A Complete Guide to Shotgun Metagenomics for Gut Microbiome Analysis: From Protocol to Clinical Application
This article provides a comprehensive guide to shotgun metagenomics for gut microbiome research, tailored for scientists and drug development professionals.
A Complete Guide to Shotgun Metagenomics for Gut Microbiome Analysis: From Protocol to Clinical Application
Abstract
This article provides a comprehensive guide to shotgun metagenomics for gut microbiome research, tailored for scientists and drug development professionals. It covers foundational principles, detailing how this culture-independent method enables comprehensive taxonomic and functional profiling of all microorganisms in a sample, surpassing the limitations of 16S rRNA amplicon sequencing. A detailed, step-by-step protocol is presented, from sample collection and DNA extraction to sequencing and bioinformatic analysis for characterizing microbial composition and function. The guide also addresses common troubleshooting and optimization challenges, including host DNA depletion and mycobiome characterization. Finally, it evaluates the validation of metagenomics against traditional diagnostic methods and its growing application in clinical and pharmaceutical contexts for pathogen detection, drug resistance profiling, and personalized medicine.
Understanding Shotgun Metagenomics: Principles and Clinical Potential of Gut Microbiome Analysis
The study of complex microbial communities, particularly the human gut microbiome, has been revolutionized by the advent of high-throughput sequencing technologies. For decades, 16S ribosomal RNA (rRNA) gene sequencing has been the cornerstone of microbial ecology, providing initial insights into the composition of bacterial communities. However, this targeted approach reveals only a fragment of the microbial picture. Shotgun metagenomic sequencing represents a paradigm shift in microbial community analysis, moving beyond mere census-taking to comprehensive functional potential assessment. This application note delineates the core principles distinguishing these methodologies and provides detailed protocols for implementing shotgun metagenomics in gut microbiome research, framed within a broader thesis on advanced metagenomic protocols.
Fundamental Methodological Differences
The foundational distinction between these techniques lies in their scope of genetic material analysis:
16S rRNA Sequencing: This amplicon-based approach targets specific hypervariable regions (V1-V9) of the 16S rRNA gene, which is present in all bacteria and archaea. Through PCR amplification and sequencing of these conserved regions, researchers can infer taxonomic composition based on sequence variation [1] [2]. This method essentially answers "who is present?" in a bacterial community, albeit with significant limitations.
Shotgun Metagenomic Sequencing: This untargeted approach involves randomly fragmenting all DNA in a sample into numerous small pieces, which are sequenced simultaneously without prior amplification of specific regions [1] [3]. These sequences are then computationally reconstructed to identify both taxonomic origins and functional elements, addressing both "who is present?" and "what are they capable of doing?" [4] [5].
Table 1: Core Methodological Comparison Between 16S rRNA and Shotgun Metagenomic Sequencing
| Parameter | 16S rRNA Sequencing | Shotgun Metagenomics |
|---|---|---|
| Genetic Target | Specific hypervariable regions of 16S rRNA gene | All genomic DNA in sample |
| Amplification Required | Yes (PCR) | No |
| Taxonomic Scope | Bacteria and Archaea only | All domains of life (Bacteria, Archaea, Fungi, Viruses) |
| Functional Profiling | Indirect prediction only | Direct assessment of functional genes |
| Bioinformatics Complexity | Beginner to Intermediate | Intermediate to Advanced |
Key Advantages of Shotgun Metagenomics
Enhanced Taxonomic Resolution and Breadth
Shotgun metagenomics provides superior taxonomic classification, enabling identification at finer phylogenetic levels:
Species and Strain-Level Discrimination: While 16S sequencing typically resolves to genus level (sometimes species), shotgun sequencing can distinguish closely related species and even strains by profiling single nucleotide variants across entire genomes [1]. This resolution is critical for identifying specific pathogenic strains or beneficial microbial variants in gut communities.
Comprehensive Taxonomic Coverage: Unlike 16S sequencing limited to bacteria and archaea, shotgun metagenomics simultaneously detects and characterizes bacteria, fungi, viruses, protozoa, and other microorganisms present in a sample [2] [6]. This comprehensive profiling is particularly valuable in gut microbiome studies where cross-domain interactions significantly impact host health.
Direct Functional Potential Assessment
The most significant advantage of shotgun metagenomics lies in its capacity to elucidate functional capabilities:
Gene Cataloging and Pathway Analysis: By sequencing all genomic material, researchers can directly identify protein-coding genes, metabolic pathways, and functional elements within microbial communities [7]. This enables construction of gene catalogs and assessment of functional diversity in gut microbiomes, revealing capabilities like carbohydrate digestion, vitamin synthesis, or inflammatory compound production [7] [5].
Antibiotic Resistance Profiling: Shotgun metagenomics enables comprehensive identification of antibiotic resistance genes (ARGs) within microbial communities, providing insights into the resistome of gut microbiota and its clinical implications [7].
Novel Gene Discovery: Functional metagenomics facilitates discovery of previously uncharacterized genes and pathways through heterologous expression in model systems like Escherichia coli [7]. This approach has revealed novel carbohydrate-active enzymes (CAZymes) and bile salt hydrolases in gut microbiomes [7].
Elimination of Amplification Biases
Shotgun metagenomics bypasses PCR amplification steps required in 16S sequencing, thereby avoiding associated biases:
Primer-Free Approach: Without dependency on primer binding sites, shotgun sequencing provides more quantitative abundance measurements and detects organisms with divergent 16S sequences that might be missed by universal primers [5] [6].
Reduced Quantitative Distortion: The absence of PCR amplification eliminates artifacts from varying gene copy numbers and amplification efficiency differences, resulting in more accurate representation of microbial abundances [5].
Experimental Design and Protocol
Sample Collection and DNA Extraction
Critical Considerations:
- Sample Preservation: Immediate freezing at -80°C is essential for fecal samples to preserve DNA integrity. For clinical studies, in-home collection with -20°C temporary storage before transfer to -80°C is recommended [6].
- Host DNA Reduction: For gut microbiome samples, consider methods to minimize host DNA contamination, which can account for >90% of sequences in some cases [5].
- Extraction Methodology: Consistent DNA extraction protocols are critical. The NucleoSpin Soil Kit (Macherey-Nagel) or PowerSoil DNA Isolation Kit (Qiagen) have demonstrated efficacy for fecal samples [6].
Library Preparation and Sequencing
Standardized Workflow:
- DNA Fragmentation: Mechanically shear DNA to 250-300bp fragments using acoustic shearing or enzymatic fragmentation [3].
- Library Construction: Utilize Illumina-compatible library prep kits with appropriate size selection (350bp insert size recommended) [3].
- Quality Control: Verify library quality and quantity using fluorometric methods (Qubit) and fragment analyzers.
- Sequencing: Illumina platforms (NovaSeq, HiSeq) with 150bp paired-end sequencing provide optimal results for metagenomic applications [3] [8].
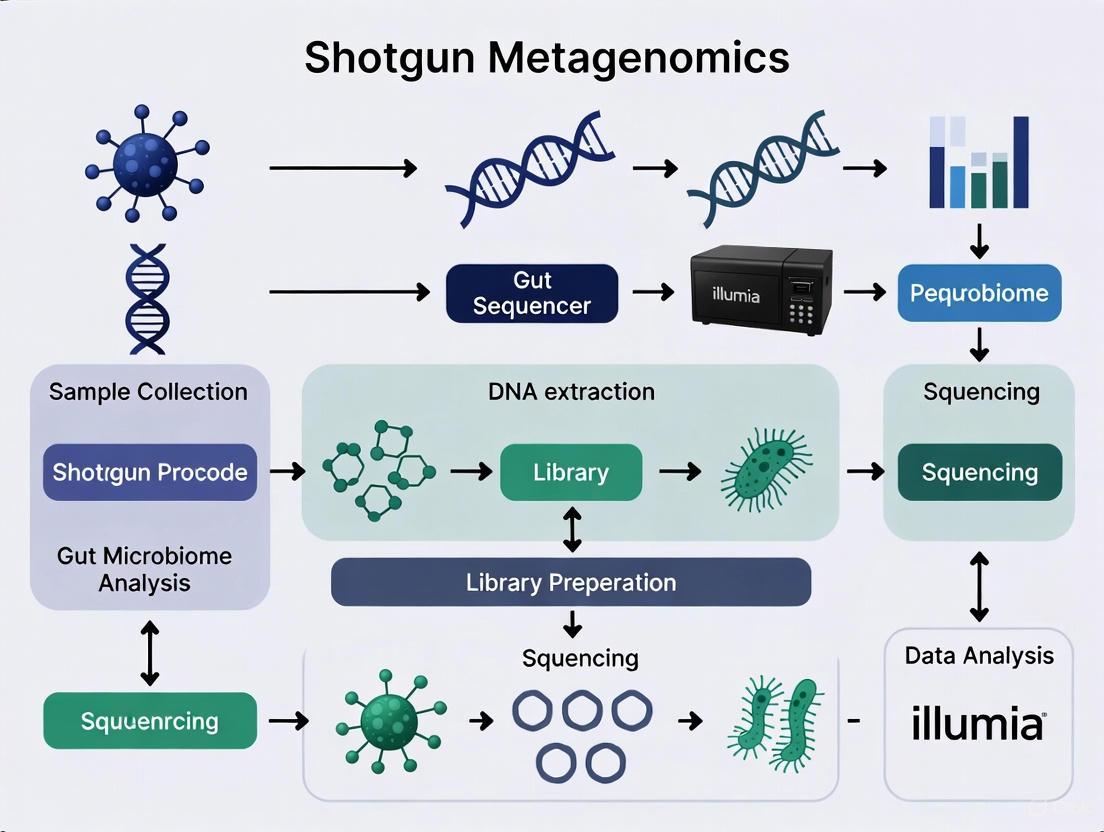
Diagram 1: Shotgun Metagenomics Workflow
Sequencing Depth Considerations
Adequate sequencing depth is critical for robust metagenomic analysis:
- Minimum Depth: 5-10 million reads per sample for basic taxonomic profiling
- Recommended Depth: 20-50 million reads per sample for functional analysis and rare species detection
- Deep Sequencing: >50 million reads for strain-level analysis and genome assembly [8]
Table 2: Sequencing Recommendations for Gut Microbiome Studies
| Analysis Type | Recommended Depth | Key Applications |
|---|---|---|
| Shallow Shotgun | 1-5 million reads | Large cohort studies, basic taxonomic profiling |
| Standard Shotgun | 10-20 million reads | Routine taxonomic and functional analysis |
| Deep Shotgun | 30-50+ million reads | Strain-level tracking, genome assembly, rare variant detection |
Bioinformatics Analysis Pipeline
Data Preprocessing and Quality Control
Essential Steps:
- Adapter Trimming: Remove sequencing adapters using tools like Cutadapt or Trimmomatic
- Quality Filtering: Eliminate low-quality reads (Phred score <20) and reads with >10% ambiguous bases [3]
- Host DNA Removal: Align reads to host genome (GRCh38 for human) using Bowtie2 and remove matching sequences [6]
Taxonomic Profiling
Reference-Based Approaches:
- Marker Gene Analysis: Tools like MetaPhlAn utilize clade-specific marker genes for efficient taxonomic classification [1]
- Whole Genome Alignment: Kraken2 and similar tools align reads to comprehensive genomic databases for maximal sensitivity
Functional Annotation
Comprehensive Workflow:
- Gene Prediction: Prodigal or FragGeneScan for identifying coding sequences in metagenomic data
- Database Alignment: Diamond or BLAST against functional databases (KEGG, COG, eggNOG)
- Pathway Reconstruction: HUMAnN2 for reconstructing complete metabolic pathways from metagenomic data [1]
Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for Shotgun Metagenomics
| Reagent/Platform | Function | Application Notes |
|---|---|---|
| PowerSoil DNA Isolation Kit (Qiagen) | DNA extraction from complex samples | Optimal for fecal samples with inhibitor removal |
| Illumina DNA Prep Kit | Library preparation | Efficient tagmentation-based library construction |
| NovaSeq 6000 System (Illumina) | High-throughput sequencing | Scalable output for large cohort studies |
| DRAGEN Metagenomics Pipeline | Bioinformatic analysis | Accelerated taxonomic classification and reporting |
| NucleoSpin Soil Kit (Macherey-Nagel) | DNA extraction | Alternative for difficult-to-lyse microorganisms |
Applications in Gut Microbiome Research
Shotgun metagenomics has enabled groundbreaking advances in understanding gut microbiome structure and function:
- Functional Dysbiosis Characterization: Moving beyond taxonomic shifts to identify functional alterations in disease states like inflammatory bowel disease, obesity, and colorectal cancer [7] [6]
- Microbiome-Host Interactome: Revealing complex interactions between microbial metabolites and host physiology through integrated multi-omics approaches [7]
- Therapeutic Monitoring: Tracking strain-level dynamics in response to interventions like probiotics, antibiotics, or fecal microbiota transplantation [9]
- Biomarker Discovery: Identifying microbial genes and pathways as diagnostic or prognostic biomarkers for gastrointestinal disorders [6]
Integration with Multi-Omics Approaches
For comprehensive understanding of gut microbiome functionality, shotgun metagenomics can be integrated with complementary approaches:
- Metatranscriptomics: RNA sequencing to assess actively expressed genes and pathways
- Metaproteomics: Mass spectrometry-based identification of expressed proteins
- Metabolomics: Profiling of microbial metabolites influencing host physiology [7]
This integrated framework provides unprecedented insights into the functional dynamics of gut microbial communities and their impact on human health and disease.
Shotgun metagenomics represents a transformative advancement over 16S rRNA sequencing, providing unparalleled resolution and functional insights into complex microbial communities like the gut microbiome. By moving beyond taxonomic census to functional potential assessment, this powerful approach enables researchers to address fundamental questions about microbiome-host interactions, disease mechanisms, and therapeutic interventions. While requiring more substantial bioinformatics resources and expertise, the depth of information obtained makes shotgun metagenomics an indispensable tool in modern microbiome research, particularly for drug development and clinical translation. As sequencing costs continue to decline and analytical methods improve, shotgun metagenomics is poised to become the gold standard for comprehensive microbiome analysis.
Shotgun metagenomics has revolutionized gut microbiome research by moving beyond taxonomic census to enable two transformative analytical dimensions: strain-level resolution and functional insights. Strain-level resolution allows researchers to distinguish between genetically distinct variants of the same microbial species, which often exhibit significant functional differences and host interactions [10]. Simultaneously, functional profiling deciphers the collective metabolic potential of the microbial community by identifying genes and pathways involved in processes like nutrient metabolism, synthesis of bioactive compounds, and antimicrobial resistance [11]. This dual capability provides a systems-level understanding of how gut microbiota influence human health and disease, forming the foundation for precision medicine approaches in microbiome research [11].
The integration of taxonomic, functional, and strain-level profiling (TFSP) represents the most advanced framework for comprehensive microbiome analysis, offering unprecedented opportunities for diagnostics, therapeutic development, and personalized treatment strategies [10]. This application note details the experimental protocols and bioinformatic tools necessary to achieve these analytical objectives within a complete shotgun metagenomics workflow.
Experimental Protocols for Comprehensive Profiling
Sample Preparation and DNA Sequencing
Protocol: Library Preparation for Shotgun Metagenomic Sequencing
- Sample Collection and Preservation: Collect fresh fecal samples in sterile containers with DNA/RNA stabilization buffer or immediately freeze at -80°C to preserve nucleic acid integrity. Avoid multiple freeze-thaw cycles.
- DNA Extraction: Use mechanical lysis (bead beating) combined with chemical lysis for maximal DNA yield from diverse microbial taxa. Recommended kits: QIAamp PowerFecal Pro DNA Kit or DNeasy PowerSoil Pro Kit. Include extraction controls to monitor contamination.
- DNA Quality Assessment: Quantify DNA using fluorometric methods (Qubit dsDNA HS Assay). Assess quality via spectrophotometry (A260/A280 ≈ 1.8-2.0) and gel electrophoresis to confirm high molecular weight DNA.
- Library Preparation: Fragment DNA to target size of 300-500 bp (covaris shearing or enzymatic fragmentation). Use Illumina-compatible library preparation kits with dual index barcodes to enable sample multiplexing. Recommended kits: Illumina DNA Prep or Nextera XT DNA Library Prep Kit.
- Sequencing: Sequence on Illumina platforms (NovaSeq 6000, NextSeq 2000) to generate 100-150 bp paired-end reads. Minimum recommended sequencing depth: 10-20 million reads per sample for human gut microbiota studies [12].
Bioinformatic Analysis for Strain-Level Resolution
Protocol: Meteor2 Workflow for Strain-Level Profiling
- Quality Control and Preprocessing:
- Taxonomic and Strain-Level Profiling:
- Install Meteor2: Available as open-source software from GitHub repository.
- Select Appropriate Database: Use environment-specific microbial gene catalogues (human gut catalogue included).
- Run Meteor2 in Full Mode: For comprehensive TFSP, use command:
meteor2 -1 sample_R1.fastq.gz -2 sample_R2.fastq.gz -d human_gut -o output_directory - Fast Mode Alternative: For rapid analysis when computational resources are limited, use the signature gene subset:
meteor2 -1 sample_R1.fastq.gz -2 sample_R2.fastq.gz -d human_gut_fast -o output_directory_fast
- Strain Tracking: Meteor2 enables strain-level analysis by tracking single nucleotide variants (SNVs) in the signature genes of metagenomic species pangenomes (MSPs), providing resolution for tracking microbial strain dissemination in applications like fecal microbiota transplantation (FMT) [10].
Functional Profiling and Pathway Analysis
Protocol: Functional Characterization of Microbial Communities
- Gene Abundance Quantification:
- Functional Annotation:
- Enzyme Annotation: Annotate carbohydrate-active enzymes (CAZymes) using dbCAN3 with default parameters [10].
- Pathway Mapping: Assign KEGG Orthology (KO) annotations using KofamScan [10].
- Antimicrobial Resistance Profiling: Identify antibiotic resistance genes (ARGs) using Resfinder with ResFinderDB and ResfinderFG databases (90% identity, 80% coverage thresholds) [10].
- Pathway Abundance Calculation:
- Aggregate gene abundances to estimate functional module abundance (Gut Brain Modules, Gut Metabolic Modules, KEGG modules).
- Normalize data using robust methods like TMM or RLE to minimize systematic variability [14].
Diagram 1: Shotgun Metagenomics Workflow for Strain and Functional Analysis
Comparative Performance of Metagenomic Tools
Tool Performance Metrics
Table 1: Performance Comparison of Metagenomic Analysis Tools
| Tool | Primary Function | Strain-Level Capacity | Functional Profiling | Key Performance Metric |
|---|---|---|---|---|
| Meteor2 | TFSP Integration | Yes (via SNVs in signature genes) | Yes (KO, CAZymes, ARGs) | 45% improved species detection sensitivity; 35% better functional abundance estimation vs. HUMAnN3 [10] |
| MetaPhlAn4 | Taxonomic Profiling | Limited | No | Uses species-specific marker genes; foundation of bioBakery suite [10] |
| HUMAnN3 | Functional Profiling | No | Yes | Comprehensive pathway analysis; outperformed by Meteor2 in abundance estimation accuracy [10] |
| StrainPhlAn | Strain-Level Profiling | Yes | No | Tracks strain populations; Meteor2 captured 9.8-19.4% more strain pairs [10] |
| CosmosID | Taxonomic Profiling | Limited | Limited | Identified 28 species in benchmark; performs well with culture-positive pathogens [15] |
| One Codex | Taxonomic Profiling | Limited | Limited | Identified 59 species in benchmark; higher detection of low-abundance organisms [15] |
Advantages Over Amplicon Sequencing
Table 2: Shotgun vs. 16S rRNA Sequencing for Gut Microbiome Studies
| Parameter | 16S rRNA Amplicon Sequencing | Shotgun Metagenomic Sequencing |
|---|---|---|
| Resolution | Genus to species level | Species to strain-level [12] |
| Functional Insights | Indirect inference only | Direct gene and pathway detection [12] |
| Bias | Primer selection bias | Minimal amplification bias |
| Pathogen Detection | Limited identification | Comprehensive detection of causative pathogens [11] |
| Antibiotic Resistance | Not available | Direct detection of ARGs [11] |
| Sequencing Depth | Lower (20,000 reads may suffice) | Higher (500,000+ reads recommended) [12] |
| Cost | Lower per sample | Higher per sample but more information |
Table 3: Key Research Reagent Solutions for Shotgun Metagenomics
| Reagent/Resource | Function | Example Products/Platforms |
|---|---|---|
| DNA Stabilization Buffers | Preserve microbial community structure during sample storage | DNA/RNA Shield, RNAlater |
| Mechanical Lysis Kits | Efficient cell wall disruption for diverse microbial taxa | QIAamp PowerFecal Pro, DNeasy PowerSoil Pro |
| Library Prep Kits | Fragment DNA and add sequencing adapters | Illumina DNA Prep, Nextera XT DNA Library Prep |
| Microbial Gene Catalogs | Reference databases for read alignment and annotation | Meteor2 databases, Integrated Gene Catalog (IGC) |
| Functional Databases | Annotate genes with functional information | KEGG, dbCAN, Resfinder, eggNOG |
| Analysis Pipelines | Integrated tools for data processing and interpretation | Meteor2, bioBakery (MetaPhlAn4, HUMAnN3, StrainPhlAn) |
| Validation Standards | Quality control and protocol standardization | NIST Stool Reference Material, ZymoBIOMICS Microbial Standards |
Applications in Clinical Research and Drug Development
The capacity for strain-level resolution and functional profiling enables critical applications in pharmaceutical research and clinical practice. Shotgun metagenomics facilitates precision antimicrobial therapy through rapid detection of antimicrobial resistance genes directly from clinical specimens, reducing dependence on empirical broad-spectrum antibiotics [11]. In one clinical implementation, researchers developed a rapid 6-hour nanopore metagenomic sequencing workflow with host DNA depletion that achieved 96.6% sensitivity for diagnosing lower respiratory infections while simultaneously identifying resistance genes for tailored therapy [11].
In drug development, microbiome-based biomarkers derived from metagenomic analyses are increasingly used for patient stratification and monitoring treatment efficacy. Large-scale multi-omics integrations encompassing over 1,300 metagenomes have identified consistent microbial signatures in inflammatory bowel disease (IBD) patients, with diagnostic models achieving high accuracy (AUROC 0.92-0.98) for distinguishing IBD from controls [11]. Similarly, metabolomic profiling of gut microbiota in type 2 diabetes has identified 111 microbial-derived metabolites with strong predictive power for disease progression [11].
For microbiome-based therapeutics such as fecal microbiota transplantation (FMT), strain-level tracking enables monitoring of donor strain engraftment and persistence. Combined metagenomic and metabolomic analyses reveal that successful FMT outcomes depend on stable engraftment and restoration of key metabolites including short-chain fatty acids, bile acid derivatives, and tryptophan metabolites [11]. This level of resolution provides crucial insights for optimizing therapeutic formulations and understanding mechanisms of action.
Implementation Considerations
Normalization Strategies for Quantitative Analysis
Proper normalization is essential for accurate differential abundance analysis in metagenomic studies. Based on systematic evaluations, Trimmed Mean of M-values (TMM) and Relative Log Expression (RLE) normalization methods demonstrate the highest overall performance for gene abundance data, maintaining high true positive rates while controlling false discovery rates [14]. These methods are particularly important when differentially abundant genes are asymmetrically distributed between experimental conditions, where simpler normalization approaches like total count scaling can produce unacceptably high false positive rates [14].
Computational Requirements and Optimization
The computational intensity of metagenomic analysis varies significantly by tool and mode. Meteor2 represents a balanced solution, requiring approximately 2.3 minutes for taxonomic analysis and 10 minutes for strain-level analysis of 10 million paired-end reads against the human microbial gene catalogue, with a modest 5 GB RAM footprint [10]. For large-scale studies or resource-constrained environments, the "fast mode" using signature gene subsets provides accelerated analysis while preserving essential profiling capabilities.
Strain-level resolution and functional profiling represent the new frontier in gut microbiome research, enabling unprecedented insights into microbial community dynamics and their impact on human health. The integration of taxonomic, functional, and strain-level profiling through platforms like Meteor2 provides a comprehensive framework for deciphering complex host-microbiome interactions. As standardization improves and computational methods advance, these approaches will increasingly drive precision medicine initiatives, therapeutic development, and personalized treatment strategies based on individual microbiome signatures.
Shotgun metagenomic sequencing has emerged as a foundational tool for gut microbiome research, enabling comprehensive analysis of microbial communities without the need for cultivation [16]. This approach involves untargeted sequencing of all microbial DNA present in a sample, providing unprecedented insights into the taxonomic composition and functional potential of the gut ecosystem [17] [18]. Unlike targeted 16S ribosomal RNA gene sequencing, which is restricted to taxonomic profiling of bacteria and archaea, shotgun metagenomics captures the full genetic repertoire, including bacteria, viruses, fungi, and archaea, while enabling strain-level resolution and functional gene annotation [9] [17].
The clinical and research applications of gut microbiome metagenomics have expanded dramatically, with demonstrated utility in inflammatory bowel disease (IBD), type 2 diabetes, colorectal cancer, and infectious diseases [11]. Recent advances have positioned metagenomics as a cornerstone of precision medicine, offering opportunities for improved diagnostics, risk stratification, and therapeutic development through robust microbial signature identification [11]. The technology now facilitates pathogen detection, antimicrobial resistance profiling, and patient stratification via enterotyping, making it an indispensable tool for both clinical and research settings [11].
Experimental Protocol: A Comprehensive Workflow
A standardized shotgun metagenomics protocol ensures reproducible and reliable results across studies. The following workflow outlines key stages from sample collection through data analysis, incorporating best practices for clinical and research applications.
Sample Collection and Preparation
Proper sample handling is critical for preserving microbial community structure and obtaining high-quality DNA:
- Sample Collection: Collect fecal samples or rectal swabs using sterile techniques. For rectal swabs, clean the perianal area with soap, water, and 70% alcohol prior to collection. Insert a sterile saline-moistened swab 4-5 cm into the anal canal and rotate gently to obtain fecal material [19]. Immediately place samples in sterile tubes and freeze at -80°C until processing [19].
- DNA Extraction: Use commercial kits designed for microbial DNA extraction, such as the MP-soil FastDNA Spin Kit for Soil or PowerSoil DNA Isolation Kit [19] [20]. The CTAB method is also preferred for certain sample types [20].
- Quality Control: Assess DNA purity, concentration, and quality using NanoDrop for purity measurements, fluorometry for concentration, and agarose gel electrophoresis for quality verification [19].
Library Preparation and Sequencing
Library construction and sequencing platform selection significantly impact data quality and resolution:
- Library Preparation: Fragment high-quality DNA to 250-300 bp lengths and construct libraries with 350bp insert sizes [20]. For PacBio HiFi sequencing, which provides long-read capabilities, SMRTbell libraries are prepared according to manufacturer specifications [21].
- Sequencing Platforms: Illumina platforms (NovaSeq, HiSeq) offer high output and accuracy for short-read sequencing [20] [16]. PacBio SMRT systems generate long reads (average up to 30 kb) that aid in assembling complex genomic regions and reconstructing closed genomes [16]. The choice between platforms depends on research goals: short-read for cost-effective profiling, long-read for resolving complex regions and achieving higher taxonomic resolution [21] [16].
Table 1: Sequencing Platform Comparison for Shotgun Metagenomics
| Platform | Read Length | Output | Key Advantages | Best Applications |
|---|---|---|---|---|
| Illumina NovaSeq | 150-250 bp | Up to 1.5Tb | High accuracy, low cost per base | Large-scale studies, taxonomic profiling |
| PacBio SMRT | Up to 30 kb | Varies | Long reads, minimal bias | Strain-level resolution, complex region assembly |
| Ion Torrent PGM | 200-400 bp | Varies | Rapid turnaround | Pathogen identification in clinical samples |
Data Processing and Quality Control
Raw sequencing data requires rigorous processing to eliminate artifacts and ensure analytical reliability:
- Quality Filtering: Remove adapter sequences, low-quality reads (average quality score <20), and short sequences (<50 bp) using tools like fastp [19].
- Host DNA Depletion: Map reads to the host genome (e.g., human) using BWA or similar alignment tools and remove matching sequences to reduce host contamination [19].
- Quality Assessment: Evaluate sequence quality scores across all bases, with particular attention to 3' end degradation common in Illumina platforms [18].
Bioinformatics Analysis Pipeline
The computational analysis of shotgun metagenomic data involves multiple steps to extract taxonomic and functional information from raw sequencing reads.
Assembly and Binning
Metagenome assembly reconstructs longer contiguous sequences from short reads:
- Assembly Approaches: Perform de novo assembly using de Bruijn graph-based tools such as metaSPAdes, MEGAHIT, or IDBA-UD [17] [16]. Reference-based assembly is preferable when closely related reference genomes are available [16].
- Binning Methods: Group contigs into putative genomes using compositional-based algorithms (S-GSOM, Phylopythia), similarity-based methods (IMG/M, MG-RAST), or hybrid approaches (PhymmBL) [16]. This process facilitates metagenome-assembled genome (MAG) construction for downstream analysis.
Taxonomic and Functional Profiling
Characterizing microbial community composition and functional capacity:
- Taxonomic Classification: Use tools like Kraken (k-mer based) or MetaPhlAn2 (clade-specific marker genes) to assign taxonomy to sequencing reads [17]. These methods compare sequences against comprehensive databases including Greengenes, SILVA, and RDP [17] [18].
- Functional Annotation: Identify protein-coding sequences using MetaGeneMark and annotate against functional databases including KEGG, eggNOG, CAZy, and CARD using BLAST or Diamond [20] [16] [19].
- Pathway Analysis: Employ the HUMAnN pipeline to determine the presence and abundance of microbial metabolic pathways [16].
Diversity and Statistical Analyses
Comparative analyses reveal differences in microbial communities across conditions:
- Alpha Diversity: Calculate within-sample diversity using Shannon Index, Observed Species, or Chao1 indices to assess community richness and evenness [17] [19].
- Beta Diversity: Evaluate between-sample differences through PCoA, NMDS, or PCA based on Bray-Curtis or other distance metrics [20] [19].
- Differential Abundance: Identify significantly different taxa or functions using statistical methods such as LEfSe, metagenomeSeq, or Wilcoxon rank-sum test [20] [19].
Research Reagent Solutions and Computational Tools
Successful implementation of shotgun metagenomics requires carefully selected reagents and bioinformatics tools. The following table outlines essential resources for conducting comprehensive gut microbiome studies.
Table 2: Essential Research Reagents and Computational Tools for Gut Metagenomics
| Category | Product/Tool | Application | Key Features |
|---|---|---|---|
| DNA Extraction Kits | PowerSoil DNA Isolation Kit | DNA extraction from complex samples | Effective for soil, sludge, and fecal samples |
| MP-soil FastDNA Spin Kit | DNA extraction from fecal samples | Comprehensive lysis of diverse microbes | |
| Sequencing Services | PacBio HiFi Sequencing | Long-read metagenomics | High accuracy, strain-level resolution |
| Illumina NovaSeq | High-throughput short-read sequencing | Cost-effective for large sample sizes | |
| Bioinformatics Tools | fastp | Quality control | Rapid adapter trimming and quality filtering |
| metaSPAdes | Metagenome assembly | De Bruijn graph approach for complex communities | |
| Kraken2 | Taxonomic classification | Ultra-fast k-mer based assignment | |
| MetaPhlAn2 | Taxonomic profiling | Clade-specific marker gene analysis | |
| HUMAnN2 | Functional profiling | Pathway abundance and coverage analysis | |
| Reference Databases | KEGG | Functional annotation | Metabolic pathways and enzyme functions |
| eggNOG | Functional annotation | Orthologous groups and functional classification | |
| CARD | Antibiotic resistance | Comprehensive resistance gene database | |
| SILVA | Taxonomic reference | Quality-checked ribosomal RNA database |
Applications and Case Studies
Shotgun metagenomics has enabled significant advances in understanding gut microbiome dynamics across various health and disease states.
Inflammatory Bowel Disease (IBD) Profiling
The "HiFi-IBD" project exemplifies the application of advanced metagenomics to inflammatory bowel disease. Researchers from Massachusetts General Hospital and Harvard University are utilizing PacBio HiFi sequencing to optimize protocols for gut metagenomics in IBD samples from the Nurses' Health Study 2 [21]. This approach enables precise functional gene profiling via HUMAnN 4 and strain-resolved analysis not achievable with short-read technologies [21]. The project aims to generate high-quality, long-read data that reveals microbial signatures specific to IBD subtypes, potentially identifying novel therapeutic targets and diagnostic biomarkers.
Acute Pancreatitis Recovery Monitoring
A 2025 study investigated gut microbiome dynamics during recovery from acute pancreatitis (AP) using shotgun metagenomics [19]. Researchers collected rectal swabs from 12 AP patients during both acute and recovery phases, conducting sequencing on the Illumina HiSeq 4000 platform [19]. Analysis revealed that during recovery from mild AP, beneficial bacteria (Bacteroidales) increased while harmful bacteria (Firmicutes) decreased [19]. However, in severe AP cases, Enterococcus abundance increased during recovery, suggesting incomplete microbial restoration [19]. Functional annotation using KEGG pathways identified specific metabolic shifts associated with clinical improvement, providing insights into microbial functions during disease recovery [19].
Colorectal Adenoma-Carcinoma Sequence
Investigators at Chulabhorn Royal Academy are applying HiFi shotgun metagenomics to study gut microbiome functional contributions to colorectal adenoma progression [21]. Previous full-length 16S rRNA sequencing revealed predicted metabolic pathways associated with polyps, but deeper metagenomic sequencing enables reconstruction of metagenome-assembled genomes (MAGs) for precise taxonomic and functional profiling [21]. This approach identifies specific microorganisms driving the adenoma-carcinoma sequence, potentially revealing novel targets for microbiome-based prevention and early intervention in colorectal carcinogenesis [21].
Technical Considerations and Challenges
Despite its powerful capabilities, shotgun metagenomics presents several technical challenges that researchers must address:
- Computational Requirements: Data analysis demands substantial computational resources and expertise in bioinformatics [16]. The large volumes of data generated (typically 10-14 Gb per sample) require high-performance computing infrastructure for efficient processing [19].
- Standardization Limitations: Lack of standardized protocols for sample collection, processing, and data analysis limits comparability across studies [22] [11]. Initiatives like the STORMS checklist and NIST reference materials aim to address these issues [11].
- Data Interpretation Complexity: Microbiome data characteristics including zero-inflation, overdispersion, and compositional effects complicate statistical analysis [23]. Methods like ANCOM, DESeq2, and metagenomeSeq have been developed to address these challenges [23].
- Reference Database Limitations: Incomplete functional annotation databases and underrepresented global populations hinder comprehensive interpretation [11]. Nearly 40-50% of sequenced genes lack definitive functional assignments, creating interpretation gaps [17].
Emerging Applications and Future Directions
Shotgun metagenomics continues to evolve with promising new applications in clinical and research settings:
- Multi-omics Integration: Combining metagenomics with metatranscriptomics, metabolomics, and metaproteomics provides more comprehensive insights into microbial community function [11] [17]. For example, integrating metagenomics with serum metabolomics has identified gut microbiota-derived metabolites associated with type 2 diabetes progression [11].
- Precision Medicine Applications: Microbial signature-based diagnostics are emerging for conditions including colorectal cancer, where machine learning frameworks integrating metagenomic data with clinical parameters show superior predictive accuracy [11].
- Therapeutic Monitoring: Metagenomics guides personalized microbiome therapies like fecal microbiota transplantation (FMT) by tracking donor strain engraftment and functional restoration [11].
- Antimicrobial Resistance Surveillance: Comprehensive antibiotic resistance gene profiling using CARD database annotations enables tracking of resistance patterns across populations and informs stewardship programs [20] [16].
Shotgun metagenomics has revolutionized our ability to study the human gut microbiome as a complex ecosystem, providing unprecedented resolution for both taxonomic classification and functional potential assessment. The comprehensive workflow outlined in this application note—from standardized sample collection through advanced bioinformatics analysis—enables researchers to generate robust, reproducible data on microbial community structure and function. As sequencing technologies continue to advance and computational methods become more sophisticated, shotgun metagenomics will play an increasingly central role in elucidating host-microbiome interactions and developing microbiome-based diagnostics and therapeutics.
The MetaHIT (Metagenomics of the Human Intestinal Tract) and the Human Microbiome Project (HMP) represent landmark initiatives that have fundamentally advanced our understanding of the human gut microbiome through shotgun metagenomics. Unlike traditional 16S rRNA sequencing that targets specific phylogenetic markers, shotgun metagenomics enables comprehensive sampling of all genes from all microorganisms present in a complex sample [7] [8]. This approach provides unparalleled insights into both the taxonomic composition and functional potential of microbial communities, allowing researchers to study unculturable microorganisms that are otherwise difficult or impossible to analyze [8]. These projects have established critical reference databases and standardized methodologies that continue to shape experimental design and analysis in gut microbiome research, paving the way for novel diagnostic and therapeutic applications [24] [25].
Key Findings and Quantitative Insights
The MetaHIT and HMP initiatives have generated substantial quantitative datasets that reveal the extraordinary complexity of the human gut microbiome. The following tables summarize core quantitative findings and methodological outputs from these projects.
Table 1: Core Quantitative Findings from Major Microbiome Initiatives
| Metric | MetaHIT Findings | Human Microbiome Project (HMP) | Significance |
|---|---|---|---|
| Gene Catalog Size | 3.3 million non-redundant genes [26] | 2 million unique genes (estimated) [24] | 150× larger than human gene complement [26] |
| Microbial Cells | 1013-1014 cells/g fecal matter [7] | Ratio of 1.3 bacterial cells per human cell [24] | Microbial cells outnumber human cells [24] |
| Bacterial Diversity | 1,000-1,150 prevalent bacterial species; ~160 species/individual [26] | 500-1,000 species in human body [24] | Individual uniqueness with shared core [26] [24] |
| Sequencing Output | 576.7 Gb from 124 individuals [26] | 541 gut samples in initial phase; >2,000 in HMP2 [25] | Unprecedented data scale enabling robust analysis |
Table 2: Methodological Advances and Technical Specifications
| Parameter | MetaHIT Protocol | Typical Shotgun Metagenomics Workflow |
|---|---|---|
| Sequencing Technology | Illumina Genome Analyser [26] | Illumina platforms (MiSeq, NovaSeq) [8] |
| Assembly Approach | SOAPdenovo de Bruijn graph-based assembly [26] | metaSPAdes, MEGAHIT [25] |
| Gene Prediction | MetaGene [26] | Prodigal, FragGeneScan |
| Data Analysis | Non-redundant gene set (95% identity over 90% length) [26] | DRAGEN Metagenomics pipeline, taxonomic classification [8] |
| Key Innovation | Establishment of minimal gut metagenome and core functions [26] | Genome-resolved metagenomics (MAGs) [25] |
Beyond these quantitative measures, MetaHIT made the crucial discovery of enterotypes—three distinct gut microbial community types dominated by Bacteroides, Prevotella, or Ruminococcus [27]. This finding suggests that human gut microbiome variation is stratified rather than continuous, with potential implications for personalized nutrition and medicine. The HMP further contributed to understanding microbial biogeography by revealing that each body site develops a specific microbial signature, with the gut exhibiting particularly high diversity compared to skin, oral, and vaginal sites [24].
Shotgun Metagenomics Protocol for Gut Microbiome Analysis
Sample Preparation and Library Construction
The following protocol provides a optimized workflow for shotgun metagenomic library construction from complex samples like fecal material, incorporating best practices from established methodologies [28].
Table 3: Reagent Formulations for Library Preparation
| Component | Specification | Purpose |
|---|---|---|
| Fx Buffer 10x | Part of QIAseq FX DNA Library Core Kit (Cat. No. 1120146) [28] | Provides optimal reaction environment for fragmentation |
| FX Enzyme Mix | Included in QIAseq FX DNA Library Core Kit [28] | Enzymatic DNA fragmentation |
| QIAseq UDI Adapters | Available in Y-Adapter Kit B (96) (Cat. No. 180314) [28] | Sample indexing and platform compatibility |
| AMPure XP Beads | Beckman Coulter (Cat. No. A63880) [28] | Size selection and purification |
| HiFi PCR Master Mix | Included in QIAseq FX DNA Library Core Kit [28] | High-fidelity library amplification |
Procedure:
DNA Fragmentation:
- Normalize DNA concentration to 1 ng/μL using Tris 10 mM or water (avoid EDTA-containing buffers) [28].
- Prepare fragmentation reaction on ice according to Table 4.
- Add 10 μL of FX Enzyme Mix to each reaction and mix thoroughly by pipetting.
- Run fragmentation program on pre-chilled thermocycler: 4°C for 1 min, 32°C for 14-24 min (time dependent on input DNA), 65°C for 30 min for enzyme inactivation, and hold at 4°C [28].
Table 4: Fragmentation Reaction Setup
Component 10 ng Input DNA 20 pg Input DNA Fx Buffer 10x 5.0 μL 5.0 μL DNA 10.0 μL 20.0 μL Nuclease-free Water 25.0 μL 12.5 μL Fx Enhancer - 2.5 μL Total Volume 40.0 μL 40.0 μL Adapter Ligation:
- Prepare adapter dilutions: 1:15 for 10 ng DNA input, 1:300 for 20 pg DNA input [28].
- Add 5 μL of diluted adapters to each fragmentation tube.
- Add 45 μL of adapter ligation mix (20 μL DNA ligase buffer 5x, 10 μL DNA ligase, 15 μL nuclease-free water) [28].
- Incubate at 20°C for 30 minutes in a thermocycler without heated lid.
Purification and Cleanup:
- Purify adapter-ligated DNA using FastGene Gel/PCR Extraction Kit following manufacturer's instructions.
- Elute DNA with 40 μL of elution buffer [28].
Library Amplification:
- Prepare library amplification mix according to Table 5.
- Run amplification program: 98°C for 2 min (initial denaturation); 10 cycles of 98°C for 20 s, 60°C for 30 s, 72°C for 30 s; final extension at 72°C for 1 min; hold at 4°C [28].
- For low input DNA (20 pg), increase to 16 cycles [28].
Table 5: Library Amplification Mix
Component Volume HiFi PCR Master Mix 2x 25.0 μL Primer Mix (10 μM) 1.5 μL Library Purified 23.5 μL Total Volume 50.0 μL Library Quantification and Pooling:
- Clean amplified library with FastGene Gel/PCR Extraction Kit.
- Quantify using qPCR with EvaGreen chemistry.
- Normalize each sample to 4 ng/μL and pool equimolar amounts.
- Perform final cleanup with AMPure XP beads (80 μL beads to 75 μL pooled library) [28].
Sequencing and Data Analysis Workflow
The following diagram illustrates the complete shotgun metagenomics workflow from sample preparation to data analysis:
Advanced Methodological Considerations
Genome-Resolved Metagenomics
A significant advancement beyond initial MetaHIT and HMP methodologies is genome-resolved metagenomics, which reconstructs microbial genomes directly from whole-metagenome sequencing data [25]. This approach involves assembling short reads into longer contigs followed by "binning" to group contigs into metagenome-assembled genomes (MAGs). The process employs two primary assembly models: Overlap-Layout-Consensus (OLC) and De Bruijn graph-based approaches, with the latter being particularly effective for high-complexity samples like gut microbiota [25]. This technique has dramatically expanded the catalog of microbial genomes from uncultured species and enables study of strain-level variation, horizontal gene transfer, and functional adaptation within gut ecosystems.
Absolute Quantitative Metagenomics
Traditional shotgun metagenomics provides relative abundance data, but emerging absolute quantitative approaches address crucial limitations by measuring actual microbial concentrations [29]. Techniques incorporating spike-in internal standards with known concentrations allow precise quantification of absolute microbial abundances, providing more accurate insights into microbial community dynamics, particularly in intervention studies [29]. This approach has revealed that drugs like berberine and metformin significantly alter absolute abundances of beneficial microbes like Akkermansia muciniphila, changes that relative quantification methods may obscure [29].
Integrated Multi-Kingdom Analysis
Recent methodologies enable simultaneous analysis of bacterial and fungal communities (mycobiome) through optimized enrichment protocols and comprehensive databases [30]. Fungal cells can be enriched from fecal samples using size-based separation techniques (centrifugation) due to their larger cell size (2-10 μm for yeast vs. 0.2-2 μm for bacteria) [30]. This integrated approach reveals interkingdom interactions and competition for nutrients, providing a more comprehensive understanding of gut ecosystem dynamics.
Applications in Drug Development and Personalized Medicine
The methodological frameworks established by MetaHIT and HMP have enabled significant advances in pharmaceutical research and development:
- Microbiome-Drug Interactions: Studies demonstrate that pharmaceutical compounds like metformin and berberine exert therapeutic effects partly through microbiome modulation, increasing abundance of beneficial taxa like Akkermansia and Bifidobacterium while decreasing conditionally pathogenic species [29].
- Microbiome-Targeted Therapeutics: The gut microbiome represents a promising therapeutic target itself, with approaches including probiotics, prebiotics, and fecal microbiota transplantation (FMT) showing efficacy for conditions like metabolic syndrome, inflammatory bowel disease, and Clostridium difficile infection [24] [31].
- Biomarker Discovery: Microbiome-based biomarkers show remarkable diagnostic potential, with gut microbial signatures classifying lean vs. obese individuals with >90% accuracy in case-control studies and providing superior diagnostic value compared to human genetic factors for conditions like C. difficile infection [24].
Table 6: Essential Research Reagents and Computational Tools
| Resource Category | Specific Tools/Reagents | Application |
|---|---|---|
| Wet Lab Reagents | QIAseq FX DNA Library Core Kit [28] | High-quality library preparation for low-input samples |
| AMPure XP Beads [28] | Size selection and purification | |
| FastDNA SPIN Kit for Soil [29] | Effective DNA extraction from complex samples | |
| Sequencing Platforms | Illumina MiSeq/NovaSeq [8] | High-throughput shotgun metagenomic sequencing |
| PacBio Sequel II [29] | Full-length 16S rRNA sequencing | |
| Bioinformatics Tools | SOAPdenovo [26] | De novo assembly of short reads |
| MetaGene [26] | ORF prediction from metagenomic sequences | |
| DRAGEN Metagenomics [8] | Taxonomic classification and analysis | |
| metaSPAdes, MEGAHIT [25] | Modern metagenome assemblers | |
| Reference Databases | Integrated Microbial Genomes [7] | Reference genome database |
| FunOMIC-T [30] | Fungal gene catalog and analysis tool |
The methodological frameworks established by MetaHIT and the Human Microbiome Project have provided the foundation for modern gut microbiome research using shotgun metagenomics. These initiatives demonstrated the unprecedented genetic diversity of human-associated microbial communities and developed standardized approaches for sample processing, sequencing, and bioinformatic analysis. Current advancements in genome-resolved metagenomics, absolute quantification, and multi-kingdom integration are building upon these foundations to enable more precise and comprehensive characterization of gut ecosystem structure and function. These protocols continue to evolve, driving discoveries in host-microbiome interactions and accelerating the development of microbiome-based diagnostics and therapeutics for human health.
Shotgun metagenomic sequencing represents a transformative approach in clinical microbiology, enabling the comprehensive analysis of all genetic material within a complex sample [8]. This culture-independent method allows researchers and clinicians to evaluate microbial diversity, detect pathogens, and profile functional genes, including those associated with antimicrobial resistance (AMR) and metabolic pathways, directly from patient specimens [11] [5]. Unlike targeted 16S rRNA sequencing, which is limited to taxonomic classification, shotgun metagenomics provides a holistic view of the microbiome's functional potential, opening new avenues for precision medicine [5] [32]. The clinical translation of this technology is now revolutionizing diagnostics, therapeutic monitoring, and patient stratification across infectious, inflammatory, metabolic, and neoplastic diseases [11] [33].
The power of shotgun metagenomics lies in its ability to generate hypotheses about microbial community functions and to identify actionable biomarkers for clinical decision-making [34]. By moving beyond correlation to causation through integrated multi-omics and mechanistic validation, researchers can now begin to unravel the complex interplay between host and microbiome in health and disease [32] [34]. This application note outlines standardized protocols and analytical frameworks to facilitate the robust implementation of shotgun metagenomics in clinical research settings, with a specific focus on gut microbiome applications in diagnostics, therapeutics, and precision medicine.
Clinical Applications and Performance Metrics
Diagnostic Applications
Shotgun metagenomics has demonstrated exceptional capabilities in clinical diagnostics, particularly in scenarios where traditional culture-based methods fail. The technology enables sensitive pathogen detection and comprehensive antimicrobial resistance profiling, providing clinicians with critical information for targeted therapeutic interventions [11].
Table 1: Clinical Diagnostic Performance of Shotgun Metagenomics Across Disease States
| Disease Area | Pathogens/Features Detected | Clinical Performance | Reference Method Comparison |
|---|---|---|---|
| Central Nervous System (CNS) Infections | Bacteria, viruses, fungi, parasites (e.g., Leptospira santarosai, Balamuthia mandrillaris) [11] | Increased diagnostic yield by 6.4% in culture-negative cases [11] | Unbiased mNGS detected unexpected pathogens missed by conventional testing [11] |
| Bone and Joint Infections | Polymicrobial and fastidious organisms [11] | ~18% higher diagnostic yield than culture alone [11] | 16S rRNA sequencing detected pathogens in patients on antimicrobial therapy [11] |
| Bloodstream Infections (Sepsis) | Diverse bacterial pathogens and AMR genes [11] | Pathogen identification up to 30 hours earlier than culture [11] | Shotgun metagenomics from blood enabled timely, targeted therapy [11] |
| Lower Respiratory Infections | Bacterial pathogens and AMR genes [11] | 96.6% sensitivity, 41.7% specificity vs. culture; 100% qPCR confirmation [11] | Rapid 6-hour nanopore sequencing with host DNA depletion [11] |
| Inflammatory Bowel Disease (IBD) | Microbial signatures (Asaccharobacter celatus, Gemmiger formicilis, Erysipelatoclostridium ramosum) [11] | AUROC 0.92-0.98 for distinguishing IBD from controls [11] | Multi-omics integration (metagenomics & metabolomics) [11] |
| Clostridioides difficile Infection | C. difficile and closely related species [11] | >99% true positive rate with minimal false positives [11] | Shotgun metagenomics combined with high-resolution 16S analysis [11] |
Therapeutic Applications and Monitoring
Beyond diagnostics, shotgun metagenomics plays a crucial role in guiding therapeutic decisions and monitoring treatment efficacy. The technology enables precision antimicrobial therapy through rapid detection of AMR genes and facilitates personalized microbiome-based interventions such as fecal microbiota transplantation (FMT) [11].
Table 2: Therapeutic Applications of Shotgun Metagenomics
| Therapeutic Area | Application | Metagenomic Assessment | Clinical Impact |
|---|---|---|---|
| Antimicrobial Stewardship | AMR gene detection directly from clinical specimens [11] | Real-time identification of resistance patterns [11] | Reduction in broad-spectrum antibiotic use; targeted therapy [11] |
| Fecal Microbiota Transplantation (FMT) | Donor selection and engraftment monitoring [11] | Strain tracking and metabolic pathway restoration assessment [11] | Correlation between donor strain engraftment and clinical improvement [11] |
| Precision Nutrition | Microbiome response to dietary interventions [32] | Functional gene shifts and metabolite production [32] | Personalized dietary recommendations based on microbial capacity [32] |
| Cancer Therapy | Modulation of immunotherapy response [33] | Taxonomic and functional profiling pre- and post-treatment [33] | Identification of microbial signatures predictive of treatment outcome [33] |
Experimental Protocols
Sample Collection and DNA Extraction Protocol
Principle: Optimal sample collection and DNA extraction are critical for obtaining high-quality, non-biased metagenomic data. The protocol must preserve microbial community structure while maximizing DNA yield and quality.
Reagents and Equipment:
- Sample collection kits (e.g., OMNIgene Gut OMR-200)
- Lysis buffer (e.g., QIAGEN PowerBead Solution)
- Proteinase K
- RNase A
- Magnetic bead-based purification beads
- Ethanol (70% and 100%)
- Elution buffer (10 mM Tris-HCl, pH 8.5)
- Thermal mixer
- Centrifuge
- Qubit fluorometer
- TapeStation or Bioanalyzer
Procedure:
- Sample Collection:
- Collect fresh stool samples in DNA-stabilizing collection kits OR immediately freeze at -80°C until processing
- For clinical specimens, process within 2 hours of collection or use preservative solutions
Cell Lysis:
- Aliquot 200 mg of stool into a tube containing 1 ml lysis buffer and 0.5 g of sterile zirconia beads
- Add 20 µl Proteinase K and 10 µl RNase A
- Vortex vigorously for 10 minutes at maximum speed
- Incubate at 65°C for 30 minutes with intermittent mixing
DNA Purification:
- Centrifuge at 13,000 × g for 5 minutes
- Transfer supernatant to a new tube
- Add 1.5 volumes of magnetic bead solution and incubate for 10 minutes
- Place on magnetic stand for 5 minutes until clear
- Discard supernatant
- Wash twice with 700 µl 70% ethanol
- Air dry for 10 minutes
- Elute DNA in 100 µl elution buffer
Quality Control:
- Quantify DNA using Qubit fluorometer
- Assess integrity via TapeStation or Bioanalyzer
- Confirm DNA concentration >5 ng/µl and fragment size >10 kb
Troubleshooting:
- Low yield: Increase starting material or extend lysis time
- DNA degradation: Reduce processing time or use different preservative
- Inhibitor carryover: Additional wash steps or dilution may be required
Library Preparation and Sequencing Protocol
Principle: Library preparation converts extracted DNA into sequencing-ready fragments with appropriate adapters. The choice of sequencing platform and depth depends on the specific research question and required resolution.
Reagents and Equipment:
- Illumina DNA Prep Kit
- IDT Unique Dual Indexes
- AMPure XP beads
- Thermal cycler
- Illumina sequencing platform (NovaSeq, HiSeq, or MiSeq)
Procedure:
- DNA Shearing:
- Dilute 100 ng DNA in 50 µl resuspension buffer
- Fragment DNA to target size of 350 bp using Covaris sonicator or enzymatic fragmentation
Library Preparation:
- Perform end repair and A-tailing according to manufacturer's protocol
- Ligate Illumina adapters with Unique Dual Indexes
- Clean up with AMPure XP beads (0.8X ratio)
- Elute in 25 µl resuspension buffer
Library Amplification:
- Amplify libraries with 8 cycles of PCR
- Clean up with AMPure XP beads (0.8X ratio)
- Elute in 25 µl resuspension buffer
Quality Control and Quantification:
- Assess library size distribution using TapeStation
- Quantify libraries by qPCR using library quantification kit
- Pool libraries at equimolar concentrations
Sequencing:
- Load pool at appropriate concentration for sequencing platform
- Sequence on Illumina platform with 2×150 bp configuration
- Target 10-20 million reads per sample for species-level resolution
Alternative Platforms:
- For long-read sequencing: Pacific Biosciences SEQUEL II or Oxford Nanopore Technologies MinION
- For rapid turnaround: Oxford Nanopore Technologies with 6-hour rapid sequencing protocols [11]
Bioinformatic Analysis Pipeline
Principle: Computational analysis transforms raw sequencing data into biologically meaningful information through quality control, taxonomic profiling, functional annotation, and association testing.
Software Requirements:
- FastQC (v0.11.9) - quality control
- Trimmomatic (v0.39) - adapter trimming
- KneadData (v0.10.0) - host decontamination
- MetaPhlAn (v4.0) - taxonomic profiling
- HUMAnN (v3.6) - functional profiling
- SHOGUN (v1.1.0) - taxonomic profiling with multiple aligners
- MaAsLin2 (v1.10.0) - association testing
Procedure:
- Quality Control:
Host DNA Depletion:
Taxonomic Profiling:
Functional Profiling:
Pathway Analysis:
Visualizing Metagenomic Data and Workflows
Clinical Metagenomic Analysis Workflow
The following diagram illustrates the comprehensive workflow from sample collection to clinical interpretation in shotgun metagenomic studies:
Multi-Omics Integration Pathway
The integration of multiple data layers is essential for advancing from correlation to causation in microbiome research:
Table 3: Essential Research Reagents and Computational Tools for Clinical Metagenomics
| Category | Resource | Specific Examples | Application |
|---|---|---|---|
| Reference Databases | SILVA [16] | SSU and LSU rRNA gene databases | Taxonomic classification and phylogenetic analysis |
| Greengenes [16] | Curated 16S rRNA gene database | Taxonomic assignment in bacterial communities | |
| KEGG [16] | Kyoto Encyclopedia of Genes and Genomes | Pathway analysis and functional annotation | |
| CARD [16] | Comprehensive Antibiotic Resistance Database | Detection and characterization of AMR genes | |
| CAZy [16] | Carbohydrate-Active enZYmes Database | Analysis of carbohydrate-active enzymes | |
| Bioinformatic Tools | HUMAnN [16] | HMP Unified Metabolic Analysis Network | Quantification of microbial pathway abundance |
| MG-RAST [16] | Metagenomics RAST server | Automated phylogenetic and functional analysis | |
| MetaPhlAn [35] | Metagenomic Phylogenetic Analysis | Profiling microbial community composition | |
| MEGAHIT [35] | Metagenome assembler | De novo assembly of metagenomic sequences | |
| Experimental Standards | NIST Reference Materials [11] | Stool microbiome reference standards | Protocol validation and inter-laboratory calibration |
| STORMS Checklist [11] | Strengthening Reporting of Microbiome Studies | Standardized reporting of microbiome research | |
| Multi-omics Integration | eggNOG [16] | Evolutionary genealogy of genes: Non-supervised Orthologous Groups | Functional annotation and orthology prediction |
| MaAsLin2 [32] | Multivariate Association with Linear Models | Identifying multivariable associations in microbiome data | |
| QIIME 2 [32] | Quantitative Insights Into Microbial Ecology | Integrated microbiome analysis platform |
Challenges and Future Directions
Despite the considerable promise of shotgun metagenomics in clinical translation, several challenges remain. Methodological variability, incomplete functional annotation of microbial "dark matter," lack of bioinformatics standardization, and underrepresentation of global populations in reference databases continue to hinder routine clinical implementation [11]. Additionally, the establishment of clinically relevant thresholds for microbial abundance and the definition of a "healthy" microbiome baseline across diverse populations present ongoing challenges [11] [33].
Future advances will require globally harmonized standards, cross-sector collaboration, and inclusive frameworks that ensure scientific rigor and equitable benefit [11]. The integration of machine learning and artificial intelligence with multi-omics data holds particular promise for unlocking complex host-microbe interactions and generating clinically actionable insights [32]. Furthermore, the development of rapid, point-of-care metagenomic sequencing technologies will accelerate the translation of microbiome science into routine clinical practice, ultimately fulfilling the promise of precision medicine guided by our microbial inhabitants [11] [8].
As standardization improves and costs decrease, shotgun metagenomics is poised to become an integral component of clinical diagnostics and therapeutic monitoring, enabling a new era of microbiome-informed personalized medicine [11] [33] [8].
A Step-by-Step Shotgun Metagenomics Protocol for Gut Microbiome Profiling
In shotgun metagenomics for gut microbiome research, the integrity of data is highly dependent on pre-analytical procedures. Sample collection and preservation methods directly impact the quantitative and qualitative measurements of microbial communities, influencing downstream taxonomic and functional analyses. Establishing standardized protocols is therefore critical for generating reliable, reproducible, and comparable data across studies, particularly in translational research and drug development. This application note details evidence-based protocols for maintaining sample integrity from stool collection through sequencing library preparation, providing researchers with a framework to minimize technical bias in gut microbiome research.
The Impact of Collection Methods on Microbiome Integrity
The choice of sample collection and preservation system introduces specific taxonomic biases that must be considered during study design. A 2025 comparative metagenomics analysis of paired fecal samples highlighted significant differences in microbial profiles between two common preservation methods: Flinters Technology Associates (FTA) cards and OMNIgene (OG) Gut tubes [36].
Key Findings from Comparative Analysis:
- Nucleic Acid Yield: OMNIgene Gut tubes consistently yielded higher nucleic acid concentrations, sequencing library concentrations, and a greater number of post-processed sequenced reads compared to FTA cards [36].
- Taxonomic Biases: At the phylum level, FTA cards showed higher relative abundances of Proteobacteria (3.07% vs. 0.96%) and Actinobacteria (14.95% vs. 6.17%), whereas OG tubes yielded higher levels of Bacteroidetes (37.02% vs. 26.34%) [36].
- Diversity Metrics: While the number of observed species and Shannon diversity were not significantly different, Simpson diversity was significantly higher in OG samples [36].
- Differential Abundance: Corynebacterium was consistently more abundant in FTA samples, while Blautia was higher in OG samples across all study groups [36].
Table 1: Comparison of Sample Collection Methods for Fecal Metagenomics
| Feature | OMNIgene Gut Tube | FTA Cards | Immediate Freezing (-70°C) |
|---|---|---|---|
| Primary Preservation Mechanism | Chelating agent-based solution for nucleic acid stabilization [36] | Cellulose-based matrix with chemicals to lyse cells and stabilize nucleic acids [36] | Halts all microbial and enzymatic activity [37] |
| Typical Nucleic Acid Yield | Higher concentrations [36] | Lower concentrations [36] | Considered the reference standard |
| Key Taxonomic Biases | Higher Bacteroidetes; Higher Blautia [36] | Higher Proteobacteria, Actinobacteria; Higher Corynebacterium [36] | Minimal, but requires cold chain |
| Best Application | Large cohort studies where cold chain is impractical; requires high biomass [36] | Field/deployment settings with extreme temperatures; low biomass targets [36] | Controlled clinical settings where cold chain is feasible [37] |
These findings underscore that consistent use of a single collection protocol within a study is paramount, as data generated using different methods should not be directly compared without appropriate normalization.
Protocols for Sample Collection and Storage
Field-Expedient Collection Protocol for Ambient Temperature Storage
This protocol is adapted for studies in remote or deployment settings where immediate freezing is not possible, based on a 2025 comparative assessment [36].
Materials:
- OMNIgene Gut OMR-200 Kit (DNA Genotek) or FTA Cards (Flinters Technology)
- Disposable gloves and sample collection paper
- Disposable spatula or spoon
- Permanent marker for labeling
- Waterproof storage bag with desiccant
Procedure:
- Preparation: Label the collection tube or FTA card with the participant ID and date prior to distribution.
- Collection: Defecate directly onto clean collection paper or a clean, dry container.
- Sampling: Using a disposable spatula, collect a representative sample from multiple areas of the stool, including the core.
- For OMNIgene Gut Tube: Fill the tube with stool to the fill line (approximately 100 mg). Ensure the stool is submerged in the stabilizing solution, close the lid tightly, and shake vigorously for 30 seconds to homogenize [36].
- For FTA Cards: Apply a pea-sized stool sample directly to the designated circle on the FTA card, ensuring it soaks into the matrix. Allow the sample to dry completely at room temperature for several hours [36].
- Storage: Store the sealed collection tube or dried FTA card at ambient temperature (20-25°C) protected from direct light. For the OMNIgene Gut tube, storage can be for up to 60 days as per manufacturer's claims. Evidence suggests FTA cards are effective for room temperature preservation for up to 8 weeks [36].
- Shipment: Package samples according to IATA regulations for biological substances and ship at ambient temperature to the processing laboratory.
Optimal Long-Term Storage Protocol for Taxonomic and Functional Stability
Long-term storage stability is crucial for multi-center trials and longitudinal studies. A 2023 study evaluated the stability of the fecal microbial community for up to 18 months under various conditions [37].
Materials:
- DNA/RNA Shield Fecal Collection Tubes (Zymo Research)
- -70°C or -80°C Freezer
- Disposable gloves and spatula
Procedure:
- Homogenization: Homogenize the entire stool sample upon collection. Sub-sampling from a non-homogenized stool can introduce significant variability [37].
- Aliquoting: Transfer a representative aliquot (e.g., 100-200 mg) into a DNA/RNA Shield Fecal Collection Tube. The preservative solution inactivates microbes and protects nucleic acids from degradation.
- Initial Storage: Immediately place the sealed tube in a -70°C to -80°C freezer. If such a freezer is not immediately available, storage at -20°C is a short-term alternative, though less ideal.
- Long-Term Stability: For studies requiring storage beyond one year, samples preserved in DNA/RNA Shield and stored at -70°C show the best preservation of taxonomic composition, alpha and beta diversity, and inferred functional pathway abundances for at least 18 months [37]. Avoid repeated freeze-thaw cycles.
Table 2: DNA Extraction Kit Performance for Shotgun Metagenomics
| DNA Extraction Kit | Lysis Method | Purification Method | Reported Performance for ONT Sequencing |
|---|---|---|---|
| QIAamp PowerFecal Pro DNA Kit (Qiagen) | Chemical & Mechanical (bead beating) [38] | Spin-column [38] | Identified all species (8/8) in Zymo Mock and (6/6) in ESKAPE Mock; best for AMR gene detection [38]. |
| QIAamp DNA Mini Kit (Qiagen) | Enzymatic (Lysozyme & Proteinase K) [38] | Spin-column [38] | Potential bias against Gram-positive species with enzymatic lysis alone [38]. |
| Maxwell RSC Cultured Cells Kit (Promega) | Enzymatic (Lysozyme) [38] | Magnetic beads [38] | Performance varies; mechanical lysis often superior for Gram-positives [38]. |
| Maxwell RSC Buccal Swab Kit (Promega) | Enzymatic (Proteinase K) [38] | Magnetic beads [38] | May be less effective for complex, tough-to-lyse gut communities. |
Wet-Lab Workflow: From DNA to Sequencer
The following diagram and protocol outline the key steps for preparing sequencing-ready libraries from stool samples.
DNA Extraction and Quantification
Procedure:
- Extraction: Use a kit that combines chemical and mechanical lysis (bead beating), such as the QIAamp PowerFecal Pro DNA kit, to ensure robust extraction from both Gram-positive and Gram-negative bacteria [38]. Follow the manufacturer's protocol for inhibitor removal to ensure high-quality DNA.
- Quantification: Quantify the extracted DNA using a fluorescence-based method like the Qubit dsDNA HS Assay Kit. Avoid spectrophotometric methods (e.g., NanoDrop) as they are sensitive to contaminants and can overestimate concentration [38] [39].
Library Preparation for Shotgun Sequencing
This protocol is adapted for Illumina platforms using the NEBNext Ultra II FS DNA Library Prep Kit [39].
Materials:
- NEBNext Ultra II FS DNA Library Prep Kit (New England BioLabs)
- Unique Dual Index Primers (e.g., Illumina)
- Qubit dsDNA HS Assay Kit (ThermoFisher Scientific)
- Agilent 2100 Bioanalyzer with High Sensitivity DNA Kit (Agilent Technologies)
- AMPure XP Beads (Beckman Coulter)
Procedure:
- End-Prep and dA-Tailing: Combine up to 100 ng of metagenomic DNA with the NEBNext Ultra II FS Enzyme Mix and Reaction Buffer. Incubate in a thermal cycler at the recommended temperatures and times (e.g., 5 minutes at 20°C, then 5 minutes at 65°C) to create blunt-ended, 5'-phosphorylated fragments with a single 'A' overhang [39].
- Adapter Ligation: Ligate the uniquely dual-indexed adapters to the 'A'-tailed DNA fragments using the NEBNext Ligation Master Mix. Incubate for 15 minutes at 20°C. Stop the reaction with EDTA if necessary.
- Size Selection and Cleanup: Purify the ligated product using AMPure XP beads. A double-sided size selection (e.g., 0.5X followed by 0.8X bead-to-sample ratio) is often performed to remove adapter dimers and select for the desired insert size.
- Library QC: Validate the final library's fragment length distribution and concentration using the Agilent Bioanalyzer High Sensitivity DNA Kit and Qubit, respectively [39]. Pool equimolar amounts of individually indexed libraries for multiplexed sequencing.
- Sequencing: Sequence on an Illumina NovaSeq or comparable platform to a depth of at least 2 Gb data per sample for robust taxonomic and functional profiling [39].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Gut Metagenomics
| Item | Function | Example Products & Notes |
|---|---|---|
| Ambient Collection Devices | Stabilizes nucleic acids at room temperature for transport. | OMNIgene Gut Kit (solution-based), FTA Cards (matrix-based). Choice depends on yield needs and field conditions [36]. |
| DNA Extraction Kits with Mechanical Lysis | Breaks down tough microbial cell walls for unbiased DNA recovery. | QIAamp PowerFecal Pro DNA Kit. Bead-beating is critical for Gram-positive bacteria [38]. |
| Fluorometric DNA Quantification Kits | Accurately measures double-stranded DNA concentration for library input. | Qubit dsDNA HS Assay Kit. Preferable over NanoDrop for accuracy with complex samples [38] [39]. |
| Ligation-Based Library Prep Kits | Prepares DNA fragments for sequencing by adding platform-specific adapters. | NEBNext Ultra II FS DNA Library Prep Kit. Provides high complexity libraries from low inputs [39]. |
| Nucleic Acid Cleanup Beads | Purifies and size-selects DNA fragments post-reaction (e.g., post-ligation). | AMPure XP Beads. Used for removing primers, adapters, and selecting insert sizes [40]. |
| Fragment Analyzer Systems | Assesses the quality and average size of final sequencing libraries. | Agilent Bioanalyzer/TapeStation. Essential QC step before sequencing to ensure library integrity [39]. |
The journey from stool to sequencer is fraught with potential biases that can compromise the integrity of gut microbiome data. This application note demonstrates that the choice of collection method (e.g., OMNIgene vs. FTA) directly influences taxonomic profiles, while long-term storage in DNA/RNA Shield preservative is optimal for functional and taxonomic stability. Furthermore, a DNA extraction protocol incorporating mechanical lysis is non-negotiable for unbiased representation of the microbial community. By adhering to these standardized, evidence-based protocols for collection, preservation, and library preparation, researchers can ensure the generation of high-fidelity, reliable metagenomic data, thereby strengthening the foundation for discoveries in human health and disease.
In shotgun metagenomic studies of the gut microbiome, the DNA extraction step is not merely a preliminary technical task; it is a fundamental determinant of data quality and biological validity. The genetic material recovered serves as the foundational lens through which the microbial community is observed. Biases introduced at this stage can distort the apparent taxonomic composition and functional potential of the microbiome, leading to inconsistent results and hampering reproducibility across studies [41] [42]. The core challenge lies in efficiently and equitably lysing a diverse range of microbial cell walls—from easily disrupted Gram-negative bacteria to tough Gram-positive species—while preserving the integrity of the DNA and minimizing co-extraction of inhibitors [43]. This application note delineates the critical steps in DNA extraction, supported by comparative data and detailed protocols, to guide researchers in obtaining representative microbial recovery for robust gut metagenomics.
The Impact of DNA Extraction on Metagenomic Profiles
The choice of DNA extraction method profoundly influences the apparent structure of the gut microbial community. Studies consistently demonstrate that different extraction protocols can alter key metrics such as microbial diversity, taxonomic abundance, and the resulting associations with host phenotypes.
A large-scale study comparing two commercially available kits on 745 paired fecal samples found significant differences in outcomes. The AllPrep DNA/RNA Mini Kit (APK), which incorporates enzymatic lysis and a bead-beating step, yielded higher DNA concentration and revealed a higher microbial diversity compared to the QIAamp Fast DNA Stool Mini Kit (FSK), which lacks mechanical lysis [41]. Critically, over 75% of bacterial species showed statistically significant differences in relative abundance between the two protocols. This technical variation directly impacted biological interpretation, as the resulting microbiome-phenotype associations for anthropometric and lifestyle factors differed remarkably depending on the kit used [41].
The omission of a mechanical lysis step, such as bead-beating, systematically leads to the underrepresentation of Gram-positive bacteria, whose robust cell walls are not efficiently disrupted by enzymatic or chemical means alone [41]. This bias was confirmed using a mock microbial community of known composition, where the APK kit (with bead-beating) provided significantly higher accuracy in recovering expected microbial abundances [41].
Table 1: Impact of DNA Extraction Method on Metagenomic Outcomes in Fecal Samples
| Metric | AllPrep DNA/RNA Mini Kit (APK) | QIAamp Fast DNA Stool Mini Kit (FSK) |
|---|---|---|
| Lysis Method | Enzymatic lysis + bead-beating | Chemical lysis (no bead-beating) |
| DNA Yield | Higher | Lower |
| Microbial Diversity | Higher | Lower |
| Gram-positive Recovery | More accurate | Underrepresented |
| Differentially Abundant Species | >75% of species showed different abundances between kits [41] | >75% of species showed different abundances between kits [41] |
| Phenotype Associations | Resulting associations with host phenotypes were markedly different [41] | Resulting associations with host phenotypes were markedly different [41] |
Comparative Performance of DNA Extraction Methods
Kit-Based Methods for Fecal Samples
Independent benchmarking studies have systematically evaluated the performance of various DNA extraction kits. One such study compared the Mag-Bind Universal Metagenomics Kit (OM) and the DNeasy PowerSoil Kit (QP) across different sample preservation states [44]. The OM kit consistently yielded a larger quantity of DNA than the QP kit. When the same library preparation protocol was controlled for, the OM kit also detected a greater average number of genes, a key metric for functional metagenomics [44].
Another evaluation focused on extracting high molecular weight (HMW) DNA suitable for long-read sequencing technologies, such as Oxford Nanopore Technologies (ONT). Among six tested methods, the Quick-DNA HMW MagBead Kit was selected as the most suitable, producing the best yield of pure HMW DNA and allowing for the accurate detection of almost all bacterial species in a complex mock community via Nanopore sequencing [43].
Table 2: Performance Comparison of Selected DNA Extraction Kits
| Extraction Kit | Key Features | Performance Findings |
|---|---|---|
| Mag-Bind Universal Metagenomics (OM) | Not specified in detail | Higher DNA quantity and higher number of genes detected compared to QP [44] |
| DNeasy PowerSoil (QP) | Widely used standard kit | Lower DNA yield and fewer genes detected compared to OM [44] |
| Quick-DNA HMW MagBead | Optimized for high molecular weight DNA | Best yield of pure HMW DNA; accurate species detection in mock community with Nanopore sequencing [43] |
| QIAamp PowerFecal Pro DNA | Chemical & mechanical lysis (bead-beating) | Identified all species in mock communities; reliable AMR gene detection with ONT [38] |
Methods for Host-Rich Samples
For samples where microbial DNA is outnumbered by host DNA, such as intestinal biopsies, host DNA depletion is a critical prerequisite. A benchmark of four commercial enrichment kits found the NEBNext Microbiome DNA Enrichment kit and the QIAamp DNA Microbiome kit to be most effective. These kits increased the proportion of bacterial DNA sequences to 24% and 28%, respectively, compared to less than 1% in untreated controls [45]. As an alternative to wet-lab depletion, Oxford Nanopore's adaptive sampling (a software-based method) can enrich microbial signals during sequencing by rejecting host DNA molecules from being sequenced, which has been shown to improve bacterial metagenomic assembly and recovery of antimicrobial resistance markers [45].
Detailed Experimental Protocol for Fecal DNA Extraction
The following protocol is adapted from methods validated in comparative studies [44] [41] [43] and is designed for the extraction of high-quality, high-molecular-weight DNA from human fecal samples suitable for shotgun metagenomic sequencing.
Materials and Equipment
Research Reagent Solutions
- Lysis Buffer: Contains guanidine thiocyanate or similar chaotropic salts to denature proteins and protect DNA from nucleases.
- Proteinase K: A broad-spectrum serine protease used to digest proteins and degrade nucleases.
- Lysozyme: An enzyme that breaks down the peptidoglycan cell walls of Gram-positive bacteria.
- Inhibitor Removal Technology: Specific resins or buffers designed to adsorb and remove common PCR inhibitors from stool (e.g., humic acids, bile salts).
- Silica-Based Magnetic Beads or Columns: Bind DNA in the presence of high-salt buffers, allowing for purification and concentration through washing steps.
- TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0): A slightly alkaline buffer used for eluting and storing purified DNA, as it chelates metal ions and stabilizes DNA.
Step-by-Step Procedure
Sample Homogenization:
- Weigh 180-220 mg of frozen fecal material into a sterile tube containing 1.0 mL of lysis buffer.
- Vortex thoroughly for 5-10 minutes until a homogeneous suspension is achieved.
Mechanical and Enzymatic Lysis:
- Add a mixture of lysozyme (final concentration ~20 mg/mL) and Proteinase K (final concentration ~20 mg/mL) to the homogenate.
- Add sterile zirconia/silica beads (0.1 mm and 0.5 mm mixture) to the tube.
- Securely cap the tube and perform bead-beating using a homogenizer (e.g., TissueLyser II or FastPrep) at 20-25 Hz for 5-10 minutes. This step is critical for disrupting tough Gram-positive bacterial cell walls.
- Incubate the lysate at 56°C for 30-60 minutes with agitation (300 rpm).
Inhibitor Removal and DNA Binding:
- Centrifuge the lysate at >13,000 × g for 5 minutes to pellet debris and beads.
- Transfer the supernatant to a new tube. Add a proprietary inhibitor removal solution (if part of the kit protocol) and vortex.
- Centrifuge again and transfer the cleaned supernatant to a fresh tube.
- Add a binding buffer containing a chaotropic salt and isopropanol to the supernatant. Then add silica magnetic beads, and mix thoroughly to allow DNA binding.
DNA Purification:
- Place the tube on a magnetic stand to separate the beads from the solution. Once clear, discard the supernatant.
- Wash the bead-bound DNA twice with 500-800 µL of a prepared wash buffer (typically ethanol-based), carefully removing all supernatant each time.
- Air-dry the beads for 5-10 minutes to ensure all ethanol has evaporated.
DNA Elution:
- Elute the pure DNA from the beads by adding 50-100 µL of pre-warmed (55°C) TE Buffer or nuclease-free water. Mix thoroughly and incubate at 55°C for 5 minutes.
- Place the tube back on the magnetic stand and transfer the eluate containing the purified DNA to a clean, labeled microcentrifuge tube.
Quality Control and Storage:
- Quantify the DNA using a fluorescence-based method (e.g., Qubit dsDNA HS Assay) for accuracy.
- Assess DNA integrity and fragment size by agarose gel electrophoresis (0.8%) or a fragment analyzer. High molecular weight DNA should appear as a tight, high-molecular-weight band with minimal smearing.
- Store the DNA at -20°C or -80°C for long-term preservation.
Workflow Visualization
Downstream Analytical Considerations
The quality of the extracted DNA directly impacts the success and interpretation of subsequent shotgun metagenomic sequencing. The choice between 16S rRNA gene sequencing and whole-genome shotgun sequencing is a primary consideration. While 16S sequencing is cost-effective for taxonomic profiling at the genus level, shotgun sequencing provides superior taxonomic resolution to the species or strain level and, crucially, enables functional gene analysis [12]. A comparative study demonstrated that when a sufficient sequencing depth is achieved (>500,000 reads per sample), shotgun metagenomics identifies a statistically significant higher number of less abundant taxa that are often missed by 16S sequencing. These less abundant genera have been shown to be biologically meaningful and capable of discriminating between experimental conditions [12].
Furthermore, the extraction protocol must be matched to the intended sequencing technology. For long-read sequencing platforms like Oxford Nanopore, the extraction of high molecular weight (HMW) DNA is paramount. Kits that combine mechanical lysis with gentle purification, such as the QIAamp PowerFecal Pro DNA kit, have been shown to effectively retrieve HMW DNA, enabling rapid and accurate taxonomic identification and antimicrobial resistance gene detection within hours of sequencing [43] [38]. The inclusion of a bead-beating step in the QIAamp PowerFecal Pro DNA kit was particularly effective in lysing Gram-positive species, ensuring their representation in the sequencing data [38].
The path to robust and reproducible gut microbiome research begins at the bench with optimized DNA extraction. Evidence unequivocally shows that the choice of extraction method is not neutral; it directly shapes the perceived microbial community and its functional capacity. To ensure representative microbial recovery, researchers should prioritize protocols that incorporate mechanical lysis, such as bead-beating, to overcome the challenge of tough Gram-positive cell walls. Furthermore, the selection of kits with proven inhibitor removal technology is essential for obtaining pure DNA compatible with sensitive downstream applications like library preparation and sequencing. As the field moves toward more complex analyses and the integration of long-read sequencing, the demand for high-quality, high-molecular-weight DNA will only increase. By standardizing and optimizing this critical first step, the scientific community can enhance the reliability of metagenomic data, thereby strengthening the conclusions drawn about the gut microbiome's role in health and disease.
Shotgun metagenomics has revolutionized gut microbiome research by enabling researchers to profile the taxonomic composition and functional potential of microbial communities in a culture-independent manner. The reliability and resolution of these studies are fundamentally dependent on two critical technical choices: the method used to prepare sequencing libraries and the selection of a sequencing platform. Library preparation involves converting extracted DNA into a format compatible with sequencing instruments, a process that includes DNA fragmentation, adapter ligation, and often amplification. The chosen method can significantly impact sequencing bias, genome coverage, and the downstream analysis of metagenomic data [46] [47]. Concurrently, the decision between short-read and long-read sequencing technologies involves balancing read accuracy, read length, and cost-effectiveness, each offering distinct advantages for specific research objectives in gut microbiome analysis [48] [49].
This application note provides a structured framework for selecting the optimal library preparation and sequencing strategies, specifically tailored for shotgun metagenomics within the context of gut microbiome research. We synthesize current methodological comparisons and performance data to guide researchers in making informed decisions that enhance the quality and biological relevance of their metagenomic studies.
Library Preparation Methods: A Comparative Analysis
The process of library preparation is a critical source of variability in metagenomic sequencing. The choice of fragmentation method—enzymatic, tagmentation, or sonication—can influence insert size, coverage uniformity, and the detection of genomic variants [46].
Fragmentation Methodologies:
- Enzymatic Fragmentation: Utilizes specific endonucleases to cleave DNA. Modern commercial kits have largely mitigated historical concerns about sequence bias and artifacts, offering quick workflows, high flexibility with DNA input amounts (as low as 1 ng), and cost-effectiveness [46] [47].
- Tagmentation: Employs a transposase enzyme to simultaneously fragment DNA and insert adapter sequences. This method significantly reduces hands-on time but may offer less control over fragment size and can exhibit sequence bias [46].
- Sonication: Uses physical shearing to produce near-random fragmentation. While it is considered a gold standard for avoiding sequence-specific bias, it requires specialized instrumentation and is less amenable to high-throughput workflows [46].
The following table summarizes the performance characteristics of several commercially available library preparation kits as demonstrated in comparative studies.
Table 1: Performance Comparison of Selected Library Preparation Kits
| Kit Name | Fragmentation Method | Input DNA Flexibility | PCR Requirement | Key Performance Characteristics |
|---|---|---|---|---|
| NEBNext Ultra II FS [46] | Enzymatic | Flexible (1 ng–1 μg) | Yes (or low-cycle) | Reproducible results, good coverage. |
| KAPA HyperPlus [46] [47] | Enzymatic | Flexible | Yes | High-quality results, low GC bias. |
| Illumina DNA Prep [50] [47] | Tagmentation | Flexible (e.g., 1-500 ng) | Yes | Streamlined workflow, cost-effective. |
| Nextera XT [50] [47] | Tagmentation | Low input (1 ng) | Yes | Significant GC bias, especially in low-GC content bacteria [47]. |
| TruSeq DNA PCR-Free [50] | Not Specified | High input (1 μg) | No | Avoids amplification biases, improved coverage in challenging genomic regions. |
Impact of Insert Size: A key finding from performance comparisons is the importance of library insert size. Libraries with DNA insert fragments longer than the cumulative sum of both paired-end reads avoid read overlap, producing more informative data. This leads to strongly improved genome coverage and consequently increased sensitivity and precision in Single Nucleotide Variant (SNV) and indel detection [46]. Furthermore, libraries prepared with minimal or no PCR generally perform better in indel detection, as PCR can introduce duplicates and amplification biases [46] [50].
Sequencing Platform Selection: Short-Read vs. Long-Read Technologies
The choice of sequencing platform dictates the fundamental nature of the data generated. The primary trade-off lies between the high accuracy of short-read platforms and the long read lengths that enable superior resolution of complex genomic regions.
Table 2: Comparative Analysis of Sequencing Platforms for Metagenomics
| Feature | Illumina (Short-Read) | Oxford Nanopore (ONT; Long-Read) | PacBio HiFi (Long-Read) |
|---|---|---|---|
| Read Length | Short (up to 2x300 bp) [48] | Long (≥1,500 bp, up to millions of bases) [49] | Long (HiFi reads: 10-25 kb) [21] |
| Typical Error Rate | Low (< 0.1%) [48] | Higher (5-15%), but improving [48] [49] | Very high single-pass error rate, but consensus accuracy >99.9% [49] |
| Strengths | High accuracy, ideal for broad microbial surveys and variant calling [48]. | Real-time sequencing, high taxonomic resolution, detects epigenetic modifications [49]. | High accuracy with long reads, excellent for de novo genome assembly and MAG reconstruction [21]. |
| Limitations | Limited species-level resolution due to short read length [48]. | Historically higher error rates can complicate variant calling [48]. | Higher DNA input requirements, currently lower throughput than Illumina. |
| Ideal for Gut Microbiome | Large-scale population studies, quantitative profiling, SNV/indel analysis [48]. | Strain-level tracking, plasmid/host association (e.g., via proximity ligation), in-field sequencing [51] [49]. | High-quality Metagenome-Assembled Genome (MAG) reconstruction, discovering complete genes and pathways [21]. |
Emerging Applications: Long-read sequencing is particularly transformative for complex functional analyses. For instance, proximity ligation shotgun metagenomics, which uses formaldehyde fixation to create physical links between mobile genetic elements (like plasmids and bacteriophages) and their bacterial hosts, has been powerfully applied using these technologies. This approach allows for the direct tracking of engraftment and host-range dynamics of donor bacteriophages in patients receiving fecal microbiota transplantation (FMT) [51].
Integrated Experimental Protocols
Protocol: Host DNA Depletion for Enhanced Urobiome or Low-Microbial-Biomass Metagenomics
Background: Samples with low microbial biomass and high host DNA background, such as urine, respiratory samples, or gut biopsies, present a significant challenge. Host DNA can overwhelm sequencing reads, reducing microbial sequencing depth [52]. This protocol is adapted from a study evaluating methods for urine but is broadly applicable.
Materials:
- QIAamp DNA Microbiome Kit (Qiagen) [52]
- MolYsis Complete5 (Molzym) [52]
- NEBNext Microbiome DNA Enrichment Kit (New England Biolabs) [52]
- HostZERO (Zymo Research) [52]
- Propidium monoazide (PMA) [52]
- QIAamp BiOstic Bacteremia DNA Kit (Qiagen; no host depletion) [52]
Method:
- Sample Input: Start with a sufficient sample volume. For liquid samples like urine, ≥3.0 mL is recommended for consistent profiling [52].
- Cell Lysis and Host DNA Depletion:
- Follow the specific workflow of the chosen commercial kit.
- The QIAamp DNA Microbiome Kit involves a multi-step process to lyse human (and other mammalian) cells and digest the released DNA, followed by microbial lysis and DNA purification. It was shown to yield the greatest microbial diversity in 16S rRNA and shotgun sequencing data and maximized MAG recovery in host-spiked samples [52].
- Alternative methods like MolYsis use selective lysis of human cells and enzymatic degradation of the released DNA.
- DNA Extraction: Proceed with the microbial DNA extraction as per the kit's instructions. Include bead-beating steps for thorough lysis of diverse microbial cells [52].
- Quality Control: Quantify DNA using a fluorometer (e.g., Qubit). Assess DNA quality, for example, with a Bioanalyzer.
Protocol: Whole-Genome Library Preparation from Minimal Bacterial Input
Background: This protocol is designed for efficient library construction from low DNA inputs, which is common when working with specific bacterial isolates or low-biomass metagenomic samples [47].
Materials:
- DNA Extraction Reagents: Glass beads (425–600 μm) for mechanical lysis [47], or a commercial kit like the EZ1 DNA Tissue Kit (Qiagen) [47].
- Library Prep Kits: NEBNext Ultra II FS, KAPA HyperPlus, or Illumina DNA Prep [46] [47].
- Agencourt AMPure XP beads (Beckman Coulter) for purification [47].
- Qubit 4 Fluorometer and Qubit dsDNA HS Assay Kit [47].
Method:
- DNA Preparation from a Single Colony:
- Glass Bead Disruption (GBD): Resuspend a single bacterial colony in a tube with glass beads and DNA-free water (bead-to-water ratio ~1:3). Vortex vigorously for 5 minutes. Centrifuge and collect the supernatant containing DNA [47].
- Note: Heat shock lysis is rapid but may be inadequate for Gram-positive or spore-forming bacteria [47].
- DNA Quantification and Normalization: Quantify the extracted DNA using the Qubit HS Assay. Dilute the DNA to a working concentration (e.g., 0.333 ng/μL) in Tris-HCl buffer. A starting quantity of 1 ng is often sufficient [47].
- Library Preparation:
- Follow the manufacturer's protocol for the selected kit (e.g., NEBNext Ultra II FS).
- Fragmentation: Perform enzymatic fragmentation. The time may need optimization for the desired insert size (e.g., 15 minutes for ~300 bp insert) [46].
- End-Repair, A-tailing, and Adapter Ligation: Perform these standard steps as per the kit instructions.
- Library Amplification: Use the minimal number of PCR cycles recommended for your input DNA to minimize bias. Some kits offer PCR-free options for higher inputs (e.g., 100 ng) [46].
- Library QC and Pooling: Purify the final library with AMPure beads. Assess library concentration (Qubit) and size distribution (e.g., Bioanalyzer). Equimolarly pool libraries for multiplexed sequencing [47].
Workflow Visualization
The following diagram illustrates the key decision points and procedural steps in a standard shotgun metagenomics workflow, from sample to data.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Kits for Shotgun Metagenomics Workflows
| Item | Function/Application | Example Products |
|---|---|---|
| Host DNA Depletion Kits | Selective removal of host DNA to increase microbial sequencing depth in low-biomass samples. | QIAamp DNA Microbiome Kit [52], MolYsis Complete5 [52], NEBNext Microbiome DNA Enrichment Kit [52] |
| DNA Extraction Kits (Mechanical Lysis) | Efficient cell wall disruption for Gram-positive and spore-forming bacteria in gut samples. | Bead-beating protocols with glass beads (425–600 μm) [47] |
| Short-Read Library Prep Kits | Construction of sequencing libraries for Illumina platforms. | NEBNext Ultra II FS [46], KAPA HyperPlus [46] [47], Illumina DNA Prep [50] [47] |
| Long-Read Library Prep Kits | Construction of sequencing libraries for PacBio or Oxford Nanopore platforms. | PacBio SMRTbell prep kits, ONT 16S Barcoding Kit (for amplicons) [48] or Ligation Sequencing Kits [49] |
| DNA Purification Beads | Size-selective cleanup and purification of DNA fragments during library preparation. | Agencourt AMPure XP beads [47] |
| DNA Quantification Kits | Accurate fluorometric quantification of DNA concentration for library normalization. | Qubit dsDNA HS Assay Kit [47] |
| Library QC Instruments | Assessment of library fragment size distribution and quality. | Agilent Bioanalyzer or TapeStation [47] |
In gut microbiome research, the journey from a raw sequencing file to robust, biologically meaningful insights hinges on the critical first stage of the bioinformatic pipeline: quality control (QC) and trimming. This initial data processing phase is responsible for ensuring the accuracy and reliability of all downstream analyses, from taxonomic profiling to functional annotation. Proper execution removes technical artifacts, mitigates sequencing errors, and enriches for genuine microbial signals, forming the foundational step in any shotgun metagenomics protocol [53] [54]. This application note provides a detailed, practical guide to implementing this essential workflow, framed within the context of a comprehensive gut microbiome study.
The following diagram illustrates the complete bioinformatic workflow from raw sequencing reads to high-quality data ready for downstream analysis. The core focus of this document, Quality Control and Trimming, is highlighted in the initial steps.
Step-by-Step Experimental Protocol
Data Integrity Assessment
Before any processing, confirm the integrity of the raw sequencing files transferred from the sequencing facility.
- Purpose: Verify that files were transferred completely and without corruption.
- Procedure:
- Compare the MD5 checksum provided by the sequencing facility with that of your local file.
- Use the command-line tool
md5sum(or equivalent) to compute the cryptographic hash.- Example Command:
md5sum sample_1.fastq.gz
- Example Command:
- Confirm that the generated hash string matches exactly the one provided. Do not proceed if the checksums do not match.
- Acceptance Criterion: An exact, character-for-character match of the MD5 checksum [54].
Initial Quality Assessment with FastQC
Evaluate the raw read quality to identify potential issues such as adapter contamination, low-quality bases, or biased sequence composition.
- Purpose: Generate a baseline quality report to inform subsequent trimming parameters.
- Procedure:
- Run FastQC on all raw FASTQ files.
- Example Command:
fastqc sample_1.fastq.gz sample_2.fastq.gz
- Example Command:
- Review the generated HTML reports. Pay close attention to:
- Per Base Sequence Quality: Identify drops in quality scores.
- Adapter Content: Determine the type and amount of adapter sequence.
- Overrepresented Sequences: Detect possible contaminants.
- Sequence Duplication Levels: Assess potential PCR bias.
- Run FastQC on all raw FASTQ files.
- Acceptance Criterion: This is a diagnostic step; no data is rejected. Findings guide the parameters for the trimming step below [54].
Adapter Trimming and Quality Filtering
Remove adapter sequences, trim low-quality bases, and exclude poor-quality reads.
- Purpose: To increase the proportion of high-quality, microbial-derived sequences for downstream analysis.
- Procedure using Trimmomatic (within KneadData):
- Use Trimmomatic via the KneadData wrapper for integrated processing.
- Set parameters based on the FastQC report. A typical command for paired-end data is:
- Example Command:
- Key Parameters Explained:
ILLUMINACLIP: Removes adapter sequences (specify file, seed mismatches, palindrome clip threshold, simple clip threshold).LEADING: Removes low-quality bases from the start of the read.TRAILING: Removes low-quality bases from the end of the read.SLIDINGWINDOW: Scans the read with a window (e.g., 4 bases), cutting when the average quality in that window drops below a threshold (e.g., Q20).MINLEN: Discards reads shorter than the specified length (e.g., 50 bp) after trimming.
- Acceptance Criterion: A minimum of 85% of bases with a Phred score ≥ Q30 (Q30) is generally recommended. GC content should fall within the expected range for the sample type [54].
Host DNA Removal
A critical step for host-associated microbiomes (e.g., gut biopsies) where the majority of sequenced DNA can be of host origin.
- Purpose: To deplete host-derived sequences, thereby increasing the relative abundance and detection sensitivity of microbial taxa [55] [54].
- Procedure using Bowtie2:
- Download a highly curated human reference genome (e.g., GRCh38 from Ensembl) [55].
- Build a Bowtie2 index for the reference genome.
- Example Command:
bowtie2-build GRCh38.fa grch38_index
- Example Command:
- Align reads to the host genome and retain the unmapped reads.
- Example Command:
- The
--un-conc-gzflag outputs the unmapped (non-host) reads in compressed FASTQ files.
- Performance Consideration: A study demonstrated that Bowtie2 alignment removed ~98% of host reads, which subsequently increased the detection sensitivity for Clostridioides difficile from 50% to 90% [54]. Recent benchmarking for dermatological samples also highlighted that Bowtie2 de-hosting, when combined with Kraken2, efficiently recovered well-established biological associations [55].
Post-QC Quality Assessment
Verify the overall success of the QC and trimming workflow.
- Purpose: To aggregate and compare quality metrics across all samples before proceeding to assembly or profiling.
- Procedure:
- Run FastQC on the final, processed FASTQ files (after host removal).
- Use MultiQC to compile results from all FastQC reports (raw and processed) into a single, interactive summary.
- Example Command:
multiqc . --filename MultiQC_Report.html
- Example Command:
- In the MultiQC report, confirm:
- An increase in the mean Per Base Sequence Quality.
- A significant reduction or elimination of adapter content.
- Successful retention of a sufficient number of reads for robust analysis.
- Acceptance Criterion: Successful remediation of issues identified in the initial FastQC report, with final quality scores meeting the pre-defined thresholds (e.g., Q30) [54].
Performance Metrics and Tool Comparison
Key Quality Metrics for Assessment
Table 1: Key quantitative metrics to assess at different stages of the QC workflow.
| Processing Stage | Key Metric | Target / Threshold | Biological / Technical Rationale |
|---|---|---|---|
| Raw Data | Q30 Score | ≥ 85% of bases [54] | Ensures base call accuracy >99.9%, minimizing false positives in downstream variant calling. |
| GC Content | Within expected range for community | Significant deviation may indicate contamination or technical bias. | |
| After Trimming | Total Reads Retained | Typically >70-80% of raw reads | Ensures sufficient data depth remains for robust statistical power. |
| Read Length | Minimum length (e.g., 50 bp) post-trimming | Overly short reads are difficult to map or assemble accurately. | |
| After De-hosting | Non-host Read Percentage | Varies by sample type (e.g., ~2% from gut biopsy [54]) | Maximizes signal from the microbial community, directly impacting sensitivity for low-abundance taxa. |
Comparison of Bioinformatics Tools
Table 2: A comparison of commonly used software tools for major steps in the QC and trimming workflow.
| Tool | Primary Function | Key Features / Strengths | Considerations |
|---|---|---|---|
| FastQC [54] | Quality Assessment | Generates comprehensive, visual HTML reports; standard in the field. | Diagnostic only; does not perform any filtering. |
| Trimmomatic [54] | Read Trimming | Highly flexible, allows precise control over trimming parameters. | Often run via wrappers like KneadData for ease of use. |
| KneadData [54] | Integrated Trimming & De-hosting | Streamlines workflow by linking Trimmomatic and Bowtie2. | A curated host reference genome must be supplied by the user. |
| Bowtie2 [55] [54] | Host DNA Removal (Alignment-based) | High accuracy; uses a well-curated, trusted host genome (e.g., GRCh38) [55]. | Can be computationally intensive for large datasets. |
| Kraken2 [54] | Host DNA Removal (k-mer based) / Taxonomy | Very fast; can be used for de-hosting if database includes host sequences. | Relies on the completeness of the k-mer database, which may be incomplete or contaminated [55]. |
| BWA [55] | Host DNA Removal (Alignment-based) | Another robust, accurate aligner for de-hosting. | Performance and resource usage are comparable to Bowtie2. |
| MultiQC [54] | Aggregate Reporting | Essential for multi-sample studies; summarizes results from many tools. | Does not perform any QC itself. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential computational "reagents" and resources for executing the shotgun metagenomics QC protocol.
| Item / Resource | Function / Purpose | Example / Specification |
|---|---|---|
| Human Reference Genome | A curated genome for alignment-based host read removal. | GRCh38 (Ensembl Release 110) [55]. Provides a high-quality, standardized reference for filtering. |
| Adapter Sequence File | Contains standard Illumina adapter sequences for trimming. | Commonly provided with tools (e.g., TruSeq3-PE-2.fa for Trimmomatic). Critical for removing sequencing artifacts. |
| Quality Control Suites | Assess read quality, nucleotide composition, and adapter content. | FastQC for individual reports; MultiQC for project-level aggregation [54]. |
| Processing Pipelines | Integrated workflows that combine trimming and de-hosting. | KneadData seamlessly integrates Trimmomatic and Bowtie2, simplifying the pipeline [54]. |
| Taxonomic Database | Required for k-mer-based classifiers if used for de-hosting or profiling. | Minikraken Database or standard Kraken2 database. Must be chosen to match the classifier and kept up-to-date [55]. |
Taxonomic profiling is a fundamental step in shotgun metagenomics that identifies and quantifies the microorganisms present within a complex community, such as the gut microbiome. This process involves classifying sequencing reads against reference databases to determine the taxonomic composition, from bacteria to fungi, at various resolution levels from phyla to strains. Advanced classifiers leverage sophisticated algorithms and comprehensive reference databases to achieve unprecedented accuracy, enabling researchers to discover novel organisms, track strains, and correlate microbial abundances with host health and disease states for therapeutic development [56] [57].
Advanced Taxonomic Classifiers and Their Applications
The evolution of taxonomic classifiers has moved from basic sequence alignment to sophisticated frameworks that integrate genomic and metagenomic data. The table below summarizes the key features of contemporary classifiers relevant to gut microbiome research.
Table 1: Advanced Taxonomic Profiling Tools for Shotgun Metagenomics
| Classifier Name | Primary Classification Method | Taxonomic Units | Notable Features | Considerations for Gut Microbiome Research |
|---|---|---|---|---|
| Meteor2 [10] | Mapping to environment-specific microbial gene catalogs | Metagenomic Species Pangenomes (MSPs) | Integrated taxonomic, functional, and strain-level profiling (TFSP); fast mode available | Excels in detecting low-abundance species; improved sensitivity in human gut microbiota |
| MetaPhlAn 4 [56] | Unique species-specific marker genes | Species-level Genome Bins (SGBs), including unknown (uSGBs) | Profiling of both known and yet-to-be-characterized species; explains significantly more reads | ~20% more reads explained in human gut microbiomes; accurate quantification of uncultivated organisms |
| FunOMIC [58] | Fungal single-copy marker genes | Fungal species and strains | Built-in taxonomic (FunOMIC-T) and functional (FunOMIC-P) databases | Specifically designed for fungal profiling (mycobiome); enables inter-kingdom interaction studies |
| MyTaxa [59] | Weighted homology of all genes in a sequence | Conventional taxa (species, genera, phyla) | Employs gene-classifying power and HGT frequency; handles novel taxa | Useful for identifying novel species from assembled contigs, such as novel Prevotella in human gut |
Protocol for Comprehensive Taxonomic Profiling of Gut Microbiome
This protocol outlines a streamlined workflow for taxonomic profiling of a human gut microbiome sample, from quality control to visualization, utilizing the strengths of multiple advanced classifiers.
Sample Preparation and Sequencing
Principle: Extract high-quality, high-molecular-weight DNA from fecal samples to construct a sequencing library that accurately represents the microbial community.
Materials:
- DNA Extraction Kit: DNeasy PowerSoil Pro Kit (Qiagen) or equivalent for efficient lysis of diverse microbes.
- Quality Control: Fluorometric methods (e.g., Qubit dsDNA HS Assay Kit) for DNA quantification and fragment analyzer for integrity check.
- Library Prep Kit: Illumina DNA Prep Kit for preparing Illumina-compatible sequencing libraries.
- Sequencing Platform: Illumina NovaSeq for deep shotgun metagenomic sequencing (e.g., 20-50 million 2x150bp paired-end reads per sample).
Bioinformatic Preprocessing
Principle: Remove low-quality sequences and host-derived reads to ensure that downstream analysis targets high-quality microbial data.
Procedure:
- Quality Trimming and Adapter Removal: Use Trimmomatic or Fastp to remove adapter sequences and trim low-quality bases.
- Host DNA Depletion: Align reads to the human reference genome (e.g., GRCh38) using Bowtie2 and discard matching reads.
- Quality Assessment: Use FastQC on the processed reads to confirm data quality.
Taxonomic Profiling with Multiple Classifiers
Principle: Apply complementary classifiers to obtain a robust and comprehensive taxonomic profile, capturing both bacterial and fungal communities.
Procedure A: Integrated Community Profiling with Meteor2
- Database Selection: Download the human gut microbial gene catalogue for Meteor2.
- Run Profiling: Execute Meteor2 in default mode for comprehensive TFSP or fast mode for rapid taxonomic and strain-level analysis.
- Output: Abundance table of MSPs, functional annotations (KEGG, CAZymes, ARGs), and strain variants.
Procedure B: Profiling Known and Unknown Taxa with MetaPhlAn 4
- Database: The built-in database containing marker genes for over 26,000 SGBs is used.
- Run Profiling: Execute MetaPhlAn 4 to profile both known (kSGBs) and unknown (uSGBs) species.
- Output: A detailed abundance profile of all detected SGBs.
Procedure C: Fungal Community Profiling with FunOMIC
- Database Setup: Download the FunOMIC-T database of fungal single-copy marker genes.
- Run Profiling: Map reads to the FunOMIC-T database using the FunOMIC.sh pipeline.
- Output: Taxonomic abundance profile of fungal species.
Data Integration and Visualization
Principle: Integrate results from different classifiers and generate publication-quality figures to interpret the community structure.
Procedure:
- Data Aggregation: Use a custom R or Python script to merge abundance tables from Meteor2, MetaPhlAn 4, and FunOMIC at the genus or species level.
- Generate Visualizations:
- Stacked Bar Chart: For comparing relative taxonomic abundance across multiple samples or groups at the phylum level using the
ggplot2package in R [60]. - Heatmap: For visualizing the abundance of top microbial taxa across all samples, often combined with hierarchical clustering.
- Principal Coordinates Analysis (PCoA) Plot: For assessing beta-diversity and visualizing sample groupings based on Bray-Curtis or other ecological distances [60] [61].
- Stacked Bar Chart: For comparing relative taxonomic abundance across multiple samples or groups at the phylum level using the
The following diagram illustrates the core bioinformatic workflow and the relationship between the different classifiers and their outputs.
Figure 1: Bioinformatic workflow for comprehensive taxonomic profiling. Processed reads are analyzed in parallel by specialized classifiers to generate integrated results.
Successful taxonomic profiling relies on a suite of bioinformatic reagents and databases. The table below lists key resources for conducting the protocols described in this application note.
Table 2: Essential Research Reagents and Resources for Metagenomic Taxonomic Profiling
| Resource Name | Type | Function in Profiling |
|---|---|---|
| Meteor2 Database [10] | Environment-specific Gene Catalogue | Provides the reference genes and pangenomes for MSP-based quantification and functional annotation. |
| MetaPhlAn 4 Marker Database [56] | Unique Marker Gene Database | Contains species-specific marker genes for accurate identification and quantification of over 26,000 SGBs. |
| FunOMIC-T Database [58] | Fungal Marker Gene Database | A comprehensive database of fungal single-copy marker genes used for precise taxonomic assignment in mycobiome analysis. |
| GTDB (Genome Taxonomy Database) [10] | Taxonomic Framework | Provides a standardized bacterial and archaeal taxonomy used by tools like Meteor2 for consistent taxonomic annotation. |
| Bowtie2 [10] | Read Mapping Tool | Aligns sequencing reads to reference gene catalogues for abundance estimation in tools like Meteor2 and FunOMIC. |
Integrating advanced classifiers like Meteor2, MetaPhlAn 4, and FunOMIC provides a powerful, multi-faceted approach to taxonomic profiling in gut microbiome research. This integrated strategy allows researchers to simultaneously profile the bacterial and fungal components of the microbiome, capture both known and unknown microbial diversity, and link taxonomy to function and strain-level dynamics [10] [56] [58].
For drug development professionals, this comprehensive profile is invaluable. It enables the identification of specific microbial biomarkers associated with disease states or therapeutic responses, paving the way for targeted interventions. The ability to track strain-level transmission, as demonstrated in faecal microbiota transplantation studies, also offers critical insights into the mechanisms of microbiome-based therapeutics [10]. As reference databases continue to expand and algorithms become more refined, taxonomic profiling will remain a cornerstone of gut microbiome research, driving discovery and innovation in human health and drug development.
Functional annotation is a critical step in shotgun metagenomics that transforms raw genomic data into biological insights by determining the roles of predicted genes. In gut microbiome research, this process reveals how microbial communities influence host health, disease states, and metabolic functions [5] [62]. Unlike 16S rRNA sequencing which primarily provides taxonomic classification, shotgun metagenomic sequencing enables comprehensive functional profiling by randomly fragmenting and sequencing all microbial DNA present in a sample [5] [62]. This approach has been shown to detect 45% more functional genes in complex samples compared to 16S methods, providing unprecedented resolution for understanding microbial contributions to human physiology and disease pathogenesis [62].
The fundamental challenge in functional annotation lies in accurately connecting gene sequences to their biological functions, which is complicated by the vast diversity of microbial genes and the incomplete nature of reference databases [63]. This application note provides detailed protocols and analytical frameworks for conducting robust functional annotation specifically within the context of gut microbiome research, with emphasis on practical implementation for drug development and clinical applications.
Core Principles of Functional Annotation
Key Conceptual Frameworks
Functional annotation in metagenomics operates on several foundational principles. First, it relies on the assumption that sequence similarity implies functional similarity, allowing researchers to infer gene function through homology searches against reference databases [63] [62]. Second, it leverages conserved protein domains and motifs to identify function even when overall sequence similarity is low [64]. Third, it employs pathway reconstruction algorithms to assemble individual gene functions into coherent metabolic networks that represent the biochemical capabilities of microbial communities [65] [66].
The complexity of functional annotation is magnified in gut microbiome studies due to the extraordinary diversity of microorganisms present and the extensive functional redundancy across different taxonomic groups [5]. Successful annotation requires integrating multiple complementary approaches to achieve comprehensive coverage of metabolic pathways, especially for non-core pathways and less-studied organisms where standard annotations are often incomplete [63].
Annotation Workflow Architecture
A robust functional annotation pipeline for gut microbiome data incorporates four interconnected stages:
- Data Preprocessing: Quality control, adapter removal, and host DNA decontamination
- Gene Prediction: Identification of coding sequences in assembled contigs or metagenome-assembled genomes (MAGs)
- Function Assignment: Homology-based assignment of gene functions using curated databases
- Pathway Reconstruction: Integration of individual gene functions into metabolic pathways and networks
This multi-stage process ensures that functional predictions are based on high-quality sequence data and are contextualized within biologically relevant pathways. The workflow must be tailored to the specific research question, as different applications (e.g., biomarker discovery versus mechanistic studies) require different levels of annotation resolution and validation [65] [62].
Computational Protocols and Methodologies
Essential Tools and Databases
Functional annotation leverages a diverse ecosystem of computational tools and reference databases, each with distinct strengths and applications in gut microbiome research.
Table 1: Key Functional Annotation Tools for Gut Microbiome Research
| Tool Name | Primary Function | Application in Gut Research | Technical Requirements |
|---|---|---|---|
| HUMAnN2 [66] | Profiling pathway abundance and coverage | Quantifying metabolic potential in communities | 4 CPU threads, ~3 hours for 100M reads |
| METABOLIC [64] | Metabolic pathway analysis & biogeochemical cycling | Modeling gut metabolite transformations | ~3 hours for 100 genomes with 40 CPU threads |
| MGS-Fast [62] | Rapid gene catalog alignment | Identifying disease-associated functional genes | Optimized for large-scale biomarker studies |
| DRAGEN [62] | Metagenomics pipeline | Species identification and abundance quantification | Hardware-accelerated for fast processing |
| Prodigal [62] | Gene prediction | Identifying coding sequences in assembled contigs | Default for prokaryotic gene prediction |
Table 2: Essential Reference Databases for Functional Annotation
| Database | Content Focus | Utility in Gut Microbiome Studies | Integration Tools |
|---|---|---|---|
| KEGG [63] [62] | Metabolic pathways and modules | Understanding microbial metabolism in gut environments | HUMAnN2, METABOLIC |
| eggNOG [62] | Orthologous groups and functional annotation | Evolutionary context of gut microbial genes | DIAMOND, BLAST+ |
| CAZy [62] | Carbohydrate-active enzymes | Studying fiber degradation and SCFA production | HMMER, dbCAN2 |
| MetaCyc [66] | Metabolic pathways and enzymes | Pathway coverage analysis | HUMAnN2, Pathway Tools |
| ChocoPhlAn [66] | Pangenome database | Species-specific gene family abundance | HUMAnN2 |
Integrated Functional Annotation Protocol
The following protocol describes a comprehensive workflow for functional annotation of gut microbiome data, integrating multiple tools for maximal coverage and accuracy.
Protocol 1: Comprehensive Functional Annotation Workflow
Sample Input: Quality-filtered metagenomic reads or assembled contigs from human gut samples
Step 1: Gene Prediction and Quantification
- For assembled contigs or MAGs, predict open reading frames using Prodigal with meta-mode:
prodigal -i contigs.fna -o genes.gff -a proteins.faa -p meta[62] - For unassembled reads, use HUMAnN2's built-in gene prediction:
humann2 --input reads.fastq --output humann2_results --threads 4[66] - Align reads to predicted genes for quantification using Bowtie2 or BWA
Step 2: Functional Annotation with Multi-Database Approach
- Annotate predicted proteins against KEGG using DIAMOND:
diamond blastp -d kegg_db -q proteins.faa -o kegg_annotations.dmnd --evalue 1e-5[62] - Simultaneously annotate against eggNOG:
emapper.py -i proteins.faa -o eggnog_annotations --db eggnog_db[62] - Run HMMER against CAZy dbCAN2 database:
hmmscan --domtblout cazy_annotations.dt dbCAN.hmm proteins.faa[64] - For comprehensive metabolic profiling, execute METABOLIC for pathway analysis:
METABOLIC.sh -i mags_dir -o metabolic_results -t 40[64]
Step 3: Pathway Reconstruction and Analysis
- Use HUMAnN2 to reconstruct MetaCyc pathways:
humann2_regroup_table --input gene_families.tsv --groups uniref50_ko --output ko_abundance.tsvfollowed byhumann2_reduce_table --input ko_abundance.tsv --output pathway_abundance.tsv --sort[66] - Apply MinPath for parsimonious pathway reconstruction to minimize false positives [66]
- Integrate annotations from multiple databases using custom scripts to resolve discrepancies and maximize coverage
Step 4: Validation and Quality Assessment
- Validate key metabolic predictions using METABOLIC's motif validation for conserved protein residues [64]
- Check pathway coverage metrics in HUMAnN2 outputs to assess completeness of detected pathways [66]
- Compare annotations across tools to identify consistent signals and potential false positives
Expected Outputs: (1) Table of gene families with abundance values; (2) Pathway abundance and coverage tables; (3) Annotated metabolic network; (4) Quality metrics for annotations
Technical Notes: This multi-tool approach increases functional coverage by approximately 40% compared to single-database methods, with even greater improvements for non-model organisms [63]. For large datasets, cloud-based solutions like the Galaxy platform can streamline execution and reproducibility [62].
Advanced Pathway-Centric Analysis
For research focused on specific metabolic processes, targeted analytical approaches provide deeper biological insights.
Protocol 2: Targeted Metabolic Pathway Analysis
Application: Deep functional analysis of specific gut-relevant pathways (e.g., SCFA production, bile acid metabolism)
Step 1: Pathway-Centric Gene Extraction
- Extract genes associated with target pathways using KEGG Module or MetaCyc Pathway identifiers
- Create custom HMM profiles for pathway-specific genes not well-covered in standard databases
Step 2: Community-Level Metabolic Modeling
- Use METABOLIC to map metabolic handoffs and potential cross-feeding relationships [64]
- Calculate microbial contributions to biogeochemical cycles relevant to gut health (e.g., sulfate reduction, methanogenesis)
Step 3: Strain-Level Functional Variation
- Identify single-amino-acid variants in key metabolic genes using SNP calling tools
- Link functional variants to strain-level abundance changes across sample conditions
Step 4: Visualization and Interpretation
- Generate metabolic Sankey diagrams showing transformations and microbial contributions [64]
- Create functional networks using MW-score (metabolic weight score) to identify key functional players [64]
Experimental Design and Visualization
Workflow Visualization
The following diagram illustrates the comprehensive functional annotation workflow for gut microbiome studies, integrating the protocols and tools described in this application note:
Functional Annotation Workflow for Gut Microbiome Data
Experimental Considerations for Gut Microbiome Studies
Successful functional annotation in gut microbiome research requires careful experimental design to address domain-specific challenges:
Sample Preparation Considerations
- Implement protocols to minimize host DNA contamination (typically human), which can overwhelm microbial signals [5] [62]
- Consider RNA sequencing approaches to distinguish active functions from dead cell DNA contamination [62]
- Standardize sample collection methods to preserve functional potential during storage and processing
Reference Database Selection
- Supplement general databases with gut-specific catalogs such as MGnify for comprehensive coverage [62]
- Consider creating custom databases for population-specific or disease-specific studies
- Regularly update database versions to incorporate newly characterized gut microbial functions
Multi-Omics Integration
- Correlate metagenomic functional potential with metatranscriptomic activity measurements
- Validate computational predictions with metabolomic profiling of expected pathway outputs
- Integrate metaproteomic data to confirm translation of predicted functions
The Scientist's Toolkit
Essential Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Resources
| Category | Specific Resource | Function in Functional Annotation | Implementation Notes |
|---|---|---|---|
| Reference Databases | KEGG, MetaCyc, eggNOG | Provide curated functional annotations | Combining multiple databases increases coverage by ~40% [63] |
| Analysis Tools | HUMAnN2, METABOLIC | Pathway reconstruction and analysis | HUMAnN2 uses ChocoPhlAn pangenome database [66] |
| Computational Infrastructure | Galaxy Platform, Docker Containers | Reproducible analysis workflows | Cloud solutions enable standardized analysis without programming [62] |
| Quality Control Tools | KneadData, FastQC | Data preprocessing and contamination filtering | Critical for removing host DNA and low-quality reads [62] [66] |
| Visualization Resources | METABOLIC plotting functions | Creating metabolic Sankey diagrams and functional networks | MW-score quantifies metabolic importance [64] |
Applications in Drug Development
Functional annotation of gut microbiome data provides valuable insights for multiple stages of drug development, from target discovery to personalized medicine approaches.
Biomarker Discovery and Validation
- Identify functional biomarkers (rather than taxonomic) for patient stratification [62]
- Detect antibiotic resistance genes with implications for treatment efficacy [62]
- Discover novel therapeutic targets within microbial metabolic pathways that influence host physiology
Mechanism of Action Studies
- Elucidate microbial contributions to drug metabolism and bioavailability
- Identify microbial functions that produce bioactive metabolites with therapeutic effects
- Understand how microbial community interactions influence treatment outcomes
Functional Annotation in Clinical Translation
A key application in drug development is using functional annotation to identify predictive biomarkers for disease detection and monitoring, as illustrated in the following protocol:
Protocol 3: Functional Biomarker Identification for Clinical Applications
Application: Developing microbiome-based diagnostic and prognostic biomarkers
Step 1: Case-Control Functional Profiling
- Perform functional annotation on metagenomic data from disease and control cohorts
- Use HUMAnN2 to quantify pathway abundances in all samples [66]
- Apply METABOLIC to identify differentially abundant metabolic functions [64]
Step 2: Machine Learning Feature Selection
- Input annotated functions into feature selection algorithms (e.g., random forest, LASSO)
- Prioritize functions with both statistical significance and biological plausibility
- Validate selected features in independent cohorts when possible
Step 3: Clinical Validation
- Build classifiers based on functional biomarkers (e.g., PDAC detection with AUROC of 0.84) [62]
- Integrate with conventional biomarkers (e.g., combining with CA19-9 increased AUROC to 0.94) [62]
- Establish clinical thresholds for functional abundance values
Implementation Example: In pancreatic ductal adenocarcinoma (PDAC) detection, Zhou et al. used functional annotation of fecal metagenomes to identify a classifier with 0.84 AUROC, which improved to 0.94 AUROC when combined with serum CA19-9 levels [62]. This demonstrates the potential for functional metagenomic annotation to contribute to non-invasive diagnostic approaches.
Troubleshooting and Quality Assurance
Common Challenges and Solutions
Table 4: Troubleshooting Guide for Functional Annotation
| Challenge | Potential Impact | Recommended Solution |
|---|---|---|
| Incomplete annotations (30-50% of genes unannotated) [63] | Limited biological insights | Combine multiple annotation tools; increases coverage by ~40% [63] |
| Host DNA contamination | Reduced microbial signal | Bioinformatic filtering (KneadData) or molecular enrichment [5] [62] |
| Database-specific biases | Inconsistent functional predictions | Use consensus approach across KEGG, RAST, EFICAz, BRENDA [63] |
| Fragmented assembly in complex communities | Incomplete gene predictions | Use complementary approaches: assembly-based and read-based annotation [62] |
| Viral sequence identification | Missed phage functions | Use specialized tools (VirSorter, geNomad) with k-mer analysis [62] |
Quality Metrics and Validation
Establishing quality metrics is essential for generating reliable functional annotations:
Annotation Completeness Assessment
- Calculate the percentage of predicted genes with functional assignments
- Benchmark against reference genomes with known functions
- Track database versions for reproducibility
Pathway Validation Measures
- Use HUMAnN2 pathway coverage scores to assess pathway completeness [66]
- Apply METABOLIC's motif validation for biochemically conserved residues [64]
- Implement positive controls with spiked-in reference communities when possible
Technical Reproducibility
- Process replicates through the entire workflow to assess technical variability
- Compare results across different database versions to identify robust findings
- Document all parameters and software versions for computational reproducibility
Functional annotation represents a powerful approach for extracting biologically meaningful insights from gut metagenomic data. By implementing the integrated protocols and methodologies described in this application note, researchers can comprehensively characterize the metabolic potential of gut microbial communities and identify functionally relevant biomarkers for drug development. The multi-tool, multi-database approach significantly enhances annotation coverage, particularly for non-model organisms and less-characterized metabolic pathways [63]. As reference databases continue to expand and computational methods evolve, functional annotation will play an increasingly central role in translating microbiome research into clinical applications and therapeutic interventions.
Solving Common Challenges: Host Depletion, Mycobiome Analysis, and Standardization
In gut microbiome research, shotgun metagenomic sequencing of intestinal biopsies and other tissue samples is often challenged by an overwhelming amount of host DNA, which can constitute over 99.99% of the total DNA [67]. This high host DNA content severely limits the detection sensitivity for microbial signals, making in-depth characterization of tissue-associated microbial communities difficult and cost-ineffective [68]. This Application Note outlines advanced wet-lab and computational techniques for depleting host DNA and enriching microbial signals, enabling more efficient and accurate shotgun metagenomic analysis of gut microbiome samples.
Table 1: Performance Comparison of Host DNA Depletion Methods
| Method | Principle | Host Depletion Efficiency | Key Advantages | Key Limitations |
|---|---|---|---|---|
| MEM (Microbial-Enrichment Methodology) [67] | Selective lysis of host cells using mechanical stress (large beads) and enzymatic degradation | ~1,600-fold host depletion in mouse intestinal scrapings [67] | Minimal microbial community perturbation; >90% of taxa show no significant difference; fast protocol (<20 min) [67] | Induces ~31% bacterial loss in stool samples [67] |
| MolYsis complete5 [69] | Selective lysis of mammalian cells with guanidinium, followed by DNase degradation | Significantly higher % microbial reads (avg. 38.31%) in milk samples vs. other methods [69] | Effective for low-volume material; no significant bias introduced in community profile [69] | Can cause drop-out of some bacterial taxa; performance varies by sample type [67] [70] |
| NEBNext Microbiome DNA Enrichment Kit [68] | Enzymatic digestion of methylated host DNA (5-mC) | 24% bacterial sequences in intestinal samples vs. <1% in controls [68] | Targeted enzymatic approach | Inefficient on tissue samples without additional optimization like detergents/bead-beating [68] [70] |
| QIAamp DNA Microbiome Kit [68] [70] | Selective lysis of cells lacking a cell wall using saponin | 28% bacterial sequences in intestinal samples vs. <1% in controls [68] | Effective host depletion shown in multiple studies | Can induce non-uniform bacterial losses; some taxa may drop >100-fold [67] |
| ONT Adaptive Sampling [68] | In silico enrichment; sequencing device rejects host reads in real-time | Increased total bacterial reads and improved metagenomic assembly [68] | No physical sample manipulation; can recover antimicrobial resistance markers and plasmids [68] | Can cause relevant shifts in observed bacterial abundance (e.g., 2-5x more E. coli reads) [68] |
Table 2: Essential Research Reagent Solutions
| Reagent / Kit Name | Primary Function | Key Application Notes |
|---|---|---|
| MEM Protocol Components [67] | Host cell lysis & microbial DNA enrichment | Uses 1.4mm beads for mechanical shearing of host cells; includes Benzonase and Proteinase K treatment. |
| MolYsis complete5 Kit [69] | Selective host DNA depletion | Optimal for low microbial biomass samples like milk; effective on bovine and human milk samples. |
| ZymoBIOMICS Spike-in Control [71] | Process control for extraction and sequencing | Spiked into samples to monitor efficiency and potential bias in host depletion workflows. |
| DNeasy PowerLyzer PowerSoil Kit (QIAGEN) [72] | Standardized DNA extraction | Often used as a base method; performance is improved with upstream stool preprocessing. |
| Stool Preprocessing Device (SPD) [72] | Standardization of fecal sample handling | Upstream device that improves DNA yield, alpha-diversity, and Gram-positive bacterial recovery for several extraction protocols. |
| Benzonase Nuclease [67] | Degradation of extracellular nucleic acids | Critical in MEM protocol to degrade host DNA released after lysis, minimizing contaminating sequences. |
| Proteinase K [67] | Host cell lysis and histone degradation | Aids in complete host cell lysis and degradation of DNA-binding proteins in the MEM protocol. |
Experimental Protocols for Key Host Depletion Methods
Protocol 1: Microbial-Enrichment Methodology (MEM) for Intestinal Biopsies
Principle: This protocol leverages the size difference between host and bacterial cells, using large beads to preferentially lyse host cells through mechanical shear stress while leaving most microbial cells intact [67].
Procedure:
- Homogenization: Process the intestinal biopsy sample using a tissue homogenizer. For optimal results with the MEM protocol, begin with a homogenized sample [67] [70].
- Host Cell Lysis: Add 1.4 mm ceramic beads to the homogenate and subject to bead-beating. This step applies high mechanical shear stress to rupture the larger host cells [67].
- Enzymatic Digestion:
- Add Benzonase to the lysate to degrade accessible extracellular nucleic acids, including DNA released from the lysed host cells.
- Add Proteinase K to further degrade host proteins and histones, ensuring complete host DNA release and degradation [67].
- Microbial DNA Extraction: Proceed with a standard microbial DNA extraction kit (e.g., DNeasy PowerLyzer PowerSoil Kit) to isolate the now-enriched microbial DNA [67] [72].
- Quality Control: Quantify DNA and assess the host depletion efficiency, for example, via qPCR targeting a host gene versus a universal bacterial gene.
Protocol 2: Comparative Evaluation of Host Depletion Kits
Principle: This protocol provides a framework for empirically testing and selecting the most suitable host depletion method for a specific sample type, such as intestinal tissue [68] [70].
Procedure:
- Sample Preparation: Aliquot homogenized intestinal tissue samples or high-host DNA content samples. Include a positive control (e.g., a mock microbial community) and negative controls (extraction blanks) [71] [69].
- Parallel Host Depletion: Extract DNA from the aliquots using different host depletion methods in parallel. Key kits to compare include:
- Library Preparation and Sequencing: Prepare shotgun metagenomic libraries for all samples and sequence on a platform of choice (e.g., Illumina) [69] [68].
- Bioinformatic Analysis:
- Use a taxonomic classifier like Kraken2 [69] to determine the percentage of reads assigned to host versus microbial databases.
- Evaluate the impact on microbial community structure by assessing alpha-diversity and beta-diversity metrics.
- For methods like Nanopore sequencing, enable Adaptive Sampling for in silico enrichment and compare the results to wet-lab methods [68].
- Decision Matrix: Select the optimal method based on the highest microbial read percentage, minimal taxonomic bias, and cost-effectiveness for your specific application.
Workflow Visualization for Host DNA Depletion
Diagram 1: A strategic workflow for overcoming host DNA contamination in gut microbiome studies, integrating wet-lab and computational enrichment techniques.
Effective host DNA depletion is a critical step for successful shotgun metagenomic analysis of gut tissue samples. No single method is universally superior; the choice depends on the sample type, research objectives, and available resources. Wet-lab methods like MEM and MolYsis offer robust physical or enzymatic depletion, while computational approaches like ONT Adaptive Sampling provide a flexible in silico alternative. By implementing the protocols and considerations outlined in this Application Note, researchers can significantly improve the yield and quality of microbial data from challenging, host-rich samples, thereby advancing our understanding of host-microbe interactions in the gut.
Within the complex ecosystem of the human gut microbiome, the fungal community, or mycobiome, represents a critical but historically overlooked component. While typically constituting only 0.1% to 1% of the entire gut microbiome, fungi exert a significant influence on host physiology, immune modulation, and disease pathogenesis [73] [74]. The characterization of the gut mycobiome has lagged substantially behind that of its bacterial counterpart, largely due to technical challenges in its capture and analysis. Shotgun metagenomics has emerged as a powerful tool for unbiased microbiome profiling, yet its effective application to fungal communities requires specialized strategies to overcome their low relative abundance and distinct cellular biology. This Application Note provides a comprehensive framework for mycobiome profiling within the broader context of shotgun metagenomics, detailing optimized wet-lab and computational protocols to reliably capture and interpret fungal community data.
The Mycobiome Profiling Challenge
The accurate profiling of the gut mycobiome using shotgun metagenomics is fraught with methodological hurdles that can compromise data fidelity.
- Low Abundance and High Background: The numerical inferiority of fungal cells compared to bacterial cells means fungal DNA can be overwhelmed by bacterial DNA in shotgun sequencing data. The ratio of fungal to bacterial cells in feces is estimated to be between 10^-9 and 10^-4, with fungal genes often comprising less than 0.08% of the total microbial metagenome [30]. This necessitates either deep sequencing, which is costly, or targeted enrichment strategies.
- Technical Limitations in Bioinformatics: A recent evaluation of bioinformatics tools for mycobiome profiling from shotgun metagenomic data revealed a limited selection of specialized software and a significant lack of comprehensive databases [75]. When six prominent tools (Kraken2, MetaPhlAn4, EukDetect, FunOMIC, MiCoP, and HumanMycobiomeScan) were evaluated on mock fungal communities, only Candida orthopsilosis was consistently identified by all tools across all communities, highlighting a concerning lack of consensus [75].
- Database Incompleteness: Many fungal genomes remain uncharacterized or are poorly represented in public databases. This incompleteness directly impacts the accuracy of taxonomic profiling, as reads from unrepresented species cannot be assigned correctly, leading to false negatives and mischaracterization of community structure.
Evaluating Bioinformatics Tools for Mycobiome Analysis
Selecting an appropriate bioinformatics pipeline is paramount for generating reliable mycobiome profiles. A 2025 benchmark study evaluated the performance of six tools on mock communities of varying richness and abundance, providing critical insights for tool selection [75].
Table 1: Performance Comparison of Mycobiome Profiling Tools on Mock Communities
| Tool | Primary Strategy | Accuracy on Species Level | Accuracy on Genus Level | Impact of Bacterial Background | Overall Strengths |
|---|---|---|---|---|---|
| FunOMIC | Marker-based / Custom DB | Recognized most species | High | Not significant | High species recognition |
| EukDetect | Marker-based (18S) | Predictions closest to correct composition | High | Not significant | High overall accuracy |
| MiCoP | Whole-genome mapping | High with same reference DB | High | Not significant | Best accuracy among whole-genome tools |
| MetaPhlAn4 | Marker-based | Variable | Accurately identified all genera | Not significant | Excellent genus-level precision |
| Kraken2 | k-mer based (LCA) | Variable; improved with richness | Variable; improved with richness | Not significant | Good precision with high richness |
| HumanMycobiomeScan | Custom database | Required code modification | Required code modification | Information not available | Specialized for human gut |
The top-performing tools for overall accuracy in both identification and relative abundance estimation were EukDetect, MiCoP, and FunOMIC, respectively [75]. It is critical to note that the addition of 90% and 99% bacterial background did not significantly impact the performance of these tools, confirming their robustness for analyzing complex metagenomic samples [75]. This evaluation underscores that no single tool provides a perfect solution, and researchers should consider the use of multiple, complementary tools to validate their findings.
Table 2: Key Research Reagent Solutions for Mycobiome Bioinformatics
| Resource Name | Type | Primary Function | Key Consideration |
|---|---|---|---|
| FunOMIC Database | Genomic Database | Comprehensive fungal genome collection for taxonomic profiling | Provides single-copy marker genes for improved quantification [30] |
| UNITE Database | Reference Database | Taxonomic assignment of fungal ITS sequences | Standard for amplicon-based studies; can complement shotgun data [76] |
| FindFungi | Analysis Pipeline | Identifies fungal species in shotgun metagenomes; uses Kraken and read distribution analysis | Reduces false positives; effective for pathogen detection [77] |
| MetaPhlAn4 | Profiling Tool | Taxonomic profiling using clade-specific marker genes | Accurate at genus level; integrates well with bacterial profiling [75] |
| Kraken2 | Classification Tool | Fast k-mer-based taxonomic classification to lowest common ancestor | Performance depends on database completeness and community richness [75] [77] |
Optimized Wet-Lab Protocols for Mycobiome Capture
To overcome the challenge of low fungal biomass, strategic wet-lab protocols are required. The following diagram and protocol outline a robust workflow for mycobiome enrichment and sequencing.
Diagram Title: Mycobiome Enrichment and Sequencing Workflow
Protocol: Fungal Cell Enrichment and Shotgun Metagenomic Sequencing
This protocol, optimized for human fecal samples, leverages the larger size of fungal cells (yeasts: 2–10 μm, hyphae: up to 40 μm) compared to bacterial cells (typically 0.2–2 µm) to enrich the fungal fraction prior to DNA extraction [30].
I. Fungal Cell Enrichment via Differential Centrifugation
- Sample Homogenization: Resuspend ~200 mg of fresh or frozen fecal sample in 10 mL of sterile Phosphate Buffered Saline (PBS). Vortex thoroughly until a homogeneous suspension is achieved.
- Low-Speed Centrifugation: Centrifuge the homogenate at 500 × g for 10 minutes at 4°C. This low-speed step pellets large debris and intact food particles, leaving microbial cells in the supernatant.
- Collection of Microbial Supernatant: Carefully transfer the supernatant to a new 15 mL conical tube, avoiding the pelleted debris.
- Enrichment Centrifugation: Centrifuge the supernatant at 2,000 × g for 15 minutes at 4°C. This intermediate speed is designed to pellet the larger fungal cells while leaving many bacterial cells in suspension.
- Pellet Collection: Discard the supernatant, which contains a large proportion of the bacterial cells. The resulting pellet is enriched for fungal biomass.
- Wash: Resuspend the fungal-enriched pellet in 1 mL of PBS to wash. Centrifuge again at 2,000 × g for 5 minutes and discard the supernatant.
II. DNA Extraction and Library Construction
- Mechanical Lysis: Extract genomic DNA from the enriched pellet using a commercial soil DNA extraction kit (e.g., MoBio PowerSoil DNA Isolation Kit). Ensure the protocol includes a rigorous mechanical lysis step (bead beating) to break down tough fungal cell walls, which is critical for high DNA yield [76] [78].
- Library Preparation: For shotgun metagenomic sequencing, use a high-throughput library preparation kit such as TruSeqNano. Benchmark studies have shown that TruSeqNano libraries performed best in recovering near-complete genome fractions from complex microbiomes compared to other kits like NexteraXT [79].
- Sequencing Parameters: Sequence the libraries on an Illumina HiSeq4000 instrument using PE150 chemistry with ~400 bp insert sizes. This configuration has been demonstrated to provide the best cost-benefit ratio and assembly contiguity for metagenomic samples [79].
Integrated Analysis: Connecting Mycobiome to Host Physiology
Once reliable mycobiome profiles are generated, the data can be integrated with host metadata to uncover biologically meaningful relationships. Rodent models have been instrumental in elucidating the causal role of the mycobiome in host health and disease.
Table 3: Insights into Mycobiome Function from Rodent Models
| Disease Context | Key Fungal Taxa | Observed Effect | Mechanistic Insight |
|---|---|---|---|
| Inflammatory Bowel Disease (IBD) | Candida spp., Aspergillus spp. | Expansion during dysbiosis; can worsen or ameliorate colitis | Antifungal treatment worsened colitis; fungal mannans may confer protection via TLR4 signaling [74] |
| Metabolic Phenotypes | Candida, Aspergillus | Correlated with adiposity, triglycerides, insulin, leptin | Vendor-specific mouse mycobiomes linked to differential metabolic responses to diet [74] |
| Antibiotic Treatment | Candida | Expanded abundance post-antibiotic therapy | Antibiotic disruption of bacteria reduced competition, exacerbating fungal colonization [74] |
| Host Genetics | Various | 33% of gut fungal variation explained by genetics x diet | Candidate genes (Taf4b, Tmc8) identified as modulators of mycobiome composition [74] |
The following diagram synthesizes the key experimental and analytical steps detailed in this Application Note, providing a logical map for a complete mycobiome study.
Diagram Title: Mycobiome Study Design Logic Map
Capturing the elusive mycobiome requires a concerted strategy that addresses its unique challenges at every stage, from bench to bioinformatics. The integration of a physical enrichment protocol, such as differential centrifugation, with deep shotgun sequencing and a consensus-based bioinformatic approach using tools like EukDetect and MiCoP, provides a robust framework for reliable fungal community profiling. As databases and tools continue to mature, standardized application of these protocols will be crucial for generating comparable and meaningful data across studies. A precise understanding of the gut mycobiome, enabled by these strategies, will open new frontiers in our comprehension of host-microbe interactions and their impact on human health and disease.
Shotgun metagenomic sequencing has revolutionized gut microbiome research by enabling comprehensive analysis of microbial community composition and function directly from stool samples, bypassing the need for cultivation [5]. However, the translation of this powerful technology from research into robust clinical applications faces significant challenges due to methodological variability across the entire workflow [11]. This variability introduces substantial inconsistencies in results, compromising reproducibility and comparability across studies.
The complexity of metagenomic data, combined with numerous analytical approaches and the lack of universally accepted protocols, has hindered the standardization necessary for clinical implementation [11]. This application note details standardized protocols and analytical frameworks designed to address these critical hurdles, providing researchers with validated methodologies to enhance reproducibility in gut microbiome studies.
Standardized Wet-Lab Protocols for Gut Microbiome Analysis
Sample Collection and DNA Extraction
Proper sample collection and processing are fundamental for obtaining reliable metagenomic data. For human gut microbiome studies, stool samples should be immediately preserved after collection using appropriate stabilization buffers or flash-freezing at -80°C to prevent microbial community shifts [80]. The protocol from MetaGenoPolis emphasizes the critical importance of ensuring proper sampling and sample conservation before proceeding to DNA extraction [80].
DNA extraction represents a significant source of technical variability. The standardized protocol employs mechanical lysis combined with commercial extraction kits designed for complex stool samples. Specifically, the use of MP-soil FastDNA Spin Kit for Soil (#6560-200, MP Biomedicals) has been demonstrated to provide high-quality metagenomic DNA from fecal samples [19]. This step is crucial for achieving sufficient microbial biomass while minimizing contamination, particularly in low-biomass samples where ultraclean reagents and "blank" sequencing controls are essential [16].
Library Preparation and Sequencing Platforms
Library preparation for shotgun metagenomics must be optimized for complex microbial communities. The Illumina platform has become dominant due to its high outputs (up to 1.5Tb per run), high accuracy (error rate of 0.1-1%), and wide availability [16]. Recent advancements in long-read technologies, particularly PacBio HiFi sequencing, offer advantages for reconstructing high-quality genomes from metagenomic samples, as demonstrated in recent grants targeting inflammatory bowel disease and colorectal cancer microbiota [21].
For typical gut microbiome studies, sequencing depth should be sufficient to capture microbial diversity, with recent clinical studies achieving 10-14 Gb per sample [19]. Paired-end sequencing (2×150 bp or 2×250 bp) on Illumina platforms provides the optimal balance between read length, throughput, and cost for most applications.
Table 1: Key Research Reagent Solutions for Shotgun Metagenomics
| Reagent/Kit | Function | Application Notes |
|---|---|---|
| MP-soil FastDNA Spin Kit for Soil | DNA extraction from complex stool samples | Effective for gram-positive and gram-negative bacteria; includes mechanical lysis [19] |
| Illumina DNA Prep Kit | Library preparation for shotgun sequencing | Compatible with various input DNA amounts; includes fragmentation and adapter ligation |
| BWA (v 0.7.17) | Removal of host DNA contamination | Maps reads to human genome for filtering; critical for clinical samples [19] |
| fastp (v 0.23.0) | Quality control and adapter trimming | Processes raw sequencing reads; removes low-quality sequences [19] |
Bioinformatics Standardization Framework
Quality Control and Read Processing
Standardized quality control is essential for reproducible metagenomic analysis. The workflow begins with adapter removal and quality filtering using tools such as fastp (v 0.23.0), eliminating low-quality reads with an average quality score below 20 and sequences shorter than 50 bp after trimming [19]. Subsequent host DNA removal through mapping to the human genome using BWA (v 0.7.17) prevents contamination from human cells in stool samples [19].
Quality-controlled reads can then be processed through two main pathways: assembly-based approaches that reconstruct longer genomic fragments, or read-based analysis that quantifies taxonomic and functional abundances directly from sequencing reads. The choice between these strategies depends on research objectives, with assembly required for obtaining full-length coding sequences or recovering microbial genomes, while read-based analysis suffices for taxonomic profiling [16].
Taxonomic and Functional Profiling
For taxonomic assignment, standardized pipelines should employ curated databases to ensure consistent annotation. The MetaGenoPolis protocol utilizes the 10.4M gut gene catalog and 8.4M oral gene catalog as comprehensive reference databases for read mapping [80]. Determination of microbial composition can be performed using tools such as MSPminer, which enables abundance-based reconstitution of microbial pan-genomes from shotgun metagenomic data [80].
Functional annotation involves mapping non-redundant genes against established databases using Diamond (v 2.0.13) with an optimized e-value cutoff of 1e-5 [19]. The KEGG database (v 94.2) provides pathway annotations that facilitate interpretation of microbial community function, while additional databases such as eggNOG, TIGRFAMs, and CAZy offer complementary functional insights [16].
Table 2: Bioinformatics Tools and Databases for Standardized Analysis
| Tool/Database | Purpose | Key Parameters |
|---|---|---|
| fastp (v 0.23.0) | Quality control and adapter trimming | Quality score: ≥20; Min length: 50 bp [19] |
| BWA (v 0.7.17) | Host DNA removal | Maps to human reference genome [19] |
| Diamond (v 2.0.13) | Functional annotation | e-value: 1e-5; database: KEGG, eggNOG [19] |
| Meteor2 | Metagenomic read mapping and quantification | References: 10.4M gut gene catalog [80] |
| MSPminer | Microbial composition analysis | Abundance-based pan-genome reconstruction [80] |
| SILVA database | Taxonomic classification | Quality-checked rRNA sequence data [16] |
Standardized Shotgun Metagenomics Workflow
Implementation in Clinical Research Settings
Validation and Quality Metrics
Implementation of standardized protocols requires rigorous validation using established quality metrics. α-diversity analysis using the Shannon index provides insight into microbial diversity within samples, while principal coordinates analysis (PCoA) reveals differences in community composition between samples [19]. These metrics should be reported consistently across studies to enable cross-cohort comparisons.
For clinical applications, enterotyping has emerged as a valuable stratification approach, grouping samples with similar structures of dominant microbiomes based on the relative abundance of microbes at the genus level using Bray-Curtis distance clustering [19]. This standardization enables population stratification and provides a global overview of inter-individual variations in gut microbial composition.
Clinical Applications and Case Studies
Standardized metagenomic protocols have enabled significant advances in clinical gut microbiome research. In inflammatory bowel disease (IBD), multi-omics integration encompassing metagenomes and metabolomes has identified consistent alterations in underreported microbial species and significant metabolite shifts, achieving high diagnostic accuracy (AUROC 0.92-0.98) in distinguishing IBD from controls [11].
In infectious disease diagnostics, standardized shotgun metagenomic sequencing has demonstrated remarkable sensitivity in detecting Clostridioides difficile directly from stool samples, with true positive diagnostic rates exceeding 99% with minimal false positives against closely related species [11]. Similarly, applications in acute pancreatitis research have revealed dynamic shifts in microbial composition during recovery phases, informing strategies for treatment and prognosis [19].
Standardization of shotgun metagenomic protocols from bench to bioinformatics is essential for advancing gut microbiome research into clinical practice. The methodologies outlined in this application note provide a framework for reducing methodological variability and enhancing reproducibility. Future efforts should focus on global harmonization of standards, cross-sector collaboration, and inclusive frameworks that ensure scientific rigor and equitable benefit from microbiome-based discoveries [11].
As sequencing technologies continue to advance and computational methods improve, the integration of metagenomic data with other omics datasets will provide deeper insights into microbial community function and host-microbe interactions. The development of internationally standardized protocols, reference materials, and analytical frameworks will be crucial for realizing the full potential of microbiome-informed precision medicine.
Shotgun metagenomics enables comprehensive analysis of the genetic material recovered directly from microbial communities, providing unprecedented insight into the functional potential of complex ecosystems like the human gut microbiome [5]. A critical step in analyzing these data is functional annotation, the process of assigning biological meaning to DNA sequences by identifying genes and predicting their functions [16]. This process relies heavily on comparing metagenomic sequences to reference databases containing experimentally validated and computationally predicted protein families and metabolic pathways [16].
However, the vast microbial diversity present in environmental and host-associated communities presents significant challenges for annotation. A substantial proportion of sequences—often 20-40% or more—routinely fail to match any known function, being classified as "hypothetical proteins" or showing no similarity to characterized sequences [5] [16]. This annotation gap fundamentally limits our ability to interpret metagenomic data and generate biologically meaningful insights, particularly for novel microbial lineages and uncharacterized gene families. This application note examines the limitations of current annotation databases and provides practical strategies to navigate these challenges in gut microbiome research.
Table 1: Major Reference Databases for Metagenomic Annotation
| Database | Primary Focus | Strengths | Limitations |
|---|---|---|---|
| KEGG | Metabolic pathways and ortholog groups [16] | Well-curated pathways; enables functional reconstruction | Limited coverage of non-model organisms |
| UniProt | Protein sequences and functional information [16] | Combination of manually annotated and computationally predicted proteins | Variable annotation depth across taxa |
| eggNOG | Orthologous groups and functional annotation [16] | Broad phylogenetic coverage; evolutionary context | Functional predictions may be incomplete |
| TIGRFAMs | Protein family classification [16] | Role-based subfamily classification | Limited to specific protein families |
| CAZy | Carbohydrate-active enzymes [16] | Specialized for carbohydrate metabolism | Narrow functional scope |
| CARD | Antibiotic resistance genes [16] | Comprehensive resistance gene coverage | Focused on specific function class |
Critical Limitations in Current Annotation Databases
Reference Database Biases and Coverage Gaps
Public databases suffer from substantial taxonomic biases toward medically and economically important microorganisms, with severe underrepresentation of environmental and host-associated taxa [5]. This creates circular limitations where poorly characterized organisms remain poorly characterized due to insufficient reference data. The functional bias in databases is equally problematic, with overrepresentation of certain well-studied metabolic pathways (e.g., central carbon metabolism) and underrepresentation of others (e.g., secondary metabolism, specialized transporters) [16]. Furthermore, technical artifacts in database construction, including propagation of existing annotation errors and inconsistent curation standards across resources, compound these fundamental limitations [5].
Impact on Gut Microbiome Research
In gut microbiome studies, database limitations manifest as high proportions of "unknown function" assignments, particularly for sequences derived from less-characterized microbial taxa [5]. This impedes our ability to connect microbial community composition to ecosystem functions, identify novel bioactive molecules, and understand host-microbe interactions. The problem is particularly acute for studies focusing on non-Western populations, where microbial diversity may be less represented in reference databases [5].
Strategic Approaches to Overcome Annotation Limitations
Database Integration and Selection Strategies
Employing multiple databases during annotation significantly improves functional coverage, as different resources have complementary strengths and taxonomic biases [16]. A tiered approach using both general and specialized databases provides the most comprehensive functional insights. Researchers should prioritize databases based on their specific research questions—for example, selecting CAZy for carbohydrate metabolism studies or CARD for antibiotic resistance profiling [16]. Implementing consensus annotation protocols that require agreement across multiple databases can improve annotation reliability, though at the potential cost of reduced annotation coverage.
Table 2: Experimental Protocols for Enhanced Functional Characterization
| Method | Application | Key Steps | Outcome |
|---|---|---|---|
| Hybrid Assembly | Improve genome reconstruction from complex communities [81] | 1. Sequence with both short-read (Illumina) and long-read (PacBio) technologies2. Perform hybrid assembly using metaSPAdes or similar tools3. Bin contigs into metagenome-assembled genomes (MAGs) | Higher quality MAGs with reduced fragmentation; improved gene prediction |
| Complementary Amplicon Sequencing | Link taxonomy with function in specific microbial groups [5] | 1. Perform 16S rRNA gene sequencing on same samples2. Conduct shotgun metagenomics3. Integrate datasets using phylogenetic placement | Connected taxonomic and functional profiles; improved interpretation |
| Targeted Gene Enrichment | Recover specific functional genes of interest [5] | 1. Design probes based on conserved regions of target gene families2. Perform hybrid capture before sequencing3. Sequence enriched libraries | Increased sequencing depth for target genes; improved detection of rare variants |
Computational and Experimental Enhancements
Assembly-based approaches that reconstruct longer contigs provide crucial context for functional prediction, enabling better gene calling and detection of operonic structures that can inform function [16]. Tiered annotation pipelines that combine rapid initial profiling (e.g., using HUMAnN2) with deeper customized analysis maximize both efficiency and depth of functional insight [16]. When database searches prove insufficient, structure-based function prediction using tools like AlphaFold2 can provide insights by identifying structural similarities to characterized proteins [5]. For high-priority targets, heterologous expression and functional screening of metagenomic clones remains the gold standard for confirming gene function, though this approach is resource-intensive [5].
Database Navigation and Functional Annotation Workflow
Integrated Protocols for Improved Functional Insights
Multi-Database Annotation Protocol
Sample Input: Quality-filtered metagenomic reads or assembled contigs
Step 1: Initial Rapid Profiling
- Run HUMAnN2 for community-wide functional profiling against pre-integrated databases
- Use Kaiju or Kraken2 for taxonomic classification
- Quality Control: Check for sufficient sequencing depth (>5M reads per sample for complex gut microbiomes)
Step 2: Custom Database Integration
- Annotate against minimum three databases with complementary focuses (e.g., KEGG, eggNOG, TIGRFAMs)
- Use parallel computing to manage computational load
- Parameter Settings: E-value cutoff ≤1e-5, minimum identity ≥60% for protein searches
Step 3: Consensus Annotation Generation
- Resolve conflicting annotations using majority voting or hierarchical evidence weighting
- Flag annotations with inconsistent assignments for manual curation
- Output: Structured annotation table with confidence scores and evidence sources
Enhanced Functional Prediction Protocol
Step 1: Genomic Context Analysis
- Identify conserved genomic neighborhoods using geNomad or similar tools
- Detect potential operonic structures and co-regulated gene clusters
- Interpretation: Genes with related functions often cluster together in microbial genomes
Step 2: Structure-Based Function Prediction
- Generate 3D protein structures using AlphaFold2 for unknown genes
- Perform structural similarity search using Foldseek against PDB
- Thresholds: Consider significant structural similarity with TM-score >0.5
Step 3: Experimental Validation Design
- Clone candidate genes into expression vectors
- Develop functional assays based on predicted activities
- Controls: Include positive and negative controls in all assays
Integrated Multi-Method Approach to Overcome Annotation Limitations
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Metagenomic Functional Characterization
| Category | Specific Product/Kit | Application Context | Critical Function |
|---|---|---|---|
| DNA Extraction | DNeasy PowerSoil Pro Kit (QIAGEN) | Low-biomass gut microbiome samples | Inhibitor removal; high DNA yield |
| Library Preparation | Nextera XT DNA Library Prep Kit (Illumina) | High-throughput metagenomic sequencing | Efficient fragmentation and adapter ligation |
| Long-read Sequencing | SMRTbell Express Template Prep Kit (PacBio) | Hybrid assembly approaches | Preparation of libraries for long-read sequencing |
| Functional Screening | pET Expression System (Novagen) | Heterologous expression of metagenomic genes | High-level protein expression in E. coli |
| Enzyme Assays | EnzCheck Ultra Amylase Assay Kit (Thermo Fisher) | Characterization of carbohydrate-active enzymes | Sensitive detection of enzyme activity |
| Cell Culture | AnaeroPack System (Mitsubishi Gas) | Cultivation of anaerobic gut microbes | Creates anaerobic conditions for functional studies |
| Bioinformatics | HUMAnN2 Pipeline | Community-wide functional profiling | Automated pathway abundance analysis |
| Structural Prediction | AlphaFold2 Software | Structure-based function prediction | Accurate protein structure prediction from sequence |
As shotgun metagenomics continues to revolutionize our understanding of gut microbiome function, acknowledging and actively addressing database limitations remains essential for generating robust biological insights. The integrated approaches outlined here—combining multi-database annotation, computational enhancements, and targeted experimental validation—provide a roadmap for navigating current annotation challenges. Future developments in database curation, long-read sequencing, and artificial intelligence-based function prediction promise to gradually close the annotation gap, ultimately enabling more complete functional characterization of complex microbial communities.
Shotgun metagenomics has revolutionized gut microbiome research by enabling comprehensive analysis of the genetic material of entire microbial communities, offering unparalleled insights into taxonomic composition, functional potential, and strain-level dynamics [5] [16]. This powerful approach bypasses the limitations of traditional culturing techniques, allowing researchers to study the vast majority of microorganisms that cannot be grown in laboratory settings [16]. However, the immense data generated and the complex nature of microbial communities introduce significant computational challenges and ethical imperatives that researchers must navigate.
The analytical process involves multiple sophisticated steps, from quality control and assembly to taxonomic and functional profiling, each requiring specialized computational tools and significant resources [5] [4]. Concurrently, as metagenomic research increasingly informs clinical diagnostics and therapeutic development, ensuring equity in participant representation, data interpretation, and benefit distribution becomes paramount. This article examines the key computational and ethical considerations in managing shotgun metagenomic data for gut microbiome research, providing structured protocols, analytical frameworks, and practical guidance for maintaining scientific rigor and ethical integrity throughout the research lifecycle.
Computational Considerations in Metagenomic Data Management
Data Generation and Processing Workflows
Shotgun metagenomic sequencing fragments all DNA from a sample, generating millions of short reads that must be computationally reconstructed and interpreted [5]. A typical analytical workflow encompasses several processing stages, each with distinct computational demands:
Quality Control and Preprocessing: Raw sequencing reads first undergo quality assessment using tools like FastQC to evaluate per-base sequence quality, GC content, and potential adapter contamination. This is often followed by trimming or filtering to remove low-quality sequences, which is crucial for downstream analysis accuracy [4].
Taxonomic and Functional Profiling: Processed reads are analyzed to determine microbial composition (taxonomic profiling) and metabolic capabilities (functional profiling). Reference-based methods align sequences to known genomic databases, while de novo approaches assemble reads without reference genomes, each with distinct computational trade-offs [5] [16]. Advanced tools like Meteor2 leverage environment-specific microbial gene catalogs to deliver integrated taxonomic, functional, and strain-level profiling (TFSP), demonstrating strong performance in detecting low-abundance species and improving functional abundance estimation accuracy by at least 35% compared to previous methods [10].
Strain-Level Analysis: High-resolution characterization of microbial strains enables tracking of microbial transmission and fine-scale community dynamics. Meteor2 accomplishes this by tracking single nucleotide variants (SNVs) in signature genes, capturing more strain pairs than established methods (an additional 9.8-19.4% in benchmark tests) [10].
Table 1: Key Computational Steps in Shotgun Metagenomic Analysis
| Processing Stage | Primary Tools/Methods | Computational Output | Key Challenges |
|---|---|---|---|
| Quality Control | FastQC, Trimmomatic | Quality-filtered reads | Handling large volume of raw data; identifying technical artifacts |
| Taxonomic Profiling | MetaPhlAn4, Meteor2, kraken2 | Taxonomic abundance table | Database completeness; ambiguous reads; low-abundance taxa |
| Functional Profiling | HUMAnN3, Meteor2, MG-RAST | Functional pathway abundances | Gene annotation accuracy; pathway inference; metabolic reconstruction |
| Strain-Level Analysis | StrainPhlAn, Meteor2 | Strain variants; population genetics | Detection sensitivity; reference bias; computational intensity |
Data Storage and Computational Resource Requirements
The volume of data generated by shotgun metagenomics presents substantial storage and processing challenges. A single metagenomic sample can produce 1-10 billion base pairs, requiring gigabytes of storage per sample [16]. Longitudinal studies compound these requirements, as evidenced by research tracking gut microbiome changes over three years [82].
Different sequencing technologies impose varying computational burdens. While Illumina platforms dominate metagenomic sequencing due to high output and accuracy, emerging long-read technologies like PacBio SMRT sequencing provide superior resolution for complex genomic regions but generate different data types requiring specialized analytical approaches [16] [21].
Table 2: Computational Resource Considerations for Metagenomic Analyses
| Analysis Type | Minimum RAM | Storage per Sample | Processing Time | Primary Bottlenecks |
|---|---|---|---|---|
| Quality Control | 4-8 GB | 1-5 GB | 30-60 minutes | I/O operations; multi-threading |
| Taxonomic Profiling | 8-16 GB | 5-15 GB | 2-6 hours | Database indexing; read mapping |
| Functional Profiling | 16-32 GB | 10-25 GB | 4-12 hours | Pathway reconstruction; annotation transfers |
| Strain-Level Analysis | 32-64 GB | 20-50 GB | 6-24 hours | Variant calling; population genetics |
| Metagenome Assembly | 64-128 GB | 50-200 GB | 12-48 hours | Graph construction; memory allocation |
Modern tools are addressing these computational challenges through optimization strategies. For instance, Meteor2 offers a "fast mode" that uses a lightweight version of gene catalogs containing only signature genes, enabling rapid taxonomic and strain profiling with modest resource requirements (5 GB RAM, 10 minutes for 10 million reads) [10].
Diagram 1: Computational Workflow for Shotgun Metagenomics
Ethical Considerations for Equitable Metagenomic Research
Population Representation and Data Equity
A critical ethical challenge in gut microbiome research lies in ensuring diverse population representation in study cohorts. Most public metagenomic databases predominantly contain data from Western, educated, industrialized, rich, and democratic (WEIRD) populations, creating blind spots in our understanding of global microbiome diversity and potentially exacerbating health disparities [83]. This representation bias can lead to:
Limited Generalizability: Microbiome-based diagnostics and therapeutics developed from narrow population subsets may have reduced efficacy or accuracy when applied to underrepresented groups. Research has demonstrated significant country-specific variations in gut microbiota, with different bacterial species accounting for similar functional deficits (like riboflavin and biotin biosynthesis) across geographical regions [83].
Perpetuation of Health Disparities: If microbiome-based interventions are primarily optimized for and accessible to already well-served populations, they risk widening existing health inequities. This is particularly concerning for conditions like Parkinson's disease, where meta-analyses have revealed consistent microbial patterns across countries but with different underlying bacterial contributors [83].
Resource Imbalances in Research: The high costs of metagenomic sequencing (ranging from hundreds to thousands of dollars per sample) can redirect research resources toward wealthy institutions and populations, further marginalizing communities facing greater health burdens [21]. Initiatives like the PacBio Microbiome SMRT Grant program attempt to address this by providing sequencing resources to researchers studying underrepresented populations [21].
Data Sharing, Privacy, and Sovereignty
Metagenomic data presents unique privacy concerns as it can reveal sensitive information about health status, dietary habits, and environmental exposures. Unlike human genomic data, microbiome data represents a complex mixture of host and microbial DNA, creating ambiguous boundaries for privacy protection. Key considerations include:
Host DNA in Metagenomic Samples: Shotgun sequencing captures all DNA in a sample, including human genetic material shed in stool. This human DNA can reveal information about an individual's ancestry, disease predispositions, and identity, yet may not be adequately protected by current ethical frameworks [5].
Microbiome as Personal Identifier: Emerging evidence suggests that microbiome profiles may be personally identifiable, raising questions about how such data should be classified and protected in research contexts and public databases.
Data Sovereignty and Community Engagement: For Indigenous and traditional communities, microbiome data may have cultural significance beyond individual privacy concerns. Respecting data sovereignty and implementing collaborative governance models are essential for ethical research practice.
Table 3: Ethical Framework for Metagenomic Research
| Ethical Dimension | Key Challenges | Recommended Practices |
|---|---|---|
| Participant Representation | WEIRD population bias; exclusion of marginalized groups | Intentional cohort diversification; community-based recruitment; resource allocation for underrepresented populations |
| Data Privacy | Host DNA in samples; microbiome as identifier; re-identification risk | Clear consent protocols; data anonymization; controlled data access; ongoing risk assessment |
| Benefit Sharing | Commercialization of microbiome findings; patenting of microbial products | Fair benefit-sharing agreements; community advisory boards; technology transfer policies |
| Clinical Translation | Equitable access to microbiome-based therapies; diagnostic accuracy across populations | Validation in diverse cohorts; affordable intervention strategies; accessible diagnostic platforms |
| Data Sovereignty | Cultural significance of microbiome; Indigenous knowledge protection | Collaborative research agreements; data governance partnerships; recognition of traditional knowledge |
Integrated Protocols for Data Management and Equity
Protocol for Comprehensive Metagenomic Analysis
This protocol outlines a standardized workflow for shotgun metagenomic analysis incorporating both computational efficiency and equity considerations, integrating best practices from recent methodological advances [10] [83] [4].
Sample Collection and Metadata Documentation
- Collect fecal samples using standardized kits to minimize technical variation
- Document comprehensive metadata including: demographic information (age, sex, ethnicity, geographical location), clinical parameters (health status, medication use, comorbidities), dietary patterns, and sample processing details
- Implement consistent storage conditions (-80°C recommended) to preserve sample integrity
DNA Extraction and Library Preparation
- Use validated extraction kits optimized for microbial lysis and human DNA depletion when appropriate
- Assess DNA quality and quantity using fluorometric methods
- Prepare sequencing libraries following manufacturer protocols, incorporating unique dual indices to enable sample multiplexing
- Consider library preparation methods that maintain representation of low-abundance taxa
Sequencing and Quality Control
- Perform shotgun sequencing on an appropriate platform (Illumina for cost-effective deep sequencing; PacBio for long-read applications requiring strain resolution)
- Conduct initial quality assessment using FastQC or similar tools
- Trim adapter sequences and low-quality bases using Trimmomatic or Cutadapt
- Remove host-derived reads if appropriate for the research question (using tools like Bowtie2 against human reference genomes)
Taxonomic, Functional, and Strain-Level Profiling
- Perform integrated TFSP using Meteor2 or similar tools against environment-specific gene catalogs
- For taxonomic profiling, use signature gene-based approaches (e.g., MetaPhlAn) or read-based methods (e.g., Kraken2) depending on research needs
- For functional profiling, annotate genes against KEGG, CAZy, and antibiotic resistance databases
- For strain-level analysis, identify single nucleotide variants in conserved genomic regions
- Normalize abundance data using appropriate statistical methods (e.g., cumulative sum scaling, relative log expression)
Data Analysis and Interpretation
- Conduct diversity analyses (alpha and beta diversity) to characterize community structure
- Perform differential abundance testing using appropriate statistical models that account for compositionality and sparsity
- Conduct correlation analyses to identify associations between microbial features and metadata variables
- Interpret findings in context of existing literature and with consideration of population-specific factors
Diagram 2: Ethical Framework for Equitable Metagenomic Research
Protocol for Implementing Equity in Metagenomic Studies
This protocol provides a structured approach to integrating equity considerations throughout the research lifecycle, from study design to result dissemination.
Equity-Focused Study Design
- Conduct a landscape analysis to identify representation gaps in existing research on similar topics
- Establish recruitment targets that ensure inclusion of populations disproportionately affected by the health condition under study
- Develop community advisory boards comprising diverse stakeholders to inform study protocols
- Design consent processes that clearly explain potential benefits and limitations of participation, including commercial applications
Culturally Responsive Participant Recruitment
- Partner with community-based organizations and healthcare providers serving diverse populations
- Translate study materials into appropriate languages and cultural contexts
- Address practical barriers to participation (transportation, childcare, compensation for time)
- Implement inclusive eligibility criteria that don't unnecessarily exclude vulnerable populations
Ethical Data Handling and Governance
- Develop clear data management plans specifying access controls, sharing policies, and destruction timelines
- Implement security measures appropriate for the sensitivity of data collected
- Establish protocols for handling incidental findings, particularly those with health implications
- Create data governance structures that include community representation for studies involving Indigenous or traditional communities
Equitable Analysis and Interpretation
- Conduct stratified analyses to identify potential population-specific effects
- Acknowledge limitations in generalizability when study populations have restricted diversity
- Avoid biological determinism in interpreting findings, considering social and environmental contexts
- Apply statistical methods appropriate for the data structure and distribution
Accessible Dissemination and Translation
- Share findings through multiple channels accessible to diverse audiences (scientific publications, community forums, plain-language summaries)
- Return individual results to participants when clinically actionable
- Consider accessibility and affordability in developing interventions or diagnostics based on research findings
- Advocate for policies that ensure benefits from microbiome research reach underserved populations
Table 4: Essential Research Reagents and Computational Resources for Shotgun Metagenomics
| Category | Specific Tools/Reagents | Function/Purpose | Equity Considerations |
|---|---|---|---|
| Sample Collection & Storage | Stool collection kits with DNA stabilizers; -80°C freezers | Preserves microbial composition; prevents DNA degradation | Choose cost-effective preservation methods; consider field conditions in resource-limited settings |
| DNA Extraction | Kits optimized for Gram-positive and Gram-negative bacteria (e.g., QIAamp PowerFecal Pro) | Comprehensive lysis of diverse microbial cell walls; removal of PCR inhibitors | Evaluate cost per sample; select protocols feasible with available laboratory infrastructure |
| Library Preparation | Illumina DNA Prep kits; PacBio SMRTbell kits | Prepares DNA for sequencing on specific platforms | Consider throughput needs and budget constraints; select methods with minimal amplification bias |
| Sequencing Platforms | Illumina NovaSeq; PacBio Revio; Oxford Nanopore | Generates sequence data with different read lengths, accuracy, and throughput | Match platform capabilities to research questions; consider cost and data storage requirements |
| Computational Tools | Meteor2, MetaPhlAn4, HUMAnN3, FastQC, Bowtie2 | Data quality control, taxonomic profiling, functional analysis | Select tools with manageable computational demands; use cloud computing for resource-intensive analyses |
| Reference Databases | KEGG, CAZy, CARD, GTDB, Meteor2 catalogues | Functional annotation; taxonomic classification | Acknowledge database limitations and population biases in interpretations |
| Data Storage Solutions | Secure servers; cloud storage (AWS, Google Cloud) | Stores raw and processed data with appropriate backup | Plan for long-term data curation costs; implement appropriate security protocols |
Shotgun metagenomics offers powerful approaches for unraveling the complexities of gut microbiome communities, but realizing its full potential requires careful attention to both computational and ethical dimensions. Effective data management demands sophisticated analytical strategies and appropriate resource allocation throughout the multi-stage workflow, from quality control through integrated taxonomic, functional, and strain-level profiling. Simultaneously, ethical implementation requires deliberate efforts to ensure diverse participant representation, equitable data governance, and fair distribution of research benefits.
The protocols and frameworks presented here provide actionable guidance for incorporating these considerations into gut microbiome research. As methodological advances like Meteor2 enhance our analytical capabilities [10], and as studies increasingly reveal population-specific patterns in microbiome-disease relationships [83], maintaining dual focus on computational rigor and ethical practice becomes ever more essential. By adopting these integrated approaches, researchers can advance our understanding of gut microbiome contributions to human health while ensuring that these benefits are accessible and relevant to diverse global populations.
Benchmarking Performance: Metagenomics vs. Traditional Methods in Clinical Scenarios
Within gut microbiome research, accurate pathogen detection is paramount for understanding microbial contributions to health and disease. Traditional culture-based methods and molecular techniques like polymerase chain reaction (PCR) have long been the standard. However, the advent of metagenomic next-generation sequencing (mNGS) represents a paradigm shift, enabling comprehensive, culture-independent analysis of microbial communities [25]. This application note provides a detailed comparison of these core pathogen detection methodologies—microbial culture, PCR, and mNGS—framed within the established workflow of a shotgun metagenomics protocol for gut microbiome research. We summarize quantitative diagnostic performance data, present standardized experimental protocols, and visualize integrated workflows to guide researchers in selecting and implementing the most appropriate method for their investigative needs.
Quantitative Comparison of Pathogen Detection Methods
The selection of a pathogen detection method involves careful consideration of performance metrics, including sensitivity, specificity, turnaround time, and cost. The following tables summarize key comparative data to inform this decision.
Table 1: Overall Diagnostic Performance of Pathogen Detection Methods
| Method | Sensitivity | Specificity | Typical Turnaround Time | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| Microbial Culture | 59.1% [84] | High (reference standard) | 22.6 ± 9.4 hours (Time to result) [84] | Allows antibiotic susceptibility testing | Low sensitivity; prior antibiotics impair growth [84] |
| Droplet Digital PCR (ddPCR) | 78.7% [84] | High | 12.4 ± 3.8 hours [84] | High sensitivity, absolute quantification without standards | Targeted; requires prior knowledge of pathogen |
| Metagenomic NGS (mNGS) | 86.6% [84] | 92% [85] | 16.8 ± 2.4 hours [84] | Unbiased detection of all microorganisms in a sample | Higher cost; complex data analysis [86] |
| Targeted NGS (tNGS) | 84% [85] | 97% [85] | Shorter than mNGS [86] | Excellent specificity; detects AMR genes/virulence factors [86] | Requires selection of target pathogens |
Table 2: Method Performance Across Different Infection Types in a Clinical Study (n=127 patients)
| Infection Type | Culture Positive Rate | mNGS Positive Detection Rate | ddPCR Positive Detection Rate |
|---|---|---|---|
| Ventriculitis, Abscess, Implant-associated Infections | Lower | Notably Higher [84] | Notably Higher [84] |
| Meningitis | Lower | Higher [84] | Higher [84] |
Detailed Experimental Protocols
Protocol 1: Shotgun Metagenomics for Gut Pathogen Detection
This protocol is designed for the comprehensive and unbiased identification of bacterial, viral, fungal, and parasitic pathogens in human fecal samples.
A. Sample Collection and DNA Isolation
- Sample Collection: Collect fresh fecal samples using a standardized kit. Alternatively, collect intestinal mucosal biopsies during endoscopy. Samples should be immediately frozen at -80°C or placed in a DNA/RNA stabilizing solution to preserve nucleic acid integrity [87].
- DNA Isolation: Use a mechanical lysis protocol (e.g., bead beating) combined with enzymatic lysis (Lysozyme, Lysostaphin, Mutanolysin) to ensure efficient disruption of diverse microbial cell walls [87]. Extract total community DNA using a kit designed for soil or stool, such as the QIAamp UCP Pathogen DNA Kit [86]. Quantify DNA using fluorometry (e.g., Qubit).
B. Library Preparation and Sequencing
- Library Preparation: Fragment purified DNA via physical or enzymatic methods. Ligate sequencing adapters to the fragments [87]. For the human gut microbiome, removal of host (human) DNA is critical and can be achieved using kits containing Benzonase [86]. Quantify the final library and ensure appropriate fragment size distribution (e.g., using a Bioanalyzer).
- Sequencing: Perform shotgun sequencing on a high-throughput platform such as the Illumina NextSeq 550Dx, aiming for a minimum of 20 million single-end or paired-end reads per sample to ensure sufficient depth for detecting low-abundance pathogens [86].
C. Bioinformatic Analysis
- Quality Control & Host Removal: Process raw reads with tools like
Fastpto remove adapters and low-quality sequences. Map reads to the human reference genome (hg38) usingBurrows-Wheeler Aligner (BWA)orBowtie2and remove aligned reads to deplete host DNA [86]. - Taxonomic Profiling: Align non-host reads to comprehensive microbial databases (e.g., GenBank, RefSeq, or specialized curated databases) using alignment tools like
SNAPorKraken2[86] [87]. For genome-resolved metagenomics, perform de novo assembly of reads into contigs using assemblers likemetaSPAdesorMEGAHIT, followed by binning into Metagenome-Assembled Genomes (MAGs) using tools likeMetaBAT2orVAMB[88] [25].
Protocol 2: Droplet Digital PCR (ddPCR) for Targeted Quantification
This protocol is for the highly sensitive and absolute quantification of a specific pathogen or antimicrobial resistance (AMR) gene once a candidate has been identified.
A. Assay Design and Sample Preparation
- Assay Design: Design primer-probe sets specific to the target pathogen's unique genomic sequence.
- Sample Preparation: Extract DNA from fecal samples as described in Protocol 1, Section A.
B. Droplet Generation and PCR Amplification
- Reaction Setup: Prepare a PCR mixture containing the extracted DNA, primers, probe, and ddPCR supermix.
- Droplet Generation: Use a droplet generator to partition the PCR reaction into thousands of nanoliter-sized water-in-oil droplets, effectively creating individual reaction chambers.
- PCR Amplification: Perform endpoint PCR on the droplet emulsion using a thermal cycler with a standardized ramp rate.
C. Droplet Reading and Data Analysis
- Droplet Reading: Transfer the PCR-amplified droplets to a droplet reader that counts each droplet as positive or negative for the fluorescent signal.
- Data Analysis: Use the manufacturer's software to apply a fluorescence amplitude threshold and calculate the absolute concentration of the target sequence (copies/μL) in the original sample using Poisson statistics.
Workflow Visualization
The following diagram illustrates the streamlined shotgun metagenomics workflow for pathogen detection, from sample to insight.
The logical relationship and comparative positioning of the three primary detection methods are shown below.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Metagenomic Pathogen Detection
| Reagent / Kit | Function / Application | Example Product |
|---|---|---|
| Pathogen DNA/RNA Kit | Simultaneous extraction of high-quality microbial DNA and RNA from complex samples. | QIAamp UCP Pathogen DNA/RNA Kit [86] |
| Host DNA Depletion Reagents | Enzymatic degradation of human host DNA to increase microbial sequencing depth. | Benzonase & Tween20 [86] |
| Ribosomal RNA Depletion Kit | Removal of bacterial and eukaryotic rRNA to enrich for mRNA and other informative RNAs in RNA-seq. | Ribo-Zero rRNA Removal Kit [86] |
| Library Preparation Kit | Fragmentation, adapter ligation, and amplification of DNA for sequencing on specific platforms. | Ovation Ultralow System V2 [86] |
| Metagenomic Assembly & Binning Software | Reconstruction of individual microbial genomes from complex metagenomic sequencing data. | metaSPAdes, MEGAHIT, MetaBAT2, VAMB [88] [25] |
Shotgun metagenomics has emerged as a powerful, culture-independent tool for pathogen detection, revolutionizing diagnostic approaches in clinical microbiology. This Application Note details the implementation and diagnostic accuracy of shotgun metagenomics protocols through focused case studies on infectious gastroenteritis and sepsis. These conditions represent significant diagnostic challenges where conventional methods often fail to identify causative pathogens. This document, framed within a broader thesis on gut microbiome research, provides validated experimental protocols, performance data, and technical workflows to guide researchers and scientists in implementing these methods for drug development and clinical diagnostics.
The protocols emphasize standardized workflows from sample collection to bioinformatic analysis, enabling comprehensive pathogen detection and functional characterization. Below are the optimized experimental workflows for metagenomic diagnosis in enteric infections and sepsis.
Figure 1: Comparative diagnostic workflows for enteric infections and sepsis using shotgun metagenomics. The enteric pathway (yellow) emphasizes direct detection from complex fecal samples, while the sepsis pathway (green) incorporates host DNA depletion and target enrichment to overcome low pathogen biomass. AMR: Antimicrobial Resistance; MAGs: Metagenome-Assembled Genomes.
Comparative Diagnostic Performance in Clinical Applications
Diagnostic Accuracy Metrics
The diagnostic performance of shotgun metagenomics varies significantly between enteric infections and sepsis, reflecting differences in sample complexity and pathogen abundance. The following table summarizes key performance metrics from recent clinical studies.
Table 1: Comparative diagnostic accuracy of shotgun metagenomics across clinical syndromes
| Clinical Syndrome | Reference Method | Sensitivity | Specificity | Additional Pathogens Detected | Key Limitations |
|---|---|---|---|---|---|
| Infectious Gastroenteritis [89] [90] | PCR (Multiplexed panels) | Lower than PCR (50% for MAGs) | Not quantified | Additional potential pathogens in most samples | Lower sensitivity for parasites; background microbiome interference |
| Sepsis [91] | Blood Culture + RT-PCR | 100% | 87.1% | Not applicable | Host DNA background (>99%); requires enrichment |
| Complex Infections [92] | Conventional Microbiology | 30.9% (9.8% exclusive to SMg) | High (No pathogens in low-suspicion cases) | Broad-spectrum detection | Optimal for high-suspicion cases only |
Clinical Impact Assessment
Beyond analytical performance, the clinical utility of metagenomic diagnostics is demonstrated through therapeutic impact and patient outcomes.
Table 2: Clinical impact and therapeutic utility of metagenomic diagnostics
| Parameter | Enteric Infections | Sepsis |
|---|---|---|
| Therapeutic Guidance | Virulence gene detection [89] | 34.8% antibiotic adjustment rate [91] |
| Patient Outcome Measure | Not assessed | 22.3% with >2-point SOFA score decrease [91] |
| Turnaround Time | Standard sequencing workflow | Same-day potential with optimized protocols [93] |
| Resistance Profiling | AMR genes detected in assemblies [89] | Simultaneous AMR gene detection [91] |
Detailed Experimental Protocols
Shotgun Metagenomics for Infectious Gastroenteritis
Sample Collection and Storage
- Sample Type: Collect fecal samples in sterile containers or rectal swabs for patients with acute diarrhea [89] [90].
- Storage: Freeze samples at -20°C or lower within 2 hours of collection until DNA extraction.
- Sample Preparation: For parasite enrichment, freeze a pea-sized amount in 200μL molecular grade water and 200μL Nuclisens easyMAG lysis buffer overnight [89].
DNA Extraction Protocol
The DNA extraction method critically influences Gram-positive versus Gram-negative bacterial representation and overall sensitivity [38].
- Homogenization: Mix 200μL liquid stool or 200mg solid stool with 1400μL ASL buffer in a Lysing Matrix A tube [89] [90].
- Mechanical Lysis: Homogenize three times for 30 seconds at speed 6.0 using a FastPrep-24 Instrument, placing samples on ice between cycles [89].
- Thermal Lysis: Incubate at 95°C for 15 minutes to enhance lysis of resistant organisms [90].
- Purification: Continue with manufacturer's instructions using QIAcube automation with QIAamp DNA Stool Kit [89].
- Elution: Elute DNA in 200μL Buffer AE [90].
- Quality Control: Measure DNA concentration using Qubit Fluorometer and purity with NanoDrop (optimal A260/A280: 1.8-2.0) [89].
Library Preparation and Sequencing
- Library Prep: Use Illumina-compatible kits following manufacturer protocols with 1-50ng DNA input [90].
- Sequencing: Perform shotgun sequencing on Illumina HiSeq 4000 or comparable platform, targeting 10-20 million reads per sample [19].
- Quality Filtering: Remove adapters and low-quality reads using fastp (v0.23.0) with quality threshold of Q20 [19].
Bioinformatic Analysis
- Host DNA Removal: Map reads to human genome (hg38) using BWA (v0.7.17) and remove matching sequences [19].
- Taxonomic Assignment: Use Kraken2 for read-based classification against standard databases [89].
- Metagenome Assembly: Perform de novo assembly using appropriate assemblers (e.g., MEGAHIT) [89].
- Binning: Recover Metagenome-Assembled Genomes (MAGs) using binning tools [89].
- Functional Annotation: Annotate virulence factors and AMR genes using CARD database and custom virulence databases [89].
Probe-Capture Metagenomics for Sepsis
Blood Sample Processing
- Sample Collection: Draw blood in EDTA tubes (6-10mL) from patients with suspected sepsis before antibiotic administration [91] [93].
- Matrix Selection: Process both whole blood and plasma, as each offers advantages for different pathogens [93].
DNA Extraction with Host Depletion
- Whole Blood Processing: Extract DNA from 10mL WB using Blood Pathogen Kit (Molzym) with add-on 10 complement for human DNA depletion [93].
- Plasma Processing: Iserve cell-free DNA from 1mL plasma using the same kit without add-on 10 complement [93].
- Automated Extraction: Use automated systems (Arrow, Diasorin) for consistency [93].
- Quality Assessment: Quantify human and bacterial DNA using ddPCR targeting RPP30 (human) and 16S rRNA (bacterial) genes [93].
Probe-Capture Enrichment
- Probe Design: Design biotinylated RNA or DNA probes targeting comprehensive pathogen panels (bacteria, fungi, viruses, parasites) [91].
- Hybridization: Incubate extracted DNA with probe library (16-24 hours) to allow specific binding to target sequences [91].
- Capture: Recover probe-target complexes using streptavidin-coated magnetic beads [91].
- Wash: Stringently wash to remove non-specifically bound DNA [91].
- Amplification: Amplify captured DNA using PCR (24 cycles) for library construction [93].
Sequencing and Analysis
- Library Preparation: Use Rapid PCR Barcoding Kit (SQK-RPB004) with Oxford Nanopore Technologies [93].
- Sequencing: Load libraries onto FLO-MIN106 R9.4 flowcells and run for up to 24 hours on MinION device [93].
- Base Calling: Perform in high-accuracy mode using Guppy (v3.6.0) [93].
- Pathogen Identification: Align reads to comprehensive pathogen databases with minimal host background [91].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Critical reagents and kits for metagenomic pathogen detection
| Reagent Category | Specific Product | Application | Performance Notes |
|---|---|---|---|
| DNA Extraction Kits | QIAamp PowerFecal Pro DNA Kit (Qiagen) | Enteric samples | Chemical + mechanical lysis; optimal for Gram-positive and Gram-negative bacteria [38] |
| DNA Extraction Kits | Blood Pathogen Kit (Molzym) | Sepsis (whole blood) | Includes human DNA depletion; better for Gram-positive bacteria [93] |
| Host Depletion | Add-on 10 complement (Molzym) | Sepsis (whole blood) | Selectively reduces human background DNA [93] |
| Library Preparation | Rapid PCR Barcoding Kit (ONT) | Rapid sequencing | 24 PCR cycles recommended for low biomass samples [93] |
| Probe Capture | Custom Panels (e.g., Illumina, Twist) | Sepsis pathogen enrichment | Significantly improves sensitivity in high-host background samples [91] |
| Quality Control | Qubit dsDNA HS Assay | DNA quantification | More accurate for low-concentration samples than spectrophotometry [89] |
| Positive Control | ZymoBIOMICS Microbial Community Standard | Process control | 8 bacterial species; validates entire workflow [38] |
Metabolic Pathways in Host-Microbe Interactions
The functional profiling of microbial communities through metagenomics reveals critical pathways involved in host-microbe interactions during infection and recovery.
Figure 2: Key microbial metabolic pathways disrupted during enteric infections and sepsis. Shotgun metagenomics enables functional profiling of these pathways through KEGG annotation, revealing mechanisms linking microbial dysbiosis to clinical disease manifestations. SCFA: Short-Chain Fatty Acids; BCAA: Branched-Chain Amino Acids; AA: Amino Acids; TCA: Tricarboxylic Acid Cycle.
Technical Considerations and Optimization Strategies
Addressing Sensitivity Limitations
The lower sensitivity of shotgun metagenomics compared to targeted PCR, particularly for low-abundance pathogens, requires specific mitigation strategies:
- Sample Enrichment: For enteric parasites, implement selective enrichment protocols before DNA extraction [89].
- Volume Optimization: Process larger sample volumes (200mg stool, 10mL blood) to increase pathogen DNA yield [89] [93].
- Sequencing Depth: Target higher read depths (15-20 million reads/sample) to detect rare pathogens [19].
- Technical Replicates: Include triplicate processing for low-biomass samples to improve detection reliability [89].
Managing Background Interference
- Kitome Characterization: Sequence extraction controls to identify reagent contaminants for subtraction from clinical samples [89].
- Host DNA Depletion: Implement selective lysis or probe-based removal of human sequences in blood samples [91] [93].
- Bioinformatic Filtering: Establish rigorous thresholds for pathogen significance based on read count and genome coverage [89].
Analytical Validation
- Reference Materials: Incorporate mock microbial communities (ZymoBIOMICS, ATCC MSA-2002) in each batch to verify sensitivity and specificity [38] [93].
- Multimodal Confirmation: Validate metagenomic findings with orthogonal methods (PCR, culture) for clinical reporting [89] [91].
- Quantitative Correlation: Establish relationship between sequencing reads and microbial load using spiked samples [89].
Shotgun metagenomics provides a powerful diagnostic platform for enteric infections and sepsis, complementing traditional methods through untargeted pathogen detection and functional characterization. While sensitivity challenges remain, particularly for low-abundance pathogens and in high-host background samples, optimized protocols incorporating appropriate DNA extraction methods, probe-based enrichment, and rigorous bioinformatic analysis enable clinically actionable results. The provided workflows and reagents offer researchers a validated foundation for implementing these methods in drug development and clinical research settings, with ongoing advancements in enrichment technologies and sequencing platforms continuing to enhance diagnostic performance.
Antimicrobial resistance (AMR) is recognized as one of the foremost global public health challenges, directly responsible for 1.27 million global deaths in 2019 and contributing to an additional 4.95 million fatalities [94] [95]. The resistome, defined as the comprehensive collection of all antibiotic resistance genes (ARGs) within a microbial community, plays a crucial role in the emergence and dissemination of AMR [96]. In the context of gut microbiome research, the gastrointestinal tract represents a significant reservoir for ARGs, where antimicrobial-resistant bacteria interact with mobile genetic elements to facilitate horizontal gene transfer [96]. This application note details integrated experimental and bioinformatic protocols for comprehensive resistome profiling using shotgun metagenomics, enabling researchers to characterize the full complement of resistance determinants within complex gut microbial communities.
Shotgun metagenomics has revolutionized AMR detection by enabling culture-independent, high-resolution identification of resistance genes and their associated mobile genetic elements [94] [11]. Unlike traditional culture-based methods that are labor-intensive, time-consuming, and lack requisite sensitivity for early resistance detection [94], shotgun metagenomics provides unparalleled capacity to identify both known and novel genetic determinants of resistance across entire microbial communities [11]. This approach is particularly valuable for gut microbiome studies, where it can detect low-abundance resistance genes that may be missed by conventional methods but nonetheless contribute to resistance dissemination through horizontal gene transfer [96].
The protocol outlined herein is framed within a broader thesis on shotgun metagenomics for gut microbiome research and is designed specifically for researchers, scientists, and drug development professionals requiring comprehensive AMR profiling. By integrating wet-lab sequencing protocols with advanced bioinformatic analysis pipelines, this application note provides an end-to-end workflow for resistome characterization that supports antimicrobial stewardship programs, drug discovery efforts, and One Health initiatives aimed at mitigating AMR transmission across human, animal, and environmental reservoirs [95].
Background
The Growing Threat of Antimicrobial Resistance
The escalating AMR crisis represents a fundamental challenge to modern medicine, complicating the treatment of infectious diseases and contributing significantly to increased morbidity and mortality rates worldwide [94]. The World Health Organization has identified AMR as one of the top global public health threats, with the Global Research on AntiMicrobial Resistance (GRAM) Project predicting that bacterial AMR will cause 39 million deaths between 2025 and 2050—equating to three deaths every minute [95]. This alarming trajectory underscores the urgent need for advanced surveillance methods that can accurately characterize resistance patterns and inform intervention strategies.
The ESKAPE pathogens—Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.—represent the most commonly isolated resistant organisms in hospital environments, highlighting the clinical significance of comprehensive resistance profiling [94]. In 2019 alone, deaths from methicillin-resistant Staphylococcus aureus (MRSA) surpassed 100,000 globally [94]. Beyond clinical settings, environmental reservoirs—including wastewater from slaughterhouses and agricultural operations—serve as critical hubs for AMR dissemination, facilitating the exchange of resistance genes between environmental and pathogenic bacteria [95].
Resistome Profiling in Gut Microbiome Research
The gut microbiome constitutes a complex ecosystem that serves as a crucial reservoir of antibiotic resistance genes, where antimicrobial-resistant bacteria interact with mobile genetic elements (MGEs) to facilitate horizontal gene transfer [96]. Recent studies of wild rodent gut microbiota have demonstrated the extensive diversity of resistomes in mammalian gastrointestinal tracts, identifying 8,119 ARGs conferring resistance to antibacterial agents across 107 drug resistance categories [96]. The most prevalent ARGs conferred resistance to elfamycin, followed by those associated with multi-class antibiotic resistance, with Enterobacteriaceae, particularly Escherichia coli, harboring the highest numbers of ARGs and virulence factor genes [96].
A strong correlation exists between the presence of mobile genetic elements, ARGs, and virulence factor genes (VFGs), highlighting the potential for co-selection and mobilization of resistance and virulence traits [96]. This relationship underscores the importance of expanded surveillance to monitor and mitigate the risk of transmission of resistant and potentially pathogenic bacteria from various reservoirs to human populations [96]. Gut microbiome metagenomics is emerging as a cornerstone of precision medicine for infectious diseases, offering exceptional opportunities for improved diagnostics, risk stratification, and therapeutic development through comprehensive resistome analysis [11].
Comparative Analysis of AMR Detection Methods
Table 1: Comparison of Antimicrobial Resistance Detection Methodologies
| Method Type | Examples | Time Required | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Conventional Methods | Disk diffusion, MIC assays [94] | 24-48 hours | Cost-effective, standardized, familiar to clinicians [94] | Labor intensive, time-consuming, lack sensitivity for early detection [94] |
| Molecular Technologies | PCR-based methods, LFIAs [94] | Several hours | Rapid detection, high sensitivity for known targets [94] | Limited to predefined targets, may miss novel mechanisms [94] |
| Next-Generation Sequencing | Illumina platforms [94] [95] | 1-3 days | High sensitivity, comprehensive profiling [94] | Higher cost, computational requirements [97] |
| Third-Generation Sequencing | Oxford Nanopore, PacBio [95] [11] | Hours to 1 day | Real-time analysis, long reads, portability [95] | Higher error rate (Nanopore), requires specialized analysis [11] |
| Shotgun Metagenomics | Illumina, PacBio HiFi [21] [11] | 1-3 days | Culture-independent, detects novel genes, functional profiling [11] | Computational complexity, host DNA contamination [11] |
Experimental Design and Workflow
The complete workflow for comprehensive resistome profiling integrates sample collection, DNA extraction, shotgun metagenomic sequencing, and bioinformatic analysis to characterize the full complement of antibiotic resistance genes within gut microbiome samples. This holistic approach enables researchers to move beyond pathogen-specific resistance detection to community-wide resistome surveillance, capturing both known and novel genetic determinants of resistance and their associated mobile genetic elements.
The critical innovation of this protocol lies in its integration of laboratory procedures with advanced computational analysis, creating a seamless pipeline from raw sample to interpretable resistome data. This end-to-end workflow is particularly valuable for tracking the dissemination of resistance genes across different ecosystems and identifying emerging resistance threats before they achieve clinical prevalence. The protocol has been optimized specifically for gut microbiome samples, addressing challenges such as high host DNA contamination, diverse microbial community composition, and variable bacterial density that can complicate resistome analysis in gastrointestinal specimens.
Figure 1: Comprehensive Workflow for Resistome Profiling in Gut Microbiome Research
Sample Collection and Preservation Protocols
Proper sample collection and preservation are critical initial steps that fundamentally impact the quality and reliability of subsequent resistome analysis. For human gut microbiome studies, rectal swabs or fecal samples represent the primary specimen types, with each offering distinct advantages depending on the clinical or research context [19]. Rectal swabs are particularly valuable in clinical settings where patients may have gastrointestinal dysfunction that complicates fecal collection, while fecal samples typically yield higher microbial biomass and are preferred for research studies [19].
The sample collection protocol should be rigorously standardized to minimize technical variability. For rectal swabs, the area around the anus should be cleaned with soap, water, and 70% alcohol, allowing the disinfectant to evaporate completely to reduce commensal skin contamination [19]. A sterile swab is then soaked in normal saline for 2 minutes before being inserted into the anus to a depth of 4-5 cm and rotated gently to obtain fecal material [19]. For direct fecal collection, samples should be obtained using sterile containers and immediately placed on ice or frozen at -80°C to preserve nucleic acid integrity and prevent microbial community shifts [80]. All samples should be stored at -80°C before shipping to the laboratory under cold chain conditions, with freeze-thaw cycles rigorously avoided to prevent DNA degradation [19].
DNA Extraction and Quality Control
High-quality DNA extraction is essential for successful shotgun metagenomic sequencing and subsequent resistome profiling. The MP-soil FastDNA Spin Kit for Soil (#6560-200, MP Biomedicals) has been specifically validated for gut microbiome samples and provides robust lysis of diverse bacterial species, including Gram-positive organisms with tough cell walls [19]. The protocol follows manufacturer's instructions with minor modifications: approximately 200 mg of fecal material is homogenized in provided lysing matrix tubes, subjected to mechanical disruption using a bead beater, followed by chemical lysis, protein precipitation, and DNA binding to a silica matrix [80] [19].
Extracted DNA must undergo rigorous quality control assessment before library preparation. DNA purity and concentration are determined using NanoDrop 2000 (Thermo Fisher Scientific), with acceptable 260/280 ratios typically ranging from 1.8-2.0 [19]. DNA quality should be further verified using a 1% agarose gel electrophoresis system to confirm high molecular weight and minimal degradation [19]. Quantitative assessment using fluorescent DNA-binding dyes such as Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) provides more accurate concentration measurements crucial for library preparation [80]. Samples failing quality thresholds should be re-extracted or excluded from downstream analysis to ensure reliable results.
Shotgun Metagenomic Sequencing
Shotgun metagenomic sequencing generates the comprehensive data required for resistome profiling by randomly fragmenting and sequencing all DNA in a sample, thereby enabling simultaneous taxonomic and functional characterization of microbial communities [11]. Both short-read (Illumina) and long-read (Oxford Nanopore, PacBio) platforms are suitable for resistome analysis, with each offering distinct advantages and limitations as summarized in Table 1.
For Illumina sequencing, the HiSeq 4000 platform provides high-throughput capacity with low error rates, typically generating 10-14 Gb of sequence data per sample to ensure adequate depth for detecting low-abundance resistance genes [19]. Library preparation involves DNA fragmentation, end-repair, adapter ligation, and PCR amplification following manufacturer protocols [80]. Alternatively, Oxford Nanopore MinION systems offer advantages of real-time analysis, long reads that facilitate assembly, and portability for field applications [95]. Recent studies have demonstrated that Nanopore systems produce similar outputs to both benchtop sequencing systems and antimicrobial susceptibility testing, validating their use for AMR tracking [95]. For the highest accuracy long-read sequencing, PacBio HiFi metagenomic sequencing enables precise functional gene profiling and strain-resolved analysis not possible with short-read approaches [21].
Bioinformatic Analysis of Resistome Data
Quality Control and Preprocessing
Raw sequencing reads require rigorous quality control and preprocessing before resistome analysis to ensure data reliability and minimize false positives in resistance gene detection. The fastp tool (v 0.23.0) provides comprehensive quality control functionality, including adapter removal, quality filtering, and length trimming in a single efficient step [19]. The recommended parameters include removing sequencing adapters, discarding low-quality reads with an average quality score below 20, and excluding sequences shorter than 50 bp after contaminant removal and trimming [19].
A critical step in gut microbiome analysis is the removal of host DNA sequences to increase microbial sequencing depth and reduce unnecessary computational overhead. The Burrows-Wheeler Aligner (BWA v 0.7.17) is commonly used to map reads to the human reference genome (hg38) for identification and removal of host-derived sequences [19]. Following host DNA removal, the proportion of bacterial reads should typically exceed 97% in fecal samples, with viral and archaeal reads accounting for approximately 1.26% and 0.01%, respectively [19]. Quality-controlled reads are then ready for assembly or direct alignment to resistance gene databases.
Resistance Gene Identification and Annotation
The core of resistome profiling involves comprehensive identification and annotation of antibiotic resistance genes using specialized databases and analysis tools. The Comprehensive Antibiotic Resistance Database (CARD) serves as the primary reference, containing 8,582 ontology terms, 6,442 reference sequences, 4,480 SNPs, and 3,354 publications covering characterized resistance determinants [97] [98]. The Resistance Gene Identifier (RGI) software, integrated with CARD, predicts resistomes based on homology and SNP models [98].
For comprehensive analysis, the sraX pipeline provides a fully automated solution for resistome profiling, offering unique features including genomic context analysis, validation of known resistance-conferring mutations, and integration of results into a single navigable HTML report [97]. sraX executes a multi-step analytical workflow that includes alignment of ARGs to analyzed genomes using DIAMOND dblastx (v0.9.29) and NCBI blastx/blastn (v2.10.0), followed by multiple-sequence alignment using MUSCLE for validating polymorphic positions conferring AMR [97]. The pipeline can incorporate additional databases including ARGminer (v1.1.1) and BacMet (v2.0) for more extensive ARG homology searches [97].
Table 2: Key Bioinformatics Tools for Resistome Analysis
| Tool Name | Type | Primary Function | Unique Features | Reference |
|---|---|---|---|---|
| sraX | Assembly-based | Comprehensive resistome analysis | Genomic context analysis, SNP validation, HTML reports | [97] |
| CARD/RGI | Database & tool | ARG identification & annotation | Curated ontology, homology & SNP models, bait capture | [98] |
| DeepARG | Read-based | ARG prediction from reads | Deep learning models, metagenome optimization | [97] |
| ARG-ANNOT | Database | ARG reference database | Manually curated resistance genes | [97] |
| MEGARes | Database | Structured ARG database | Hierarchical annotation, resistome analysis | [97] |
Analysis of Mobile Genetic Elements and Virulence Factors
The comprehensive characterization of resistomes requires analysis beyond simple ARG identification to include associated mobile genetic elements (MGEs) and virulence factor genes (VFGs) that contribute to resistance dissemination and pathogenicity. Mobile genetic elements, including transposases, ISCR elements, and integrases, play crucial roles in facilitating the horizontal transfer of ARGs within and between bacterial populations [96]. Understanding their distribution and association with ARGs is essential to elucidate how antibiotic resistance spreads through microbial communities.
Analysis of wild rodent gut microbiota has revealed 1,196 MGE-associated open reading frames across 12,255 genomes, corresponding to 370 MGEs classified into 15 types [96]. Transposable elements marked by transposase genes were the most abundant MGE type, accounting for 49% of identified elements [96]. A strong correlation exists between the presence of MGEs, ARGs, and VFGs, highlighting the potential for co-selection and mobilization of resistance and virulence traits under antibiotic selective pressure [96]. This relationship underscores the importance of integrated analysis that captures the genetic context of resistance genes to assess their transmission potential and clinical relevance.
Research Reagent Solutions
Table 3: Essential Research Reagents and Tools for Resistome Profiling
| Category | Product/Kit | Manufacturer | Primary Function | Protocol Notes |
|---|---|---|---|---|
| DNA Extraction | MP-soil FastDNA Spin Kit for Soil | MP Biomedicals | Microbial DNA extraction from fecal samples | Effective for Gram-positive and Gram-negative bacteria [19] |
| Quality Control | NanoDrop 2000 | Thermo Fisher Scientific | DNA purity & concentration | 260/280 ratio: 1.8-2.0 indicates pure DNA [19] |
| Sequencing Platform | HiSeq 4000 | Illumina | High-throughput sequencing | 10-14 Gb recommended depth per sample [19] |
| Portable Sequencer | MinION | Oxford Nanopore | Real-time sequencing in field applications | Validated for AMR tracking [95] |
| Reference Database | Comprehensive Antibiotic Resistance Database (CARD) | McMaster University | ARG annotation & analysis | Includes RGI tool for resistome prediction [98] |
| Analysis Pipeline | sraX | GitHub Repository | Comprehensive resistome analysis | Automated pipeline with HTML reports [97] |
| Quality Control Tool | fastp | GitHub Open Source | Sequencing data QC | Adapter removal, quality filtering, length trimming [19] |
Results Interpretation and Data Analysis
Quantitative Resistome Profiling
Comprehensive resistome analysis generates complex datasets requiring systematic approaches to interpretation and visualization. Quantitative assessment begins with determining the richness and diversity of ARGs within samples, followed by comparative analysis across sample groups to identify differentially abundant resistance determinants. In wild rodent gut microbiota studies, analysis of 12,255 genomes identified 8,119 putative ARG open reading frames conferring resistance to antibacterial agents across 107 drug resistance categories [96]. Among these, 5,817 (71.65%) conferred resistance to a single drug class, while 2,302 (28.35%) showed resistance to multiple classes [96].
The most prevalent resistance mechanisms observed in gut microbiome studies include antibiotic target alteration (78.93%), followed by target protection (7.47%), multitype resistance mechanisms (6.50%), and antibiotic efflux (5.65%) [96]. The distribution of resistance categories typically shows predominance of multidrug resistance genes (39.19%), followed by those targeting peptide antibiotics (7.14%) and tetracyclines (7.14%) [96]. Understanding these patterns helps researchers prioritize resistance threats and identify clinically relevant resistance mechanisms that may impact treatment efficacy.
Figure 2: Resistome Data Analysis Workflow
Statistical Analysis and Visualization
Robust statistical analysis is essential for drawing meaningful conclusions from resistome data and identifying significant patterns across sample groups. Non-parametric methods such as the Wilcoxon rank-sum test (P < 0.05) are commonly used to identify species with significant abundance differences between sample groups based on read abundance data [19]. For multivariate analysis, principal coordinates analysis (PCoA) visualizes differences and distances between samples by analyzing the community composition, with similar mean values indicating compositional similarity between groups [19].
Visualization of resistome data typically includes heatmaps displaying ARG abundance across samples, bar plots showing resistance mechanism proportions, and circular plots illustrating the distribution of dominant microbial taxa associated with resistance genes [97] [19]. The sraX pipeline automatically generates comprehensive graphical outputs, including proportions of drug classes and types of mutated loci, and integrates results into a fully navigable HTML report file [97]. For studies incorporating functional profiling, alignment against the KEGG database (v 94.2) using Diamond (v 2.0.13) with an optimized e-value cutoff of 1e-5 enables pathway analysis and functional annotation of resistance-associated metabolic pathways [19].
Clinical and Epidemiological Correlation
The ultimate value of resistome profiling lies in its ability to inform clinical decision-making and public health interventions through correlation with epidemiological and clinical outcome data. Metagenomic sequencing has demonstrated particular utility for precise antimicrobial therapy by enabling rapid detection of AMR genes and pathogen identification directly from clinical specimens, thereby reducing use of unnecessary broad-spectrum antibiotics [11]. This approach is especially valuable in culture-negative or polymicrobial infections where conventional methods fail [11].
Studies have shown that real-time nanopore metagenomic sequencing with host DNA depletion can diagnose lower respiratory bacterial infections with 96.6% sensitivity compared to culture, while simultaneously enabling identification of AMR genes to facilitate early, tailored therapy adjustments [11]. Similarly, application of shotgun metagenomics directly to blood samples from critically ill patients with sepsis has achieved pathogen identification up to 30 hours earlier than traditional cultures, while simultaneously detecting resistance genes to enable timely, targeted antimicrobial therapy [11]. These advances demonstrate the transformative potential of comprehensive resistome profiling for improving patient outcomes and supporting antimicrobial stewardship programs.
Applications in Drug Development and Precision Medicine
Informing Antibiotic Drug Discovery
Comprehensive resistome profiling provides invaluable insights for antibiotic drug discovery and development by identifying emerging resistance mechanisms and characterizing their distribution across different populations and environments. Understanding the full complement of resistance genes within microbial communities helps drug developers identify vulnerable targets, design compounds that evade existing resistance mechanisms, and prioritize development candidates with lower susceptibility to prevalent resistance determinants. The deep characterization of resistance mechanisms, including target alteration, antibiotic efflux, and enzyme inactivation, provides critical information for structure-based drug design and mechanism-of-action studies.
The pharmaceutical industry is increasingly incorporating resistome data into early-stage discovery programs to derisk development pipelines and ensure new antibiotics retain efficacy against clinically relevant resistance mechanisms. The CARD database and related resources provide comprehensive information on resistance determinants that can guide medicinal chemistry efforts and target selection [98]. Additionally, the Antibiotic Resistance Platform (ARP)—a cell-based array of mechanistically distinct individual resistance elements in an identical genetic background—enables direct antibiotic susceptibility testing spanning 18 classes of antibiotics and over 100 antibiotic resistance genes, providing valuable data for evaluating novel compounds [98].
Enabling Precision Medicine Approaches
Gut microbiome metagenomics is emerging as a cornerstone of precision medicine, offering exceptional opportunities for patient stratification and personalized therapeutic interventions based on individual resistome profiles [11]. Enterotyping, which stratifies individuals by microbiome composition, adds a valuable dimension for precision diagnostics and tailored treatment selection, particularly for infectious diseases where resistome profile may significantly impact therapeutic efficacy [11]. This approach enables clinicians to select antimicrobial regimens based on the specific resistance genes present in a patient's microbiome, potentially improving outcomes and reducing unnecessary antibiotic exposure.
Metagenomic analysis also critically informs personalized microbiome therapies like fecal microbiota transplantation (FMT), where comprehensive resistome profiling of both donors and recipients ensures safe microbial transfer without inadvertently transmitting resistance genes [11]. Studies have demonstrated that successful FMT depends on stable donor strain engraftment and restoration of key metabolites, with donor-recipient compatibility influencing these outcomes [11]. Longitudinal metagenomic monitoring post-intervention facilitates early detection of engraftment failures or adverse microbial shifts, allowing timely clinical interventions that improve patient management through clearer, more causally informative insights into engraftment trajectories [11].
This application note has detailed comprehensive methodologies for antimicrobial resistance profiling to uncover the full resistome within gut microbiome research. The integrated protocol spanning sample collection, DNA extraction, shotgun metagenomic sequencing, and advanced bioinformatic analysis enables researchers to move beyond targeted resistance detection to comprehensive characterization of the entire resistance gene repertoire in complex microbial communities. The standardized workflows, reagent specifications, and analysis pipelines provide a robust foundation for implementing resistome profiling in both research and clinical settings.
The critical importance of resistome surveillance is underscored by the escalating AMR crisis, with bacterial antimicrobial resistance projected to cause 39 million deaths between 2025 and 2050 [95]. As resistome profiling technologies continue to advance, their integration into routine clinical practice and public health surveillance represents a crucial strategy for mitigating AMR transmission and preserving antibiotic efficacy. The protocols outlined herein support this transition by providing detailed, actionable methodologies that researchers and clinicians can implement to enhance AMR detection, track resistance dissemination, and inform therapeutic decision-making within a One Health framework that recognizes the interconnectedness of human, animal, and environmental health [95].
Longitudinal multi-omics analysis represents a powerful framework in gut microbiome research, moving beyond single-time-point snapshots to capture the dynamic interplay between the host and its microbial community over time. This approach is crucial for understanding the functional mechanisms underlying host adaptation and complex gastrointestinal disorders [99] [100]. Integrating shotgun metagenomics with metatranscriptomics and metabolomics allows researchers to move from correlative observations to causative insights, revealing how microbial genetic potential translates into active function and influences host physiology through metabolic output. This Application Note details the protocols and analytical strategies for validating shotgun metagenomic findings through robust multi-omic correlation, providing a structured pathway to mechanistic discovery.
Application Note: Protocol for Multi-Omic Correlation
Experimental Design and Longitudinal Sampling
Objective: To capture temporal dynamics and establish causality in host-microbe interactions. Rationale: Cross-sectional sampling often fails to account for the inherent variability in chronic gastrointestinal conditions and may miss critical fluctuations related to disease activity. Longitudinal sampling overcomes individual heterogeneity and reveals consistent, person-specific microbial patterns that are obscured in single-time-point analyses [100].
Protocol:
- Cohort Selection: Recruit cohorts based on specific criteria (e.g., health status, disease subtype, environmental exposure). Match participants for gender, age, and BMI to minimize confounding factors [100].
- Sampling Frequency: Collect samples at regular intervals (e.g., weekly, monthly) over an extended period. The specific frequency and duration should be tailored to the research question, such as studying acclimatization phases or disease flare-ups [99] [100].
- Sample Types: Concurrently collect multiple sample types at each time point:
- Stool Samples: For shotgun metagenomics (microbial DNA), metatranscriptomics (microbial RNA), and metabolomics (luminal metabolites).
- Blood Samples: For plasma/serum metabolomics and clinical indices (e.g., via hematology analyzers like Cobas 6000) [99].
- Tissue Biopsies: (If applicable) For host transcriptomics, methylomics, and mucosa-associated microbiota analysis [100].
- Metadata Collection: Systematically record detailed metadata at each visit, including dietary recalls, medication use, symptom severity scores (e.g., IBS Symptom Severity Score), and clinical measurements [100].
Wet-Lab Processing Protocols
A. Sample Collection and Storage
- Stool: Immediately freeze fresh stool samples in liquid nitrogen and store at -80°C until processing [99].
- Blood: Collect fasting venous blood with EDTA-K2. Centrifuge at 4,000 rpm for 15 min to separate plasma, then store aliquots at -80°C [99].
B. Nucleic Acid Extraction
- Metagenomic DNA from Stool: Use a magnetic stool DNA kit with special grinding beads for effective lysis. Perform quality control via spectrophotometry (e.g., Nanodrop) and fluorometry (e.g., Qubit) [99].
- Total RNA from Stool (for Metatranscriptomics): Use a kit designed for complex samples that efficiently removes DNA and stabilizes RNA. Verify RNA integrity using an RNA Integrity Number (RIN) on a bioanalyzer.
C. Sequencing and Metabolomic Profiling
- Shotgun Metagenomic Sequencing: Conduct shotgun paired-end sequencing on a platform such as Illumina HiSeq 2500, generating 150 bp reads. This sequences all DNA fragments, enabling reconstruction of microbial genomes and functional potential [99] [7].
- Metatranscriptomic Sequencing: Prepare RNA libraries from total RNA after ribosomal RNA depletion. Sequence on an Illumina platform to profile the actively transcribed genes within the microbiome.
- Metabolomic Profiling:
- Sample Preparation: For plasma, add pre-chilled methanol containing an internal standard (e.g., 2-chlorophenylalanine) to 50 µL of sample. Vortex and centrifuge to deproteinize and collect the supernatant for analysis [99].
- Instrumental Analysis: Perform widely targeted metabolomic profiling using Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS). Separate metabolites on a column like an ACQUITY UPLC HSS T3 C18 (1.8 µm, 2.1 × 100 mm) with a mobile phase gradient of water and acetonitrile, both containing 0.04% acetic acid [99].
Computational and Bioinformatic Analysis
A. Primary Data Processing
- Metagenomics/Metatranscriptomics:
- Quality Control: Filter raw reads to remove low-quality bases, adapter contamination, and host (human) sequences using tools like Readfq and Bowtie2 [99].
- Assembly and Gene Prediction: Assemble high-quality reads into scaftigs using SOAPdenovo. Predict genes from scaftigs with MetaGeneMark. Create a non-redundant gene catalog by clustering genes with CD-HIT [99].
- Taxonomic/Functional Profiling: Align reads to reference databases (e.g., NCBI NR for taxonomy, KEGG for function) using DIAMOND to assign taxonomy and functional annotations (e.g., KEGG Orthology terms) [99].
- Metabolomics:
- Process raw MS data using software (e.g., Analyst 1.6.3) for peak picking, alignment, and relative quantification against a targeted metabolite database [99].
B. Multi-Omic Integration and Correlation Analysis
- Data Normalization: Normalize sequence count data (e.g., using CSS, TSS) and metabolomic data (e.g., Pareto scaling) before integration.
- Longitudinal Averaging: For each subject, average the abundance of microbial species, functions, and metabolites across all time points to identify stable, person-specific signals and reduce noise [100].
- Association Mapping: Perform pairwise correlation analyses (e.g., Spearman or Pearson correlation) between:
- Metagenomic species abundance vs. Plasma metabolite levels.
- Metatranscriptomic gene expression vs. Metabolite levels.
- Microbial functional pathways (from KEGG) vs. Host clinical indices.
- Statistical Validation: Adjust for multiple hypothesis testing using False Discovery Rate (FDR) control. Prioritize correlations that are statistically significant (e.g., FDR < 0.25) and biologically plausible [99] [100].
- Pathway Analysis: Integrate significant correlations to reconstruct active host-microbe metabolic pathways, such as purine metabolism, which has been implicated in gastrointestinal function [100].
The following workflow diagram summarizes the core multi-omic integration process.
Key Findings from Multi-Omic Studies
The following table summarizes quantitative results from published longitudinal multi-omic studies, illustrating the type of data and correlations this protocol can yield.
Table 1: Summary of Key Quantitative Findings from Longitudinal Multi-Omic Studies
| Study Focus | Altered Microbial Species (Example) | Associated Metabolite/Pathway | Correlated Host Physiological Change | Key Statistical Result |
|---|---|---|---|---|
| High-Altitude Adaptation [99] | Prevotella copri (enriched) | Lactic acid, Sphingosine-1-phosphate, Taurine, Inositol (elevated) | Altered purine metabolism; changes in clinical indices | 41 plasma metabolites significantly elevated; changes in microbiota explained significant variation in metabolome. |
| Irritable Bowel Syndrome (IBS) [100] | Streptococcus spp. (enriched in IBS-C/D) | Purine metabolism pathway | Gastrointestinal motility, visceral hypersensitivity | Purine metabolism identified as a novel host-microbial metabolic pathway in IBS with translational potential. |
| Irritable Bowel Syndrome (IBS) [100] | Lactobacillus spp. (>20 species enriched in severe IBS-D) | Alcohol dehydrogenase (ADH) KEGG Orthology terms | Abdominal pain intensity (primary IBS symptom) | 74 and 44 KO terms associated with severe IBS-C and IBS-D, respectively (FDR < 0.1). |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for Multi-Omic Gut Microbiome Research
| Item | Function/Application | Example Product/Catalog |
|---|---|---|
| Magnetic Stool DNA Kit | Efficient extraction of high-quality microbial DNA from complex fecal samples for metagenomic sequencing. | TIANGEN Magnetic Stool DNA Kit (with special grinding beads) [99] |
| DNA/RNA Stabilization Solution | Preserves nucleic acid integrity during sample storage and transport, critical for accurate metatranscriptomics. | RNAlater or similar products |
| UPLC-MS/MS Grade Solvents | High-purity solvents for metabolomic profiling to minimize background noise and ion suppression. | Acetonitrile and Water (e.g., Optima LC/MS Grade) with additive like 0.04% Acetic Acid [99] |
| Internal Standards for Metabolomics | Corrects for analytical variability during sample preparation and MS analysis, enabling semi-quantification. | 2-chlorophenylalanine; stable isotope-labeled compounds [99] |
| Hematology Analyzer | Measures clinical host indices (e.g., from plasma) that can be correlated with multi-omic data. | Cobas 6000 (Roche) or similar [99] |
| KEGG Database | Functional annotation of metagenomic and metatranscriptomic sequences into pathways and modules. | Kyoto Encyclopedia of Genes and Genomes [99] [7] |
The integration of longitudinal shotgun metagenomics with metatranscriptomics and metabolomics provides a robust protocol for validating the functional role of the gut microbiome. This multi-omic approach moves beyond cataloging microbial taxa to uncovering the active biochemical dialogue between the host and microbiota, offering profound insights into mechanisms of health, disease, and environmental adaptation. The detailed methodologies and analytical frameworks presented here provide a validated roadmap for researchers aiming to derive causal, mechanistically driven hypotheses from complex microbiome data.
Assessing Reproducibility and Reliability in Multi-Center Studies
Ensuring reproducibility and reliability is a fundamental premise of scientific research, yet it presents distinct challenges in multi-center studies utilizing shotgun metagenomics for gut microbiome analysis [101]. The re-use of complex, high-dimensional data from multiple institutions necessitates rigorous standardization to support meaningful comparative effectiveness research and observational studies [101]. Reproducibility in this context means that a second study should arrive at the same conclusions—similar in both direction and magnitude—when following the same protocols, a challenging goal given the technical variability inherent in sequencing technologies, bioinformatics processing, and cross-site methodological differences [101] [102]. The interdisciplinary nature of microbiome research, spanning epidemiology, biology, bioinformatics, and translational medicine, creates substantial reporting heterogeneity that can compromise reproducibility and hamper comparative analysis of published results [102]. This document outlines specific protocols and application notes to address these challenges within the context of a broader thesis on shotgun metagenomics protocols for gut microbiome research.
Reproducibility Requirements Framework for Multi-Center Studies
Based on analysis of large research initiatives, specific requirements for supporting reproducibility of multi-center studies have been identified. The requirements are driven by whether data change after the researcher receives them, whether and how the data grow throughout the study, and whether and how data move between institutions [101].
Table 1: Core Reproducibility Requirements for Multi-Center Microbiome Studies
| Requirement Category | Description | Impact on Reliability |
|---|---|---|
| Data Definition | Specific definition for each data element, including origin and processing history | How and where data originate impacts availability and meaning; essential for cross-site consistency [101] |
| Data Access | Ethics/institutional approvals; access to personnel for data extraction | Determines which data can be used and in what way; impacts data accessibility [101] |
| Data Transfer | Documentation of data receipt history and original values | Marks receipt by research team; necessary to reconstruct study and preserve data as received [101] |
| Data Transformation | History of all data changes, standardization, and mapping operations | Transformations can cause information loss; essential for complete traceability [101] |
| Reporting Standards | Adherence to standardized reporting checklists (e.g., STORMS) | Facilitates manuscript preparation, peer review, and reader comprehension [102] |
Shotgun Metagenomics Protocol for Multi-Center Studies
Sample Collection and Preservation
Standardized protocols across all participating centers are critical for reliable results.
Table 2: Standardized Sample Collection Protocol Across Centers
| Protocol Step | Standardization Requirement | Quality Control Check |
|---|---|---|
| Participant Criteria | Detailed inclusion/exclusion criteria; antibiotics/treatment history | Document recent antibiotic use, medications affecting microbiome [102] |
| Sample Collection | Identical collection kits, stabilization buffers, time-to-preservation | Record time from collection to preservation/freezing |
| Sample Preservation | Uniform temperature conditions (-80°C), identical cryovials | Monitor freezer temperatures with continuous logging |
| Shipping Protocol | Standardized shipping conditions, temperature monitoring | Use data loggers; establish chain-of-custody documentation |
Laboratory Processing and Sequencing
Laboratory processing introduces significant potential for batch effects, requiring meticulous standardization [102].
DNA Extraction Protocol:
- Use identical extraction kits across all centers
- Include positive and negative controls in each extraction batch
- Standardize equipment (e.g., centrifuges, thermomixers) specifications
- Document lot numbers for all reagents and kits
Library Preparation and Sequencing:
- Employ identical library preparation kits with standardized protocols
- Utilize the same sequencing platforms (e.g., Illumina MiSeq, HiSeq, NovaSeq) across centers
- Standardize sequencing depth (minimum 10 million reads per sample)
- Implement cross-center calibration using shared reference standards
Bioinformatics Processing Pipeline
Pre-processing to eliminate uninformative data is essential for effective analysis [103].
Statistical Analysis and Data Integration
Statistical analysis of sparse, unusually distributed, high-dimensional data requires specialized approaches [102].
Compositional Data Analysis:
- Apply proper compositional data transformations (e.g., centered log-ratio)
- Address sparsity through appropriate modeling techniques
- Implement cross-site normalization procedures
Batch Effect Correction:
- Use ComBat or other cross-site batch effect correction methods
- Include technical replicates across centers to assess variability
- Implement randomized processing orders where feasible
STORMS Reporting Framework for Enhanced Reproducibility
The STORMS (Strengthening The Organization and Reporting of Microbiome Studies) checklist provides comprehensive reporting guidance tailored to microbiome studies [102].
Table 3: Essential STORMS Reporting Elements for Multi-Center Studies
| STORMS Section | Key Reporting Elements | Multi-Center Considerations |
|---|---|---|
| Abstract | Study design, sequencing methods, body sites sampled | Specify number of centers, cross-site standardization approach [102] |
| Introduction | Background, hypothesis, or pre-specified objectives | Justify multi-center design; state primary multi-center research question [102] |
| Methods: Participants | Eligibility criteria, demographics, temporal context | Document center-specific recruitment strategies; report dates for all centers [102] |
| Methods: Laboratory | DNA extraction, library preparation, sequencing protocols | Detail any center-specific protocol adaptations; report QC metrics by center [102] |
| Methods: Bioinformatics | Processing pipeline, software versions, database references | Specify computational resources at each center; document software standardization [102] |
| Methods: Statistics | Data normalization, batch correction, hypothesis testing | Describe methods for handling center effects in statistical models [102] |
Research Reagent Solutions and Essential Materials
Table 4: Key Research Reagent Solutions for Reproducible Shotgun Metagenomics
| Reagent/Material | Function | Standardization Requirement |
|---|---|---|
| DNA Extraction Kits | Cell lysis and DNA purification from complex samples | Use identical kits across centers; document lot numbers [102] |
| Library Preparation Kits | Fragment end-repair, adapter ligation, index addition | Standardize kits and protocols; validate performance across centers |
| Quantitation Standards | DNA concentration measurement accuracy | Use fluorometric methods (Qubit) rather than spectrophotometric |
| Positive Control Materials | Process monitoring and cross-center calibration | Implement shared reference standards across all participating centers |
| Negative Control Reagents | Contamination detection and background subtraction | Include extraction and amplification negative controls in each batch |
Quality Assurance and Validation Protocol
Cross-Center Quality Control Measures
Data and Metadata Documentation Standards
Comprehensive documentation supports the traceability aspect of reproducibility [101].
Essential Metadata Categories:
- Clinical/demographic participant data with standardized coding
- Sample collection and processing parameters across all centers
- Sequencing run metrics and quality thresholds
- Bioinformatics software versions and parameters
- Statistical analysis scripts and computational environment details
Data Quality Assessment:
- Implement pre-specified quality thresholds for inclusion
- Document all data transformations and processing steps
- Report data quality assessment results alongside research findings [101]
Achieving reproducibility in multi-center shotgun metagenomics studies requires systematic attention to standardization at every stage, from study design through sample collection, wet laboratory procedures, bioinformatics processing, and statistical analysis. The frameworks and protocols outlined here provide actionable guidance for researchers aiming to enhance the reliability of their multi-center microbiome studies. By implementing these standardized approaches and comprehensive reporting standards, the field can advance toward more reproducible, comparable, and clinically relevant insights into the human gut microbiome.
Conclusion
Shotgun metagenomics has fundamentally transformed our ability to decode the complex ecosystem of the gut microbiome, providing unparalleled resolution for both taxonomic classification and functional potential. The robust protocols and advanced bioinformatic tools now available make it an powerful tool for researchers and drug development professionals, enabling applications from discovering novel therapeutic targets to tracking antimicrobial resistance. However, for its full potential to be realized in clinical practice, key challenges such as protocol standardization, computational bottlenecks, and the development of globally representative databases must be addressed. Future progress hinges on cross-disciplinary collaboration and the integration of multi-omics data, paving the way for microbiome-based diagnostics and personalized therapies to become a mainstream reality in biomedical research and clinical care.
References
- 1. 16S rRNA Gene Sequencing vs. Shotgun Metagenomic ... [https://blog.microbiomeinsights.com/16s-rrna-sequencing-vs-shotgun-metagenomic-sequencing]
- 2. Unravel Your Metagenomic Sample - Shotgun Sequencing ... [https://cegat.com/unravel-your-metagenomic-sample-shotgun-sequencing-vs-16s-sequencing/]
- 3. A Beginner's Guide to Microbial Shotgun Metagenomic ... [https://www.novogene.com/us-en/resources/blog/a-beginners-guide-to-microbial-shotgun-metagenomic-sequencing-2/]
- 4. Data Processing and Visualization for Metagenomics [https://gwu-libraries.github.io/metagenomics-analysis/aio/index.html]
- 5. An introduction to the analysis of shotgun metagenomic data [https://pmc.ncbi.nlm.nih.gov/articles/PMC4059276/]
- 6. Comparison between 16S rRNA and shotgun sequencing in ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10621-7]
- 7. Application of metagenomics in the human gut microbiome [https://pmc.ncbi.nlm.nih.gov/articles/PMC4299332/]
- 8. Shotgun Metagenomic Sequencing [https://www.illumina.com/areas-of-interest/microbiology/microbial-sequencing-methods/shotgun-metagenomic-sequencing.html]
- 9. Human gut microbiome study through metagenomics [https://www.sciencedirect.com/science/article/abs/pii/S2529993X25001972]
- 10. Accurate profiling of microbial communities for shotgun ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-025-02249-w]
- 11. Gut microbiome metagenomics in clinical practice [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562794/]
- 12. Comparison between 16S rRNA and shotgun sequencing ... [https://www.nature.com/articles/s41598-021-82726-y]
- 13. Analysis and Interpretation of metagenomics data: an approach [https://pmc.ncbi.nlm.nih.gov/articles/PMC9675974/]
- 14. Comparison of normalization methods for the analysis of ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4637-6]
- 15. Comparison of Three Commercial Tools for Metagenomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7041569/]
- 16. Introduction to Shotgun Metagenomics, from Sampling ... [https://www.cd-genomics.com/introduction-to-shotgun-metagenomics-from-sampling-to-data-analysis.html]
- 17. Tools for Analysis of the Microbiome - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7598837/]
- 18. Characterization of the Gut Microbiome Using 16S or ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2016.00459/full]
- 19. Metagenomics reveals functional profiles of gut microbiota ... [https://www.nature.com/articles/s41598-025-05127-5]
- 20. A Beginner's Guide to Microbial Shotgun Metagenomic ... [https://genesmart.vn/en/a-beginners-guide-to-microbial-shotgun-metagenomic-sequencing/]
- 21. 2025 Microbiome SMRT Grant Awardees [https://www.pacb.com/blog/2025-microbiome-smrt-grant-awardees/]
- 22. Human gut microbiome study through metagenomics [https://pubmed.ncbi.nlm.nih.gov/41058431/]
- 23. A Survey of Statistical Methods for Microbiome Data Analysis [https://www.frontiersin.org/journals/applied-mathematics-and-statistics/articles/10.3389/fams.2022.884810/full]
- 24. Current understanding of the human microbiome - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7043356/]
- 25. Genome-resolved metagenomics: a game changer for ... [https://www.nature.com/articles/s12276-024-01262-7]
- 26. A human gut microbial gene catalogue established by ... [https://www.nature.com/articles/nature08821]
- 27. MetaHIT [https://www.gutmicrobiotaforhealth.com/metahit/]
- 28. Protocol of shotgun metagenomics library construction [https://www.protocols.io/view/protocol-of-shotgun-metagenomics-library-construct-dmmh4436.html]
- 29. Absolute quantitative metagenomic analysis reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403899/]
- 30. Shotgun metagenomics reveals interkingdom ... - Microbiome [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-023-01693-w]
- 31. Hypertension-gut microbiota research trends: a bibliometric ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1543258/full]
- 32. Microbiome Based Precision Medicine through Integrated ... [https://www.sciencedirect.com/science/article/abs/pii/S094450132500343X]
- 33. The human microbiome in clinical translation: from bench ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1632435/full]
- 34. From big data and experimental models to clinical trials [https://www.sciencedirect.com/science/article/pii/S0092867425001072]
- 35. Development of a time-series shotgun metagenomics ... [https://www.nature.com/articles/s41598-021-91615-3]
- 36. Field expedient stool collection methods for gut microbiome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12188722/]
- 37. Long-term taxonomic and functional stability of the gut ... [https://www.nature.com/articles/s41598-022-27033-w]
- 38. Evaluation of DNA extraction kits for long-read shotgun ... [https://www.nature.com/articles/s41598-024-80660-3]
- 39. Library Preparation for Shotgun Metagenomic and Single ... [https://bio-protocol.org/exchange/minidetail?id=9891584&type=30]
- 40. Shotgun Metagenomic Sequencing of Bacterial ... [https://www.protocols.io/view/shotgun-metagenomic-sequencing-of-bacterial-enrich-hazjb2f4p.html]
- 41. Choice of DNA extraction method affects stool microbiome ... [https://www.nature.com/articles/s41598-024-54353-w]
- 42. Wet Lab Protocols Matter: Choice of DNA Extraction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550470/]
- 43. Comparison of 6 DNA extraction methods for isolation of high ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09537-5]
- 44. Comparative Analysis of Sample Extraction and Library ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7271268/]
- 45. Benchmarking microbial DNA enrichment protocols from ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.1184473/full]
- 46. a comparison of library preparation methods for Illumina ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8805253/]
- 47. Rapid, user-friendly, cost-effective DNA and library ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11598-7]
- 48. Comparative analysis of illumina and oxford nanopore ... [https://www.nature.com/articles/s41598-025-18768-3]
- 49. Why Are Long-Read Sequencing Methods Revolutionizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388134/]
- 50. Which NGS DNA Library Prep Kit should you choose? [https://frontlinegenomics.com/which-ngs-dna-library-prep-kit-should-you-choose/]
- 51. Use of proximity ligation shotgun metagenomics to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12439552/]
- 52. Evaluating urine volume and host depletion methods to ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-025-02191-x]
- 53. diversity detected by high-coverage 16S and... Gut microbiome [https://www.nature.com/articles/s41597-020-0427-5?error=cookies_not_supported&code=517c5119-cf59-4891-ba04-79748bed656c]
- 54. Metagenomic Sequencing Data: Step-by-Step Guide From ... [https://www.cd-genomics.com/resource-how-analyze-metagenomic-sequencing-data.html]
- 55. Dermatological implications of alignment-based de-hosting and... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07246-z]
- 56. Extending and improving metagenomic taxonomic profiling ... [https://www.nature.com/articles/s41587-023-01688-w]
- 57. Metagenomics for taxonomy profiling: tools and approaches [https://pmc.ncbi.nlm.nih.gov/articles/PMC7161568/]
- 58. FunOMIC: Pipeline with built-in fungal taxonomic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9293737/]
- 59. MyTaxa: an advanced taxonomic classifier for genomic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4005636/]
- 60. Choosing the Perfect Way to Visualize Your Microbiome Data [https://bitesizebio.com/78637/visualize-microbiome-data/]
- 61. Bacterial and fungal community taxonomic profiling [https://bio-protocol.org/exchange/minidetail?id=18895828&type=30]
- 62. How to Annotate Genes from Metagenomic Shotgun ... [https://www.cd-genomics.com/resource-metagenomic-shotgun-sequencing-annotate-genes.html]
- 63. Combining multiple functional annotation tools increases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6299973/]
- 64. METABOLIC: high-throughput profiling of microbial genomes ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-021-01213-8]
- 65. Ten simple rules for creating reusable pathway models for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8375987/]
- 66. Functional Analysis [https://learn.gencore.bio.nyu.edu/metgenomics/shotgun-metagenomics/functional-analysis/]
- 67. Microbial-enrichment method enables high-throughput ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10885704/]
- 68. Benchmarking microbial DNA enrichment protocols from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10169731/]
- 69. Evaluation of methods for the reduction of contaminating ... [https://www.nature.com/articles/s41598-020-78773-6]
- 70. Host DNA depletion efficiency of microbiome ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701219310693]
- 71. Deciphering the impact of contaminating microbiota in DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12502690/]
- 72. Comparison of DNA extraction methods for 16S rRNA gene ... [https://www.nature.com/articles/s41598-023-33959-6]
- 73. Global hotspots and trends in gut mycological research [https://pmc.ncbi.nlm.nih.gov/articles/PMC11479889/]
- 74. The role of the mycobiome in host physiology and disease [https://www.nature.com/articles/s41684-025-01620-6]
- 75. Challenges in capturing the mycobiome from shotgun ... [https://pubmed.ncbi.nlm.nih.gov/40055808/]
- 76. A comprehensive assessment of fungal communities in ... [https://environmentalmicrobiome.biomedcentral.com/articles/10.1186/s40793-022-00450-0]
- 77. Identification of fungi in shotgun metagenomics datasets [https://pmc.ncbi.nlm.nih.gov/articles/PMC5812651/]
- 78. Fungal Community Analysis by Large-Scale Sequencing of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1214672/]
- 79. Optimizing sequencing protocols for leaderboard ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1834-9]
- 80. Protocol for whole shotgun metagenomics pipeline for the ... [https://www.protocols.io/view/protocol-for-whole-shotgun-metagenomics-pipeline-f-dvbk62kw.html]
- 81. Metagenomics: Tools and Insights for Analyzing Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4426941/]
- 82. Long-Term Dietary Effects on Human Gut Microbiota Composition Employing Shotgun Metagenomics Data Analysis - PubMed [https://pubmed.ncbi.nlm.nih.gov/35760036/]
- 83. Meta-analysis of shotgun sequencing of gut microbiota in ... [https://www.nature.com/articles/s41531-024-00724-z]
- 84. Comparison of microbial culture, metagenomic next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12343261/]
- 85. Comparative Diagnostic Accuracy of Metagenomic Next ... [https://www.sciencedirect.com/science/article/pii/S0163445325002610]
- 86. Comparative diagnostic performance of metagenomic and ... [https://www.nature.com/articles/s41598-025-11834-w]
- 87. Analysis and Interpretation of metagenomics data: an approach [https://biologicalproceduresonline.biomedcentral.com/articles/10.1186/s12575-022-00179-7]
- 88. Metaphor—A workflow for streamlined assembly and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10388702/]
- 89. Evaluation of shotgun metagenomics as a diagnostic tool for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12404398/]
- 90. Evaluation of shotgun metagenomics as a diagnostic tool for ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0331288]
- 91. Clinical implement of Probe‐Capture Metagenomics in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11968419/]
- 92. Shotgun metagenomics for the diagnosis of infections [https://www.sciencedirect.com/science/article/pii/S0163445325002191]
- 93. Critical Steps in Shotgun Metagenomics‐Based Diagnosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11730497/]
- 94. Frontiers | Detection of antimicrobial resistance via state-of-the-art technologies versus conventional methods [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1549044/full]
- 95. Tracking Antimicrobial Resistance Using New Methods. - MDPI Blog [https://blog.mdpi.com/2025/08/22/tracking-antimicrobial-resistance/]
- 96. Comprehensive genome catalog analysis of the resistome ... [https://www.nature.com/articles/s41522-025-00746-2]
- 97. sraX: A Novel Comprehensive Resistome Analysis Tool [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.00052/full]
- 98. The Comprehensive Antibiotic Resistance Database [https://card.mcmaster.ca/]
- 99. Longitudinal multi-omics analysis uncovers the altered ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-024-01781-5]
- 100. Longitudinal Multi-omics Reveals Subset-Specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8109273/]
- 101. Research Reproducibility in Longitudinal Multi-Center ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5001777/]
- 102. Reporting guidelines for human microbiome research [https://www.nature.com/articles/s41591-021-01552-x]
- 103. Characterization of the Gut Microbiome Using 16S or ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4837688/]