Beyond Culturability: A Modern Guide to Assessing Bacterial Viability Through Metabolic Activity and Membrane Integrity
This guide provides a contemporary framework for assessing bacterial viability, moving beyond the traditional reliance on culturability.
Beyond Culturability: A Modern Guide to Assessing Bacterial Viability Through Metabolic Activity and Membrane Integrity
Abstract
This guide provides a contemporary framework for assessing bacterial viability, moving beyond the traditional reliance on culturability. Tailored for researchers and drug development professionals, it details the three established pillars of viability—culturability, metabolic activity, and membrane integrity—and explores the challenges of viable but non-culturable (VBNC) states. The article offers a critical comparison of traditional and novel methodologies, from plate counts and flow cytometry to viability PCR and AI-driven modeling. It further delivers practical strategies for troubleshooting common issues, optimizing preservation protocols, and validating method performance to ensure accurate, reproducible, and fit-for-purpose results in biomedical research and therapeutic development.
The Three Pillars of Bacterial Viability: Defining Culturability, Metabolic Activity, and Membrane Integrity
Core Principles of Bacterial Viability
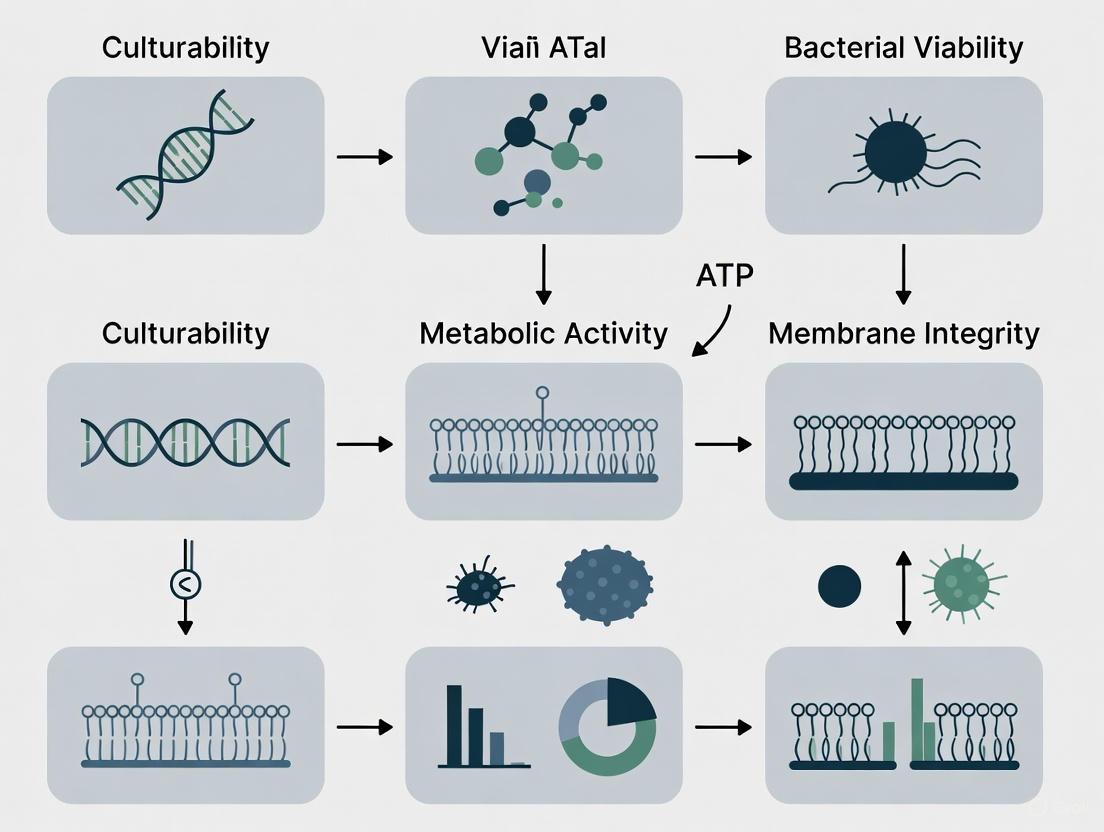
Viability assessment is a critical practice in microbiology, essential for infectious disease diagnosis, drug development, food safety, and the evaluation of microbial therapeutics [1]. The viability of bacterial pathogens is currently defined by three widespread and accepted criteria: culturability, metabolic activity, and membrane integrity [2] [3].
Culturability refers to the ability of a bacterial cell to reproduce and form a visible colony on appropriate solid media. This has been the gold standard for over a century, as a visible colony originates from a single viable mother cell [2]. However, a significant limitation is that bacteria can enter a viable but non-culturable (VBNC) state under stress, where they remain alive but cannot form colonies on routine media [2] [3].
Metabolic activity serves as an alternative criterion, detecting vital biochemical processes like enzyme function or substrate uptake. This approach can often detect VBNC bacteria, as they may still maintain active metabolism [2] [3]. However, some VBNC cells can enter a dormant state with silenced metabolism, evading this detection method [2].
Membrane integrity is a fundamental indicator of cell viability, based on the principle that a live bacterium has an intact membrane, while a dead bacterium has a disrupted or broken membrane [2] [3]. This criterion is particularly useful because membrane disruption is considered a definitive event in cell death [4].
Table 1: The Three Core Criteria for Bacterial Viability Assessment
| Criterion | Fundamental Principle | Key Advantage | Primary Limitation |
|---|---|---|---|
| Culturability | Ability to reproduce and form colonies on solid media [2]. | Considered the historical "gold standard"; provides definitive proof of viability [5]. | Cannot detect VBNC bacteria [2] [3]. |
| Metabolic Activity | Presence of active biochemical processes (e.g., enzyme activity, substrate uptake) [2]. | Can often detect VBNC bacteria that are metabolically active [2]. | Dormant cells with silenced metabolism may not be detected [2]. |
| Membrane Integrity | Structural intactness of the cell membrane [2] [3]. | Considers membrane disruption a definitive marker of cell death [4]. | May not detect viable cells with transient membrane damage [4]. |
Established and Emerging Assessment Methods
A wide array of techniques has been developed based on the three core viability criteria, each with specific methodologies, applications, and limitations.
Culturability-Based Methods
The plate culture method is the traditional approach for assessing bacterial viability [2]. This technique involves spreading a sample on an agar plate, incubating it under appropriate conditions, and counting the resulting colonies [2]. Each colony-forming unit (CFU) signifies one viable progenitor cell. Recent advancements focus on automating this process with instruments like spiral platers and automated colony counting systems that use image analysis to reduce time and manual effort [2]. Despite its longevity, the primary drawback is its inability to detect VBNC bacteria, and the process can take from 2 days up to a week to obtain final results [2].
An innovative alternative to traditional plating is the Geometric Viability Assay (GVA). This method calculates viable counts based on the distribution of colonies growing inside a pipette tip or similar cone. The probability of a colony forming at a point along the cone's axis is proportional to the cross-sectional area at that point, allowing for the accurate computation of the original viable cell concentration over 6 orders of magnitude. GVA significantly reduces the time and consumables required compared to standard CFU assays [6].
Metabolic Activity-Based Methods
These methods leverage the biochemical activity of living cells to determine viability.
- Dye Reduction Assays: Reagents like PrestoBlue HS and alamarBlue HS contain resazurin, which viable bacteria reduce to highly fluorescent resorufin. This "add-and-read" format is ideal for high-throughput screening (HTS) of biofilm viability, requiring no washing or lysis steps. Measurements can be performed in endpoint or kinetic mode using a fluorescence microplate reader [7].
- Fluorescein Diacetate (FDA) Uptake: FDA is a non-fluorescent, lipophilic dye that passively crosses bacterial membranes. Inside viable cells, non-specific intracellular esterases hydrolyze FDA into fluorescein, a polar, fluorescent compound that accumulates intracellularly, providing a measurable signal [2] [3].
- Glucose Uptake Assays: These measure a cell's consumption of glucose, a primary energy source. One strategy uses an artificial fluorescent glucose analog, 2-NBDG, which is taken up and metabolized by viable cells. Alternatively, enzymatic assays measure the depletion of glucose from the medium using glucose oxidase [2] [3].
Membrane Integrity-Based Methods
Techniques in this category distinguish live and dead cells based on the barrier function of the cell membrane.
- Live/Dead Staining with Fluorophores: This is a common and powerful approach. A classic combination uses SYTO 9, a green fluorescent nucleic acid stain that labels all cells, and propidium iodide (PI), a red fluorescent stain that only penetrates cells with damaged membranes. Thus, cells with intact membranes fluoresce green, while those with compromised membranes fluoresce red [8]. This staining is widely used in conjunction with Confocal Laser Scanning Microscopy (CLSM) to study biofilm viability in 3D [8].
- Enzyme Release Assays: These assays detect the leakage of cytoplasmic enzymes, such as lactate dehydrogenase (LDH), into the culture supernatant upon membrane rupture. The amount of enzyme released correlates with the number of dead cells [4].
- Molecular Methods with Viability Markers: Newer molecular techniques, like droplet digital PCR (ddPCR), are being developed to quantify only viable cells. This is achieved by pre-treating samples to prevent the amplification of DNA from dead cells with leaky membranes, thereby ensuring that the signal originates predominantly from cells with intact membranes [1].
Table 2: Technical Comparison of Key Viability Assessment Methods
| Method | Underlying Principle | Typical Readout | Throughput | Key Applications |
|---|---|---|---|---|
| Plate Culture | Culturability [2] | Colony count (CFU/mL) | Low (days) | Gold standard for culturable bacteria; drug efficacy testing [2]. |
| Geometric Viability Assay (GVA) | Culturability [6] | Colony distribution in a cone | Very High (~1200/day) | High-throughput drug screens; checkerboard assays [6]. |
| Dye Reduction (e.g., PrestoBlue) | Metabolic activity (resazurin reduction) [7] | Fluorescence/ Absorbance | High | HTS of biofilm viability; kinetic studies of antimicrobials [7]. |
| Live/Dead Staining + CLSM | Membrane integrity [8] | Fluorescence (Green/Red) | Medium | 3D architecture and viability of biofilms; biomaterial testing [8]. |
| FDA Hydrolysis | Metabolic activity (esterase activity) [2] | Fluorescence | Medium | Detection of metabolically active VBNC cells [2]. |
| ddPCR with Viability Markers | Membrane integrity [1] | DNA copy number | High | Quantifying viable but non-culturable (VBNC) pathogens [1]. |
Detailed Experimental Protocols
Biofilm Viability Assay Using Metabolic Dyes
This protocol is adapted for a 96-well plate format to assess the viability of Staphylococcus aureus biofilms after treatment with investigational compounds, using PrestoBlue HS or alamarBlue HS reagent [7].
Workflow:
- Inoculation and Biofilm Formation: Prepare an exponential-phase culture of S. aureus in tryptic soy broth. Add the bacterial suspension to a 96-well microplate. For treatment studies, add the antimicrobial compounds at this stage. Include medium-only wells as negative controls. Seal the plate and incubate for 18 hours at 37°C with shaking (e.g., 220 rpm).
- Removal of Planktonic Cells: After incubation, carefully remove the media containing non-adherent (planktonic) cells by pipetting. Gently wash the adhered biofilms once with phosphate-buffered saline (PBS).
- Viability Staining and Measurement: Add PrestoBlue HS or alamarBlue HS reagent directly to the washed biofilms. Incubate at room temperature for 40 minutes. Measure the fluorescence of the reduced product, resorufin, using a microplate reader with excitation/emission wavelengths of 560/590 nm.
- Data Analysis: Calculate the signal-to-background ratio by comparing the fluorescence in biofilm-containing wells to that in medium-only control wells. The percentage of viability inhibition by antimicrobial compounds can be calculated relative to untreated biofilm controls.
Diagram 1: Workflow for Biofilm Viability Assay Using Metabolic Dyes
Live/Dead Staining and Confocal Microscopy for Biofilms
This protocol details the use of SYTO 9 and propidium iodide (PI) to assess the viability and 3D structure of a biofilm via Confocal Laser Scanning Microscopy (CLSM) [8].
Workflow:
- Biofilm Growth and Staining: Grow biofilms on a suitable surface (e.g., a glass coverslip in a 6-well plate). After the desired growth period, carefully wash the biofilm with a gentle buffer like PBS to remove non-adherent cells. Prepare a working solution of the LIVE/DEAD stain by mixing SYTO 9 and PI as per manufacturer's instructions. Add the stain to the biofilm and incubate in the dark for a specified period (e.g., 15-30 minutes).
- Microscopy and Image Acquisition: After staining, gently rinse the biofilm to remove excess dye. Mount the sample for imaging. Use a CLSM to acquire Z-stack images of multiple, random fields of view for each sample. Set the appropriate laser wavelengths and detection filters for SYTO 9 (e.g., excitation 488 nm, emission ~500-550 nm) and PI (e.g., excitation 561 nm, emission ~570-620 nm).
- Automated Image Analysis: Use open-source software like Fiji/ImageJ for automated, objective quantification. The analysis macro should process the green (SYTO 9, live) and red (PI, dead) channels separately. Steps typically include:
- Applying a filter to reduce noise.
- Using an automated thresholding algorithm (e.g., Otsu, Triangle) to create a binary mask of the bacterial cells.
- Performing morphological operations to separate touching objects.
- Measuring the area or volume of the green and red fluorescence to quantify the relative proportions of live and dead cells in the biofilm.
Diagram 2: Workflow for Live/Dead Staining and CLSM Analysis of Biofilms
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Bacterial Viability Assessment
| Reagent / Material | Function / Application | Specific Example |
|---|---|---|
| PrestoBlue HS / alamarBlue HS | High-sensitivity resazurin-based reagents for measuring metabolic activity in biofilms via add-and-read microplate assays [7]. | PrestoBlue HS Cell Viability Reagent (Thermo Fisher Scientific) for HTS of S. aureus biofilm inhibitors [7]. |
| SYTO 9 & Propidium Iodide | Fluorescent nucleic acid stains for simultaneous labeling of live (green) and dead (red) cells based on membrane integrity [8]. | FilmTracer LIVE/DEAD Biofilm Viability Kit (Invitrogen) for confocal microscopy of biofilms [8]. |
| Fluorescein Diacetate (FDA) | A non-fluorescent dye converted to fluorescent fluorescein by intracellular esterases in metabolically active cells [2] [3]. | Assessing metabolic activity in viable but non-culturable (VBNC) bacteria [2]. |
| Triphenyl Tetrazolium Chloride (TTC) | A colorless compound reduced to a red formazan pigment by metabolically active bacteria, used to enhance colony contrast [6]. | Adding TTC to low-concentration agarose for visualizing embedded colonies in the Geometric Viability Assay (GVA) [6]. |
| Crystal Violet | A simple stain that binds to cells and polysaccharides in the biofilm matrix, used for quantifying total biofilm biomass [9]. | Staining adherent biomass in a 96-well plate format, followed by solubilization and OD measurement [9]. |
| Mueller-Hinton Broth/Agar | A standardized medium recommended for antimicrobial susceptibility testing (AST) [9]. | Culturing Campylobacter jejuni for biofilm formation inhibition assays [9]. |
Advanced Concepts and Future Perspectives
The field of microbial viability assessment is evolving to address existing challenges. A major frontier is the accurate detection and quantification of Viable But Non-Culturable (VBNC) cells. While methods based on metabolic activity and membrane integrity can sometimes detect these cells, they are not infallible, as dormant cells may have inactive metabolism [2]. Emerging technologies aim to overcome these limitations. Fluorescence Lifetime Imaging Microscopy (FLIM) shows promise by measuring the nanosecond-scale decay of fluorescence from membrane potential probes, a parameter that is independent of intensity and more robust against confounding effects. This provides a quantitative measure of membrane potential, a key indicator of viability, even in VBNC cells [1].
Furthermore, there is a strong drive towards standardization and automation. The development of open-source image analysis tools, like the macro for Fiji/ImageJ described for CLSM analysis, aims to ensure reproducibility and reduce user subjectivity in quantifying viability [8]. The search for higher throughput and lower waste has also led to innovative methods like the Geometric Viability Assay (GVA), which dramatically increases efficiency while maintaining a wide dynamic range [6]. These advances, combined with the ongoing development of molecular techniques like viability-ddPCR, are expanding the toolbox available to researchers and clinicians, enabling more precise and reliable viability measurements in complex biomedical and clinical contexts.
For over a century, the ability to culture microorganisms on nutrient media has been the undisputed gold standard for determining bacterial viability. This paradigm is fundamentally challenged by the viable but non-culturable (VBNC) state, a dormancy strategy adopted by numerous bacterial species in response to environmental stress. VBNC cells are metabolically active and possess an intact cell membrane but cannot form colonies on routine media, leading to a significant underestimation of viable cell counts and potential health risks. This review examines the critical limitations of culturability as a sole viability criterion, synthesizes current understanding of the VBNC state—including its inducing conditions, characteristic biomarkers, and detection methods—and discusses the implications for public health and industrial disinfection practices.
The Culturability Paradigm and the VBNC Challenge
The plate count method, developed in the late 19th century, fundamentally shaped microbiology by providing a simple, quantitative measure of viable bacteria. Its premise is straightforward: one viable cell will grow and divide to form a visible colony. This method remains the cornerstone of microbiological testing in clinical, food, and water safety contexts. However, its exclusive reliance on the ability to proliferate on artificial media presents a significant blind spot.
The VBNC state was first identified in 1982 when Escherichia coli and Vibrio cholerae were observed to lose culturability while maintaining viability [10] [11]. Since this discovery, over 100 bacterial species across 50 genera have been reported to enter this dormant state [12] [11]. The VBNC state is now recognized as a survival strategy employed by bacteria to withstand prolonged exposure to adverse environmental conditions, distinct from cell death and sporulation [10].
Defining Characteristics of VBNC Cells
VBNC cells are defined by three primary characteristics:
- Non-culturability: Failure to form colonies on standard media that normally support growth.
- Metabolic Activity: Maintenance of low-level metabolic processes, demonstrable through various assays.
- Membrane Integrity: Possession of an intact cell membrane, distinguishing them from dead cells.
Key Differences Between VBNC, Culturable, and Dead Cells
The following table summarizes the fundamental distinctions between these cellular states, highlighting why conventional detection methods fail to identify VBNC cells.
Table 1: Characteristics of VBNC, Culturable, and Dead Cells
| Characteristic | VBNC Cells | Viable, Culturable Cells | Dead Cells |
|---|---|---|---|
| Culturability on Standard Media | No | Yes | No |
| Membrane Integrity | Intact [10] | Intact | Damaged [10] |
| Metabolic Activity | Low but detectable [10] [13] | High | None [10] |
| Gene Expression & Protein Synthesis | Continued (low level) [10] | Active | None |
| Morphology | Reduced cell size, rounding [10] | Normal | May be lysed |
| Response to Environmental Cues | Can resuscitate [10] | Grows and divides | No response |
| Virulence Potential | May be retained or reduced [10] | Present | None |
Conditions Inducing the VBNC State and Methodologies for Study
A wide array of environmental stresses can trigger the VBNC state. Understanding these inducers is crucial for designing experiments and interpreting the presence of VBNC cells in various settings.
Common Inducers of the VBNC State
Table 2: Common Stressors Inducing the VBNC State and Exemplary Species
| Inducing Condition | Examples of Affected Species |
|---|---|
| Nutrient Starvation [10] [11] | E. coli, Shigella dysenteriae, Vibrio parahaemolyticus, Klebsiella pneumoniae |
| Temperature Extremes (especially low temperature) [10] [11] | V. vulnificus, Listeria monocytogenes, E. coli O157:H7 |
| Oxidative Stress [11] | |
| Osmotic Stress [11] | Aeromonas hydrophila |
| Chlorination/Disinfectants [11] [14] [15] | Listeria monocytogenes, Pseudomonas aeruginosa |
| UV Radiation [11] [13] | Aeromonas sp., Pseudomonas sp., E. coli, Staphylococcus aureus |
| High Pressure [11] | |
| Extreme pH [11] | Staphylococcus aureus, E. coli O157:H7 |
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Key Reagents and Methodologies for VBNC Research
| Reagent / Method | Function / Principle | Application in VBNC Research |
|---|---|---|
| Propidium Monoazide (PMA/PMAxx) [14] | DNA intercalating dye; penetrates only cells with compromised membranes, inhibiting PCR amplification. | Used in viability qPCR (v-qPCR) to selectively detect cells with intact membranes (viable/VBNC). |
| Ethidium Monoazide (EMA) [14] | Similar to PMA but can penetrate some intact membranes via efflux pumps. | Often combined with PMAxx in v-qPCR for improved discrimination in complex matrices. |
| 5-Cyano-2,3-Ditolyl Tetrazolium Chloride (CTC) [13] | Tetrazolium salt; reduced to fluorescent formazan by active electron transport chain. | Measures respiratory activity at population and single-cell levels (e.g., via flow cytometry). |
| SYTO 9 & Propidium Iodide (PI) [11] | Fluorescent nucleic acid stains; SYTO9 labels all cells, PI labels only dead cells. | Differentiates live/dead cells based on membrane integrity (e.g., in Live/Dead staining). |
| D2O (Deuterium Oxide) [13] | Stable isotope; incorporated into newly synthesized biomolecules during metabolic activity. | Used with Raman spectroscopy to measure metabolic activity at the single-cell level. |
| Nalidixic Acid [11] | Antibiotic that inhibits DNA synthesis. | Used in the Direct Viable Count (DVC) method to prevent cell division, allowing elongation of viable cells. |
Advanced Detection and Quantification Methodologies
Overcoming the limitations of plating requires a multi-faceted approach to viability assessment. The diagram below illustrates the logical relationship and application of the primary methods used to detect and quantify VBNC cells.
Diagram: A multi-method approach is essential for accurately profiling VBNC cells, combining membrane integrity checks with metabolic activity assays.
Experimental Protocol: Differentiating VBNC Cells in Complex Water Matrices
Research on Listeria monocytogenes in Process Wash Water (PWW) from the fresh-cut produce industry provides a validated protocol for detecting VBNC cells in a challenging, real-world matrix [14].
Optimized Viability qPCR (v-qPCR) Protocol:
- Sample Preparation: Treat water samples with a combination of 10 μM EMA and 75 μM PMAxx.
- Incubation: Incubate the mixture in the dark at 40°C for 40 minutes.
- Photoactivation: Expose the samples to light for 15 minutes using a dedicated PMA-Lite device to crosslink the dyes to DNA from dead cells.
- DNA Extraction and qPCR: Proceed with standard DNA extraction and quantitative PCR.
- Quantification: The signal from qPCR corresponds to DNA from viable and VBNC cells with intact membranes, while DNA from dead cells is suppressed.
Key Validation Note: This specific EMA/PMAxx combination was found to be more effective than flow cytometry for complex water matrices, as the organic matter in PWW can cause interference and overestimation of dead cells [14]. The number of VBNC cells can be calculated as: VBNC count = Viable count (from v-qPCR) - Culturable count (from plating) [16].
Metabolic and Molecular Characteristics of VBNC Cells
Upon entering the VBNC state, cells undergo profound physiological and molecular changes.
Metabolic Reprogramming
Metabolomic studies on Pseudomonas aeruginosa induced into the VBNC state by chlorine stress reveal a distinct metabolic profile [17]:
- Downregulation: Nucleotide, amino acid, peptidoglycan, and glutathione metabolism are suppressed as part of a general downscaling of energy-intensive processes.
- Upregulation: Essential phospholipid synthesis and the glyoxylate cycle pathway are upregulated, potentially to provide necessary carbon sources and maintain basal metabolic flow in a non-growing state.
- Fatty Acid Metabolism: Increased fatty acid metabolic activity and lipid accumulation suggest a shift towards energy storage.
Transcriptional Changes
Transcriptome analysis of VBNC Bacillus subtilis cells highlights a robust stress response [18]:
- Induction of ICEBs1 conjugative element genes, suggesting a response to antibiotic-induced DNA damage and oxidative stress.
- Upregulation of the queuosine (Q) biosynthesis pathway, which may help minimize translation errors and contribute to antibiotic tolerance.
- Activation of operons related to detoxification and oxidative stress, a common hallmark of the VBNC state across species.
The Critical Limitations of Relying on Culturability
The failure to account for VBNC cells has direct and serious consequences:
- Public Health Risk: Pathogens in the VBNC state can retain virulence and pose a significant threat. For example, VBNC V. vulnificus can resuscitate in a suitable host and cause fatal infections [10]. Their presence in drinking water distribution systems [16] and food processing lines [14] represents a hidden risk that routine monitoring misses entirely.
- Ineffective Disinfection Monitoring: Common disinfection methods like chlorination and UV irradiation can efficiently induce the VBNC state rather than kill cells. One study found that chlorination induced ~10⁵–10⁶ cells/mL of VBNC bacteria while reducing culturable counts to zero [15]. This leads to a false sense of security and underestimation of the required disinfectant dose.
- Misleading Microbial Quality Assessments: Environmental, food, and clinical samples may be declared safe based on negative culture results, while potentially harboring a significant population of viable VBNC pathogens.
The VBNC state represents a fundamental challenge to the long-standing gold standard of microbiological viability. Relying solely on culturability is inadequate for modern public health and microbiological research, as it fails to detect a physiologically distinct and clinically relevant population of bacteria. A paradigm shift is necessary, moving towards a multi-parameter assessment of viability that integrates measures of membrane integrity, metabolic activity, and genetic content. The development of robust, standardized, and accessible methods for detecting VBNC cells is crucial for accurate risk assessment in clinical diagnostics, food safety, and water treatment, ultimately leading to more effective public health protection.
Assessing metabolic activity is a fundamental strategy for evaluating bacterial viability, providing critical insights into cellular function that complement other criteria like culturability and membrane integrity. Metabolic activity assays function by detecting key biochemical processes within live cells, including enzymatic activity, redox potential, or ATP production, which serve as reliable indicators of cellular health and functionality [19]. Since a decline in metabolic function is often an early marker of cell stress or death, these methods generally offer higher sensitivity for detecting viability changes compared to traditional membrane integrity assays [19]. This approach is particularly valuable for identifying viable but nonculturable (VBNC) bacteria—cells that remain metabolically active despite losing the ability to form colonies on standard culture media, a common limitation of traditional plate counting methods [2] [3].
These assays measure the catalytic activity of cellular enzymes or the uptake and utilization of specific substrates. Viable cells with active metabolism can convert non-fluorescent or non-chromogenic compounds into easily detectable products, providing a direct correlation between signal intensity and the number of viable cells present [2]. This technical guide explores the core principles, methodologies, and applications of dye and substrate uptake assays, providing researchers with a comprehensive framework for implementing these powerful tools in bacterial viability research.
Core Principles and Mechanisms of Action
Metabolic activity assays for bacterial viability assessment operate on several well-established biochemical principles, each targeting different aspects of cellular metabolism.
Enzymatic Conversion of Fluorogenic Substrates
A common mechanism involves the use of fluorogenic substrates that penetrate bacterial cells and are hydrolyzed by intracellular enzymes. Fluorescein diacetate (FDA) is a prime example of this approach. FDA is a non-polar, non-fluorescent compound that readily crosses intact bacterial membranes through passive transport [3]. Once inside viable cells, nonspecific intracellular enzymes—including esterases, lipases, and proteases—hydrolyze FDA to release fluorescein, a polar fluorescent compound that accumulates intracellularly due to its inability to cross lipid membranes [2] [3]. The resulting fluorescence signal provides a direct measure of enzymatic activity, which correlates with cell viability. A significant advantage of this technology is that extracellular FDA produces no background signal, enhancing assay sensitivity [3]. However, the method has limitations, including potential signal quenching at high intracellular fluorescein concentrations and sensitivity to pH fluctuations, as the hydrolysis reaction produces acetic acid that can lower intracellular pH and affect enzyme activity [3].
Glucose Uptake and Metabolism Monitoring
Another strategy assesses viability by measuring bacterial uptake and utilization of glucose, a primary energy source for most organisms. This approach can be implemented through two main methodologies:
Artificial Fluorescent Glucose Analogs: Compounds like 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG) serve as glucose analogs that are transported into viable bacterial cells via glucose transport systems [3]. Once incorporated, metabolically active bacteria decompose 2-NBDG into non-fluorescent compounds, while dead bacteria retain the fluorescent signal. However, a significant limitation is that not all bacterial species can transport and process 2-NBDG, with studies showing inability for uptake in species including Vibrio mimicus, Bacillus cereus, Plesiomonas shigelloides, Aeromonas hydrophila, and certain E. coli strains [3].
Enzymatic Glucose Consumption Assays: This method measures the depletion of glucose from the culture medium using enzymatic reactions. Glucose oxidase converts remaining glucose to D-gluconic acid and H₂O₂, followed by a colorimetric reaction with o-dianisidine catalyzed by peroxidase, resulting in a color change that can be quantified [3].
Specialized Fluorescent Probes for Bacterial Typing
Recent advances have led to the development of specialized bio-orthogonal fluorescent dyes that enable both viability assessment and bacterial differentiation. For instance, researchers have designed probes that discriminate between Gram-positive and Gram-negative bacteria through metabolic engineering approaches [3]. These compounds selectively label metabolically active bacteria based on their cell wall composition—for example, targeting peptidoglycan in Gram-positive bacteria or lipopolysaccharide in Gram-negative species—allowing for simultaneous viability assessment and bacterial classification [3].
Figure 1: Three primary mechanisms for assessing bacterial viability through metabolic activity assays. Each pathway utilizes different biochemical principles to differentiate metabolically active cells.
Experimental Protocols and Methodologies
Fluorescein Diacetate (FDA) Assay Protocol
The FDA hydrolysis assay provides a straightforward method for assessing bacterial viability based on intracellular esterase activity. The following protocol can be adapted for both planktonic cells and biofilm samples:
Reagent Preparation:
- Prepare FDA stock solution by dissolving fluorescein diacetate in acetone or DMSO at 1-2 mg/mL.
- Dilute stock solution in appropriate buffer (e.g., phosphate-buffered saline) to achieve a working concentration of 10-100 μg/mL immediately before use.
- Note: FDA solution should be prepared fresh for each experiment as it hydrolyzes spontaneously in aqueous solution.
Assay Procedure:
- Sample Preparation: Harvest bacterial cells and wash twice with sterile buffer to remove residual culture medium. Adjust cell density to approximately 10⁶-10⁷ CFU/mL for optimal signal detection.
- Dye Incubation: Add FDA working solution to bacterial suspension at 1:10 (v/v) ratio. Mix gently and incubate in the dark at optimal growth temperature for 15-60 minutes.
- Signal Measurement: Terminate reaction by placing samples on ice. Measure fluorescence intensity using appropriate instrumentation:
- Flow Cytometry: Analyze 10,000-50,000 events per sample with fluorescein detection settings (excitation ~488 nm, emission ~530 nm).
- Fluorescence Microscopy: Examine wet mounts immediately under blue light excitation.
- Plate Reader: Transfer samples to black-walled microplates and measure fluorescence with bottom reading mode.
Critical Considerations:
- Include proper controls: unstained cells, heat-killed cells, and reagent blank.
- Optimize incubation time for each bacterial strain to balance signal intensity and dye toxicity.
- Account for potential pH sensitivity, as fluorescein fluorescence intensity is pH-dependent [3].
2-NBDG Glucose Uptake Assay Protocol
This protocol monitors glucose transport activity as an indicator of metabolic function in bacterial cells:
Reagent Preparation:
- Prepare 2-NBDG stock solution in DMSO or sterile water according to manufacturer instructions.
- Dilute to working concentration (typically 50-200 μM) in appropriate bacterial growth medium without glucose.
Assay Procedure:
- Sample Preparation: Harvest bacterial cells during mid-logarithmic growth phase. Wash twice with PBS or minimal medium to remove residual carbon sources.
- Glucose Starvation: Resuspend cells in carbon-free medium and incubate for 30-60 minutes to deplete intracellular energy reserves.
- Dye Loading: Add 2-NBDG working solution to bacterial suspension and incubate at growth temperature for 15-30 minutes.
- Signal Measurement: Wash cells twice with ice-cold PBS to remove extracellular dye. Measure fluorescence immediately using:
- Flow Cytometry: Analyze bacterial population with FITC settings.
- Fluorescence Microscopy: Visualize using standard FITC filter sets.
- Microplate Reader: Quantify fluorescence in clear-bottom black-walled plates.
Data Interpretation:
- Viable, metabolically active cells typically show decreased fluorescence due to 2-NBDG metabolism.
- Dead or metabolically inactive cells retain strong fluorescence signal.
- Note: Not all bacterial species transport 2-NBDG effectively; preliminary validation is essential [3].
2-Aminobenzamide (2-AB) Glycan Uptake Protocol
For studying substrate specificity in microbial communities, particularly in gut microbiome research, fluorescent glycan labeling provides valuable insights:
Glycan Labeling Procedure:
- Substrate Generation: Label oligosaccharides with 2-AB using reductive amination with 2-picoline-borane in methanol or water [20].
- Purification: Remove excess label and reaction byproducts using chromatography or precipitation methods.
- Validation: Confirm labeling efficiency and structural integrity using HILIC chromatography with fluorescence detection.
Uptake Assay:
- Fermentation Setup: Inoculate bacterial cultures in appropriate medium supplemented with 2-AB-labeled glycans as sole carbon source.
- Incubation: Grow cultures under optimal conditions for 12-48 hours.
- Analysis: Monitor glycan uptake using multiple complementary techniques:
- Flow Cytometry: Detect 2-AB fluorescence in bacterial cells to identify substrate-utilizing populations.
- Chromatography: Quantify residual labeled glycans in supernatant over time.
- Mass Spectrometry: Identify metabolic products of glycan degradation.
- Microscopy: Visualize intracellular accumulation of fluorescent metabolites.
Applications: This approach is particularly valuable for investigating competitive substrate utilization in complex microbial communities and identifying specialized metabolic capabilities of different bacterial strains [20].
Quantitative Comparison of Metabolic Activity Assays
Table 1: Technical specifications and performance characteristics of different metabolic activity assays for bacterial viability assessment
| Assay Type | Detection Mechanism | Key Reagents | Dynamic Range | Time Required | Key Advantages | Major Limitations |
|---|---|---|---|---|---|---|
| FDA Hydrolysis | Enzymatic conversion to fluorescent product | Fluorescein diacetate | 10³-10⁸ cells/mL | 30-90 minutes | Simple protocol; No extracellular background; Broad bacterial applicability | pH-sensitive; Potential signal quenching; Variable hydrolysis rates |
| 2-NBDG Uptake | Glucose analog transport and metabolism | 2-NBDG | 10⁴-10⁷ cells/mL | 45-120 minutes | Direct measure of glucose metabolism; Can distinguish metabolic activity levels | Limited bacterial uptake spectrum; Requires starvation pre-treatment |
| 2-AB Glycan Uptake | Substrate-specific utilization | 2-AB labeled glycans | 10⁵-10⁹ cells/mL | 12-48 hours | Reveals substrate preferences; Works in complex communities | Specialized application; Requires glycan synthesis expertise |
| Tetrazolium Reduction (MTT/XTT) | Reduction to colored formazan | MTT, XTT, WST-1 | 10⁴-10⁷ cells/mL | 2-6 hours | Works with eukaryotic and prokaryotic cells; No washing steps | Redox interference; Formazan precipitation; Cytotoxic at high concentrations |
Table 2: Compatibility of metabolic activity assays with different detection platforms and sample types
| Assay Type | Flow Cytometry | Microplate Reader | Microscopy | Planktonic Cells | Biofilms | VBNC Detection |
|---|---|---|---|---|---|---|
| FDA Hydrolysis | Excellent | Good | Excellent | Excellent | Moderate | Good |
| 2-NBDG Uptake | Good | Good | Good | Good | Limited | Moderate |
| 2-AB Glycan Uptake | Excellent (with FACS) | Good (with HPLC) | Good | Good | Good | Excellent |
| Tetrazolium Reduction | Limited | Excellent | Limited | Good | Moderate | Limited |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents and materials for implementing metabolic activity assays in bacterial viability research
| Reagent/Material | Function/Application | Key Considerations | Example Commercial Sources |
|---|---|---|---|
| Fluorescein Diacetate (FDA) | Fluorogenic substrate for esterase activity | Prepare fresh solutions; pH-sensitive detection | Sigma-Aldrich, Thermo Fisher, Millipore |
| 2-NBDG | Fluorescent glucose analog for uptake studies | Validate transport capability for target bacteria | Cayman Chemical, Invitrogen |
| 2-Aminobenzamide (2-AB) | Fluorescent tag for glycan labeling | Use picoline-borane for safer reductive amination | Sigma-Aldrich, TCI America |
| Tetrazolium Salts (MTT, XTT, WST-1) | Redox indicators for metabolic activity | Potential cytotoxicity at high concentrations | Abcam, Roche, Dojindo Molecular |
| HILIC Columns | Separation of fluorescently labeled glycans | Essential for analyzing 2-AB labeled metabolites | Waters, Agilent, Thermo Scientific |
| Microfluidic Chips | Single-cell analysis of metabolic activity | Enables high-resolution studies of heterogeneity | Dolomite, Micronit, Fluidigm |
Advanced Applications and Integration with Other Viability Assessment Methods
Metabolic activity assays find particular utility in challenging research scenarios where traditional culturability methods fail. Their application is especially valuable for detecting viable but nonculturable (VBNC) bacteria, which retain metabolic activity despite being unculturable on standard media [2] [3]. This state can be induced by various environmental stresses, including nutrient limitation, extreme temperatures, and exposure to antibiotics or biocides [3]. While metabolic activity assays can detect VBNC cells, it's important to note that some VBNC bacteria may enter a dormant state with minimal metabolic activity, potentially limiting detection sensitivity in these cases [3].
In biofilm research, metabolic activity assays provide crucial information about the physiological heterogeneity within these structured microbial communities. Biofilms exhibit gradients of metabolic activity due to differential access to nutrients and oxygen, creating distinct microenvironments where cells range from highly active to dormant [21]. This heterogeneity significantly impacts antibiotic efficacy and biofilm resilience, making metabolic activity measurements essential for comprehensive biofilm characterization.
For a complete viability assessment, researchers should combine metabolic activity assays with other approaches targeting different viability criteria:
- Membrane Integrity Assays: Use dyes like propidium iodide (PI) or SYTOX Green that are excluded by intact membranes but penetrate compromised cells [19] [1].
- Culturability Methods: Employ plate counts or most probable number (MPN) techniques to assess reproductive capability [2] [3].
This multi-parameter approach provides a more comprehensive understanding of bacterial physiological status, especially when investigating complex phenomena like antibiotic tolerance or stress response mechanisms.
Figure 2: Integrated approach to bacterial viability assessment combining multiple criteria and methodologies for a comprehensive understanding of cellular status.
Metabolic activity assays using dye and substrate uptake provide powerful, sensitive approaches for assessing bacterial viability, particularly for detecting physiologically active but nonculturable cells. When properly selected and optimized based on research objectives and bacterial species, these methods yield invaluable insights into cellular function that complement data from culturability and membrane integrity assessments. As microbial research increasingly addresses complex communities and challenging environments, these metabolic probes will continue to be essential tools in the microbiologist's arsenal, especially when integrated with emerging technologies like microfluidics, single-cell analysis, and advanced imaging.
Within the framework of bacterial viability criteria—encompassing culturability, metabolic activity, and membrane integrity—the integrity of the cell membrane stands out as the most definitive indicator of cell death [2]. While culturability indicates the ability to reproduce and metabolic activity signals functional physiology, the loss of membrane integrity represents a point of no return, irrevocably compromising the fundamental barrier that separates the cell from its environment [22] [23]. This irreversible breakdown distinguishes membrane integrity from other viability parameters, which may indicate temporary, non-lethal states such as the viable but non-culturable (VBNC) state, where bacteria maintain an intact membrane and metabolic activity but cannot proliferate on standard media [2] [24]. The critical nature of membrane integrity arises from its essential function in maintaining cellular homeostasis, facilitating energy production, and preserving vital concentration gradients. When this barrier fails, the resulting uncontrolled flux of ions and molecules, dissipation of proton motive force, and leakage of essential cellular components lead directly and irreversibly to cell death [22] [23]. Consequently, assessing membrane integrity provides a direct and reliable method for differentiating between live cells, which possess an intact membrane, and dead cells, where membrane integrity has been permanently lost.
Comparative Analysis of Viability Criteria
The three primary criteria for assessing bacterial viability each probe different aspects of cellular physiology, with distinct advantages and limitations. Table 1 provides a systematic comparison of these criteria, highlighting why membrane integrity serves as the most definitive marker for cell death.
Table 1: Comparison of Bacterial Viability Assessment Criteria
| Viability Criterion | What It Measures | Key Advantages | Major Limitations | Correlation with True Viability |
|---|---|---|---|---|
| Culturability | Ability to form colonies on standard growth media [2]. | Simple, well-established; provides live isolates for further study [2]. | Fails to detect VBNC cells; time-consuming (days) [2] [24]. | Indirect; can significantly underestimate viable population [24]. |
| Metabolic Activity | Presence of active enzyme systems, substrate uptake, or ATP production [2] [25]. | Can detect VBNC cells; faster than culturability (minutes to hours) [2]. | Dormant cells have low activity; results sensitive to pH, substrate concentration [2] [25]. | Indirect; can be reversible; may underestimate (dormancy) or overestimate (residual enzyme activity) [2]. |
| Membrane Integrity | Physical integrity and selective permeability of the cytoplasmic membrane [22] [25]. | Directly measures a fundamental property of life; rapid (minutes); distinguishes live/dead unequivocally [22] [23]. | May not identify early stages of stress before membrane compromise [22]. | Direct; loss is irreversible and incompatible with continued life [22] [23]. |
As illustrated in Table 1, membrane integrity assessment directly probes a fundamental characteristic of a living cell. The loss of membrane integrity is a terminal event, making it a definitive endpoint for confirming cell death, whereas disruptions in culturability and metabolic activity can be transient and reversible.
Membrane Integrity as a Definitive Marker of Cell Death
The Irreversible Nature of Membrane Integrity Loss
The cell membrane is a phospholipid bilayer that functions as a selective barrier, crucial for maintaining the internal ionic composition, preserving the proton motive force essential for ATP synthesis, and preventing the leakage of essential cellular components [22] [26]. A viable cell must actively maintain its membrane potential and selective permeability. When a cell undergoes death, one of the final, irreversible steps in the cascade is the permanent loss of this membrane barrier function [22]. This loss leads to depolarization (the collapse of the electrical potential across the membrane), uncontrolled exchange of ions with the extracellular environment, and the leakage of cytoplasmic contents [22] [23]. Unlike a transient change in metabolic rate or a temporary loss of culturability, this comprehensive membrane failure is a point of no return. A cell cannot repair widespread membrane disintegration and, therefore, cannot recover viability once this stage is reached [22].
Advantages in Detecting VBNC and Stressed Populations
Relying solely on culturability for viability assessment can be profoundly misleading, as many bacteria exposed to sub-lethal environmental stresses (e.g., nutrient starvation, extreme temperatures, or biocides) can enter the Viable But Non-Culturable (VBNC) state [2] [24]. In this state, cells fail to grow on standard laboratory media but remain alive, as confirmed by their metabolic activity and, most importantly, their intact membranes [2] [24]. Studies have shown that pathogens like Listeria monocytogenes, Escherichia coli, and Salmonella enterica can retain an intact membrane and metabolic activity after exposure to stressors like household cleaners and salts, even when they are no longer culturable [24]. This poses a significant public health risk, as these VBNC cells may resuscitate and cause infection under favorable conditions. Membrane integrity assays, therefore, provide a more accurate and comprehensive risk assessment in food safety, water treatment, and clinical diagnostics by detecting this hidden, yet viable and potentially dangerous, microbial population.
Key Methodologies and Experimental Protocols
A range of well-established techniques is available to assess membrane integrity, from simple dye-based assays to advanced electrophysiological measurements.
Fluorescent Staining and Microscopy
This is one of the most common and accessible methods. It utilizes fluorescent dyes that differentiate between intact and compromised membranes.
Common Dyes and Mechanisms:
- Propidium Iodide (PI): A small, positively charged molecule that is excluded by intact membranes. It only enters cells with damaged membranes, where it binds to nucleic acids and emits a strong red fluorescence [24] [27].
- SYTO/Green Nucleic Acid Stains: Cell-permeable dyes that label all nucleic acids, staining all cells (live and dead) green. They are often used in combination with PI for a clear live/dead contrast [24].
- FluoVolt: A dye whose fluorescence intensity changes in response to changes in membrane potential, allowing for the direct measurement of membrane depolarization, an early indicator of integrity loss [22].
Experimental Protocol (Basic Live/Dead Staining):
- Sample Preparation: Harvest bacterial culture by gentle centrifugation and wash with an appropriate buffer (e.g., phosphate-buffered saline) [24].
- Staining: Resuspend the cell pellet in a buffer containing a mixture of a membrane-permeant green dye (e.g., SYTO 9) and a membrane-impermeant red dye (e.g., PI) [24].
- Incubation: Incubate the sample in the dark for 15-20 minutes at room temperature.
- Analysis: Apply a small aliquot to a microscope slide and visualize using a fluorescence microscope with appropriate filter sets. Cells with intact membranes will fluoresce green, while cells with compromised membranes will fluoresce red [24].
Enzymatic Release Assays
These assays measure the release of intracellular enzymes upon membrane rupture.
Lactate Dehydrogenase (LDH) Release Assay:
- Principle: The cytoplasmic enzyme LDH is released into the extracellular medium only when the plasma membrane is damaged [25] [27].
- Protocol: Cells are exposed to a treatment, and the supernatant is collected. The supernatant is incubated with a reaction mixture containing lactate, NAD+, and a dye. LDH catalyzes the conversion of lactate to pyruvate, generating NADH. NADH then reduces a tetrazolium salt (INT) in a second step to a red formazan product, which is measured spectrophotometrically. The amount of formazan is directly proportional to the number of dead cells [25].
Dead-Cell Protease Assay:
- Principle: Upon losing membrane integrity, dead cells release proteases into the surrounding environment [25].
- Protocol: A fluorogenic or luminogenic substrate for a dead-cell protease is added directly to the cell culture. The substrate cannot cross intact membranes. Only in wells containing dead cells will the substrate be cleaved by the released proteases, generating a fluorescent or luminescent signal that can be quantified [25].
Membrane Potential Measurement
This method directly assesses the physiological function of the intact membrane.
- Principle: A viable cell maintains a stable resting membrane potential (negative inside) through the active regulation of ion gradients. The collapse of this potential (depolarization) is a key indicator of membrane failure and a definitive marker of cell death [22].
- Protocol (Using Potential-Sensitive Dyes):
- Dye Loading: Cells are loaded with a membrane potential-sensitive dye like FluoVolt [22].
- Exposure: Cells are exposed to a toxicant or stressor.
- Measurement: Fluorescence intensity is monitored in real-time using a plate reader or flow cytometer. A significant and sustained increase in fluorescence indicates membrane depolarization and, thus, a loss of viability [22].
Table 2: Key Research Reagents for Membrane Integrity Assessment
| Reagent / Assay Name | Detection Method | Principle / Function | Key Applications |
|---|---|---|---|
| Propidium Iodide (PI) | Fluorescence Microscopy / Flow Cytometry | Nucleic acid intercalation in membrane-compromised cells [24]. | Standard live/dead staining; endpoint cytotoxicity [24] [27]. |
| LIVE/DEAD BacLight | Fluorescence Microscopy | Dual staining with SYTO 9 (green, all cells) and PI (red, dead cells) [24]. | Differentiating live/dead bacterial populations [24]. |
| FluoVolt Membrane Potential Kit | Fluorometry / Live-Cell Imaging | Fluorescence change in response to membrane potential [22]. | Real-time, direct measurement of membrane integrity loss [22]. |
| CytoTox-Glo Cytotoxicity Assay | Luminescence | Measures dead-cell protease activity released after membrane rupture [25]. | Specific, non-lytic quantification of dead cells; multiplexing with viability assays [25]. |
| CytoTox 96 Non-Radiocative Assay | Absorbance Spectrophotometry | Measures LDH release via conversion of tetrazolium salt to red formazan [25]. | Colorimetric, high-throughput assessment of cytotoxicity [25] [27]. |
Diagram 1: A workflow for classifying bacterial viability states based on three criteria, highlighting that membrane integrity loss is the definitive indicator of death.
Advanced Research and Novel Applications
Membrane Integrity in Drug Development and Delivery
Understanding membrane interactions is critical in pharmaceutical research. Model cell membranes are used to study how drug molecules and Drug Delivery Systems (DDS) interact with and cross this critical barrier [26]. Furthermore, the principle of membrane integrity is harnessed in advanced therapeutics. Biomimetic nanoparticles coated with natural cell membranes (e.g., from red blood cells or immune cells) leverage the surface proteins of the source cells to evade the immune system, thereby achieving longer circulation times and improved targeted delivery to diseased tissues [28] [29]. The functionality of these platforms depends entirely on the integrity and correct orientation of the coating membrane.
Differentiating Biocide Mechanisms of Action
Membrane integrity assays are pivotal in classifying antimicrobial agents and understanding their mechanism of action. Studies exposing Pseudomonas fluorescens to different biocides demonstrate this clearly:
- Lytic Biocides (e.g., Benzalkonium Chloride - BAC): These cationic agents directly target and disrupt the lipid bilayer, leading to immediate loss of membrane integrity, cell lysis, and rapid death [23].
- Electrophilic Biocides (e.g., DBNPA): These agents primarily act on intracellular targets, such as thiol groups in proteins, inhibiting metabolism and enzyme function. Membrane integrity is often lost as a secondary, downstream consequence of metabolic shutdown, not the primary cause of death [23].
This distinction is crucial for developing effective disinfection strategies and understanding the potential for microbial tolerance.
Within the triad of bacterial viability criteria, membrane integrity stands as the most definitive marker for cell death. Its loss represents an irreversible event that is fundamentally incompatible with life, distinguishing it from the potentially reversible states indicated by loss of culturability or metabolic activity. Robust and versatile methodologies, from fluorescent staining to enzymatic assays, provide researchers with powerful tools to accurately assess this parameter. As research continues to advance, the application of membrane integrity principles is expanding into cutting-edge fields like targeted drug delivery and precise antimicrobial development, solidifying its status as a cornerstone of cell viability research.
Understanding the Viable But Non-Culturable (VBNC) State and Its Clinical Implications
The Viable But Non-Culturable (VBNC) state is a survival strategy adopted by bacteria facing environmental stress. In this state, cells are alive and metabolically active but cannot form colonies on conventional growth media, the standard method for detecting viable bacteria [30] [31]. This phenomenon challenges the century-old paradigm that equates bacterial viability with cultivability [2].
The VBNC state holds significant clinical importance as it allows pathogenic bacteria to evade routine diagnostic detection and resist antimicrobial treatments. Cells in the VBNC state can maintain virulence potential and resume infections when conditions become favorable, contributing to chronic, recurrent, and unresolved infections [30] [32]. This guide examines the VBNC state within the framework of bacterial viability criteria—culturability, metabolic activity, and membrane integrity—providing researchers and drug development professionals with the technical foundation to address this microbial survival strategy.
Defining the VBNC State and Distinguishing It from Other Dormancy Phenomena
Core Definition and Diagnostic Criteria
A bacterial cell is considered to be in the VBNC state when it meets three essential criteria [31]:
- Loss of Culturability: It fails to form colonies on solid media that normally support its growth, resulting in zero colony-forming units (CFU).
- Metabolic Activity: It maintains measurable metabolic processes, including energy generation, membrane potential, and transcriptional/translational activity.
- Resuscitation Potential: It can revert to a culturable state upon removal of the inducing stress or application of specific resuscitation signals.
VBNC State Versus Persister Cells and Dormancy
The VBNC state is one of several bacterial survival strategies. It is crucial to differentiate it from other phenomena such as persister cells and general dormancy.
Table 1: Key Differences Between VBNC State and Persister Cells
| Feature | VBNC State | Persister Cells |
|---|---|---|
| Culturability | Non-culturable (CFU=0) [31] | Culturable (though not growing under stress) [31] |
| Induction | Moderate, long-term stresses (starvation, temperature, salinity) [31] | Specific, often antibiotic stress [31] |
| Metabolic Activity | Low but measurable [30] [31] | Very low or dormant [31] |
| Resuscitation | Requires a change in conditions (e.g., temperature shift, nutrient addition) [31] | Resumes growth upon simple stress removal [31] |
| Population Size | Can comprise most of the population [32] | Typically a small subpopulation (≈1%) [31] |
Furthermore, while "VBNC" and "dormant" are sometimes used interchangeably, a key distinction exists: dormant cells have a metabolic activity below the detection limit, whereas VBNC cells maintain measurable metabolic activity [31]. Some researchers hypothesize that persister cells and VBNC cells may represent different stages along a dormancy continuum, where active cells under stress first become persisters, which may then develop into VBNC state cells [30].
The following diagram illustrates this hypothesized relationship and the defining features of the VBNC state:
Environmental Triggers for VBNC State Entry
A wide range of environmental stresses can induce the VBNC state. These are frequently encountered in both natural habitats and clinical settings [31]:
- Nutrient starvation (e.g., in water systems or on hospital surfaces)
- Temperature extremes (both low and high temperatures)
- High osmotic pressure/salinity
- Oxidative stress
- pH fluctuations
- UV irradiation
- Presence of heavy metals
- Microbial competition (e.g., in multi-species biofilms)
Notably, processes designed to eliminate bacteria, such as chlorination of wastewater, pasteurization of milk, and the use of food preservatives, have also been documented to induce the VBNC state instead of causing death [33].
Resuscitation from the VBNC state involves a reversal of the inducing conditions or specific molecular signals. Common resuscitation triggers include:
- Temperature upshift (e.g., moving from low environmental temperature to host body temperature) [30]
- Introduction of nutrients [32]
- Co-culture with host cells or other microorganisms [30]
- Removal of the initial stressor (e.g., dilution of an antibiotic or disinfectant) [32]
The timeframe for successful resuscitation can vary. Studies on Acinetobacter baumannii showed that VBNC cells could be resuscitated efficiently for up to three months, with some capacity for revival observed even after ten months [32].
Clinical Implications and Risks
The VBNC state poses substantial challenges in clinical microbiology and patient care:
- Diagnostic Failures: Routine clinical diagnosis relies heavily on culture-based methods. VBNC pathogens evade these detection attempts, leading to false-negative results and missed diagnoses [30] [31].
- Chronic and Recurrent Infections: VBNC cells can persist in the host during antimicrobial therapy. Following treatment cessation, these cells may resuscitate, causing disease relapse. This has been associated with chronic oral infections, persistent wound infections, and recurrent urinary tract infections [30] [32].
- Antimicrobial Resistance: VBNC cells exhibit increased tolerance to antibiotics and antimicrobial agents due to their extremely low metabolic activity, as many antibiotics target active cellular processes [30] [34].
- Retention of Virulence: Some pathogens continue to express virulence factors in the VBNC state. For example, VBNC E. coli O157 and beer-spoilage Lactobacillus can produce shiga toxins and lactic acid, respectively, while in the state [31]. Legionella pneumophila in the VBNC state can infect and replicate within amoebae and human macrophages [31].
Oral pathogens such as Porphyromonas gingivalis (associated with periodontitis and systemic diseases), Enterococcus faecalis (prominent in endodontic infections), and the transient oral resident Helicobacter pylori have all been observed to enter the VBNC state, complicating the treatment of oral and gastrointestinal diseases [30] [34].
Detection and Assessment Methodologies
Overcoming the limitations of culture-based methods requires a multi-parametric approach to viability assessment. The following table summarizes the core strategies and specific techniques for detecting VBNC cells.
Table 2: Viability Assessment Strategies for Detecting VBNC Cells
| Viability Criterion | Detection Method | Principle | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Culturability | Plate Culture [2] | Ability to form colonies on solid media. | Standard method; allows identification. | Fails to detect VBNC cells by definition. |
| Metabolic Activity | Dye Uptake (e.g., FDA) [2] | Non-fluorescent substrate enters cell and is hydrolyzed by enzymes (esterases, lipases) into a fluorescent product. | Indicates enzymatic activity; can use fluorescence microscopy/flow cytometry. | Sensitivity to pH; potential for dye efflux; may not detect deeply dormant cells. |
| Tetrazolium Reduction (e.g., CTC) [32] [35] | Colorless CTC is reduced to insoluble, fluorescent formazan by active electron transport systems. | Indicates respiratory activity. | Some bacteria lack reduction capability; dye can be toxic. | |
| qRT-PCR of mRNA [36] | Quantifies messenger RNA (mRNA) of essential genes (e.g., 16S rDNA, rpoS). | High sensitivity; targets molecules with short half-life, strongly correlating with viability. | Technically complex; requires mRNA stabilization and reverse transcription. | |
| Membrane Integrity | LIVE/DEAD Staining (e.g., SYTO9/PI) [32] [36] | SYTO9 stains all cells; Propidium iodide (PI) only enters cells with damaged membranes, quenching SYTO9. | Directly assesses cell envelope integrity. | Can overestimate dead cells; cells with intact membranes may not be metabolically active. |
| Propidium Monoazide (PMA) qPCR [31] | PMA preferentially enters dead cells with compromised membranes and covalently binds DNA, inhibiting its PCR amplification. | Selectively amplifies DNA from cells with intact membranes; distinguishes viable from dead. | Requires optimization for different bacterial species/samples. |
The standard experimental workflow to conclusively demonstrate the VBNC state integrates these methods, as outlined below:
The Scientist's Toolkit: Essential Reagents for VBNC Research
Table 3: Key Research Reagent Solutions for VBNC Studies
| Reagent / Kit | Primary Function | Key Considerations |
|---|---|---|
| LIVE/DEAD BacLight Bacterial Viability Kit [32] [36] | Differentiates cells with intact (green) vs. damaged (red) membranes using SYTO9 and Propidium Iodide (PI). | Requires fluorescence microscopy or flow cytometry. Can overestimate dead cells; proper controls are essential. |
| CTC (5-cyano-2,3-ditolyl tetrazolium chloride) [32] [35] | Measures respiratory activity. Reduced to red-fluorescent formazan by metabolically active cells. | Can be toxic to some bacteria; not all viable cells may reduce it effectively. |
| PMA (Propidium Monoazide) Dye [31] | Used in PMA-qPCR to selectively inhibit DNA amplification from dead cells with compromised membranes. | Critical for differentiating DNA from live/dead cells in molecular assays; requires protocol optimization. |
| 2-NBDG (Fluorescent Glucose Analog) [2] | Assesses glucose uptake as a measure of metabolic activity. Incorporated and degraded by viable cells. | Not all bacterial species can transport and metabolize 2-NBDG. |
| FDA (Fluorescein Diacetate) [2] [35] | Assesses general enzymatic (esterase) activity. Hydrolyzed inside cells to produce fluorescent fluorescein. | Sensitive to pH; fluorescein can leak out of cells if membrane potential is low. |
| qRT-PCR Reagents for mRNA detection [36] | Detects and quantifies gene expression (e.g., of 16S rRNA or stress response genes) as a viability marker. | Targets the labile mRNA pool; requires careful RNA handling and reverse transcription steps. |
The VBNC state represents a fundamental survival strategy for a vast range of bacteria, with profound implications for clinical microbiology, infectious disease management, and antimicrobial drug development. Its study necessitates a paradigm shift from relying solely on culturability to employing a multi-faceted assessment of viability based on metabolic activity and membrane integrity. Understanding the triggers, molecular mechanisms, and resuscitation pathways of the VBNC state is crucial for developing novel diagnostic techniques that can detect these elusive cells and for designing therapeutic strategies that either prevent entry into the state or effectively eradicate VBNC populations, thereby addressing a significant source of persistent and recurrent infections.
From Lab Benches to Pipelines: A Practical Guide to Viability Assays
Culture-dependent methods remain the cornerstone for assessing bacterial viability, providing the definitive standard for quantifying living microorganisms capable of replication. The Colony Forming Unit (CFU) assay, often called the standard plate count, has maintained its status as the gold standard in microbiology for over a century due to its direct measurement of cellular proliferation [6]. This method's fundamental principle is simple yet powerful: each viable bacterial cell that can divide and form a visible colony originates from a single cultivable unit in the original sample. Despite the emergence of rapid alternative techniques, culture-based quantification continues to be indispensable across diverse fields including pharmaceutical development, food safety testing, clinical diagnostics, and environmental monitoring [37] [38].
Within the broader context of bacterial viability criteria, culturability represents a stringent definition of life—the capacity for cellular division and population growth under specific laboratory conditions. While other viability indicators such as metabolic activity and membrane integrity can be assessed through faster, culture-independent assays, they do not necessarily correlate with replicative potential [4]. Microbes may maintain metabolic functions or intact membranes while losing the ability to divide, creating a subpopulation of "viable but non-culturable" (VBNC) cells. Therefore, standard plate counts provide a conservative yet practically essential measure of viability, particularly when therapeutic efficacy or product safety depends on eliminating proliferating pathogens [39] [40].
Recent advancements have transformed traditional culture methods through automation and computational approaches, addressing long-standing limitations while preserving the core principles of microbial cultivation. Automated colony counting systems now leverage advanced imaging and artificial intelligence to dramatically improve the speed, accuracy, and consistency of CFU enumeration [37] [41]. These innovations are particularly valuable in high-throughput screening environments where pharmaceutical companies and research institutions must process thousands of samples efficiently while maintaining data integrity and regulatory compliance [42] [43].
Theoretical Foundations and Methodological Principles
The Colony Forming Unit (CFU) Concept
The Colony Forming Unit represents the fundamental unit of culturability in microbiology, defined as a single viable microbial cell or group of closely associated cells capable of producing a distinct visible colony through repeated divisions on or in a semisolid culture medium. This concept is operationalized through the standard plate count method, which involves serially diluting a bacterial suspension, plating onto appropriate agar media, incubating under optimal conditions, and counting the resulting colonies [6]. The critical assumption underpinning this method is that each visible colony arises from one proliferative unit, enabling backward calculation of the original viable cell concentration. The dynamic range of traditional CFU assays typically spans from 1 to 100,000,000 viable cells per sample, though this requires laborious dilution series to obtain countable plates (typically containing 25-250 discrete colonies) [6] [42].
The CFU assay directly measures the aspect of viability most relevant to infectious disease transmission, bioburden assessment, and antimicrobial efficacy—the retained capacity for population expansion. However, this method inherently selects for microorganisms capable of growth under the specific laboratory conditions provided, including medium composition, incubation temperature, atmosphere, and time [39]. This selectivity creates discrepancies between culturability and other viability indicators, as sublethally injured cells may maintain membrane integrity and metabolic activity while temporarily losing divisional capacity, and certain bacterial species may enter VBNC states in response to environmental stresses [4].
Methodological Evolution and Innovation
The fundamental methodology of culture-based enumeration has evolved significantly from its origins in late 19th-century microbiology. Traditional approaches involve manual dilution and plating techniques that remain widely used but present substantial challenges in terms of time investment (often requiring 24-48 hours incubation), labor intensity, and inter-operator variability [37]. Modern innovations have focused on addressing these limitations while preserving the core principle of measuring replicative potential.
Recent methodological advances include the Geometric Viability Assay (GVA), which represents a paradigm shift in culture-based quantification. This approach replicates CFU measurements over 6 orders of magnitude while reducing time and consumable requirements by more than 10-fold compared to traditional methods [6]. GVA operates by embedding serially diluted samples in a conical geometry (typically a standard pipette tip) containing agar medium, incubating, and then calculating the original viable cell concentration based on the distribution of colonies along the cone's axis. The probability of colony formation at any point is proportional to the cross-sectional area at that position, enabling precise estimation of viable counts from the spatial distribution of colonies without requiring complete enumeration [6].
Simultaneously, automated digital colony counting systems have transformed traditional plate reading through enhanced imaging capabilities and sophisticated image analysis algorithms. Systems like the Neogen Petrifilm Plate Reader Advanced combine high-resolution imaging with fixed artificial intelligence to detect and count colonies in approximately 6 seconds per plate [37]. These systems employ algorithmic analysis that differentiates colonies based on size, color, and other defining characteristics, standardizing microbial enumeration across operators and facilities—a critical advantage in regulated industries [37] [41].
Current Technologies in Automated Colony Counting
Algorithmic Approaches and Performance
Advanced computational methods have emerged to address the persistent challenge of accurately counting merged colonies in high-throughput applications. MCount represents a significant innovation as the first solution that incorporates both contour information and regional algorithms for colony counting [42]. By optimizing the pairing of contours with regional candidate circles, MCount can accurately infer the number of merged colonies that traditional region-based algorithms would count as single units. When evaluated on a precisely labeled Escherichia coli dataset of 960 images containing 15,847 segments, MCount achieved an average error rate of just 3.99%, significantly outperforming established solutions including NICE (16.54%), AutoCellSeg (33.54%), and OpenCFU (50.31%) [42].
The enhanced performance of modern algorithms is particularly valuable in high-density plating scenarios common in pharmaceutical screening and functional genomics, where colonies frequently merge due to spatial constraints. These advanced tools employ sophisticated image processing techniques including concave point detection, Hough transforms for circular object recognition, and watershed segmentation adapted for microbial colonies [42]. The implementation of such algorithms in user-friendly platforms with minimal hyperparameter requirements (MCount requires only two hyperparameters) facilitates broader adoption across microbiology laboratories without specialized computational expertise [42].
Commercial Systems and Implementation
Commercial automated colony counting systems have evolved into sophisticated instruments that combine robust hardware with intelligent software. Leading systems such as the Scan Ai series (Scan 3000 Ai and Scan 5000 Ai) leverage artificial intelligence to deliver counting accuracy reported to be 25% higher than conventional automated counters [41]. These systems can process up to 400 plates per hour while automatically discriminating between colonies and common artifacts such as labels on plate bottoms, air bubbles, condensation, and particulates [41]. The AI models can further distinguish between different microorganism types including bacteria, yeasts, and molds based on their distinct morphological characteristics [41].
Modern systems like the Neogen Petrifilm Plate Reader Advanced demonstrate how automation addresses the limitations of manual counting. These instruments provide fixed algorithmic analysis that standardizes interpretation across operators and sites, eliminating human subjective variability [37]. The implementation of "locked AI" in systems such as Scan Ai ensures data security through local storage and offline operation while allowing controlled updates that maintain established qualifications and regulatory compliance [41]. This balance of technological sophistication and practical implementation considerations makes contemporary automated colony counters suitable for both research and quality control environments where method validation and audit readiness are essential [37] [43].
Table 1: Comparative Analysis of Colony Counting Methods
| Method | Time Requirement | Accuracy | Throughput | Key Applications |
|---|---|---|---|---|
| Traditional Manual Counting | 45 minutes per plate [43] | ~80% [43] | Low | Research labs, educational use |
| Semi-Automated Method | 25 minutes per plate [43] | ~85% [43] | Moderate | Small-scale testing facilities |
| Digital Imaging Method | 15 minutes per plate [43] | ~92% [43] | Medium-High | Medium-throughput screening |
| Full Automation (AI-Powered) | 10 minutes per plate [43] | ~95% [43] | High (400 plates/hour) [41] | Pharmaceutical QC, high-throughput labs |
| Geometric Viability Assay (GVA) | Up to 30x faster than drop CFU [6] | High correlation with CFU (r=0.98) [6] | 1,200 viability measurements per researcher daily [6] | Checkerboard assays, drug screens, time-courses |
Table 2: Commercial Automated Colony Counting Systems
| System | Key Features | Counting Speed | AI Capabilities | Typical Use Cases |
|---|---|---|---|---|
| Neogen Petrifilm Plate Reader Advanced | Enhanced imaging with fixed AI, specific Petrifilm plate optimization | 6 seconds per plate [37] | Algorithmic colony differentiation | Food safety testing, quality control labs |
| Scan Ai Series (3000/5000) | Artifact discrimination, microorganism classification | 400 plates per hour [41] | Locked AI, local data processing | Regulatory environments, high-volume testing |
| MCount Software | Merged-colony resolution, contour and regional algorithms | Variable based on computational resources | Statistical hyperparameter optimization | Research institutions, high-density plating applications |
Experimental Protocols and Methodologies
Standard Plate Count Protocol
The traditional colony forming unit assay follows a standardized protocol that requires meticulous technique to ensure accurate quantification. The following protocol describes the essential steps for performing manual standard plate counts:
Sample Preparation: Begin with thorough homogenization of the bacterial suspension to ensure uniform distribution of cells. For solid samples, appropriate preliminary processing such as stomaching or blending is required to achieve a homogeneous suspension [38].
Serial Dilution Preparation: Prepare a logarithmic dilution series in sterile diluent (typically buffered peptone water or physiological saline). Use wide-bore pipettes to avoid shearing effects and transfer volumes accurately between dilution tubes. Common dilution factors range from 1:10 to 1:100 at each step, depending on the expected bacterial concentration [6] [38].
Plating Technique: Transfer a precise volume (typically 0.1-1.0 mL) of appropriate dilutions onto the surface of pre-poured agar plates or into empty sterile plates followed by pouring of molten agar (pour plate method). For spread plating, distribute the aliquot evenly across the agar surface using a sterile spreader. Multiple dilutions should be plated in replicates to ensure statistical reliability and obtain countable plates (25-250 colonies) [38].
Incubation: Invert plates and incubate under conditions optimal for the target microorganisms (typically 24-48 hours at appropriate temperature and atmosphere). The incubation time must be standardized as some slow-growing organisms or injured cells may require extended periods to form visible colonies [39].
Enumeration and Calculation: Count colonies manually using a colony counter with Quebec grid or digitally via automated systems. Calculate the CFU concentration in the original sample using the formula: CFU/mL = (number of colonies counted) / (dilution factor × volume plated). Only plates with 25-250 discrete colonies should be considered statistically valid [38].
Geometric Viability Assay Protocol
The Geometric Viability Assay offers a innovative approach to viability testing that dramatically reduces time and consumable requirements:
Sample Embedding: Completely mix the bacterial sample with melted LB agarose cooled to ≤55°C to a final agarose concentration of 0.5%. For enhanced contrast, include triphenyl tetrazolium chloride (TTC) in the melted agarose at manufacturer-recommended concentrations [6].
Tip Loading: Aspirate the agarose-sample mixture into a standard pipette tip. Ensure no air bubbles are introduced during loading as these may disrupt colony distribution patterns [6].
Solidification and Ejection: Allow the agarose to solidify completely at room temperature or refrigerated conditions. Eject the solidified agarose column from the tip into an empty tip rack or specialized holding apparatus [6].
Incubation: Incubate the agarose columns overnight at the appropriate temperature for the target microorganisms (e.g., 37°C for mesophilic bacteria). Maintain humidity to prevent desiccation during incubation [6].
Imaging and Analysis: Image the incubated agarose columns using a standardized optical setup. For research applications, a custom-built system with a mirrorless Canon camera has been described, though commercial imaging systems may be adapted [6]. Analyze the distribution of colonies along the cone's axis using the probability density function: PDF(x) = 3x²/h³, where x is the perpendicular distance from the tip and h is the total cone length. Calculate the CFU concentration using: CFUs/mL = N(x) / (V × ∫PDF(x)dx), where N(x) represents the number of colonies between positions x₁ and x₂, and V is the cone volume [6].
Automated Counting with AI-Based Systems
Implementation of automated colony counting systems requires standardized protocols to ensure consistent performance:
Sample Preparation and Plating: Follow standard plating procedures appropriate for the sample type and target microorganisms. Ensure even colony distribution and avoid over-crowding that exceeds the system's resolution capabilities for merged colonies [41].
Image Acquisition: Place plates according to manufacturer specifications on the imaging stage. Modern systems typically include standardized lighting configurations and focal distances to ensure consistent image quality. Some systems incorporate multiple imaging angles or focal planes to enhance colony detection [37] [41].
Algorithm Processing: Initiate the automated counting sequence. AI-based systems typically apply pre-processing filters to correct for background variations, plate artifacts, and condensation effects before colony detection [42] [41].
Validation and Verification: Review automated counts against quality control samples with known values. For regulated environments, maintain validation records demonstrating system performance characteristics including precision, accuracy, and linearity over the operational range [37].
Data Export and Documentation: Export results to laboratory information management systems (LIMS) for permanent record keeping. Automated systems typically provide digital images with counted colonies marked, creating an audit trail for regulatory compliance [37] [41].
Standard Plate Count Workflow
Automated Counting Process
Essential Research Reagent Solutions
The implementation of culture-dependent methods requires specific reagents and materials standardized for reliable performance. The following table details essential research reagents and their applications in viability assessment:
Table 3: Essential Research Reagents for Culture-Dependent Viability Assessment
| Reagent/Material | Function | Application Examples | Technical Considerations |
|---|---|---|---|
| Selective Agar Media | Supports growth of specific microorganisms while inhibiting others | MacConkey Agar (gram-negative selection), Baird-Parker Agar (Staphylococcus selection) [38] | Composition must be validated for target organisms; may require supplementation |
| Differential Agar Media | Distinguishes microorganisms based on biochemical characteristics | Petrifilm Aerobic Count (AC) Plates with red colonies indicating general aerobic bacteria [37] | Colorimetric changes typically require 24-48 hours incubation for full expression |
| Tetrazolium Salts (TTC, XTT) | Redox indicators visualize metabolically active colonies | Triphenyl tetrazolium chloride (TTC) in GVA for enhanced colony contrast [6] | Concentration must be optimized to avoid growth inhibition |
| 2,6-Dichlorophenolindophenol (DCIP) | Redox dye for colorimetric viability assessment | Rapid quantification of oral bacteria viability in antimicrobial testing [40] | Spectrophotometric measurement required; correlates with metabolic activity |
| Lactate Dehydrogenase (LDH) Assay | Measures enzyme release from damaged cells | Cytotoxicity assessment in co-culture systems [4] | Background levels may vary; requires cell-free supernatant |
| Membrane Integrity Dyes | Distinguishes cells with compromised membranes | Propidium iodide exclusion assay for dead cell identification [4] | Incubation time critical to avoid false positives from dye penetration |
Applications in Pharmaceutical and Biomedical Research
Antimicrobial Efficacy Testing
Culture-dependent methods provide critical data for evaluating antimicrobial compound efficacy throughout the drug development pipeline. In checkerboard assays for synergistic interactions and time-kill studies for bactericidal kinetics, automated colony counting enables precise quantification of viable pathogens following antimicrobial exposure [6]. The geometric viability assay has demonstrated particular utility in labor-intensive antimicrobial testing scenarios, simplifying workflow while maintaining correlation with traditional CFU measurements [6]. These applications depend on accurate discrimination between live and dead bacterial populations following treatment with experimental therapeutics, requiring methods that specifically measure replicative potential rather than indirect viability indicators.
In pharmaceutical quality control, culture methods remain essential for sterility testing of non-sterile products and bioburden assessment of raw materials. Automated systems like the Scan Ai series provide the throughput necessary for manufacturing environments while delivering the documentation required for regulatory submissions [41]. The implementation of fixed algorithmic analysis in systems such as the Neogen Petrifilm Plate Reader Advanced standardizes interpretation across facilities and operators, a critical factor in multisite pharmaceutical operations [37].
Probiotic Quality Control and Label Accuracy
Culture-dependent methods are indispensable for verifying label claims in probiotic products, where viability directly correlates with therapeutic efficacy. Recent investigations of commercial probiotics sold in the United Kingdom demonstrated that poultry products largely met or exceeded their labeled claims, while human products showed greater variability, with one product containing no detectable viable bacteria despite label claims [38]. These assessments combine selective plate counts with identification technologies such as MALDI-TOF MS to simultaneously quantify and confirm microbial identity, ensuring product integrity throughout shelf life.
Standard plate counts in probiotic quality control typically employ a practical acceptance range of ±0.5 log CFU from declared values, acknowledging the inherent variability in biological systems while maintaining consumer protection standards [38]. The integration of automated colony counting systems in quality control laboratories enhances throughput and consistency while reducing operator-dependent variability in enumeration. This application highlights the continued relevance of culture-based methods in verifying commercial products where metabolic activity or membrane integrity assessments alone would be insufficient to guarantee proliferative capacity at the site of action.
Environmental and Public Health Monitoring
Culture-dependent methods continue to play essential roles in environmental monitoring and public health protection, particularly in assessing airborne bacterial concentrations and composition. Studies of ambient bacterial aerosols using surrogate lung fluid collection media have revealed how bacterial features change upon deposition in respiratory airways, providing insights into inhalation health risks [39]. These investigations combine culturing-based methods with molecular techniques to characterize culturable numbers, total numbers, and metabolic activity, creating comprehensive profiles of microbial populations in various environments.
In food safety testing, automated colony counting on platforms such as Petrifilm has transformed microbial enumeration by reducing hands-on time while improving data traceability [37]. The standardization afforded by automated systems ensures consistent interpretation of results across multiple technicians and testing locations, essential for compliance with food safety regulations and industry standards. These applications demonstrate how technological advances in culture-based methods continue to enhance their utility in protecting public health despite the availability of more rapid alternative methods.
Culture-dependent methods incorporating both standard plate counts and automated colony counting remain indispensable tools for assessing bacterial viability based on the fundamental criterion of replicative potential. While newer techniques based on metabolic activity or membrane integrity offer speed advantages, they cannot replace the definitive measurement of cellular division provided by CFU assays [4]. The ongoing innovation in this field—from geometric viability assays that dramatically reduce resource requirements to AI-powered colony counting systems that enhance accuracy and throughput—ensures that culture methods will maintain their relevance in pharmaceutical development, clinical diagnostics, and quality control applications.
The integration of automation and artificial intelligence addresses historical limitations of culture-based methods while preserving their gold standard status for viability assessment. Systems capable of processing hundreds of plates per hour with minimal operator intervention make comprehensive testing feasible even in high-volume environments [41]. Meanwhile, advanced algorithms that accurately resolve merged colonies enable reliable enumeration in high-density plating scenarios essential for modern screening applications [42]. These technological advances, combined with the intrinsic biological relevance of measuring divisional capacity, secure the position of culture-dependent methods as essential components in the microbial viability assessment toolkit for the foreseeable future.
Evaluating bacterial viability is a cornerstone of microbiological research, particularly in drug development and environmental monitoring. The contemporary framework for this assessment rests on three established criteria: culturability, metabolic activity, and membrane integrity [2]. While culturability, demonstrated by the ability to form colonies on solid media, has been the traditional gold standard, it is now widely recognized that a significant population of bacteria can enter a viable but non-culturable (VBNC) state [2]. In this state, cells fail to grow on conventional media but maintain metabolic activity and membrane integrity, remaining capable of causing infections [2] [44]. This limitation has propelled the adoption of techniques that directly probe cellular physiology.
Among the most informative physiological probes are those measuring metabolic activity. This technical guide focuses on two pivotal techniques: the Fluorescein Diacetate (FDA) Hydrolysis assay and the 2-NBDG Glucose Uptake assay. The FDA hydrolysis assay measures the activity of non-specific intracellular enzymes, such as esterases, lipases, and proteases, providing a broad indicator of cellular metabolic vigor [2] [45]. In contrast, the 2-NBDG uptake assay specifically interrogates the function of the glucose transport system, a critical pathway in cellular energy metabolism [46] [47]. Together, these methods offer powerful, complementary tools for researchers to accurately determine the viability and metabolic status of bacterial cells, bypassing the limitations of culture-based methods and providing deeper insights into cellular function in real-time.
Theoretical Foundations of the Techniques
The Principle of FDA Hydrolysis
The Fluorescein Diacetate (FDA) Hydrolysis assay serves as a robust indicator of total enzymatic activity in bacterial cells. The underlying principle involves the use of FDA, a non-polar, non-fluorescent molecule that readily permeates the intact lipid bilayer membranes of viable cells via passive transport [2]. Once inside the cell, FDA acts as a substrate for a wide spectrum of non-specific intracellular enzymes, including esterases, lipases, and proteases. The hydrolysis of FDA by these enzymes removes the acetate groups, converting it into fluorescein [2] [45].
This product, fluorescein, is a polar molecule that is impermeant to the cell membrane, leading to its accumulation within cells with intact membranes. Crucially, fluorescein is highly fluorescent, exhibiting strong light absorption at 490 nm and emitting a yellow-green light [45]. Therefore, the intensity of the fluorescent signal is directly proportional to the hydrolytic enzyme activity within the cell, which in turn reflects the metabolic activity and viability of the cell. A key advantage of this method is that extracellular FDA does not produce a background signal, as it remains non-fluorescent until hydrolyzed [2]. This assay has been successfully adapted for various formats, including bulk samples and, with specific protocol adjustments, even for biofilms formed on immobilization carriers, allowing for the measurement of total enzymatic activity without disrupting the biofilm structure [45].
The Principle of 2-NBDG Glucose Uptake
The 2-NBDG Glucose Uptake assay provides a specific means to investigate a critical metabolic function: the transport of glucose. This technique utilizes 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose, or 2-NBDG, a fluorescently labeled analog of 2-deoxyglucose [47]. The molecular structure of 2-NBDG is designed to mimic natural glucose, allowing it to be recognized and transported into the cell by the same glucose transporter (GLUT) proteins that shuttle glucose across the cell membrane [46] [48].
Upon entering the cytoplasm, 2-NBDG is phosphorylated by hexokinase, the first enzyme in the glycolytic pathway, into 2-NBDG-6-phosphate [47]. Similar to its natural counterpart, this phosphorylated form is not a significant substrate for other cellular enzymes and becomes trapped within the cell, leading to intracellular accumulation [46]. The attached nitrobenzoxadiazole (NBD) fluorophore allows for the direct detection and quantification of this accumulated compound using flow cytometry, fluorescence microscopy, or microplate readers with filters suitable for FITC (Ex/Em ~485/535 nm) [47] [48]. The resulting fluorescent signal thus serves as a direct measure of glucose transporter activity. It is important to note that this method directly measures glucose uptake, which occurs on a rapid timescale (minutes), and is distinct from measuring long-term glucose consumption, which involves a multitude of metabolic pathways [46].
Comparative Analysis of Metabolic Assays
A critical step in selecting an appropriate viability assay is understanding its strengths, limitations, and optimal applications. The table below provides a structured comparison of the FDA Hydrolysis and 2-NBDG Glucose Uptake assays against other common methods for assessing bacterial metabolic activity.
Table 1: Comparison of Metabolic Activity Assays for Bacterial Viability
| Assay Method | Principle of Detection | Key Advantages | Key Disadvantages / Limitations |
|---|---|---|---|
| FDA Hydrolysis | Hydrolysis of non-fluorescent FDA to fluorescent fluorescein by intracellular enzymes [2] [45] | - Measures broad metabolic activity [45]- Works on intact biofilms [45]- No extracellular background signal [2] | - Signal sensitive to intracellular pH [2]- Fluorescein efflux can occur in acidic conditions [2] |
| 2-NBDG Uptake | Uptake and accumulation of a fluorescent glucose analog via glucose transporters [46] [47] | - Direct measure of glucose transport activity [46]- Amenable to real-time imaging and flow cytometry [47] | - Not all bacterial species transport 2-NBDG [2]- Large molecular size may alter transport kinetics [46] |
| Dye Uptake (e.g., CTC/INT) | Reduction of tetrazolium salts to colored formazan by electron transport chain [2] | - Indicates respiratory activity | - May not detect cells with inactive respiration |
| Enzymatic Glucose Assay | Measurement of remaining glucose in medium via glucose oxidase [2] | - Measures bulk glucose consumption | - Indirect measure; does not distinguish between uptake and other metabolic fates |
The following diagram illustrates the logical decision-making process for selecting an appropriate bacterial viability assessment method based on the specific research goals, contextualizing where FDA and 2-NBDG assays fit within the broader toolkit.
Diagram 1: A decision pathway for selecting bacterial viability assessment methods, highlighting the context for using FDA Hydrolysis and 2-NBDG Uptake assays.
Detailed Experimental Protocols
Protocol for FDA Hydrolysis Assay in Biofilms
This protocol is optimized for measuring the Total Enzymatic Activity (TEA) of a whole biofilm formed on a carrier, such as polyurethane foam (PUR), without detaching cells, thus preserving the native biofilm structure and function [45].
- Step 1: Pre-incubation. Place the biofilm-immobilized carrier into a suitable vessel containing an appropriate buffer, such as phosphate buffer (pH 7.6). Incubate on an orbital shaker for 15 minutes to equilibrate the system.
- Step 2: Substrate Application. Slowly inject the FDA substrate solution directly into the middle of the immobilized carrier to ensure efficient delivery into the biofilm matrix.
- Step 3: Hydrolysis Reaction. Incubate the carrier with the substrate on an orbital shaker at 130 rpm and 30°C for 1 hour. The shaking facilitates mass transfer of the substrate and product within the biofilm.
- Step 4: Reaction Termination & Measurement. After incubation, remove an aliquot of the buffer solution. Measure the fluorescence of the generated fluorescein spectrophotometrically (absorption at 490 nm).
- Step 5: Data Normalization. Determine the dry mass of the biofilm by comparing the dried weight of the immobilized carrier with that of an unimmobilized carrier. Normalize the fluorescence data to the biofilm dry mass to express TEA.
- Critical Considerations:
- Carrier Adsorption: Prior to the experiment, conduct a control test to determine if the carrier material adsorbs fluorescein. For example, PUR has been shown to adsorb up to 7.7% of fluorescein at higher concentrations (>2.5 µg/mL), and this should be accounted for in final calculations [45].
- pH Dependence: The assay is sensitive to pH, as an acidic environment can lead to protonation and efflux of fluorescein, reducing the detected signal [2].
Protocol for 2-NBDG Glucose Uptake Assay
This protocol outlines the steps for using 2-NBDG to measure glucose uptake in cell cultures, suitable for analysis with a fluorometric plate reader, fluorescent microscope, or flow cytometer [48].
- Step 1: Pre-equilibration. Culture and treat cells according to experimental design. Prior to the assay, equilibrate cells in a glucose-free medium for a period (e.g., two hours) to deplete endogenous glucose and upregulate glucose transport systems [48].
- Step 2: Probe Incubation. Add 2-NBDG to the culture medium at the desired concentration (e.g., 10 µM). Incubate for a short, defined period (e.g., 10-30 minutes) at 37°C to allow for active uptake and phosphorylation [47] [48].
- Step 3: Termination and Washing. Remove the cell culture supernatant and wash the cells thoroughly with a cold cell-based assay buffer to remove any extracellular 2-NBDG.
- Step 4: Viability Staining (Optional for Flow Cytometry). To exclude dead cells (which may take up the probe passively due to compromised membranes), resuspend the cell pellet in buffer containing a viability dye like propidium iodide (PI) [48].
- Step 5: Analysis. Resuspend cells in assay buffer and analyze immediately. For a plate reader, use excitation/emission filters of 485/535 nm. For flow cytometry or microscopy, use the appropriate filter set for FITC.
- Critical Considerations:
- Cell Line Variability: Uptake kinetics can vary significantly. For instance, in MCF-7 cells, uptake is rapid for the first 1-5 minutes and reaches a maximum around 20-30 minutes [47].
- Species Specificity: A significant limitation is that not all bacterial species can transport 2-NBDG. Studies show that species like Vibrio mimicus, Bacillus cereus, and some E. coli strains cannot take in 2-NBDG effectively [2].
- Probe Characteristics: The large size of the fluorescent tag may alter transport kinetics compared to natural glucose, which is a concern for accurate measurement of transporter activity [46].
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of the FDA and 2-NBDG assays requires specific reagents and materials. The following table details the key components of the research toolkit for these techniques.
Table 2: Essential Research Reagent Solutions for Metabolic Probing
| Item Name | Function / Description | Example Application Notes |
|---|---|---|
| Fluorescein Diacetate (FDA) | A non-fluorescent pre-fluorophore that passively diffuses into cells and is hydrolyzed by intracellular enzymes [2] [45]. | Used as a general marker for total enzymatic activity and metabolic activity in viability studies [45]. |
| 2-NBDG (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose) | A fluorescent glucose analog transported into cells via glucose transporters and phosphorylated for intracellular trapping [47]. | Serves as a direct tracer for glucose uptake; used in studying glucose metabolism in various cell types and disease models [47]. |
| Propidium Iodide (PI) | A membrane-impermeant fluorescent nucleic acid stain that only enters cells with damaged membranes [49] [48]. | Used as a viability dye to exclude dead cells during flow cytometry analysis of 2-NBDG uptake [48]. |
| Glucose-Uptake Inhibitor (e.g., Apigenin) | A compound that specifically inhibits cellular glucose uptake [48]. | Serves as a negative control in 2-NBDG assays to confirm the specificity of the uptake signal [48]. |
| Cell-Based Assay Buffer | A balanced salt solution, often provided in tablet form, for washing cells and maintaining physiological pH during assays [48]. | Used to wash away extracellular 2-NBDG and to resuspend cells for final analysis [48]. |
| Polyurethane Foam (PUR) Cubes | A common, non-toxic carrier material with a large surface area for bacterial immobilization and biofilm formation [45]. | Used as a support structure for growing biofilms in the FDA hydrolysis assay to study immobilized biocatalysts [45]. |
Data Interpretation and Technical Considerations
Quantification and Standardization
Accurate quantification is paramount for reliable results. For the FDA hydrolysis assay, the fluorescence or absorbance readings must be normalized to a biomass parameter. When working with immobilized biofilms, the most straightforward method is to use the dry weight of the biomass, obtained by comparing the weight of the carrier before and after biofilm growth and drying [45]. The resulting data can be expressed as Total Enzymatic Activity (TEA) per unit of dry mass. For the 2-NBDG assay, data normalization typically involves correlating the fluorescent signal to the cell number. In flow cytometry, this is intrinsic, as data is collected per event (cell). For plate reader assays, normalization to total protein content or using a parallel cell viability assay (e.g., AlamarBlue) is common practice. The use of internal controls is critical. This includes a negative control without the substrate (FDA or 2-NBDG) to account for autofluorescence, and for 2-NBDG, a control with a glucose uptake inhibitor (e.g., apigenin) to confirm the specificity of the signal to transporter-mediated uptake [48].
Troubleshooting and Limitation Analysis
A sophisticated understanding of the limitations of these techniques is necessary for correct data interpretation. The FDA assay, while excellent for measuring broad metabolic activity, is inherently sensitive to the intracellular pH of the cells. A drop in pH can lead to fluorescein protonation and subsequent efflux from the cell, thereby underreporting the true enzymatic activity [2]. For the 2-NBDG assay, the primary consideration is biological relevance. The 2-NBDG molecule is substantially larger than natural glucose due to its fluorescent tag, raising valid concerns that it may not be a perfect substrate for all glucose transporters, potentially leading to inaccurate reflections of true glucose transporter activity [46]. Furthermore, its utility is limited in bacterial studies, as a number of species, including Vibrio mimicus and Bacillus cereus, have been shown to be incapable of taking up 2-NBDG, making the assay unsuitable for these organisms [2]. Researchers must therefore validate that their model system is appropriate for the chosen probe. Finally, for both assays, kinetic parameters (incubation time, substrate concentration) must be empirically determined for each new cell type or experimental condition to ensure measurements are taken within the linear range of the assay and not at saturation points [47] [45].
In microbiological research, accurately determining cell viability is fundamental, and membrane integrity is widely regarded as one of the most definitive proofs of cell viability. A intact cytoplasmic membrane indicates that a cell can potentially maintain electrochemical gradients and exhibit metabolic activity, though it does not, in isolation, guarantee cell replication. Cells with damaged or compromised membranes are considered dead, as they cannot maintain or generate the essential electrochemical gradient, leading to the exposure of cellular structures to the environment and eventual lysis [49]. The assessment of membrane integrity is particularly suitable for detecting starved, dormant, or injured cells, which may require a reactivation step to recover other cellular functions [49].
The principle behind membrane integrity-based viability assays is primarily founded on dye exclusion methods [49]. These methods leverage the fact that live cells with intact membranes are impermeable to certain charged dyes. In contrast, dead cells with compromised membranes allow these dyes to enter, where they typically bind to intracellular components like nucleic acids and emit fluorescence. This fundamental difference provides a straightforward and reliable means to distinguish between live and dead cell populations using techniques such as flow cytometry and fluorescence microscopy [50] [51].
LIVE/DEAD viability kits are fluorescent-based assays designed for easy and sensitive differentiation of live, dead, and, in some cases, injured cell populations. These kits are versatile and can be used to evaluate viability in a wide range of organisms, including mammalian cells, bacteria, fungi, and yeast [51]. The underlying mechanisms vary by kit but are predominantly based on membrane integrity, esterase activity, or metabolic activity [51].
A key advantage of some specific kits, such as the LIVE/DEAD Fixable Dead Cell Stain series, is their compatibility with formaldehyde fixation. This allows the staining pattern to be preserved following fixation, enabling intracellular staining procedures and the neutralization of pathogens without losing viability information [50] [51]. The following table summarizes several key LIVE/DEAD kits and their characteristics.
Table 1: Selection of LIVE/DEAD Cell Viability Assay Kits
| Kit Name | Platform(s) | Fluorescent Dyes | Mechanism of Action | Live Cell Signal | Dead Cell Signal |
|---|---|---|---|---|---|
| LIVE/DEAD Viability/Cytotoxicity | FC, FM, M | Calcein AM, Ethidium Homodimer-1 | Esterase Activity, Membrane Integrity | Green (Calcein) | Red (EthD-1) |
| LIVE/DEAD BacLight Bacterial Viability | FC, FM, M | SYTO 9, Propidium Iodide (PI) | Membrane Integrity | Green (SYTO 9) | Red (PI) |
| LIVE/DEAD Fixable Dead Cell Stains | FC | Fixable Viability Dyes (e.g., Green, Violet) | Membrane Integrity (Amino-reactive) | N/A (Labels dead cells) | Green, Violet, etc. |
| LIVE/DEAD Cell Viability Assay (C12 Resazurin/SYTOX Green) | FC, FM, M | C12-Resazurin, SYTOX Green | Metabolic Activity, Membrane Integrity | Red (C12-Resorufin) | Green (SYTOX Green) |
| LIVE/DEAD FungaLight Yeast Viability | FC | SYTO 9, Propidium Iodide (PI) | Membrane Integrity | Green (SYTOX 9) | Red (PI) |
FC = Flow Cytometry; FM = Fluorescence Microscopy; M = Microplate Assay
Detailed Experimental Protocols
Protocol for Fixable Viability Stains in Flow Cytometry
The LIVE/DEAD Fixable Dead Cell Stains are designed to covalently bind to amines, both intracellular and extracellular. When a cell's membrane is compromised, the dye enters and binds to internal amines, resulting in intense staining. In live cells with intact membranes, the dye only accesses external amines, resulting in minimal staining. This pattern is preserved after fixation [50].
Table 2: Emission Specifications for Select LIVE/DEAD Fixable Stains
| LIVE/DEAD Dye | Common Excitation Source | Excitation Max (nm) | Emission Max (nm) |
|---|---|---|---|
| Violet stain | 405 nm laser | 416 | 451 |
| Aqua stain | 405 nm laser | 367 | 526 |
| Green stain | 488 nm laser | 495 | 520 |
| Yellow stain | 405 nm laser | 400 | 575 |
| Red stain | 488 nm or 561 nm lasers | 595 | 615 |
| Far Red stain | 633/635 nm laser | 650 | 665 |
Step-by-Step Staining Protocol [50]:
- Preparation: Thaw the vial of dye and dilute it by adding 50 µL of anhydrous DMSO.
- Cell Suspension: Prepare cells in a protein-free buffer at a concentration between 1x10⁴ and 1x10⁶ cells per mL. The protein concentration should be <1% to prevent quenching.
- Staining: Add 1 µL of the diluted stain to 1 mL of the cell suspension.
- Incubation: Mix thoroughly and incubate for 30 minutes at room temperature, protected from light.
- Wash: Wash the cells once to remove unbound dye. This step is optional but recommended.
- Fixation (Optional): If required, fix the cells with formaldehyde. The staining pattern will be preserved.
- Analysis: Analyze the cells on a flow cytometer using the appropriate laser and filter settings for the dye used.
Protocol Tips:
- The kit is specifically intended for flow cytometry and should not be used for fluorescence microscopy [50].
- Washing after staining is optional but can reduce background signal.
- Cell staining is preserved after fixation, but fixation is not a mandatory step for analysis [50].
Protocol for Dual-Color Viability/Cytotoxicity Kits in Microscopy
Kits like the LIVE/DEAD Viability/Cytotoxicity Kit (containing Calcein-AM and Ethidium Homodimer-1) or the Live-Dead Cell Staining Kit (containing Calcein-AM and Propidium Iodide) are well-suited for fluorescence microscopy. These kits employ a two-mechanism approach: live cells are identified by enzymatic activity, while dead cells are identified by compromised membrane integrity [51] [52].
Step-by-Step Staining Protocol [52]:
- Solution Preparation: Prepare the working solution by mixing Calcein-AM and Propidium Iodide (PI) in a buffer such as PBS.
- Staining: Incubate the cell sample with the working solution for a designated time (typically 15-30 minutes) at room temperature, protected from light.
- Wash (Optional): Gently wash the cells to remove excess dye, if necessary for image clarity.
- Imaging: Immediately observe the cells under a fluorescence microscope using standard FITC (green) and TRITC (red) filter sets.
Mechanism Details:
- Live Cells (Green): Calcein-AM, a non-fluorescent, cell-permeant dye, enters all cells. In live cells, intracellular esterases hydrolyze Calcein-AM into Calcein, which is fluorescent and green, and is retained within the cell due to its charged nature [51] [52].
- Dead Cells (Red): Propidium Iodide (PI) is a membrane-impermeant dye that can only enter cells with damaged membranes. It intercalates with nuclear DNA, producing a bright red fluorescence [52].
The workflow and underlying mechanisms for this type of assay are summarized in the diagram below.
The Scientist's Toolkit: Essential Research Reagents
Successful viability staining relies on a set of key reagents and instruments. The table below outlines essential materials and their functions in membrane integrity assays.
Table 3: Essential Reagents and Tools for Viability Staining
| Item | Function / Principle | Example Application |
|---|---|---|
| Fixable Viability Dyes | Amine-reactive dyes that covalently bind to proteins; intensity indicates membrane damage. Preserved after fixation. | Distinguishing live/dead cells in fixed samples for flow cytometry [50]. |
| Calcein-AM | Cell-permeant substrate for intracellular esterases; conversion to fluorescent calcein indicates enzymatic activity (viability). | Paired with PI/EthD-1 in LIVE/DEAD kits for microscopy/flow cytometry [51] [52]. |
| Propidium Iodide (PI) | Membrane-impermeant nucleic acid stain; enters only cells with compromised membranes. | Standard dead cell stain for bacteria and mammalian cells in flow cytometry and microscopy [51] [49]. |
| SYTO 9 Green Stain | Cell-permeant nucleic acid stain that labels all bacteria. Used in combination with PI. | In BacLight kit, labels all bacteria; PI quenches its fluorescence in membrane-compromised cells [51]. |
| Flow Cytometer | Instrument for multi-parameter analysis of single cells in suspension based on light scatter and fluorescence. | Immunophenotyping and viability analysis of thousands of cells per second [53]. |
| Fluorescence Microscope | Instrument for visualizing fluorescence in cells and tissues, providing spatial context. | Direct observation and imaging of live/dead cell distributions in a population or biofilm [51]. |
Membrane integrity staining with LIVE/DEAD kits provides a robust and widely accessible methodology for assessing cell viability. The techniques outlined in this guide, leveraging flow cytometry and fluorescence microscopy, offer researchers powerful tools to quantify and visualize live and dead populations based on the fundamental biological principle of membrane integrity. The availability of diverse kits, with varying mechanisms and fluorescent colors, allows for flexibility in experimental design, enabling integration with other staining protocols and adaptation to specific research needs, from basic microbiological studies to advanced drug discovery programs.
The accurate determination of bacterial viability is a cornerstone of microbiology research, with critical implications for public health, drug development, and food safety. Viability extends beyond mere cellular presence to encompass functional capacity, traditionally defined by cultivability on growth media. However, the discovery of viable but non-culturable (VBNC) states has necessitated more sophisticated assessment criteria based on metabolic activity and membrane integrity. This technical guide examines three advanced methodologies—PMA-qPCR, rRNA-Targeted Flow-FISH, and Microfluidics—that address these complex viability criteria through molecular and engineering approaches. These techniques enable researchers to move beyond traditional culture methods, which often fail to detect VBNC organisms and require days to yield results [54] [55]. The integration of these advanced methods provides a multi-parameter assessment of bacterial physiology, offering unprecedented resolution for differentiating live from dead cells and characterizing microbial function at the single-cell level.
PMA-qPCR for Viability Assessment Based on Membrane Integrity
Principle and Mechanism
Propidium Monoazide quantitative PCR (PMA-qPCR) is a molecular technique that selectively amplifies DNA from viable cells with intact membranes while excluding DNA from membrane-compromised cells. The method exploits the fundamental physiological difference between live and dead cells: membrane integrity. PMA, a photo-activatable DNA-intercalating dye, penetrates only cells with damaged membranes. Upon exposure to bright light, PMA covalently cross-links to the DNA, forming modifications that inhibit subsequent PCR amplification. Consequently, DNA from viable cells with intact membranes remains unmodified and is exclusively amplified and quantified during the qPCR process [56] [57]. This elegant mechanism allows researchers to overcome a significant limitation of conventional qPCR, which cannot distinguish between DNA from live and dead cells, potentially leading to substantial overestimation of viable populations in samples containing substantial numbers of inactivated organisms [54].
Experimental Protocol and Workflow
The following diagram illustrates the standardized workflow for the PMA-qPCR method:
Critical Steps and Optimization Parameters:
- Sample Preparation: Homogenize samples thoroughly to ensure representative sampling and eliminate cell clumps that might impede PMA penetration.
- PMA Concentration Optimization: Titrate PMA concentrations (typically 1-100 µM) to determine the optimal level that completely suppresses DNA amplification from heat-killed control cells without affecting signals from viable cells [56] [57].
- Photoactivation Conditions: Ensure uniform and adequate light exposure using dedicated PMA-Lite devices or high-intensity halogen lamps. Inadequate light exposure results in incomplete DNA cross-linking and false positives.
- Primer Specificity Validation: Design primers targeting single-copy core genes identified through whole-genome sequence analysis. For Lacticaseibacillus paracasei, the Alkaline shock protein 23 gene has been successfully utilized. Validate primer inclusivity against all target strains and exclusivity against non-target species [56].
- Standard Curve Construction: Generate standard curves using serial dilutions of viable target cells (e.g., 10³ to 10⁸ CFU/mL) to establish correlation between quantification cycle (Cq) values and cell numbers [56].
Applications and Performance Data
PMA-qPCR has been rigorously validated for quantifying viable bacteria in complex matrices. The table below summarizes performance characteristics from recent studies:
Table 1: PMA-qPCR Performance Metrics for Viable Bacterial Quantification
| Species/Matrix | Linear Range (CFU/mL) | Limit of Quantification | Accuracy (Bias) | Key Target Gene | Reference |
|---|---|---|---|---|---|
| Lacticaseibacillus paracasei (Probiotics) | 10³ - 10⁸ | 7.30 × 10³ CFU/mL | ±0.5 Log₁₀ units | Alkaline shock protein 23 | [56] |
| Multispecies Oral Biofilm (after CHX treatment) | N/A | 5-50 GE/reaction (species-dependent) | 1-1.6 log₁₀ reduction vs culture | 16S rDNA / single-copy genes | [57] |
| Streptococcus mutans (Oral Biofilm) | 6 log₁₀ concentrations | 5 GE/reaction | Correlated with CFU counts | 16S rDNA (multiple copies) | [57] |
PMA-qPCR demonstrates particular utility in scenarios requiring rapid assessment of treatment efficacy. When applied to five-species oral biofilms treated with 0.2% chlorhexidine (CHX), PMA-qPCR showed 1 to 1.6 log₁₀ reduction in PCR counts, closely matching traditional CFU counts for total bacteria and most individual species [57]. However, performance limitations emerge with certain disinfectants like sodium hypochlorite (NaOCl), which directly damages DNA and inhibits PCR amplification, complicating result interpretation [57].
rRNA-Targeted Flow-FISH for Simultaneous Identification and Viability Assessment
Principle and Mechanism
rRNA-Targeted Flow-FISH (Fluorescence In Situ Hybridization coupled with Flow Cytometry) combines molecular hybridization with single-cell analysis to detect and quantify microorganisms based on ribosomal RNA content. The technique employs fluorescently labeled oligonucleotide probes complementary to species-specific rRNA sequences. Following hybridization, flow cytometry enables rapid quantification and characterization of individual cells based on their fluorescent signals [58] [59]. Since ribosomal RNA is abundant in metabolically active cells (typically 10³-10⁴ copies per cell) and degrades rapidly upon cell death, rRNA detection serves as a reliable indicator of metabolic activity and cellular integrity [58]. This correlation between rRNA content and metabolic status makes Flow-FISH particularly valuable for identifying viable microorganisms without culturing.
Technical Approaches and Probe Design
Three principal Flow-FISH assay formats have been developed for microbiological applications:
Table 2: Comparison of Flow-FISH Technical Approaches
| Assay Type | Probe Characteristics | Advantages | Limitations | Applications |
|---|---|---|---|---|
| Single-Probe FISH | LNA or PNA probes (15-25 bp); directly labeled | High specificity; stable hybridization; simplified protocol | Lower signal intensity; limited multiplexing capability | Telomere length assessment; high-abundance target detection [58] |
| Multiple-Probe FISH | 20-nt DNA oligos (multiple per target); custom-designed | Signal amplification via multiple labeling; compatible with protein staining | More complex probe design; potential for non-specific binding | Microbial gene expression; multiplexed detection [58] |
| Branched Signal Amplification | DNA probes with overhangs; sequential hybridization | Exceptional sensitivity (>100x signal amplification); low-abundance target detection | Time-consuming; higher cost; complex optimization | Detection of low-copy RNA targets; microRNA analysis [58] |
Probe Design Considerations:
- Target Selection: Identify hypervariable regions of 16S or 23S rRNA that provide sufficient sequence divergence for species-specific detection.
- Specificity Validation: Perform in silico analysis against rRNA databases to ensure probe binding is exclusive to target organisms.
- Accessibility Mapping: Consider rRNA secondary structure, as some target regions may be structurally obscured and require optimized hybridization conditions.
- Fluorophore Selection: Choose fluorophores compatible with flow cytometer laser lines and emission filters, considering potential autofluorescence in certain sample types.
Experimental Protocol and Applications
The standard Flow-FISH protocol involves these critical stages:
Flow-FISH has been successfully applied to diverse microbiological scenarios:
- Pathogen Detection: Identification and quantification of intracellular pathogens like Mycobacterium species and Legionella in clinical and environmental samples [58].
- Mixed Culture Analysis: Simultaneous detection of multiple bacterial species (Escherichia coli, Clostridium species) and fungal strains (Candida albicans, Saccharomyces carlsbergensis) in complex microbiota without cultivation [58].
- Physiological Status Assessment: Correlation of rRNA content with metabolic activity and antibiotic susceptibility in bacterial populations [58] [59].
- Live Cell Sorting: Isolation of specific microbial populations based on gene expression patterns for downstream culture or omics analysis, enabling studies on bacterial virulence, antibiotic resistance, and metabolite utilization [58].
Microfluidic Systems for Integrated Molecular Analysis
Principles and Advantages
Microfluidic technology miniaturizes and integrates laboratory processes onto compact chips, typically handling small fluid volumes (nanoliter to microliter range). These systems leverage unique physical phenomena at the microscale, including laminar flow, rapid heat transfer, and short diffusion lengths, to achieve faster reaction times and higher analytical sensitivity [60] [61]. For microbiological applications, microfluidic devices enable unprecedented capabilities in single-cell analysis, rapid pathogen detection, and automated processing of complex samples. The key advantages include minimal reagent consumption, high surface-to-volume ratios enhancing reaction efficiency, portability for field-deployable diagnostics, and potential for full integration of sample preparation, amplification, and detection modules [60].
Microfluidic Platform Configurations
Multiple microfluidic architectures have been developed for molecular detection of microorganisms:
Table 3: Microfluidic Platforms for Microbial Detection
| Platform Type | Working Principle | Key Features | Applications | References |
|---|---|---|---|---|
| Continuous-Flow PCR Microdevices | Sample flows through fixed temperature zones in serpentine channels | Rapid thermal cycling; 30-60 minute analysis time; high throughput | Detection of RNA viruses (Ebola, Zika); bacterial pathogen identification | [60] [61] |
| Droplet-Based Microfluidics | Samples partitioned into thousands of nanoliter droplets | Digital quantification; single-cell analysis; reduced cross-contamination | Absolute quantification of pathogens; digital PCR; single-cell microbiology | [60] |
| Loop-Mediated Isothermal Amplification (LAMP) Chips | Isothermal amplification in microchambers | Simplified temperature control; compatibility with point-of-care applications | Waterborne pathogen detection; field-deployable environmental monitoring | [60] [55] |
Implementation for Microbial Detection
Continuous-Flow qRT-PCR System for RNA Virus Detection: This system incorporates a disposable microfluidic chip with a long microfluidic channel that directs the PCR solution through areas heated to different temperatures. The sample first passes through a reverse transcription zone (for RNA-to-cDNA conversion), then through a thermal cycling area for amplification, with real-time fluorescence monitoring throughout the process [61]. This design has demonstrated detection of RNA viruses like Ebola, with potential application for other viral pathogens including Zika and chikungunya viruses [61].
Integrated Sample-to-Answer Systems: Advanced microfluidic platforms aim to combine sample preparation, nucleic acid extraction, amplification, and detection in a single automated device. These systems are particularly valuable for applications requiring rapid results in field settings, such as water quality monitoring [55] or point-of-care clinical diagnostics [60]. The implementation of isothermal amplification methods like LAMP in microfluidic formats further simplifies instrumentation requirements while maintaining high sensitivity and specificity for pathogen detection [55].
Research Reagent Solutions and Essential Materials
Table 4: Essential Research Reagents for Advanced Microbial Viability Assessment
| Reagent/Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Viability Markers | Propidium Monoazide (PMA); Ethidium Monoazide (EMA) | Selective DNA modification in membrane-compromised cells | PMA exhibits better membrane selectivity than EMA; optimize concentration for each bacterial species [56] [57] |
| Nucleic Acid Probes | LNA/DNA mixmers; PNA probes; DNA oligos with branched amplification | Target-specific hybridization for detection | LNA/PNA probes offer higher affinity and specificity; branched systems provide superior sensitivity [58] |
| Polymerase Enzymes | Taq DNA polymerase; Bst polymerase for LAMP | DNA amplification in PCR and isothermal methods | Bst polymerase enables isothermal amplification but has temperature sensitivity [60] |
| Microfluidic Chip Materials | PDMS; PMMA; Cyclic Olefin Copolymer (COC) | Device fabrication for microfluidic applications | COC offers excellent optical properties and low autofluorescence for detection [60] |
| Sample Preparation Reagents | DNA extraction kits; inhibitor removal resins; lysis buffers | Nucleic acid isolation and purification from complex matrices | Compatibility with downstream molecular applications is critical; inhibitor removal essential for environmental samples [55] [56] |
| qPCR Reagents | Species-specific TaqMan probes/primers; SYBR Green master mixes | Target amplification and detection | TaqMan assays offer superior specificity; validate primers for inclusivity/exclusivity [56] [57] |
Comparative Analysis and Implementation Guidance
Method Selection Criteria
Each advanced method offers distinct advantages for specific research scenarios:
PMA-qPCR is ideally suited for:
- Quantitative assessment of viable target organisms in complex mixtures (probiotics, biofilms)
- Applications requiring correlation with traditional CFU counts
- Scenarios where membrane integrity is the primary viability criterion
- High-throughput quality control in industrial settings [56]
rRNA-Targeted Flow-FISH excels in:
- Single-cell analysis of metabolic activity and gene expression
- Detection and characterization of unculturable microorganisms
- Multispecies communities analysis without cultivation bias
- Applications requiring physical sorting of viable subpopulations [58]
Microfluidic Systems provide optimal solutions for:
- Rapid, point-of-care pathogen detection
- Applications with limited sample volumes
- Automated, integrated sample-to-answer workflows
- Digital quantification at single-molecule resolution [60] [55]
Technical Integration and Future Perspectives
The convergence of these methodologies represents the future of microbial viability assessment. Integrated systems combining microfluidic sample processing with PMA-qPCR or Flow-FISH detection are emerging as powerful tools for comprehensive microbial characterization [60]. Similarly, the combination of viability staining with advanced molecular detection in lab-on-a-chip formats enables unprecedented resolution in analyzing complex microbial communities. As these technologies continue to evolve, they will increasingly address current limitations in sensitivity, multiplexing capability, and field-deployment robustness, ultimately providing researchers and drug development professionals with increasingly sophisticated tools for elucidating bacterial viability, function, and pathogenicity.
The accurate assessment of bacterial viability is a cornerstone of microbiology, with critical implications across clinical diagnostics, pharmaceutical development, and public health. Traditional viability criteria—culturability, metabolic activity, and membrane integrity—have long served as the foundation for microbial analysis [2]. However, these conventional methods often face significant limitations, including the inability to detect viable but non-culturable (VBNC) bacteria, lengthy processing times, and inherent biases that restrict their scalability and accuracy [2] [62].
The emergence of artificial intelligence (AI) and machine learning (ML) is fundamentally transforming this landscape. By leveraging advanced computational power, these technologies can extract subtle, complex patterns from multifaceted biological data that often elude conventional analysis. This technical guide explores the pioneering role of AI and ML in developing next-generation bacterial viability prediction methods, focusing on their operational frameworks, performance metrics, and practical implementation within modern research environments.
AI-Driven Methodologies for Viability Assessment
Nanopore Sequencing and Deep Learning
A groundbreaking approach utilizes nanopore sequencing technology combined with deep neural networks to assess microbial viability directly from raw electrical signal data (squiggles) [63]. This fully computational framework bypasses the need for DNA basecalling and capitalizes on the hypothesis that DNA from dead microorganisms accumulates detectable squiggle signatures due to factors like external damage or the lack of functional DNA repair mechanisms.
- Experimental Protocol: The model was trained on controlled data from viable and UV-killed Escherichia coli cultures. Native DNA was sequenced using nanopore technology, and the raw signal data was used to train the deep learning model. The key differentiator is the use of raw nanopore signals rather than basecalled nucleotide sequences [63].
- AI Framework: A deep neural network architecture was designed to learn the distinguishing features between viable and non-viable cells directly from the squiggle data. The model's decision-making process was then interpreted using explainable AI (XAI) tools, which pinpoint the specific signal patterns most predictive of viability [63].
- Validation and Applications: The model demonstrated high prediction accuracy in a real-world application estimating the viability of obligate intracellular Chlamydia suis, indicating its ability to generalize across taxonomic boundaries. However, the study also highlighted a limitation: when a different killing method (antibiotic exposure) was used, a new, method-specific model had to be trained for accurate predictions, underscoring that model generalizability can be context-dependent [63].
Generative AI for Antimicrobial Discovery
While not a direct viability assay, generative artificial intelligence represents a powerful adjacent technology for designing novel antimicrobial peptides (AMPs) that target viable, drug-resistant bacteria [64]. This approach addresses the viability criterion by creating compounds effective against persistent, viable pathogens.
- Model Architecture (ProteoGPT): Researchers developed a protein large language model (LLM) pre-trained on the manually curated Swiss-Prot database. This foundational model, comprising over 124 million parameters, was further refined through transfer learning into specialized sub-models for specific tasks [64]:
- AMPSorter: A classifier that identifies potential AMPs from non-AMPs with high precision (AUC=0.99).
- BioToxiPept: A classifier that predicts peptide cytotoxicity to minimize toxic risks.
- AMPGenix: A generator that creates novel, theoretically viable AMP sequences.
- Experimental Workflow: The generative pipeline involved high-throughput screening of hundreds of millions of peptide sequences. The top candidates were validated in vitro and in vivo against clinical superbugs like carbapenem-resistant Acinetobacter baumannii (CRAB) and methicillin-resistant Staphylococcus aureus (MRSA). The generated AMPs exhibited mechanisms of action involving membrane disruption and depolarization, directly affecting bacterial viability [64].
Machine learning models can predict fundamental physiological traits like optimal growth temperature (OGT) from genomic data, which is intrinsically linked to viability and cultivation potential [65].
- Methodology: A Random Forest model was trained on a dataset of 1,498 bacterial genomes. The input features were protein domain frequencies (derived from Pfam annotations) for each genome. The model learned the complex relationships between the presence and abundance of specific protein domains and the organism's OGT [65].
- Performance: The model achieved a high coefficient of determination (R² = 0.853) on test data, with 82.4% of predictions falling within a ±10°C error margin of the actual OGT. This provides a practical tool to guide cultivation strategies for previously "unculturable" microbes by informing appropriate growth conditions [65].
- Biological Insight: Analysis of the model identified key protein domains signaturely associated with thermal adaptation. For example, domains related to polyamine metabolism, tRNA methyltransferases, and CRISPR-Cas systems were enriched in thermophiles, while domains involved in redox homeostasis and transport were more abundant in psychrophiles [65].
Quantitative Performance of Featured AI Models
Table 1: Performance metrics of key AI models for predicting bacterial viability and related traits.
| Model/AI Approach | Primary Function | Input Data Type | Key Performance Metrics |
|---|---|---|---|
| Nanopore Deep Learning [63] | Viability classification (live/dead) | Raw nanopore signal data | High accuracy in controlled settings; applicable across taxa (Chlamydia) |
| ProteoGPT/AMPSorter [64] | AMP identification & generation | Protein sequences | AUC=0.99; AUPRC=0.99; >90% precision on stringent benchmark |
| Random Forest for OGT [65] | Prediction of Optimal Growth Temperature | Protein domain frequencies | R²=0.853; 82.4% of predictions within ±10°C error |
| Geometric Viability Assay (GVA) [6] | High-throughput viability counting | Colony distribution in a cone | Correlation with CFU: Pearson r=0.98; 30x time reduction vs. standard CFU |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key reagents, technologies, and their functions in AI-enhanced viability research.
| Reagent / Technology | Function in Research | Example Application / Note |
|---|---|---|
| Nanopore Sequencer | Generates raw electrical signal data (squiggles) from DNA/RNA strands. | Enables real-time sequencing; raw data used for viability inference [63]. |
| Triphenyl Tetrazolium Chloride (TTC) | Colorimetric redox indicator; reduces to red formazan in metabolically active cells. | Used in GVA to enhance colony contrast within agarose [6]. |
| Protein Domains Database (Pfam) | Curated database of protein families and domains. | Used as feature set for ML models predicting physiological traits like OGT [65]. |
| Generative AI Models (LLMs) | In silico generation and screening of novel peptide sequences. | Accelerates discovery of antimicrobial peptides (AMPs) [64]. |
| Low-Melt Agarose | Used to create solid growth medium that solidifies at low temperatures. | Essential for embedding cells in GVA without heat stress [6]. |
Workflow and Architecture Diagrams
The integration of AI and machine learning into microbial viability science marks a profound shift from traditional, labor-intensive methods toward high-throughput, computational, and often more insightful predictive frameworks. Technologies like nanopore signal analysis, generative AI for antimicrobial discovery, and genome-based trait prediction are not merely incremental improvements but represent fundamentally new paradigms for assessing and exploiting microbial viability.
These emerging technologies demonstrate a common strength: the ability to decipher complex, high-dimensional biological data that is intractable to manual analysis. While challenges remain—such as ensuring model generalizability across diverse conditions and integrating these tools into standardized clinical workflows—the trajectory is clear. AI-powered viability assessment is poised to accelerate drug discovery, enhance diagnostic precision, and deepen our fundamental understanding of microbial life, ultimately strengthening the global response to the mounting challenge of antimicrobial resistance.
Solving Common Pitfalls and Enhancing Viability in Research and Formulations
The Viable but Non-Culturable (VBNC) state represents a significant survival strategy adopted by numerous bacterial species when confronted with sub-lethal environmental stresses. In this physiological state, cells maintain metabolic activity and viability but lose the ability to form colonies on conventional growth media, the very foundation of standard microbiological detection [66]. This fundamental discrepancy creates a critical blind spot in risk assessment across clinical, food safety, and pharmaceutical fields, as pathogens in the VBNC state can evade routine culture-based detection while retaining virulence potential and the capacity to resuscitate when conditions improve [2] [67]. Overcoming the VBNC challenge requires a paradigm shift from relying solely on culturability to an integrated approach that incorporates multiple viability criteria: culturability, metabolic activity, and membrane integrity [2].
This guide provides researchers and drug development professionals with advanced strategies and detailed methodologies for the accurate detection of VBNC cells, their resuscitation, and the precise quantification of viable bacterial populations, thereby enabling a more realistic assessment of microbial risk and therapeutic efficacy.
VBNC State Induction and Underlying Molecular Mechanisms
The VBNC state is a survival response triggered by various environmental stresses. Understanding these triggers is essential for predicting and managing VBNC formation.
Common Inducers of the VBNC State
- Physical Stresses: Temperature extremes (particularly cold shock), high hydrostatic pressure, and UV irradiation [66] [67].
- Chemical Stresses: Exposure to reactive oxygen species (ROS), bile salts, heavy metals, and antibiotics [66] [67].
- Nutritional Stresses: Starvation conditions, such as incubation in artificial seawater or other nutrient-poor environments [67].
- Food & Beverage Processing: Stresses encountered during manufacturing and storage, including low pH, high osmotic pressure, and hop acids in beer [66].
Hallmarks of the VBNC Cell
Upon entering the VBNC state, bacterial cells undergo significant physiological and morphological transformations [66]:
- Metabolic Downshift: A severe reduction in metabolic activity and cessation of cell division.
- Morphological Changes: Conversion from a typical rod shape to a smaller, spherical coccoid form.
- Biochemical Adaptations: Increased envelope rigidity due to changes in cell wall and membrane composition, and retention of basal metabolism, rRNA, and ATP pools.
- Genetic Reprogramming: Transcriptional shifts that favor stress tolerance genes and suppress replication-associated pathways.
Advanced Viability Assessment: A Three-Pillar Framework
Accurate viability assessment must move beyond culturability to integrate three established criteria. The following table summarizes the core principles, key methods, and limitations of each approach.
Table 1: Core Criteria for Bacterial Viability Assessment
| Viability Criterion | Underlying Principle | Key Methods | Primary Limitations |
|---|---|---|---|
| Culturability | Ability of a cell to divide and form a visible colony on standard media [2]. | • Plate counts (CFU) [2] | Fails to detect VBNC cells; time-consuming (2-7 days) [2]. |
| Metabolic Activity | Presence of active enzyme systems or substrate uptake [2]. | • Fluorescein diacetate (FDA) hydrolysis [2]• 2-NBDG glucose uptake assay [2]• Flow cytometry (Active Fluorescent Units, AFUs) [66] | May miss dormant cells with silenced metabolism; FDA is pH-sensitive [2]. |
| Membrane Integrity | An intact cytoplasmic membrane, a hallmark of live cells [2]. | • Propidium Monoazide (PMA) dye exclusion coupled with qPCR/dPCR [67]• Flow cytometry with live/dead stains [66] | Requires specialized equipment; PMA optimization is critical to avoid false positives [67]. |
Culture-Independent Detection and Quantification
Nucleic Acid-Based Detection (PMA-qPCR/dPCR)
This method selectively amplifies DNA only from cells with intact membranes. Propidium Monoazide (PMA) is a dye that penetrates only compromised membranes, intercalates with DNA, and forms a covalent crosslink upon light exposure, thereby inhibiting PCR amplification [67].
Detailed Protocol: PMA-dPCR for Absolute Quantification of VBNC Cells [67]
- Application: Absolute quantification of viable Klebsiella pneumoniae cells without a standard curve.
- Key Reagents: PMA (Biotium), primers/probes for single-copy genes (KP, rpoB, adhE).
- Procedure:
- PMA Treatment Optimization: Test final PMA concentrations from 5-200 μM. Incubate in the dark for 5-30 minutes, followed by a 15-minute photoactivation using a 650W halogen light source at a 20 cm distance.
- DNA Extraction: Extract genomic DNA from PMA-treated and control samples.
- Droplet Digital PCR (ddPCR): Partition the DNA sample into thousands of nanodroplets. Perform PCR amplification in each droplet.
- Endpoint Reading and Analysis: Count the positive (fluorescent) and negative droplets. Use Poisson statistics to calculate the absolute copy number per mL of the target genes, providing a direct count of viable cells.
Flow Cytometry and Viability Staining
Flow cytometry allows for the rapid counting and differentiation of cells based on viability stains, reported as Active Fluorescent Units (AFUs), providing a complementary metric to CFU [66].
Experimental Workflow:
- Staining: Stain the bacterial suspension with a combination of fluorescent dyes (e.g., SYBR Green for total cells and Propidium Iodide for dead cells).
- Analysis: Pass the cells through the flow cytometer in a single-file stream.
- Detection & Discrimination: Lasers excite the dyes, and detectors measure the emitted fluorescence, distinguishing and counting subpopulations (intact/live, compromised/dead) in real-time.
Diagram 1: Flow Cytometry Viability Workflow
Resuscitation is the process of reversing the VBNC state, defined by the recovery of colony-forming ability without population growth. True resuscitation must be distinguished from the outgrowth of a few residual culturable cells. Key evidence includes a measurable increase in CFU without a concurrent increase in total cell count (from flow cytometry) and recovery that occurs only under specific rescue conditions [66].
Table 2: Documented Resuscitation Methods for VBNC Bacteria
| Bacterial Species | VBNC Inducing Stress | Resuscitation Method | Proposed Mechanism |
|---|---|---|---|
| Lactobacillus brevis [66] | Cold storage (0°C), hop acids | Supplementing culture media with catalase (1000 IU/mL) | Scavenging of reactive oxygen species (ROS) |
| Lactobacillus plantarum [66] | Prolonged cold storage (4°C) | Supplementing recovery media with catalase | Relief from oxidative stress |
| Bifidobacterium spp. [66] | Low pH in yoghurt | Neutralization and anaerobic pre-incubation in nutrient-rich media | Removal of low-pH stress and nutrient upshift |
| Lacticaseibacillus paracasei [66] | Low temperature, acidity | Transfer to nutrient-rich media (MRS) or skim milk with yeast extract | Nutrient upshift and temperature adjustment |
| HiAlc K. pneumoniae [67] | Prolonged incubation in artificial seawater at 4°C | Resuspension in fresh, nutrient-rich YPD media and removal of antibiotic pressure | Nutrient upshift and removal of stressor |
A generalized resuscitation protocol involves the following steps:
- Stress Removal: Centrifuge the VBNC-inducing sample and resuspend the cell pellet in a fresh, nutrient-rich resuscitation medium.
- Condition Optimization:
- Temperature: Incubate at the optimal growth temperature for the target bacterium (e.g., 37°C for human pathogens).
- Nutrient Supplementation: Add specific resuscitation-promoting factors. As evidenced in Table 2, catalase (1000 IU/mL) is highly effective for lactic acid bacteria exposed to oxidative stress [66].
- Antioxidants: Include sodium pyruvate or other antioxidants in the medium to mitigate ROS.
- Monitoring and Validation: Monitor culturability (CFU) over time. Use culture-independent methods (e.g., flow cytometry for total viable count) in parallel to confirm that the increase in CFU is due to true resuscitation and not the outgrowth of a small number of residual cells [66].
Diagram 2: VBNC Resuscitation Pathway
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for VBNC Research
| Reagent / Material | Function / Application | Example Usage & Notes |
|---|---|---|
| Propidium Monoazide (PMA) [67] | DNA-binding dye excluded by intact membranes; used to selectively detect viable cells via qPCR/dPCR. | Critical for PMA-ddPCR; optimal concentration (e.g., 5-200 μM) and incubation time must be determined for each bacterial strain. |
| Catalase [66] | Enzyme that decomposes hydrogen peroxide; used as a supplement in resuscitation media. | Effective at 1000 IU/mL for resuscitating VBNC Lactobacillus and Bifidobacterium induced by oxidative stress. |
| Single-Copy Gene Primers/Probes [67] | Target genes for precise quantification in qPCR/dPCR assays. | For K. pneumoniae, rpoB, KP (hemolysin), and adhE are reliable single-copy targets. Using multiple genes averages out potential errors. |
| Fluorescein Diacetate (FDA) [2] | Cell-permeant substrate hydrolyzed by esterases to fluorescent fluorescein in metabolically active cells. | Used for enzymatic activity assays; results are pH-sensitive, and high product concentration can cause quenching. |
| Artificial Sea Water (ASW) [67] | A defined, nutrient-poor medium for inducing the VBNC state in the laboratory. | Contains 40 g/L sea salt, sterilized by 0.22 μm filtration. Used to study starvation-induced VBNC state. |
The limitations of traditional plate counts necessitate a more sophisticated framework for assessing bacterial viability, particularly in the context of the VBNC state. The path forward lies in an integrated quantification approach that synergistically combines the strengths of culture-dependent and culture-independent methods [66]. For instance, coupling resuscitation-enhanced CFU counts with viability data from PMA-dPCR or flow cytometry provides a comprehensive and accurate picture of the true viable population [66] [67].
While methods based on metabolic activity and membrane integrity are powerful for research, it is crucial to recognize that most international regulations for product label claims (e.g., probiotic counts in foods and supplements) still mandate CFU enumeration [66]. Therefore, integrating a controlled resuscitation step prior to plating is a practical and powerful strategy to improve the accuracy of CFU counts for regulatory compliance, ensuring that VBNC cells are accounted for and that health benefit claims are substantiated by a true measure of viable dose [66].
Optimizing Cryoprotectants and Storage Conditions for Long-Term Stability
The long-term stability of biological materials, including bacterial cells, liposomal nanoparticles, and other biologics, is a cornerstone of reproducible scientific research and effective therapeutic development. Maintaining viability and functional integrity during storage is paramount, as cell death during storage is inevitable but must be minimized as much as possible [68]. The stability of these biological systems is critically dependent on two key factors: the formulation of cryoprotective agents (CPAs) and the physical storage conditions employed. Cryoprotectants function through multiple mechanisms to preserve cellular integrity during freeze-drying and frozen storage, protecting cells from damage caused by ice crystal formation, osmotic stress, and solute concentration effects [69] [70].
This technical guide provides an in-depth examination of cryoprotectant optimization and storage parameter selection within the context of bacterial viability research. For researchers and drug development professionals, we synthesize current experimental data, elucidate fundamental protection mechanisms, and provide standardized protocols for evaluating key viability metrics—culturability, metabolic activity, and membrane integrity. By integrating theoretical principles with practical applications, this resource aims to enhance reproducibility and stability in biological preservation systems.
Fundamental Mechanisms of Cryoprotection
Cryoprotectants safeguard biological materials through interconnected physical and chemical mechanisms that mitigate the damaging effects of freezing and dehydration. Understanding these mechanisms is essential for rational CPA selection and formulation optimization.
Penetrating vs. Non-Penetrating Cryoprotectants
CPAs are categorized based on their ability to cross cellular membranes. Penetrating cryoprotectants, including dimethyl sulfoxide (DMSO), glycerol, and ethylene glycol, cross cell membranes and act inside cells. They reduce the amount of free water available for ice crystal formation and lower the freezing point of water, thereby decreasing the likelihood of intracellular ice formation that can rupture membranes and organelles [70] [71]. However, these agents can exhibit toxicity at excessive concentrations and may adversely affect cell genetics, limiting their widespread use [70].
Non-penetrating cryoprotectants, primarily sugars such as sucrose, trehalose, and glucose, do not enter cells but exert protective effects in the extracellular space. These compounds form highly viscous, glassy matrices during freezing that physically restrict ice crystal growth and minimize mechanical cellular damage [69] [72]. According to the water replacement theory, sugars bind to exposed polar head groups of membrane lipids through hydrogen bonding, stabilizing membrane structure and preventing phase transitions during dehydration [69] [70].
Molecular Interactions and Hydrogen Bonding
The protective efficacy of cryoprotectants, particularly sugars, is governed by their molecular interactions with water and biological structures. Density functional theory (DFT) calculations reveal that sucrose exhibits particularly effective cryoprotective properties by forming stable hydrogen bonds with water molecules. The electron density around oxygen atoms in the hydroxyl groups of sucrose creates "hot spots" for interaction with hydrogen atoms of water, leading to the formation of short, strong hydrogen bonds with energies higher than those in pure water [70].
These interactions create a dynamic hydrate shell around cells and biomolecules, which prevents the rearrangement of water molecules into the ice crystal lattice. Cryoprotectants with multiple polar groups, such as hydroxyl (-OH) or carbonyl (-C=O) groups, create a dense network of hydrogen bonds with water, directly affecting the stability of these protective shells [70].
Table 1: Classification and Mechanisms of Common Cryoprotectants
| Cryoprotectant | Type | Common Concentrations | Primary Mechanism | Considerations |
|---|---|---|---|---|
| Glycerol | Penetrating | 5-15% (v/v) [68] | Lowers freezing point, reduces intracellular ice formation | Well-tolerated by most bacterial cells; common for bacterial stocks |
| DMSO | Penetrating | 5-15% (v/v) [68] [73] | Penetrates cells, prevents intracellular ice formation | Can be toxic at higher concentrations; may affect cell genetics |
| Sucrose | Non-penetrating | 5-12% (w/v) [69] [72] | Forms glassy matrix, water replacement, vitrification | Excellent for lyophilization; forms stable hydrogen bonds |
| Trehalose | Non-Penetrating | 10-20% (w/v) [72] | Water replacement, membrane stabilization | Naturally found in stress-tolerant organisms |
| Skim Milk | Non-Penetrating | 7-10% (w/v) [69] | Forms protective film, buffers osmotic changes | Contains proteins and sugars; complex protective action |
Optimizing Cryoprotectant Formulations
The efficacy of cryoprotectant solutions depends not only on the selection of individual components but also on their combinations, concentrations, and interactions with specific biological systems.
Cryoprotectant Synergy in Bacterial Preservation
Research demonstrates that combining penetrating and non-penetrating cryoprotectants often yields superior results compared to single-agent formulations. A study on probiotic strains from chicken gut found that a combination of 5% glucose, 5% sucrose, 7% skim milk powder, and 2% glycine provided optimal protection during lyophilization and storage. This formulation effectively reduced oxidative and gastrointestinal stress while preserving key probiotic traits, including adhesion potential, antimicrobial activity, and metabolic stability [69].
Similarly, research on Enterobacterales strains revealed significant differences in survival rates based on cryoprotectant composition after 12 months of storage at -20°C. A formulation containing 70% glycerol with nutrient supplements (peptone and yeast extract) achieved the highest survival rate (88.87%), significantly outperforming glycerol alone (44.81%) [74]. This underscores the importance of nutritional components in maintaining viability during long-term storage.
Concentration-Dependent Effects and Toxicity
Cryoprotectant efficacy follows a concentration-dependent response, with optimal ranges varying by organism and agent. A study on amphibian sperm cryopreservation demonstrated that low concentrations of permeating cryoprotectants improved membrane integrity viability, whereas high concentrations led to high toxicity, particularly with DMSO [73]. Similar concentration-dependent effects have been observed in bacterial systems, where excessive CPA concentrations can denature proteins and disrupt membrane integrity [68].
Table 2: Optimized Cryoprotectant Formulations for Different Biological Systems
| Biological System | Optimal Cryoprotectant Formulation | Storage Conditions | Reported Efficacy | Source |
|---|---|---|---|---|
| Probiotic Strains (Bacillus, Lactobacillus, Staphylococcus) | 5% glucose, 5% sucrose, 7% skim milk powder, 2% glycine [69] | -80°C for 12 months | Preserved viability, stress resistance, and probiotic functionality | Sardar et al., 2025 |
| Enterobacterales Strains | 70% glycerin, nutrient supplements (peptone, yeast extract), 8% glucose [74] | -20°C for 12 months | 88.87% survival rate | Scientific Reports, 2024 |
| Lipid Nanoparticles (LNPs) | 10-12% sucrose [72] | -80°C for 1 month | Maintained physicochemical characteristics and transfection efficiency | PMC, 2023 |
| Amphibian Sperm (D. suweonensis) | 15% DMSO + 0.6 M sucrose [73] | Vapor phase LN2 (10 cm height) | 86.5% membrane integrity viability | Animals, 2025 |
| Limbal Stem Cells | 5% propylene glycol [75] | Vapor phase liquid nitrogen for 1 week | Maintained membrane integrity, metabolism, and colony-forming efficiency | Cryobiology, 2018 |
Storage Conditions and Temperature Optimization
Temperature parameters during freezing, storage, and thawing significantly impact the long-term stability and functional integrity of preserved biological materials.
Temperature-Dependent Stability
The relationship between storage temperature and viability follows a general principle: the viable storage period increases as the storage temperature decreases [68]. However, different biological systems exhibit specific temperature optima. For bacterial cultures, storage at -80°C provides viability for 1-10 years, while freeze-dried cultures stored at ≤4°C can remain viable for 15 years or more [68].
Research on probiotic strains demonstrated that ultra-low temperature storage (-80°C) combined with optimized cryoprotectant formulations provided optimal protection, effectively preserving key probiotic traits during 12 months of storage [69]. Similarly, a study on lipid-based nanocarriers showed that storage at -80°C with sucrose cryoprotection maintained nanoparticle stability and efficacy for up to one month [72].
Cooling Rate Optimization
The cooling rate during freezing represents a critical parameter that must be carefully controlled. According to the "two-factor hypothesis" of cryoinjury, excessively slow cooling exposes cells to solution effects (high solute concentration and dehydration stress), while excessively rapid cooling causes intracellular ice formation [73]. An optimal cooling rate window exists that balances these competing damaging mechanisms.
In amphibian sperm cryopreservation, cooling rate optimization demonstrated that samples frozen at 10 cm above liquid nitrogen (slower cooling) exhibited greater viability than those frozen at 5 cm, reflecting the balance between intracellular ice formation during rapid cooling and solution effects during slow cooling [73].
Table 3: Storage Temperature Guidelines for Biological Materials
| Storage Condition | Temperature | Approximate Storage Duration | Typical Applications | Key Considerations |
|---|---|---|---|---|
| Refrigeration | 4°C | 4-6 weeks (agar plates); 3 weeks-1 year (stab cultures) [68] | Working bacterial stocks, short-term storage | Simple but limited duration; culture-specific viability loss |
| Standard Freezer | -20°C | 1-3 years [68] | Bacterial glycerol stocks, some formulations | Requires cryoprotectants; temperature fluctuations risk |
| Ultra-Low Freezer | -80°C | 1-10 years [68] | Long-term bacterial storage, lipid nanoparticles, sensitive biologics | Maximum stability for most non-freeze-dried materials |
| Cryogenic Storage | -150°C (liquid nitrogen) | 10+ years [68] | Cell lines, primary cells, sensitive microorganisms | Maximum viability preservation; higher equipment costs |
| Freeze-Dried (Refrigerated) | ≤4°C | 15 years+ [68] | Stable bacterial strains, reference materials | Long-term storage; requires specialized equipment |
Assessing Bacterial Viability and Functionality
Comprehensive evaluation of preserved bacterial samples extends beyond simple survival metrics to encompass functional properties that validate maintained biological activity.
Viability Metrics and Assessment Methods
Multiple complementary assessment methods are required to fully characterize bacterial viability and functionality post-preservation:
Membrane Integrity: Frequently assessed using fluorescent viability stains such as SYBR-14/propidium iodide assays, which distinguish between cells with intact and compromised membranes [73]. This method provides rapid quantification of structural integrity but does not confirm cellular functionality.
Culturability: Determined through standard plate counting methods, where serial dilutions of bacterial suspensions are plated on appropriate media and incubated to quantify colony-forming units (CFU) [69] [74]. This approach confirms reproductive capacity but may underestimate viability in stressed cells that enter viable-but-non-culturable states.
Metabolic Activity: Measured using assays such as alamarBlue (resazurin reduction), which quantifies cellular metabolic function through the conversion of non-fluorescent resazurin to fluorescent resorufin [75]. This method provides insight into functional viability but may be influenced by metabolic quiescence.
Functional Property Retention
For probiotic and industrial strains, preservation of functional characteristics is as important as viability maintenance. Research on lyophilized probiotic strains evaluated stress resistance under simulated gastric and intestinal conditions, adhesion potential, antimicrobial activity, and metabolic stability [69]. These functional assessments ensure that preserved strains maintain their therapeutic or industrial utility after storage.
Similarly, a study on Enterobacterales strains noted that despite high survival rates, the biochemical properties of tested strains changed after 12 months of cryopreservation, with alterations in biochemical profiles potentially related to environmental adaptation and cold stress responses [74]. This highlights the importance of evaluating functional stability beyond basic viability metrics.
Experimental Protocols for Cryopreservation Assessment
Standardized protocols ensure reproducible evaluation of cryoprotectant efficacy and storage stability. The following workflow outlines key experimental procedures for assessing bacterial cryopreservation:
Diagram 1: Experimental workflow for cryopreservation assessment. This flowchart outlines the key steps in evaluating cryoprotectant efficacy, from culture preparation through functional validation.
Protocol: Bacterial Cryopreservation and Viability Assessment
Materials:
- Late log-phase bacterial culture
- Cryoprotectant solutions (filter-sterilized)
- Centrifuge with cooling capability
- Cryogenic vials
- Controlled-rate freezer (optional)
- Water bath (37°C)
- Appropriate culture media
Procedure:
Culture Preparation and Harvest
Cryoprotectant Addition and Equilibration
Freezing and Storage
- For controlled-rate freezing: Use a programmed freezer with optimal cooling rate for specific bacterial strain [75].
- For manual freezing: Place vials at -80°C or in vapor phase of liquid nitrogen [68] [73].
- Store frozen vials at target temperatures (-20°C, -80°C, or liquid nitrogen) for stability testing.
Thawing and Recovery
- Rapidly thaw cryopreserved samples by gently agitating in a 37°C water bath for 3-5 minutes [74].
- Immediately transfer thawed suspensions to pre-warmed growth media for recovery.
- Perform serial dilutions in sterile PBS or appropriate diluent for viability assessment.
Viability and Functionality Assessment
- Culturability: Spread plate appropriate dilutions on selective or non-selective media. Incubate under optimal conditions and enumerate CFU after 18-48 hours [69] [74].
- Membrane Integrity: Use fluorescent viability stains (SYBR-14/propidium iodide) according to manufacturer protocols. Quantify using fluorescence microscopy or flow cytometry [73].
- Metabolic Activity: Incubate recovered cells with alamarBlue (10% v/v) for 6-12 hours. Measure fluorescence at 544 nm excitation/585 nm emission [75].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Reagents for Cryopreservation Research
| Reagent/Category | Specific Examples | Function in Cryopreservation | Application Notes |
|---|---|---|---|
| Penetrating CPAs | Glycerol, DMSO, Propylene Glycol, Ethylene Glycol [75] [68] | Reduce intracellular ice formation; lower freezing point | Concentration-dependent toxicity; typically 5-15% (v/v) |
| Non-Penetrating CPAs | Sucrose, Trehalose, Glucose, Skim Milk [69] [72] | Form glassy matrix; water replacement; membrane stabilization | Sucrose at 5-12% effective for lyophilization and storage |
| Nutrient Supplements | Peptone, Yeast Extract [74] | Support cellular repair and metabolism during stress | Enhance survival in combination with CPAs |
| Buffering Systems | Phosphate-Buffered Saline (PBS) [74] | Maintain pH and osmolarity during processing | Critical for pH-sensitive strains |
| Viability Assays | alamarBlue, SYBR-14/PI, Plate Count Methods [75] [73] | Quantify metabolic activity, membrane integrity, and culturability | Use multiple methods for comprehensive viability assessment |
Optimizing cryoprotectants and storage conditions for long-term biological stability requires a systematic approach that integrates cryoprotectant chemistry, temperature parameters, and appropriate viability assessment methods. The most effective preservation strategies typically combine penetrating and non-penetrating cryoprotectants at optimized concentrations, employ ultra-low temperature storage (-80°C or below), and utilize controlled freezing and thawing rates to minimize cryoinjury.
Future directions in cryopreservation research include developing strain-specific preservation protocols, leveraging computational methods like DFT to predict cryoprotectant efficacy, and exploring novel CPA combinations that minimize toxicity while maximizing stability. By applying the principles and protocols outlined in this technical guide, researchers and drug development professionals can significantly enhance the long-term stability and functional integrity of biological materials, ultimately supporting reproducible research and effective therapeutic development.
Addressing Stress-Induced Viability Loss in Probiotics and Live Biotherapeutics
The therapeutic efficacy of probiotics and Live Biotherapeutic Products (LBPs) is fundamentally contingent on the delivery of viable microorganisms to the host. The conventional definition of viability, anchored in the ability of a cell to form a colony on a culture plate, is increasingly recognized as insufficient for capturing the full spectrum of bacterial survival states, particularly after exposure to stressors. Stresses encountered during manufacturing, storage, and gastrointestinal (GI) transit can induce a viable but non-culturable (VBNC) state, where cells maintain metabolic activity and membrane integrity but fail to proliferate on standard media, leading to a significant underestimation of true viability [66]. This state represents a critical adaptive survival strategy but poses a substantial challenge for quality control and efficacy assessment.
Understanding and mitigating stress-induced viability loss is therefore not merely a technical hurdle but a prerequisite for developing reliable and effective probiotic products and LBPs. This guide provides a comprehensive technical overview for researchers and drug development professionals, focusing on the mechanisms of stress-induced viability loss, advanced methods for its detection, and robust strategies to enhance microbial resilience.
Key Stressors and Their Impact on Probiotic Viability
Probiotics face a cascade of stressors throughout their product life cycle, from production to their site of action in the host. A detailed understanding of these stressors is the first step in developing effective mitigation strategies. The table below summarizes the primary stressors and their documented effects on probiotic cells.
Table 1: Key Stressors Affecting Probiotic Viability and Functionality
| Stress Category | Specific Stressors | Impact on Cells | Consequences for Viability & Function |
|---|---|---|---|
| Production & Processing | Freeze-drying, Spray-drying, High-Pressure Processing, Ultrasound [76] [77] | Dehydration, membrane damage, sublethal injury, metabolic attenuation [76] | Loss of culturability, entry into VBNC state, reduced acidification capacity [76] [66] |
| Storage | Temperature fluctuations, Oxygen, Time [77] | Oxidative damage, cumulative metabolic shutdown [78] | Decline in CFU counts over shelf-life, increased VBNC population [77] [66] |
| GI Transit | Low gastric pH, Bile salts, Pancreatic enzymes [79] | Acid-induced protein denaturation, membrane disruption by detergents [79] | Cell death, loss of culturability, failure to colonize or exert beneficial effects [79] |
| Oxidative Stress | Reactive Oxygen Species (ROS) in airborne droplets, during processing, or in the gut [78] | Damage to DNA, proteins, and lipids [78] | Rapid loss of culturability, as demonstrated in E. coli and various Lactobacilli [78] [66] |
The Viable But Non-Culturable (VBNC) State
The VBNC state is a pivotal concept in understanding the gap between culturability and true viability. Cells in the VBNC state exhibit metabolic activity, membrane integrity, and stress tolerance genes but cannot form colonies on the media typically used for their cultivation [77] [66]. This state can be induced by multiple stresses common in probiotic production, including oxidative stress, refrigeration, and high-pressure processing [66]. For instance, Lacticaseibacillus paracasei can enter a VBNC state under low temperature and acidity, during which gene expression shifts from replication to stress tolerance and substrate-use efficiency [66]. Critically, VBNC cells are capable of resuscitation under favorable conditions, regaining culturability and functionality, which underscores the importance of detection methods that go beyond plate counting [66].
Advanced Methodologies for Assessing Viability and Stress Tolerance
Moving beyond the Colony-Forming Unit (CFU) count requires a multiparametric approach that assesses different aspects of cellular integrity and function. The following diagram illustrates the logical relationship and workflow for integrating these advanced methods.
Culture-Independent Viability Enumeration
These methods are essential for detecting cells that are viable but not culturable (VBNC).
- Flow Cytometry (FCM): This is a powerful technique for multi-parameter analysis at the single-cell level. By using specific fluorescent dyes, FCM can distinguish subpopulations based on:
- Membrane Integrity: Staining with cell-permeant (e.g., SYTO 24) and cell-impermeant (e.g., Propidium Iodide, PI) nucleic acid dyes differentiates cells with intact (viable) from damaged (dead) membranes [76] [77].
- Metabolic Activity: Staining with carboxyfluorescein diacetate (cFDA) detects intracellular esterase activity, identifying metabolically active cells [76]. FCM can reveal how stressors like ultrasound generate heterogeneous populations of viable, culturable, and VBNC cells [76].
- Viability PCR (vPCR): This method uses DNA-intercalating dyes like propidium monoazide (PMA) or PMAxx that penetrate only cells with compromised membranes. The dye covalently cross-links to DNA upon light exposure, preventing its amplification in PCR. Consequently, qPCR or dPCR selectively amplifies DNA from cells with intact membranes, providing a culture-independent count of viable cells [80] [66].
- Flow-FISH: This technique combines FCM with fluorescence in situ hybridization (FISH), using rRNA-targeted probes. It allows for species-specific quantification and viability assessment in multi-strain products, overcoming the limitations of plating for complex blends [80].
Functional and Stress Tolerance Assays
- Simulated Gastrointestinal (GI) Transit Assay: This is a critical functional test to predict in vivo survival.
- Protocol: A standardized protocol involves suspending probiotics in a simulated gastric juice (e.g., pH 2.0-3.0 with pepsin) for a defined period (e.g., 2 hours) at 37°C, followed by exposure to a simulated intestinal fluid (e.g., with bile salts like oxgall at 0.3-1.0%) for a further period (e.g., 4 hours) [79]. Viability is assessed before, during, and after exposure using both plate counts and a culture-independent method like FCM.
- Isothermal Microcalorimetry (IMC): IMC measures the heat flow produced by metabolic processes in a culture. It can be used to monitor the growth dynamics and metabolic activity of probiotics, providing a real-time profile that correlates with viability and can detect changes induced by stress [80].
- Metabolic Profiling: Using systems like Biolog phenotype microarrays, the carbon metabolism profiles of probiotics can be characterized. This helps identify strains with robust and versatile metabolic capabilities, which may correlate with better stress tolerance and survival in diverse environments [79].
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Probiotic Viability and Stress Tolerance Research
| Reagent / Kit | Primary Function | Technical Context & Application |
|---|---|---|
| SYTO 24 & Propidium Iodide (PI) [76] | Dual staining for membrane integrity in Flow Cytometry. | SYTO 24 labels all cells; PI labels only cells with compromised membranes. Cells stained with SYTO 24 only are considered intact/viable. |
| Carboxyfluorescein diacetate (cFDA) [76] | Fluorescent probe for metabolic activity (esterase). | Converted to fluorescent carboxyfluorescein (cF) by intracellular esterases in metabolically active cells. |
| PMA/PMAxx dye [80] [66] | Viability dye for PCR-based enumeration (vPCR). | Selectively enters dead cells with damaged membranes and cross-links DNA, inhibiting its amplification in subsequent qPCR/dPCR. |
| Catalase [66] | Enzyme used in resuscitation protocols. | Added to recovery media (e.g., 1000 IU/mL) to quench oxidative stress (H₂O₂), allowing resuscitation of VBNC cells induced by cold or hop acids in Lactobacilli. |
| Cell Counting Kit-8 (CCK-8) [80] | Tetrazolium-based colorimetric assay for cell viability. | Used to optimize the concentration of viable cells in co-culture experiments, e.g., with cancer cell lines. |
| Biolog Phenotype Microarrays [79] | High-throughput metabolic profiling. | Tests the ability of a probiotic strain to utilize hundreds of different carbon sources, indicating metabolic versatility and robustness. |
Strategies for Enhancing Probiotic Stress Tolerance
Strain Selection and Pre-Adaptation
The foundational step is the selection of intrinsically robust strains. Comprehensive screening should include:
- Inherent Stress Tolerance: Evaluating strains for natural resistance to acid, bile, heat, and oxygen [79].
- Pre-Adaptation: Subjecting strains to sub-lethal levels of a specific stress (e.g., gradual acidification, mild heat shock) can induce adaptive responses, training the cells to better withstand subsequent, more severe stresses during processing and GI transit [79].
Formulation and Encapsulation Technologies
The product matrix and physical protection are crucial for maintaining viability.
- Microencapsulation: Encapsulating probiotics within biopolymer matrices (e.g., alginate, chitosan) provides a physical barrier against environmental stressors like low pH and bile salts, enhancing survival through GI transit and during storage [66].
- Protective Matrices: The inclusion of cryoprotectants (e.g., trehalose, glycerol) during freeze-drying or lyoprotectants in final formulations can significantly reduce cellular damage during dehydration and rehydration [77].
- Attenuation Technologies: Emerging techniques like controlled ultrasound treatment can be used to modulate probiotic metabolism intentionally. For example, attenuating sugar metabolism can prevent acid production in the food matrix, improving product stability without killing the cells, as demonstrated in Lacticaseibacillus casei [76].
- Resuscitation Before Enumeration: For accurate quality control, incorporating a resuscitation step prior to standard plate counting can recover VBNC cells. This involves pre-incubating samples in a nutrient-rich, stress-relieving medium (e.g., supplemented with catalase to counteract oxidative stress or at a neutral pH to recover from acid stress) before plating [66].
Addressing stress-induced viability loss in probiotics and LBPs requires a paradigm shift from a culture-based to a function-based viability concept. By integrating advanced, culture-independent enumeration methods with a deep understanding of microbial physiology and robust mitigation strategies, researchers and developers can ensure that their products deliver therapeutically relevant doses of viable and functional microorganisms. This integrated approach is key to unlocking the full clinical and commercial potential of live biotherapeutic products.
Accurately determining bacterial viability is a cornerstone of microbiological research, clinical diagnostics, and drug development. The scientific community largely relies on three established criteria for viability assessment: culturability, metabolic activity, and membrane integrity [2]. Culturability, determined by the ability of a single cell to form a colony on an appropriate solid medium, has been the historical gold standard for over a century [2]. However, a significant limitation is its inability to detect bacteria that have entered the viable but non-culturable (VBNC) state due to environmental stress [2] [1].
To overcome this limitation, researchers often turn to dye-based assays that probe the other two criteria: metabolic activity and membrane integrity. These assays utilize fluorescent or colorimetric probes that react with specific intracellular components or processes. However, a critical and often overlooked confounder in these assays is oxidative stress. Under conditions of oxidative stress, the dramatic increase in reactive oxygen species (ROS) can directly interfere with dye chemistry, leading to the generation of method-specific artifacts that compromise data validity [81] [82]. This guide details the sources of these artifacts and provides strategies for their management within the framework of bacterial viability criteria.
Bacterial Viability Criteria: A Framework for Assay Selection
A comprehensive understanding of what constitutes a "viable" bacterium is essential before selecting an assay. The following table summarizes the three core criteria, their underlying principles, and key limitations.
Table 1: Fundamental Criteria for Assessing Bacterial Viability
| Viability Criterion | Fundamental Principle | Common Detection Methods | Key Limitations |
|---|---|---|---|
| Culturability | Ability of a single cell to proliferate and form a visible colony on a solid medium [2]. | Plate counts, automated colony counters [2]. | Cannot detect VBNC cells; lengthy incubation (2-7 days) [2]. |
| Metabolic Activity | Presence of active enzyme systems or substrate conversion processes [2] [35]. | Tetrazolium reduction (CTC, INT), fluorescein diacetate (FDA) hydrolysis, substrate uptake assays [2] [35]. | Dormant cells have silenced metabolism; signal depends on metabolic rate, not just cell number [2] [35]. |
| Membrane Integrity | Physical intactness of the cell membrane, a hallmark of live cells [2]. | Membrane-impermeant nucleic acid stains (e.g., propidium iodide), ddPCR with sample pre-treatment [2] [1]. | Does not confirm reproductive capacity; can be transiently compromised [2]. |
The following diagram illustrates the logical relationship between these viability criteria and the associated detection techniques, highlighting how they converge to define a viable cell.
Oxidative Stress and Its Interference with Viability Assays
Fundamentals of Oxidative Stress
Oxidative stress results from an imbalance between the production of reactive oxygen species (ROS) and the cell's ability to detoxify them [81] [83]. Common ROS include the superoxide anion (•O₂⁻), hydrogen peroxide (H₂O₂), and the highly reactive hydroxyl radical (•OH) [83]. In bacteria, ROS can be generated as byproducts of normal aerobic respiration or through exposure to external stressors like antibiotics or environmental toxins [82]. These reactive molecules can oxidize vital cellular components like lipids, proteins, and nucleic acids [81].
Mechanisms of Assay Interference
ROS can artificially inflate or diminish signals from viability dyes through several mechanisms, leading to both false-positive and false-negative results.
- Direct Dye Oxidation: Many redox-sensitive dyes are designed to produce a fluorescent signal upon oxidation by specific cellular enzymes. However, elevated levels of general ROS can non-specifically oxidize these dyes, generating a fluorescence signal that is independent of the intended enzymatic activity or membrane potential. For instance, the reduced, non-fluorescent form of Dihydroethidium (DHE) is specifically oxidized by superoxide to a fluorescent product, but it can also react with other ROS to a lesser extent, reducing specificity [82].
- Alteration of Cellular Redox State: The intracellular environment becomes highly reducing under severe oxidative stress. This can alter the reduction potential of electron transport chains and the activity of dehydrogenases, which are the primary targets of tetrazolium salts like CTC and INT [35]. This may lead to an overestimation of metabolic activity or, conversely, to toxic effects that suppress the signal [35].
- Membrane Damage: While membrane damage is a marker of cell death, sub-lethal oxidative stress can cause transient and subtle changes in membrane permeability. This can allow dyes that are normally membrane-impermeant to leak into cells prematurely, or cause efflux of activated dyes, distorting measurements of both membrane integrity and metabolic function [81].
Managing Artifacts in Key Assay Types
Metabolic Activity Assays
These assays measure the activity of bacterial enzyme systems, such as dehydrogenases in the electron transport chain or non-specific esterases.
Tetrazolium Salts (e.g., CTC, XTT, INT): These salts are reduced by active enzymes to colored, fluorescent formazan products [35].
- Oxidative Artifacts: Superoxide and other ROS can directly reduce some tetrazolium salts abiotically. Furthermore, bacterial production of secondary metabolites with reducing capacity can also cause non-specific reduction, leading to false positives [35].
- Management Strategies:
- Include Proper Controls: Perform a blank measurement with a formaldehyde-fixed sample (e.g., 1.5-4.0% final concentration) to account for abiotic reduction [35].
- Optimize Concentration and Time: Use the minimum effective dye concentration and shortest incubation time to reduce toxicity and non-specific signal [35].
- Validate with Complementary Methods: Correlate results with a membrane integrity assay to confirm viability.
Fluorescein Diacetate (FDA) Hydrolysis: Non-fluorescent FDA passively crosses the membrane and is hydrolyzed by intracellular esterases to release fluorescent fluorescein [2].
- Oxidative Artifacts: The hydrolysis reaction produces acetic acid, which can lower the intracellular pH. This acidic environment can protonate fluorescein, enhancing its passive efflux from the cell and resulting in an underestimation of activity (false negative) [2].
- Management Strategies:
- pH Buffering: Use adequately buffered assay media to stabilize the intracellular pH.
- Short Incubation: Limit incubation time to minimize acid-induced efflux.
Membrane Integrity Assays
These assays distinguish between intact and compromised membranes using dyes that are excluded by live cells.
- Nucleic Acid Stains (e.g., Propidium Iodide): These dyes are typically impermeant to intact membranes and only stain cells with compromised envelopes.
- Oxidative Artifacts: Sub-lethal oxidative stress can cause transient pore formation or damage to efflux pumps, allowing the dye to enter cells that are still metabolically active and potentially culturable, leading to a false-positive death signal [81].
- Management Strategies:
- Combine with Metabolic Probe: Use a membrane-permeant metabolic dye (e.g., a tetrazolium salt) in a multiplexed assay to identify cells that are PI-positive but still metabolically active.
- Avoid Over-fixation: If using fixation, standardize the protocol as harsh fixatives can damage membranes.
Direct ROS Detection Assays
While used to measure oxidative stress itself, these assays are prone to cross-reactivity and require careful interpretation.
- Dihydroethidium (DHE) and MitoSOX Red: DHE is used to detect cellular superoxide, while MitoSOX is a derivative targeted to mitochondria in eukaryotic cells [82].
- Artifacts and Specificity: DHE is not absolutely specific for superoxide and can react with other ROS to produce a similar fluorescent signal, complicating quantification [82].
- Management Strategies:
- Use Specific Inhibitors and Inducers: Include controls like menadione (ROS inducer) and MnTBAP (SOD mimetic, superoxide scavenger) to validate the signal source [82].
- Advanced Detection: For eukaryotic cells, HPLC can be used to differentiate the specific superoxide product (2-hydroxyethidium) from other oxidation products [82].
Table 2: Research Reagent Solutions for Managing Oxidative Stress Artifacts
| Reagent / Assay | Primary Target | Function in Managing Artifacts | Key Considerations |
|---|---|---|---|
| Formaldehyde | N/A (Control) | Fixative for abiotic reduction control in tetrazolium assays [35]. | Use at 1.5-4.0% final concentration for blank measurement. |
| Menadione | Cellular ROS | Pharmacological inducer of superoxide generation; serves as a positive control [82]. | Validates the responsiveness of ROS-sensitive dyes like DHE. |
| MnTBAP | Superoxide | Cell-permeant superoxide dismutase (SOD) mimetic; scavenges superoxide [82]. | Used as a negative control to confirm superoxide-specific signal. |
| CellROX Reagents | General ROS | Fluorogenic dyes that become fluorescent upon oxidation; some are fixable and detergent-resistant [81]. | CellROX Green is compatible with fixation, allowing cell sorting. |
| MitoSOX Red | Mitochondrial Superoxide | Cationic derivative of DHE targeted to mitochondria in live cells [81] [82]. | Specific for mitochondrial superoxide in eukaryotic cells. |
| Image-iT Lipid Peroxidation Kit | Lipid Peroxidation | Ratiometric probe (BODIPY 581/591 C11) shifts fluorescence upon lipid oxidation [81]. | Provides a ratiometric readout, which is less susceptible to artifacts. |
Integrated Experimental Protocol for Artifact Mitigation
The following workflow provides a systematic approach for designing viability experiments that account for oxidative stress.
Step 1: Define the Viability Goal Clearly determine which aspect of viability is most relevant to the research question. Is it reproductive capacity (culturability), general metabolic health, or irreversible membrane rupture? This dictates the primary assay choice [2].
Step 2: Select Complementary Assays No single assay is perfect. Use an orthogonal approach by combining assays based on different principles. A powerful combination is a membrane integrity stain (e.g., propidium iodide) with a metabolic activity dye (e.g., a tetrazolium salt or FDA) [35]. This can help identify stressed cells with compromised membranes that retain metabolic activity.
Step 3: Design a Rigorous Control Strategy Controls are critical for interpreting results and identifying artifacts.
- For Metabolic/Tetrazolium Assays: Include a sample fixed with formaldehyde (1.5-4.0% final concentration) to measure and subtract any abiotic (non-biological) reduction of the dye [35].
- For ROS Detection Assays: Use a positive control (e.g., menadione to induce superoxide) and a negative control (e.g., MnTBAP, a superoxide scavenger) to confirm the assay's specificity and dynamic range [82].
- For All Assays: Include a vehicle control to account for any effects of the solvent used for the dyes.
Step 4: Optimize Assay Conditions Empirically
- Dye Concentration and Toxicity: Perform a dose-response curve to find the lowest concentration that gives a robust signal, as some dyes (e.g., CTC, INT) can be toxic to bacteria at high concentrations [35].
- Incubation Time: Determine the shortest incubation time that yields a measurable signal to minimize stress on the cells and the potential for artifact generation.
- Buffer Conditions: Ensure the assay medium is properly buffered to prevent pH shifts that can affect dye fluorescence and cellular efflux (critical for FDA hydrolysis) [2].
Step 5: Execute and Correlate Data Run the multiplexed assays and analyze the results. Correlate the signals from the different assays. For example, cells that are positive for metabolic activity but negative for membrane integrity may represent a stressed, sub-population. This correlated data provides a more nuanced and accurate picture of the physiological state of the bacterial population than any single assay alone.
The accurate assessment of bacterial viability is complex and must be approached with an understanding of its multidimensional nature. Dye-based assays for metabolic activity and membrane integrity are powerful tools but are highly susceptible to interference from oxidative stress, leading to method-specific artifacts. By understanding the principles of these assays and the mechanisms of interference, researchers can implement robust mitigation strategies. These include using defined viability criteria, selecting orthogonal assays, incorporating comprehensive controls, and carefully optimizing experimental conditions. Adopting this systematic and critical approach is essential for generating reliable and meaningful data in microbiology and drug development.
Protocol Optimization for Robust and Reproducible Results Across Sample Types
Protocol optimization represents a fundamental requirement in microbiological research, particularly in the critical field of bacterial viability assessment. The evaluation of bacterial viability relies on three established criteria: culturability, metabolic activity, and membrane integrity [2]. Each of these criteria offers distinct advantages and suffers from specific limitations, making method selection and optimization essential for obtaining accurate, reproducible results. The growing recognition of Viable But Non-Culturable (VBNC) bacterial states has further complicated traditional assessment methods and underscored the necessity for robust, optimized protocols [2]. VBNC bacteria maintain metabolic activity and membrane integrity while losing culturalbility on standard laboratory media, creating significant challenges for pathogen detection in clinical, food, and environmental settings.
The consequences of non-optimized protocols extend beyond academic concerns, directly impacting public health and safety. For instance, in Listeria monocytogenes detection, false positives from non-optimized culture-based methods can trigger substantial economic losses, while false negatives pose genuine threats to public health [84]. Similarly, membrane integrity tests used in water reclamation and wine filtration industries require careful optimization to prevent misinterpretation that could compromise product safety [85] [86]. This technical guide provides a comprehensive framework for optimizing bacterial viability protocols to enhance robustness, reproducibility, and translational applicability across diverse sample matrices and research objectives.
Foundational Principles: The Three Pillars of Viability Assessment
Defining Viability Criteria and Their Limitations
Culturability: This traditional gold standard assesses the ability of bacteria to proliferate and form colonies on appropriate solid media. While providing definitive evidence of viability, this method fundamentally fails to detect VBNC bacteria that have entered a dormant state due to environmental stressors such as low temperatures, nutrient deprivation, or antibiotic exposure [2]. Optimization approaches include automated colony counting systems that reduce time consumption and manual steps, though the inherent limitation regarding VBNC states remains [2].
Metabolic Activity: This criterion assesses viability through biochemical processes, including substrate uptake (e.g., fluorescent dyes, glucose) and enzymatic activity. Methods based on metabolic activity can detect VBNC bacteria but may fail when bacteria enter deeply dormant states with minimal metabolic activity [2]. Optimization challenges include pH sensitivity, dye efflux issues, and species-specific variability in substrate utilization [2].
Membrane Integrity: This approach distinguishes viable cells based on their intact membranes, which exclude certain dyes and molecules. While capable of detecting dormant cells with intact membranes, these methods can be technically complex, often require multiple processing steps, and may involve specialized equipment [2]. Recent advances in viability PCR (vPCR) using propidium monoazide (PMA) have significantly enhanced the applicability of membrane integrity assessments in complex samples [87].
Table 1: Core Viability Assessment Methods with Applications and Limitations
| Viability Criterion | Example Methods | Detects VBNC? | Key Limitations |
|---|---|---|---|
| Culturability | Plate culture, Automated colony counting | No | Cannot detect VBNC states; lengthy incubation (2-7 days) |
| Metabolic Activity | Fluorescein diacetate (FDA) hydrolysis, 2-NBDG glucose uptake, ATP assays | Yes | pH sensitive; species-specific substrate uptake; dormant cells not detected |
| Membrane Integrity | Propidium monoazide (PMMA) viability PCR, Flow cytometry with viability dyes | Yes | Multiple steps required; may need specialized equipment; optimization critical for complex samples |
The Impact of Sample Matrices on Viability Assessment
The sample matrix profoundly influences method selection and optimization requirements. Complex matrices such as whole blood, food products, or environmental samples introduce interfering substances that compromise assay sensitivity and specificity. For example, in vPCR detection of Escherichia coli in whole blood, the red color and host DNA content necessitated protocol optimization through the addition of a eukaryotic-specific lysis step prior to PMA exposure to achieve accurate quantification [87]. Similarly, wine filtration membrane integrity tests require optimization that accounts for instrumental and operational uncertainties to prevent false positives and negatives [85].
Optimization Strategies for Core Viability Methods
Enhancing Culturability-Based Detection
While culturability methods cannot detect VBNC states, they remain essential for regulatory compliance and isolation of viable pathogens. Optimization strategies focus on improving selectivity, reducing labor, and minimizing false results:
Selective Media Optimization: For Listeria monocytogenes detection, comparative studies demonstrate that chromogenic media like Brilliance Listeria Agar offer superior specificity compared to Oxford and ALOA agars, significantly reducing false positives from competing species like Listeria innocua and Enterococcus spp. [84].
Confirmation Test Selection: Biochemical confirmation tests vary in reliability. The CAMP test with S. aureus exhibits perfect specificity (100%) for L. monocytogenes identification, while the CAMP test with R. equi and L-rhamnose fermentation show higher false-positive rates (10.5% and 15.8% respectively) [84].
Automated Systems Integration: Automated colony counting systems based on image analysis algorithms can process samples in 11-21 seconds with minimal average relative error (0.2%), significantly enhancing throughput and reproducibility while reducing manual labor [2].
Advancing Metabolic Activity Assessments
Metabolic activity assays benefit from optimization approaches that address substrate specificity, detection limitations, and signal stability:
Dye-Based Assay Optimization: Fluorescein diacetate (FDA) hydrolysis assays require careful pH optimization and incubation time standardization. The acetic acid byproduct of FDA hydrolysis decreases intracellular pH, potentially quenches fluorescence signals, and affects intracellular enzyme activity [2].
Glucose Uptake Measurements: The artificial fluorescent glucose analog 2-NBDG enables direct assessment of glucose transport activity but exhibits species-specific limitations. Notably, several species including Vibrio mimicus, Bacillus cereus, and some E. coli strains cannot transport 2-NBDG, necessitating alternative assessment methods [2].
ATP Quantification Protocols: Direct ATP measurement via luminescence-based assays provides a high-throughput, cost-effective approach to metabolic profiling. Optimization includes cell normalization strategies and integration with metabolic inhibitors to dissect pathway-specific contributions to energy metabolism [88].
Table 2: Optimization Strategies for Metabolic Activity Assays
| Assay Type | Key Parameters to Optimize | Common Challenges | Optimization Approaches |
|---|---|---|---|
| FDA Hydrolysis | pH, incubation time, dye concentration | Signal quenching, pH-dependent efflux | pH buffering, concentration titration, time course studies |
| 2-NBDG Uptake | Substrate concentration, bacterial species | Species-specific transport limitations | Preliminary validation for target species, alternative substrates |
| ATP Measurement | Cell lysis efficiency, normalization method | Signal stability, cell number effects | Internal standards, optimized lysis protocols, cell counting methods |
Optimizing Membrane Integrity Approaches
Membrane integrity assessments, particularly vPCR, require meticulous optimization for reliable results in complex matrices:
PMA Exposure Protocol: Effective vPCR requires optimization of PMA concentration (typically 25 μM), incubation conditions (15 minutes with rotation at room temperature), and photoactivation parameters (20 minutes using dedicated light exposure systems) [87]. Protocol efficiency is validated by calculating ΔCt values between samples with and without PMA exposure, targeting <1 cycle difference for live cells and >4 cycles for dead cells.
Matrix Interference Mitigation: For complex samples like whole blood, adding a eukaryotic-specific lysis step prior to PMA exposure significantly improves assay performance. This involves treatment with commercial red blood cell lysis solution (15 minutes at room temperature) followed by host DNA depletion to reduce background interference [87].
Detection Limit Validation: Rigorous determination of limits of detection (LOD) using spiked samples confirms assay sensitivity. For E. coli in blood, an optimized vPCR protocol achieved an LOD of 10² CFU/mL in samples containing only live cells and equivalent sensitivity in samples containing mixed live and heat-killed cells [87].
The following workflow diagram illustrates the optimized vPCR protocol for bacterial detection in complex matrices like whole blood:
Quality Control and Validation Frameworks
Establishing Robust Quality Control Measures
Effective protocol optimization requires comprehensive quality control measures to ensure reproducibility and reliability:
Control Sample Inclusion: Every experimental run should include live cells, heat-killed cells, and sample processing controls. Heat-killing efficiency must be validated through plate counts confirming 0 CFU/mL [87].
Quantification Standards: For vPCR, establish a linear range of quantification through serial dilution studies. Optimized protocols typically demonstrate linearity from 10² to 10⁸ CFU/mL (R² > 0.997) [87].
Method Comparison Studies: Compare optimized methods with standard approaches using statistical analyses like Bland-Altman plots. For vPCR, expect consistent overestimation compared to plate counts (approximately 1.85-1.98 Log10 CFU/mL bias), establishing a reproducible correction factor [87].
Addressing False Positives and Negatives
Systematic approaches to reducing erroneous results are essential for protocol robustness:
False Positive Reduction: In culture-based L. monocytogenes detection, combining chromogenic media with specific confirmation tests (CAMP test with S. aureus) reduces false positives from 15.8% to 0% [84].
False Negative Prevention: For membrane integrity tests in industrial settings, multi-objective optimization models that explicitly incorporate instrumental and operational uncertainties can eliminate false negatives that might otherwise compromise product safety [85].
Threshold Optimization: Establish statistically derived thresholds through repeated testing of intact and compromised systems. Industrial validation of 40 intact and 40 defective filters achieved 100% classification accuracy through rigorous threshold optimization [85].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents for Viability Assessment Protocols
| Reagent/Kit | Primary Function | Application Notes |
|---|---|---|
| PMA Real-Time PCR Bacterial Viability Kit | Selective detection of live bacteria via membrane integrity | Critical for vPCR; requires optimization of concentration and light exposure; effective for Gram-negative and Gram-positive pathogens [87] |
| Chromogenic Media (e.g., Brilliance Listeria Agar) | Selective isolation and differentiation of target pathogens | Reduces false positives; provides species differentiation based on colony appearance [84] |
| Zymo HostZERO Microbial DNA Kit | Host DNA depletion for complex samples | Essential for blood and tissue samples; reduces eukaryotic background in bacterial detection assays [87] |
| Fluorescein Diacetate (FDA) | Metabolic activity assessment through enzymatic hydrolysis | pH sensitive; requires standardization; passive transport into cells [2] |
| 2-NBDG (Fluorescent Glucose Analog) | Glucose uptake measurement | Species-specific limitations; not transported by some bacterial species [2] |
| Luminescent ATP Detection Assay | Cellular energy metabolism quantification | High-throughput capability; direct energy measurement; cost-effective [88] |
Protocol optimization for bacterial viability assessment is not a luxury but a necessity for generating robust, reproducible, and clinically relevant data. The increasing recognition of complex bacterial states like VBNC cells demands methodological sophistication that transcends traditional cultural approaches. Successful optimization requires systematic attention to sample matrix effects, comprehensive validation against established standards, and implementation of rigorous quality control measures. By adopting the optimization strategies outlined in this technical guide, researchers can significantly enhance the reliability of their viability assessment data, ultimately contributing to improved public health outcomes, more effective therapeutic development, and safer food and water supplies. The future of bacterial viability assessment lies in integrated approaches that combine multiple criteria to overcome the limitations of any single method, providing a comprehensive understanding of bacterial physiology across diverse research and applied settings.
Ensuring Accuracy: A Critical Comparison of Viability Methods and Metrics
This whitepaper provides a comprehensive technical guide for establishing robust quality metrics—proportionality, variability, and linearity—in bacterial viability research. With the emerging challenges in assessing viable but nonculturable (VBNC) bacteria and the critical need for accurate viability measurements in drug development, this document outlines standardized methodological frameworks, detailed experimental protocols, and quantitative assessment criteria. Focusing on the three established pillars of bacterial viability—culturability, metabolic activity, and membrane integrity—we present a structured approach for researchers to validate their assessment techniques, ensuring reliability and reproducibility in antimicrobial efficacy testing, sterility validation, and therapeutic development.
The accurate assessment of bacterial viability is fundamental to public health microbiology, antimicrobial drug development, and infectious disease research. Viability is traditionally defined through three measurable criteria: culturability, the ability to reproduce and form colonies on media; metabolic activity, the presence of ongoing biochemical processes; and membrane integrity, the structural and functional state of the cellular envelope [89]. A significant challenge in the field is the viable but nonculturable (VBNC) state, where bacteria are metabolically active and possess an intact membrane but cannot be cultured using standard laboratory methods, leading to potential underestimation of viable populations in clinical and environmental samples [89].
Establishing quality metrics is therefore paramount for interpreting viability data correctly. Proportionality ensures that the measured signal scales directly with the number of viable cells. Variability quantifies the precision and reproducibility of the measurement technique. Linearity confirms that this proportional response is consistent across the intended dynamic range of the assay. This guide details the experimental and analytical frameworks for evaluating these metrics within the context of the core viability criteria, providing scientists with the tools to critically validate their methods and instruments.
Core Viability Criteria and Assessment Techniques
Culturability
Culturability, assessed through colony-forming unit (CFU) counts, is the historical gold standard for viability. It directly measures reproductive capacity.
- Experimental Protocol:
- Sample Preparation: Serially dilute the bacterial suspension in a suitable buffer (e.g., phosphate-buffered saline, PBS).
- Plating: Spread plate or pour plate appropriate dilutions onto nutrient-rich agar media.
- Incubation: Incubate plates at the optimal temperature for the species until colonies are visible (typically 24-48 hours).
- Enumeration: Count colonies and calculate the CFU/mL using the formula: ( CFU/mL = (\text{Number of colonies}) / (\text{Dilution factor} \times \text{Volume plated}) ).
- Inherent Limitations: This method fails to detect VBNC cells, as demonstrated in studies where metagenomic sequencing revealed a significant portion of the seed microbiome was nonculturable [90].
Metabolic Activity
Metabolic assays detect ongoing biochemical processes, such as enzyme activity or respiration, providing a broader view of viability that includes VBNC cells.
- Experimental Protocol (Example: Flow Cytometry with a Fluorescent Probe):
- Staining: Incubate the bacterial suspension with a fluorogenic substrate, such as 2',7'-Dichlorofluorescin diacetate (DCFH-DA). This cell-permeable compound is cleaved by intracellular esterases to produce a fluorescent signal upon oxidation [91].
- Incubation: Allow the reaction to proceed for a defined period (e.g., 15-30 minutes) at room temperature, protected from light.
- Analysis: Analyze the cells using a flow cytometer or fluorescence microplate reader. The fluorescence intensity is proportional to the metabolic activity.
- Advanced Applications: Techniques like metatranscriptomics can track dynamic metabolic changes by measuring microbial gene expression in real-time, offering unparalleled insight into functional viability [92].
Membrane Integrity
Assays based on membrane integrity distinguish between cells with intact and compromised cytoplasmic membranes, a clear indicator of cell death.
- Experimental Protocol (Fluorescence Staining for Microscopy/Flow Cytometry):
- Dual Staining: Prepare a staining solution containing two dyes: a membrane-impermeant nucleic acid stain like Propidium Iodide (PI) and a membrane-permeant stain like SYTO 9.
- Incubation: Add the stain mixture to the bacterial pellet, resuspend, and incubate for 15 minutes in the dark.
- Detection: Cells with intact membranes will fluoresce green (SYTO 9), while cells with compromised membranes will fluoresce red (PI). Analyze via flow cytometry or confocal microscopy [91].
- Physical Disruption Studies: Research on electrical stimulation and microwave plasma provides quantitative data on force-induced membrane disruption. For instance, electrical stimulation significantly inhibits growth by disrupting the cell membrane integrity of E. coli and S. aureus [93]. Similarly, microwave plasma treatment causes membrane impairment, leading to the leakage of intracellular proteins, lipids, and nucleic acids, which can be quantified spectrometrically [91].
Comparative Analysis of Viability Criteria
Table 1: Comparison of Core Bacterial Viability Assessment Criteria
| Criterion | What It Measures | Key Techniques | Detects VBNC? | Primary Limitation |
|---|---|---|---|---|
| Culturability | Reproductive capacity | Colony Forming Unit (CFU) counts | No | Fails to detect viable but nonculturable (VBNC) cells [89] |
| Metabolic Activity | Biochemical processes | Flow cytometry, tetrazolium salts, metatranscriptomics [92] | Yes | Dormant cells may show low activity [89] |
| Membrane Integrity | Structural state of cell envelope | Fluorescence staining (PI/SYTO 9), intracellular leakage assays [91] | Yes (VBNC cells have intact membranes) | May overestimate viability if membrane is intact but cell is irreversibly damaged |
Establishing Quality Metrics
Proportionality
Proportionality confirms that the output signal from an assay is directly proportional to the number of viable cells in the sample. This is a fundamental requirement for any quantitative method.
- Experimental Validation:
- Prepare a series of samples with known, varying concentrations of viable cells. The gold standard for the "true" count is often established via CFU for culturable populations.
- Measure each sample using the viability assay under evaluation (e.g., fluorescence intensity from a metabolic probe).
- Plot the measured signal against the reference viable count. A proportional relationship is indicated by a strong linear correlation (R² > 0.98) with a line that passes through, or near, the origin.
Variability
Variability metrics assess the precision and reproducibility of an assay, distinguishing between intra-assay (within-run) and inter-assay (between-run) variability.
- Experimental Protocol for Quantitative Analysis:
- Sample Preparation: Create a homogeneous bacterial suspension with a mid-range concentration of viable cells.
- Replication: For intra-assay variability, analyze the same sample multiple times (n ≥ 10) in a single run. For inter-assay variability, analyze aliquots of the same sample across different runs (different days, different operators; n ≥ 5).
- Calculation: Calculate the Coefficient of Variation (CV) for the results. The formula is: ( CV = (\text{Standard Deviation} / \text{Mean}) \times 100\% ).
- Acceptance Criteria: A CV of < 10% is generally considered acceptable for biological assays, with < 5% indicating excellent precision. High variability in membrane integrity assays, for example, could signal inconsistent staining or flow cytometer instability.
Linearity
Linearity evaluates whether the proportional relationship between the measured signal and the cell concentration holds across the assay's entire working range. It defines the upper and lower limits of quantitation.
- Experimental Protocol:
- Prepare a wide range of cell concentrations, spanning from below the expected limit of detection to above the expected limit of quantitation.
- Measure the signal for each concentration.
- Perform linear regression analysis on the data. The linear range is the concentration interval over which the relationship is linear (R² > 0.98) and the residuals are randomly distributed.
- Data Interpretation: Non-linearity at high concentrations can indicate signal saturation or probe depletion, while non-linearity at low concentrations may be due to background interference falling below the limit of detection.
Quantitative Metrics from Recent Research
Table 2: Exemplary Quantitative Data from Viability and Inactivation Studies
| Study Focus | Agent/Method | Target Bacteria | Key Metric & Result | Implication for Quality Metrics |
|---|---|---|---|---|
| Microbial Culturability [90] | Culture on two media types | Seed microbiome | 65-90% of abundant bacteria were culturable, vs. lower % for fungi. | Highlights inherent proportionality gap between total and culturable populations. |
| Membrane Disruption [93] | Electrical Stimulation (ES) | E. coli, S. aureus | ES application significantly inhibited growth for all strains; model predicted inhibition zones. | Provides a measurable outcome (zone diameter) for assessing variability in membrane integrity assays. |
| Membrane Disruption [91] | Microwave Plasma | S. aureus, S. abony | Achieved >88% cell death via flow cytometry; 6-log reduction in CFU in 300s. | Demonstrates strong linear correlation between membrane damage (PI uptake) and culturability loss (CFU). |
| Metabolic Prediction [94] | Machine Learning Model | L. pneumophila in macrophages | Model predicted bacterial replication with 83% accuracy based on early metabolic shifts. | Supports the proportionality between early metabolic signals and subsequent viability outcomes. |
Experimental Workflows and Signaling Pathways
The following diagrams, generated with Graphviz, illustrate the logical relationships between viability criteria and a generalized experimental workflow for metric validation.
Diagram 1: Viability criteria relationship map, highlighting the limitation of culturability.
Diagram 2: Core workflow for validating viability assessment quality metrics.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Bacterial Viability Research
| Reagent / Material | Function / Application | Example Use-Case |
|---|---|---|
| Propidium Iodide (PI) | Membrane-impermeant nucleic acid stain. Binds to DNA in cells with compromised membranes. | Flow cytometry and microscopy to quantify dead populations in membrane integrity assays [91]. |
| DCFH-DA | Cell-permeant fluorescent probe for reactive oxygen species (ROS). Measures general metabolic activity. | Detecting oxidative stress in bacteria or assessing metabolic activity in viability assays [91]. |
| SYTO 9 | Cell-permeant green fluorescent nucleic acid stain. Labels all bacteria in a population. | Used in combination with PI (e.g., in LIVE/DEAD kits) to differentiate live/dead cells via fluorescence [91]. |
| Nutrient Agar/Broth | Complex media providing nutrients for bacterial growth. | Standard culturing technique for determining colony-forming units (CFUs) as a measure of culturability [90]. |
| Phosphate Buffered Saline (PBS) | Isotonic buffer for washing cells, preparing dilutions, and resuspending samples. | Used as a suspension medium in electrical stimulation and plasma treatment studies [93] [91]. |
| 3,3′-Diethyloxacarbocyanine Iodide (DiOC₂(3)) | Fluorescent dye for monitoring membrane potential. | Assessing bacterial membrane depolarization in metabolic activity studies under stress conditions [91]. |
| Peptide Nucleic Acid (PNA) Clamps | Oligomers that block amplification of host DNA in PCR. | Used in 16S rRNA amplicon sequencing to selectively analyze bacterial DNA in host-associated samples [90]. |
The rigorous establishment of quality metrics is not merely a procedural formality but a critical component of robust and reliable bacterial viability research. By systematically evaluating proportionality, variability, and linearity across the triad of culturability, metabolic activity, and membrane integrity assays, researchers and drug development professionals can ensure their data accurately reflects the biological reality of their samples. This is especially crucial in an era defined by the challenges of VBNC bacteria, antimicrobial resistance, and the development of novel non-antibiotic therapies. The frameworks, protocols, and metrics outlined in this whitepaper provide a pathway to standardize practices, enhance data comparability across studies, and ultimately, strengthen the scientific conclusions that underpin public health and therapeutic interventions.
The quantification of viable bacteria is fundamental to microbiological research, drug development, and public health. However, the most common enumeration methods—Colony Forming Unit (CFU) assays, flow cytometry, and Polymerase Chain Reaction (PCR)—measure fundamentally different cellular properties, leading to agreements and divergences that are both predictable and context-dependent. This technical guide synthesizes current research to demonstrate that these methods converge when the targeted cellular property (culturability, metabolic activity, membrane integrity, or nucleic acid presence) is aligned with the experimental question. Conversely, they diverge significantly when bacterial populations are stressed, injured, or in a viable but non-culturable (VBNC) state. Understanding the principles and limitations of each method is critical for selecting a fit-for-purpose viability assay and for the accurate interpretation of data in research and development.
A critical challenge in microbiology is that "bacterial viability" is not a single, defined state but a complex concept assessed through different proxies. The three widespread and accepted criteria are culturability, metabolic activity, and membrane integrity [2]. Different methods target these different criteria:
- CFU Assays measure culturability—the ability of a cell to proliferate and form a colony on a solid medium.
- Flow Cytometry often measures metabolic activity (e.g., via enzymatic probes) or membrane integrity (e.g., via dye exclusion).
- PCR detects the presence of specific nucleic acid sequences, which may originate from both live and dead cells.
In an ideal, homogeneous population of healthy cells, these methods may show good agreement. However, bacteria exposed to environmental stresses, antibiotics, or food processing can enter states where these criteria are no longer aligned, most notably the viable but non-culturable (VBNC) state [2] [66]. In the VBNC state, cells retain metabolic activity and membrane integrity but lose the ability to form colonies on standard media, leading to a severe underestimation of viability by CFU counts [66]. It is this dissociation of viability criteria that primarily drives the divergence between methods.
Methodological Foundations and Measurands
A clear understanding of what each method actually measures is the first step in comparing them.
Colony Forming Units (CFU)
The CFU assay is the traditional gold standard for assessing viability. It is a functional test that quantifies the number of culturable cells capable of replication under a given set of growth conditions [2]. Its major limitation is its inability to detect VBNC cells, sublethally damaged cells with extended lag phases, or cells that require specific nutrient conditions not provided in the growth medium [2] [44].
Flow Cytometry
Flow cytometry is a high-throughput, single-cell analysis technique that uses light scattering and fluorescence to characterize cells. When used for viability assessment, it relies on fluorescent probes that target specific cellular functions. The entire measurement process, including sample staining, instrument acquisition settings, and data analysis, influences the results [95] [96]. Key approaches include:
- Membrane Integrity: Using a combination of membrane-permeant (e.g., SYTO9, SYTO family dyes) and membrane-impermeant (e.g., Propidium Iodide - PI) nucleic acid stains. Viable cells with intact membranes stain green, while dead cells with compromised membranes stain red [76] [97].
- Metabolic Activity: Using fluorogenic substrates that are converted into fluorescent products by intracellular enzymes. A common probe is carboxyfluorescein diacetate (cFDA), which is hydrolyzed to fluorescent carboxyfluorescein (cF) by intracellular esterases in metabolically active cells [76].
Polymerase Chain Reaction (PCR)
PCR amplifies specific DNA sequences to detect and sometimes quantify a target organism. Standard PCR does not distinguish between DNA from live or dead cells. Viability PCR attempts to overcome this by using sample pre-treatment with DNA-intercalating dyes like propidium monoazide (PMA) or ethidium monoazide (EMA). These dyes penetrate cells with compromised membranes and covalently bind to DNA upon light exposure, thereby inhibiting the amplification of DNA from dead cells [2].
Table 1: Fundamental Principles of Bacterial Viability Methods
| Method | Primary Measurand | Viability Criterion | Key Output |
|---|---|---|---|
| CFU | Ability to proliferate | Culturability | Colony Forming Units per mL (CFU/mL) |
| Flow Cytometry | Membrane integrity / Metabolic activity | Structural integrity / Enzymatic function | Percentage of stained cells / Active Fluorescent Units (AFU) |
| PCR | Presence of target DNA | Nucleic acid presence (Live/Dead distinction requires pre-treatment) | Gene copies / Cycle threshold (Ct) |
A Framework for Agreement and Divergence
The agreement between CFU, flow cytometry, and PCR is not random but follows predictable patterns based on the physiological state of the bacterial population. The following diagram illustrates the decision-making workflow for method selection and interpretation based on expected cell states.
Conditions Leading to Agreement
Methods will generally agree when analyzing healthy, unstressed, and log-phase bacterial cultures. In this state, cells are culturable, metabolically active, and have intact membranes. CFU counts and flow cytometry counts (targeting membrane integrity or metabolism) will show strong correlation. Standard PCR will still diverge as it cannot distinguish live from dead cells, but viability PCR (PMA-treated) may align with the other two methods.
Conditions Leading to Divergence
Divergence is the rule rather than the exception when cells are exposed to stress. The most common discrepancy is CFU counts being significantly lower than counts from flow cytometry or viability PCR, indicating the presence of a non-culturable population.
- VBNC State Induction: Stresses like nutrient starvation, extreme temperatures, oxidative stress, high pressure, and exposure to biocides can induce the VBNC state [2] [66] [98]. For example, studies on Lacticaseibacillus casei exposed to ultrasound showed that while CFU counts dropped, flow cytometry using viability stains revealed cells that were metabolically active and maintained membrane integrity but had entered the VBNC state [76].
- Sublethal Injury: Cells with sublethal damage may have compromised membranes that allow entry of small molecules but still retain the potential for repair and culturability on specially formulated media. This can cause flow cytometry (using membrane integrity dyes) to overestimate death compared to CFU on non-selective media [97].
- PCR-Specific Limitations: Even with viability dyes, PCR can diverge. The efficiency of PMA/EMA dyes can be affected by factors like the degree of membrane damage, the presence of inhibitors, and the physical clumping of cells, potentially leading to false-positive or false-negative signals [2].
Table 2: Typical Patterns of Method Agreement and Divergence Under Different Conditions
| Physiological Condition | CFU vs. Flow Cytometry | CFU vs. PCR | Flow Cytometry vs. PCR | Primary Reason for Divergence |
|---|---|---|---|---|
| Healthy, Log-Phase Cells | Good agreement | PCR > CFU | Flow Cytometry ≈ Viability-PCR* | Standard PCR detects DNA from all cells |
| VBNC State Induced | CFU < Flow Cytometry | CFU < PCR | Flow Cytometry ≈ Viability-PCR* | Loss of culturability while membrane & metabolism persist |
| Early Stage Cell Death | CFU > Flow Cytometry | CFU > Viability-PCR* | Varies | Membrane integrity lost before loss of culturability |
| Late Stage / Sterilized | All methods approach zero | PCR >> CFU | PCR >> Flow Cytometry | DNA persists long after loss of viability |
Note: *Viability-PCR refers to methods using pre-treatment with dyes like PMA.
Experimental Protocols for Comparative Studies
To systematically compare these methods, researchers can adopt standardized experimental designs. A robust approach, modified from the ISO 20391-2:2019 standard, involves testing a dilution series of a bacterial sample to assess the proportionality, linearity, and variability of each counting method [95] [96].
Protocol: Modified ISO 20391-2 Design for Microbial Cell Counting
This protocol is designed to generate quantitative quality metrics for comparing different cell counting methods across a wide concentration range [95].
Sample Preparation:
- Begin with a concentrated stock of the bacterial strain of interest (e.g., Escherichia coli NIST0056).
- Serially dilute the stock to create a dilution series spanning a log-scale range of concentrations (e.g., from ~5 x 10⁵ cells/mL to 2 x 10⁷ cells/mL). Prepare multiple sample replicates (e.g., n=3) for each dilution factor.
Blinded Analysis:
- Code the samples and provide them to operators in a blinded manner to minimize bias.
- Analyze each sample replicate using the methods to be compared (e.g., CFU, flow cytometry with appropriate stains, PCR).
Data Analysis and Quality Metrics:
- For each method and dilution level, calculate the mean and coefficient of variation (CV) of the measured concentration.
- Plot the measured concentration against the expected concentration (based on dilution factor) for each method.
- Calculate key quality metrics:
- Proportionality: The slope of the line of best fit. An ideal method has a slope of 1, indicating that measured values decrease proportionally with dilution.
- Linearity (R²): How well the data fits a linear model.
- Precision (CV): The variability of measurements across replicates.
Protocol: Integrated Viability Assessment Using Flow Cytometry and Staining
This protocol details a multi-parametric flow cytometry assay to differentiate subpopulations, including VBNC cells [76].
Cell Staining for Viability and Metabolic Activity:
- Viability/Membrane Integrity Staining: Resuspend the bacterial pellet in a buffer like PBS. Add a combination of cell-permeant (e.g., SYTO 24) and cell-impermeant (e.g., Propidium Iodide - PI) nucleic acid stains. Incubate in the dark for a standardized time (e.g., 15 minutes).
- Metabolic Activity Staining: In a separate tube, stain the cells with a fluorogenic enzyme substrate such as carboxyfluorescein diacetate (cFDA). cFDA is non-fluorescent and passively diffuses into cells. Intracellular esterases in metabolically active cells cleave the diacetate groups, producing fluorescent carboxyfluorescein (cF), which is retained by cells with intact membranes.
Flow Cytometry Acquisition:
- Analyze the stained samples using a flow cytometer. Set thresholds on forward scatter (FSC-H) and side scatter (SSC-H) to gate on the bacterial population.
- Collect fluorescence signals in the appropriate channels (e.g., FL1 for SYTO/cF, FL3 for PI).
- Use unstained controls and single-stained controls to set photomultiplier tube voltages and compensation.
Data Analysis and Gating:
- Create a dot plot of PI (y-axis) vs. cF (x-axis) or SYTO (x-axis).
- Identify distinct subpopulations:
- Viable & Culturable: PI-negative, cF-high (or SYTO-high).
- Viable but Non-Culturable (VBNC): PI-negative, cF-low (or SYTO-low; metabolically active but potentially dormant).
- Dead/Necrotic: PI-positive.
The workflow for this integrated, multi-parametric analysis is summarized below.
The Scientist's Toolkit: Key Reagents and Materials
The following table details essential reagents and their functions for the viability assessment methods discussed.
Table 3: Research Reagent Solutions for Bacterial Viability Assessment
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| SYTO9 / Propidium Iodide (PI) [97] | Dual fluorescent stain for flow cytometry to assess membrane integrity. SYTO9 stains all cells; PI stains only cells with damaged membranes. | PI has a stronger affinity for DNA than SYTO9 and will displace it. Critical to account for SYTO9 bleaching and species-specific staining differences (e.g., dead P. aeruginosa stains more intensely with SYTO9 than live cells). |
| Carboxyfluorescein diacetate (cFDA) [76] | Fluorogenic substrate used to measure esterase activity, a marker of metabolic activity, in flow cytometry. | Converted to fluorescent carboxyfluorescein (cF) by intracellular esterases. Accumulation of cF indicates metabolic activity and an intact membrane to retain the product. |
| Propidium Monoazide (PMA) [2] | DNA-intercalating dye used in viability PCR. Penetrates only dead cells with compromised membranes and covalently binds DNA upon photoactivation, preventing its amplification. | Efficiency depends on the extent of membrane damage. Optimization of dye concentration and light exposure is required for different bacterial species and sample matrices. |
| Catalase / Sodium Pyruvate [66] | Resuscitation supplements added to culture media to recover VBNC cells. Scavenge reactive oxygen species (ROS) that may prevent colony formation. | Used to confirm VBNC state and improve accuracy of CFU counts for stressed populations (e.g., in high-pressure processed L. rhamnosus or beer-spoilage Lactobacilli). |
| Defined Culture Media (e.g., MRS, TSB) | For CFU assays and cell cultivation. | The choice of medium and incubation conditions is critical. Some VBNC cells may only resuscitate on specialized media supplemented with specific resuscitation factors. |
The comparative analysis of CFU, flow cytometry, and PCR reveals that no single method provides a complete picture of bacterial viability. Their agreement is a function of the physiological homogeneity of the population under study. The following best practices are recommended for researchers and drug development professionals:
- Align Method with Application: Select a viability assay based on the biological question. For assessing infectious dose or potency, CFU may be required. For rapid process monitoring or detecting stressed populations, flow cytometry is superior.
- Adopt a Multi-Parametric Approach: Relying on a single method is insufficient for complex samples. An integrated approach, combining CFU with a culture-independent method like flow cytometry, provides a more accurate assessment of total viable populations, including VBNC cells [76] [66].
- Validate and Standardize: Employ standardized protocols, such as the modified ISO design, to generate quantitative quality metrics (proportionality, linearity, CV) for your methods in your specific context [95]. This allows for robust performance comparisons.
- Understand Method Limitations: Acknowledge that CFU can underestimate, flow cytometry requires careful interpretation of staining, and PCR (even viability-PCR) detects a molecular target that may not correlate with the ability to replicate.
In conclusion, the divergence between CFU, flow cytometry, and PCR is not a methodological failure but a valuable data point that reveals the physiological complexity of bacterial populations. By understanding the principles behind each technique, researchers can make informed decisions, select fit-for-purpose methods, and accurately interpret the nuanced story of bacterial viability.
ISO Standards and Fit-for-Purpose Validation in Pharmaceutical and Diagnostic Development
In the development of pharmaceuticals and diagnostics, ensuring the reliability and relevance of biological data is paramount. The concepts of ISO standards and "fit-for-purpose" validation represent two complementary pillars supporting this endeavor. ISO standards provide the formal, internationally recognized framework for quality management and analytical procedures, while the fit-for-purpose approach offers a flexible, scientific rationale for validating methods based on their specific intended use, particularly for complex biological analyses [99] [100]. This guide explores the integration of these paradigms, with a specific focus on their application in research involving bacterial viability—a field where defining and measuring "viability" itself requires a multi-faceted approach based on culturability, metabolic activity, and membrane integrity [2].
This document provides a technical guide for researchers, scientists, and drug development professionals, framing the discussion within the context of a broader thesis on bacterial viability research. It details the relevant ISO standards, explains the principles and implementation of fit-for-purpose validation, and provides detailed methodologies for assessing bacterial viability, complete with data presentation and essential research tools.
ISO Standards in Pharmaceutical and Diagnostic Development
The International Organization for Standardization (ISO) develops voluntary, internationally recognized standards that ensure the quality, safety, and efficacy of products and services across industries, including pharmaceuticals and medical devices [99] [101]. These standards are foundational for regulatory compliance and global market access.
Key ISO and IEC Standards
For developers of pharmaceuticals, diagnostics, and related software, several standards are particularly critical. The table below summarizes the most relevant ones.
Table 1: Key ISO and IEC Standards for Pharmaceutical and Diagnostic Development
| Standard Number | Title & Focus | Primary Application & Significance |
|---|---|---|
| ISO 9001 [99] | Quality Management Systems | Provides a framework for standardizing processes, improving operational efficiency, and ensuring quality across all operations. |
| ISO 13485:2016 [101] | Medical Devices - Quality Management Systems - Requirements for Regulatory Purposes | Mandatory for medical device manufacturers. Requires a certified Quality Management System (QMS) for tracking changes to devices and processes. |
| ISO 14971:2019 [101] | Application of Risk Management to Medical Devices | Guides the classification of medical device risks, consideration of potential harms, and implementation of risk mitigation strategies. |
| IEC 62304:2006+A1:2015 [101] | Medical Device Software - Software Life Cycle Processes | Defines processes for validating and testing software to ensure proper integration and function within medical systems. |
| ISO 17025 [99] | General Requirements for the Competence of Testing and Calibration Laboratories | Accredits testing and calibration laboratories, ensuring they perform tests reliably and accurately. |
| ISO 19344 [76] | Microbiology of the food chain — Flow cytometry for the quantification of microorganisms | Provides standardized protocols for using flow cytometry and fluorescent staining to evaluate microbial viability and metabolic activity. |
| ISO 8871-5:2025 [102] | Elastomeric parts for parenteral use and for devices for pharmaceutical use - Part 5: Functional requirements and testing | Specifies updated testing requirements for components like rubber stoppers and seals in injectable drug delivery systems. |
The ISO Certification Process
Achieving ISO certification is a multi-stage process that ensures thorough implementation and long-term adherence to the standard's requirements [99]:
- Initial Analysis: Evaluation of existing processes to identify gaps against the standard's requirements.
- Planning: Development of an action plan to address the identified gaps.
- Implementation: Adaptation of processes, personnel training, and establishment of new procedures.
- Internal Audit: Verification of compliance with the standard's requirements through self-assessment.
- Certification Audit: An independent audit conducted by an accredited certification body.
- Certification: Issuance of the ISO certificate upon successful completion of the audit.
- Continuous Monitoring: Maintenance of compliance through regular internal audits and periodic re-evaluations.
The Fit-for-Purpose Validation Framework
In biomarker and biological assay development, a rigid, one-size-fits-all approach to validation is often impractical. The "fit-for-purpose" approach has therefore been developed and accepted by regulatory bodies to provide scientific flexibility and rigor [100] [103].
Core Principles and Definition
The benchmark definition of method validation from the ISO is "the confirmation by examination and the provision of objective evidence that the particular requirements for a specific intended use are fulfilled" [100] [104]. The fit-for-purpose approach builds on this by directly linking the level of validation stringency to the assay's Context of Use (COU) [103]. The COU is a detailed description of how the biomarker data will be used in decision-making, ranging from exploratory research to pivotal safety or efficacy assessments [103]. The fundamental principle is that the validation should be appropriate for the intended use of the data and the associated regulatory requirements [103].
Assay Classification and Validation Parameters
Biomarker assays are categorized into five general classes based on their ability to quantify an analyte. The validation parameters required for each class differ, as outlined in the consensus table below.
Table 2: Fit-for-Purpose Validation Parameters by Assay Category [100] [104]
| Performance Characteristic | Definitive Quantitative | Relative Quantitative | Quasi-Quantitative | Qualitative (Categorical) |
|---|---|---|---|---|
| Accuracy / Trueness (Bias) | + | + | ||
| Precision | + | + | + | |
| Reproducibility | + | |||
| Sensitivity | + | + | + | + |
| Specificity | + | + | + | + |
| Dilution Linearity | + | + | ||
| Parallelism | + | + | ||
| Assay Range | + | + | + | |
| Lower Limit of Quantification (LLOQ) | + | + | ||
| Upper Limit of Quantification (ULOQ) | + | + |
Abbreviations: LLOQ=lower limit of quantitation; ULOQ=upper limit of quantitation.
- Definitive Quantitative: Uses fully characterized calibrators to determine absolute concentrations (e.g., mass spectrometric analysis) [100] [104].
- Relative Quantitative: Uses a response-concentration calibration with reference standards not fully representative of the biomarker (e.g., many Ligand Binding Assays - LBAs) [104].
- Quasi-Quantitative: No calibration standard; reports a continuous numerical response as a sample characteristic (e.g., quantitative RT-PCR) [104].
- Qualitative: Results are categorical, such as ordinal (discrete scoring scales) or nominal (yes/no) (e.g., immunohistochemistry) [100] [104].
The Phases of Fit-for-Purpose Validation
Validation is an iterative process that proceeds through discrete stages [100] [104]:
- Stage 1: Definition of Purpose and Assay Selection: The most critical phase, where the COU is defined, and a candidate assay is selected.
- Stage 2: Method Development and Validation Planning: Reagents are assembled, a method validation plan is written, and the final assay classification is determined.
- Stage 3: Experimental Performance Verification: The assay's performance parameters are tested experimentally, leading to the evaluation of its fitness-for-purpose.
- Stage 4: In-Study Validation: The assay's robustness is assessed in the clinical or research context, identifying real-world variables like sample collection and stability issues.
- Stage 5: Routine Use and Continuous Monitoring: The assay enters routine use, with quality control (QC) monitoring, proficiency testing, and ongoing evaluation.
Application to Bacterial Viability Research
A critical application of these principles is in the field of bacterial viability assessment. Traditional methods relying solely on culturability have significant limitations, primarily the inability to detect bacteria in the Viable But Non-Culturable (VBNC) state [2]. A comprehensive assessment requires a multi-parameter approach.
The Three Pillars of Viability Assessment
Table 3: The Three Accepted Criteria for Bacterial Viability Assessment [2]
| Criterion | Definition | Traditional Method | Key Limitation |
|---|---|---|---|
| Culturability | The ability of a bacterial cell to reproduce and form a colony on a solid growth medium. | Plate Culture / Colony Forming Units (CFU) Counting | Cannot detect VBNC cells, which are alive but cannot form colonies under standard conditions [2]. |
| Metabolic Activity | The presence of active enzymatic processes or substrate consumption within the cell. | Dyes Uptake (e.g., FDA, cFDA), Glucose Uptake Assays | Dormant cells may have silenced metabolism, leading to false negatives. Assays can be pH-sensitive [2]. |
| Membrane Integrity | The state of having an intact and functional cell membrane. | Fluorescent Staining with membrane-permeant (SYTO) and -impermeant (Propidium Iodide) dyes. | A cell with an intact membrane may still be dead or dying; does not confirm full physiological activity [2]. |
Integrated Experimental Protocol: Assessing Ultrasound-Induced Attenuation in Probiotics
The following detailed protocol, adapted from a 2025 study on Lacticaseibacillus casei, exemplifies the integration of traditional methods with advanced, standardized techniques (like flow cytometry) for a comprehensive viability assessment [76].
Objective: To comprehensively evaluate the effects of ultrasound attenuation on the viability and metabolic activity of probiotic bacteria.
Materials and Bacterial Strain:
- Strain: Lacticaseibacillus casei ATCC 393 [76].
- Growth Medium: deMan, Rogosa, and Sharpe (MRS) broth [76].
- Staining Reagents:
- SYTO 24: Cell-permeant green fluorescent nucleic acid stain, labels all cells.
- Propidium Iodide (PI): Cell-impermeant red fluorescent nucleic acid stain, labels only cells with compromised membranes.
- Carboxyfluorescein diacetate (cFDA): Non-fluorescent compound converted to green fluorescent carboxyfluorescein (cF) by intracellular esterases, indicating metabolic activity [76].
Methodology:
- Culture Preparation: Grow L. casei in MRS broth for 18 h at 37°C. Centrifuge the culture and resuspend the pellet in sterile deionized water to a final density of ~9 Log CFU/mL [76].
- Ultrasound Treatment: Subject the bacterial suspension to sonication (e.g., 20 kHz, 57 W) for varying durations (e.g., 6 and 8 minutes). Keep samples on ice to prevent heat inactivation [76].
- Multi-Parameter Analysis:
- Culturability (Plate Count): Serially dilute the sonicated and control suspensions. Spread on MRS agar plates and incubate for 24-48 hours. Count Colony Forming Units (CFU) to determine cultivable population [76].
- Flow Cytometry Analysis (Viability & Metabolic Activity):
- Viability/Membrane Integrity Assay (ISO 19344 Protocol): Stain diluted samples with both SYTO 24 and PI. Analyze with a flow cytometer. Cells staining only with SYTO are considered viable with intact membranes; cells staining with both SYTO and PI have compromised membranes [76].
- Metabolic Activity Assay (ISO 19344 Protocol): Stain diluted samples with cFDA. Analyze with a flow cytometer. Cells exhibiting fluorescence are metabolically active [76].
- Functional Assessment (Acidification): Monitor the pH change of the sonicated bacteria when introduced into a medium containing sugar over time (e.g., 24 hours) to assess metabolic functionality [76].
- Growth Recovery: Construct growth curves for sonicated and non-sonicated cultures over an extended period to assess the ability to restore normal physiology [76].
Interpretation of Results: This multi-parametric approach allows for the discrimination of different physiological subpopulations:
- Viable and Culturable: CFU+ / SYTO+ PI- / cFDA+
- Viable But Non-Culturable (VBNC): CFU- / SYTO+ PI- / cFDA+ (or cFDA±)
- Injured/Dormant: CFU- / SYTO+ PI- / cFDA-
- Dead: CFU- / SYTO+ PI+ / cFDA- [76]
The study demonstrated that 6 minutes of sonication similarly reduced all subpopulations, while 8 minutes generated a more heterogeneous population, inducing a VBNC state in a portion of the cells [76].
The Scientist's Toolkit: Key Research Reagents for Viability Assessment
Table 4: Essential Reagents for Bacterial Viability and Metabolic Activity Assays
| Research Reagent / Solution | Function / Principle | Application in Viability Assessment |
|---|---|---|
| SYTO 24 / SYTO 9 Stains [76] | Cell-permeant green fluorescent nucleic acid dyes. | Labels all bacterial cells in a sample, used to determine total cell count in conjunction with membrane-impermeant dyes. |
| Propidium Iodide (PI) [2] [76] | Cell-impermeant red fluorescent nucleic acid dye. | Penetrates only cells with damaged membranes. Used to discriminate dead cells from live ones (with intact membranes). |
| Carboxyfluorescein Diacetate (cFDA) [76] | Non-fluorescent substrate hydrolyzed by intracellular esterases to green fluorescent carboxyfluorescein (cF). | Acts as a marker for general metabolic activity. Accumulates in cells with active enzyme systems. |
| Fluorescein Diacetate (FDA) [2] | Non-polar, non-fluorescent dye hydrolyzed by nonspecific intracellular enzymes (esterases, lipases) to fluorescein. | Indicates broad metabolic activity via passive uptake and enzymatic conversion. |
| 2-NBDG [2] | Fluorescently labeled glucose analog (2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose). | Used to measure glucose uptake activity, a specific indicator of metabolic function in viable cells. |
| Physiological Solution (0.9% NaCl) [98] | Isotonic saline solution. | Used for washing and resuspending bacterial pellets without causing osmotic shock, maintaining cell integrity. |
| Phosphate Buffered Saline (PBS) [76] | A balanced salt solution maintaining a stable pH. | Used for diluting samples for flow cytometry to avoid noise interference during analysis. |
The integration of robust ISO standards with the flexible, science-driven fit-for-purpose validation framework provides a powerful approach for ensuring data quality and relevance in pharmaceutical and diagnostic development. This is particularly critical in complex biological fields like bacterial viability research, where relying on a single criterion like culturability is insufficient. By adopting a multi-parameter strategy that assesses culturability, metabolic activity, and membrane integrity, and by leveraging standardized techniques like flow cytometry, researchers can generate a comprehensive and accurate understanding of cellular physiology. This rigorous, context-aware methodology is essential for advancing drug development, validating diagnostic tools, and ultimately ensuring product safety and efficacy.
Accurately determining bacterial viability is a cornerstone of public health, clinical microbiology, and pharmaceutical development. Traditional methods, which often rely on a single criterion, can significantly underestimate viable cell numbers and misrepresent infectious risks. This is largely due to the phenomenon where bacteria enter a viable but nonculturable (VBNC) state under stress, rendering them undetectable by conventional culture-based methods yet potentially pathogenic [2] [44]. This case study advocates for a multi-parametric approach, correlating the three established viability criteria—culturability, metabolic activity, and membrane integrity—to achieve high-confidence assessments. By integrating these methods, researchers can overcome the limitations inherent to any single technique and obtain a more accurate and comprehensive understanding of bacterial physiology and survival [2] [62].
The Three Pillars of Bacterial Viability
Bacterial viability assessment is predicated on three widespread and accepted criteria, each with distinct advantages and limitations [2].
- Culturability: This traditional gold standard measures a bacterium's ability to replicate and form colonies on a solid growth medium. It implies that the cell is metabolically active, capable of reproduction, and has an intact membrane. However, its primary limitation is the inability to detect VBNC cells, which are alive and potentially virulent but have lost the capacity to form colonies on media normally supporting their growth [2] [44].
- Metabolic Activity: This criterion assesses whether a cell is physiologically active through mechanisms like the uptake and hydrolysis of fluorescent dyes (e.g., Fluorescein diacetate, FDA) or the reduction of tetrazolium salts. It can detect VBNC cells but may fail to identify dormant cells that have temporarily silenced their metabolism [2] [62].
- Membrane Integrity: This criterion differentiates between live and dead cells based on the physical status of the cell membrane. A dead bacterium typically has a disrupted or broken membrane, while a live one has an intact membrane that can exclude certain dyes. This method can detect cells that are no longer metabolically active but is considered a late-stage indicator of cell death [2] [105].
Table 1: Core Criteria for Bacterial Viability Assessment
| Viability Criterion | Underlying Principle | Key Advantage | Key Limitation |
|---|---|---|---|
| Culturability | Ability to reproduce and form visible colonies on solid media [2] | Considered the historical gold standard; allows for bacterial identification [2] | Cannot detect VBNC bacteria; time-consuming (days to weeks) [2] |
| Metabolic Activity | Presence of active enzyme systems or respiration (e.g., esterases, dehydrogenases) [2] [62] | Can detect VBNC bacteria; faster than culture methods [2] | May miss dormant cells with silenced metabolism; signal can be pH-sensitive [2] [62] |
| Membrane Integrity | Physical intactness of the cytoplasmic membrane [2] [105] | Direct physical assessment; not reliant on cellular activity [2] | A late marker of cell death; does not distinguish between healthy and stressed cells [105] |
Experimental Protocols for Multi-Parametric Assessment
To execute a correlated viability study, standardized protocols for each criterion are essential. The following methods can be performed in parallel on the same bacterial sample.
Assessing Culturability: Viable Plate Count
The viable plate count method determines the number of culturable bacteria in a sample by relying on their ability to form colonies [106].
- Serial Dilution: Aseptically prepare a serial dilution (e.g., 10-fold dilutions) of the bacterial sample in a sterile buffer or saline solution to obtain a countable number of colonies (typically 30-300) [106].
- Plating: Spread a fixed volume (e.g., 100 µL) of appropriate dilutions onto the surface of nutrient-rich agar plates.
- Incubation: Incubate the plates at the optimal temperature for the specific bacterium for a prescribed time (e.g., 24-48 hours).
- Enumeration: Count the number of colonies on plates with well-separated colonies. Calculate the colony-forming units per milliliter (CFU/mL) using the formula: CFU/mL = (Number of colonies) / (Dilution factor × Volume plated in mL) [106].
Assessing Metabolic Activity: Tetrazolium Salt Reduction Assay
Tetrazolium salts (e.g., MTT, XTT) are colorless compounds that are reduced to brightly colored formazan derivatives by metabolically active cells with a functional electron transport system [62].
- Sample Preparation: Prepare a cell suspension in an appropriate buffer. For a negative control, include a sample fixed with formaldehyde (e.g., 1.5-4.0% final concentration) to account for any abiotic reduction [62].
- Dye Incubation: Add the tetrazolium salt to the sample at a predetermined, non-toxic concentration (e.g., 0.2 - 1 mg/mL for MTT). Incubate in the dark for a specific time (e.g., 30 minutes to 4 hours) [62].
- Signal Measurement:
- For insoluble formazan products (e.g., MTT), solubilize the cells and the formed crystals with an organic solvent like ethanol or DMSO.
- Measure the absorbance of the solubilized formazan at its specific wavelength (e.g., 570 nm for MTT).
- For soluble formazan products (e.g., XTT), measure the absorbance of the supernatant directly after centrifugation [62].
- Analysis: The metabolic activity is proportional to the formazan concentration, indicated by the absorbance value.
Assessing Membrane Integrity: Live/Dead Staining with Fluorescent Dyes
This assay uses a combination of fluorescent dyes to distinguish between cells with intact and damaged membranes [105].
- Staining Solution: Prepare a working solution containing two dyes:
- SYTO 9: A membrane-permeant green fluorescent nucleic acid stain that labels all cells.
- Propidium Iodide (PI): A membrane-impermeant red fluorescent nucleic acid stain that only enters cells with compromised membranes, where it quenches the SYTO 9 fluorescence [105].
- Staining: Add the dye mixture to the bacterial pellet and incubate in the dark for 15-20 minutes.
- Analysis by Microscopy/Flow Cytometry:
- Fluorescence Microscopy: Visualize the cells under a fluorescence microscope with appropriate filter sets. Cells with intact membranes will fluoresce green, while those with damaged membranes will fluoresce red.
- Flow Cytometry: For a quantitative assessment, analyze the stained suspension with a flow cytometer. This allows for the precise enumeration of the subpopulations of live (green) and dead (red) cells [105].
Correlative Workflow and Data Interpretation
The power of a multi-criteria assessment lies in the integrated analysis of data from the parallel protocols. The following workflow and decision matrix guide this correlation.
Table 2: Interpreting Correlated Viability Data
| Culturability | Metabolic Activity | Membrane Integrity | Interpreted Physiological State |
|---|---|---|---|
| Positive | Positive | Intact | Fully Viable: Healthy, culturable, and metabolically active cells. The gold standard for viability [2]. |
| Negative | Positive | Intact | VBNC State: Live cells that are metabolically active but cannot be cultured by standard methods. A high-risk state for infectious disease [2]. |
| Negative | Negative | Intact | Dormant or Stressed: Cells may be in a deep dormant state or early in the death process where metabolism is silenced but the membrane is not yet compromised. Viability is indeterminate [2] [105]. |
| Negative | Negative | Compromised | Non-Viable: Dead cells. The loss of membrane integrity is considered a definitive indicator of cell death [2] [105]. |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Viability Assessment
| Reagent / Material | Function / Principle | Example Application |
|---|---|---|
| Nutrient Agar Plates | Solid growth medium to support bacterial replication and colony formation. | Viable plate count for culturability assessment [2] [106]. |
| Tetrazolium Salts (e.g., MTT, XTT) | Colorimetric indicators of metabolic activity. Reduced by active dehydrogenases in live cells to colored formazan [62]. | Metabolic activity assay; often used in spectrophotometric microplate assays. |
| Fluorescein Diacetate (FDA) | A non-fluorescent, lipophilic compound that crosses intact membranes. Hydrolyzed by intracellular esterases to fluorescent fluorescein in metabolically active cells [2]. | Fluorescence-based metabolic activity staining (often used with microscopy or flow cytometry). |
| SYTO 9 / Propidium Iodide (PI) | A dual fluorescent stain for membrane integrity. SYTO 9 labels all cells; PI labels only cells with damaged membranes and quenches SYTO 9 [105]. | Live/Dead BacLight staining for fluorescence microscopy or flow cytometry. |
| Spectrophotometer / Microplate Reader | Instrument to measure the optical density (OD) of bacterial cultures or the absorbance of colorimetric products. | Measuring culture growth (OD600) or quantifying formazan dye production in metabolic assays [107]. |
| Fluorescence Microscope | Microscope equipped with specific light sources and filters to excite and detect fluorescent signals from stained cells. | Visualizing and qualitatively assessing results from fluorescent viability stains (e.g., FDA, SYTO 9/PI) [105]. |
| Flow Cytometer | Instrument that counts and analyzes individual cells in a fluid stream as they pass by lasers, detecting light scattering and fluorescence. | Quantitative, high-throughput analysis of cell populations stained with fluorescent viability probes [105]. |
Relying on a single method for bacterial viability assessment, particularly one based solely on culturability, is an insufficient strategy that can lead to a significant underestimation of viable pathogens and a misunderstanding of microbial risks. This case study demonstrates that a correlative framework, which systematically integrates data from culturability, metabolic activity, and membrane integrity assays, provides a far more robust and high-confidence assessment. This multi-parametric approach is crucial for accurately detecting VBNC and dormant cells, which are invisible to traditional culture methods but critically relevant in clinical, pharmaceutical, and public health contexts. By adopting this comprehensive strategy, researchers and drug development professionals can make more informed decisions, ultimately enhancing product safety and patient outcomes.
Accurately determining bacterial viability is a cornerstone of public health, clinical diagnostics, and drug development. However, reliance on any single method can severely underestimate viable bacterial populations due to the phenomenon of viable but non-culturable (VBNC) cells and dormant states. This whitepaper outlines an integrated framework for bacterial viability assessment, arguing that a multi-parameter approach—simultaneously evaluating culturability, metabolic activity, and membrane integrity—is essential for a comprehensive profile. By synthesizing traditional and emerging methodologies, this guide provides researchers and drug development professionals with a robust, standardized strategy to overcome the limitations of singular techniques and accurately quantify bacterial viability across diverse applications.
The Critical Need for a Multi-Parameter Approach
Bacterial viability assessment is fundamental to infectious disease diagnosis, antibiotic development, food safety, and water quality monitoring. Historically, viability has been synonymous with culturability—the ability of a bacterial cell to form a colony on a solid medium. However, this century-old paradigm is insufficient, as it fails to detect bacterial subpopulations that remain alive but resist cultivation under standard laboratory conditions.
When faced with environmental stresses such as nutrient deprivation, extreme temperatures, or antibiotic exposure, many bacterial pathogens enter a Viable But Non-Culturable (VBNC) state [2] [44]. In this state, cells undergo a metabolic shutdown or dormancy, ceasing reproduction while maintaining baseline life functions and the potential to resuscitate under favorable conditions [44]. Consequently, culture-dependent methods alone can dramatically underestimate viable counts and infectious risks. For instance, live/dead staining with confocal laser scanning microscopy (CLSM) has detected large amounts of viable cells in culture-negative samples, with one study reporting 76.7% of samples being culture-negative but viability-positive [44]. This discrepancy underscores the critical need for an integrated framework that combines assessments of:
- Culturability: The ability to reproduce and form colonies.
- Metabolic Activity: The presence of ongoing biochemical processes.
- Membrane Integrity: The structural intactness of the cellular envelope.
These three criteria form the foundational pillars of a comprehensive viability assessment, enabling researchers to capture the entire spectrum of bacterial life states [2] [1].
Deconstructing the Three Pillars of Viability Assessment
Pillar 1: Culturability
Principle and Methodology Culturability, typically assessed via the Plate Culture Method, is the traditional gold standard. It measures a bacterium's capacity to reproduce and form visible colonies on appropriate solid media, implying functionality across all essential cellular processes [2] [44]. Automated systems like spiral platers and automated colony counters (e.g., Scan 500) have improved efficiency, reducing counting times to seconds with minimal error [2].
Limitations and the VBNC State The primary limitation is its inability to detect VBNC cells. Furthermore, the method is time-consuming, requiring 2-3 days for isolation and up to a week for final results, and it cannot detect sublethally damaged or fastidious uncultivable bacteria [2] [44].
Table 1: Viability Assessment Based on Culturability
| Method | Measurand | Key Advantage | Primary Limitation | Time to Result |
|---|---|---|---|---|
| Plate Culture | Ability to form colonies | Considered the historical "gold standard"; provides isolate for further study | Cannot detect VBNC or dormant cells | 2 days to 1 week |
Pillar 2: Metabolic Activity
Principle and Methodology This approach detects viable cells based on their biochemical activities, particularly the activity of intracellular enzymes or the uptake and utilization of substrates. It can detect VBNC cells that remain metabolically active [2].
- Dyes Uptake Assay: Uses fluorogenic substrates like Fluorescein diacetate (FDA). Non-fluorescent FDA passively diffuses into cells, where active intracellular esterases hydrolyze it into fluorescent fluorescein, which accumulates inside cells with intact membranes [2].
- Glucose Uptake Assay: Measures consumption of glucose or its fluorescent analog, 2-NBDG. Viable cells actively transport and metabolize these compounds. Alternative enzymatic assays measure glucose depletion by coupling glucose oxidase reactions to a colorimetric readout [2].
- Membrane Potential Indicators: Techniques like Fluorescence Lifetime Microscopy (FLIM) measure the fluorescence lifetime of membrane potential probes, which is a more robust measure of metabolic energy status than intensity-based measurements [1].
Limitations Metabolic activity is highly sensitive to environmental conditions like pH, which can affect enzyme activity and dye efflux [2]. Furthermore, dormant VBNC cells may exhibit extremely low or undetectable metabolic activity, leading to false negatives [2]. Not all bacteria can uptake artificial substrates like 2-NBDG [2].
Table 2: Viability Assessment Based on Metabolic Activity
| Method | Key Reagent/Probe | Mechanism of Action | Key Advantage | Key Limitation |
|---|---|---|---|---|
| Dyes Uptake | Fluorescein Diacetate (FDA) | Hydrolyzed by intracellular esterases to fluorescent product | Can detect some VBNC cells | Sensitive to pH; quenching effects |
| Substrate Uptake | 2-NBDG (Glucose analog) | Transported & metabolized by active cells | Probes specific metabolic pathways | Not universal; some bacteria don't uptake it |
| Membrane Potential | FLIM voltage probes | Fluorescence lifetime changes with membrane potential | Quantitative; less prone to intensity artifacts | Requires specialized instrumentation |
Pillar 3: Membrane Integrity
Principle and Methodology This strategy uses the integrity of the cell membrane as a proxy for viability, as a compromised membrane is a definitive indicator of cell death [2] [1]. It often employs fluorescent dyes that differentiate between intact and compromised membranes.
- Nucleic Acid Staining: Dyes like propidium iodide (PI) are membrane-impermeant and only enter cells with damaged membranes, binding to DNA and producing fluorescence. Conversely, membrane-permeant dyes like SYTO9 stain all cells. Used in combination, they allow for live/dead differentiation in techniques like flow cytometry or CLSM [44].
- Molecular Methods with Selective DNA Amplification: Techniques like droplet digital PCR (ddPCR) can be modified to preferentially amplify DNA only from cells with intact membranes. Samples are pre-treated with DNA-intercalating agents like propidium monoazide (PMA) that penetrate dead cells; upon light exposure, these agents cross-link to the DNA, preventing its amplification in subsequent PCR steps [1].
Limitations While highly specific for cell death, a dormant cell with an intact membrane may be classified as viable even if it is not metabolically active or culturable [2]. The methods can require multiple steps and specialized instrumentation [2].
Table 3: Viability Assessment Based on Membrane Integrity
| Method | Technology | Principle | Key Advantage | Key Limitation |
|---|---|---|---|---|
| Fluorescent Staining | Flow Cytometry, CLSM | Dyes (e.g., PI, SYTO9) distinguish intact vs. damaged membranes | High-throughput; can be quantitative | Cannot detect dormant cells with intact membranes |
| Viability-ddPCR | Droplet Digital PCR | PMA dye inhibits DNA amplification from membrane-compromised cells | Links viability to molecular detection (PCR) | Requires optimization for different species/matrices |
An Integrated Workflow for Comprehensive Viability Profiling
No single method can provide a complete picture of bacterial viability. The following workflow diagram and integrated protocol illustrate a synergistic approach to overcome this challenge.
Experimental Protocol for Integrated Assessment
This protocol is designed for a well-equipped microbiology laboratory and can be adapted for specific bacterial species.
I. Sample Preparation
- Harvest and Wash: Harvest bacterial cells from culture by centrifugation (e.g., 5,000 x g for 10 minutes). Wash the pellet twice in a sterile, non-reactivity buffer like Phosphate Buffered Saline (PBS) to remove residual media and metabolites.
- Standardize Concentration: Adjust the cell suspension to a standardized optical density (e.g., OD600 ≈ 0.1) in PBS to ensure consistent loading across different assays. For stressor studies, subdivide the suspension and apply the relevant stressor (e.g., antibiotic, heat, nutrient starvation).
II. Parallel Viability Assays Conduct the following assays in parallel from the same prepared sample.
A. Membrane Integrity via Flow Cytometry
- Staining: Combine 300 µL of bacterial suspension with 2 µL of SYTO9 stain and 2 µL of Propidium Iodide (PI) stain from a commercial live/dead kit (e.g., BacLight). Vortex gently.
- Incubation: Incubate the mixture in the dark at room temperature for 15 minutes.
- Acquisition: Analyze the sample on a flow cytometer. Use unstained and single-stained controls to set up compensation and gating.
- SYTO9+ / PI-: Population with intact membranes (viable).
- SYTO9+ / PI+: Population with compromised membranes (dead).
B. Metabolic Activity via Fluorescein Diacetate (FDA) Hydrolysis
- Dye Preparation: Prepare a stock solution of FDA in acetone (e.g., 5 mg/mL) and dilute in PBS to a working concentration (e.g., 10 µg/mL) immediately before use.
- Reaction: Mix 500 µL of bacterial suspension with 500 µL of FDA working solution.
- Incubation and Measurement: Incubate in the dark for 30-60 minutes. Transfer 200 µL to a black 96-well plate. Measure fluorescence using a microplate reader (excitation ~485 nm, emission ~535 nm). Include a negative control (buffer only) and a killed-cell control for background subtraction.
C. Culturability via Plate Count
- Serial Dilution: Perform a 10-fold serial dilution of the bacterial sample in PBS.
- Plating: Spread plate 100 µL of appropriate dilutions (e.g., 10^-4 to 10^-7) onto nutrient agar plates, in triplicate.
- Incubation and Counting: Incubate plates at the optimal temperature for the species (e.g., 37°C for E. coli) for 24-48 hours. Count colonies and calculate Colony Forming Units per mL (CFU/mL).
III. Data Integration and Analysis
- Normalize Data: Express results from all methods as cells/mL or relative percentage.
- Cross-Compare Populations:
- Culturable Population (C): From plate count.
- Metabolically Active Population (M): From FDA assay, normalized to cell count.
- Membrane-Intact Population (I): From flow cytometry.
- Infer VBNC State: The subpopulation that is membrane-intact and/or metabolically active but non-culturable can be inferred: VBNC ≈ (I ∪ M) - C. This provides a more accurate estimate of the true viable population.
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for Viability Assessment
| Reagent/Material | Function | Example Application |
|---|---|---|
| SYTO9 & Propidium Iodide | Fluorescent nucleic acid stains for distinguishing cells with intact (SYTO9+) and damaged (PI+) membranes. | Live/Dead staining for flow cytometry or confocal microscopy [44]. |
| Fluorescein Diacetate (FDA) | Fluorogenic substrate hydrolyzed by non-specific intracellular esterases in metabolically active cells. | Metabolic activity assay measured by fluorometry [2]. |
| 2-NBDG | Fluorescent analog of glucose transported by active glucose transporters. | Assessing metabolic activity via specific sugar uptake pathways [2]. |
| Propidium Monoazide (PMA) | DNA-intercalating dye that penetrates only dead cells; upon photoactivation, it cross-links DNA and inhibits PCR. | Viability-ddPCR; selective amplification from membrane-intact cells [1]. |
| Phosphate Buffered Saline (PBS) | Isotonic, non-reactivity buffer for washing and resuspending bacterial cells without inducing osmotic shock. | Sample preparation and dilution for all assays. |
| General Nutrient Agar | Solid growth medium containing essential nutrients to support bacterial growth and colony formation. | Plate count method for assessing culturability [2]. |
Validation and Quality Metrics for Integrated Counting
Implementing an integrated framework requires rigorous method validation. Research from the National Institute of Standards and Technology (NIST) demonstrates the use of quality metrics, adapted from the ISO 20391-2:2019 standard, to evaluate cell counting method performance [95]. Key metrics include:
- Proportionality: The degree to which measured values decrease in direct proportion to sample dilution. An ideal method is perfectly proportional.
- Coefficient of Variation (CV): A measure of the method's precision and variability.
- Linearity (R²): How well the dilution series data fits a linear model.
Applying this framework to E. coli samples analyzed by CFU, Coulter counting, fluorescence flow cytometry, and impedance flow cytometry revealed that while total cell count methods were in good agreement, viable cell count methods showed significantly more variability [95]. This finding powerfully reinforces the need for a multi-method approach, as no single "viable count" is perfect. By calculating these metrics for each pillar of their integrated framework, researchers can objectively select the most fit-for-purpose methods for their specific application, be it probiotic potency testing, antimicrobial efficacy studies, or microbiome research.
The complex physiological states of bacteria, particularly the VBNC and dormant states, render any single-parameter viability assessment incomplete and potentially misleading. This whitepaper establishes that a comprehensive viability profile is only achievable through an integrated framework that concurrently probes culturability, metabolic activity, and membrane integrity. The synergistic workflow and validation strategies presented herein provide researchers and drug development professionals with a robust, standardized pathway to overcome the limitations of historical methods. By adopting this multi-faceted approach, the scientific community can enhance the accuracy of infectious risk assessments, improve the development of antimicrobials and probiotics, and advance our fundamental understanding of bacterial survival in challenging environments.
Conclusion
Accurate bacterial viability assessment is no longer a one-method-fits-all endeavor. A modern approach requires a nuanced understanding of the three core criteria—culturability, metabolic activity, and membrane integrity—and a recognition of their respective strengths and blind spots, particularly concerning the VBNC state. For researchers and drug developers, the future lies in adopting integrated, fit-for-purpose strategies that combine traditional and culture-independent methods, validated with robust quality metrics. This holistic framework is crucial for advancing critical areas, including the development of stable probiotics and live biotherapeutic products (LBP), accurate infectious disease diagnostics, and the evaluation of novel anti-microbial therapies. Future progress will be driven by standardization of new methods, the commercialization of rapid, high-throughput technologies, and a deeper molecular understanding of bacterial dormancy and resuscitation.
References
- 1. Methods to Quantify Microbial Viability | NIST [https://www.nist.gov/programs-projects/methods-quantify-microbial-viability]
- 2. Recent Methods for the Viability Assessment of Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9500772/]
- 3. Recent Methods for the Viability Assessment of Bacterial ... [https://www.mdpi.com/2076-0817/11/9/1057]
- 4. An Overview of the Current State of Cell Viability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11719996/]
- 5. Emerging Technologies for Viability Enumeration of Live ... [https://www.frontiersin.org/research-topics/52945/emerging-technologies-for-viability-enumeration-of-live-microorganisms/magazine]
- 6. A high-throughput and low-waste viability assay for microbes [https://www.nature.com/articles/s41564-023-01513-9]
- 7. Assess the Cell Viability of Staphylococcus aureus Biofilms [https://www.thermofisher.com/us/en/home/references/newsletters-and-journals/bioprobes-journal-of-cell-biology-applications/bioprobes-79/microplate-viability-assays-staphylococcus-aureus-biofilms.html]
- 8. Biofilm viability checker: An open-source tool for automated ... [https://www.nature.com/articles/s41522-021-00214-7]
- 9. A Guideline for Assessment and Characterization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10632153/]
- 10. The importance of the viable but non-culturable state in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4040921/]
- 11. Overview of VBNC, a survival strategy for microorganisms [https://pmc.ncbi.nlm.nih.gov/articles/PMC9526772/]
- 12. The Viable but Non-Culturable (VBNC) State, a Poorly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10818521/]
- 13. Population and single cell metabolic activity of UV-induced ... [https://pubmed.ncbi.nlm.nih.gov/31229870/]
- 14. Detection and Quantification Methods for Viable but Non ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.00673/full]
- 15. Nanowire-assisted electroporation via inducing cell ... [https://www.sciencedirect.com/science/article/abs/pii/S0043135424006778]
- 16. Occurrence and quantification of culturable and viable but ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969720363816]
- 17. Metabolic characteristics and markers in viable but ... [https://www.sciencedirect.com/science/article/abs/pii/S0013935122014384]
- 18. Transcriptome analysis and prediction of the metabolic ... [https://www.nature.com/articles/s41598-022-21102-w]
- 19. From dye exclusion to high-throughput screening: A review ... [https://www.sciencedirect.com/science/article/pii/S0734975025002502]
- 20. Technical pipeline for screening microbial communities as ... [https://www.nature.com/articles/s42003-022-03383-z]
- 21. Biofilms: structure, resistance mechanism, emerging control ... [https://pubs.rsc.org/en/content/articlehtml/2025/pm/d5pm00094g]
- 22. A Promising Method for the Determination of Cell Viability [https://pmc.ncbi.nlm.nih.gov/articles/PMC9367465/]
- 23. Frontiers | A Multi-Purpose Approach to the Mechanisms of Action of... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.842414/full]
- 24. Induction of the viable but non-culturable state in bacterial pathogens... [https://www.nature.com/articles/s41598-018-33595-5?error=cookies_not_supported]
- 25. How to Measure Cell Viability [https://www.promega.com/resources/guides/cell-biology/cell-viability/]
- 26. Membrane interactions in drug delivery: Model cell ... [https://www.sciencedirect.com/science/article/abs/pii/S0001868619303598]
- 27. Guidelines for Regulated Cell Death Assays: A Systematic ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.634690/full]
- 28. Cell or Cell Membrane-Based Drug Delivery Systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4440443/]
- 29. Application of targeted drug delivery by cell membrane-based ... [https://eurjmedres.biomedcentral.com/articles/10.1186/s40001-024-02124-8]
- 30. Viable but non-cultivable state in oral microbiota [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1533768/full]
- 31. Viable but nonculturable (VBNC) state, an underestimated ... [https://www.sciencedirect.com/science/article/abs/pii/S0966842X23001476]
- 32. a fundamental survival strategy of Acinetobacter baumannii [https://pmc.ncbi.nlm.nih.gov/articles/PMC10653857/]
- 33. The Viable but Non-Culturable (VBNC) State, a Poorly ... [https://www.mdpi.com/2076-2607/12/1/39]
- 34. Viable but non-cultivable state in oral microbiota [https://pmc.ncbi.nlm.nih.gov/articles/PMC11959090/]
- 35. A Review of Methods to Determine Viability, Vitality, and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.547458/full]
- 36. "Viable But Non-Culturable (VBNC)": Zombies of the ... [https://bitesizebio.com/41416/viable-but-non-culturable-vbnc-zombies-of-the-bacterial-world/]
- 37. Automation in Colony Counting | Modern Microbial ... [https://www.neogen.com/en/usac/neocenter/blog/colony-counting-how-automation-is-transforming-microbial-enumeration/]
- 38. Assessing Bacterial Viability and Label Accuracy in Human ... [https://pubmed.ncbi.nlm.nih.gov/40871437/]
- 39. Culturability, metabolic activity and composition of ambient ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969719331195]
- 40. Assessment of Oral Microbial Viability by 2,6- ... [https://www.mdpi.com/2079-6382/14/6/590]
- 41. New Scan Ai automatic colony counters [https://www.rapidmicrobiology.com/news/ai-powered-precision-for-colony-counting]
- 42. MCount: An automated colony counting tool for high ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0311242]
- 43. Exploring Innovations in Automatic Colony Counters at the ... [https://www.zryqsw.com/blog/innovations-in-automatic-colony-counters-2025-china-import-and-export/]
- 44. Bacterial Viability - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/bacterial-viability]
- 45. Fluorescein Diacetate Hydrolysis Using the Whole Biofilm ... [https://www.mdpi.com/2073-4344/8/10/434]
- 46. Comparison of Glucose Uptake Assay Methods [https://www.promega.com/resources/pubhub/comparison-of-glucose-uptake-assay-methods/]
- 47. 2-NBDG - Fluorescent Glucose Uptake Probe [https://www.apexbt.com/2-nbdg.html]
- 48. 2-NBDG Glucose Uptake Assay Kit (ab235976) [https://www.abcam.com/en-us/products/assay-kits/2-nbdg-glucose-uptake-assay-kit-ab235976]
- 49. Membrane Integrity - an overview [https://www.sciencedirect.com/topics/engineering/membrane-integrity]
- 50. LIVE/DEAD Fixable Dead Cell Stains Protocol [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/live-dead-fixable-dead-cell-stains.html]
- 51. LIVE/DEAD Cell Viability Assays [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/cell-viability/live-dead-cell-viability-assays.html]
- 52. Live-Dead Cell Staining Kit - Cell Viability Detection [https://www.apexbt.com/live-dead-cell-staining-kit.html]
- 53. Flow Cytometry: The Complete Guide [https://www.antibodies.com/applications/flow-cytometry]
- 54. Recent and Advanced DNA-Based Technologies for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10606304/]
- 55. Advances in point-of-care and molecular techniques to ... [https://www.nature.com/articles/s41545-024-00368-9]
- 56. Development and validation of a PMA-qPCR method for ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1456274/full]
- 57. PMA-qPCR to quantify viable cells in multispecies oral ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12051517/]
- 58. Flow-FISH as a Tool for Studying Bacteria, Fungi and Viruses [https://www.mdpi.com/2673-6284/10/4/21]
- 59. Potential of Flow Cytometric Approaches for Rapid ... [https://www.mdpi.com/2304-8158/10/12/3112]
- 60. Microfluidic-Based Nucleic Acid Amplification Systems in ... [https://www.mdpi.com/2072-666X/10/6/408]
- 61. Continuous-flow, microfluidic, qRT-PCR system for RNA ... [https://pubmed.ncbi.nlm.nih.gov/29116351/]
- 62. A Review of Methods to Determine Viability, Vitality, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7705206/]
- 63. Nanopore- and AI-empowered microbial viability inference [https://pmc.ncbi.nlm.nih.gov/articles/PMC12405693/]
- 64. A generative artificial intelligence approach for the ... [https://www.nature.com/articles/s41564-025-02114-4]
- 65. Machine learning prediction of bacterial optimal growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12553261/]
- 66. Probiotic Viability Reconsidered: Integrating VBNC ... [https://www.mdpi.com/2076-2607/13/11/2479]
- 67. Absolute quantification of VBNC state formation and ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1627943/full]
- 68. Storing Bacterial Samples for Optimal Viability [https://www.thermofisher.com/us/en/home/industrial/microbiology/microbiology-learning-center/storing-bacterial-samples-for-optimal-viability.html]
- 69. Optimization of cryoprotectants and storage temperatures for ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0328216]
- 70. DFT-based evaluation of cryoprotectants and their role in ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224025000884]
- 71. The Role of Cryoprotective Agents in Liposome ... [https://www.mdpi.com/1422-0067/23/20/12487]
- 72. The Effect of Cryoprotectants and Storage Conditions on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11468535/]
- 73. Optimizing Sperm Cryopreservation from Four Endangered ... [https://www.mdpi.com/2076-2615/15/20/3013]
- 74. Efficacy assessment of different cryoprotectants for ... [https://www.nature.com/articles/s41598-024-71529-6]
- 75. Defining the optimal cryoprotectant and concentration for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6167250/]
- 76. Exploring ultrasound-induced metabolic attenuation in ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1589054/full]
- 77. Assessing Viability and Stress Tolerance of Probiotics—A ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.818468/full]
- 78. Oxidative Stress Contributes to Bacterial Airborne Loss ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10101003/]
- 79. Stress tolerance and metabolism profiling of selected ... [https://www.sciencedirect.com/science/article/abs/pii/S2212429225000951]
- 80. Editorial: Emerging technologies for viability enumeration ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1546438/full]
- 81. Oxidative Stress Detection | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/oxidative-stress.html]
- 82. Measurement of Oxidative Stress Markers In Vitro Using ... [https://www.ncbi.nlm.nih.gov/books/NBK566442/]
- 83. Oxidative stress: from molecular studies to clinical intervention ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12443558/]
- 84. Optimization of a validated labor-efficient culture-based ... [https://www.sciencedirect.com/science/article/abs/pii/S095671352500605X]
- 85. Multi-objective optimization for pressure hold integrity ... [https://www.sciencedirect.com/science/article/abs/pii/S0263224125027101]
- 86. Evaluation of Membrane Integrity Monitoring Methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10714397/]
- 87. Optimization and Testing of a Commercial Viability PCR ... [https://www.mdpi.com/2076-2607/12/4/765]
- 88. Protocol for analyzing energy metabolic pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10960080/]
- 89. Recent Methods for the Viability Assessment of Bacterial Pathogens: Advances, Challenges, and Future Perspectives - PubMed [https://pubmed.ncbi.nlm.nih.gov/36145489/]
- 90. The diversity, dynamics, and culturability of bacterial ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1621463/full]
- 91. Investigating the effects of microwave plasma on bacterial ... [https://www.nature.com/articles/s41598-025-02312-4]
- 92. Tracking Microbial Rhythms Reveals New Target for Treating ... [https://today.ucsd.edu/story/tracking-microbial-rhythms-reveals-new-target-for-treating-metabolic-diseases]
- 93. Analysis of disruptive action of electrical current on cell ... [https://pubmed.ncbi.nlm.nih.gov/41085669/]
- 94. Backtracking metabolic dynamics in single cells predicts ... [https://www.nature.com/articles/s41467-025-64225-0]
- 95. Measurement quality metrics to improve absolute microbial ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1631377/full]
- 96. Measurement quality metrics to improve absolute microbial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12401475/]
- 97. Critical aspects of using bacterial cell viability assays with the... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-015-0376-x]
- 98. Viability studies of bacterial strains exposed to nitrogen ... [https://www.nature.com/articles/s41598-025-94898-y]
- 99. The ISO standards in pharmaceuticals: a complete guide - SFAM [https://www.sfamgroup.com/en/iso-standards-pharmaceutical-industry-guide/]
- 100. Fit-for-purpose biomarker method validation for application ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2990602/]
- 101. The ISO standards you need to know for SaMD - 2025 update [https://www.hardianhealth.com/insights/iso-standards-for-software-medical-device]
- 102. ISO 8871-5:2025 – Revised Testing for Injectable ... [https://www.cormica.com/updates-to-iso-8871-5-2025/]
- 103. Best practices for the development and fit-for-purpose ... [https://aapsopen.springeropen.com/articles/10.1186/s41120-021-00050-1]
- 104. Fit-for-purpose biomarker method validation in anticancer ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644610002606]
- 105. Membrane Integrity Test - an overview [https://www.sciencedirect.com/topics/engineering/membrane-integrity-test]
- 106. 10.3: Measurement of Bacterial Growth [https://bio.libretexts.org/Courses/City_College_of_San_Francisco/Introduction_to_Microbiology_(Liu_et_al.)/10%3A_Microbial_Growth/10.03%3A_Measurement_of_Bacterial_Growth]
- 107. An Easy Guide for Researchers Using Bacterial Cultures [https://bitesizebio.com/41100/is-your-bacterial-culture-still-growing/]