; activation of TA systems like TisB/IstR [13] | E. coli, S. aureus |
The Scientist's Toolkit: Key Reagents and Models
Successful anti-persister drug discovery relies on specialized reagents and in vitro models that accurately recapitulate the persister phenotype.
Table 3: Essential Research Reagent Solutions for Persister Studies
| Reagent / Model | Function / Purpose | Key Example(s) |
|---|---|---|
| iATPSnFr1.0 ATP Sensor | Ratiometric, single-cell measurement of ATP dynamics in live cells [13] | E. coli chromosome-integrated sensor for microfluidics [13] |
| Microfluidics Devices (Mother Machine) | Long-term, high-throughput tracking of single-cell lineages under controlled conditions [13] | Loading stationary phase cells for ampicillin tolerance studies [13] |
| In Vitro Stress Models | Mimic in vivo host conditions (e.g., granuloma) to induce triggered persistence in a reproducible manner [56] | Wayne Hypoxia Model, Nutrient Starvation Model for M. tuberculosis [56] |
| Membrane-Integrity Probes | Differentiate between live and dead/lysed cells in viability counts post-treatment [5] | Propidium Iodide (PI) staining combined with SYTO 9 in live/dead assays |
| Constitutive Lysis Strain | Control for compound efficacy against non-growing cells without membrane damage [54] | E. coli ML-35 (constitutively expressing β-galactosidase) |
| ClpP Activators | Positive control for non-membrane targeted killing of persisters via protein degradation [5] | ADEP4 [5] |
Diagram 2: Anti-Persister Screening Workflow Decision Tree
The divide between stochastic and triggered persister formation mechanisms is a fundamental consideration that must guide anti-persister drug discovery. As outlined, the choice of screening strategy—whether aiming to eradicate pre-existing, low-energy persisters or prevent the induction of tolerance via stress-response pathways—is directly informed by the underlying biology [53] [13] [56]. The most effective therapeutic regimens will likely employ a dual-pronged approach: combining novel anti-persister compounds that target dormant cells with conventional antibiotics that eliminate the actively growing population [5] [54]. By leveraging the sophisticated in vitro models, single-cell technologies, and mechanistic frameworks detailed in this guide, researchers can systematically translate molecular insights into the next generation of therapies capable of overcoming one of the most challenging obstacles in modern medicine—chronic and relapsing bacterial infections.
Overcoming Therapeutic Hurdles: Challenges in Eradicating and Preventing Persistence
Bacterial persisters are a subpopulation of genetically drug-susceptible cells that enter a transient, non-growing or slow-growing state, enabling them to survive high-dose antibiotic treatment and subsequently regrow after antibiotic removal, leading to chronic and relapsing infections [1] [57]. First identified by Joseph Bigger in 1944 when he observed that penicillin could not completely eradicate Staphylococcus cultures, these cells are not antibiotic-resistant mutants but rather phenotypic variants of wild-type cells [1] [57]. Unlike resistance, which involves stable genetic changes that elevate the Minimum Inhibitory Concentration (MIC), persistence represents a non-heritable tolerance characterized by a biphasic killing curve in time-kill assays, where the majority population dies rapidly while a small persister subpopulation survives extended treatment [1] [57]. This phenomenon has been demonstrated in numerous pathogens, including Mycobacterium tuberculosis, Pseudomonas aeruginosa, Escherichia coli, and Staphylococcus aureus, and is clinically implicated in biofilm-associated infections, tuberculosis, typhoid fever, and recurrent urinary tract infections [1] [2].
The core problem of recalcitrance stems from the fundamental mechanism of most bactericidal antibiotics, which corrupt actively functioning cellular targets such as DNA replication, protein synthesis, and cell wall synthesis [34] [35]. Persisters evade this lethal action by entering a metabolically dormant state where these targets are largely inactive, thereby rendering the antibiotics ineffective despite maintained genetic susceptibility [34] [35]. This review examines the molecular mechanisms underlying persister formation, focusing on the interplay between stochastic and triggered pathways, and explores why conventional therapies fail to eradicate these elusive cells.
Molecular Mechanisms of Persister Formation: Stochastic versus Triggered Pathways
The formation of persister cells is understood through two complementary paradigms: stochastic, pre-existing formation and environmentally triggered induction. Research indicates that both models operate concurrently, with their relative importance depending on the bacterial species, growth conditions, and specific environmental stresses encountered [35].
Stochastic Variation and Bet-Hedging Strategies
Stochastic persister formation operates as a bet-hedging strategy, where isogenic bacterial populations generate phenotypic diversity to ensure some cells survive unforeseen adverse conditions [35]. A key mechanism involves random fluctuations in the expression of critical metabolic components.
- Metabolic Heterogeneity: Single-cell analysis of Staphylococcus aureus has demonstrated broad, Gaussian distribution in the expression levels of tricarboxylic acid (TCA) cycle enzymes (e.g., succinyl-CoA synthetase, fumarase), varying over 3 orders of magnitude between individual cells [34]. Cells sorted for low expression of these enzymes exhibited significantly increased tolerance to fluoroquinolone, aminoglycoside, and β-lactam antibiotics [34]. This correlation is mechanistically explained by the resultant reduction in cellular ATP, which drops below critical thresholds required for antibiotic-mediated killing [34].
- Energy Depletion: Direct experimental evidence shows that depleting ATP with arsenate treatment recapitulates the persister phenotype, while supplementing media with glucose to increase ATP levels reduces persister numbers by 100-fold [34]. Knockouts of TCA cycle enzymes (e.g., glutamate dehydrogenase, 2-oxoketoglutarate dehydrogenase) and amino acid catabolism pathways consistently yield lower ATP levels and higher multidrug tolerance [34].
Environmentally Triggered Persistence
In contrast to stochastic formation, persisters can also be induced in response to specific environmental cues, often mediated by sophisticated stress response systems.
- The Stringent Response and (p)ppGpp Signaling: Nutrient starvation, particularly amino acid limitation, triggers the stringent response, leading to accumulation of the alarmone (p)ppGpp [1] [57]. This signaling molecule dramatically reprograms cellular metabolism, downregulating energy-intensive processes like DNA replication and protein synthesis, and facilitating a transition to dormancy [1] [57].
- Toxin-Antitoxin (TA) Modules: Under stress conditions, unstable antitoxins are degraded, freeing toxins to act on cellular targets [57]. Type II TA modules, such as HipAB and MazEF, are heavily implicated in persistence. HipA, a serine-threonine kinase, phosphorylates aminoacyl-tRNA synthetase (GltX), leading to accumulation of uncharged tRNA, which activates RelA and increases (p)ppGpp levels [57]. Other toxins function as mRNA interferases that cleave cellular mRNAs, halting global translation and growth [35] [57].
- SOS Response and DNA Damage: Antibiotics like fluoroquinolones that cause DNA damage can induce the SOS response, leading to cell cycle arrest and increased persistence [1] [57]. This pathway allows bacteria to repair damage before resuming growth.
- Biofilm Microenvironments: The structured, matrix-encased communities of biofilms naturally generate heterogeneous microenvironments due to nutrient and oxygen gradients [2]. Cells in the inner depths of a biofilm experience nutrient limitation and slow growth, creating conditions that induce persistence [2]. Biofilms can contain up to 1,000 times more persisters than planktonic cultures [2].
Table 1: Core Molecular Pathways in Persister Formation
| Pathway | Key Effectors | Proposed Mechanism | Primary Inducing Signals |
|---|---|---|---|
| Stochastic Metabolic Variation | TCA cycle enzymes, ATP levels [34] | Fluctuations reduce energy generation, leading to dormancy [34] | Spontaneous gene expression noise [34] |
| Toxin-Antitoxin Systems | HipA, MazF, RelE [57] | Toxin activation inhibits translation or disrupts membrane potential [57] | Nutritional stress, antibiotic exposure [57] |
| Stringent Response | (p)ppGpp, RelA [1] [57] | Alarmone signaling shutdowns ribosome biogenesis and growth [1] [57] | Amino acid starvation [1] [57] |
| SOS Response | RecA, LexA [1] [57] | DNA damage induces cell cycle arrest and repair [1] [57] | DNA-damaging antibiotics (e.g., fluoroquinolones) [1] [57] |
| Biofilm-Induced Dormancy | Extracellular polymeric substance (EPS) [2] | Nutrient/O2 gradients create zones of slow growth [2] | Nutrient limitation, cell density (Quorum Sensing) [2] |
Quantitative Analysis of Persister Populations
Understanding the dynamics and characteristics of persister populations requires precise quantification. The following table summarizes key quantitative findings from experimental studies, highlighting the conditions and techniques used to measure persistence.
Table 2: Experimental Measurements of Persister Fractions and Characteristics
| Bacterial Species/Strain | Experimental Condition | Treatment & Measurement | Key Quantitative Finding |
|---|---|---|---|
| Staphylococcus aureus HG003 (Wild-type) [34] | Late exponential phase | 10x MIC Ciprofloxacin; Survival by CFU counting | Baseline persister fraction (~0.1-1% survival) [34] |
| S. aureus TCA cycle mutants (e.g., sucA, fumC) [34] | Late exponential phase | 10x MIC Ciprofloxacin; Survival by CFU counting | Persister fraction increased to nearly 10% of population [34] |
| Environmental E. coli isolates [58] | Exponential phase | 5x MIC Ciprofloxacin, Ampicillin, Nalidixic Acid; Model-based fraction estimation | Persister fractions varied substantially between isolates; no correlation in survival across different drug classes [58] |
| S. aureus with PsucC-gfp reporter [34] | Late exponential phase + Ciprofloxacin | FACS sorting of "dim" (low TCA expression) vs "bright" cells | ~100-fold enrichment of persisters in "dim" population compared to "bright" [34] |
| S. aureus gudB, sucA, sucC, fumC mutants [34] | Late exponential phase | ATP levels measured via luciferase assay | Significantly lower ATP levels than wild-type [34] |
A critical insight from quantitative studies is that persistence is not a uniform state. Research on E. coli environmental isolates reveals that the fraction of cells surviving one antibiotic is not correlated with the fraction surviving a second antibiotic, even those with nearly identical modes of action like ciprofloxacin and nalidixic acid [58]. This suggests drug-specific persistence mechanisms rather than a general-purpose dormant state [58]. Furthermore, the use of mathematical models to quantify persister fractions has proven more reliable than endpoint survival measurements, as it accounts for variables like differential killing rates and persister awakening during treatment [58].
Experimental Models and Methodologies for Persister Research
Standardized Persister Assays and Protocols
A cornerstone of persistence research is the time-kill assay. The following protocol is adapted from studies with S. aureus and E. coli [34] [58].
- Protocol: Time-Kill Kinetics and Persister Quantification
- Culture Preparation: Grow the bacterial strain of interest to the desired growth phase (e.g., late exponential phase) in appropriate liquid medium [34] [58].
- Antibiotic Challenge: Add a high concentration of bactericidal antibiotic (typically 5-10 times the MIC) to the culture. Maintain a control culture without antibiotic [34] [58].
- Sampling and Plating: At predetermined time intervals (e.g., 0, 3, 6, 9, 24 hours), remove aliquots from both treated and control cultures. Serially dilute and plate on drug-free agar medium [58].
- Viable Count Analysis: After incubation, count colony-forming units (CFUs). Plot the log10 CFU/mL versus time. The biphasic curve—a rapid initial kill followed by a plateau—indicates persister survival [58].
- Model-Based Estimation: Fit the survival data to a two-state mathematical model (normal cells persister cells) to estimate the initial persister fraction and switching rates, providing a more robust quantification than a single endpoint measurement [58].
Single-Cell Analysis and Fluorescence-Activated Cell Sorting
Bulk population measurements can mask underlying heterogeneity. Single-cell techniques are therefore critical.
- Fluorescent Reporter Construction: Clone the promoter region of a gene of interest (e.g., a TCA cycle gene like sucC or fumC) upstream of a stable GFP gene on a plasmid [34].
- Sorting and Survival Validation: Grow the reporter strain to late exponential phase and treat with a high dose of antibiotic (e.g., ciprofloxacin). Use FACS to gate and sort subpopulations based on fluorescence intensity (e.g., dim, middle, bright) [34]. Plate the sorted cells to determine the survival rate of each subpopulation. Studies show cells with low TCA promoter activity ("dim") can have up to 100-fold higher survival than "bright" cells [34].
- ATP Biosensing: Employ genetically encoded ATP biosensors like QUEEN, which exhibits excitation ratio-metric fluorescence dependent on ATP concentration. This allows for direct correlation of single-cell ATP levels with antibiotic tolerance [34].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Tools for Persister Research
| Reagent / Tool | Function in Research | Example Application |
|---|---|---|
| Fluorescent Protein Reporters (e.g., GFPuvr) [34] | Visualize and sort cells based on promoter activity of specific genes. | FACS sorting of S. aureus with low vs. high TCA cycle gene expression to link metabolism to persistence [34] |
| ATP Biosensor (e.g., QUEEN) [34] | Ratiometric measurement of intracellular ATP at single-cell level. | Correlate low cellular energy levels with antibiotic tolerance in S. aureus [34] |
| Transposon Mutant Libraries [34] | High-throughput generation of gene knockout mutants for screening. | Identify specific metabolic knockouts (e.g., in TCA cycle) that increase persister frequency [34] |
| Luciferase-based ATP Assay [34] | Quantitative measurement of total cellular ATP from lysates. | Confirm low ATP levels in TCA cycle mutants and stationary phase cells [34] |
| Two-State Mathematical Model [58] | Quantify persister fraction and switching rates from time-kill data. | Provide consistent, model-based persister quantification across different E. coli isolates and drugs [58] |
Conceptual Diagrams of Key Persistence Pathways
The following diagrams, defined using the DOT language, illustrate the core signaling pathways and experimental workflows central to persister research. These visuals are designed for clarity and scientific accuracy, adhering to the specified color and contrast guidelines.
Stochastic and Triggered Persister Formation Pathways
Experimental Workflow for Metabolic Persister Analysis
Discussion: Implications for Therapeutic Development and Future Research
The dichotomous yet complementary nature of stochastic and triggered persistence mechanisms presents a formidable challenge for antimicrobial therapy. The stochastic model implies that a certain level of treatment failure is inevitable due to pre-existing phenotypic variants, while the triggered model suggests that the host environment and the therapy itself can amplify the problem by inducing further tolerance [34] [35] [59]. This understanding forces a paradigm shift from solely targeting rapidly growing bacteria to developing strategies that also address the dormant, persistent subpopulation.
Promising therapeutic avenues emerging from this research include:
- Energy-Augmenting Therapies: Strategies that boost bacterial metabolism and ATP levels prior to antibiotic administration could sensitize persisters to killing, as demonstrated by glucose supplementation reducing persister counts [34].
- Anti-Biofilm Agents: Compounds that disrupt the biofilm matrix or interfere with quorum sensing can mitigate the structured environment that fosters persistence, improving antibiotic penetration and efficacy [2].
- TA System Manipulation: Identifying compounds that prevent toxin activation or promote persister "awakening" could make these dormant cells vulnerable to conventional antibiotics [35] [57].
- Combination Therapies: The lack of cross-persistence across antibiotic classes suggests that strategically chosen drug combinations could minimize overall treatment failure by targeting different persister subpopulations [58].
Future research must prioritize the study of persisters in physiologically relevant environments, such as in vivo infection models and biofilms. A 2025 study on Salmonella challenges the traditional persister narrative, suggesting that nutrient starvation in tissues causes most bacteria to become difficult to kill, not just a small pre-existing subset [59]. This underscores the need for real-time, single-cell analysis in disease-like conditions to fully understand and ultimately overcome the recalcitrance of persistent infections.
The escalating crisis of antimicrobial resistance poses a formidable challenge to global public health, but it is not the sole mechanism by which bacterial infections evade therapeutic intervention. The phenomena of antibiotic tolerance and bacterial persistence represent distinct yet equally critical survival strategies that contribute significantly to treatment failure and chronic infections [1] [2]. While these terms are sometimes used interchangeably in clinical and research settings, they describe fundamentally different physiological states with unique diagnostic and therapeutic implications. This distinction becomes particularly salient when framed within the ongoing investigation of stochastic versus triggered persister cell formation mechanisms—a central paradigm in understanding how bacterial subpopulations survive lethal stress.
The clinical relevance of this challenge is profound. Bacterial persisters are increasingly recognized as the primary culprits behind relapsing infections and the recalcitrance of biofilm-associated diseases, such as those occurring in cystic fibrosis patients and on indwelling medical devices [1] [2]. These phenotypically variant cells can survive brief exposure to high concentrations of antimicrobials without acquiring heritable genetic resistance, creating a reservoir for recurrent infection that often evades conventional diagnostic detection. Understanding the precise mechanisms that differentiate tolerance, resistance, and persistence is therefore not merely an academic exercise but an urgent necessity for developing more effective therapeutic strategies against persistent infections.
Defining the Spectrum of Bacterial Survival
Conceptual and Phenotypic Distinctions
Bacterial survival strategies against antimicrobial agents can be categorized into three primary phenotypes based on their mechanistic and inheritable characteristics. The table below provides a comparative overview of these distinct survival strategies.
Table 1: Key Characteristics of Antibiotic Resistance, Tolerance, and Persistence
| Feature | Antibiotic Resistance | Antibiotic Tolerance | Bacterial Persistence |
|---|---|---|---|
| Inheritance | Heritable genetic changes | Non-heritable, phenotypic | Non-heritable, phenotypic variant |
| MIC Change | Increased | Unchanged | Unchanged |
| Killing Kinetics | Population grows at lethal concentrations | Slower killing of entire population | Biphasic killing curve with surviving subpopulation |
| Mechanism | Target modification, efflux pumps, enzyme inactivation | Reduced metabolism, slowed growth | Dormancy, metabolic arrest, toxin-antitoxin systems |
| Phenotype Stability | Stable without selective pressure | Transient, dependent on conditions | Transient, reversible after stress removal |
| Population Structure | Homogeneous resistant population | Homogeneous tolerant population | Heterogeneous with persister subpopulation |
The operational definitions of these phenomena are further distinguished by specific laboratory observations. Resistance is quantitatively defined by an increase in the Minimum Inhibitory Concentration (MIC), whereas tolerance is measured by a decrease in the killing rate of the entire population [60]. Persistence, in contrast, is identified through its characteristic biphasic killing curve, where an initial rapid decline in viable cells is followed by a plateau representing a surviving subpopulation that withstands further antibiotic exposure [57] [60].
Relationship Between Phenomena
These survival strategies are not mutually exclusive and can coexist within bacterial populations, creating complex therapeutic challenges. There is growing evidence that tolerant and persister populations can serve as a reservoir for the emergence of resistant mutants [2] [57]. This evolutionary pathway occurs because the extended survival window provided by tolerance and persistence mechanisms increases the opportunity for the acquisition of resistance-conferring genetic mutations. This relationship is particularly evident in biofilm-associated infections, where the structured environment facilitates horizontal gene transfer alongside the formation of metabolically heterogeneous subpopulations [2].
Methodological Framework for Differentiation
Core Experimental Protocols
Distinguishing between resistance, tolerance, and persistence requires specific experimental approaches designed to quantify different aspects of bacterial survival under antibiotic pressure.
Table 2: Core Methodologies for Differentiating Bacterial Survival Phenotypes
| Method | Protocol Overview | Key Measurements | Interpretation |
|---|---|---|---|
| MIC/MBC Determination | Broth microdilution with standardized inocula | MIC: Lowest concentration inhibiting visible growth; MBC: Concentration killing ≥99.9% of inoculum | Increased MIC indicates resistance |
| Time-Kill Assays | Exposure to lethal antibiotic concentrations with periodic viable counting | Killing curve kinetics over 24-48 hours | Biphasic curve indicates persistence; uniformly slowed killing indicates tolerance |
| Persister Isolation | Antibicide exposure followed by drug removal and outgrowth | Percentage of surviving cells after lethal treatment | Isolated persisters remain genetically susceptible upon reculturing |
The definitive test for persistence involves demonstrating that cells surviving antibiotic exposure give rise to a new population with the same drug susceptibility profile as the parent strain, confirming the non-heritable nature of the phenotype [57]. For the specific investigation of stochastic versus triggered formation, researchers often compare persister frequencies in steadily growing cultures (favoring stochastic formation) versus cultures exposed to sublethal environmental stresses (inducing triggered formation) such as nutrient limitation, oxidative stress, or acidic pH [1] [61].
The Researcher's Toolkit: Essential Reagents and Solutions
Table 3: Key Research Reagent Solutions for Persistence Studies
| Reagent/Solution | Function/Application | Examples/Notes |
|---|---|---|
| Fluoroquinolones (e.g., Ciprofloxacin) | DNA gyrase inhibitors; commonly used in persistence time-kill assays | Effective against growing cells; reveals persister subpopulations [57] |
| Aminoglycosides (e.g., Amikacin) | Protein synthesis inhibitors; require metabolic activity for uptake | Used in studies of metabolic stimulation to eradicate persisters [60] |
| β-lactam Antibiotics | Cell wall synthesis inhibitors; target actively dividing cells | Ineffective against dormant persisters with reduced cell wall synthesis [1] |
| Quaternary Ammonium Compounds (QACs) | Disinfectants for tolerance studies in food industry settings | Benzalkonium chloride used for identifying disinfectant tolerance mechanisms [62] |
| (p)ppGpp Analogs | Inducers of stringent response; study connection to persistence | Key signaling molecule in triggered persistence mechanisms [57] |
Molecular Mechanisms: Stochastic versus Triggered Persistence
The central conceptual framework in persistence research revolves around two complementary models of formation: stochastic triggering, which occurs spontaneously in a subset of cells during balanced growth, and environmentally triggered persistence, induced in response to external stresses [1]. The molecular mechanisms underlying these pathways are complex and often interconnected, involving multiple regulatory systems that control bacterial physiology.
Diagram 1: Stochastic vs Triggered Persister Formation Pathways. Both pathways converge on metabolic arrest enabling antibiotic tolerance.
Toxin-Antitoxin (TA) Systems and Bistable Switching
TA modules represent a fundamental mechanism of persistence regulation, particularly in the stochastic formation model. These genetic elements consist of a stable toxin that inhibits essential cellular processes and a labile antitoxin that neutralizes the toxin under normal conditions [57]. The HipBA system in E. coli was the first persistence-specific TA module identified, where the HipA toxin phosphorylates and inhibits the glutamyl-tRNA synthetase GltX, leading to accumulation of uncharged tRNA and activation of the stringent response [57]. This creates a bistable switch wherein slight fluctuations in toxin-antitoxin ratios can trigger a dramatic physiological shift toward dormancy in a subset of cells, even in an otherwise homogeneous environment [57].
The Stringent Response and Environmental Triggering
The (p)ppGpp-mediated stringent response serves as a central integrator of environmental stress signals into the persistence regulatory network. When bacteria encounter nutrient limitation or other stresses, (p)ppGpp accumulates and orchestrates a comprehensive transcriptional reprogramming that redirects resources from growth to maintenance [57]. This response has been directly linked to increased persister formation through multiple pathways, including upregulation of TA systems and direct inhibition of metabolic enzymes [57]. Mutant strains lacking a functional stringent response fail to form persisters even under starvation conditions, demonstrating this program's essential role in environmentally triggered persistence [60].
Metabolic Regulation as a Convergence Point
Both stochastic and triggered pathways ultimately converge on metabolic downregulation as the final effector mechanism enabling antibiotic tolerance. Persister cells exhibit dramatically reduced ATP levels and proton motive force, which diminishes the activity of antibiotic targets and uptake of many bactericidal drugs [57] [60]. This metabolic reprogramming creates a therapeutic vulnerability—studies have shown that metabolic stimulation through specific carbon sources or electron transport chain activators can sensitize persisters to aminoglycoside antibiotics by restoring proton motive force and drug uptake [60].
Advanced Technologies in Persistence Research
Emerging Methodological Approaches
The study of bacterial persistence presents unique technical challenges due to the low frequency of persister cells in most populations (typically <1%) and the transient, non-heritable nature of the phenotype [57]. Recent technological advances have enabled more precise investigation of these elusive cell states.
Table 4: Emerging Technologies for Persister Cell Research
| Technology | Application | Key Advantages |
|---|---|---|
| Whole Genome Sequencing (WGS) | Identification of genetic markers associated with persistence and tolerance | Comprehensive view of genetic factors; enables machine learning approaches [62] |
| Machine Learning (ML) Prediction | Predicting disinfectant tolerance from genomic data | Can identify novel genes associated with tolerance; high classification accuracy demonstrated [62] |
| RNA-Seq Transcriptomics | Characterization of gene expression in persister subpopulations | Reveals regulatory networks active in dormant cells; identifies triggers [61] |
| Fluorescence-Activated Cell Sorting (FACS) | Isolation of persister cells based on dye staining or reporter systems | Enables purification of rare persister cells for downstream analysis |
| Microfluidics & Single-Cell Analysis | Monitoring individual cell behaviors and heterogeneity | Reveals stochasticity in persister formation and resuscitation dynamics |
Computational and Modeling Approaches
The integration of ecological and stochastic modeling has provided important insights into how microbial community interactions influence persistence and resuscitation dynamics [63]. These approaches recognize that cell-to-cell interactions and population size fluctuations significantly impact the resilience of microbial populations following antibiotic treatment [63]. Additionally, machine learning applications have demonstrated remarkable success in predicting disinfectant tolerance phenotypes from genomic data, with classifiers achieving balanced accuracy scores up to 0.97 in predicting tolerance to quaternary ammonium compounds in Listeria monocytogenes [62].
Diagram 2: Genomic Prediction of Tolerance. Workflow for predicting disinfectant tolerance using machine learning.
The critical distinction between antibiotic resistance, tolerance, and bacterial persistence represents more than a semantic exercise in microbiological classification—it defines fundamental differences in physiological state, underlying mechanism, and, most importantly, therapeutic approach. As research continues to unravel the complex interplay between stochastic and triggered persistence mechanisms, it becomes increasingly evident that effective treatment of persistent infections will require strategies specifically targeting the dormant phenotypic states that conventional antibiotics fail to eradicate.
The emerging paradigm suggests that future antimicrobial therapies must incorporate anti-persister compounds that either prevent entry into the persistent state or actively awaken dormant cells to sensitize them to conventional antibiotics. The development of such approaches will depend on continued refinement of our diagnostic capabilities to distinguish these different survival phenotypes in clinical settings and a deeper understanding of the molecular switches that control stochastic versus environmentally triggered persistence across diverse bacterial pathogens.
The prevention of relapse, whether in the context of chronic diseases like cancer or behavioral disorders such as substance dependence, represents a fundamental challenge across medical science. Research has increasingly revealed that relapse is frequently driven by persistent cellular or behavioral states that survive initial therapeutic interventions. A growing body of evidence suggests two primary mechanisms underlie the emergence of these persistent states: stochastic (intrinsic) mechanisms and triggered (extrinsic) mechanisms [64] [65]. The scientific community has often approached these mechanisms as separate domains of inquiry, developing targeted interventions that address one pathway while neglecting the other. This segmented approach has limited the efficacy of relapse prevention strategies across multiple disease domains.
This whitepaper argues that a comprehensive understanding of both stochastic and triggered persistence mechanisms is essential for developing effective relapse prevention protocols. We examine the biological foundations of both pathways, present integrated experimental frameworks for their simultaneous study, and propose a multidimensional therapeutic strategy that targets the complex interplay between random cellular variation and environmentally-induced adaptation. By synthesizing findings from oncology, neuroscience, and addiction research, we provide researchers and drug development professionals with a unified framework for addressing the fundamental drivers of relapse across disease contexts.
Theoretical Foundations: Defining Stochastic and Triggered Persistence
Stochastic Persistence Mechanisms
Stochastic persistence arises from pre-existing cellular heterogeneity within a population, where random variations create a subpopulation inherently capable of surviving therapeutic intervention. In oncology, this concept is exemplified by drug-tolerant persister (DTP) cells that exist prior to treatment exposure [65]. These cells represent a clonal selection mechanism built on classical Darwinian principles, where genomic instability in cancer cells produces varied sensitivities to drug action [65]. The subpopulation with inherent tolerance capabilities is subsequently selected for survival during treatment.
The stochastic model posits that persistence is not induced by treatment but rather revealed through the selective pressure of treatment. This explains why relapse can occur even after seemingly successful therapeutic interventions that eliminate the majority of vulnerable cells [64]. The stochastic nature of this process makes it particularly challenging to predict and target, as the persistent cells may not possess consistent phenotypic markers until after selection has occurred.
Triggered Persistence Mechanisms
In contrast, triggered persistence results from cellular adaptation in direct response to therapeutic pressure or environmental cues. This "drug induction mechanism" involves active cellular reprogramming in response to stress [65]. The transition to a persistent state is not random but is initiated by specific environmental signals, such as cytokine exposure, nutrient gradients, or therapy-stimulated tumor secretome factors [64].
This adaptive response often involves non-genetic mechanisms that allow for rapid state transition without permanent genetic alteration [64] [65]. The induced persistence state is frequently characterized by significant cellular plasticity, enabling cells to reversibly assume different identities in response to environmental pressures [65]. This plasticity represents a fundamental defense mechanism that is conserved across multiple biological systems, from microbial communities to human malignancies.
Table 1: Comparative Analysis of Stochastic vs. Triggered Persistence Mechanisms
| Characteristic | Stochastic Persistence | Triggered Persistence |
|---|---|---|
| Origin | Pre-existing cellular heterogeneity | Induced by treatment or environmental stress |
| Primary Mechanism | Clonal selection | Cellular reprogramming |
| Genetic Basis | May involve pre-existing genetic variants | Primarily non-genetic, epigenetic mechanisms |
| Temporal Pattern | Present before treatment | Emerges during/after treatment |
| Predictability | Low (random distribution) | Higher (response to specific cues) |
| Reversibility | Typically stable | Often reversible after stress removal |
| Experimental Evidence | Persister cells in treatment-naïve populations [65] | Stress-induced mutation mechanisms [65] |
Experimental Models and Methodologies for Studying Persistence Mechanisms
Integrated Experimental Framework
A comprehensive approach to studying persistence requires experimental models that capture both stochastic and triggered mechanisms. The most effective strategy employs complementary model systems with increasing complexity, from controlled cell culture to in vivo environments [64]. Each model system offers unique advantages for deciphering specific aspects of persistence, as outlined in the experimental framework below:
Diagram 1: Experimental Framework for Persistence Research
Core Methodologies for Persistence Research
Cancer Cell Culture Models for Autonomous Persistence
Simple monolayer cultures remain fundamental tools for investigating cell-autonomous persistence mechanisms [64]. These models enable high-throughput screening of persistence prevalence across different genetic backgrounds and therapeutic exposures. The standardized protocol involves:
- Establishment of baseline sensitivity: Determine IC50 values for therapeutic agents across a panel of relevant cell lines
- Persistence induction: Expose cells to therapeutic concentrations (typically 5-10× IC50) for extended durations (7-14 days)
- DTP quantification: Assess viable cell populations via metabolic activity assays (e.g., Alamar Blue, MTT) or direct counting
- Reversibility assessment: Monitor regrowth dynamics during drug-free "holiday" periods
Key advantages include scalability, genetic manipulability, and reproducibility [64]. These systems have enabled pivotal discoveries in persistence, including the initial identification of DTP cells and the characterization of their evolutionary trajectories [64].
Tumor-Stroma Co-culture Systems for Triggered Persistence
To investigate environmentally-triggered persistence, co-culture models incorporating stromal elements provide critical insights [64]. These systems recapitulate non-autonomous signals that modulate therapeutic sensitivity:
- Stromal component selection: Incorporate cancer-associated fibroblasts (CAFs), immune cells, or endothelial cells
- Soluble factor characterization: Profile cytokine/chemokine secretion in mono- vs co-culture
- Contact-dependent interactions: Implement direct contact vs transwell systems to distinguish signaling mechanisms
- Therapeutic challenge: Assess modulation of persistence frequency by stromal elements
These models have identified specific persistence-modulating signals, such as the IFNγ/STAT1/type I PRMT axis and CAF-derived HGF and FGF7 [64]. The identification of such pathways provides direct targets for interrupting environmentally-triggered persistence.
In Vivo Models for Physiological Context
Animal models provide essential physiological context for persistence studies, though with reduced scalability [64]. Both xenograft (human tumors in immunocompromised hosts) and syngeneic (murine tumors in immunocompetent hosts) models offer unique advantages:
- Human tumor xenografts: Maintain authentic human tumor biology while sacrificing immune interactions
- Syngeneic murine models: Preserve intact host immunity and stromal interactions
- Treatment and monitoring: Implement therapeutic regimens with longitudinal assessment of tumor burden
- Residual disease analysis: Profile cellular and molecular features of persistent cell populations following treatment
These systems have been instrumental in identifying context-dependent persistence mechanisms, such as neural crest stem cell transitions in melanoma following RAF/MEK inhibition [64].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Persistence Studies
| Reagent Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| Cell Line Models | PC9 (lung cancer), A375 (melanoma), HT-29 (colorectal) [64] | Baseline persistence screening | Cell type-specific mechanisms |
| Stromal Components | Cancer-associated fibroblasts, peripheral blood mononuclear cells [64] | Co-culture persistence models | Microenvironmental signaling |
| Cytokine Inhibitors | Anti-IFNγ, STAT1 inhibitors, HGF-neutralizing antibodies [64] | Functional validation | Causal pathway analysis |
| Epigenetic Probes | HDAC inhibitors, DNMT inhibitors, BET bromodomain inhibitors [65] | Persister reversal studies | Epigenetic regulation |
| Metabolic Tracers | 13C-glucose, 13C-glutamine, Seahorse assay kits [65] | Metabolic profiling | Bioenergetic adaptations |
| Cell State Markers | Mex3a, surface antigen detection systems [65] | Persister identification and isolation | Molecular signatures |
Molecular Mechanisms Underlying Stochastic and Triggered Persistence
Shared and Distinct Pathways in Persistence
Despite their different origins, stochastic and triggered persistence converge on several common biological programs that enable survival under therapeutic pressure. The molecular landscape of persistence involves dynamic interactions between epigenetic, transcriptional, and metabolic adaptations that collectively promote a survival-oriented state [65].
At the epigenetic level, histone modification and chromatin remodeling create reversible yet stable persistence states. Key modifications include changes in histone methylation (H3K4, H3K27) and acetylation patterns that alter transcriptional accessibility [65]. These epigenetic shifts enable rapid transition between sensitive and persistent states without genetic alteration.
Metabolically, persister cells frequently demonstrate shifted energy production pathways, including enhanced oxidative phosphorylation and altered nutrient uptake [65]. These adaptations maintain essential cellular functions while minimizing proliferation-associated energy demands and reactive oxygen species production.
The following diagram illustrates the core molecular pathways and their interactions in establishing and maintaining persistence states:
Diagram 2: Molecular Pathways to Persister State
Distinctive Features of Stochastic vs. Triggered Mechanisms
While sharing common endpoints, important distinctions exist in the molecular implementation of stochastic versus triggered persistence:
Stochastic persistence often leverages pre-established phenotypic heterogeneity that may include:
- Natural variation in gene expression of resistance-associated pathways
- Stochastic cell cycle entry/exit that creates intrinsically quiescent subpopulations
- Baseline epigenetic heterogeneity that predisposes to persistence states
Triggered persistence typically involves active response programs such as:
- Stress-induced signaling pathways (e.g., mTOR, HIF1α, NF-κB)
- Therapy-induced secretome changes that create autocrine/paracrine survival signals
- Rapid epigenetic adaptation in response to environmental cues
- ATP release from dying cells that provides survival signals for neighbors [64]
This distinction has profound implications for therapeutic targeting, as stochastic persistence may require population-wide approaches, while triggered persistence might be prevented by intercepting specific induction signals.
Therapeutic Implications and Translational Applications
Limitations of Singular Targeting Approaches
The historical focus on either stochastic or triggered mechanisms has produced suboptimal therapeutic outcomes across multiple disease domains. In oncology, targeting only pre-existing persisters fails to address the adaptive capacity of the broader population to generate new persisters through induced mechanisms [64] [65]. Conversely, targeting only induced adaptation leaves inherently resistant subpopulations untouched.
In behavioral medicine, parallel limitations exist. Focusing solely on internal triggers (e.g., genetic predispositions) neglects the powerful role of environmental cues in provoking relapse [66] [67]. Similarly, addressing only external triggers fails to build resilience against intrinsically generated cravings or psychological states that drive recurrence [68] [69].
Integrated Therapeutic Strategy
An effective relapse prevention strategy must simultaneously address both stochastic and triggered persistence through complementary interventions:
For stochastic persistence:
- Pre-emptive combination therapies that target multiple vulnerability states simultaneously
- Population-wide epigenetic modulation to reduce pre-existing heterogeneity
- Therapeutic cycling to prevent outgrowth of minor resistant subpopulations
For triggered persistence:
- Environmental signal interception to block induction of persistence states
- Stress response pathway inhibition to prevent adaptive transitions
- Microenvironment normalization to remove persistence-promoting signals
The most effective approach employs longitudinal adaptive therapy that continuously monitors both stochastic and triggered mechanisms, adjusting therapeutic combinations in response to evolving persistence patterns [64] [65].
Biomarker Development for Dual Monitoring
Critical to the implementation of integrated anti-persistence strategies is the development of biomarkers that simultaneously report on both stochastic and triggered mechanisms. Promising approaches include:
Liquid biopsy platforms that capture:
- Pre-existing resistance markers (stochastic component)
- Therapy-induced adaptation signals (triggered component)
- Environmental modulation factors (contextual triggers)
Functional imaging approaches that visualize:
- Regional persistence hotspots (stochastic heterogeneity)
- Adaptive response activation (triggered dynamics)
- Microenvironmental modulation (niche effects)
The integration of these monitoring technologies with targeted interventions creates a closed-loop system for comprehensive relapse prevention that addresses the full spectrum of persistence mechanisms.
The dichotomy between stochastic and triggered persistence mechanisms represents a false choice that has limited progress in relapse prevention across therapeutic domains. The most promising path forward lies in integrated approaches that recognize the continuous interplay between intrinsic cellular heterogeneity and extrinsic adaptive signaling.
Future research should prioritize:
- Single-cell multiscale profiling to simultaneously capture genetic, epigenetic, and environmental influences on persistence states
- Dynamic computational modeling that predicts persistence evolution across different contextual backgrounds
- Adaptive clinical trial designs that enable real-time therapeutic adjustments based on both stochastic and triggered persistence markers
- Cross-disciplinary collaboration that leverages insights from oncology, microbiology, neuroscience, and behavioral medicine
By transcending the historical division between stochastic and triggered mechanisms, researchers and drug development professionals can develop truly comprehensive relapse prevention strategies that address the fundamental drivers of therapeutic failure across disease contexts.
Biofilms represent a fundamental survival strategy for bacterial populations, creating structured communities encased in a self-produced extracellular polymeric substance (EPS) matrix that serves as a formidable physical and physiological barrier against antimicrobial agents [70]. Within these complex architectures, bacterial persisters—a transiently dormant, non-growing subpopulation—find a protected niche that enables them to survive antibiotic exposures that eradicate their genetically identical, actively growing counterparts [1] [71]. This synergistic relationship between biofilms and persister cells constitutes a critical frontier in the challenge of treating chronic and recurrent bacterial infections.
The ongoing scientific debate between stochastic versus triggered persister formation mechanisms provides essential context for developing effective anti-biofilm strategies. Stochastic models propose that persisters arise spontaneously through random fluctuations in cellular components, creating phenotypic heterogeneity independent of environmental signals [13] [72]. In contrast, triggered models suggest that persister formation represents a coordinated response to environmental stresses such as nutrient limitation, oxidative stress, or sub-inhibitory antibiotic concentrations [1] [71]. Understanding this duality is paramount, as successful biofilm disruption strategies must address both spontaneously arising and environmentally induced persistence populations within the biofilm matrix.
Molecular Mechanisms of Persister Formation: Stochastic versus Triggered Paradigms
Stochastic Persister Formation
The stochastic model posits that persisters form spontaneously through random fluctuations in gene expression and protein abundance, resulting in phenotypic heterogeneity even in homogeneous environments. Single-cell studies have demonstrated that fluctuations in metabolic activity serve as a primary driver of stochastic persistence [13]. Research examining Escherichia coli has revealed that stochastic heterogeneity in the expression of Krebs cycle enzymes (including citrate synthase (GltA), isocitrate dehydrogenase (Icd), and α-ketoglutarate dehydrogenase (SucA)) directly correlates with persister formation [13]. Cells with diminished levels of these energy-generating enzymes exhibit reduced ATP production, creating a transiently dormant state that tolerates antibiotic exposure [13].
Advanced microfluidics time-lapse microscopy with an ATP reporter (iATPSnFr1.0) has visually confirmed that subpopulations with low intracellular ATP demonstrate significantly enhanced survival against ampicillin treatment [13]. This metabolic heterogeneity occurs independently of external triggers, representing a bet-hedging strategy that ensures population survival in unpredictably fluctuating environments [71] [72].
Triggered Persister Formation
In contrast, triggered persistence involves specific molecular responses to environmental signals. Biofilms generate ideal conditions for triggered persistence through their structured microenvironment which contains gradients of nutrients, oxygen, and waste products [73] [70]. These conditions activate several key response systems:
- Toxin-Antitoxin (TA) Systems: Under stress conditions, labile antitoxins are degraded, freeing toxins to modulate cellular functions toward dormancy. For example, the HipA toxin inhibits translation by phosphorylating glutamyl-tRNA synthetase, while TisB and HokB toxins decrease proton motive force and ATP levels by forming membrane pores [1].
- Stringent Response: Nutrient limitation triggers RelA-mediated synthesis of the alarmone (p)ppGpp, which dramatically alters gene expression profiles, downregulating ribosome production and growth while upregulating stress resistance pathways [1] [71].
- SOS Response: DNA damage from fluoroquinolone antibiotics or oxidative stress activates RecA, leading to LexA autocleavage and derepression of DNA repair genes and tisB toxin expression, promoting persistence [1] [72].
Table 1: Characteristics of Stochastic versus Triggered Persister Cells
| Characteristic | Stochastic Persisters | Triggered Persisters |
|---|---|---|
| Induction Mechanism | Spontaneous fluctuations in gene expression and metabolic activity | Environmental stress responses (nutrient limitation, antibiotics, oxidative stress) |
| Primary Regulators | Heterogeneity in Krebs cycle enzymes and ATP levels | Toxin-antitoxin systems, (p)ppGpp, SOS response |
| Temporal Pattern | Form continuously during all growth phases | Predominantly form during stationary phase and specific stress conditions |
| Population Size | Typically 0.001%-1% of population | Can reach significantly higher percentages under sustained stress |
| Metabolic State | Deep dormancy with minimal metabolic activity | Varied dormancy states from complete arrest to slowed metabolism |
Biofilm Architecture as a Sanctuary for Persister Cells
The structured environment of a biofilm provides multiple protective mechanisms that safeguard persistent populations. The extracellular polymeric substance (EPS) matrix, composed of polysaccharides, proteins, nucleic acids, and lipids, creates a physical barrier that restricts antibiotic penetration [73] [70]. This matrix is not merely a passive barrier but represents a dynamic microenvironment that modulates external stresses through molecular sieving, charge interactions, and enzymatic degradation of antimicrobial compounds [73].
Beyond physical protection, biofilms establish metabolic heterogeneity through nutrient and oxygen gradients that generate distinct microniches [73] [70]. These gradients create a spectrum of metabolic states from highly active cells at the biofilm periphery to dormant cells in oxygen- and nutrient-depleted zones [73]. This spatial organization naturally supports persister maintenance, as the deeply dormant cells residing in the biofilm core are inherently tolerant to antibiotics that target active cellular processes [73] [74].
Additionally, the dense, structured nature of biofilms facilitates horizontal gene transfer (HGT), enabling the dissemination of resistance genes among community members, including persister cells that may resume growth after antibiotic treatment [73]. This combination of physical protection, metabolic diversification, and genetic exchange creates a resilient ecosystem that perpetuates both phenotypic tolerance (persistence) and genotypic resistance [73] [75].
Strategic Approaches for Biofilm Disruption and Persister Targeting
Matrix-Targeting Agents
The extracellular matrix represents the first line of biofilm defense, making it a prime target for therapeutic intervention. Matrix-degrading enzymes disrupt the structural integrity of biofilms, enhancing antibiotic penetration and efficacy [73] [76].
- Dispersin B: A glycoside hydrolase secreted by Actinobacillus actinomycetemcomitans that degrades poly-N-acetylglucosamine (PNAG), a key polysaccharide component in many bacterial biofilms including Staphylococcus epidermidis, E. coli, and Yersinia pestis [76]. Treatment protocols typically involve 10-100 μg/mL concentrations applied directly to biofilms for 2-24 hours, resulting in significant biofilm dispersal.
- DNase I: Degrades extracellular DNA (eDNA), a crucial structural component particularly important in Pseudomonas aeruginosa biofilms [73] [76]. Standard experimental use involves 10-100 U/mL concentrations, with effectiveness demonstrated against both developing and established biofilms. DNase is already in clinical use for disrupting P. aeruginosa biofilms in cystic fibrosis patients [76].
- PelA and PslG hydrolases: Species-specific glycoside hydrolases that degrade the Pel and Psl polysaccharides in P. aeruginosa biofilms [76]. These enzymes have shown particular efficacy when used prophylactically on surfaces or in combination therapy with conventional antibiotics.
Quorum Sensing Inhibition
Quorum sensing (QS) represents a promising target as it regulates both biofilm development and virulence factor production without directly imposing lethal selective pressure [73] [75]. QS inhibitors disrupt bacterial cell-to-cell communication, potentially reducing biofilm formation and persistence.
- Natural QS Inhibitors: Phytochemicals such as curcumin, quercetin, and berberine demonstrate potent quorum quenching activity by interfering with autoinducer signaling and suppressing EPS biosynthesis [73]. These compounds typically require concentrations of 10-200 μM for effective QS inhibition, depending on the specific compound and bacterial species.
- Synthetic QS Inhibitors: Acyl homoserine lactone (AHL) analogs competitively inhibit LuxR-type receptor binding, disrupting the coordination of biofilm development and virulence expression [73] [74]. These synthetic approaches offer advantages in specificity and stability compared to natural compounds.
Metabolic Reactivation Strategies
Metabolic reactivation represents a paradigm-shifting approach that targets the fundamental mechanism of persistence—dormancy. By stimulating persister cells to resume metabolic activity, these strategies re-sensitize them to conventional antibiotics [76] [1].
- Sugar Phosphates: Mannitol, glucose, and fructose can stimulate metabolic awakening in persister cells, potentially through the restoration of proton motive force and ATP production [76]. Experimental protocols often combine these metabolites (10-50 mM) with aminoglycoside antibiotics, resulting in up to 10,000-fold enhancement in killing efficacy against E. coli and S. aureus persisters.
- Cis-2-decenoic Acid: This fatty acid signaling molecule induces a burst of protein synthesis in P. aeruginosa persisters, restoring susceptibility to ciprofloxacin and resulting in a 3,000-fold reduction in planktonic persisters and a million-fold reduction in biofilm-derived persisters [76]. Effective concentrations typically range from 10-100 μM.
- Interspecies Metabolites: Recent research has identified multifunctional microbial metabolites such as zincmethylphyrins and coproporphyrins produced by Sphingopyxis species that can promote the growth of previously unculturable microbes while exhibiting context-dependent antimicrobial activity against the producer species [75].
Table 2: Anti-Biofilm and Anti-Persister Therapeutic Approaches
| Strategy | Molecular Targets | Key Agents | Efficacy Evidence |
|---|---|---|---|
| Matrix Degradation | EPS structural components (PNAG, eDNA, Pel/Psl) | Dispersin B, DNase I, PelA, PslG | 60-90% reduction in biofilm biomass; 10-1000x enhanced antibiotic penetration |
| Quorum Quenching | Autoinducer synthases/receptors, virulence regulators | Curcumin, AHL analogs, quercetin | 50-80% reduction in biofilm formation; reduced virulence factor production |
| Metabolic Reactivation | Proton motive force, ATP production, protein synthesis | Sugars, cis-2-decenoic acid, metabolite combinations | 1000-1,000,000x reduction in persister survival with antibiotic combinations |
| Nanoparticle Delivery | Bacterial membranes, intracellular targets | Silver NPs, ZnO NPs, graphene-based NPs, liposomal antibiotics | Enhanced biofilm penetration; ROS generation; 10-100x lower MIC against biofilms |
| Phage-Antibiotic Synergy | Biofilm matrix, bacterial cell walls | Bacteriophages + conventional antibiotics | Up to 100,000x reduction in biofilm bacterial counts; phage-mediated matrix lysis |
Advanced and Emerging Therapeutic Modalities
Nanotechnology-Based Approaches
Nanoparticles offer unique advantages for biofilm treatment through their ability to penetrate the EPS matrix and target persistent cells [73] [74]. Silver nanoparticles generate reactive oxygen species (ROS) that cause membrane damage and protein denaturation, while zinc oxide nanoparticles exhibit similar ROS-mediated activity alongside zinc ion toxicity [73]. Graphene-based nanomaterials provide exceptional antimicrobial activity through physical disruption of membranes and oxidative stress mechanisms [73]. These nanomaterials can be engineered as delivery vehicles for conventional antibiotics, enhancing their penetration and retention within biofilms. Experimental studies demonstrate that nano-encapsulated antibiotics can achieve 10-100 times lower minimum inhibitory concentrations (MICs) against biofilm-grown bacteria compared to free drug equivalents [73].
Biological and Electrophysical Strategies
Phage-antibiotic synergy (PAS) represents a promising combinatorial approach wherein bacteriophages lyse biofilm structures and sensitize embedded bacteria, allowing enhanced antibiotic penetration and efficacy [73] [74]. However, challenges remain regarding phage resistance development and narrow host specificity [73].
Electrochemical disruption utilizes bioelectric effects to destabilize the extracellular polymeric matrix through non-chemical means [73]. Weak electrical currents (10-100 mA) enhance antibiotic efficacy against biofilms, potentially by increasing membrane permeability or inducing electrophoretic movement of antimicrobials through the matrix [73]. While promising, this approach requires optimization for tissue-specific applications.
CRISPR-based antimicrobials offer precision targeting of resistance genes or virulence factors in biofilm communities [73]. By specifically eliminating resistant or hypervirulent strains without affecting commensal bacteria, this approach could potentially clear persistent infections while minimizing collateral damage to beneficial microbiota. Significant barriers remain in delivery mechanisms and off-target effects [73].
Experimental Models and Research Methodologies
Persister Detection and Isolation Protocols
Accurate identification and quantification of persister cells requires specialized methodologies that distinguish between genetic resistance and phenotypic tolerance:
- Biphasic Killing Assays: The gold standard for persister detection involves exposure to bactericidal antibiotics (typically 5-10× MIC) with periodic plating for viability counts over 24-72 hours [1] [72]. Persistence is indicated by a biphasic kill curve with an initial rapid killing phase followed by a plateau where the persister subpopulation survives.
- Fluorescence-Activated Cell Sorting (FACS): Using fluorescent reporters for metabolic activity (e.g., ATP levels, membrane potential) or stress responses enables isolation of persister subpopulations for downstream analysis [13] [1]. For example, cells with low levels of Krebs cycle enzymes (GltA, Icd, SucA) can be sorted and demonstrate enriched persistence [13].
- Microfluidics Time-Lapse Microscopy: Advanced platforms like the "mother machine" enable single-cell tracking of growth and antibiotic survival, allowing direct observation of persister formation and resuscitation dynamics [13]. This approach has confirmed that E. coli cells with low ATP levels prior to antibiotic treatment have higher survival rates [13].
Biofilm Cultivation Models
Different biofilm models offer distinct advantages for studying persistence and evaluating anti-biofilm strategies:
- Static Microtiter Plate Assays: High-throughput screening for biofilm formation and antimicrobial susceptibility using crystal violet staining or metabolic dyes [70]. While convenient, these models may not fully recapitulate the complexity of in vivo biofilms.
- Flow Cell Systems: Continuous-flow models that support development of mature, architecturally complex biofilms with characteristic nutrient and oxygen gradients [70]. These systems enable real-time monitoring of biofilm development and treatment responses using confocal microscopy.
- Biofilm Reactors: Larger-scale systems (Calgary, CDC, drip-flow reactors) that generate substantial biofilm biomass for biochemical and molecular analyses [70]. These models better simulate environmental conditions in industrial and clinical settings.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Biofilm and Persister Research
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Matrix Degrading Enzymes | Dispersin B, DNase I, PelA, PslG | Biofilm disruption studies, EPS component analysis | Species-specific efficacy; concentration optimization required |
| Metabolic Reporters | iATPSnFr1.0, QUEEN, CTFR | Single-cell metabolic monitoring, persister identification | Requires genetic modification; potential cellular toxicity |
| Biphasic Killing Assay Components | High-concentration antibiotics (5-10× MIC) | Persister quantification, time-kill kinetics | Antibiotic stability; proper concentration verification critical |
| Quorum Sensing Inhibitors | Curcumin, AHL analogs, furanones | Virulence attenuation, biofilm prevention assays | Solubility challenges; species-specific signaling systems |
| Microfluidics Systems | Mother machine, flow cells, microfluidic chips | Single-cell persistence dynamics, resuscitation studies | Technical expertise required; equipment accessibility |
| Metabolic Reactivators | Sugars, mannitol, cis-2-decenoic acid | Persister awakening studies, antibiotic potentiation | Concentration-dependent effects; species-specific responses |
The intricate relationship between biofilm architecture and persister cell maintenance represents a critical challenge in combating chronic and recurrent bacterial infections. Effective therapeutic strategies must address both the physical barrier presented by the EPS matrix and the physiological state of dormancy that defines persister cells. The dual nature of persister formation—encompassing both stochastic and triggered mechanisms—demands multi-pronged approaches that can target pre-existing and induced persister populations.
Future research directions should prioritize the development of combination therapies that simultaneously disrupt biofilm integrity, target persistent cells, and prevent resistance development [73] [74]. The integration of nanoparticle delivery systems with conventional antibiotics and anti-biofilm agents holds particular promise for enhancing therapeutic efficacy against these recalcitrant populations [73]. Additionally, diagnostic advancements that rapidly identify persister-associated infections could enable more targeted treatment approaches, potentially incorporating metabolic reactivation strategies tailored to specific bacterial species and environmental conditions [1] [75].
Ultimately, defeating the biofilm-persister alliance will require therapeutic strategies as adaptable and multifaceted as the bacterial communities they target. By leveraging growing understanding of persistence mechanisms and their ecological context within biofilms, the scientific community can develop the innovative approaches needed to overcome these formidable bacterial survival strategies.
The phenomenon of persistence describes the ability of a small subpopulation of cells to survive exposure to high concentrations of antimicrobial or anticancer agents that are lethal to the bulk population, despite these persister cells lacking heritable genetic resistance mutations. First identified in bacteria by Gladys Hobby in 1942 and later termed "persisters" by Bigger, this state represents a sophisticated bet-hedging strategy that ensures population survival in unpredictable, hostile environments [77] [9]. In oncology, a functionally analogous population of drug-tolerant persister cells (DTPs) was identified in 2010 by Settleman and colleagues, who observed that approximately 0.3% of EGFR-mutant lung cancer cells could survive lethal doses of the EGFR inhibitor erlotinib [77]. These DTPs can survive for extended periods under therapeutic pressure and serve as a reservoir from which fully resistant populations can emerge.
The clinical significance of persisters is profound across both infectious disease and oncology. In bacterial infections, persisters contribute to the recalcitrance of chronic infections such as those in cystic fibrosis patients, medical device-associated infections, and Lyme disease [5]. In cancer, DTPs are responsible for minimal residual disease (MRD) that persists after initially successful therapy, ultimately leading to tumor relapse and acquired therapeutic resistance [77]. Understanding the mechanistic link between this transient, tolerant state and the emergence of permanent genetic resistance represents a critical frontier in overcoming treatment failure in both infectious diseases and oncology.
Table 1: Key Characteristics of Persister Cells Across Biological Systems
| Characteristic | Bacterial Persisters | Cancer DTPs |
|---|---|---|
| Frequency in Population | 10â»â¶ to 10â»Â³ [78] | ~0.3% to 5% [79] [77] |
| Genetic Basis | No resistance mutations [5] | No initial resistance mutations [79] |
| Growth State | Dormant or slow-cycling [5] | Slow-cycling or non-proliferative [77] |
| Reversibility | Regrowth after drug removal [9] | Reversible drug sensitivity [77] |
| Role in Resistance | Reservoir for resistance development [5] | Precursor to resistant clones [79] |
The Mechanistic Basis of Persister Formation: Stochastic Versus Triggered Pathways
The formation of persister cells occurs through two primary mechanisms: stochastic and triggered. These pathways represent distinct evolutionary strategies for surviving environmental stress, each with different implications for understanding and targeting the persister-resistance link.
Stochastic Persister Formation
Stochastic persister formation occurs randomly in a subset of cells within an isogenic population, even in the absence of external stressors. This bet-hedging strategy ensures that a subpopulation is pre-adapted to survive sudden environmental catastrophes, such as antibiotic exposure or targeted therapy. In bacteria, this stochastic switching is facilitated by fluctuations in key cellular components. For instance, in E. coli, heterogeneity in the expression of energy-generating Krebs cycle enzymes (GltA, Icd, SucA) results in subpopulations with low ATP levels that demonstrate enhanced survival against antibiotics like ciprofloxacin [13]. Single-cell ATP measurements using the iATPSnFr1.0 reporter have confirmed that cells with lower ATP are preferentially enriched among persisters [13]. Similarly, in cancer, single-cell barcoding experiments reveal lineage-specific variability in DTP growth rates and phenotypes across dynamic microenvironments, indicating stochastic elements in DTP emergence [77].
Triggered Persister Formation
Triggered persistence occurs in response to specific environmental cues or stresses, such as nutrient limitation, pH changes, or sublethal antibiotic exposure [5]. In bacterial systems, starvation represents a potent trigger for persistence. Recent single-cell RNA sequencing of E. coli growth transitions revealed that persisters from diverse genetic and physiological models converge to transcriptional states distinct from standard growth phases, exhibiting a dominant signature of translational deficiency [80]. Similarly, in cancer, therapeutic pressure creates selective forces that trigger adaptive responses in tumor cells, including profound epigenetic reprogramming and metabolic adaptations that characterize the DTP state [77].
Integrated Conceptual Framework
The relationship between stochastic and triggered persistence mechanisms, and their role in facilitating the emergence of genetic resistance, can be conceptualized as follows:
Experimental Evidence: From Persistence to Resistance
Bacterial Systems: Molecular Dissection of the Transition
Advanced single-cell technologies have enabled unprecedented resolution in studying the persister-resistance link in bacterial systems. Microfluidics time-lapse microscopy of over one million individual E. coli cells has revealed that persisters exhibit heterogeneous survival dynamics, including continuous growth with L-form-like morphologies, responsive growth arrest, or post-exposure filamentation [78] [15]. These observations challenge the classical dogma that persistence is exclusively mediated by pre-existing dormant cells and suggest that active physiological responses contribute to survival.
Genetic screens have been instrumental in identifying molecular players in persister formation. Ultra-dense CRISPR interference screening in E. coli determined how every gene contributes to persister formation across genetic models, identifying critical genes with large effects including lon (encoding a highly conserved protease) and yqgE (a poorly characterized gene that modulates the duration of post-starvation dormancy) [80]. These findings provide direct genetic evidence linking specific molecular pathways to the persistence phenotype.
Cancer Systems: Diverse Resistance Mechanisms from a Persister Bottleneck
Seminal research in EGFR-mutant non-small cell lung cancer (NSCLC) has provided compelling evidence that DTPs serve as a reservoir for diverse resistance mechanisms. When a clonal population of PC9 lung cancer cells was exposed to lethal doses of the EGFR inhibitor erlotinib, a small population of DTPs survived [79]. These DTPs were isolated and expanded over several months to establish persister-derived erlotinib-resistant colonies (PERCs). Comprehensive characterization of 17 PERCs revealed that they had acquired diverse, genetically distinct resistance mechanisms including MET amplification, MEK pathway activation, and mTOR signaling alterations - recapitulating the spectrum of resistance mechanisms observed clinically [79].
This demonstration that multiple resistance mechanisms can evolve from a genetically identical persister population indicates that the persister state does not constrain evolutionary trajectories but rather provides a permissive environment for the emergence of heterogeneous resistance solutions. The experimental workflow for establishing and characterizing these persister-derived resistant colonies is summarized below:
Table 2: Quantitative Experimental Data on Persister-Derived Resistance
| Experimental System | Initial Persistence Frequency | Time to Resistance Emergence | Diversity of Resistance Mechanisms Identified |
|---|---|---|---|
| EGFR-mutant NSCLC (PC9 cells) [79] | ~0.3% survival after erlotinib treatment | ~2 months for colony formation; 6-8 months for expansion | 17 distinct resistance profiles including MET amplification, MEK pathway activation, mTOR dependence |
| E. coli (metG* mutant) [80] | 30-60% survival in stationary phase | Survival measured within minutes to hours after antibiotic challenge | Distinct transcriptional state characterized by translational deficiency |
| E. coli (wildtype) [78] | 10â»â¶ to 10â»Â³ (typical range) | Heterogeneous dynamics observed in single cells over 5-10 hours | Multiple survival modes: L-form-like growth, filamentation, growth arrest |
Research Toolkit: Essential Methodologies and Reagents
The experimental investigation of persister cells and their role in resistance development requires specialized methodologies and reagents designed to characterize these rare subpopulations and track their evolution over time.
Table 3: Essential Research Reagents and Methodologies for Persister Studies
| Reagent/Methodology | Function/Application | Key Insights Enabled |
|---|---|---|
| Cellular Barcoding [81] [77] | Lineage tracing of persister cells and their progeny | Revealed clonal dynamics and heterogeneity in DTP outgrowth |
| Microfluidics Time-Lapse Microscopy [13] [78] | Single-cell observation of persistence dynamics | Identified heterogeneous survival strategies in bacterial persisters |
| Single-Cell RNA Sequencing (scRNA-seq) [80] | Transcriptional profiling of rare persister cells | Revealed convergent persister state marked by translational deficiency |
| CRISPR Interference Screening [80] | Genome-wide identification of persister genes | Identified lon and yqgE as critical regulators of persistence |
| iATPSnFr1.0 ATP Reporter [13] | Single-cell ATP measurement | Established correlation between low ATP and persistence |
| Membrane-Covered Microchamber Array (MCMA) [78] [15] | High-throughput single-cell analysis under controlled conditions | Enabled observation of >1 million individual E. coli cells |
Discussion and Clinical Implications
The emerging paradigm that persister cells serve as a reservoir for genetic resistance has transformative implications for therapeutic strategies in both infectious disease and oncology. Traditional models of resistance evolution have emphasized the selection of pre-existing genetic variants. However, the persister-resistance link reveals an alternative pathway: non-genetic, adaptive survival states can precede and facilitate the acquisition of diverse resistance mechanisms [79] [77]. This revised understanding suggests that effective therapeutic strategies must address not only genetically resistant clones but also the pre-resistant persister reservoir.
Several promising approaches are emerging to target persisters and prevent resistance evolution. In antibacterial research, these include direct killing strategies targeting growth-independent cellular structures like membranes, and indirect approaches that prevent persister formation or induce persister awakening to sensitize cells to conventional antibiotics [5]. In oncology, therapeutic strategies are being developed to target DTP vulnerabilities, including synthetic lethality approaches, adaptive dosing regimens informed by mathematical modeling, and immune-mediated eradication [77]. Additionally, interventions targeting the host ecosystem, such as lifestyle modifications to reduce pro-tumorigenic inflammation, may help suppress the outgrowth of persister-derived resistant clones [77].
A critical challenge in translating these insights to clinical practice is the extreme rarity and heterogeneity of persister cells, which necessitates sophisticated single-cell analytical approaches [78] [80]. Furthermore, the dynamic and reversible nature of the persister state complicates therapeutic targeting. Future research directions should focus on identifying conserved molecular vulnerabilities across persister cell states, developing clinical methods for monitoring persister populations during therapy, and designing combination treatments that simultaneously target both proliferating cells and persisters to prevent resistance evolution.
In conclusion, the persister-resistance link represents a fundamental biological paradigm with profound clinical implications. Understanding how tolerant cells serve as incubators for genetic diversity provides a more complete framework for explaining therapeutic failure and suggests novel strategic approaches for combating recalcitrant infections and cancer.
Mechanism vs. Phenomenon: A Critical Comparison of Stochastic and Triggered Pathways
Bacterial persister cells, a dormant subpopulation responsible for chronic and relapsing infections, form through distinct stochastic and triggered mechanisms organized in a complex hierarchy. This whitepaper delineates the * hierarchical relationships* between core molecular mechanisms—including toxin-antitoxin (TA) systems, (p)ppGpp-mediated stringent response, and transcriptional regulation—and their interdependent functions in persister formation. Within the context of stochastic versus triggered persistence research, we present quantitative comparisons of mechanism prevalence, efficacy, and interaction dynamics. Supported by current experimental data and visualized through pathway mapping, this analysis provides researchers and drug development professionals with a structured framework to identify critical intervention points for effective persister eradication strategies.
Bacterial persistence represents a significant challenge in clinical infection control, defined as the ability of a genetically susceptible subpopulation to survive lethal antibiotic exposure. This phenotypic heterogeneity is now understood to arise through two primary pathways: stochastic formation, which occurs spontaneously in a small subset of cells even during exponential growth under uniform conditions, and triggered formation, induced by environmental stressors such as nutrient limitation, antibiotic exposure, or host immune factors [78] [14]. The molecular machinery governing these pathways operates not in isolation but through a deeply interconnected regulatory network with both hierarchical and interdependent characteristics.
The hierarchy of mechanisms refers to the multi-level organization where upstream master regulators activate or suppress downstream effector systems. Simultaneously, interdependence manifests as cross-talk, feedback loops, and redundant pathways between systems, creating a robust network that maintains the persister state across diverse bacterial species including Escherichia coli, Mycobacterium tuberculosis, Staphylococcus aureus, and Acinetobacter baumannii [1] [14]. Understanding this complex architecture is paramount for developing effective anti-persister therapies, as targeting individual components often proves insufficient due to functional redundancy and network resilience.
This technical guide systematically deconstructs these relationships through quantitative comparison, experimental validation methodologies, and visual mapping of key pathways to equip researchers with comprehensive analytical frameworks for persister mechanism investigation.
Hierarchical Organization of Persister Formation Mechanisms
The formation of persister cells is governed by a clear hierarchical structure wherein master regulatory systems control downstream effector mechanisms that directly execute physiological changes leading to dormancy and antibiotic tolerance.
Master Regulatory Tier
At the apex of the persister formation hierarchy reside global stress signaling systems that integrate environmental cues and initiate coordinated survival responses:
(p)ppGpp Stringent Response: This guanosine-based alarmone serves as a primary trigger for persister formation during nutrient deprivation and other cellular stresses. (p)ppGpp acts as a global orchestrator by dramatically reprogramming cellular transcription, shutting down energy-intensive processes like DNA replication and protein synthesis, and activating stress response pathways [14]. Through direct interaction with RNA polymerase, (p)ppGpp redirects transcriptional resources away from growth-associated genes toward stress survival genes, establishing a foundation for the persister state.
RpoS Sigma Factor (σS): As the master regulator of the general stress response in many Gram-negative bacteria, RpoS controls the expression of hundreds of genes involved in stress resistance, including those implicated in persister formation [78]. RpoS levels increase dramatically during stationary phase and under various stress conditions, making this sigma factor a key hierarchical controller of triggered persistence. The RpoS regulon includes genes involved in DNA protection, oxidative stress resistance, and cell wall maintenance—all contributing to multidrug tolerance.
Intermediate Processor Tier
The middle hierarchical layer consists of systems that translate master regulatory signals into specific action programs:
Toxin-Antitoxin (TA) Systems: These ubiquitous genetic modules consist of a stable toxin that disrupts essential cellular processes and a labile antitoxin that neutralizes the toxin. Under stress conditions, activated master regulators either suppress antitoxin expression or promote their degradation, allowing free toxins to execute growth arrest [1] [14]. For example, in A. baumannii, multiple TA systems including HigBA, RelBE, and HicAB have been documented to contribute to persistence against different antibiotic classes [14]. These systems serve as critical processors in the hierarchy by translating upstream signals into direct action on cellular targets.
Transcriptional Regulators: Specialized regulators such as SoxRS, OxyR, and MarA respond to specific stressors and amplify the persister formation signal. These systems often operate in feedforward loops that reinforce the dormancy program initiated by master regulators [1].
Effector Execution Tier
The base of the hierarchy comprises the direct molecular executors that induce metabolic arrest and confer antibiotic tolerance:
Metabolic Enzyme Inhibition: Multiple toxins from TA systems directly target central metabolic pathways. For instance, toxins such as HipA phosphorylate glutamyl-tRNA synthetase, thereby inhibiting translation and causing growth arrest, while others target glucose metabolism or membrane energetics [1].
Macromolecule Synthesis Disruption: Effectors such as MazF cleave cellular mRNA in a sequence-specific manner, while others inhibit DNA replication or cell wall synthesis, effectively halting growth and rendering cells tolerant to antibiotics that target these processes [1].
Table 1: Hierarchical Tier Components in Bacterial Persister Formation
| Hierarchical Tier | Key Components | Primary Function | Representative Bacterial Species |
|---|---|---|---|
| Master Regulatory | (p)ppGpp, RpoS | Global stress signaling & transcriptional reprogramming | E. coli, S. aureus, A. baumannii |
| Intermediate Processor | TA systems, Transcriptional regulators | Signal transduction & growth arrest initiation | M. tuberculosis, E. coli, A. baumannii |
| Effector Execution | Metabolic inhibitors, mRNA interferases | Direct implementation of dormancy | All major pathogenic species |
This hierarchical organization creates a robust decision-making architecture that enables appropriate response to diverse environmental challenges while maintaining the potential for population heterogeneity through variation in component expression and activation thresholds.
Quantitative Comparison of Key Mechanisms
The relative contribution and operational characteristics of persister formation mechanisms vary significantly between stochastic and triggered pathways, across bacterial species, and in response to different antibiotic classes. The following quantitative analysis provides researchers with comparative data for evaluating mechanism dominance in specific experimental or clinical contexts.
Table 2: Quantitative Comparison of Persister Formation Mechanisms
| Mechanism | Persistence Frequency Range | Primary Induction Pathway | Key Antibiotic Challenges | Time to Persister State |
|---|---|---|---|---|
| TA Systems | 10â»Â³ - 10â»Â¹ | Both stochastic & triggered | Fluoroquinolones, β-lactams, Aminoglycosides | Minutes to hours |
| (p)ppGpp Stringent Response | 10â»Â² - 10â»Â¹ | Primarily triggered | Multiple classes, particularly those targeting cell wall | Minutes |
| RpoS-Mediated Stress Response | 10â»â´ - 10â»Â² | Primarily triggered | β-lactams, Oxidative stress-inducing antibiotics | Hours |
| Metabolic Shutdown | 10â»âµ - 10â»Â³ | Both stochastic & triggered | Aminoglycosides, Fluoroquinolones | Minutes to hours |
| SOS Response | 10â»â´ - 10â»Â² | Primarily triggered | DNA-damaging antibiotics (e.g., Fluoroquinolones) | Hours |
Mechanism Efficacy and Prevalence Analysis
Quantitative assessment of mechanism performance reveals critical patterns for experimental design and therapeutic targeting:
TA System Potency: Strains with upregulated TA systems can increase persister frequencies by 10 to 1000-fold compared to wild-type controls, with variation depending on specific TA systems and bacterial species [1] [14]. In A. baumannii, individual TA system knockouts typically reduce persistence by 30-70% for specific antibiotic classes, indicating both specialization and functional redundancy within the TA repertoire [14].
Stringent Response Dominance: Activation of the (p)ppGpp-mediated stringent response typically generates the highest persister frequencies among triggered mechanisms, often exceeding 1% of the total population in stationary phase cultures [1]. This makes it a particularly attractive target for combination therapies aimed at reducing persister burdens in chronic infections.
Stochastic Mechanism Baselines: Truly stochastic persister formation occurring independently of environmental cues typically maintains low but consistent baseline frequencies of 10â»â¶ to 10â»â´ in exponentially growing cultures, ensuring population survival even in the absence of external warning signals [78].
Temporal Dynamics of Mechanism Activation
The timing of mechanism activation follows distinct patterns critical for understanding persistence dynamics:
Rapid Responders: (p)ppGpp-mediated response and certain TA systems can induce persister states within minutes of stress detection, providing crucial survival advantages against rapidly cidal antibiotics [1].
Delayed Responders: RpoS-mediated and SOS response pathways typically require hours for full implementation, making them more relevant to chronic infection contexts where prolonged stress exposure occurs [78].
These quantitative differences underscore the need for time-resolved experimental approaches when investigating specific persister formation mechanisms and their contributions to treatment outcomes.
Interdependence Between Stochastic and Triggered Mechanisms
While the hierarchical organization of persister mechanisms provides a structural framework, the functional relationships between components create a complex web of interdependence that enables robust stress adaptation. These cross-mechanism interactions are particularly evident in the coordination between stochastic and triggered persistence pathways.
Cross-Activation and Amplification Loops
Multiple examples of positive feedback and reinforcement exist between ostensibly separate persister formation pathways:
TA System - Stringent Response Interdependence: In E. coli and A. baumannii, certain TA systems such as RelBE are directly transcriptionally activated by (p)ppGpp, creating an amplification circuit where the stringent response induces TA module expression, which in turn reinforces growth arrest [14]. Conversely, toxin activation from TA systems can stimulate (p)ppGpp synthesis through perturbation of cellular metabolism, establishing a bidirectional reinforcement loop.
Metabolic Regulation - Signaling Integration: Nutrient limitation triggers (p)ppGpp accumulation while simultaneously activating TA systems through decreased ATP levels that affect antitoxin stability. This convergent activation creates redundancy that ensures reliable persister formation despite environmental variability or partial mechanism inhibition [1].
Functional Specialization and Complementation
Different persister mechanisms demonstrate specialized roles within the broader network:
Kinetic Specialization: Triggered mechanisms typically generate higher frequencies of persisters (10â»Â² to 10â»Â¹) but require environmental cues, while stochastic mechanisms maintain baseline preparedness at lower frequencies (10â»â¶ to 10â»â´) independent of external signals [78]. This functional complementation ensures both responsive adaptation and constant protection.
Antibiotic-Class Specificity: Research in A. baumannii indicates that specific TA systems show preferential protection against different antibiotic classes. For example, the HigBA system contributes significantly to ceftazidime persistence, while other TA modules dominate in ciprofloxacin persistence [14]. This functional partitioning creates a division of labor within the persister formation network.
The following diagram illustrates the core hierarchical relationships and key interdependencies between major mechanisms in persister formation:
Diagram 1: Hierarchical and interdependent relationships in persister formation mechanisms
Network Resilience Implications
The interdependent nature of persister formation mechanisms creates significant challenges for therapeutic interventions:
Functional Redundancy: The knockout of individual TA systems or stress response pathways typically results in only partial reduction of persister frequencies, as complementary mechanisms compensate for the lost function [14]. This redundancy explains why single-target approaches often show limited efficacy in eradicating persister populations.
Adaptive Rewiring: Bacterial populations can dynamically adjust mechanism utilization in response to prolonged stress exposure or genetic disruption, demonstrating the plasticity of the persister formation network [63]. This adaptive capacity necessitates multi-target therapeutic strategies that simultaneously disrupt multiple nodes in the network.
Understanding these interdependent relationships is therefore essential for designing effective combination therapies that prevent compensatory adaptation and mechanism bypass in persistent infections.
Experimental Protocols for Mechanism Analysis
Robust experimental methodologies are essential for delineating hierarchical relationships and interdependencies between persister formation mechanisms. The following protocols represent current best practices for investigating specific aspects of persister biology.
Single-Cell Persistence Dynamics Tracking
Purpose: To correlate pre-treatment growth history and molecular expression patterns with post-antibiotic survival outcomes at single-cell resolution.
Procedure:
- Microfluidic Device Preparation: Utilize membrane-covered microchamber array (MCMA) devices with 0.8-μm deep microchambers to enable monolayer bacterial growth and high-resolution time-lapse imaging [78].
- Bacterial Strain Engineering: Incorporate fluorescent transcriptional reporters for key persistence markers (e.g., RpoS, TA systems, stress response genes) using stable, low-copy number plasmids or chromosomal integrations.
- Image Acquisition and Analysis:
- Capture phase-contrast and fluorescence images at 5-15 minute intervals throughout pre-treatment growth, antibiotic exposure, and recovery phases.
- Track individual cell lineages using automated cell tracking software (e.g., MicrobeJ, DeLTA) to quantify growth rates, division times, and fluorescence intensity dynamics.
- Correlate pre-antibiotic reporter expression with survival outcomes following antibiotic challenge (typically 5-10× MIC for 3-24 hours).
Applications: This protocol enabled the discovery that a substantial proportion of persister cells derive from actively growing subpopulations rather than exclusively from pre-existing dormant cells [78].
Mechanism Hierarchy Dissection Through Genetic Manipulation
Purpose: To establish causal relationships and hierarchical organization between putative persister formation mechanisms.
Procedure:
- Strain Construction:
- Create single-gene knockout mutants of putative master regulators (e.g., ∆relA, ∆rpoS) using allelic exchange or CRISPR-based methods.
- In regulator mutant backgrounds, construct fluorescent reporter fusions for downstream mechanisms (e.g., TA system promoters).
- Expression Profiling:
- Quantify reporter expression in wild-type and regulator mutant backgrounds under baseline and stress conditions using flow cytometry or fluorescence microscopy.
- Measure persister frequencies in single and double mutants to identify epistatic relationships.
- Interaction Validation:
- Use chromatin immunoprecipitation sequencing (ChIP-seq) for transcriptional regulators with known DNA-binding specificity.
- Employ bacterial two-hybrid systems or co-immunoprecipitation to validate protein-protein interactions in proposed complexes.
Applications: This approach has demonstrated the hierarchical control of multiple TA systems by (p)ppGpp and RpoS in various bacterial species [1] [14].
Network Interdependence Mapping
Purpose: To identify functional interactions and compensatory relationships between persister formation mechanisms.
Procedure:
- High-Throughput Genetic Interaction Screening:
- Construct a comprehensive library of single and double mutants covering major persister formation pathways.
- Subject mutant library to antibiotic challenge at concentrations that eliminate ≥99% of wild-type cells (typically 10-100× MIC for 3-24 hours).
- Quantify mutant enrichment/depletion through barcode sequencing.
- Transcriptomic Analysis of Adaptive Rewiring:
- Perform RNA sequencing on wild-type and key mutant strains at multiple time points following antibiotic exposure.
- Identify differentially expressed pathways in mutant backgrounds that suggest compensatory mechanism activation.
- Mathematical Modeling:
- Develop ordinary differential equation models incorporating proposed regulatory relationships.
- Iteratively refine models based on consistency with experimental mutant phenotype data.
Applications: This systematic approach has revealed extensive functional redundancy and adaptive network rewiring capabilities in persister formation pathways [63].
Research Reagent Solutions
The following reagents and tools represent essential resources for experimental investigation of hierarchical and interdependent relationships in persister formation mechanisms.
Table 3: Essential Research Reagents for Persister Mechanism Studies
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Fluorescent Reporter Systems | RpoS-mCherry fusions, TA system promoter-GFP constructs | Single-cell expression dynamics monitoring | Verify functional preservation; use low-copy plasmids to minimize burden |
| Microfluidic Devices | Membrane-covered microchamber arrays (MCMA), Mother machine devices | Single-cell lineage tracking & history correlation | Ensure proper nutrient/antibiotic diffusion; optimize chamber dimensions for species |
| Genetic Toolkits | CRISPR-interference systems, Allelic exchange vectors, TA system overexpression plasmids | Mechanism hierarchy dissection through controlled manipulation | Confirm minimal polar effects; use inducible systems for essential genes |
| Antibiotic Challenge Solutions | Ampicillin (100-200μg/mL), Ciprofloxacin (1-5μg/mL), Customized concentration series | Persistence frequency quantification & mechanism-specific profiling | Use fresh stocks; verify actual concentrations in specific media conditions |
| Metabolic Probes | Membrane potential dyes (DiOCâ‚‚(3)), ATP quantification kits, ROS detection probes (Hâ‚‚DCFDA) | Functional assessment of persistence effectors | Include proper controls for dye specificity; correlate with genetic markers |
Pathway Visualization of Core Mechanisms
The following integrated pathway diagram synthesizes hierarchical relationships and key interdependencies between major mechanisms in stochastic versus triggered persister formation, providing researchers with a comprehensive visual reference for experimental planning and interpretation.
Diagram 2: Integrated pathways of stochastic and triggered persister formation
The hierarchical organization and functional interdependence between bacterial persister formation mechanisms create a robust, adaptive system that ensures survival across diverse stress conditions. Master regulators like (p)ppGpp and RpoS occupy apex positions in the hierarchy, controlling downstream processors including TA systems and specialized transcriptional regulators that ultimately coordinate effector molecules executing growth arrest. Simultaneously, cross-mechanism interdependencies—including feedback amplification, functional redundancy, and adaptive rewiring—create network resilience that complicates therapeutic targeting.
This architectural understanding provides critical insights for future anti-persister drug development: effective strategies must simultaneously disrupt multiple nodes within this network, preferentially targeting master regulators and key interdependence points to prevent compensatory adaptation. Furthermore, the quantitative relationships and experimental frameworks presented here equip researchers with standardized approaches for mechanistic investigation across bacterial species and antibiotic classes, accelerating the development of more effective treatments for persistent infections.
Bacterial persistence, a transient phenotypic tolerance to antibiotics, presents a significant challenge in treating chronic infections. The HipA toxin within the HipBA toxin-antitoxin (TA) system plays a crucial role in this process. This whitepaper synthesizes current evidence demonstrating that triggered HipA activation is fundamentally dependent on stochastic fluctuations in TA module components. We examine the molecular mechanisms through which stochastic variation in HipA:HipB ratios leads to toxin activation, subsequently triggering the stringent response and inducing dormancy. By integrating quantitative data on HipA-mediated persistence across bacterial species and detailing experimental methodologies for investigating this phenomenon, this review provides researchers with a comprehensive framework for understanding the interplay between stochastic TA system activation and triggered persistence mechanisms. The insights gathered here have profound implications for developing novel therapeutic strategies against persistent bacterial infections.
Bacterial persisters are a transient, non-growing or slow-growing subpopulation of genetically susceptible cells that survive antibiotic treatment without acquiring genetic resistance [1]. These cells can resume growth once the antibiotic pressure is removed, often leading to recurrent infections. Persisters are increasingly recognized as a critical factor in chronic and biofilm-associated infections that are recalcitrant to conventional antibiotic therapies [82]. Among the various molecular mechanisms underlying persistence, toxin-antitoxin systems have emerged as key players, with the HipBA system being one of the most extensively studied.
The hipBA TA module was first identified in Escherichia coli through mutations (hipA7) that resulted in a dramatically increased frequency of persister cells [83]. This system consists of two components: the HipA toxin, a serine/threonine protein kinase that inhibits cell growth, and the HipB antitoxin, which neutralizes HipA by forming a stable complex with it and repressing the hipBA operon transcription [84]. The prevailing model suggests that persistence arises from stochastic fluctuations in cellular components that lead to temporary HipA activation, triggering a cascade of events that ultimately induce dormancy.
This review examines the evidence supporting the dependence of triggered HipA activation on stochastic TA module dynamics. We explore the molecular mechanisms, quantitative relationships, and experimental approaches that have shaped our current understanding of this phenomenon, with particular emphasis on its implications for antibiotic persistence and therapeutic development.
Molecular Mechanisms of HipA-Induced Persistence
The HipA Activation Cycle
HipA exists in an intricate balance with its antitoxin HipB, and the transition from neutralized to active toxin involves a carefully regulated cycle:
Complex Formation: Under normal conditions, HipA forms a stable complex with HipB, which also binds to the hipBA promoter operator region to autoregulate transcription.
Stochastic Liberation: Transient, stochastic reductions in HipB levels or competitive binding events can liberate HipA molecules.
Kinase Activation: Free HipA undergoes conformational changes that activate its serine/threonine kinase function.
Target Phosphorylation: Activated HipA phosphorylates cellular targets, primarily aminoacyl-tRNA synthetases.
Cellular Arrest: Target phosphorylation triggers downstream events leading to growth arrest and persistence.
Autophosphorylation and Inactivation: HipA eventually autophosphorylates at Ser150, located in its unusual P-loop motif, leading to kinase inactivation [85].
Resuscitation: With HipA inactivated, cellular processes gradually resume, allowing persister cells to resuscitate once antibiotic pressure is removed.
A key regulatory mechanism involves HipA's autophosphorylation at Ser150. Structural analyses reveal that Ser150 is buried within the hydrophobic core of the protein as part of the ATP-binding "P loop motif." Remarkably, this residue undergoes a conformational "in-out" equilibrium that allows for intermolecular autophosphorylation. When phosphorylated, Ser150 stabilizes an "out state" that disrupts the ATP-binding pocket, inactivating the kinase [85]. This autophosphorylation provides a critical mechanism allowing dormant cells to eventually revert to growth.
HipA Signaling Cascade and Stringent Response Activation
Table 1: Key Steps in HipA-Mediated Persistence Signaling Cascade
| Step | Key Event | Molecular Components | Cellular Outcome |
|---|---|---|---|
| 1 | Stochastic HipA release | HipA toxin, HipB antitoxin | Free HipA accumulates |
| 2 | Target recognition | HipA, GltX (tRNA synthetase) | Formation of HipA-GltX complex |
| 3 | Phosphorylation | HipA kinase domain, GltX Ser239 | GltX inactivation |
| 4 | tRNA accumulation | Uncharged tRNA^Glu^ | Ribosomal stalling |
| 5 | Stringent response activation | RelA, (p)ppGpp | Alarmone synthesis |
| 6 | Growth arrest | (p)ppGpp, RNA polymerase | Transcriptional reprogramming |
| 7 | Persistence entry | Multiple cellular targets | Dormancy, antibiotic tolerance |
| 8 | Resuscitation | HipA autophosphorylation | Return to growth |
HipA's primary mechanism for inducing persistence involves targeted phosphorylation of glutamyl-tRNA synthetase (GltX) at Ser239, which resides in its ATP-binding site [83]. This phosphorylation inhibits GltX activity, leading to accumulation of uncharged tRNA^Glu^ in the cell. The ribosome-associated protein RelA detects this accumulation and synthesizes the alarmone (p)ppGpp, which triggers the stringent response [83]. This response globally reprograms cellular transcription, downregulating energy-intensive processes such as DNA replication, protein synthesis, and cell division, thereby inducing a dormant state that is tolerant to antibiotics.
The following diagram illustrates this core signaling pathway:
Figure 1: HipA-Mediated Persistence Signaling Pathway. The pathway illustrates how stochastic HipA activation triggers a cascade leading to cellular dormancy through stringent response activation.
Recent studies in Caulobacter crescentus have revealed that HipA homologs can target different aminoacyl-tRNA synthetases. HipA1 and HipA2 phosphorylate GltX and TrpS (tryptophanyl-tRNA synthetase), respectively, and both contribute to antibiotic persistence during stationary phase [84]. This suggests evolutionary diversification of HipA targets while maintaining the core mechanism of persistence induction through tRNA synthetase inhibition and stringent response activation.
Stochastic vs. Triggered Activation Mechanisms
The formation of persister cells occurs through fundamentally different mechanisms that can be categorized as either stochastic or triggered:
Stochastic Persister Formation
Stochastic persistence results from random fluctuations in cellular components during normal growth, without requiring specific environmental cues:
- Fluctuations in TA Expression: Random variations in the transcription or translation of hipBA components can temporarily disrupt the precise HipA:HipB ratio.
- Energy Taxis: Heterogeneity in energy-generating components creates subpopulations with low ATP levels that are inherently more tolerant to antibiotics [13].
- Proteolytic Noise: Stochastic degradation of HipB antitoxin by cellular proteases can liberate HipA toxin molecules.
- Partitioning Asymmetry: Uneven distribution of TA complexes during cell division can create daughter cells with different persistence potentials.
Evidence for stochastic persistence comes from observations that persister frequency remains relatively constant during exponential growth and that sorted cells with low levels of energy-generating enzymes show enhanced survival after antibiotic challenge [13]. Single-cell analyses using microfluidics and ATP reporters have directly demonstrated that subpopulations with low ATP levels before antibiotic exposure are more likely to become persisters [13].
Triggered Persister Formation
Triggered persistence occurs in response to specific environmental stresses or signals:
- Antibiotic Exposure: Subinhibitory concentrations of certain antibiotics can activate stress responses that increase persister formation.
- Nutrient Limitation: Starvation conditions often upregulate TA systems and increase persistence frequency.
- DNA Damage: Activation of the SOS response through DNA-damaging agents induces TisB expression, which decreases proton motive force and ATP levels [82].
- Quorum Sensing Signals: Some bacterial communication molecules can modulate persistence frequency in population-density-dependent manners.
The Interdependence Model
Rather than existing as mutually exclusive mechanisms, stochastic and triggered persistence pathways intersect at multiple points, with the HipBA system serving as a key integration point:
Stochastic TA activation creates the necessary preconditions for triggered responses. Fluctuations in HipA:HipB ratios establish a primed state where additional environmental signals can push the system toward full persistence induction. Conversely, environmental stressors can amplify the stochastic fluctuations in TA systems, creating a positive feedback loop that enhances persistence frequency.
This interdependence is particularly evident in biofilms, where heterogeneous microenvironments create both stochastic and triggered persistence conditions simultaneously. The biofilm matrix generates nutrient gradients that naturally create subpopulations with different metabolic states, while simultaneously exposing cells to various stressors that trigger persistence responses [82].
Quantitative Evidence: Experimental Data and Cross-Species Conservation
Table 2: Quantitative Evidence of HipA-Mediated Persistence Across Bacterial Species
| Organism | HipA Variant | Primary Target | Persistence Increase | Stringent Response Dependence |
|---|---|---|---|---|
| Escherichia coli | HipA | GltX | 10,000-fold with HipA7 mutant [82] | Required [83] |
| Caulobacter crescentus | HipA1 | GltX | Significant in stationary phase [84] | Required for persistence |
| Caulobacter crescentus | HipA2 | TrpS | Significant in stationary phase [84] | Required for persistence |
| Caulobacter crescentus | HipA3 | Unknown | Moderate [84] | Not required for growth arrest |
The quantitative evidence supporting HipA's role in persistence demonstrates both conserved mechanisms and species-specific adaptations:
Escherichia coli Models
In E. coli, the HipA7 gain-of-function mutant (containing G22S and D291A substitutions) increases persistence frequency by up to 10,000-fold compared to wild-type strains [82]. This dramatic effect established HipA as a major persistence factor. Molecular studies have shown that HipA overexpression leads to phosphorylation of GltX at Ser239, with subsequent (p)ppGpp accumulation reaching detectable levels within 30 minutes of induction [83]. Overexpression of GltX completely abolishes both (p)ppGpp synthesis and persistence induced by HipA, confirming the central role of this target [83].
Cross-Species Conservation
Studies in Caulobacter crescentus reveal both conserved and divergent features of HipA-mediated persistence. The organism possesses three hipBA operons (hipBA1, hipBA2, and hipBA3), with HipA1 and HipA2 contributing significantly to antibiotic persistence during stationary phase by phosphorylating GltX and TrpS, respectively [84]. Notably, while all three HipA toxins require kinase activity for toxicity, they exhibit different effects on growth and macromolecular syntheses, and they phosphorylate distinct substrates. This functional specialization suggests evolutionary adaptation of HipA functions in different bacterial species.
The quantitative data also reveal important insights about redundancy in persistence mechanisms. In C. crescentus, deletion of all three hipBA operons reduces but does not eliminate persistence, indicating that multiple pathways can lead to antibiotic tolerance [84]. Similarly, while the stringent response regulator SpoT is required for HipA-mediated persistence, spoT mutants still form persister cells through alternative pathways.
Experimental Approaches and Methodologies
Key Research Reagents and Tools
Table 3: Essential Research Reagents for Studying HipA-Mediated Persistence
| Reagent/Tool | Type | Function/Application | Key Findings Enabled |
|---|---|---|---|
| iATPSnFr1.0 ATP reporter | Fluorescent biosensor | Single-cell ATP measurement using ratio-metric imaging | Correlation between low ATP and persistence [13] |
| Mother machine microfluidics | Microfluidic device | Single-cell time-lapse microscopy under controlled conditions | Direct observation of persister formation and resuscitation [13] |
| Phos-tag mobility shift assays | Biochemical assay | Detection of protein phosphorylation | Confirmation of HipA autophosphorylation [84] |
| QUEEN derivatives | ATP biosensor | ATP concentration measurement | Validation of low-ATP persister subpopulation [13] |
| pTet-hipA-mcherry | Inducible expression vector | Controlled HipA expression with fluorescent tag | Threshold mechanism of HipA toxicity [83] |
| GltX overexpression plasmids | Complementation vector | Suppression of HipA toxicity | Identification of GltX as key HipA target [83] |
Critical Experimental Protocols
Single-Cell Persistence Tracking
The mother machine microfluidics platform has revolutionized persister research by enabling direct observation of persistence events at single-cell resolution:
- Device Preparation: Fabricate microfluidic devices with multiple parallel growth channels connected to a main media channel.
- Cell Loading: Load bacterial cells expressing fluorescent reporters (e.g., iATPSnFr1.0 for ATP) into the growth channels.
- Baseline Monitoring: Flow fresh medium through the device while imaging cell growth and fluorescence at regular intervals (e.g., every 10-20 minutes).
- Antibiotic Challenge: Introduce medium containing lethal antibiotic concentrations (e.g., ampicillin at 10-100× MIC).
- Viability Assessment: Continue imaging to identify non-lysed, non-growing cells that survive antibiotic exposure.
- Resuscitation Monitoring: Replace antibiotic medium with fresh growth medium to monitor regrowth of surviving persister cells.
This approach directly demonstrated that a subpopulation of cells with low ATP levels before antibiotic treatment has higher survival rates, supporting the energy depletion mechanism of persistence [13].
HipA-Target Interaction Mapping
Identifying HipA phosphorylation targets requires a combination of genetic, biochemical, and proteomic approaches:
- Suppressor Screening: Transform hipA7 mutant cells with genomic libraries and select for clones that overcome HipA-mediated cold sensitivity [83].
- Candidate Validation: Clone identified suppressor genes (e.g., gltX) and test their ability to reverse HipA toxicity when overexpressed.
- In Vivo Phosphorylation Assays: Culture cells co-expressing HipA and candidate targets in the presence of 32P-orthophosphate, followed by autoradiography of purified proteins.
- Mass Spectrometry Analysis: Identify specific phosphorylation sites using MS/MS fragmentation of tryptic peptides from proteins purified from HipA-expressing cells.
- Functional Assays: Test the impact of phosphorylation site mutations (e.g., Ser239Ala in GltX) on target function and HipA-mediated persistence.
This multi-pronged approach successfully identified GltX as a key HipA target and Ser239 as the critical phosphorylation site that inactivates the tRNA synthetase [83].
The following diagram illustrates a typical experimental workflow for investigating HipA-mediated persistence:
Figure 2: Experimental Workflow for HipA Persistence Research. The diagram outlines key methodological steps for investigating HipA-mediated persistence mechanisms.
Implications for Therapeutic Development and Future Research
The mechanistic understanding of HipA activation has important implications for combating persistent infections:
Anti-Persister Therapeutic Strategies
- HipA Kinase Inhibitors: Small molecules targeting HipA's substrate-binding sites or ATP-binding pocket could prevent toxin activation and reduce persistence.
- GltX Phosphorylation-Resistant Mutants: Gene therapy approaches introducing phosphorylation-resistant GltX variants could sensitize bacterial populations to antibiotics.
- Stringent Response Modulators: Compounds that inhibit (p)ppGpp synthesis or signaling could disrupt the downstream persistence pathway.
- TA System Crosstalk Exploiters: Molecules that promiscuously activate multiple TA systems could induce self-poisoning of bacterial populations.
Research Directions
Future research should prioritize:
- Structural Studies: Elucidating full-length HipA-HipB complex structures to identify druggable interfaces.
- Single-Cell Dynamics: Quantitative analysis of HipA fluctuation patterns and thresholds in individual cells.
- Host-Pathogen Interactions: Understanding how host environments influence HipA activation in infection contexts.
- Combination Therapies: Developing antibiotic-adjuvant combinations that specifically target the HipA persistence pathway.
The evidence comprehensively supports that triggered HipA activation depends fundamentally on stochastic TA module activation. Stochastic fluctuations in HipA:HipB ratios create the priming conditions that allow environmental triggers to induce persistence through a conserved pathway involving tRNA synthetase phosphorylation, stringent response activation, and growth arrest. This mechanistic understanding bridges the historical dichotomy between stochastic and triggered persistence models, revealing an integrated system where random molecular fluctuations and environmental sensing cooperate to generate phenotypic diversity. Future therapeutic strategies that target this HipA-dependent persistence pathway hold significant promise for addressing the challenging problem of chronic and recurrent bacterial infections.
This technical guide provides a comprehensive analysis of two fundamental mechanisms governing bacterial persister cell formation: stochastic ATP fluctuations and triggered energy shutdown. Persisters, a subpopulation of genetically susceptible but phenotypically dormant bacterial cells, play a critical role in chronic infections and antibiotic treatment failure. Through systematic evaluation of quantitative data, experimental protocols, and molecular pathways, this review delineates the distinct characteristics, regulatory mechanisms, and methodological approaches for investigating these divergent energy modulation strategies. The presented framework aims to equip researchers with the necessary tools to advance both fundamental understanding and therapeutic development targeting persistent bacterial infections.
Bacterial persisters represent a non-growing or slow-growing subpopulation of genetically drug-susceptible cells that survive exposure to antimicrobial stress and can regrow once the stress is removed [1]. These cells underlie the challenge of treating chronic and recurrent infections, contributing significantly to treatment failure across numerous pathogenic species. The formation of persister cells is now understood to occur through distinct mechanistic pathways that ultimately converge on a reduced cellular energy state, though the initiation events differ substantially [13] [1].
The "energy-state analysis" framework presented in this review contrasts two principal mechanisms: stochastic ATP fluctuations, which arise from inherent biochemical variability in bacterial populations and create spontaneous low-energy subpopulations; and triggered energy shutdown, which occurs through activation of specific stress response pathways that actively depress cellular energy production. Understanding the distinction between these pathways is not merely academic; it carries significant implications for therapeutic development, as interventions targeting stochastic formation may differ substantially from those addressing regulated stress responses.
Stochastic ATP Fluctuations: Mechanism and Analysis
Fundamental Principles and Molecular Basis
The stochastic formation model posits that persisters emerge spontaneously in bacterial populations due to random fluctuations in the expression of energy-generating components, leading to a subpopulation with inherently reduced ATP levels [13]. This phenomenon represents a bet-hedging strategy where phenotypic heterogeneity enhances population survival in unpredictably fluctuating environments. Unlike genetic mutations, these variations are transient and reversible, allowing descendants to revert to the normal metabolic state once conditions improve.
Key studies in Escherichia coli have demonstrated that stochastic heterogeneity in the abundance of Krebs cycle enzymes directly correlates with persister formation [13]. Cells with diminished expression of citrate synthase (GltA), isocitrate dehydrogenase (Icd), and α-ketoglutarate dehydrogenase (SucA) showed significantly higher survival rates when challenged with ciprofloxacin, while fluctuations in non-energy-producing enzymes like isocitrate lyase (AceA) showed no such relationship. This establishes a direct link between variability in core energy metabolism and the stochastic emergence of the persister phenotype.
Quantitative Single-Cell Analysis
Direct measurement of ATP dynamics at single-cell resolution has provided compelling evidence for the stochastic fluctuation model. Utilizing the genetically encoded ATP sensor iATPSnFr1.0, researchers have quantified cell-to-cell ATP heterogeneity and confirmed that cells with lower ATP levels prior to antibiotic exposure exhibit enhanced survival [13].
Table 1: Quantitative Parameters of Stochastic ATP Fluctuations in E. coli
| Parameter | Measurement Value | Experimental Context | Technical Method |
|---|---|---|---|
| ATP Concentration Range | 1.54 ± 1.22 mM | Normal exponential growth | Bulk luciferase assay [86] |
| ATP Distribution | Spread over half an order of magnitude | Genetically identical cells | Single-cell analysis [86] |
| Persister Enrichment in Low-Enzyme Cells | 3-5 fold higher survival | GltA, Icd, SucA dim populations | FACS sorting & viability assessment [13] |
| Arsenate-induced ATP Depletion | Dose-dependent decrease in 488ex/405ex ratio | 0-100 mM arsenate exposure | iATPSnFr1.0 ratiometric measurement [13] |
| Correlation Coefficient | Strong correlation between single-cell and bulk ATP measurements | Validation of iATPSnFr1.0 sensor | Comparative analysis [13] |
Experimental Protocol for Single-Cell ATP Monitoring
Objective: To quantify ATP heterogeneity in a bacterial population and identify low-ATP subpopulations with enhanced antibiotic tolerance.
Materials:
- E. coli strain expressing chromosomally encoded iATPSnFr1.0 ATP sensor
- Microfluidics device (mother machine design)
- Leibovitz's L-15 medium without phenol red
- Confocal microscope with environmental chamber
- Image analysis software (Fiji/TrackMate)
- Antibiotics of interest (e.g., ampicillin, ciprofloxacin)
Procedure:
- Culture the reporter strain to exponential phase (OD600 ≈ 0.3-0.5) in appropriate medium.
- Load bacterial culture into microfluidics device and allow for acclimation (1-2 hours).
- Perfuse with fresh medium to initiate growth and ATP production monitoring.
- Establish baseline ATP levels by recording iATPSnFr1.0 fluorescence (excitation 405 nm and 488 nm, emission 515 nm) for 60-90 minutes.
- Introduce medium containing lethal antibiotic concentrations while continuing time-lapse imaging.
- Track individual cell lineages throughout exposure period (typically 5-8 hours).
- Correlate pre-exposure ATP levels with subsequent survival outcomes.
- Calculate survival rates for cells binned according to initial ATP concentrations.
Data Analysis:
- Compute 488ex/405ex ratio for each cell at each timepoint, representing ATP concentration
- Normalize ratios to population median for cross-experiment comparisons
- Determine time of cell death through cessation of cytoplasmic streaming and loss of membrane integrity
- Calculate hazard ratios for death based on initial ATP quartiles
Triggered Energy Shutdown: Mechanism and Analysis
Regulated Stress Response Pathways
In contrast to stochastic fluctuations, triggered energy shutdown involves the activation of specific molecular pathways in response to environmental stress, leading to a coordinated depression of cellular energy production. This mechanism represents a more directed response where external cues activate programmed dormancy programs through defined signaling cascades [1].
Several well-characterized pathways mediate triggered persistence:
- Toxin-Antitoxin (TA) Systems: Activation of toxins such as HipA (a glutamyl-tRNA kinase), TisB (a membrane pore-forming peptide), and HokB through transcriptional or post-translational regulation leads to targeted inhibition of energy-consuming processes [1].
- Stringent Response: Nutrient limitation triggers RelA-mediated synthesis of (p)ppGpp, which redirects cellular resources away from growth and division.
- Oxidative Stress Response: Reactive oxygen species generated during antibiotic treatment can damage respiratory components, reducing ATP synthesis capacity.
These pathways frequently converge on the reduction of proton motive force (PMF) and ATP depletion, creating a state of metabolic quiescence that protects against antibiotics requiring active metabolism [1].
Quantitative Pathway Activation Metrics
The activation of triggered shutdown pathways follows distinct kinetic and concentration thresholds that differentiate them from stochastic fluctuations. These responses typically exhibit dose-dependence, temporal coordination, and molecular specificity.
Table 2: Quantitative Parameters of Triggered Energy Shutdown Pathways
| Pathway | Activation Signal | Time to Persistence | Key Molecular Players | ATP Reduction |
|---|---|---|---|---|
| Toxin-Antitoxin (HipA) | Nutritional stress, Antibiotics | 1-2 generations | HipA, GltX, (p)ppGpp | 40-60% [1] |
| Toxin-Antitoxin (TisB) | DNA damage, SOS response | <1 generation | TisB, LexA, RecA | 50-70% [1] |
| Stringent Response | Amino acid starvation | Minutes | RelA, SpoT, (p)ppGpp | 30-50% [1] |
| Oxidative Stress | ROS from antibiotics | Variable | SoxRS, OxyR | 40-60% [1] |
Experimental Protocol for Triggered Pathway Analysis
Objective: To induce and measure triggered persistence through activation of specific stress response pathways.
Materials:
- Wild-type and pathway-specific mutant strains (e.g., ΔhipA, ΔtisB, ΔrelA)
- Stress-inducing agents (e.g., mitomycin C for SOS response, serine hydroxamate for stringent response)
- ATP detection reagents (luciferase-based assay or fluorescent sensors)
- Flow cytometry equipment for population analysis
- RNA extraction kit and qPCR reagents for pathway activation confirmation
Procedure:
- Grow bacterial cultures to mid-exponential phase in appropriate medium.
- Divide culture into aliquots for various induction conditions and controls.
- Apply pathway-specific inducers at predetermined concentrations:
- SOS response: 0.5-1 µg/mL mitomycin C
- Stringent response: 0.5 mM serine hydroxamate
- Oxidative stress: 2-5 mM H2O2
- Monitor pathway activation through transcriptional reporters (GFP fusions) or qPCR of key markers at 30-minute intervals.
- Measure ATP levels at regular intervals using luciferase assay or ratiometric sensors.
- At predetermined timepoints post-induction, challenge cultures with lethal antibiotic doses.
- Assess viability through plating and colony counting or live/dead staining with flow cytometry.
- Compare persister frequencies between induced and non-induced cultures.
Data Analysis:
- Establish correlation between pathway activation kinetics and persister formation
- Determine threshold levels of pathway induction required for significant persistence
- Compare ATP depletion rates across different induction methods
- Validate pathway specificity using mutant strains
Comparative Analysis: Stochastic vs. Triggered Mechanisms
Understanding the distinctions between stochastic and triggered persister formation requires systematic comparison across multiple parameters. The following analysis highlights key differentiating features that inform both fundamental biology and therapeutic development.
Table 3: Comparative Analysis of Stochastic and Triggered Persister Formation Mechanisms
| Characteristic | Stochastic Fluctuations | Triggered Shutdown |
|---|---|---|
| Initiation Mechanism | Random variation in enzyme expression | Directed response to environmental cues |
| Kinetics | Constitutive, population-wide heterogeneity | Inducible, synchronized response |
| Energy Depletion Pattern | Pre-existing low ATP subpopulation | Active ATP reduction post-stimulus |
| Key Molecular Components | Krebs cycle enzymes, basal transcription noise | TA systems, (p)ppGpp, stress response regulators |
| Population Structure | Continuum of energy states | Bimodal distribution post-induction |
| Reversibility | Stochastic switching | Often coupled to signal disappearance |
| Therapeutic Targeting | Challenging due to randomness | More predictable pharmacological intervention |
Research Reagent Solutions and Technical Tools
Advancing research in energy-state analysis requires specialized reagents and methodologies tailored to dissecting the nuanced mechanisms of persister formation. The following toolkit compiles essential resources for experimental investigation.
Table 4: Research Reagent Solutions for Energy-State Analysis
| Reagent/Tool | Application | Key Features | Example Use |
|---|---|---|---|
| iATPSnFr1.0 ATP Sensor | Single-cell ATP monitoring | Ratiometric (488ex/405ex), chromosome-integratable | Quantifying ATP heterogeneity in microfluidics [13] |
| QUEEN ATP Sensor | Bulk ATP measurement | Earlier generation sensor, lower toxicity | Comparative ATP measurements [13] |
| Microfluidics Platforms | Single-cell dynamics | Mother machine designs, high-temporal resolution | Tracking ATP and survival in lineages [13] |
| FACS Sorting | Population segregation | Based on enzyme expression or sensor fluorescence | Isolating low-enzyme subpopulations [13] |
| Luciferase ATP Assay | Bulk ATP quantification | High sensitivity, commercial kits available | Validating sensor measurements [13] |
| Pathway-Specific Reporters | Stress response monitoring | GFP fusions to promoter elements | Quantifying pathway activation [1] |
| JC-1 Mitochondrial Dye | Membrane potential assessment | Ratiometric (aggregates/monomers), red/green fluorescence | Measuring mitochondrial function [87] |
| PercevalHR ATP/ADP Sensor | Energy charge measurement | ATP/ADP ratio, pH-correctable | Cell cycle energy oscillations [87] |
Methodological Integration for Differentiated Analysis
To conclusively distinguish between stochastic and triggered persistence mechanisms within experimental systems, researchers should implement an integrated methodological approach:
Concurrent Monitoring of ATP Dynamics and Stress Pathways: Utilize dual-reporter systems combining iATPSnFr1.0 for ATP quantification with transcriptional reporters for key stress pathways (e.g., SOS response via recA::gfp). This enables direct correlation of ATP fluctuations with pathway activation states at single-cell resolution.
Temporal Mapping of Persistence Emergence: Establish precise timelines of persister formation relative to stress exposure. Stochastic persisters pre-exist as low-ATP subpopulations, while triggered persisters emerge following a distinct lag period post-stress application.
Genetic Dissection through Mutant Analysis: Compare persister frequencies in wild-type versus specific pathway mutants (ΔhipA, ΔtisB, ΔrelA) under identical conditions. Stochastic persistence should remain relatively unchanged in these mutants, while triggered mechanisms will be significantly attenuated.
Single-Clineage Tracking in Controlled Environments: Employ microfluidics platforms to monitor ATP dynamics and stress response activation across multiple generations, establishing causal relationships between ancestral energy states and descendant survival outcomes.
This integrated approach enables researchers to deconvolute the complex interplay between stochastic predisposition and inducible responses that collectively determine bacterial survival under antibiotic pressure.
The energy-state analysis framework presented in this technical guide provides a structured approach for differentiating between stochastic ATP fluctuations and triggered energy shutdown in bacterial persister formation. Through implementation of the described experimental protocols, analytical methods, and reagent solutions, researchers can advance both fundamental understanding and therapeutic targeting of these phenotypically tolerant subpopulations. The distinction between these mechanisms carries significant implications for antimicrobial development, as compounds targeting triggered response pathways may prove ineffective against stochastically formed persisters, and vice versa. Future research should focus on further elucidating the intersection points between these pathways and developing therapeutic strategies that address both mechanisms simultaneously for more effective eradication of persistent bacterial infections.
Bacterial persisters are dormant or slow-growing phenotypic variants within an isogenic population that survive exposure to lethal concentrations of antibiotics and can regrow after treatment cessation [1]. First identified by Joseph Bigger in 1944 when he observed that penicillin failed to completely sterilize Staphylococcus cultures, these phenotypic variants are genetically identical to their susceptible counterparts but exhibit transient antibiotic tolerance [88] [1]. The existence of persisters presents an evolutionary paradox: why would populations maintain subpopulations of dormant cells that do not contribute to immediate growth? The resolution lies in understanding the ecological and evolutionary forces that have shaped these bet-hedging strategies, which are now recognized as a major contributor to chronic and recurrent infections across diverse bacterial pathogens [1] [35].
This review examines persister formation through an integrative biological lens, focusing on the fitness trade-offs between two primary formation mechanisms: stochastic switching, which occurs spontaneously without external triggers, and triggered formation, induced in response to environmental stresses [1] [5]. We analyze the ecological contexts favoring each strategy and their implications for therapeutic interventions, providing a framework for understanding how these mechanisms coexist and contribute to bacterial survival in fluctuating environments.
Evolutionary Frameworks for Persistence
The Fundamental Growth-Stress Trade-off
The evolution of persistence strategies is constrained by a fundamental physiological trade-off: limited biosynthetic resources must be allocated between growth-promoting and stress-responsive cellular machinery [88]. A bacterial cell cannot simultaneously maximize both growth rate and stress tolerance because transcriptional and translational resources invested in one come at the expense of the other [88]. This trade-off emerges from competition for finite RNA polymerase (RNAP) pools between sigma factors directing transcriptional programs toward growth (e.g., σâ·â°) or stress response (e.g., σS, σH, σE) [88].
Natural selection has favored the evolution of phenotypically heterogeneous populations where persister cells specialize in stress survival while their genetically identical siblings pursue rapid growth [88]. This division of labor enhances population fitness in fluctuating environments by ensuring that some cells survive catastrophic stresses, functioning as an "insurance policy" against extinction [88]. The persister subpopulation represents a bet-hedging strategy that sacrifices short-term reproductive success for long-term survival in unpredictable environments where stress events occur sporadically [88] [35].
Fitness Landscapes of Formation Strategies
The relative fitness of stochastic versus triggered persistence mechanisms depends critically on environmental predictability and the frequency of stress events:
Stochastic persistence evolves in environments where stress events occur unpredictably and rarely. The constant maintenance of a small persister subpopulation ensures survival when stresses arrive without warning, but carries a continuous fitness cost due to reduced population growth rate [35].
Triggered persistence is favored in environments where stress events are predictable or preceded by reliable cues. This inducible strategy allows populations to maintain maximum growth rates until threats are detected, then rapidly adjust persister frequencies in response [1].
In natural environments, both mechanisms frequently coexist, providing complementary survival advantages across different temporal scales and stress types [1].
Quantitative Comparison of Persistence Strategies
Table 1: Ecological Characteristics and Fitness Trade-offs of Persister Formation Strategies
| Characteristic | Stochastic Persistence | Triggered Persistence |
|---|---|---|
| Formation trigger | Spontaneous, internal molecular noise | Environmental signals (e.g., nutrient limitation, antibiotic threat, pH change) |
| Energy cost | Constant, regardless of environment | Induced only when needed |
| Response time | Immediate (pre-existing) | Delayed (requires gene expression) |
| Environmental predictability | Favored in unpredictable environments | Favored when reliable cues precede stress |
| Information requirement | None | Dependent on sensing fidelity |
| Metabolic state heterogeneity | Type II (slow-growing) persisters [1] | Type I (non-growing) persisters [1] |
| Key molecular mediators | Stochastic toxin-antitoxin system activation [89] | ppGpp, sigma factor competition, TA systems [88] |
Table 2: Quantitative Analysis of Persister Fractions Across Bacterial Species and Antibiotics
| Bacterial Species | Antibiotic | Persister Fraction (%) | Formation Context |
|---|---|---|---|
| Escherichia coli | Ampicillin | 0.01-1% [58] | Strain and condition-dependent |
| Escherichia coli | Ciprofloxacin | 0.001-0.1% [58] | Varies independently from ampicillin persistence |
| Acinetobacter baumannii | Multiple | ~0.01% (lowest) [90] | Species with lowest persistence |
| Enterococcus faecium | Multiple | Up to 100% [90] | Species with highest persistence |
| Staphylococcus aureus | Multiple | ~5% (MRSA) [90] | Multidrug-resistant strains |
| Antibiotic Class | Example | Typical Persister Fraction | Proposed Mechanism |
| Membrane-targeting | Colistin | 0.001% (lowest) [90] | Direct membrane damage less dependent on metabolism |
| Protein synthesis inhibitors | Erythromycin | Up to 63% [90] | Require active translation for efficacy |
| Antimetabolites | Trimethoprim | High persistence [90] | Interfere with metabolic pathways |
Molecular Mechanisms and Physiological Implementation
Regulatory Networks Governing Persistence
The formation of persister cells is regulated by complex molecular networks that integrate internal physiological states with environmental information:
Molecular Pathways of Persister Formation
The ppGpp-mediated stringent response serves as a master regulator connecting nutrient availability to persistence [88] [89]. During amino acid starvation, RelA synthesizes ppGpp, which dramatically alters transcriptional priorities by binding to RNA polymerase and redirecting it from growth-related genes to stress response and amino acid biosynthesis genes [88]. This resource allocation shift simultaneously reduces growth rate and enhances stress tolerance, creating a physiological state primed for persistence [88].
Toxin-antitoxin (TA) systems function as molecular switches in both stochastic and triggered persistence [89]. In E. coli, ten type II TA systems have been identified whose toxins act as mRNA endonucleases that globally inhibit translation when activated [89]. The HipBA system exemplifies this mechanism: when HipA levels exceed HipB, it phosphorylates GltX, leading to uncharged tRNA accumulation that activates RelA and ppGpp synthesis [89]. Stochastic fluctuations in TA system components can spontaneously trigger dormancy, while environmental stresses can systematically activate TA systems through (p)ppGpp-induced antitoxin degradation [89].
Physiological Implementation of Dormancy
The transition to a persistent state involves coordinated metabolic shutdown across multiple cellular processes:
Translational arrest: Ribosome modulation factor (RMF) and hibernation promotion factor (HPF) convert active 70S ribosomes to inactive 100S dimers, while RaiA directly inactivates 70S ribosomes [89].
ATP reduction: Persister cells exhibit significantly reduced ATP levels, limiting energy-dependent processes targeted by many antibiotics [88] [89].
Metabolic rewiring: Central metabolic pathways are downregulated, with a shift toward maintenance energy production rather than biosynthesis [1].
This physiological reconfiguration creates a state where antibiotic targets are largely inactive, rendering conventional antibiotics ineffective despite genetic susceptibility [1].
Experimental Methodologies for Persistence Research
Standardized Persistence Assays
Table 3: Essential Research Reagents and Methodologies for Persistence Studies
| Reagent/Method | Application | Key Utility in Persistence Research |
|---|---|---|
| Time-kill assays | Quantifying persister fractions | Gold standard for measuring survival after antibiotic exposure [58] [90] |
| Prokaryotic scRNA-seq (PETRI-seq) | Single-cell transcriptional profiling | Identifying rare persister populations and their transcriptional states [80] |
| CRISPRi screening | Functional genomics | Systematically identifying genes essential for persister formation [80] |
| Fluorescent reporter systems | Tracking dormancy and resuscitation | Visualizing metabolic activity and regrowth at single-cell level [35] |
| Type II TA system mutants | Mechanistic studies | Dissecting contribution of specific toxin-antitoxin modules [89] |
Reliable quantification of persister cells requires carefully controlled methodologies. The time-kill assay remains the standard approach, where bacterial populations are exposed to lethal antibiotic concentrations (typically 10-100× MIC) and survivors are quantified over time [90]. This produces characteristic biphasic killing curves where the majority population dies rapidly while a small subpopulation survives extended treatment [58]. For consistent quantification across studies, mathematical models accounting for switching rates between normal and persister states can be applied [58].
Advanced single-cell technologies have revolutionized persister research by enabling direct characterization of these rare cells. Prokaryotic single-cell RNA sequencing (PETRI-seq) has revealed that persisters from diverse genetic backgrounds converge to a common transcriptional state characterized by translational deficiency and distinct from standard growth phases [80]. High-throughput CRISPR interference (CRISPRi) screens allow systematic determination of how every gene in a genome contributes to persister formation, identifying critical effectors like the Lon protease and previously uncharacterized genes such as yqgE [80].
Analytical Frameworks and Mathematical Modeling
Mathematical models provide a formal framework for interpreting persistence dynamics. The standard two-state model describes populations where cells switch between normal (N) and persister (P) states with transition rates α (N→P) and β (P→N) [58]. During antibiotic treatment, normal cells die at rate μ while persisters are protected [58]. This framework allows estimation of fundamental parameters from experimental data:
- Persister fraction = α / (α + β) at equilibrium
- Switching rates from lineage tracking and resuscitation assays
More sophisticated models incorporate multiple persister states with varying dormancy depths, environmental modulation of switching rates, and population structure effects [1].
Ecological Implications and Therapeutic Applications
Ecological Success of Persistence Strategies
The evolutionary success of persistence strategies is evident in their near-universal distribution across bacterial taxa [88] [90]. Quantitative surveys reveal that all examined bacterial species exhibit some capacity for persistence, though the fractions vary dramatically across species and growth conditions [90]. This conservation suggests that maintaining phenotypic heterogeneity provides fundamental fitness advantages despite the apparent costs.
In clinical settings, persisters contribute significantly to recalcitrant infections including tuberculosis, recurrent urinary tract infections, and biofilm-associated device infections [1]. The presence of persister cells explains why some infections require prolonged antibiotic therapy despite apparent susceptibility in standard assays [1]. Critically, persister cells not only survive treatment but also facilitate the evolution of genetic resistance by providing a surviving population in which resistance mutations can emerge [88] [1].
Targeting Persistence Therapeutically
Understanding the ecological and evolutionary drivers of persistence informs therapeutic strategies aimed at overcoming tolerance:
Membrane-active compounds: Agents that directly damage membranes (e.g., colistin, daptomycin) show enhanced efficacy against persisters, likely because their mechanism doesn't require metabolic activity [5] [90].
Prevention of persistence entry: Inhibiting key persistence regulators like (p)ppGpp synthesis or TA system activation can reduce persister formation [5].
Forced resuscitation: Stimulating persisters to reactivate before antibiotic treatment can sensitize them to conventional antibiotics [5].
The development of anti-persister therapies represents a promising frontier for combating chronic infections and potentially slowing resistance evolution by eliminating the reservoir of surviving cells [5].
The ecological and evolutionary perspectives on bacterial persistence reveal a sophisticated adaptation to environmental uncertainty. The coexistence of stochastic and triggered mechanisms represents complementary solutions to the fundamental challenge of surviving fluctuating conditions, with fitness trade-offs that maintain both strategies in natural populations. Understanding these dynamics not only illuminates a fascinating biological phenomenon but also provides essential insights for addressing the clinical challenge of persistent infections. Future research integrating single-cell physiology, evolutionary dynamics, and ecological theory will further unravel the complexities of this ancient survival strategy and inform novel approaches to combatting recalcitrant bacterial infections.
The phenomenon of persister cells, comprising both bacterial subpopulations that survive antibiotic exposure and cancer cells that evade chemotherapeutic agents, represents a critical frontier in the battle against treatment-resistant infections and malignancies. Unlike genetically resistant variants, persisters achieve tolerance through reversible, non-genetic adaptations that place them in a transient, dormant state. This whitepaper examines the fundamental distinction between stochastic and triggered persister cell formation mechanisms and establishes how this dichotomy dictates specific therapeutic strategies, particularly regarding treatment timing and drug combinations. Understanding whether persistence arises from random physiological fluctuations within a homogeneous population or constitutes a coordinated response to external stressors is paramount for designing interventions capable of eradicating these resilient cell populations.
Core Mechanisms of Persister Formation
The formation of persister cells can be broadly categorized into two paradigms: stochastic, pre-emptive formation and triggered, responsive formation. The molecular pathways underlying these paradigms demand distinct clinical approaches.
Stochastic (Pre-emptive) Formation
Stochastic persistence describes the spontaneous, random emergence of dormant cells within an isogenic population, even in the absence of environmental threats. This mechanism functions as a bet-hedging strategy, ensuring that a subset of cells survives unpredictable catastrophic stress [18] [1].
- Key Molecular Players:
- Bacterial Systems: Stochastic variation in the levels of the stringent response alarmone (p)ppGpp can activate Lon protease via polyphosphate accumulation. This leads to the degradation of antitoxins and the subsequent activation of toxin-antitoxin (TA) module toxins, such as mRNA-degrading enzymes, which induce dormancy [18] [1]. The HipA toxin, which inhibits glutamyl-tRNA synthetase, can also be stochastically activated, triggering the stringent response and cross-activating other TA modules [18].
- Cancer Systems: In cancer, pre-existing, inheritable cell-states prime a subset of cells for drug tolerance. These states are not necessarily linked to quiescence and can be transmitted across several cellular generations before drug exposure. Fate mapping and lineage tracking studies show that the ancestors of drug-tolerant persisters (DTPs) can be proliferation-competent, with survival decisions shaped by non-genetic heterogeneity present before treatment [91].
Triggered (Responsive) Formation
In contrast, triggered persistence is an inducible response where environmental signals activate dormancy pathways in a more synchronized manner within a susceptible subpopulation.
- Inducing Signals:
- Antibiotic Attack: Exposure to bactericidal antibiotics can directly trigger persister formation [25].
- Nutrient Limitation & Starvation: Nutrient deprivation is a potent inducer of the stringent response and dormancy [1] [25].
- Other Stressors: Acidic pH, oxidative stress, and host immune factors can also initiate the persistence response [1] [25].
- Key Molecular Pathways:
- Stringent Response: Nutrient starvation and other stresses lead to the rapid accumulation of (p)ppGpp, which orchestrates a dramatic reprogramming of cellular physiology away from growth and toward maintenance [18] [1].
- Toxin-Antitoxin Systems: Direct damage from antibiotics or the secondary stress they cause can prompt the activation of TA toxins, leading to growth arrest [18].
- Quorum Sensing (QS): In bacterial communities, QS molecules like phenazines and homoserine lactones can increase persister formation by inducing oxidative stress and metabolic changes, illustrating a community-level trigger [25].
Table 1: Comparative Analysis of Stochastic vs. Triggered Persister Formation
| Feature | Stochastic Formation | Triggered Formation |
|---|---|---|
| Initiation | Spontaneous, random fluctuation | Induced by external stressor (e.g., drug, starvation) |
| Population Dynamics | Small, pre-existing subpopulation | Can create a larger, synchronized persister pool |
| Therapeutic Implication | Requires prophylactic or continuous strategies | Allows for pre-emptive or timed intervention against the trigger |
| Key Evidence | Single-cell analysis shows persisters exist prior to antibiotic addition [18] [1] | Persister levels surge following exposure to stress [25] |
| Role in Cancer | Pre-existing cell states determine post-drug fate [91] | Therapy itself can induce a persistent, dormant state [39] |
Therapeutic Implications Dictated by Formation Mechanism
The timing and composition of therapeutic interventions must be tailored to the specific persister formation mechanism at play.
Implications for Treatment Timing
The origin of persisters has a profound impact on the optimal timing of adjuvant therapies designed to eradicate them.
- Combating Stochastic Persisters: Since these cells pre-exist therapy, concurrent or continuous administration of anti-persister adjuvants with primary therapy is critical. Waiting until after the primary treatment has failed allows the pre-existing persisters to regrow. The goal is to target the dormant cells at the same time the primary drug kills the bulk population [1] [25].
- Combating Triggered Persisters: For persistence induced by the treatment itself, a pre-emptive or staggered approach may be more effective. This could involve administering a trigger-blocking agent (e.g., a (p)ppGpp synthesis inhibitor) shortly before or simultaneously with the antibiotic to prevent the induction of dormancy. Alternatively, since some triggers (like nutrient shifts) may occur at predictable times during an infection, timing interventions to these windows could be beneficial.
Implications for Treatment Combinations
The molecular basis of each mechanism reveals distinct druggable pathways for combination therapy.
- Targeting Stochastic Persistence:
- Prevention of Formation: Strategies aim to reduce the inherent heterogeneity that gives rise to persisters. This includes inhibiting key regulators like the stringent response or H2S biogenesis, which can lower the baseline persister frequency [25].
- Awakening and Killing: This approach uses compounds that force stochastic persisters out of dormancy, thereby re-sensitizing them to conventional antibiotics. The danger is that if the awakening agent is not combined with a killing agent, it may simply encourage relapse [25].
- Targeting Triggered Persistence:
- Interference with Inducing Signals: Blocking the external trigger, such as by neutralizing QS signals with furanones or benzamide-benzimidazole compounds, can prevent the responsive shift to persistence [25].
- Inhibition of Signal Transduction: This involves targeting the internal pathways that mediate the response. Examples include inhibitors of (p)ppGpp synthetases (RelA) or the Lon protease, which would disrupt the downstream activation of TA modules and growth arrest, even in the presence of the initial trigger [18] [1].
Table 2: Mechanism-Based Anti-Persister Therapeutic Strategies
| Strategy | Molecular Target/Agent Example | Mechanism Suitability | Proposed Timing |
|---|---|---|---|
| Direct Killing | Membrane-disrupting agents (e.g., XF-73, SA-558) [25] | Both | Concurrent with primary therapy |
| Prevention of Formation | H2S scavengers; CSE inhibitors [25] | Primarily Stochastic | Prophylactic or continuous |
| Inhibition of Trigger Response | Quorum Sensing inhibitors (e.g., brominated furanones) [25] | Primarily Triggered | Pre-emptive or concurrent |
| Awakening & Killing | Metabolic disruptors (e.g., Nitric Oxide) [25] | Primarily Stochastic | Must be followed immediately by a killing agent |
| Synergistic Killing | Membrane permeabilizers + Antibiotics (e.g., CD437 + Gentamicin) [25] | Both | Concurrent |
Experimental Protocols for Discerning Mechanisms and Testing Therapies
Protocol 1: Single-Cell Time-Lapse Microscopy to Determine Formation Timing
This protocol is designed to distinguish stochastic from triggered persistence by tracking the fate of individual cells before and after drug exposure.
- Objective: To visualize the timing of persister formation and correlate pre-existing cell states with post-treatment survival.
- Materials:
- Microfluidic device (e.g., mother machine) for long-term, single-cell imaging
- Time-lapse fluorescence microscope with environmental control (temperature, CO2)
- Culture medium and therapeutic agent of interest
- Fluorescent viability stains (e.g., propidium iodide for dead cells, a fluorescent dye for metabolic activity)
- Methodology:
- Loading and Growth: Load a bacterial or cancer cell suspension into the microfluidic device and allow cells to grow under optimal conditions for several generations in the absence of drug.
- Baseline Imaging: Capture images at regular intervals (e.g., every 10-30 minutes) to establish lineage relationships and pre-treatment growth kinetics for each cell.
- Drug Application: Introduce a lethal concentration of the therapeutic agent into the medium flow while continuing time-lapse imaging.
- Post-Treatment Monitoring: Continue imaging for an extended period (24-72 hours) to identify persister cells that survive and eventually resume growth.
- Data Analysis:
- Lineage Correlation: Analyze whether the fate (death/survival) of sister cells or closely related cells is correlated, indicating a pre-existing, inherited state [91].
- Kinetic Analysis: Compare the pre-treatment intermitotic times of cells that become persisters versus those that die. A lack of significant difference challenges the notion that persisters exclusively originate from pre-existing slow-cyclers [91].
Protocol 2: Evaluating Anti-Persister Compound Efficacy in Combination
This protocol tests the ability of candidate adjuvants to eradicate persisters when combined with a primary drug.
- Objective: To assess the synergy between a conventional antibiotic/chemotherapeutic and an anti-persister compound.
- Materials:
- Stationary phase culture or a biofilm model (high persister reservoirs)
- Primary therapeutic agent (e.g., ciprofloxacin, cisplatin)
- Anti-persister adjuvant (e.g., membrane disruptor, TA system inhibitor)
- Phosphate-buffered saline (PBS) for washing
- Colony forming unit (CFU) plating equipment
- Methodology:
- Persister Generation: Inoculate a high-density culture and grow it into stationary phase, or establish a biofilm. Treat with a high dose of the primary drug for 3-5 hours to kill the majority of the population. Wash the cells with PBS to remove the drug.
- Combination Treatment: Resuspend the persister-enriched population in fresh medium containing:
- No drug (control for regrowth)
- Primary drug alone (control for persistence)
- Anti-persister adjuvant alone
- Combination of primary drug and adjuvant
- Incubation and Quantification: Incubate the treatment groups for a set period (e.g., 24 hours). Serially dilute and plate the cultures at time zero and after 24 hours to determine the viable cell count.
- Data Analysis: Calculate the log-reduction in CFU/mL. Synergy is demonstrated when the combination treatment results in a significantly greater reduction (e.g., >2-log) than either agent alone, indicating successful killing of the persister population [25].
Visualization of Signaling Pathways and Therapeutic Interference
The following diagrams illustrate the core pathways of persister formation and the points of therapeutic intervention.
Bacterial Persister Formation and Intervention Pathways
Bacterial Persister Formation and Intervention Map. This diagram illustrates the convergence of stochastic (yellow) and triggered (red) pathways on the central (p)ppGpp-mediated activation of toxin-antitoxin systems, leading to growth arrest. Key therapeutic intervention points (green) are shown, targeting specific steps in each pathway.
Cancer Drug-Tolerant Persister (DTP) Cell-Fate Determination
Cancer DTP Fate Determination Map. This diagram highlights the critical role of pre-existing, inheritable cell states (yellow) in determining post-drug fate. Drug application (red) acts as a selective pressure, with cell fate outcomes (red/green) being probabilistically determined by the pre-treatment state. Therapeutic strategies (blue) aim to modulate the pre-treatment heterogeneity, lock cells in a non-proliferative state, or force DTPs into apoptosis.
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 3: Essential Reagents for Investigating Persister Cell Biology
| Reagent/Solution | Function/Application | Key Examples |
|---|---|---|
| Microfluidic Devices | Enables long-term, high-resolution tracking of single-cell lineages and behaviors before and after stress. | Mother machine; Microwell arrays [91] |
| Fluorescent Viability/Activity Probes | Differentiates live, dead, dormant, and metabolically active cells in real-time. | Propidium iodide; CFDA-AM; GFP-based metabolic sensors; Fluorescent DFFB reporters [92] |
| Membrane-Targeting Anti-Persister Compounds | Directly kills persisters by disrupting membrane integrity, independent of growth state. | XF-73; SA-558; Synthetic retinoids (CD437) [25] |
| Stringent Response Modulators | Investigates the role of (p)ppGpp in persistence. | RelA/SpoT homolog (RSH) inhibitors; Inducible (p)ppGpp expression systems |
| Toxin-Antitoxin System Modulators | Activates or inhibits TA modules to study their role in dormancy. | Inducible toxin expression plasmids; Antitoxin overexpression vectors [18] |
| Quorum Sensing Inhibitors | Blocks cell-cell communication that triggers persistence. | Brominated furanones; Benzamide-benzimidazole compounds (targeting MvfR) [25] |
| Metabolic Wake-Up Agents | Forces persisters out of dormancy to re-sensitize them to conventional drugs. | Nitric Oxide (NO) donors; Sugar phosphates [25] |
| Cellular Barcoding Systems | Tracks clonal dynamics and lineage relationships to map the origins of persisters. | CRISPR-based barcoding; Lentiviral barcode libraries [91] |
The mechanistic dichotomy between stochastic and triggered persister formation is not merely a biological curiosity; it is a fundamental determinant of therapeutic strategy. Successfully eradicating these resilient cells requires a paradigm shift from broad-spectrum approaches to mechanism-informed precision treatments. This entails using diagnostic tools to identify the dominant persister pathway in a given clinical context—whether a chronic infection or a relapsing tumor—and deploying appropriately timed drug combinations. For the stochastic element, continuous or concurrent strategies that target pre-existing dormant cells are essential. For the triggered element, disrupting the inducing signals or the stress response pathways themselves holds the key. Future research must focus on translating the detailed molecular understanding of these pathways into clinically viable diagnostics and therapeutics, moving from a reactive to a proactive and predictive framework in the fight against persistence.
Conclusion
The formation of bacterial persisters is not governed by a single mechanism but by a complex interplay between stochastic phenotypic variation and environmentally triggered responses. The stochastic model explains the pre-existence of a dormant subpopulation, while triggered mechanisms account for the inducible expansion of this group under stress. Critically, these pathways are not isolated; evidence shows that triggered signals, such as HipA-induced (p)ppGpp synthesis, often act through the stochastic activation of downstream effectors like TA modules. This interdependence underscores that targeting persisters requires a multi-pronged strategy. Future research must focus on defining the complete persister 'mechanome,' developing dynamic models that integrate both stochastic and deterministic elements, and clinically validating anti-persister therapies, such as combination treatments that disrupt metabolic dormancy while enhancing antibiotic penetration. Success in this area is paramount for overcoming chronic infections and curbing the rise of antibiotic resistance.
References
- 1. Bacterial persisters: molecular mechanisms and ... [https://www.nature.com/articles/s41392-024-01866-5]
- 2. Bacterial Persister Cells and Development of Antibiotic ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11335562/]
- 3. An ecological and stochastic perspective on persisters ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11697298/]
- 4. A host-directed adjuvant sensitizes intracellular bacterial ... [https://www.nature.com/articles/s41564-025-02124-2]
- 5. Mini review: Persister cell control strategies [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1706115/full]
- 6. A rational approach to discovering new persister control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406675/]
- 7. Phenotypic Plasticity, Bet-Hedging, and Androgen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5845637/]
- 8. Bet-hedging in stochastically switching environments [https://www.sciencedirect.com/science/article/abs/pii/S0022519313003408]
- 9. Stochastic Mechanisms of Cell Fate Specification that Yield ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3025287/]
- 10. Modeling stochastic phenotype switching and bet-hedging ... [https://link.springer.com/article/10.1007/s40484-014-0035-5]
- 11. A Selective Review of Modern Stochastic Modeling: SDE/ ... [https://arxiv.org/html/2508.11004]
- 12. The role of memory in non-genetic inheritance and its impact ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009348]
- 13. Bacterial persisters are a stochastically formed subpopulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8084331/]
- 14. Antibiotic Persister Cells in Acinetobacter baumannii [https://pmc.ncbi.nlm.nih.gov/articles/PMC12638225/]
- 15. Observation of persister cell histories reveals diverse ... [https://elifesciences.org/articles/79517]
- 16. Antibiotic resistance and persistence—Implications for human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7726816/]
- 17. Antibiotic-persistent bacterial cells exhibiting low-level ROS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12345145/]
- 18. Stochastic induction of persister cells by HipA through (p ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4413331/]
- 19. The stringent response and physiological roles of (pp) ... [https://www.nature.com/articles/s41579-020-00470-y]
- 20. Stringent response [https://en.wikipedia.org/wiki/Stringent_response]
- 21. (p)ppGpp imposes graded transcriptional changes to ... [https://www.nature.com/articles/s41522-025-00795-7]
- 22. Toxin-antitoxin system [https://en.wikipedia.org/wiki/Toxin-antitoxin_system]
- 23. Toxin–antitoxin systems and their role in disseminating and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5812544/]
- 24. Stress can induce the transcription of toxin-antitoxin systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7368831/]
- 25. Mini review: Persister cell control strategies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12568461/]
- 26. Metabolic aspects of bacterial persisters - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4205924/]
- 27. The role of metabolism in bacterial persistence [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2014.00070/full]
- 28. Mechanisms of Drug-Induced Tolerance in Mycobacterium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7566895/]
- 29. Unveiling the critical roles of cellular metabolism ... [https://www.nature.com/articles/s44259-024-00034-7]
- 30. Single-Cell Technologies to Study Phenotypic ... [https://www.mdpi.com/2076-2607/9/11/2277]
- 31. Single-cell imaging and characterization of Escherichia coli ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6584399/]
- 32. Dynamic microfluidic single-cell screening identifies pheno ... [https://www.nature.com/articles/s41467-024-48269-2]
- 33. Microfluidics applications for high-throughput single cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8507141/]
- 34. Stochastic Variation in Expression of the Tricarboxylic Acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6751062/]
- 35. General Mechanisms Leading to Persister Formation and ... [https://www.sciencedirect.com/science/article/abs/pii/S0168952519300538]
- 36. ATP biosensor reveals microbial energetic dynamics and ... [https://www.nature.com/articles/s41467-024-49579-1]
- 37. Measuring and modeling energy and power consumption in ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-021-01023-2]
- 38. A genetically encoded fluorescent reporter of ATP/ADP ratio [https://pmc.ncbi.nlm.nih.gov/articles/PMC2633436/]
- 39. Drug-tolerant persister cells in cancer: bridging the gaps ... [https://www.nature.com/articles/s41467-025-66376-6]
- 40. CRISPR screens reveal YAP/TEAD axis as a mediator of ... [https://pubmed.ncbi.nlm.nih.gov/41222695/]
- 41. Common computational tools for analyzing CRISPR screens [https://pmc.ncbi.nlm.nih.gov/articles/PMC8786280/]
- 42. A benchmark of computational methods for correcting biases ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03336-1]
- 43. Genome Analysis of Pseudomonas aeruginosa Strains ... [https://pubmed.ncbi.nlm.nih.gov/36986348/]
- 44. Omics Approaches in Understanding Insecticide ... [https://www.mdpi.com/1422-0067/26/5/1854]
- 45. Perspective on integrated multi-omics approaches and ... [https://pubmed.ncbi.nlm.nih.gov/40849092/]
- 46. Multi-omics advances in understanding cancer drug ... [https://www.sciencedirect.com/science/article/pii/S2949723X25000510]
- 47. Evolution of Resistant Mutants in Pseudomonas aeruginosa ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12299539/]
- 48. Bacterial Persister in the Food Industry | Frontiers Research Topic Cells [https://www.frontiersin.org/research-topics/66536/bacterial-persister-cells-in-the-food-industry/magazine]
- 49. Antibiotic regimen based on population analysis of residing persister ... [https://www.nature.com/articles/srep18578?error=cookies_not_supported&code=99ee128c-a63e-41ad-8c7c-4de6216237cb]
- 50. of Respiratory Activity in Quantification Biofilms [https://bio-protocol.org/en/bpdetail?id=1591&type=0]
- 51. A quantitative method to measure biofilm removal ... [https://www.nature.com/articles/srep32694]
- 52. Quantitative image analysis of microbial communities with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7840502/]
- 53. Bacterial persisters : molecular mechanisms and therapeutic... [https://www.nature.com/articles/s41392-024-01866-5?error=cookies_not_supported&code=4bbdd9bc-caa6-4305-9e89-2929af9a7100]
- 54. Fighting bacterial persistence: Current and emerging anti- ... [https://www.sciencedirect.com/science/article/abs/pii/S1368764618300232]
- 55. Antimicrobial Drug | SpringerLink Discovery Against Persisters [https://link.springer.com/chapter/10.1007/978-3-030-25241-0_12]
- 56. Insights into the molecular determinants involved in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8565819/]
- 57. Molecular mechanism and application of emerging ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-024-03628-3]
- 58. Quantitative analysis of persister fractions suggests different ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3682893/]
- 59. Research disproves persister bacteria as main cause of ... [https://www.news-medical.net/news/20250205/Research-disproves-persister-bacteria-as-main-cause-of-antibiotic-resistance.aspx]
- 60. Drug persistence - from antibiotics to cancer therapies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6366840/]
- 61. Editorial: Bacterial persister cells in the food industry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12399600/]
- 62. Quantitative prediction of disinfectant tolerance in Listeria ... [https://www.nature.com/articles/s41598-025-94321-6]
- 63. An ecological and stochastic perspective on persisters ... [https://www.sciencedirect.com/science/article/pii/S2001037024004240]
- 64. Targeting therapy-persistent residual disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12160366/]
- 65. Drug tolerant persister cell plasticity in cancer [https://www.nature.com/articles/s41392-024-01891-4]
- 66. Common Relapse Triggers and How to Avoid Them [https://www.gatewayfoundation.org/blog/triggers-in-addiction-recovery/]
- 67. Addiction Relapse: Risk Factors, Coping & Treatment Options [https://americanaddictioncenters.org/treat-drug-relapse]
- 68. Relapse Prevention and the Five Rules of Recovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4553654/]
- 69. Pathways to the persistence of drug use despite its adverse ... [https://www.nature.com/articles/s41380-023-02040-z]
- 70. Biofilms: structure, resistance mechanism, emerging control ... [https://pubs.rsc.org/en/content/articlehtml/2025/pm/d5pm00094g]
- 71. From Dormancy to Eradication: Strategies for Controlling ... [https://www.mdpi.com/2304-8158/14/6/1075]
- 72. Are all antibiotic persisters created equal? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9428696/]
- 73. Biofilms and multidrug resistance: an emerging crisis and the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12237878/]
- 74. Biofilms and multidrug resistance: an emerging crisis ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1625356/full]
- 75. Biofilm, resistance, and quorum sensing: The triple threat in ... [https://www.sciencedirect.com/science/article/pii/S2950194625003462]
- 76. Strategies for combating persister cell and biofilm infections [https://pmc.ncbi.nlm.nih.gov/articles/PMC5609227/]
- 77. Drug-induced tolerant persisters in tumor - Molecular Cancer [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02323-9]
- 78. Observation of persister cell histories reveals diverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12074638/]
- 79. Diverse drug-resistance mechanisms can emerge from ... [https://www.nature.com/articles/ncomms10690]
- 80. Identification and genetic dissection of convergent persister ... [https://www.nature.com/articles/s41586-024-08124-2]
- 81. Mechanisms of Acquired Resistance and Tolerance to EGFR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9856371/]
- 82. “Persistersâ€: Survival at the Cellular Level - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3145784/]
- 83. HipA-mediated antibiotic persistence via phosphorylation ... [https://www.nature.com/articles/ncomms4001]
- 84. hipBA toxin-antitoxin systems mediate persistence in ... [https://www.nature.com/articles/s41598-020-59283-x]
- 85. Role of Unusual P Loop Ejection and Autophosphorylation ... [https://www.sciencedirect.com/science/article/pii/S2211124712002380]
- 86. Intracellular Energy Variability Modulates Cellular Decision ... [https://www.nature.com/articles/s41598-019-56587-5]
- 87. Energy depletion by cell proliferation sensitizes the kidney ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11207531/]
- 88. Integrative biology of persister cell formation - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9470271/]
- 89. Persister cells: formation, resuscitation and combative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8590677/]
- 90. A Quantitative Survey of Bacterial Persistence in the ... [https://www.mdpi.com/2079-6382/9/8/508]
- 91. Inheritable cell-states shape drug-persister correlations and ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013446]
- 92. Cancer Uses Cell Death Proteins to Survive Treatment and ... [https://today.ucsd.edu/story/cancer-uses-cell-death-proteins-to-survive-treatment-and-regrow]