Defining Gold Standards in Microbial Viability: From Classic CFU Assays to Cutting-Edge Technologies
This article provides a comprehensive analysis of gold-standard methods for assessing microbial viability, tailored for researchers, scientists, and drug development professionals.
Defining Gold Standards in Microbial Viability: From Classic CFU Assays to Cutting-Edge Technologies
Abstract
This article provides a comprehensive analysis of gold-standard methods for assessing microbial viability, tailored for researchers, scientists, and drug development professionals. We explore the foundational principles defining microbial life, from the established colony-forming unit (CFU) assay to modern criteria based on metabolic activity and membrane integrity. The scope covers traditional and emerging methodological applications, including high-throughput and culture-independent techniques. Practical guidance on troubleshooting common issues and optimizing protocols is provided, alongside a critical comparative analysis validating method selection for diverse research and clinical applications, including live biotherapeutic products and pathogen detection.
What Does 'Viable' Really Mean? Foundational Concepts and Definitions in Microbial Viability
Viability assessment is a critical step in evaluating bacterial pathogens to determine infectious risks to public health and in ensuring the potency of live microbial products [1] [2]. Based on three accepted viable criteria—culturability, metabolic activity, and membrane integrity—current viability assessments are categorized into three main strategies, each with distinct applications, advantages, and limitations [1]. The gold standard for measuring the potency of live microbial products has historically been colony-forming units (CFU) enumeration, which relies on culturability [2]. However, the phenomenon of viable but nonculturable (VBNC) bacteria and the need for rapid, precise methods have driven the development and adoption of techniques based on metabolic activity and membrane integrity [1] [3]. This guide provides an in-depth technical overview of these three criteria, detailing the principles, gold-standard methods, experimental protocols, and current challenges, framed within the context of modern microbial research and drug development.
The Three Viability Criteria: Core Principles and Gold-Standard Methods
The following table summarizes the core principles, associated gold-standard methods, and key challenges for each of the three accepted viability criteria.
Table 1: Core Principles and Gold-Standard Methods for Viability Assessment
| Viability Criterion | Core Principle | Gold-Standard Method(s) | Primary Challenge |
|---|---|---|---|
| Culturability | Measures the ability of a single bacterial cell to reproduce and form a visible colony on an appropriate solid medium [1]. | Plate Culture Method (CFU enumeration) [1] [2]. | Cannot detect Viable But Nonculturable (VBNC) cells, which are metabolically active but have lost reproductive capacity [1]. |
| Metabolic Activity | Assesses the biochemical processes of viable cells, such as enzyme activity or substrate uptake [1] [4]. | Tetrazolium Reduction Assays (e.g., MTT, WST-1) and Resazurin Reduction [5] [4]. | Dormant cells may exhibit negligible metabolic activity, leading to false negatives [1]. |
| Membrane Integrity | Evaluates the physical status of the cell membrane; a dead bacterium typically has a disrupted or broken membrane [1] [3]. | Fluorescent Staining (e.g., Propidium Iodide) combined with membrane-impermeant dyes [1] [3]. | Requires multiple steps and specific equipment (e.g., fluorometer), and can be less popular for some applications [1]. |
Detailed Methodologies and Experimental Protocols
Viability Assessment Based on Culturability
The plate culture method, discovered by Robert Koch in 1881, is the traditional method for detecting bacterial viability based on culturability [1]. A sample is plated on an agar plate and incubated under conditions appropriate for the target bacteria. Viable, culturable cells form visible colonies, whereas nonviable cells do not [1]. The result is expressed as Colony Forming Units per milliliter (CFU/mL).
Protocol: Standard Plate Count (CFU Enumeration) [1]
- Sample Preparation: Serially dilute the bacterial sample in a sterile, physiologically balanced solution (e.g., saline or phosphate-buffered saline).
- Plating: Spread a measured volume (e.g., 100 µL) of appropriate dilutions onto the surface of nutrient-rich agar plates. Alternatively, use an automated spiral plater.
- Incubation: Invert plates and incubate at the optimal temperature for the specific bacterium for 24-48 hours or longer, as required.
- Enumeration: Count the number of distinct colonies on plates that contain between 30 and 300 colonies. Calculate the CFU/mL using the formula: CFU/mL = (Number of colonies counted) / (Dilution factor × Volume plated in mL)
- Automation (Optional): Automated systems can capture images of plates and count colonies using software, reducing time and manual error [1].
Viability Assessment Based on Metabolic Activity
This approach measures the metabolic processes of viable cells. A common method involves using tetrazolium salts, which are reduced by metabolically active cells to colored formazan products [4].
Protocol: WST-1 Assay for Bacterial Metabolic Activity [5] The WST-1 assay is a colorimetric method that quantitatively assesses cell viability by measuring cellular metabolic activity via mitochondrial dehydrogenases.
- Reagent Preparation: Use a commercially available WST-1 assay reagent.
- Cell Seeding and Treatment: Seed bacterial cells into the wells of a 96-well plate. Incubate the cells under standard conditions, with or without exposure to test compounds.
- Addition of WST-1 Reagent: Add WST-1 reagent directly to each well. A volume of 10 µL per 100 µL of culture medium is typically recommended.
- Control Wells Setup:
- Blank Control: Culture medium and WST-1 reagent only (no cells).
- Untreated Control: Cells and culture medium without test compounds.
- Positive Control: Cells treated with a known cytotoxic agent.
- Incubation and Measurement: Incubate the plate under standard conditions for 0.5–4 hours. Monitor color development. Measure the absorbance of each well using a microplate reader at 440–450 nm, with a reference wavelength above 600 nm.
The biochemical reaction is: WST-1 (Tetrazolium Salt) + Mitochondrial Dehydrogenases + Electron Carriers → Formazan Dye (Soluble). The amount of formazan dye produced is directly proportional to the number of viable, metabolically active cells in the sample [5].
Alternative Protocol: DCIP Reduction Assay [6] 2,6-Dichlorophenolindophenol (DCIP) is a redox dye that acts as a substitute for NADP+. Viable cells reduce blue, oxidized DCIP to a colorless form.
- Reagent Preparation: Prepare a DCIP solution in an appropriate buffer.
- Assay Setup: In a microplate, mix bacterial suspension with the DCIP solution.
- Incubation and Measurement: Incubate the plate at room temperature and monitor the loss of blue color by measuring the decrease in absorbance at 600 nm spectrophotometrically over time. The rate of absorbance reduction is proportional to the number of viable cells.
Viability Assessment Based on Membrane Integrity
This criterion discriminates live and dead cells based on the integrity of the cell membrane. It often employs fluorescent dyes that can or cannot penetrate intact membranes.
Protocol: Membrane Integrity Assessment using Fluorescent Stains [3] A common approach involves using a combination of membrane-permeant and membrane-impermeant fluorescent dyes.
- Stain Preparation: Prepare working solutions of stains, such as SYTO 9 (labels all cells) and propidium iodide (PI), which only penetrates cells with damaged membranes.
- Staining: Mix the bacterial sample with the fluorescent dyes and incubate in the dark for 15-20 minutes.
- Analysis: Analyze the stained cells using fluorescence microscopy or flow cytometry.
- Viable cells with intact membranes will fluoresce green (SYTO 9).
- Non-viable cells with compromised membranes will fluoresce red (PI), as PI enters the cell and quenches SYTO 9 fluorescence.
This method is highly effective for distinguishing between live and dead bacterial populations and can be used to confirm the lethal action of lytic biocides like Benzalkonium Chloride (BAC), which disrupts the cell membrane [3].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Viability Assessment
| Reagent / Material | Function in Viability Assessment |
|---|---|
| Nutrient Agar Plates | Provides a solid growth surface for culturable bacteria to form colonies for CFU enumeration [1]. |
| Tetrazolium Salts (e.g., MTT, WST-1) | Compounds reduced by metabolically active cells to colored formazan products, enabling colorimetric quantification of viability [5] [4]. |
| Resazurin | A redox dye that changes from blue to pink/colorless (fluorescent) upon reduction by metabolically active cells [4]. |
| 2,6-Dichlorophenolindophenol (DCIP) | A redox dye that changes from blue to colorless when reduced by viable cells, serving as an electron acceptor in metabolic pathways [6]. |
| Fluorescent Dyes (e.g., Propidium Iodide, SYTO 9) | Used in combination to differentiate cells based on membrane integrity; impermeant dyes like PI only stain dead cells [3]. |
| Universal Neutralizer | Used to quench the activity of biocides at the end of an exposure period in antimicrobial efficacy tests, preventing carry-over effect during plating [3]. |
Experimental Workflow and Logical Relationships
The following diagram illustrates the decision-making workflow for selecting a viability assessment method based on the three core criteria and the phenomenon of VBNC cells.
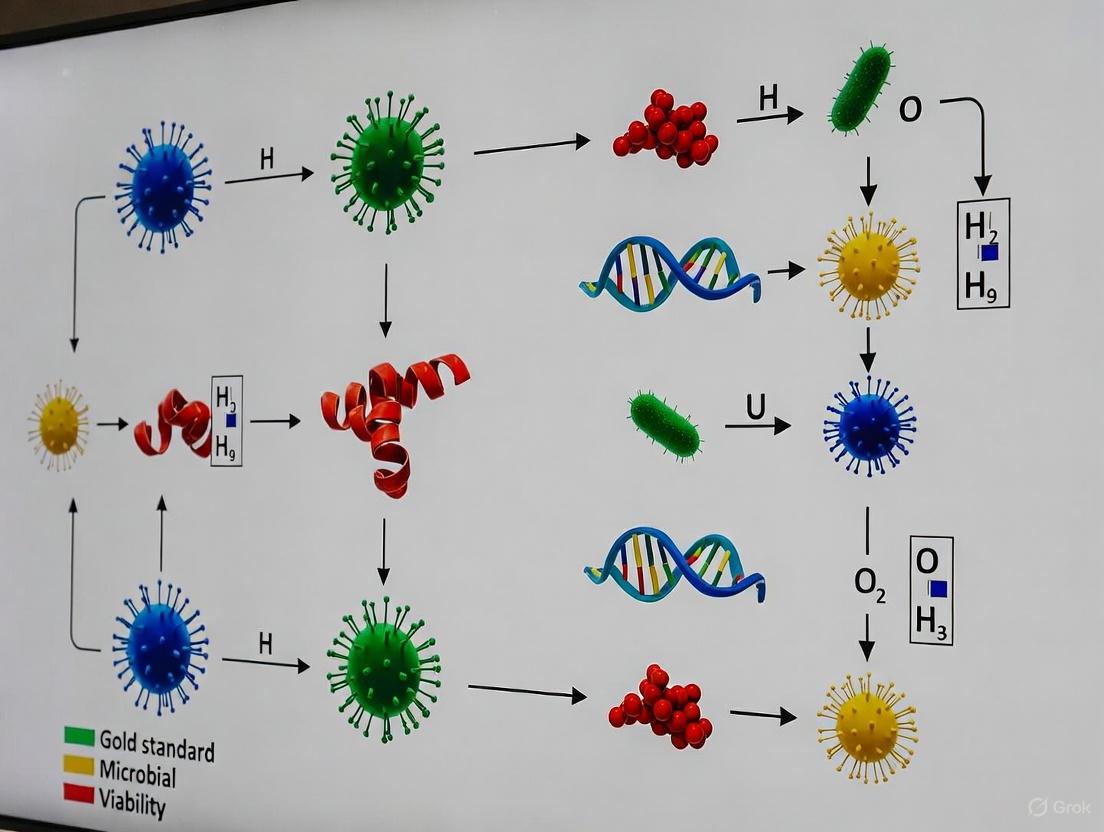
Diagram 1: A workflow for viability assessment based on the three core criteria. A 'No' to culturability but 'Yes' to metabolic activity indicates a Viable But Nonculturable (VBNC) state [1].
The triad of culturability, metabolic activity, and membrane integrity provides a robust framework for assessing microbial viability. While the plate count method remains the historical gold standard for culturable cells, its limitation in detecting VBNC states necessitates the use of complementary techniques based on metabolism and membrane integrity. Emerging technologies, including microfluidics and advanced staining protocols, continue to improve the speed, accuracy, and accessibility of these methods [1] [2]. For researchers and drug development professionals, selecting the appropriate viability criterion and corresponding assay is paramount and must be guided by the specific research question, the nature of the microbial sample, and an awareness of the physiological states—such as dormancy and the VBNC state—that can significantly impact the interpretation of results.
For decades, the colony-forming unit (CFU) assay has remained the undisputed gold standard for quantifying viable microorganisms in both research and industrial applications. This technical guide explores the historical foundation of the CFU assay, its continued relevance in modern microbiology, and the emerging technologies that seek to address its limitations. We examine the assay's fundamental principles, detailing standardized protocols for its execution across diverse microbial species. Furthermore, we present a comprehensive analysis of innovative approaches like the Geometric Viability Assay (GVA) that build upon the CFU concept while dramatically improving efficiency. Through comparative performance data and detailed methodological frameworks, this review provides researchers with the practical knowledge to select appropriate viability enumeration methods for their specific applications, from basic research to drug discovery and quality control in live biotherapeutic products.
The colony-forming unit (CFU) assay represents one of the most fundamental and enduring techniques in microbiology, providing the critical link between visible colony formation and the presence of viable, replicating cells in a sample. Despite its simplicity, this method has maintained its status as the regulatory and scientific gold standard for viability enumeration across diverse fields including clinical microbiology, pharmaceutical development, food safety, and functional genomics [7] [8]. The assay's enduring relevance stems from its direct measurement of the defining characteristic of microbial viability—the ability to undergo cellular division and form a visible colony. While emerging technologies offer advantages in speed and scalability, they are invariably validated against the CFU benchmark, cementing its foundational role in microbiology.
The historical significance of the CFU methodology extends beyond microbial applications. The seminal work by Dr. James Till and Dr. Ernest McCulloch in 1961 established the spleen colony-forming unit (CFU-S) assay, which provided the first functional evidence of clonal hematopoietic stem cells at the single-cell level [9] [10]. Their pioneering approach demonstrated that transplanted bone marrow cells could form macroscopic nodules in the spleens of irradiated mice, with each nodule arising from a single multipotent progenitor cell capable of both self-renewal and differentiation [9]. This foundational principle—that a single viable cell can give rise to a visible colony—underpins all modern CFU applications across biological disciplines.
Fundamental Principles and Methodological Framework
Core Principles of Colony Formation
The CFU assay operates on several fundamental principles that define its utility and limitations. Each visible colony theoretically originates from a single viable cell capable of undergoing sufficient divisions to form a macroscopic entity. This clonal origin provides both quantitative information (through colony counting) and qualitative information (through colony morphology analysis). The assay inherently distinguishes between viable and non-viable cells based on reproductive capability, excluding those cells that may be metabolically active but unable to divide (the viable but non-culturable state). The linear relationship between the number of colonies counted and the dilution factor applied enables back-calculation of the original viable cell concentration in a sample, typically expressed as CFU per milliliter or gram.
Standardized Experimental Protocol
The execution of a reliable CFU assay requires meticulous attention to protocol across several critical phases:
Sample Preparation and Dilution:
- Purpose: To achieve an optimal colony count (typically 30-300 colonies per plate for statistical reliability) through serial dilution.
- Procedure:
- Prepare serial 10-fold dilutions of the sample in appropriate buffer or growth medium.
- Mix each dilution thoroughly to ensure uniform cell distribution.
- Select 2-3 appropriate dilution levels for plating based on expected cell density.
Plating and Incubation:
- Spread Plate Method:
- Transfer a fixed volume (typically 100 μL) of diluted sample onto the surface of pre-poured agar plates.
- Spread evenly using a sterile spreader and allow liquid to absorb.
- Pour Plate Method:
- Mix sample dilution with molten agar (cooled to approximately 45°C).
- Pour mixture into sterile plates and allow to solidify.
- Incubation: Incubate plates under optimal conditions (temperature, atmosphere, duration) for the target microorganism.
Colony Counting and Calculation:
- Manual Counting: Count colonies visually, using a colony counter if available.
- Automated Counting: Utilize imaging systems like the STEMvision instrument for standardized analysis [11].
- Calculation:
- CFU/mL = (Number of colonies counted × Dilution factor) / Volume plated
- Report results as an average of countable plates with appropriate significant figures.
Essential Research Reagent Solutions
Table 1: Key reagents and materials for CFU assays
| Item | Function | Application Notes |
|---|---|---|
| Agar Plates | Solid support for colony growth | Select medium composition based on nutritional requirements of target microbe |
| Dilution Blanks | Sterile buffer for serial dilutions | Phosphate-buffered saline or 0.85% saline commonly used |
| MethoCult Media | Specialized semisolid media for hematopoietic progenitors | Contains cytokines for specific lineage support [11] |
| Triphenyl Tetrazolium Chloride (TTC) | Visualization aid | Colorless compound turns red upon microbial reduction, enhancing colony contrast [7] |
| STEMvision System | Automated colony imaging and analysis | Reduces subjectivity in counting and classification [11] |
Emerging Innovations: The Geometric Viability Assay
Theoretical Foundation and Protocol
A significant innovation in viability testing, the Geometric Viability Assay (GVA), addresses multiple limitations of traditional CFU methods while maintaining their fundamental principle [7] [8]. The GVA leverages the geometric properties of a cone (specifically, a standard pipette tip) to create an inherent dilution series in a single step. The probability of a colony forming at any point along the cone's axis is proportional to the cross-sectional area at that point, defined by the probability density function (PDF): PDF(x) = 3x²/h³, where x is the perpendicular distance from the tip and h is the total length of the cone [7].
The GVA protocol comprises these critical steps:
- Sample Preparation: Mix the microbial sample with melted agarose (0.5% final concentration) cooled to ≤55°C.
- Loading: Transfer the mixture into a standard pipette tip.
- Solidification: Allow agarose to solidify at room temperature or appropriate temperature for sensitive samples.
- Incubation: Eject tip into a rack and incubate under optimal conditions for target microorganisms.
- Imaging and Analysis: Capture images using custom optical setup or smartphone system, then calculate CFU concentration based on colony positions using the formula: CFUs/mL = N(x) / (V × ∫PDF(x)dx), where N(x) represents colonies between positions x₁ and x₂, and V is the cone volume [7] [8].
Performance Comparison with Traditional Methods
Table 2: Quantitative comparison of viability enumeration methods
| Parameter | Traditional CFU | Geometric Viability Assay | Flow Cytometry |
|---|---|---|---|
| Dynamic Range | 1 - 100,000,000 CFU/mL [8] | 1 - 1,000,000 CFU/mL [7] | Varies with staining method |
| Time Investment | ~3 hours for 96 samples [12] | ~5 minutes for 96 samples [12] | ~1-2 hours sample preparation |
| Plastic Consumption | High (multiple plates/tubes) | 15-fold reduction [12] | Moderate (tubes only) |
| Throughput | Limited by dilution series | 1,200 measurements/researcher/day [7] | High with automation |
| Organism Compatibility | Culturable organisms only | Gram-positive/-negative bacteria, yeast, biofilms [7] | All microorganisms |
| VBNC Detection | No | No | Yes |
Comparative Analysis of Viability Enumeration Technologies
Limitations of Traditional CFU Methods
While the CFU assay provides the benchmark for viability assessment, several significant limitations impact its utility in modern applications. The method is notoriously time-intensive and resource-consuming, requiring multiple dilution steps, substantial plasticware, and extended incubation periods [7] [8]. Perhaps most critically, CFU enumeration fails to detect viable but non-culturable (VBNC) cells—those that maintain metabolic activity but cannot replicate under standard laboratory conditions [13]. This limitation is particularly problematic for complex samples like probiotic blends, where microorganisms with varying growth requirements may not form colonies under standardized conditions [13]. Additionally, the CFU assay provides limited information about cellular heterogeneity within populations and cannot distinguish between specific strains in multi-strain products without additional methodological modifications.
Emerging Alternative Technologies
Molecular Approaches:
- PMA-qPCR: Propidium monoazide quantitative PCR selectively penetrates membrane-compromised cells and covalently binds DNA upon photoactivation, preventing its amplification. This allows quantification of intact, potentially viable cells with species-specificity [13]. Recent developments have enabled species-specific enumeration of probiotics including Lactobacillus acidophilus, Bifidobacterium bifidum, and Lacticaseibacillus rhamnosus, with applications in quality control of multi-strain products [13].
Cytometry-Based Methods:
- Flow Cytometry: This culture-independent approach assesses viability based on cellular parameters like membrane integrity (using fluorescent dyes) and metabolic activity, providing results within hours rather than days [13]. Recent ring tests demonstrated method robustness across equipment and operators, with good agreement between fluorescence flow cytometry and impedance flow cytometry techniques [13].
- Flow-FISH: Combining flow cytometry with fluorescence in situ hybridization enables species-specific quantification in blended products, outperforming plate counting in repeatability and uncertainty [13].
Novel Detection Platforms:
- Isothermal Microcalorimetry (IMC): This label-free method measures heat production from metabolic processes, establishing correlations between time-to-peak heat detection and CFU/mL while providing insights into microbial activity beyond mere viability [13].
- Cell Counting Kit-8 (CCK-8): Originally developed for eukaryotic cells, tetrazolium-based colorimetric assays have been adapted for probiotics, enabling rapid viability assessment during co-culture experiments [13].
Applications and Future Perspectives
The CFU assay maintains its critical role across diverse research and industrial applications. In drug discovery, particularly for antibiotics against persistent cells, reliable viability assessment is paramount for evaluating compound efficacy [7] [8]. The emergence of high-throughput methods like GVA has enabled previously impractical screening scales, facilitating discoveries such as the ROS-mediated bactericidal mechanism of diphenyliodonium [14]. In regenerative medicine, hematopoietic CFU assays remain indispensable for quantifying stem and progenitor cells, with automated systems like STEMvision standardizing analysis across laboratories [11].
For the live microorganism industry—including probiotics and live biotherapeutic products—accurate potency measurement through viability enumeration is essential for quality assurance and regulatory compliance [13] [2]. While the CFU method remains the regulatory benchmark, emerging technologies offer solutions for strain-specific enumeration in complex matrices, addressing critical gaps in current methodology [13]. The future of viability assessment likely involves method-specific integration based on application requirements, with traditional CFU providing validation standards, while high-throughput, culture-independent methods enable scalable quality control and advanced research applications.
The colony-forming unit assay maintains its position as the unchallenged gold standard in microbial viability assessment due to its direct measurement of cellular replication, extensive validation history, and methodological simplicity. While emerging technologies address specific limitations regarding throughput, VBNC detection, and strain specificity, they invariably require correlation to CFU standards for validation. Innovations like the Geometric Viability Assay demonstrate that fundamental principles of colony formation can be enhanced through mathematical modeling and geometric optimization, bridging traditional microbiology with contemporary scalability needs. As the field advances, the integration of these complementary approaches will expand our understanding of microbial viability beyond binary cultivation-based assessments, ultimately enhancing both fundamental research and applied microbiological applications across diverse sectors.
The Viable But Non-Culturable (VBNC) state is a survival strategy employed by many bacterial species when faced with environmental stress. Cells in this state are metabolically active and possess an intact membrane but cannot form colonies on routine culture media, the gold standard in traditional microbiology. This state poses significant challenges for public health, food safety, and pharmaceutical drug development, as VBNC pathogens retain virulence and can resuscitate, leading to recalcitrant infections and false-negative diagnostic results. This whitepaper delves into the characteristics and induction of the VBNC state, critically evaluates current and emerging detection methodologies against the benchmark of microbial viability assessment, and discusses its profound implications for ensuring product safety and efficacy in the pharmaceutical industry.
The VBNC state, first reported in Escherichia coli and Vibrio cholerae in 1982, is a dormant condition distinct from cell death and sporulation [15]. It is recognized as a general stress response enabling bacteria to survive adverse conditions, with over 100 bacterial species, including major human pathogens like Staphylococcus aureus, Listeria monocytogenes, and Mycobacterium tuberculosis, documented to enter this state [16] [17] [15]. The core paradox of VBNC cells—being alive yet unculturable—renders them invisible to conventional, growth-dependent detection methods, creating a critical blind spot in microbiological risk assessment [17] [18].
For pharmaceutical research and development, this blind spot is particularly concerning. Critical processes such as raw material testing, water system monitoring, in-process bioburden control, and finished product sterility testing predominantly rely on culture-based techniques [18]. The potential presence of VBNC organisms in input materials or their induction via sublethal sterilization processes can lead to an inaccurate assessment of microbial quality, potentially compromising patient safety and product stability [18] [19]. Furthermore, the retained pathogenicity and resuscitation potential of VBNC cells underscore the necessity for robust, non-culture-based viability assays in disinfectant efficacy studies and antimicrobial drug development [15] [19].
Characteristics and Induction of the VBNC State
Defining Characteristics
VBNB cells are defined by several key characteristics that differentiate them from both culturable and dead cells.
- Culturability: A definitive loss of the ability to proliferate and form colonies on routine culture media, even after supplementation with standard nutrients [15].
- Metabolic Activity: Continued, albeit reduced, metabolic processes, including respiration, ATP synthesis, gene expression, and substrate uptake [1] [15].
- Membrane Integrity: Possession of an intact and often modified cell membrane, a hallmark of viability [1] [15].
- Morphological Changes: Cells typically undergo significant size reduction (dwarfing) and changes in shape, such as a transition from rod-shaped to coccoid [16] [15].
- Biochemical Alterations: The state involves major biomolecular remodeling, including changes in protein profiles, fatty acid composition, and increased peptidoglycan cross-linking, which contributes to enhanced resistance [16] [15].
Table 1: Contrasting Features of Bacterial States
| Feature | Culturable Cells | VBNC Cells | Dead Cells |
|---|---|---|---|
| Growth on Media | Yes | No | No |
| Metabolic Activity | High | Low to Detectable | None |
| Membrane Integrity | Intact | Intact | Compromised |
| Gene Expression | Active | Modified, but active | None |
| Response to Stimuli | Normal | Requires resuscitation | None |
| Virulence Potential | Yes | Retained (in some pathogens) | None |
The transition into the VBNC state is typically triggered by the imposition of sublethal environmental stresses. Common induction factors include:
- Nutrient starvation (e.g., in low-nutrient media or water) [20] [15].
- Temperature shifts, particularly cold shock [15].
- Oxidative stress from disinfectants like hydrogen peroxide or hypochlorite [21] [22].
- Osmotic stress [15].
- Antibiotic pressure, as demonstrated by the induction of staphylococci using gentamycin [20].
- UV light and other physical stresses [22].
Once conditions become favorable, VBNC cells can undergo resuscitation, reverting to a metabolically active and culturable state. This process can be triggered by removing the stressor, temperature upshift, or adding specific nutrients or resuscitation-promoting factors. For instance, Pseudomonas aeruginosa induced into the VBNC state by UV radiation can resuscitate when introduced into a rich LB medium [22]. Similarly, the addition of siderophores like Ferrioxamine E has been shown to significantly improve the resuscitation and recovery of pathogens like Salmonella and Staphylococcus aureus from various samples [19].
Gold Standard and Emerging Methods for VBNC Detection
The "gold standard" for microbial viability in pharmaceutical quality control has historically been culturability. However, the VBNC phenomenon necessitates a re-evaluation of this paradigm, shifting the focus to criteria that are independent of growth, primarily metabolic activity and membrane integrity [17] [1]. A tiered, multi-method approach is often required for definitive identification.
Established Detection Methodologies
The following methods form the cornerstone of modern VBNC detection and quantification.
Table 2: Key Methodologies for Detecting VBNC Bacteria
| Method | Target Principle | Key Reagents / Techniques | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Live/Dead Staining & Fluorescence Microscopy | Membrane Integrity | SYTO 9 & Propidium Iodide (e.g., BacLight kit) [20] [23] | Direct visualization, distinguishes live (green) from dead (red) | Qualitative/semi-quantitative; can overestimate viability if membrane is intact but metabolism halted |
| Viability PCR (v-PCR) | Membrane Integrity | Propidium Monoazide (PMA) or EMA; qPCR [23] [22] | Specific, quantitative, detects specific pathogens | Requires optimization for complex matrices; may not detect cells with intact membrane but no metabolism |
| Direct Viable Count (DVC) | Metabolic Activity | Nalidixic acid, incubation, staining [16] | Confirms elongation capacity, a sign of viability | Requires incubation and microscopy |
| Adenosine Triphosphate (ATP) Assay | Metabolic Activity | Luciferase enzyme reaction [1] | Rapid, indicates active metabolism | Low signal in dormant cells with minimal metabolism |
| Flow Cytometry | Membrane Integrity / Physiology | SYTO 9 & PI; automated analysis [23] | High-throughput, multi-parameter analysis | Can be unreliable in complex matrices like process wash water [23] |
Advanced and Emerging Detection Techniques
Innovative technologies are pushing the boundaries of VBNC detection, offering new avenues for rapid and precise analysis.
- ATR-FTIR Spectroscopy: This technique detects biochemical changes in VBNC cells by measuring infrared absorption. Studies on E. coli have identified specific spectral biomarkers, such as the 995 cm⁻¹ RNA band, which consistently changes across different stress conditions, presenting a powerful label-free diagnostic tool [16].
- Hyperspectral Microscopy Imaging (HMI) with AI: This cutting-edge approach combines high-resolution imaging that captures spatial and spectral data with deep learning. A 2024 study demonstrated that an AI model (EfficientNetV2) trained on HMI data could classify VBNC E. coli with 97.1% accuracy, far surpassing the performance of models using standard RGB images [21].
- Optimized Molecular Combinations: To improve accuracy, researchers are refining existing methods. For example, a viability-qPCR protocol combining EMA and PMAxx dyes was developed to better suppress DNA amplification from dead cells, providing a more reliable quantification of VBNC Listeria monocytogenes in complex food process wash water [23].
The following diagram illustrates a generalized experimental workflow for inducing and confirming the VBNC state, integrating multiple detection methods to ensure robust results.
The Scientist's Toolkit: Essential Reagents and Materials
Successful research into the VBNC state relies on a suite of specialized reagents and materials. The following table details key solutions used in the featured experiments and the broader field.
Table 3: Key Research Reagent Solutions for VBNC Studies
| Reagent / Material | Function in VBNC Research | Example Application |
|---|---|---|
| Propidium Monoazide (PMA/PMAxx) | DNA intercalating dye; penetrates compromised membranes of dead cells and covalently cross-links to DNA upon photoactivation, inhibiting its amplification in PCR. | Used in viability qPCR (v-PCR) to selectively detect viable/VBNC cells with intact membranes in water [22] and food samples [23]. |
| SYTO 9 & Propidium Iodide (PI) | Fluorescent nucleic acid stains for live/dead differentiation. SYTO 9 stains all cells; PI penetrates only dead cells with damaged membranes, quenching SYTO 9 fluorescence. | Used in the BacLight viability kit for direct visualization and quantification of VBNC cells via fluorescence microscopy [20] [23]. |
| Ferrioxamine E | A siderophore that provides the essential micronutrient iron (III). Acts as a growth factor and resuscitation promoter for stressed bacteria. | Supplementation in pre-enrichment broths to improve recovery and resuscitation of VBNC Salmonella, Cronobacter spp., and S. aureus [19]. |
| Gentamycin (antibiotic) | An aminoglycoside antibiotic used as a selective stressor to induce the VBNC state in experimental settings. | Used at concentrations of 4x, 8x, and 16x the MIC to induce the VBNC state in staphylococcal biofilms [20]. |
| Chlorine-based Disinfectants | Oxidizing biocides (e.g., sodium hypochlorite) used for disinfection that can sublethally injure cells, inducing the VBNC state. | Studied for their efficacy in inactivating P. aeruginosa in drinking water and their potential to induce the VBNC state [22]. |
Molecular Mechanisms and Regulatory Pathways
The transition into the VBNC state is an active, genetically regulated process. While the full picture is still emerging, several key molecular mechanisms have been identified.
A primary regulatory system involves the Stringent Response and Toxin-Antitoxin (TA) Modules. Under stress, the alarmone (p)ppGpp accumulates, triggering a drastic downregulation of energy-consuming processes like replication and translation. Concurrently, specific TA systems are activated. In these modules, stable toxin proteins are released through the degradation of their cognate, unstable antitoxins. These toxins further inhibit vital cellular processes, promoting dormancy. This coordinated shutdown allows the cell to preserve energy and integrity [17].
Global stress regulators, such as RpoS (the stationary phase sigma factor), also play a critical role by modulating the expression of a large suite of genes involved in stress resistance. Biochemically, this is accompanied by a remodeling of the cell envelope, including increased peptidoglycan cross-linking and changes in membrane fatty acid profiles, which explains the enhanced resistance of VBNC cells to subsequent physical and chemical challenges [16] [15].
Implications for Pharmaceutical Research and Drug Development
The VBNC state presents multifaceted challenges and considerations for the pharmaceutical sector, impacting areas from quality control to antimicrobial drug development.
- Risk to Patient Safety: While the direct risk from VBNC cells in finished products is considered low due to their inability to replicate in the package and the denaturation of toxins during aggressive manufacturing processes, the potential for resuscitation in the patient environment cannot be entirely disregarded for some opportunistic pathogens [18] [19].
- Compromised Quality Control: The reliance on growth-based methods like Microbial Limit Tests and Sterility Tests means that VBNC organisms constitute a "hidden bioburden," leading to an underestimation of microbial contamination in raw materials, water for injection (WFI), and manufacturing environments [18]. This is critical as water systems are common reservoirs for VBNC bacteria [22] [18].
- Challenges in Disinfectant and Antimicrobial Efficacy Testing: Standard tests often evaluate efficacy against logarithmic-phase cultures. However, bacteria in the VBNC state exhibit drastically increased tolerance to antimicrobials, including antibiotics, biocides, and physical stresses [17] [15]. This can lead to an overestimation of a treatment's effectiveness. Evaluating disinfectants and novel anti-infective drugs against VBNC populations is essential for developing more robust contamination control strategies and effective therapies for persistent infections.
The VBNC state represents a critical adaptation in the bacterial life cycle that fundamentally challenges the historical gold standard of microbial viability—culturability. Its implications for diagnostic accuracy, public health risk assessment, and antimicrobial efficacy are profound. For pharmaceutical scientists and drug development professionals, acknowledging this state is no longer optional but a necessity for advancing microbial quality control and developing next-generation antimicrobials. The future lies in adopting a more comprehensive definition of viability, one that integrates culturability with measures of metabolic activity and membrane integrity. The ongoing development of advanced, culture-independent detection technologies, such as AI-enhanced hyperspectral imaging and refined molecular assays, promises to illuminate this hidden microbial world, ultimately leading to safer pharmaceutical products and more effective therapeutic interventions.
In microbial research, defining cell death is far more complex than observing the cessation of visible growth. A cell is considered viable if it can perform essential functions, whereas death is often linked to the irreversible loss of the plasma membrane's barrier function, the formation of apoptotic bodies, or engulfment by phagocytes [24]. However, this definition is complicated by physiological states such as the viable but non-cultivable (VBNC) state, where cells remain metabolically active and possess membrane integrity but cannot form colonies on standard growth media—a traditional indicator of viability [25]. This state represents a survival strategy under stress, allowing microbes to evade conventional detection methods and antimicrobial treatments [25].
This complexity necessitates a multifaceted approach to viability assessment. No single assay can fully capture the spectrum of microbial life and death, making the choice of method critical. This guide provides a technical framework for researchers, contextualizing methods within a broader thesis on establishing gold standards for microbial viability research in pharmaceutical and biotechnological applications.
Core Concepts: The Spectrum of Microbial Viability States
Key Physiological States
Microbial populations can exist in several distinct physiological states, each with defining characteristics:
- Viable and Cultivable: Cells are metabolically active, have intact membranes, and can proliferate on standard culture media.
- Viable but Non-Cultivable (VBNC): Cells are alive and metabolically active but cannot be cultured on media normally supporting their growth. They retain membrane integrity and the potential to resuscitate under favorable conditions [25]. This state is induced by environmental stresses and is characterized by low metabolic activity and altered gene expression [25].
- Dead: Cells have undergone irreversible membrane damage, leading to a loss of barrier function and the release of intracellular contents. This state is terminal and non-reversible [24].
The Dormancy Continuum
The VBNC state is part of a broader survival strategy that includes persister cells. A proposed model suggests that active cells under stress first transition into persisters (a transient, tolerant state), which may further develop into the more stable VBNC state [25]. This dormancy continuum underscores the need for viability assays that go beyond culturability.
Gold-Standard Methods for Assessing Microbial Viability and Death
Viability assays are categorized based on the fundamental cellular properties they measure. Orthogonal validation using multiple methods from different categories is essential for accurate conclusions [26].
Table 1: Categorization of Core Viability Assessment Methods
| Category | Principle | Example Assays | What It Measures | Key Limitations |
|---|---|---|---|---|
| Membrane Integrity | Ability of the cell membrane to exclude dyes or retain enzymes. | Trypan Blue, Propidium Iodide (PI), SYTOX, Lactate Dehydrogenase (LDH) release [26] [24] | Loss of membrane integrity; a marker for necrotic death. | Can miss early-stage apoptosis or VBNC cells; false positives from viable cell stress [24]. |
| Metabolic Activity | Measurement of key biochemical processes (enzymatic activity, redox potential, ATP levels). | Tetrazolium salts (MTT, XTT), Resazurin, ATP assays (BacTiter-Glo) [26] [27] [28] | Cellular metabolism and redox capacity. | Measures activity, not death itself; can be influenced by metabolic shifts not linked to viability [26]. |
| Culturability & Proliferation | Ability to grow and divide on standard laboratory media. | Plate Count (CFU/PFU), Growth Curves (OD) [29] | Reproductive capacity and population growth. | Fails to detect VBNC cells; time-consuming; low throughput [30] [25]. |
| Genetic & Molecular | Detection of biomarkers associated with viability or specific death pathways. | ddPCR (with viability dyes), qPCR, Annexin V (apoptosis), Caspase assays [30] [24] | Membrane integrity (via DNA accessibility), apoptosis markers. | May not differentiate DNA from live/dead cells without specific modifications; complex and costly [30] [29]. |
Table 2: Comparative Analysis of Common Viability Assays
| Assay | Detection Mode | Throughput | Time to Result | Sensitivity | Cost |
|---|---|---|---|---|---|
| Plate Count (CFU) | Visual colony counting | Low | Days | High (for cultivable cells) | Low |
| Flow Cytometry | Fluorescence | Medium-High | Minutes to Hours | High | High |
| ATP Assay | Luminescence | High | Minutes | Very High | Medium |
| Tetrazolium (XTT) | Colorimetric / Fluorometric | High | Hours | Medium | Low |
| qPCR/ddPCR | Fluorescence | Medium | Hours | Very High | High |
Advanced and Emerging Techniques
- Flow Cytometry (FCM): FCM is a powerful tool for multiparametric, single-cell analysis. It can distinguish populations based on size, granularity, and fluorescence from multiple viability dyes simultaneously (e.g., SYBR Green for total cells and PI for dead cells) [29] [31]. It is rapid, accurate, and reproducible, making it suitable for studying dynamic population changes [29].
- Droplet Digital PCR (ddPCR): Modified ddPCR protocols are being developed to quantify only DNA from membrane-intact cells. Samples are treated prior to analysis to prevent amplification from "leaky" dead cells, providing an absolute count of viable microbes [30].
- Fluorescence Lifetime Imaging Microscopy (FLIM): FLIM measures the nanosecond decay time of a fluorophore, which is independent of intensity and photobleaching. This makes it highly quantitative for assessing parameters like membrane potential, a key indicator of viability, especially for VBNC cells [30].
Experimental Protocols for Key Viability Assays
Flow Cytometry for Bacterial Viability and Enumeration
This protocol, adapted from a study on Bdellovibrio bacteriovorus, enables the distinction and counting of mixed microbial populations [29].
Title: Flow Cytometry Viability Workflow
Detailed Protocol:
- Sample Preparation: Harvest bacterial culture and dilute in an appropriate buffer (e.g., PBS) to a recommended concentration of ~10^5 - 10^7 cells/mL.
- Staining: Add nucleic acid stain(s).
- For Total Cell Count: Add SYBR Green I to a final concentration of 1X. Incubate in the dark for 15-30 minutes.
- For Viability (Membrane Integrity): Combine SYBR Green I with a membrane-impermeant dye like Propidium Iodide (PI). PI enters only cells with damaged membranes and quenches SYBR Green fluorescence [29].
- Flow Cytometry Analysis: Analyze samples using a flow cytometer. Establish trigger parameters on the green fluorescence channel (SYBR Green). Use a slow flow rate for accuracy.
- Data Acquisition and Gating:
- Collect a minimum of 10,000 events per sample.
- Use a scatter plot of Side Scatter (SSC) vs. Forward Scatter (FSC) to gate on the bacterial population and exclude debris.
- Use a density plot of Green Fluorescence (e.g., FITC channel) vs. Red Fluorescence (e.g., PE-Texas Red channel for PI).
- Gating Strategy: SYBR Green-positive / PI-negative events are intact/viable cells. SYBR Green-positive / PI-positive events are membrane-compromised/dead cells [29].
- Validation: Validate the FCM counts against a standard method like plaque-forming units (PFU) or quantitative PCR (qPCR) for correlation [29].
Metabolic Activity Assay Using Resazurin
The resazurin assay is a sensitive, fluorometric method for measuring the metabolic activity of a population, commonly used for biofilm viability [32] [33].
Detailed Protocol:
- Reagent Preparation: Prepare a fresh resazurin solution (e.g., 0.15 mg/mL in sterile PBS or culture medium). Filter-sterilize.
- Sample Setup: For biofilms, grow cells in a microtiter plate. Gently wash to remove non-adherent cells. For planktonic cells, seed cells directly in the plate.
- Dye Addition and Incubation: Replace the medium with a fresh medium containing the resazurin solution (typically a 1:10 v/v ratio). Include a negative control (medium with resazurin but no cells) and a blank (medium only). Seal the plate to prevent evaporation and incubate under optimal growth conditions protected from light.
- Kinetic Measurement: Monitor the fluorescence (Excitation: 560 nm, Emission: 590 nm) periodically using a microplate reader. The reduction of blue, non-fluorescent resazurin to pink, highly fluorescent resorufin is proportional to the metabolic activity of the cells.
- Data Analysis: Subtract the background fluorescence of the negative control. Plot fluorescence versus time to determine the metabolic reduction rate. The rate or the endpoint fluorescence can be used as a proxy for viable cell mass [32] [28].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Viability Testing
| Reagent / Kit | Function / Target | Principle of Operation | Example Applications |
|---|---|---|---|
| Propidium Iodide (PI) | Membrane integrity / Dead cells | Nucleic acid intercalator; excluded by intact membranes. Fluoresces red. | Flow cytometry, fluorescence microscopy to identify necrotic populations [26] [31]. |
| SYBR Green I | Nucleic acid / Total cells | Permeant DNA dye; stains all cells. Fluoresces green. | Total cell counting by flow cytometry, often combined with PI [29]. |
| BacTiter-Glo Assay | Metabolic activity / ATP | Luciferase reaction quantifies ATP, present in metabolically active cells. Produces luminescence. | High-throughput screening of antimicrobials; rapid viability assessment [27]. |
| Resazurin (AlamarBlue) | Metabolic activity | Viable cells reduce blue resazurin to fluorescent pink resorufin. | Biofilm viability, drug susceptibility testing, cytotoxicity screens [32] [33]. |
| Annexin V | Apoptosis / Early cell death | Binds phosphatidylserine (PS) exposed on the outer leaflet of apoptotic cells. | Differentiating apoptosis from necrosis, often used with PI [26] [24]. |
| Tetrazolium Salts (XTT, MTT) | Metabolic activity / Redox potential | Reduced by metabolically active cells to colored formazan crystals. | Mitochondrial activity assays (eukaryotes), bacterial redox activity [26] [28]. |
Defining microbial death requires moving beyond a single-parameter approach. The gold standard lies in a holistic strategy that integrates multiple, orthogonal methods to capture the complexity of microbial physiology—from complete lysis to membrane-intact VBNC cells. The future of microbial viability research points toward:
- Multimodal Assays: Combining, for example, membrane integrity staining with metabolic activity measurements in a single, high-throughput workflow [26].
- Advanced Imaging: Techniques like FLIM that provide quantitative, environment-independent measurements of physiological parameters like membrane potential [30].
- Molecular Probes: Development of novel probes that target specific biomarkers of viability beyond culturability, enabling the detection and study of VBNC populations in complex environments like biofilms [25].
By critically selecting assays based on biological relevance rather than convenience and embracing orthogonal validation, researchers can generate robust, reproducible data that truly reflects the state of microbial life and death, ultimately accelerating discovery and development in pharmaceuticals and biotechnology.
A Practical Guide to Viability Methods: From Classic Plating to High-Throughput Innovation
In microbial viability research, the colony-forming unit (CFU) assay has remained the undisputed gold standard for enumerating viable cells across diverse disciplines including clinical diagnostics, drug discovery, and food safety [7] [34]. This culture-dependent technique combines conceptual simplicity with readily available reagents to achieve an enormous dynamic range, commonly measuring between 1 and 100,000,000 viable cells in a sample [7]. The fundamental principle underpinning this method is that one viable cell of a cultivable microorganism, when inoculated onto an appropriate solid nutrient medium and incubated under suitable conditions, will give rise to one visible colony [34]. Despite the emergence of sophisticated culture-independent molecular techniques that analyze microbial communities without cultivation [35], the CFU assay maintains its preeminent position for applications requiring quantitative assessment of cultivable microorganisms, particularly in therapeutic development and antimicrobial efficacy testing [36] [34].
The persistence of plate count methodologies as reference standards reflects their direct measurement of microbial replicative capacity—a functional endpoint that correlates directly with therapeutic efficacy for live biotherapeutic products [37] and with infectious potential in clinical contexts [34]. While culture-independent methods provide comprehensive community profiling capabilities that reveal the vast uncultivated microbial diversity [35] [38], they cannot distinguish between viable and non-viable cells or provide isolates for further characterization and therapeutic use [35]. This technical guide examines both established and emerging plate count methodologies, detailing their protocols, performance characteristics, and appropriate applications within modern microbiological research and drug development contexts.
Fundamental Principles of Culture-Dependent Methods
Culture-dependent methods encompass techniques that grow microorganisms in laboratory conditions, allowing for the isolation and identification of individual microbial species [35]. These approaches are characterized by their ability to enable detailed analysis of microbial physiology, metabolism, and replicative potential—attributes essential for assessing the viability and functionality of microbial agents [35]. The core strength of culture-dependent methods lies in their capacity to isolate and preserve pure microbial strains for further study and potential biotechnological applications, including the development of probiotics, antibiotics, and other bioactive compounds [35] [36].
Despite their foundational role in microbiology, traditional culture-dependent techniques face significant limitations, most notably the "great plate count anomaly" where only a small fraction of microorganisms (typically <1%) in most environments can be cultured under laboratory conditions [35] [36]. This fundamental constraint introduces selection bias by favoring fast-growing or easily cultivable microorganisms, potentially misrepresenting true community structure in complex samples [35]. Additionally, these methods are time-consuming and labor-intensive, making them less suitable for large-scale ecological studies or rapid diagnostics where high-throughput processing is essential [35].
Table 1: Core Characteristics of Culture-Dependent and Culture-Independent Approaches
| Characteristic | Culture-Dependent Methods | Culture-Independent Methods |
|---|---|---|
| Basis of detection | Microbial growth in laboratory conditions | Direct analysis of genetic material |
| Cultivable fraction | Typically <1% of total diversity | Nearly 100% of community diversity |
| Viability assessment | Direct measurement via replication | Indirect inference from markers |
| Throughput potential | Low to moderate | High to very high |
| Information obtained | Physiological characteristics, metabolism | Genetic composition, community structure |
| Strain isolation | Enables pure culture isolation | Does not provide isolates |
| Key applications | Antimicrobial testing, therapeutic development | Community profiling, diversity studies |
Established Plate Count Methodologies
Manual Colony Counting: The Reference Standard
Manual colony counting represents the foundational approach for quantifying viable microorganisms and serves as the reference standard against which all alternative methods are validated [34]. The conventional protocol involves serial dilution of the sample in appropriate buffer, plating onto selective or non-selective solid media, incubation under optimized conditions, and visual enumeration of developed colonies [7] [34]. The accuracy of this method depends critically on achieving appropriate colony densities (typically 30-300 colonies per plate) to minimize confounding factors like colony overlap while maximizing counting precision [34].
Recent studies have reaffirmed the critical importance of manual verification in specific clinical contexts. A 2022 comparative study demonstrated that automated platelet counts significantly underestimated platelet levels in thrombocytopenic patients (mean automated count 58 ± 28×10⁹/L versus manual count 117 ± 13×10⁹/L, p < 0.001), with pseudo-thrombocytopenia observed in 52% of patients primarily due to platelet clumps (42%) and giant platelets (39%) [39]. This systematic underestimation highlights the necessity of manual verification as a gold standard in situations where accurate quantification directly impacts clinical decision-making.
The manual CFU assay protocol involves the following key steps:
- Sample Preparation: Serial decimal dilution in appropriate physiological buffer.
- Plating: Spread plating or pour plating onto appropriate solid media.
- Incubation: Under optimal temperature, atmosphere, and duration for target microorganisms.
- Enumeration: Visual counting of discrete colonies and calculation of CFU concentration.
Despite its resource-intensive nature, manual counting remains the reference method due to its proven reliability, minimal equipment requirements, and adaptability to diverse microbial species and growth characteristics [34].
Automated Colony Counting Systems
Automated colony counting systems have been developed to address the labor-intensive nature of manual counting, particularly in high-volume laboratory settings [34] [40]. These systems typically utilize digital imaging combined with image analysis algorithms to detect and enumerate colonies on standard Petri dishes. Modern platforms can image up to 15 Petri dishes per minute, equivalent to 600 plates per hour, significantly exceeding manual processing capacity [40]. Advanced systems incorporate machine learning models specifically designed for high accuracy counting across diverse plate types, organism morphologies, and plating densities [40].
A comprehensive 2023 evaluation of a commercially available automated system (UVP ColonyDoc-It Imaging Station) revealed important considerations regarding automated counting accuracy [34]. When compared to manual counting across multiple bacterial species (Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, Enterococcus faecium) and the yeast Candida albicans, fully automatic counting without visual correction showed an overall mean difference of 59.7% from manual counts for bacterial species and 71.4% for C. albicans [34]. Performance was particularly compromised at extreme colony densities, with overestimation or underestimation occurring in 29% and 45% of isolates, respectively, and only a moderate relationship (R² = 0.77) with manual counting [34].
Table 2: Performance Comparison of Automated Colony Counting With and Without Visual Correction
| Parameter | Automated Count Without Visual Correction | Automated Count With Visual Correction |
|---|---|---|
| Overall mean difference from manual count | 59.7% (bacteria), 71.4% (yeast) | 1.8% (bacteria), 2.8% (yeast) |
| Proportion with overestimation | 29% | 2% |
| Proportion with underestimation | 45% | 42% |
| Relationship with manual counting (R²) | 0.77 | 0.99 |
| Mean counting time per plate | 30 seconds | 104 seconds |
| Optimal colony density range | Limited performance at extremes | Improved accuracy across range |
When the same system was used with visual correction of automatically generated results on a computer display, the concordance with manual counts improved dramatically, showing an overall mean difference of only 1.8% for bacteria and 2.8% for yeast, with a strong relationship (R² = 0.99) to manual counting [34]. However, this enhanced accuracy came at the cost of increased processing time (104 seconds per plate versus 70 seconds for manual counting), eliminating the time-saving advantage of full automation [34]. These findings underscore that while automated systems offer throughput advantages, manual verification remains essential for applications demanding high quantitative accuracy.
Emerging Methodologies in Viability Assessment
High-Throughput Screening Approaches
Recent innovations in viability assessment methodologies have focused on increasing throughput while maintaining the quantitative rigor of traditional CFU assays. A notable advancement is the Geometric Viability Assay (GVA), which replicates CFU measurements over 6 orders of magnitude while reducing time and consumable requirements by over 10-fold compared to conventional methods [7]. This approach computes a sample's viable cell count based on the distribution of embedded colonies growing inside a pipette tip, leveraging the probability of colony formation at different positions along the cone's axis [7].
The GVA protocol involves:
- Sample Embedding: Fully mixing the microbial sample with melted agarose cooled to ≤55°C.
- Tip Loading: Transferring the mixture to a standard pipette tip.
- Solidification: Allowing the agarose to solidify within the tip.
- Incubation: Ejecting tips into sterile racks and incubating under appropriate conditions.
- Imaging and Analysis: Capturing images and calculating CFU concentration based on colony distribution.
The probability density function governing colony distribution in the cone is defined as PDF(x) = 3x²/h³, where x is the perpendicular distance from the tip along the axis and h is the total length of the cone [7]. The total CFU concentration can be estimated using the formula CFUs/mL = N/(V×∫PDF(x)dx), where N represents the number of colonies in the counted sub-volume and V is the volume of the cone [7]. This approach has demonstrated strong correlation with traditional drop CFU assays (Pearson r = 0.98, P = 4×10⁻¹⁶) across multiple Gram-positive and Gram-negative bacterial species, biofilms, and eukaryotic yeast cells [7].
Another automated approach for viability testing involves the Most-Probable-Number (MPN) technique implemented in systems like TEMPO, which automates the MPN method to estimate populations of total aerobes, coliforms, and Escherichia coli [41]. Studies comparing this automated MPN technique with traditional plating methods for analyzing freshly processed broiler chicken carcasses showed very high correlation (correlation coefficient 0.972) for prechill drip samples, though lower correlation (0.710) for postchill whole carcass rinse samples with lower microbial loads [41]. These automated systems demonstrate the potential for maintaining methodological accuracy while significantly increasing processing capacity.
Application in Live Biotherapeutic Product Development
High-throughput viability assessment has found particular relevance in the development of Live Biotherapeutic Products (LBPs), where preservation of cell viability during manufacturing, transportation, and storage is critical for therapeutic efficacy [37]. Traditional lyophilization processes can compromise cell viability, necessitating protective excipients known as lyoprotectants [37]. While current research centers on disaccharides like trehalose and sucrose, there is growing need to screen broader lyoprotectant alternatives tailored to specific microbial strains [37].
A recently developed high-throughput screening method based on well-plates evaluates lyoprotectants for representative LBPs by lyophilizing various formulations on metal 96-well plates, followed by washing to remove lyoprotectants, dilution, and inoculation into nutritional medium for growth monitoring [37]. This approach demonstrated a significant inverse relationship between log₁₀(cell viability) and growth lag time (Pearson's coefficient = -0.9862), indicating that higher post-lyophilization viability correlates with shorter lag times during recovery growth [37]. Such methods accelerate formulation development for microbial therapeutics by enabling rapid screening of protective compounds under standardized conditions.
Diagram 1: Methodological evolution in microbial viability assessment, showing the progression from manual techniques through automated systems to innovative approaches that balance accuracy with efficiency.
Experimental Protocols for Gold Standard Viability Assessment
Standardized Manual CFU Protocol
Principle: Viable microorganisms are quantified by serially diluting samples, plating onto appropriate media, and counting discrete colonies after incubation.
Materials:
- Appropriate liquid broth medium
- Solid agar plates (non-selective or selective based on application)
- Sterile physiological buffer (e.g., phosphate-buffered saline)
- Sterile dilution tubes
- Automated pipettes and sterile tips
- Spreading devices
- Incubator set to optimal temperature
Procedure:
- Prepare serial decimal dilutions (typically 10⁻¹ to 10⁻⁸) in sterile buffer.
- Pipette 100μL from appropriate dilutions onto center of agar plates.
- Spread inoculum evenly over surface using sterile spreader.
- Invert plates and incubate at optimal temperature until colonies are visible (typically 24-48 hours).
- Count plates containing 30-300 discrete colonies.
- Calculate CFU/mL using formula: CFU/mL = (number of colonies) / (dilution factor × volume plated)
Validation Parameters:
- Perform in triplicate for statistical reliability
- Include positive and negative controls
- Verify typical colony morphology for target microorganism
- Document incubation conditions and duration
Geometric Viability Assay Protocol
Principle: Viable cells are quantified based on spatial distribution of colonies grown in pipette tips, using geometric probability to calculate concentration.
Materials:
- Sterile pipette tips (standard 200μL)
- Low-melt agarose (0.5% final concentration in appropriate medium)
- Triphenyl tetrazolium chloride (TTC) for colony contrast
- Custom imaging setup or mirrorless camera
- Appropriate liquid growth medium
- Empty tip racks for incubation
Procedure:
- Mix microbial sample with melted LB agarose containing TTC, cooled to ≤55°C.
- Aspirate mixture into pipette tips and allow to solidify.
- Eject agarose-containing tips into empty tip rack.
- Incubate overnight at optimal temperature.
- Image tips using custom optical setup.
- Measure position of colonies along tip axis.
- Calculate CFU concentration using probability density function.
Calculation: The probability density function is PDF(x) = 3x²/h³, where x is perpendicular distance from tip and h is total cone length. CFU concentration = N/(V×∫PDF(x)dx), where N is colonies in counted sub-volume and V is cone volume.
Essential Research Reagent Solutions
Table 3: Key Research Reagents for Culture-Dependent Viability Assessment
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Triphenyl tetrazolium chloride (TTC) | Colony contrast enhancement | Incorporated into agarose at 0.5% final concentration for improved visualization [7] |
| Low-melt agarose | Embedding matrix for 3D assays | Maintained at 37°C before embedding for temperature-sensitive samples [7] |
| R2A agar | Cultivation of slow-growing bacteria | Used for environmental isolates with reduced nutrient requirements [38] |
| Soil extract agar | Isolation of common soil bacteria | Captures diversity from complex environmental samples [38] |
| Actinomycetes isolation agar | Selective cultivation of Actinomycetes | Formulated with specific inhibitors to reduce background flora [38] |
| Lyoprotectants (trehalose, sucrose) | Cell preservation during lyophilization | Protective excipients for Live Biotherapeutic Products [37] |
| Cycloheximide | Fungal inhibitor | Added at 60μg/ml to bacterial media to suppress fungal contamination [38] |
| Specialized media (YEM, YMA) | Isolation of specific taxa | Formulations targeting Rhizobium, Agrobacterium, and related species [38] |
Culture-dependent plate count techniques maintain their status as gold standards in microbial viability assessment due to their direct measurement of replicative capacity and proven reliability across diverse applications. While manual methods provide reference-standard accuracy essential for clinical validation and regulatory submissions, emerging automated and high-throughput approaches offer compelling alternatives for screening applications and formulation development. The strategic researcher recognizes that method selection involves balancing accuracy, throughput, and resource constraints, with manual verification remaining essential in contexts where quantitative precision directly impacts therapeutic decisions or clinical outcomes. As microbial biotechnology continues to evolve, particularly in the development of Live Biotherapeutic Products and antimicrobial agents, culture-dependent viability assessment will maintain its foundational role in translating microbial science into therapeutic applications.
Diagram 2: Decision framework for selecting appropriate viability assessment methodologies based on research objectives, sample characteristics, and resource constraints.
Within the broader context of microbial viability research, the gold standard for quantifying viable cells has historically been the colony-forming unit (CFU) assay, which measures culturability [7] [42]. However, a significant limitation of this approach is its inability to detect bacteria in the viable but nonculturable (VBNC) state, a condition induced by environmental stress where cells remain metabolically active but cease to divide on standard media [43] [1]. This critical gap has driven the adoption of culture-independent methods that assess cell viability through alternative, direct physiological criteria.
Assays based on metabolic activity represent a powerful class of culture-independent techniques. These methods function on the principle that viable cells maintain core metabolic functions, such as enzyme activity and membrane transport, even when they are not actively replicating [44] [43]. By measuring these functions, metabolic assays can provide a snapshot of viability that includes VBNC populations, offering a more comprehensive picture of microbial activity in environmental, clinical, and industrial samples. This guide focuses on two foundational strategies within this class: dye reduction assays using compounds like tetrazolium salts and fluorescein diacetate (FDA), and substrate uptake assays.
Theoretical Foundation: Metabolic Activity as a Viability Criterion
The Link Between Metabolism and Viability
Metabolism is defined as the sum of all biochemical reactions in a living organism aimed at maintenance, development, and reproduction [44]. Measurements of metabolic activity serve as proxies for viability because they indicate an active attempt by the cell to sustain itself. Techniques that monitor electron transport system (ETS) activity, membrane transport, and enzymatic hydrolysis provide indirect but reliable correlates of cellular energy production and overall physiological state [44]. It is crucial, however, to distinguish between metabolic activity (a rate) and the mere presence of a metabolic product. True assessment of metabolism requires measuring changes over time with sufficient resolution [44].
Advantages and Limitations
The primary advantage of metabolic assays is their ability to detect the VBNC state, a capability beyond the reach of traditional plating methods [43] [1]. Furthermore, many metabolic assays can be formatted for high-throughput screening, providing rapid results compared to the days-long incubations required for CFU assays [37] [7].
A key limitation is that metabolic activity does not always equate to replicative capacity—the strictest definition of viability. Moreover, some VBNC cells can enter a dormant state with minimal metabolic activity, potentially evading detection by these methods [43] [1]. The reliability of certain assays, particularly tetrazolium reduction, can also be compromised by compounds that influence cellular redox states or by abiotic reduction, leading to potential false positives or negatives [44] [45] [46]. Consequently, the scientific community often recommends using multiple tests that assess different viability criteria (e.g., metabolic activity combined with membrane integrity) for a robust assessment [42].
Tetrazolium Salt Reduction Assays
Biochemical Principle and Workflow
Tetrazolium salt assays are based on the reduction of these compounds by metabolically active cells. Colorless, water-soluble tetrazolium salts readily penetrate the cell membranes of many bacteria. Inside the cell, they act as artificial electron acceptors that are reduced by the electron transport system (ETS), particularly by enzymes linked to NADH or NADPH, as well as dehydrogenases [44]. This reduction process converts the soluble tetrazolium salt into an intensely colored, often insoluble, product called formazan (Figure 1), which accumulates within the cell [44]. The amount of formazan produced is proportional to the metabolic activity of the cell population.
Figure 1. Biochemical pathway of tetrazolium salt reduction. The process involves diffusion of the tetrazolium salt into the cell, its enzymatic reduction by the active electron transport system, and the formation of measurable formazan crystals.
Key Tetrazolium Salts and Their Properties
A variety of tetrazolium salts are used in microbiology, each with distinct chemical properties that make them suitable for different applications (Table 1). These salts differ in their solubility, the solubility of their corresponding formazan products, and their specific spectral properties.
Table 1: Common Tetrazolium Salts and Their Characteristics [44]
| Short Name | Full Name | Tetrazolium Solubility | Formazan Solubility | Absorbance Coefficient (λ_max) | Redox Intermediate Required |
|---|---|---|---|---|---|
| TTC | Triphenyl Tetrazolium Chloride | High (50 mg/mL) | Insoluble | 14,320 cm⁻¹·M⁻¹ | No |
| MTT | Thiazolyl Blue Tetrazolium Bromide | Moderate (5 mg/mL) | Insoluble | 13,000-16,900 cm⁻¹·M⁻¹ (578 nm) | No |
| INT | Iodonitrotetrazolium Chloride | Moderate (4 mg/mL) | Insoluble | 12,000 cm⁻¹·M⁻¹ (480-490 nm) | No |
| XTT | 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide | Low (2.5 mg/mL) | Soluble | 21,600-23,800 cm⁻¹·M⁻¹ (470-475 nm) | Recommended |
| WST-8 | 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium | High (50 mg/mL in H₂O) | Soluble | 30,700 cm⁻¹·M⁻¹ (460 nm) | Required |
Detailed Experimental Protocol: MTT Assay
The MTT assay is one of the most commonly used tetrazolium-based methods, though it requires steps to solubilize the insoluble formazan product for measurement [44] [45].
Materials:
- MTT reagent: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, dissolved in PBS or saline to a typical stock concentration of 5 mg/mL. Filter sterilize and store protected from light.
- Solubilization solution: Acidified isopropanol (0.04 N HCl in isopropanol) or DMSO.
- Multi-well plates and a spectrophotometric plate reader.
Procedure:
- Sample Preparation: Prepare a bacterial suspension in an appropriate buffer or growth medium. It is critical to include a blank control (medium without cells) and a negative control (e.g., formaldehyde-fixed cells) to account for abiotic reduction [44].
- Incubation with MTT: Add the MTT stock solution to the sample to achieve a final concentration typically between 0.2 - 1 mg/mL. Incubate the mixture for a predetermined time (e.g., 1-4 hours) at the optimal temperature for the microorganism, protected from light.
- Solubilization: Carefully remove the supernatant (if the formazan is insoluble). Add the solubilization solution (e.g., acidified isopropanol) to dissolve the formazan crystals. Mix thoroughly until all crystals are dissolved.
- Measurement: Transfer the solution to a cuvette or read directly in the plate well. Measure the absorbance against the blank control at the peak wavelength for formazan (typically 560-570 nm for MTT formazan).
Critical Considerations:
- Toxicity: Some tetrazolium salts, like INT and CTC, can be toxic to certain bacterial strains, potentially killing them during the assay and leading to underestimation of viability [46].
- Abiotic Reduction: The presence of reducing agents in the sample (e.g., plant extracts, flavonoids) can cause non-biological reduction of the tetrazolium salt, yielding false positive signals. A control with fixed cells (e.g., with 1.5-4% formaldehyde) is essential to correct for this [44].
- Assay Limitations: Compounds that affect mitochondrial function or increase reactive oxygen species (ROS) can disrupt the MTT reduction rate, leading to false results. For instance, a study comparing MTT with a metabolism-independent assay showed that MTT could indicate antagonism between two compounds, while the other assay showed synergy, highlighting the potential for misinterpretation [45].
Fluorescein Diacetate (FDA) Hydrolysis Assay
Biochemical Principle and Workflow
The FDA assay measures the activity of non-specific intracellular esterases, lipases, and proteases [43]. FDA is a non-polar, non-fluorescent compound that passively diffuses across the intact cell membrane. Once inside a viable cell, active intracellular enzymes hydrolyze FDA, removing the acetate groups and converting it into fluorescein, a polar and highly fluorescent molecule (Figure 2). Because fluorescein is charged, it is trapped inside cells with intact membranes, leading to an accumulation of fluorescence that can be quantified.
Figure 2. Biochemical pathway of Fluorescein Diacetate (FDA) hydrolysis. The non-fluorescent FDA diffuses into the cell, where it is hydrolyzed by nonspecific intracellular enzymes to yield fluorescent fluorescein, which is retained in cells with intact membranes.
Detailed Experimental Protocol
Materials:
- FDA stock solution: Dissolve FDA in acetone or DMSO to a typical concentration of 1-10 mg/mL. Store aliquoted at -20°C protected from light and moisture.
- Buffer: An appropriate physiological buffer, such as phosphate-buffered saline (PBS) or a sodium phosphate buffer (e.g., 50 mM, pH 7.4).
- Fluorescence plate reader or fluorometer.
Procedure:
- Sample Preparation: Suspend bacterial cells in the chosen buffer. The cell density should be optimized to ensure the fluorescence signal is within the linear range of the detector.
- Incubation with FDA: Add the FDA stock solution to the sample to achieve a final concentration typically between 5-20 µg/mL. Mix immediately and incubate at the desired temperature for a specific time (e.g., 10-60 minutes), protected from light.
- Termination and Measurement: The reaction can be stopped by diluting the sample with cold buffer or, in some protocols, by adding a quenching agent. Measure the fluorescence intensity using an excitation wavelength of ~490 nm and an emission wavelength of ~520 nm.
Critical Considerations:
- pH Sensitivity: The fluorescence intensity of fluorescein is highly dependent on pH. An acidic environment can protonate fluorescein, allowing it to leak out of cells via passive efflux and reducing the measured signal. The hydrolysis reaction itself produces acetic acid, which can lower the intracellular pH and potentially inhibit enzyme activity [43] [1].
- Efflux and Quenching: If the intracellular concentration of fluorescein becomes too high, a self-quenching effect can occur, leading to a non-linear relationship between cell number and fluorescence [43]. Furthermore, some cells may actively export fluorescein.
- Enzyme Specificity: The assay relies on non-specific enzymes, whose optimal activity can vary greatly with pH and other conditions, making universal optimization challenging [43].
Substrate Uptake Assays
Glucose Uptake Assays
Glucose uptake is a fundamental metabolic activity in many viable bacteria. This can be measured using modified glucose analogs or by monitoring glucose consumption.
2-NBDG Uptake: A common method uses the fluorescent glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG). Viable bacteria with an active glucose transport system import 2-NBDG. Once inside, it is metabolized into a non-fluorescent compound. The decrease in extracellular fluorescence over time, measured with a fluorometer, is thus proportional to metabolic activity [43] [1]. A major limitation is that not all bacterial species can transport 2-NBDG; for example, strains of Bacillus cereus, Aeromonas hydrophila, and some E. coli cannot take it up [1].
Enzymatic Glucose Consumption: This approach measures the depletion of glucose from the medium. A sample is incubated with bacteria, and the supernatant is recovered. The remaining glucose is quantified using an enzymatic assay, typically involving glucose oxidase, which produces hydrogen peroxide (H₂O₂). The H₂O₂ then reacts with a chromogen (e.g., o-dianisidine) in a peroxidase-catalyzed reaction to produce a colored compound that can be measured colorimetrically [1]. A decrease in glucose concentration in the test sample compared to a cell-free control indicates glucose consumption by the viable cells.
Advanced Metabolic Labeling
Recent advances involve the use of bio-orthogonal chemistry and metabolic labeling. For instance, probes like TPEPy-d-Ala can be incorporated into bacterial cell walls during active growth and remodeling. Once incorporated into peptidoglycan, the probe's mobility is restricted, leading to a fluorescence "turn-on" signal that indicates metabolic activity [43] [1]. Similarly, other dyes can distinguish between Gram-positive and Gram-negative bacteria by targeting specific cell wall components (e.g., Kdo-N3 in Gram-negative lipopolysaccharide vs. D-Ala-N3 in Gram-positive peptidoglycan) that are metabolically incorporated [43].
Comparative Analysis and Practical Implementation
Comparison of Method Characteristics
The choice of assay depends on the specific application, required throughput, and available instrumentation. The table below summarizes the key features of the discussed methods.
Table 2: Comparison of Metabolic Activity Assays for Microbial Viability
| Assay | Target / Principle | Key Advantage | Key Limitation | Throughput | Detection Method |
|---|---|---|---|---|---|
| Tetrazolium (e.g., MTT) | ETS Activity / Reductases | Measures core energy metabolism; various salt options | Toxicity of some salts; abiotic reduction; false results with metabolic modulators [44] [45] [46] | Medium-High | Absorbance |
| FDA Hydrolysis | Non-specific Esterase Activity | Simple; indicates general enzymatic activity | Highly sensitive to pH; potential for dye efflux and quenching [43] [1] | Medium | Fluorescence |
| 2-NBDG Uptake | Glucose Transport & Metabolism | Direct measure of a central carbon source uptake | Not applicable to all bacterial species [43] [1] | Medium | Fluorescence (Loss) |
| Enzymatic Glucose Assay | Glucose Consumption | Measures depletion of a natural substrate | Indirect measure; requires separation of cells; background glucose in samples [1] | Low-Medium | Absorbance |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for Metabolic Assays
| Reagent / Material | Function in the Assay | Example & Notes |
|---|---|---|
| Tetrazolium Salts (MTT, XTT, CTC) | Electron acceptors reduced by active ETS to colored formazan. | MTT for absorbance; CTC for fluorescence microscopy and flow cytometry [44]. |
| Fluorescein Diacetate (FDA) | Substrate for intracellular hydrolases, producing fluorescent fluorescein. | Requires optimization of pH and incubation time to prevent efflux [43]. |
| 2-NBDG | Fluorescent glucose analog taken up by active glucose transporters. | Check for compatibility with the bacterial species under study [1]. |
| Triphenyl Tetrazolium Chloride (TTC) | Tetrazolium salt forming red, insoluble formazan. | Often used in solid media or agarose tips for colony staining [7]. |
| Solubilization Solution (DMSO, Isopropanol) | Dissolves insoluble formazan crystals for absorbance reading. | Required for MTT and other assays producing insoluble formazan [44] [45]. |
| Selection Media (Antibiotics) | Inhibits growth of non-target bacteria in viability assays. | Used in WST-8 assays to differentiate Gram-positive and Gram-negative bacteria [46]. |
Integrated Experimental Workflow
A generalized workflow for implementing these assays, from sample preparation to data interpretation, is outlined below.
Figure 3. Generalized workflow for metabolic activity assays. This flowchart outlines the common steps involved in performing dye reduction and substrate uptake assays, emphasizing the critical need for appropriate controls.
Metabolic assays based on dye reduction and substrate uptake are indispensable tools in the modern microbiology toolkit. They provide a rapid, culture-independent means to assess microbial viability, crucially extending detection to include VBNC cells that are invisible to traditional CFU counts. However, no single assay is perfect. Factors such as the microbial population being studied, the sample matrix, and the presence of metabolic inhibitors must be carefully considered. As the field advances, the integration of these methods with other viability criteria—such as membrane integrity and direct activity measurements—along with the development of more robust, high-throughput formats like the geometric viability assay [7], will continue to enhance our ability to accurately and comprehensively understand microbial life and death. For critical applications, the consensus is clear: relying on a single method is risky, and a multi-faceted approach to viability assessment is the most scientifically sound strategy [45] [42].
Within the framework of microbial viability research, the ability to rapidly and accurately distinguish between live and dead bacterial cells is paramount across diverse fields, including clinical diagnostics, pharmaceutical development, and food and water safety. While traditional culture-based methods, which rely on bacterial culturability, have long been considered a gold standard, they possess significant limitations, most notably their inability to detect bacteria in the viable but nonculturable (VBNC) state [1]. This has driven the adoption of culture-independent assays that probe fundamental aspects of cell physiology.
Among these, viability assessments based on membrane integrity represent a crucial strategy. An intact cytoplasmic membrane is a fundamental characteristic of a viable cell, and its compromise is often considered a "point of no return" in the cell death pathway [47]. This technical guide focuses on a widely used fluorescence-based assay that employs the nucleic acid stains SYTO 9 and propidium iodide (PI) to assess bacterial viability by reporting on the integrity of the cellular membrane [48].
The Principle of Membrane Integrity Staining
The SYTO 9/PI assay operates on a straightforward principle: differential membrane permeability. SYTO 9 is a green-fluorescent nucleic acid stain that is membrane-permeant and can label all cells in a population, regardless of their viability status. In contrast, propidium iodide (PI) is a red-fluorescent stain that is membrane-impermeant and can only enter cells with compromised membrane barriers [48] [49].
When used in combination, and with PI in sufficient excess, a competitive interaction occurs. In live cells with intact membranes, only SYTO 9 enters, resulting in green fluorescence. In dead cells with damaged membranes, both dyes enter, but PI's higher affinity for nucleic acids causes it to displace SYTO 9, leading to a reduction in green fluorescence and a dominant red fluorescence signal [47] [50]. This interaction is further enhanced by Förster Resonance Energy Transfer (FRET), where the emission from SYTO 9 is absorbed by PI, further muting green fluorescence and amplifying red fluorescence in dead cells [47].
Detailed Experimental Protocols
The application of the SYTO 9/PI assay can be adapted for different downstream detection methods. The following protocols provide detailed methodologies for microscopy and flow cytometry.
Protocol for Fluorescence Microscopy
This protocol is adapted from the standard LIVE/DEAD BacLight Bacterial Viability Kit for microscopy and is ideal for visualizing the spatial distribution of live and dead cells within a population, such as in biofilms [48].
1. Culture Conditions and Preparation of Bacterial Suspensions
- Grow a 25 mL bacterial culture to late log-phase in an appropriate nutrient broth (e.g., LB Broth).
- Centrifuge the culture at 10,000 × g for 10 minutes to pellet the cells.
- Carefully remove the supernatant and resuspend the pellet in 2 mL of wash buffer (e.g., 0.85% NaCl). Phosphate buffers are not recommended as they may decrease staining efficiency.
- Dilute 1 mL of this suspension into 20 mL of wash buffer and incubate at room temperature for 1 hour, mixing every 15 minutes.
- Repeat the centrifugation and resuspension steps twice more, finally resuspending the pellet in 10 mL of wash buffer [48].
2. Staining and Imaging
- Combine equal volumes of the SYTO 9 and PI stock solutions from the viability kit in a microfuge tube.
- Add 3 µL of the combined dye mixture to each milliliter of the prepared bacterial suspension.
- Incubate the stained suspension at room temperature in the dark for 15 minutes.
- Pipette 5 µL of the stained suspension onto a glass microscope slide and cover with a coverslip.
- Image the cells using a fluorescence microscope with standard FITC (for SYTO 9) and Texas Red (for PI) filter sets [48].
Protocol for Flow Cytometry
Flow cytometry allows for the high-throughput, quantitative analysis of cell populations. This protocol, optimized for yeast but applicable to bacteria with validation, details a standardized approach [47].
1. Sample Preparation and Stress Treatment
- Grow cells to the mid-log phase in an appropriate medium.
- Pellet cells by centrifugation (e.g., 3,000 × g for 5 min) and apply the desired stressor or drug treatment in a defined volume.
- Incubate the plate for the required time (e.g., 120 min at 30°C with shaking).
2. Staining for Flow Cytometry
- Prepare a sterile staining buffer. Note: 0.85% saline buffer is recommended as it produces minimal staining artifacts compared to water or growth media [47].
- After treatment, pellet the cells and carefully remove the supernatant by aspiration.
- Resuspend the cells in the sterile staining buffer to a standardized optical density (e.g., OD600 = 1).
- Prepare a working stock of PI (e.g., 0.2 mM) and a fresh working stock of SYTO 9 (e.g., 33.4 µM).
- Add the dyes to the cell suspension at the optimized concentration and incubate in the dark for 15-30 minutes.
- Analyze the samples on a flow cytometer, using unstained and single-stained controls to set up the instrument and define the live (green) and dead (red) populations [47].
Key Experimental Parameters
Table 1: Critical Experimental Parameters for SYTO 9/PI Staining
| Parameter | Microscopy Protocol [48] | Flow Cytometry Protocol [47] | Significance |
|---|---|---|---|
| Staining Buffer | 0.85% NaCl or other wash buffer (phosphate not recommended) | 0.85% saline buffer (minimizes artifacts) | Buffer composition affects staining efficiency and dye interaction. |
| Dye Incubation | 15 minutes at room temperature in the dark | 15-30 minutes in the dark | Ensures sufficient dye uptake and interaction; protects dyes from photobleaching. |
| Dye Ratio | Equal volumes of SYTO 9 and PI stock solutions | Defined working stocks (e.g., 33.4 µM SYTO 9, 0.2 mM PI) | A sufficient excess of PI is critical for the competitive displacement of SYTO 9 in dead cells. |
| Cell Concentration | Washed, concentrated suspension | Standardized to OD600 = 1 | Prevents over- or under-staining and ensures accurate fluorescence measurements. |
Quantitative Data and Spectral Information
The accurate interpretation of SYTO 9/PI staining requires an understanding of the dyes' spectral profiles and the quantitative outcomes of the assay under different conditions.
Table 2: Spectral Properties and Staining Outcomes of SYTO 9 and Propidium Iodide
| Property | SYTO 9 | Propidium Iodide (PI) |
|---|---|---|
| Excitation/Emission | 480/500 nm [48] | 490/635 nm [48] |
| Standard Microscope Filter | FITC [48] | Texas Red [48] |
| Membrane Permeability | Permeant (enters all cells) [49] | Impermeant (enters only cells with compromised membranes) [49] |
| Fluorescence in Live Cells | Green | None |
| Fluorescence in Dead Cells | Quenched (displaced by PI) | Red |
| Storage Conditions | ≤20°C, protect from light [48] | ≤20°C, protect from light [48] |
Applications and Validation in Research
The SYTO 9/PI assay is extensively used in microbial research to rapidly assess cell viability under various conditions.
- Antibiotic Susceptibility Testing: The assay has been successfully applied to detect the killing action of lytic antibiotics (e.g., ampicillin, polymyxin B) against E. coli in near real-time. The change in fluorescence signal provides a rapid readout of bactericidal activity, potentially informing treatment decisions faster than culture-based methods [49].
- Quantifying Post-Stress Survival: In yeast models like Candida glabrata, an optimized SYTO 9/PI protocol coupled with flow cytometry provides a fast and scalable alternative to Colony Forming Unit (CFU) assays. It can distinguish between live, damaged, and dead cell populations immediately after stress, offering complementary information to culturability, which measures the ability to recover and reproduce [47].
A key validation step involves comparing the results of membrane integrity staining with other viability criteria. For instance, a study on E. coli and Staphylococcus epidermidis biofilms showed that while a high percentage of cells stained PI-positive (suggesting death), a majority were still metabolically active, as determined by fluorescein diacetate (FDA) staining [50]. This discrepancy underscores the importance of using orthogonal methods to confirm viability, especially in complex structures like biofilms.
Limitations and Critical Considerations
Despite its utility, the SYTO 9/PI assay has several important limitations that researchers must consider when interpreting data.
- Overestimation of Death in Biofilms: A significant limitation is the potential for false-positive dead signals in adherent cells and biofilms. This is often due to the presence of extracellular nucleic acids (eNA) in the biofilm matrix. PI can bind to this eNA, creating a red fluorescent "coating" that can be mistaken for staining from within a dead cell, leading to a substantial overestimation of cell death [50]. In some 24-hour biofilms, over 75% of cells stained red with PI despite 68% being metabolically active and over 80% being cultivable [50].
- The Viable But Nonculturable (VBNC) State: Bacteria in the VBNC state have an intact membrane but are dormant and non-culturable. The SYTO 9/PI assay will correctly identify these cells as "live" based on membrane integrity, which is a key advantage over culture-based methods [1]. However, this also means the assay cannot distinguish between actively growing and dormant viable cells.
- Dependence on Membrane Integrity as a Viability Marker: While membrane rupture is a definitive marker of cell death, some bactericidal antibiotics operate via non-lytic mechanisms (e.g., ciprofloxacin). In these cases, killing may not be immediately detectable by the SYTO 9/PI assay, as the membrane can remain intact for some time after the cells are irreversibly committed to death [49].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents and Materials for SYTO 9/PI Assays
| Item | Function / Description | Example / Comment |
|---|---|---|
| LIVE/DEAD BacLight Kit | Commercial kit containing optimized concentrations of SYTO 9 and PI. | L7012 (for microscopy) [48]. Components can also be purchased separately. |
| SYTO 9 Stain | Green-fluorescent nucleic acid stain; labels all cells. | 3.34 mM stock solution; store at ≤20°C, protected from light [48] [47]. |
| Propidium Iodide (PI) | Red-fluorescent nucleic acid stain; labels dead cells. | 20 mM stock solution; store at ≤20°C, protected from light [48] [47]. |
| Staining Buffer | Isotonic solution for washing and resuspending cells. | 0.85% NaCl is commonly used and recommended over phosphate buffers [48] [47]. |
| Nutrient Broth | For culturing bacteria prior to assay. | e.g., LB Broth, Tryptic Soy Broth (TSB) [48] [49]. |
| Fluorescence Microscope | For visualizing stained cells. | Requires FITC and Texas Red filter sets [48]. |
| Flow Cytometer | For high-throughput, quantitative analysis of cell populations. | Allows distinction of live, dead, and damaged subpopulations [47]. |
The accurate assessment of microbial viability remains a cornerstone of microbiology, with profound implications for drug development, public health, and fundamental research. The colony-forming unit (CFU) assay has long been considered the gold standard for enumerating viable cells, combining simplicity with a dynamic range covering 1 to 100,000,000 viable cells per sample [7]. This method relies on the fundamental principle that a viable cell will proliferate to form a visible colony when provided with appropriate nutrients and conditions. However, the CFU assay presents significant limitations in modern research contexts: it is notoriously time-intensive, resource-consuming, and generates substantial plastic waste [51] [7]. These constraints become particularly problematic in drug discovery campaigns, high-throughput screening environments, and situations requiring rapid results.
In response to these challenges, three innovative approaches have emerged that either enhance or circumvent traditional plating methods: the Geometric Viability Assay (GVA), which reimagines the physical format of colony growth; flow cytometry, which enables rapid single-cell analysis; and microcalorimetry, which directly measures metabolic heat production. These methods represent a paradigm shift in viability testing, offering researchers sophisticated tools that provide not just viability counts, but additional layers of biological information. This technical guide examines these innovative platforms, detailing their principles, methodologies, and applications within the context of modern microbial research demands.
Methodological Deep Dive: Principles and Protocols
Geometric Viability Assay (GVA): Geometric Principles in Viability Assessment
The Geometric Viability Assay (GVA) represents a fundamental rethinking of the colony counting paradigm by leveraging geometric principles to eliminate the need for serial dilutions. This method utilizes the conical shape of a standard pipette tip to create a natural gradient of colony density, with the probability of colony formation varying predictably along the cone's axis [52] [7].
Theoretical Foundation and Mathematical Model The core principle of GVA hinges on the relationship between a cone's cross-sectional area and the axial position. The probability density function (PDF) governing colony formation is expressed as:
$${\rm{PDF}}\left(x\right)=\frac{3{x}^{2}}{{h}^{3}}$$
where x represents the perpendicular distance from the tip along the axis and h is the total length of the cone [7]. This mathematical relationship means colonies are less likely to form near the tip where the cross-sectional area is smaller, effectively creating an inherent dilution series within a single tip. The total CFU concentration can then be estimated by measuring the positions of a subset of colonies within a defined sub-volume, using the formula:
$${\rm{CFUs/mL}}=\frac{N\left(x\right) | {x}{1}\le x < {x}{2}}{V {\int }{{x}{1}}^{{x}_{2}}{\rm{PDF}}\left(x\right){dx}}$$
where N(x) represents the number of colonies counted between positions x1 and x2, and V is the volume of the cone [7]. Remarkably, simulations demonstrate that the CFU estimate typically falls within a factor of 2 of the true value in 97% of cases based on the positions of just the first 10 colonies, even when the tip contains over 10,000 colonies [7].
Experimental Protocol The GVA procedure consists of several key steps that can be completed with standard laboratory equipment:
- Sample Preparation: Cells are suspended in an appropriate liquid medium. For anaerobic organisms like Clostridium perfringens, this step must occur in an anaerobic environment [53].
- Hydrogel Embedding: The sample is mixed with molten agarose (0.5-0.66% final concentration) cooled to ≤55°C (or 37°C for temperature-sensitive samples) [52] [7]. The medium is typically supplemented with nutrients supporting growth of the target microbe (e.g., LB for bacteria, YEPD for yeast) [7].
- Solidification: The cell-agarose mixture is aspirated into a pipette tip and allowed to solidify at room temperature or in a refrigerator [52].
- Incubation: Tips are ejected into a tip rack and incubated under appropriate conditions (temperature, atmosphere) until colonies form. For anaerobes, the entire tip rack is placed in an anaerobic jar system [53].
- Imaging and Analysis: Colonies are counted and their positions measured. This can be achieved through several approaches:
Visualization of GVA Workflow The following diagram illustrates the complete GVA procedure from sample preparation to data analysis:
GVA has been successfully validated with diverse microorganisms including Gram-negative (Escherichia coli, Pseudomonas aeruginosa) and Gram-positive bacteria (Bacillus subtilis), fungi (Saccharomyces cerevisiae), biofilms, and environmental samples [7]. Recent adaptations have extended its utility to anaerobic pathogens like Clostridium perfringens by incorporating anaerobic jars and alternative stains like Bromocresol Green for better contrast [53]. The method reduces operator time by over 30-fold and consumable usage by 10-fold compared to traditional CFU assays while maintaining a dynamic range of 1 to 1,000,000 viable cells [7].
Flow Cytometry: Single-Cell Viability Assessment
Flow cytometry offers a radically different approach to viability assessment by analyzing individual cells based on physiological properties rather than reproductive capacity. This method provides rapid results (minutes to hours) and can distinguish subpopulations within heterogeneous samples, making it invaluable for detecting mixed populations with varying viability states [54] [55].
Physiological Principles and Stain Mechanisms Flow cytometry-based viability assessment relies on fluorescent dyes that report on different aspects of cellular physiology:
- Membrane Integrity: Viability kits like the LIVE/DEAD BacLight Bacterial Viability Kit utilize nucleic acid stains with different membrane permeability properties. SYTO 9 penetrates all cells, while propidium iodide (PI) only enters cells with compromised membranes, causing a spectral shift that distinguishes live (green) from dead (red) populations [55].
- Metabolic Activity: The BacLight RedoxSensor kits detect the activity of bacterial oxidases and reductases, providing information about cellular vitality beyond mere membrane integrity. Metabolically active cells reduce the dye, resulting in enhanced fluorescence [55].
- Membrane Potential: The BacLight Bacterial Membrane Potential Kit uses DiOC₂(3), which exhibits green fluorescence in all cells but becomes concentrated in cells maintaining a membrane potential, causing a shift to red fluorescence in healthy cells [55].
Experimental Protocol A typical flow cytometry viability protocol involves these key steps:
- Sample Preparation: Bacterial suspensions are diluted to appropriate concentrations (typically 10⁵–10⁶ cells/mL) in buffer to avoid coincidence detection [56].
- Staining: Samples are incubated with viability dyes for 5-15 minutes. Recent safety-focused protocols have explored replacing hazardous dyes like PI (carcinogenic, mutagenic, and reprotoxic) with safer alternatives such as eFluor Fixable Viability Dyes, which maintain fluorescence after fixation [54].
- DNA Staining (if needed): Since bacteria have size and granularity similar to background particles, a DNA staining step with SYTO or DRAQ5 is often added to differentiate true bacterial events from debris [54].
- Flow Cytometric Analysis: Samples are run through the flow cytometer with appropriate laser excitation and detection filters matched to the dyes used. For volumetric counting, no counting beads are needed as modern instruments can determine concentration based on the sampled volume [56].
- Data Analysis: Populations are gated based on forward versus side scatter (FSC-H vs. SSC-H) to identify bacterial cells, followed by analysis on fluorescence plots (e.g., FL1 vs. FL3) to distinguish live and dead subpopulations [56].
Visualization of Flow Cytometry Viability Assessment The diagram below outlines the key steps and decision points in flow cytometry-based viability analysis:
This method has been validated across multiple bacterial species (both Gram-positive and Gram-negative) and instrument platforms, demonstrating reliability when properly optimized for specific bacterial strains and dye combinations [54]. The ability to fix samples after staining with certain dyes further enhances safety when working with pathogens [54].
Microcalorimetry: Metabolic Activity as a Viability Proxy
Isothermal microcalorimetry (IMC) takes a label-free approach to viability assessment by directly measuring the heat produced by microbial metabolic activity. This method operates on the fundamental principle that all metabolic processes are exothermic, with the heat flow (μW/s) directly proportional to the metabolic rate of microorganisms [56] [57].
Theoretical Foundation in Bioenergetics The heat production in microbial systems stems from the totality of biochemical reactions, which can be simplified as:
Substrate₁ + ... Substrateₙ → Biomass + Byproduct₁ + ... Byproductₘ + Heat
The thermal power (heat flow per unit time) serves as a proxy for metabolic activity, while the total heat integrated over time relates to the extent of metabolic reactions [57]. IMC is exceptionally sensitive, capable of detecting as few as 10⁴ active microbial cells, and in theory, can detect a single microbe given sufficient incubation time [57]. This sensitivity, combined with its non-invasive nature, makes it particularly valuable for monitoring slow-growing organisms, assessing viability in turbid or opaque samples, and detecting metabolically active but non-culturable (VBNC) cells [56] [57].
Experimental Protocol A standard IMC viability protocol involves these key steps:
- Instrument Preparation: The isothermal microcalorimeter (e.g., calScreener) is calibrated and stabilized at the desired temperature (e.g., 37°C for many pathogens) [56].
- Sample Loading: Bacterial suspensions (30-300 μL) are added to sterile ampoules or vials, often with nutritional media. For viability enumeration, serial dilutions may be prepared similarly to CFU assays [56].
- Measurement: Sealed ampoules are placed in the instrument, and heat flow is monitored continuously using dedicated software (e.g., calView 2.0) typically for 24-48 hours [56].
- Data Analysis: Heat flow curves are analyzed to extract parameters such as time to detection peak, maximum heat flow, and total heat production. These parameters can be correlated with viable counts from other methods (e.g., flow cytometry, plate counts) to establish calibration curves [56] [57].
- Quantification: In proof-of-concept studies, a significant inverse relationship has been observed between log₁₀(cell viability) and growth lag time (Pearson's coefficient = -0.9862), enabling viability estimation from metabolic kinetics [56] [37].
Visualization of Microcalorimetry Workflow The following diagram illustrates the process of metabolic viability assessment using isothermal microcalorimetry:
IMC has proven particularly valuable in probiotic and Live Biotherapeutic Product (LBP) development, where it helps characterize strain metabolism under different formulation conditions and can detect metabolic activity even when cells fail to form colonies on standard agar plates [56]. The method's ability to handle diverse media matrices without interference from turbidity or viscosity further expands its application potential [56].
Comparative Analysis of Methodologies
Technical Specifications and Performance Metrics
The table below provides a systematic comparison of the key technical parameters across the three high-throughput methods and the traditional CFU assay:
Table 1: Performance Comparison of Microbial Viability Assessment Methods
| Parameter | Traditional CFU | Geometric Viability Assay (GVA) | Flow Cytometry | Isothermal Microcalorimetry (IMC) |
|---|---|---|---|---|
| Measurement Principle | Reproductive capacity | Reproductive capacity & geometric distribution | Membrane integrity, metabolic activity, & physiology | Metabolic heat production |
| Dynamic Range | 1–10⁸ CFU/mL [7] | 1–10⁶ CFU/mL [7] | 10⁴–10⁸ cells/mL [55] | 10⁴–10⁸ cells/mL [57] |
| Time to Results | 24–48 hours [56] | ~24 hours [7] | 5–30 minutes (+ incubation if needed) [55] | 2–48 hours (real-time monitoring) [56] |
| Throughput Capacity | Low (manual intensive) | High (1,200 samples/researcher/day) [7] | Very High (96-well plate format) | Medium (multiple channels simultaneous monitoring) |
| Key Advantages | Gold standard, simple, versatile | 30× faster than CFU, low waste, cost-effective [7] | Rapid, subpopulation resolution, multiple parameters | Label-free, detects VBNC cells, works with opaque samples [56] [57] |
| Primary Limitations | Labor intensive, high plastic waste, slow | New method, requires specialized analysis | Cannot distinguish culturable vs non-culturable viable cells | Indirect measurement, requires correlation with other methods [56] |
| Optimal Applications | Low-throughput validation, regulatory testing | High-throughput screening, antibiotic susceptibility testing [53] | Rapid screening, population heterogeneity studies | Stress response monitoring, formulation optimization [56] |
Research Reagent Solutions and Essential Materials
Successful implementation of these methods requires specific reagents and materials optimized for each platform:
Table 2: Essential Research Reagents and Materials for High-Throughput Viability Assays
| Method | Key Reagents/Materials | Function/Purpose | Examples/Specifications |
|---|---|---|---|
| GVA | Low-melt agarose | Hydrogel matrix for 3D colony growth | 0.5–0.66% in appropriate growth medium [7] [53] |
| Tetrazolium chloride (TTC) | Colony contrast enhancement | Visualizes colonies as red formazan products [7] | |
| Bromocresol Green (BG) | Alternative stain for anaerobes | pH-sensitive dye turns colonies blue against colorless background [53] | |
| Pipette tips | Micro-scale culturing vessel | Standard P200 tips [52] | |
| Flow Cytometry | LIVE/DEAD BacLight Kit | Membrane integrity assessment | SYTO 9 & PI stains, 488 nm laser [55] |
| BacLight RedoxSensor Kit | Metabolic activity measurement | Detects reductase activity, compatible with fixation [55] | |
| Bacterial Membrane Potential Kit | Membrane potential assessment | DiOC₂(3) stain, red/green fluorescence shift [55] | |
| DNA stains (SYTO, DRAQ5) | Background discrimination | Differentiates bacteria from debris [54] | |
| Micro-calorimetry | Culture media | Supports metabolic activity | MRS for lactobacilli, TGB for anaerobes [56] [53] |
| Calorimeter ampoules | Sample containment | Titanium vials with plastic inserts [56] | |
| Reference standards | Instrument calibration | Chemical reactions with known enthalpy [57] |
The evolution of microbial viability assessment from the traditional CFU assay to sophisticated high-throughput platforms represents significant progress in microbiological methodology. Each innovative approach—GVA, flow cytometry, and microcalorimetry—offers unique advantages that address specific limitations of gold standard methods while introducing new capabilities for advanced research applications.
GVA stands out for its ability to maintain the biological relevance of colony formation while dramatically improving efficiency and reducing waste. Its recent adaptation for anaerobic bacteria [53] and compatibility with antibiotic susceptibility testing [53] demonstrates its expanding utility in challenging research contexts. Flow cytometry provides unparalleled speed and the ability to resolve heterogeneous subpopulations, making it ideal for time-sensitive applications and studies of population dynamics. Microcalorimetry offers a truly label-free approach that captures metabolic activity even from viable but non-culturable cells, providing insights that complement reproduction-based assays.
Rather than viewing these methods as replacements for traditional CFU assays, the most powerful research approach involves strategic integration of multiple platforms. CFU remains essential for validation and in contexts where reproductive capacity is the primary endpoint. However, incorporating these high-throughput innovations enables researchers to address more complex biological questions, screen larger compound libraries, and obtain results with unprecedented speed and efficiency. As these technologies continue to mature and become more accessible, they promise to accelerate discovery across microbiology, pharmaceutical development, and clinical diagnostics.
Accurately determining microbial viability is fundamental to multiple fields, including clinical diagnostics, pharmaceutical development, food safety, and probiotic research. Traditional culture-based methods, while considered a historical gold standard for viability assessment, are time-consuming, unable to detect viable-but-nonculturable (VBNC) cells, and problematic for mixed-species communities like biofilms [58]. Molecular techniques have emerged as powerful alternatives, with quantitative PCR (qPCR) offering speed and sensitivity. However, conventional qPCR cannot distinguish DNA from live and dead cells, limiting its utility for viability assessment [59].
Two advanced technologies now address this critical limitation. Viability PCR (v-PCR), particularly using propidium monoazide (PMA), couples membrane integrity—a key viability criterion—with molecular detection. Simultaneously, Droplet Digital PCR (ddPCR) provides absolute quantification without standard curves and demonstrates enhanced resilience to PCR inhibitors [60]. This whitepaper provides an in-depth technical guide to these methods, framing them within the ongoing evolution of gold-standard approaches for microbial viability research. We will explore their principles, optimized protocols, and performance characteristics, supported by structured data and experimental workflows.
Viability PCR (PMA-qPCR)
Viability PCR (v-PCR) is a culture-independent method that rapidly discriminates viable microbes based on membrane integrity. The core mechanism involves propidium monoazide (PMA) or its superior alternative PMAxx, a photoactive, membrane-impermeant dye that selectively enters dead cells with compromised membranes [61]. Upon entering, PMA intercalates into DNA and forms covalent cross-links upon exposure to intense visible light. This modification permanently inhibits PCR amplification [58] [61]. Consequently, in a subsequent qPCR reaction, DNA from dead cells shows delayed amplification (higher Cq) or is completely suppressed, while DNA from viable cells with intact membranes is amplified normally [61] [62].
The technique is validated for dozens of bacteria, fungi, and viruses and is suitable for complex sample types, including soil, feces, water, biological specimens, and food [61]. A significant advantage is its ability to detect VBNC cells, which possess intact membranes but cannot form colonies on culture media [59].
Digital PCR (ddPCR)
Droplet Digital PCR (ddPCR) is a third-generation PCR technology that enables absolute quantification of nucleic acids without the need for a standard curve. The principle involves partitioning a single PCR sample into thousands of nanoliter-sized droplets, effectively creating numerous individual reactions [63] [64]. After end-point PCR amplification, each droplet is analyzed for fluorescence. Droplets containing the target sequence are scored as positive, while those without are negative [63]. The absolute copy number of the target DNA molecule in the original sample is then calculated using binomial Poisson statistics based on the ratio of positive to negative droplets [64] [60].
This partitioning confers two major advantages for viability studies: superior sensitivity for detecting low-abundance targets and increased tolerance to PCR inhibitors commonly found in complex matrices like feces [64] [60]. However, ddPCR has a narrower dynamic range and can suffer from reaction saturation at high target concentrations (>10⁶ copies/µL) [63] [60].
Quantitative Performance Comparison
The table below summarizes key performance characteristics of PMA-qPCR and ddPCR as reported in recent studies, providing a direct comparison of their capabilities.
Table 1: Performance Comparison of PMA-qPCR and ddPCR
| Parameter | PMA-qPCR | Droplet Digital PCR (ddPCR) |
|---|---|---|
| Principle of Quantification | Relative (based on standard curve) | Absolute (via Poisson statistics) |
| Viability Discrimination | Yes (based on membrane integrity) | No (quantifies total DNA, live and dead) |
| Linear Range | Typically 10³ to 10⁸ CFU/mL [62] | Up to 10⁵ copies/µL (saturation at high concentrations) [63] [60] |
| Limit of Detection (LOD) | ~10³ CFU/mL [62] [60] | 10-100 times more sensitive than qPCR in some studies [63] |
| Sensitivity in Complex Matrices | High, but can be affected by sample opacity and inhibitors | Excellent; partitioning reduces inhibitor effects [64] [60] |
| Key Advantage | Rapid viability assessment for specific strains | Absolute quantification, high precision, and inhibitor tolerance |
| Key Limitation | Potential overestimation of live cells; requires optimization [58] | Cannot differentiate between live and dead cells without a pre-treatment (e.g., PMA) |
The following table illustrates the quantitative results achievable with these methods in practical research scenarios.
Table 2: Representative Experimental Data from Recent Studies
| Application | Method | Key Quantitative Result | Reference |
|---|---|---|---|
| Photodynamic Inactivation of Mixed Biofilm (C. albicans-S. aureus) | PMA-qPCR | Genome copies of viable S. aureus reduced from 1.65 × 10⁸ to 3.19 × 10⁷; C. albicans from 4.39 × 10⁷ to 1.91 × 10⁷ (P < 0.01). | [58] |
| Quantification of L. paracasei in Probiotics | PMA-qPCR | Linear range 10³ to 10⁸ CFU/mL (R² = 0.994). Accuracy bias within ±0.5 Log₁₀ units. | [62] |
| Detection of Lactiplantibacillus plantarum | ddPCR vs qPCR | ddPCR showed a 10-fold lower limit of detection than qPCR. Good linearity for both (R² ≥ 0.996). | [63] |
| Quantification of Bacterial 16S in Lung Tissue | ddPCR vs qPCR | ddPCR negative controls had a smaller standard deviation (0.28) vs qPCR (0.70), indicating higher precision at low concentrations. | [64] |
| STEC Quantification in Bovine Feces | ddPCR vs qPCR | Both showed a limit of quantification of ~2.75-3.06 log copies/g feces. ddPCR was less prone to PCR inhibition. | [60] |
Detailed Experimental Protocols
Protocol 1: PMA-qPCR for ViableLacticaseibacillus paracaseiin Probiotics
This validated protocol ensures accurate quantification of viable probiotic cells [62].
1. Species-Specific Primer Design:
- Step: Identify a target gene unique to the species. For L. paracasei, the Alkaline shock protein 23 gene was used.
- Method: Perform a gene presence/absence analysis on all available whole-genome sequences (WGS) of the target species using software like CD-HIT. Verify nucleotide specificity via BLASTN against public databases.
- Primer Design: Use software (e.g., Primer Premier v6.0) adhering to standard qPCR principles (amplicon size 80-200 bp, Tm ~60°C).
2. PMA Treatment Optimization:
- Dye Concentration: Test a range of PMA or PMAxx concentrations (e.g., 10-100 µM) to find the level that fully suppresses amplification from dead cells without affecting viable cell signals.
- Procedure:
- a. Mix sample with PMA dye to the final optimal concentration.
- b. Incubate in the dark for 5-10 minutes with occasional mixing.
- c. Place samples on ice and expose to high-intensity light for 15-30 minutes using a dedicated photolysis device (e.g., PMA-Lite 2.0) to activate the dye.
3. DNA Extraction:
- Step: Extract genomic DNA from PMA-treated samples using a commercial kit. The choice of kit should be optimized for the sample matrix (e.g., feces, food, pure culture) to ensure high yield and purity [62].
4. qPCR Amplification and Quantification:
- Reaction Setup: Use a master mix like Forget-Me-Not EvaGreen qPCR Master Mix. Include no-template controls and a standard curve.
- Standard Curve: Construct using serial dilutions of genomic DNA from a reference strain with a known concentration (e.g., determined by fluorometry) and convert to cell equivalents based on genome size.
- Cycling Conditions: Standard qPCR cycling conditions are used (e.g., 95°C for 2 min, followed by 40 cycles of 95°C for 5 sec and 60°C for 30 sec).
Protocol 2: ddPCR for Absolute Quantification of Bacterial Load
This protocol adapts a qPCR assay for the absolute quantification of bacterial 16S rRNA genes in complex samples like lung tissue [64].
1. Sample Preparation and DNA Extraction:
- Step: Extract total DNA from the sample (e.g., tissue, feces) using a robust kit (e.g., Qiagen DNeasy kit). Accurately measure DNA concentration.
2. ddPCR Reaction Setup:
- Probe-Based Assay: Adapt an existing qPCR assay (e.g., for 16S rRNA, stx, or eae genes) for ddPCR. The probe and primer concentrations may require re-optimization.
- Partitioning: Mix the sample DNA with the ddPCR supermix and load into a DG8 cartridge. Use the QX200 Droplet Generator to partition the sample into ~20,000 nanoliter droplets.
3. PCR Amplification:
- Thermal Cycling: Transfer droplets to a 96-well plate and run PCR amplification on a thermal cycler. A sample protocol: 1 cycle at 95°C for 5-10 minutes, 40 cycles of 94°C for 30 seconds and 60°C for 60 seconds, followed by enzyme deactivation at 98°C for 10 minutes. A ramp rate of 2°C/second is standard.
4. Droplet Reading and Analysis:
- Reading: Place the plate in the QX200 Droplet Reader, which automatically aspirates droplets from each well and reads the fluorescence in each droplet.
- Threshold Setting: Use the manufacturer's software (e.g., Quantasoft) to set a fluorescence amplitude threshold that clearly separates positive and negative droplet populations.
- Quantification: The software provides an absolute concentration in copies/µL of the input reaction, which can be back-calculated to copies per gram of original sample.
Essential Research Reagent Solutions
The following table catalogs key reagents and equipment essential for implementing PMA-qPCR and ddPCR, as highlighted in the search results.
Table 3: Research Reagent Solutions for Viability and Digital PCR
| Item | Function | Example Products / Notes |
|---|---|---|
| PMAxx Dye | Next-generation viability dye; provides superior live/dead discrimination compared to PMA. | Biotium Cat. No. 40069 [61] |
| PMA Dye | The original viability dye; validated in hundreds of publications. | Biotium Cat. No. 40019 [61] |
| PMA Enhancer | Improves live/dead discrimination in gram-negative bacteria when used with PMA/PMAxx. | Biotium Cat. No. 31038 [61] |
| Photoactivation Device | Provides consistent, uniform illumination for optimal PMA photolysis. Critical for reliable results. | PMA-Lite 2.0 (for tubes), Glo-Plate 2.0 (for plates) [61] |
| Viability PCR Starter Kits | Contain PMA/PMAxx, qPCR master mix, and enhancer for easy adoption with any cell type. | Biotium Cat. No. 31075-X (with PMAxx) [61] |
| Strain-Specific v-PCR Kits | Include viability dye, master mix, and validated primers for common bacterial pathogens. | e.g., PMA Real-Time PCR Kits for S. aureus, L. monocytogenes, E. coli O157:H7 [61] |
| ddPCR System | Integrated system for droplet generation, thermal cycling, and droplet reading. | Bio-Rad QX200 Droplet System [64] [60] |
| Evaporation Seal | Prevents well-to-well contamination and sample evaporation during thermal cycling. | ddPCR Sealing Foil [60] |
Workflow and Signaling Pathways
The diagram below illustrates the core workflows for PMA-qPCR and ddPCR, highlighting their fundamental operational principles and differences.
PMA-qPCR and ddPCR represent significant advancements in the molecular toolbox for microbial analysis, each addressing distinct challenges beyond the capabilities of traditional gold standards. PMA-qPCR fulfills the critical need for rapid viability assessment based on membrane integrity, proving invaluable in probiotic quality control [62], antimicrobial efficacy testing [58], and food safety [59]. Meanwhile, ddPCR excels in scenarios requiring absolute quantification with high precision, superior sensitivity for low-abundance targets, and robust performance in inhibitor-rich complex matrices [63] [64] [60].
The future of gold-standard viability research lies not in a single technology, but in the strategic selection and integration of these methods. For comprehensive analysis, researchers can even combine them into a PMA-ddPCR workflow, marrying the viability discrimination of PMA with the absolute, inhibitor-tolerant quantification of ddPCR. As these protocols continue to be optimized and validated across diverse sample types, they are redefining the benchmarks for accurate, reliable, and informative microbial viability research.
Troubleshooting Viability Assays: Overcoming Common Pitfalls and Optimizing Protocols
In microbial viability research, the colony-forming unit (CFU) count has long been regarded as the regulatory gold standard for quantifying viable microorganisms. This method estimates the number of cells that are alive and capable of dividing to form visible colonies on standard culture media [65]. However, a significant challenge emerges when bacterial cells enter a physiological state known as viable but non-culturable (VBNC), where they remain metabolically active and potentially pathogenic but fail to grow on the very media traditionally used for their detection [66]. This discrepancy creates a critical blind spot in microbiological risk assessment, as CFU-based methods inevitably underestimate true viability, potentially compromising public health safety, therapeutic efficacy, and regulatory compliance.
The VBNC state represents a survival strategy triggered by environmental stresses commonly encountered during food processing, storage, and transit through the gastrointestinal tract. Bacteria in this state maintain membrane integrity, basal metabolism, and specific enzymatic activities while exhibiting changes in morphology and gene expression [66]. Consequently, the scientific community is increasingly recognizing the need to complement traditional CFU counts with culture-independent methods that can detect this elusive microbial population, thereby refining our understanding of what constitutes a "gold standard" in viability assessment.
Detection and Quantification: Bridging the Culturability Gap
Accurately quantifying total viable populations requires a dual approach that integrates both culture-dependent and culture-independent methodologies. The following table summarizes the key techniques available for detecting and enumerating VBNC cells, highlighting their respective principles and limitations.
Table 1: Methods for Detecting and Quantifying VBNC Cells
| Method | Principle | Target | Key Advantage | Main Limitation |
|---|---|---|---|---|
| CFU Count [65] | Growth on standard culture media | Culturable cells | Regulatory gold standard; simple | Misses VBNC cells entirely |
| Viability PCR/dPCR [66] [67] | Selective DNA intercalation (e.g., PMA) from dead cells | Intact DNA from viable cells | Specific detection of live/dead cells; quantitative | Does not confirm metabolic activity |
| Flow Cytometry (AFU) [66] | Membrane-permeant fluorescent viability stains | Cells with intact membranes & enzymatic activity | Rapid; counts metabolically active cells | Requires specialized equipment |
| Flow-FISH [66] | Fluorescent in situ hybridization of rRNA | Ribosomal RNA content | Species-level identification in mixed cultures | Complex protocol |
| Catalase Resuscitation [66] | Neutralization of oxidative stress on agar | Resuscitable VBNC cells | Recovers culturability | Strain-specific effectiveness |
The integration of these methods is crucial for a comprehensive assessment. For instance, ISO-standardized flow cytometry quantifies active fluorescent units (AFUs), providing a count of metabolically active cells that includes both culturable and VBNC populations [66]. Similarly, viability PCR, which uses DNA-intercalating dyes like propidium monoazide (PMA) to selectively cross-link DNA from membrane-compromised (dead) cells, allows for the quantification of "live" genomic equivalents that correlate with viability but are distinct from CFU counts [67]. When CFU counts are significantly lower than AFUs or live genomic equivalents, the presence of a substantial VBNC population is indicated.
Experimental Protocols for VBNC Research
Protocol for Viability qPCR with PMA Treatment
This protocol, validated for Campylobacter in food samples, allows for the quantification of live bacterial cells by distinguishing DNA from intact (live) and membrane-compromised (dead) cells [67].
- Sample Preparation: Homogenize the sample (e.g., food rinse, probiotic suspension) in an appropriate buffer.
- ISPC Addition: Add a known number of dead cells from a closely related but distinguishable species (e.g., C. sputorum) as an Internal Sample Process Control (ISPC). This controls for the efficiency of PMA treatment and corrects for DNA losses during processing.
- PMA Treatment: Add PMA dye to the sample to a final concentration of ~50 µM. Mix thoroughly and incubate in the dark for 5-10 minutes.
- Photoactivation: Place the sample on ice and expose it to bright light from a halogen lamp or a dedicated PMA-Lite device for 15-20 minutes. This step cross-links PMA into the DNA of dead cells, preventing its amplification.
- DNA Extraction: Proceed with standard genomic DNA extraction from the PMA-treated sample.
- qPCR Analysis: Perform triplex qPCR to simultaneously quantify:
- The target pathogen (e.g., thermophilic Campylobacter spp.)
- The ISPC (e.g., C. sputorum)
- An Internal Amplification Control (IAC) to detect PCR inhibition
- Quantification: Calculate the number of live target bacteria based on the standard curve, with the ISPC signal ensuring the PMA treatment was effective.
This method details the recovery of VBNC cells to make them detectable again by plate counting, using catalase supplementation as a key intervention [66].
- Induction of VBNC State: Subject the bacterial culture (e.g., Lactobacillus brevis or Lactobacillus plantarum) to inducing stresses such as prolonged cold storage (e.g., 0-4°C for months) or repeated sub-culturing in a stressful medium like beer.
- Confirm VBNC State: Verify the loss of culturability by plating on standard MRS agar, while confirming continued membrane integrity and metabolic activity via flow cytometry with viability stains.
- Resuscitation Plating:
- Prepare MRS agar plates supplemented with catalase at a concentration of 1000 IU/mL.
- Spread the sample containing VBNC cells onto the catalase-supplemented agar.
- Incubate under optimal atmospheric conditions and temperature for the target strain.
- Control Plating: In parallel, plate the same sample on non-supplemented MRS agar. Recovery of colonies only on the catalase-supplemented media is evidence of true resuscitation from the VBNC state.
- CFU Enumeration: Count the colonies formed on the catalase plates after the standard incubation period. The difference between counts on supplemented and non-supplemented media indicates the initial VBNC population.
Diagram 1: Catalase-based VBNC resuscitation workflow.
Essential Research Reagent Solutions
Successful VBNC research relies on a specific toolkit of reagents and materials. The table below outlines key solutions required for the experiments described in this guide.
Table 2: Key Research Reagent Solutions for VBNC Studies
| Reagent/Material | Function | Example Application |
|---|---|---|
| Propidium Monoazide (PMA) [67] | DNA intercalating dye; selectively penetrates dead cells with compromised membranes and cross-links DNA upon photoactivation, preventing its PCR amplification. | Viability qPCR for Campylobacter; differentiating live/dead cells without culture. |
| Catalase [66] | Enzyme that decomposes hydrogen peroxide into water and oxygen; rescues VBNC cells from oxidative stress, enabling resuscitation on solid media. | Supplementation of MRS agar to recover VBNC Lactobacillus brevis and L. plantarum. |
| Lyophilized ISPC [67] | Internal Sample Process Control; a known count of dead, distinguishable cells added to monitor PMA treatment efficiency and normalize DNA recovery. | Lyophilized dead C. sputorum cells used in Campylobacter viability qPCR validation. |
| Selective Media [66] | Culture media designed to support the growth of specific bacteria while inhibiting others; can be modified with resuscitating agents. | MRS agar for lactic acid bacteria, with or without catalase supplementation. |
| Viability Stains [66] | Fluorescent dyes (e.g., for membrane integrity, enzymatic activity) used to stain metabolically active cells for flow cytometry. | Flow cytometry quantification of Active Fluorescent Units (AFUs) in probiotics. |
Molecular Pathways and Mechanisms of the VBNC State
Understanding the VBNC phenomenon requires insight into the molecular triggers and mechanisms that lead to its induction and subsequent resuscitation. The transition is not a random event but a regulated response to environmental pressures.
Diagram 2: Molecular pathways in VBNC induction and resuscitation.
The entry into the VBNC state is typically triggered by sublethal environmental stresses, including extreme temperatures, oxidative stress (e.g., from hop acids in beer), high acidity, and nutrient starvation [66]. In response, cells undergo a profound genetic and metabolic shift. As observed in Lacticaseibacillus paracasei, this involves a transcriptional reprogramming that favors genes for substrate-use efficiency and stress tolerance, while downregulating those associated with replication [66]. Physiologically, this manifests as a reduction in cell size, slowed metabolism, increased envelope rigidity, and the retention of rRNA and enzymatic activity, hallmarks of a viable but non-growing phenotype [66].
Resuscitation from the VBNC state is a distinct process from population growth. It refers to the recovery of the ability to form colonies without cell division. This reversal is often dependent on the specific stress that induced the state. For instance, oxidative stress induced by cold storage in Lactobacillus brevis and L. plantarum can be countered by supplementing culture media with catalase, which breaks down inhibitory peroxides [66]. Other resuscitation triggers include nutrient upshifts, adjustments to optimal temperature and pH, and the presence of signaling molecules like resuscitation-promoting factors [66]. Evidence of true resuscitation, as opposed to the outgrowth of a few residual culturable cells, includes a stable total viable count (from flow cytometry) alongside a rising CFU count, or recovery that occurs only on specific rescue media but not on standard media.
The evidence is clear that reliance on CFU counts alone provides an incomplete and potentially misleading picture of microbial viability, particularly in stressed populations. The VBNC state is a prevalent and functionally significant phenomenon that cannot be ignored in fields ranging from public health to probiotic development. Addressing this challenge necessitates a paradigm shift from a single-method "gold standard" to an integrated, multi-metric framework.
The path forward involves the synergistic use of culture-dependent and culture-independent methods. Protocols that incorporate a resuscitation step, such as catalase supplementation, before plating can significantly enhance the accuracy of CFU counts [66]. Meanwhile, techniques like viability qPCR and flow cytometry provide complementary, quantitative data on the total viable population, including VBNC cells [66] [67]. For regulatory bodies and industry, embracing this integrated approach is crucial for accurately assessing the efficacy of probiotics, the risk of pathogenic bacteria, and the true shelf-life of microbial products. By reconciling data from both methodological streams, the scientific community can develop a more robust and truthful understanding of microbial viability, ultimately leading to safer products and more effective therapeutic interventions.
Metabolic assays are a cornerstone of microbial viability research, providing critical insights into cell health, function, and proliferation by quantifying the activity of key cellular enzymes. These assays are indispensable in fields ranging from drug discovery and toxicology screening to environmental microbiology [68] [69]. Despite their widespread use, their accuracy and reliability are frequently compromised by three persistent technical challenges: dye toxicity, pH sensitivity, and non-cellular abiotic reduction [70] [69]. Overcoming these hurdles is essential for improving the predictive power of preclinical models, which currently fail to detect toxicity in approximately 30% of drug candidates that later fail in human clinical trials [68]. This guide provides an in-depth analysis of these interference mechanisms and offers evidence-based strategies for assay optimization, framing these solutions within the context of gold-standard methodologies for microbial viability research.
Understanding Core Challenges in Metabolic Assays
Dye Toxicity and Its Impact on Cellular Viability
Many metabolic assays rely on reagents that can themselves alter cell physiology or cause death, leading to artificially low viability readings. This is particularly problematic in long-term incubation studies or those involving sensitive cell types.
- Tetrazolium Salts (MTT): The MTT assay, while widely used, requires careful handling. The formazan crystals produced are insoluble and require a dissolution step with solvents like DMSO. Furthermore, MTT itself is known to be cytotoxic, and its light-sensitive nature mandates that the assay be performed in the dark, typically as an endpoint measurement [69].
- Intermediate Electron Acceptors: Newer tetrazolium salts like MTS, XTT, and WST-1 produce soluble formazan products, eliminating the dissolution step. However,因为他们不能轻易穿透细胞膜, they require an intermediate electron acceptor reagent (e.g., phenazine ethyl sulfate). This intermediate can be toxic to cells, and its use requires careful optimization of reaction conditions to minimize this cytotoxicity while ensuring adequate signal generation [69].
pH Sensitivity and Fluctuations in the Microenvironment
The activity of cellular dehydrogenases, the enzymes central to many dye reduction assays, is highly sensitive to the pH of the surrounding environment. Fluctuations in pH can drastically alter enzymatic reaction rates, leading to significant signal drift and unreliable data.
- Culture Medium Composition: The use of growth medium supplemented with serum can sometimes mask or alter the effect of a drug, as seen with the proteasome inhibitor bortezomib [70]. This has led some researchers to opt for serum-free media, which can in turn affect cellular metabolism and pH.
- Physiological Variations: In vivo or ex vivo applications present even greater challenges. For instance, in sepsis, the physiological microenvironment is characterized by significant pH disturbances, primarily manifesting as metabolic acidosis (lactic acidosis) and respiratory acidosis [71]. Implementing real-time monitoring, as demonstrated with advanced fiber-optic sensors, reveals the dynamic and complex interplay between pH and cellular glucose metabolism, creating a "self-reinforcing vicious cycle" that can invalidate static assay readings [71].
Abiotic (Non-Cellular) Reduction and False Positives
A critical, often overlooked source of error is the non-enzymatic, chemical reduction of assay dyes by compounds in the experimental system. This abiotic reduction generates a signal that is indistinguishable from that produced by live, metabolically active cells, leading to false positive viability signals.
- Chemical Interference: The MTT assay is particularly susceptible to chemical interference from compounds commonly used in labware or biological samples [69]. Reducing agents and other reactive molecules can directly reduce the tetrazolium salt, artificially inflating the viability count.
- Culture Medium Components: Certain components of culture media can also participate in redox reactions with assay dyes. Background absorbance readings for assays like MTS, XTT, and WST-1 are generally higher than for MTT, and these readings are dependent on the specific culture medium and its pH [69].
Table 1: Core Challenges and Their Impact on Metabolic Assays
| Challenge | Primary Effect | Common Assays Affected | Consequence for Data |
|---|---|---|---|
| Dye Toxicity | Compromises cell membrane integrity and metabolic function | MTT (cytotoxic), MTS/XTT (via intermediate reagent) | Underestimation of true viability; time-dependent signal decay |
| pH Sensitivity | Alters cellular dehydrogenase enzyme activity | All dehydrogenase-based assays (Tetrazolium, Resazurin) | Signal drift; reduced accuracy in dynamic or stressed environments |
| Abiotic Reduction | Non-cellular chemical reduction of the dye | MTT (highly susceptible), Resazurin | False positive signals; overestimation of viable cell count |
Optimization Strategies and Experimental Protocols
Managing Dye Toxicity and Artifacts
Proactive management of dye-related toxicity is paramount for obtaining accurate results.
- Strategy 1: Assay Selection. For live-cell analysis or time-course experiments, transition from toxic, endpoint assays like MTT to more benign alternatives. Resazurin assays are relatively inexpensive, non-toxic, and allow for multiple readings from the same plate, though extended incubation times (>4 hours) should be avoided [69]. Calcein-AM and similar dyes are cleaved by intracellular esterases in live cells and are also non-toxic, making them excellent for kinetic studies [69].
- Strategy 2: Protocol Optimization. When use of a toxic dye is unavoidable, rigorously optimize the protocol. This includes minimizing incubation times with the dye and carefully titrating the concentration of any intermediate electron acceptor required for assays like MTS to find the balance between signal generation and cell health [69].
- Strategy 3: Leverage High-Throughput Alternatives. Consider adopting modern, low-toxicity, high-throughput methods. The Geometric Viability Assay (GVA), for instance, replicates the gold standard colony-forming unit (CFU) count over 6 orders of magnitude while reducing time and consumables by over 10-fold. It embeds cells in a soft agar matrix within a pipette tip, eliminating the need for soluble metabolic dyes and their associated toxicity [7].
Protocol: Resazurin Assay for Non-Toxic, Kinetic Viability Measurement
- Prepare a working solution of resazurin sodium salt in PBS or culture medium (e.g., 0.15 mg/mL). Protect from light.
- Add the working solution to your cell samples in culture medium. A 1:10 ratio (v/v) of resazurin solution to total media volume is typical.
- Incubate at the appropriate culture conditions (e.g., 37°C, 5% CO2) for 1-4 hours. Protect the plate from light.
- Measure fluorescence using a plate reader with excitation at 530–560 nm and emission at 580–610 nm.
- Note: The incubation time must be optimized for each cell type. A standard curve should be established to ensure the signal is within the linear range. Avoid sterile filtering the solution as it may reduce the resazurin [70] [69].
Controlling for pH Sensitivity
Stabilizing the pH of the assay environment is critical for reproducible results.
- Strategy 1: Use Buffered Media. Ensure the culture medium is supplemented with an adequate buffering system, such as HEPES, especially for assays extending beyond a few hours or conducted outside a controlled CO2 environment.
- Strategy 2: Account for Physiological Variability. In complex biological environments, pH cannot always be controlled but must be monitored. Integrated sensing systems, like the multifunctional fiber sensor (PGFs) that simultaneously monitor pH and glucose, can provide critical contextual data for interpreting metabolic assay results, revealing correlations between acid-base imbalance and metabolic dysfunction [71].
- Strategy 3: Validate with pH Controls. When developing a new assay, include control experiments to determine the sensitivity of the signal to pH variations within the expected physiological range for your model system.
Mitigating Abiotic Reduction and Chemical Interference
Eliminating non-cellular sources of signal is key to improving specificity.
- Strategy 1: Include No-Cell Controls. The most critical step is to run parallel "no-cell" or "background" controls containing all assay components (culture medium, drugs, dye) except the cells. The signal from these controls should be subtracted from the experimental readings.
- Strategy 2: Use Purified Reagents and Validated Materials. Source high-purity chemicals and use labware that has been tested for compatibility with viability assays to minimize leaching of reducing agents.
- Strategy 3: Select Less Susceptible Assays. Resazurin assays are generally less prone to abiotic interference than tetrazolium-based assays like MTT [69]. Luminescence-based assays, such as those detecting ATP content, offer even higher sensitivity and are less susceptible to chemical interference, as the signal is generated by a specific enzyme-substrate reaction (luciferase-luciferin) [69].
Protocol: Testing for Abiotic Reduction in Your System
- Set up experimental wells containing your standard culture medium, any test compounds, and the metabolic dye (e.g., MTT or resazurin).
- Create a matrix of controls:
- Background Control: Medium + dye only (no cells, no compounds).
- Compound Control: Medium + dye + the highest concentration of each test compound (no cells).
- Vehicle Control: Medium + dye + the vehicle (e.g., DMSO) at the highest concentration used (no cells).
- Incubate the control plates under the same conditions and for the same duration as your experimental plates.
- Measure the signal. Any signal significantly above the background control in the compound or vehicle controls indicates abiotic reduction. This value must be subtracted from all experimental readings involving that compound.
Table 2: Comparative Analysis of Common Metabolic Viability Assays
| Assay | Mechanism | Toxicity & Interference | Key Optimization Parameters | Best Use Cases |
|---|---|---|---|---|
| MTT | Reduction to insoluble formazan by cellular dehydrogenases [69] | High (cytotoxic, requires solubilization with DMSO); Susceptible to chemical interference [69] | Incubation time; solubilization method; protect from light | Endpoint measurements; historical data comparison |
| MTS/XTT/WST-1 | Reduction to soluble formazan via intermediate electron acceptor [69] | Moderate (toxicity from intermediate reagent); Higher background [69] | Titration of intermediate reagent; incubation time (<4h) [69] | Kinetic measurements (multiple reads); higher throughput |
| Resazurin | Reduction to fluorescent resorufin by metabolically active cells [69] | Low (non-toxic); Risk of fluorescence interference [69] | Incubation time (1-4h); filter excitation/emission wavelengths to avoid crosstalk [69] | Kinetic, non-toxic measurements; multiplexing with other assays |
| ATP Luminescence | ATP from viable cells fuels luciferase reaction to produce light [69] | Very Low; Minimal chemical interference; Highly sensitive [69] | Cell lysis efficiency; compatibility with test compounds | High-sensitivity applications; toxicology screens; measuring very low cell numbers |
| GVA (Geometric Viability) | Direct colony growth in micro-cones (pipette tips), not a metabolic dye assay [7] | None (bypasses dye chemistry); High-throughput and low-waste [7] | Agarose concentration; cell embedding density; image analysis | Replacing CFU assays; high-throughput screening; slow-growing or sensitive microbes |
The Scientist's Toolkit: Essential Research Reagents and Materials
Selecting the right tools is fundamental to a robust metabolic assay.
Table 3: Key Research Reagent Solutions for Optimized Metabolic Assays
| Item / Reagent | Function | Critical Considerations |
|---|---|---|
| HEPES-Buffered Medium | Maintains stable pH during extended incubations outside a CO2 incubator. | Prevents acidification of medium, which can alter dehydrogenase activity and assay signal. |
| DMSO (Cell Culture Grade) | Solubilizes hydrophobic compounds and dissolves MTT formazan crystals. | Inherent cytotoxicity requires use of matched vehicle controls for every drug concentration [70]. |
| Phenazine Ethyl Sulfate (PES) | Intermediate electron acceptor for MTS, XTT, and WST-1 assays. | Must be titrated to find a balance between signal generation and cellular toxicity [69]. |
| Resazurin Sodium Salt | Non-toxic, cell-permeable blue dye reduced to pink fluorescent resorufin. | Incubation times must be optimized to prevent over-reduction and signal saturation. |
| Cell-Tested Culture Plates | Housing for cells during assay. | Use plates designed to minimize evaporation and "edge effects," which can cause well-to-well variability [70]. |
| Bleach Decontamination Solution | For killing pathogens on the exterior of assay vessels (e.g., GVA pipette tips) [7]. | Essential for safe handling of pathogenic strains in non-standard formats. |
| Low-Melt Agarose | For embedding cells in 3D viability assays like GVA [7]. | Allows for cell immobilization and colony growth without affecting cell viability at low concentrations. |
Visualizing the Pathways and Workflows
The following diagrams illustrate the core mechanisms and optimized workflows for metabolic viability testing.
Diagram 1: Metabolic Assay Interference Mechanisms
Diagram 2: Optimized Assay Selection and Validation Workflow
Optimizing metabolic assays by systematically addressing dye toxicity, pH sensitivity, and abiotic reduction is not merely a technical exercise—it is a fundamental requirement for generating reliable and biologically meaningful data. The gold standard in microbial viability research is evolving from a single, perfect assay to a rigorous, method-aware approach. This involves selecting the most appropriate assay for the biological question, implementing a suite of controls to validate the system, and being willing to adopt innovative, dye-free technologies like the Geometric Viability Assay [7] that circumvent traditional limitations. By integrating these strategies, researchers can significantly improve the replicability and reproducibility of their work, thereby strengthening the bridge between preclinical findings and clinical success.
Determining microbial viability is a cornerstone of microbiology, infectious disease research, and drug development. The "gold standard" has historically been the culture method, which provides direct evidence of microbial growth and reproduction under laboratory conditions [72]. However, this method is labor-intensive, time-consuming, and cannot provide real-time results or differentiate the viability of individual bacteria within a population [72]. Modern research increasingly relies on fluorescence-based techniques, such as flow cytometry and fluorescence microscopy, which offer rapid, quantitative, and single-cell level analysis of microbial viability. These methods leverage the power of fluorescent dyes to distinguish live from dead cells based on physiological differences, most commonly membrane integrity.
Despite their advantages, fluorescence-based methods present significant technical challenges that can compromise data accuracy. Three hurdles are particularly pervasive: spectral bleed-through (or overlap) in multiplex fluorescence detection, variable lysis efficiency during sample preparation, and high background signals leading to poor live/dead discrimination. This whitepaper provides an in-depth technical guide to understanding and overcoming these hurdles, ensuring that viability data is reliable, reproducible, and truly reflective of the biological reality.
Overcoming Spectral Bleed-Through (Compensation)
Spectral bleed-through occurs when the emission signal of one fluorophore is detected in the channel of another, causing inaccurate data interpretation and false positives. This is a critical issue in multicolor viability panels.
Understanding the Core Problem
In a perfect experiment, each fluorophore would be excited by its specific laser and emit light that is only detected by its designated detector. In reality, fluorophores have broad emission spectra that often overlap with the detection windows of other channels. For example, the emission of a green fluorescent dye like SYTO9 might be partially detected in the yellow channel, and the signal from a red fluorescent protein like mCherry can spill into the far-red detector [73]. This phenomenon is not merely a minor inconvenience; it can lead to the misidentification of cell populations and erroneous conclusions, particularly when detecting antigens with low expression levels or rare cell populations [74].
Strategic Solutions and Technologies
a) Careful Panel Design: The first line of defense is intelligent fluorophore selection. Choose dyes with well-separated emission spectra. The extensive selection of LIVE/DEAD Fixable Viability Dyes, with excitations from UV to 808 nm, provides the flexibility to fit dyes into complex panels without overlapping with other markers [75]. Always consult a dye's excitation/emission (Ex/Em) spectra before experimental design.
b) Spectral Flow Cytometry: A powerful technological solution is spectral flow cytometry. Unlike conventional cytometers that use a series of dichroic mirrors and bandpass filters to direct light to specific detectors, spectral analyzers measure the entire emission spectrum for each fluorophore in a sample to create a unique "spectral fingerprint" [76]. During analysis, sophisticated unmixing algorithms deconvolute these overlapping signals, providing a pure signal for each fluorochrome and effectively eliminating bleed-through [76].
c) Proper Compensation Controls: For conventional flow cytometers, compensation is the mathematical correction for spectral overlap. This requires running single-stained controls for every fluorophore in the panel. The cytometer's software then calculates a compensation matrix to subtract the bleed-through signal from each channel. This is a non-negotiable step for any multicolor experiment.
Experimental Protocol: Establishing a Compensation Matrix
- Prepare Single-Stained Controls: For each fluorescent marker in your panel (including the viability dye), prepare an individual sample stained with only that one marker. Use cells or compensation beads as recommended for the specific antibody or dye.
- Acquire Data: Run each single-stained control on the flow cytometer using the same instrument settings (voltages, gains) as your experimental samples.
- Calculate Compensation: In the flow cytometry software, use the single-stain controls to set the compensation matrix. The software will quantify the amount of spillover into other channels and generate correction values.
- Apply and Verify: Apply the compensation matrix to your experimental data. Verify the compensation by visualizing the compensated data; properly compensated populations should appear negative in channels where the fluorophore does not emit.
Table: Quantitative Spectral Overlap Considerations for Common Viability Dyes
| Viability Dye (Example) | Excitation Laser (nm) | Emission Peak (nm) | Key Spectral Overlap Considerations |
|---|---|---|---|
| LIVE/DEAD Fixable Violet | 405 | 451 | Pacific Blue, CellTrace Violet, BV421 [75] |
| LIVE/DEAD Fixable Green | 488 | 520 | NB510, NB530 dyes [75] |
| Propidium Iodide (PI) | 488/561 | ~617 | PE-Texas Red, NY610, NY590 channels [75] [74] |
| LIVE/DEAD Fixable Far Red | 633/635 | 665 | NR660 dyes [75] |
| SYTOX Green | 488 | 523 | FITC, GFP channels [77] |
Figure 1: Spectral Bleed-Through Pathway and Solutions. This diagram visualizes the cause of spectral bleed-through, where a fluorophore's signal is detected in the wrong channel, and outlines the three primary strategies to resolve it.
Optimizing Lysis Efficiency for Intracellular Bacteria Studies
A critical application of viability staining is assessing the survival of bacteria inside host cells. The accuracy of these assays hinges on a lysis protocol that efficiently removes extracellular bacteria without harming the often more fragile intracellular bacteria.
The Technical Challenge
The goal is to achieve selective lysis of the eukaryotic host cell membrane while leaving both live and dead bacterial membranes intact. Inefficient lysis leads to an underestimation of intracellular bacterial load, as extracellular bacteria remain attached to host cell debris. Over-lysing, however, can damage the bacterial membranes themselves, killing live bacteria and causing dead bacteria to lose their cytoplasmic contents, which ultimately leads to an overestimation of viability [77]. This balance is delicate and must be empirically determined for each host-pathogen system.
A Standardized Protocol for Differentiating Viability of Internal vs. External Bacteria
The following protocol, adapted for fluorescence microscopy, allows for the simultaneous discrimination of bacterial location (internal vs. external) and viability [77].
Workflow Overview: The process involves infecting host cells, labeling extracellular bacteria with a specific marker, permeabilizing the host cells, and then staining all bacteria with a viability dye pair to distinguish live from dead.
Figure 2: Viability and Localization Staining Workflow. This protocol enables simultaneous discrimination of bacterial location and viability status.
Materials:
- Host cells adherent to glass coverslips.
- Bacteria of interest.
- MOPS/MgCl2 buffer.
- Alexa Fluor 647-coupled antibody or lectin specific to the bacterium.
- Live/Dead Staining Solution: 5 μM SYTO9, 30 μM propidium iodide, 0.1% saponin in MOPS/MgCl2.
- Fluorescence microscope with capabilities for green, red, and far-red imaging.
Detailed Steps [77]:
- Infect and Label Extracellular Bacteria: Infect cells and gently rinse. Incubate with the Alexa Fluor 647-conjugated reagent for 10 minutes in the dark. This reagent will bind only to extracellular bacteria, labeling them blue.
- Permeabilize and Stain for Viability: Rinse cells to remove unbound reagent. Aspirate the media and add the Live/Dead Staining Solution. Incubate for 15 minutes at room temperature in the dark. The saponin permeabilizes the host cell membrane, allowing the dyes access to intracellular bacteria. SYTO9, being membrane-permeant, stains all bacteria green. Propidium iodide (PI), being membrane-impermeant, only stains bacteria with compromised membranes (dead bacteria) red.
- Mount and Image: Rinse the coverslips and mount them on slides. Acquire images within 30 minutes to prevent dye leakage.
- Analysis: Count bacteria based on their fluorescence profiles:
- External Viable: Blue (extracellular marker) + Green (SYTO9).
- External Nonviable: Blue (extracellular marker) + Red (PI).
- Internal Viable: Green (SYTO9) only.
- Internal Nonviable: Red (PI) only.
Key Controls: It is essential to validate that the viability dyes function correctly. A pure culture of mid-logarithmic phase bacteria should be >95% SYTO9-positive (viable). A population of heat-killed bacteria should be predominantly PI-positive [77].
Minimizing Background Signal and Improving Live/Dead Discrimination
High background signal and poor discrimination between live and dead cell populations are among the most common failures in viability assays. This often stems from non-specific dye binding or the use of inappropriate dyes for the target microbe.
The Mechanism of Amine-Reactive Viability Dyes
Fixable amine-reactive dyes, such as the LIVE/DEAD Fixable Viability Stains, operate on a different principle than DNA-binding dyes like PI. These dyes are fluorescent, cell-impermeant, and contain a reactive group that covalently binds to free amine groups on cellular proteins [75] [74].
- In Live Cells: The dye cannot penetrate the intact membrane, so it only labels surface amines, resulting in dim staining.
- In Dead Cells: The compromised membrane allows the dye to enter and label the abundant intracellular amines, resulting in very bright staining.
The difference in fluorescence intensity between live and dead populations is typically greater than 50-fold, allowing for excellent discrimination [75]. A key advantage is that the covalent binding is preserved after fixation, enabling intracellular staining protocols, which is not possible with non-fixable dyes like PI [75].
The Gram-Positive Challenge and Specialized Dyes
Traditional DNA-binding dyes like propidium iodide (PI) are efficiently excluded by live gram-negative bacteria due to their protective outer membrane. However, live gram-positive bacteria, which lack this outer membrane, often uptake these dyes, resulting in high background in the "live" population and poor separation [78]. This necessitates the use of specialized dyes.
Solution: Novel membrane-impermeant DNA binding dyes, such as the BactoView Dead Stains, have been chemically engineered to be efficiently excluded by both gram-positive and gram-negative strains. This provides highly selective staining of dead cells and clear live/dead discrimination for gram-positive bacteria [78].
Experimental Protocol: Using Fixable Viability Dyes
This protocol is optimized for flow cytometry and can be incorporated into surface and intracellular antibody staining workflows.
Detailed Steps [75]:
- Prepare a Single-Cell Suspension: Create your cell suspension in a protein-free buffer (e.g., PBS). The absence of protein is critical, as any free protein (e.g., from serum) will compete for the dye and quench the signal.
- Stain with Viability Dye: Add the recommended amount of LIVE/DEAD Fixable Viability Dye to the cell suspension. Incubate on ice for 20-30 minutes. Keeping the cells cold helps minimize active dye efflux.
- Quench the Reaction: After incubation, add a large volume of a protein-containing buffer (e.g., PBS with 1-5% FBS or BSA). The free amines in the serum will bind to and quench any unreacted dye, preventing it from staining cells later in the protocol.
- Wash and Proceed: Centrifuge the cells to remove the quench buffer. The viability stain is now fixed to the cells. You can proceed with surface antibody staining, fixation, and permeabilization for intracellular staining as required by your protocol.
Table: Research Reagent Solutions for Microbial Viability Studies
| Reagent Category | Specific Examples | Function & Key Characteristics |
|---|---|---|
| DNA-Binding Dyes | Propidium Iodide (PI), 7-AAD, SYTOX Green [77] [74] | Membrane-impermeant; stain nucleic acids in dead cells. Inexpensive, but not fixable. |
| Amine-Reactive Dyes | LIVE/DEAD Fixable Viability Stains, Zombie Dyes [75] [74] | Covalently bind to cellular amines. Provide bright dead cell signal and are fixable, ideal for intracellular staining. |
| Gram-Positive Optimized Dyes | BactoView Dead Stains [78] | Novel dyes engineered for low background in live gram-positive bacteria. |
| Membrane-Permeant Counterstains | SYTO9, SYTO16, DAPI [77] | Stain all bacteria (live and dead) and are used in conjunction with a membrane-impermeant dead stain. |
| Extracellular Labeling | Alexa Fluor 647-conjugated Antibodies or Lectins [77] | Used prior to permeabilization to differentially label bacteria outside of host cells. |
The journey to robust and reliable microbial viability data is paved with technical challenges, but these are not insurmountable. By understanding the principles behind spectral bleed-through, researchers can employ strategic panel design and leverage spectral cytometry to achieve clean, multi-parametric data. By meticulously optimizing lysis protocols, the fate of intracellular pathogens can be accurately unraveled. Finally, by selecting the appropriate viability dye for the application—whether it be a fixable amine-reactive dye for complex immunophenotyping or a specialized dye for gram-positive bacteria—background signal can be minimized, and the clear distinction between life and death can be brought into sharp focus. Mastering these techniques ensures that viability assessment remains a pillar of rigorous and reproducible microbiological research.
Microbial viability research is fundamental to advancements in public health, pharmaceutical development, and antimicrobial discovery. The gold standard for assessing viability has traditionally been the colony-forming unit (CFU) assay, which measures the ability of a microbial cell to proliferate and form a visible colony [79]. However, the application of this gold standard becomes profoundly complex when dealing with structurally and compositionally heterogeneous samples such as biofilms, environmental swabs, and probiotic formulations. These samples present unique challenges, including the need to dislodge and disaggregate cells without causing lethality, the presence of mixed microbial communities, and the requirement to preserve the viability of delicate strains during processing and storage. This guide details optimized protocols for these complex samples, framing them within the context of gold standard viability assessment to ensure data accuracy and reproducibility.
Gold Standard Viability Methods and Complex Sample Challenges
The cornerstone of microbial viability assessment is the culture-based method, which confirms metabolic activity and the capacity for replication. For planktonic (free-floating) cells, this is straightforward. However, its application to complex samples requires careful consideration of the specific sample matrix.
- Biofilms: Biofilms are structured communities of microorganisms enclosed in a self-produced polymeric matrix and adherent to a surface [80]. This matrix acts as a barrier, making it difficult for disinfectants to penetrate and protecting resident cells. Sessile bacteria within a biofilm can be 10 to 10,000 times more resistant to antimicrobial agents than their planktonic counterparts [80]. Standard viability protocols often fail to effectively disrupt this matrix without killing a significant proportion of the cells, leading to a severe underestimation of the true viable population.
- Environmental Swabs: Swabs collected from hospitals, industrial surfaces, or natural environments are characterized by low biomass and the potential presence of inhibitory substances. The gold standard here requires efficient elution of cells from the swab material into a suitable buffer, followed by culturing. Inefficient elution is a major source of error, and the diversity of microorganisms present may necessitate the use of multiple culture media to capture the true viability of the community.
- Probiotic Formulations: These products often contain lyophilized (freeze-dried) live microorganisms. The gold standard for viability is the post-lyophilization survival rate. The process of lyophilization introduces severe stresses, including osmotic and oxidative damage, which can drastically reduce cell viability. Optimizing this process with protective excipients, known as lyoprotectants, is critical to maintaining therapeutic efficacy [79].
The following section provides a toolkit of reagents and materials essential for addressing these challenges.
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Essential Reagents and Materials for Protocol Optimization
| Item | Function/Application | Specific Examples |
|---|---|---|
| Lyoprotectants | Protect microbial cells during freeze-drying by stabilizing cell membranes and proteins [79]. | Trehalose, Sucrose, Glycerol, Skim Milk, GBSS formulation (Glycerinum, Brain Heart Infusion, Sucrose, Sodium Glutamate) [79] [81]. |
| Biofilm Disruption Agents | Physically or enzymatically break down the extracellular polymeric substance (EPS) of biofilms to release embedded cells for viability counting. | Neutral pH Dithiothreitol (DTT), DNase I, Dispersin B, Mucolyse. |
| Elution Buffers | Maximize the release of microorganisms from swab tips into a liquid medium for subsequent plating. | Buffers with surfactants (e.g., Tween 80), mild alkali solutions, or proprietary elution solutions. |
| High-Throughput Screening Tools | Enable rapid evaluation of hundreds of lyoprotectant formulations or culture conditions using a small footprint [79]. | 96-well or 384-well plates, automated liquid handlers, plate readers. |
| Specialized Growth Media | Support the recovery of stressed or damaged cells after processing (resuscitation) and selectively grow target organisms from mixed communities. | Tryptic Soy Agar (TSA), Brain Heart Infusion (BHI), deMan, Rogosa and Sharpe (MRS) agar, reinforced clostridial medium (RCM). |
| Microporous Filter Membranes | Serve as a carrier for bacterial tablets in standardized quality control, allowing for uniform cell dispersion and facilitating accurate colony counting [81]. | Mixed cellulose ester membranes, polycarbonate track-etch membranes. |
Optimized Protocols for Complex Samples
Probiotic Formulations: Lyophilization and Viability Assessment
Maintaining high viability in lyophilized probiotic products is a major industrial and pharmaceutical challenge. The following optimized protocol leverages a high-throughput screening (HTS) method to efficiently identify optimal lyoprotectant formulations.
Detailed Protocol:
- Culture Preparation: Inoculate the probiotic strain (e.g., Lactiplantibacillus plantarum or Saccharomyces boulardii) in an appropriate liquid medium and incubate until the mid- to late-exponential growth phase.
- Formulation with Lyoprotectants: Harvest cells by gentle centrifugation and resuspend in a range of candidate lyoprotectant solutions. Promising candidates include trehalose, sucrose, and the GBSS formulation, which has demonstrated high survival rates for E. coli [81].
- High-Throughput Lyophilization: Dispense the cell-lyoprotant suspensions into the wells of a metal 96-well plate. Perform lyophilization using a standardized freeze-drying cycle.
- Post-Lyo Viability Assessment: Rehydrate the lyophilized cakes with a defined volume of sterile recovery medium. Serially dilute the resuspended cells and plate on appropriate solid media for CFU counting. Alternatively, use a growth-based HTS assay where the lag time of the recovery growth is measured; a strong inverse correlation (Pearson's coefficient up to -0.9862) has been established between log10(viability) and growth lag time, allowing for rapid, indirect viability assessment [79].
- Validation: The viability results from the well-plate lyophilization should be compared against a traditional bulk lyophilization process to ensure scalability and reliability.
The workflow for this HTS method is summarized in the diagram below.
Biofilm Samples: Processing for Accurate Viability Counts
Accurately determining the viability of cells within a biofilm requires a protocol that effectively removes the biofilm from its substrate and disperses it into single cells or small aggregates without causing significant cell death.
Detailed Protocol:
- Biofilm Growth: Grow biofilms on a relevant substrate (e.g., stainless steel, titanium, silicone) under conditions that mimic the target environment [82].
- Gentile Rinse: Gently rinse the biofilm substrate with a neutral buffer (e.g., PBS) to remove loosely attached planktonic cells.
- Biofilm Harvesting and Dispersal:
- Physical Disruption: Place the substrate in a tube containing buffer and vortex vigorously with glass or stainless-steel beads. Alternatively, use sonication in a water bath sonicator. Note: optimize power and time to maximize dispersal while minimizing heat-induced death.
- Enzymatic Disruption: For robust biofilms, incubate the substrate with an enzymatic cocktail. A solution of neutral pH Dithiothreitol (DTT) with or without DNase I is highly effective at breaking down the matrix without harming most bacterial cells.
- Viability Quantification: Perform serial dilutions of the dispersed biofilm suspension in a neutralizing diluent and plate on appropriate agar media for CFU enumeration. The resulting count represents the number of viable biofilm-derived cells.
The key stages of biofilm development that these protocols aim to disrupt are illustrated below.
Environmental Swabs: Elution and Culture
The reliability of environmental monitoring hinges on the efficient recovery of microorganisms from collection swabs.
Detailed Protocol:
- Sampling: Use a pre-moistened (with a suitable elution buffer) swab to sample a defined surface area using a standardized pattern and pressure.
- Elution: Aseptically place the swab tip into a tube containing 1-10 mL of elution buffer. Vortex the tube vigorously for 30-60 seconds, or use a mechanical vortexer with a tube adapter for consistency. Roll the swab against the inner wall of the tube during vortexing to express the liquid and cells.
- Concentration (if needed): For surfaces with expected low contamination, the eluate can be filtered through a membrane filter. The membrane is then placed aseptically on the surface of a nutrient agar plate.
- Culture and Identification: Plate the eluate (or serial dilutions) onto non-selective and selective media to obtain a total viable count and to isolate specific pathogens of interest (e.g., ESKAPEE pathogens). Incubate under appropriate conditions and count CFUs.
Data Presentation and Analysis
Quantitative data from viability assays must be presented clearly to allow for comparison and informed decision-making. The following tables provide templates for summarizing key results.
Table 2: Viability Data for Probiotic Formulations with Different Lyoprotectants
| Lyoprotectant Formulation | Post-Lyophilization Viability (CFU/mL) | Viability Relative to Control (%) | Growth Lag Time (hours) |
|---|---|---|---|
| Trehalose (10% w/v) | 5.2 x 10^8 | 85.5% | 2.1 |
| Sucrose (10% w/v) | 4.8 x 10^8 | 79.0% | 2.4 |
| GBSS Formulation | 5.8 x 10^8 | 95.4% | 1.8 |
| Skim Milk (10% w/v) | 3.9 x 10^8 | 64.2% | 3.0 |
| Control (No protectant) | 6.1 x 10^7 | 10.0% | 8.5 |
Table 3: Efficacy of Biofilm Disruption Methods on Stainless Steel Coupons
| Disruption Method | Viable Count (CFU/cm²) | Log Reduction vs. Untreated Control | Coefficient of Variation (%) |
|---|---|---|---|
| Vortexing (Glass Beads) | 1.5 x 10^6 | 0.5 | 15.2 |
| Sonication (15 min) | 3.2 x 10^6 | 0.2 | 22.8 |
| DTT + DNase Incubation | 8.9 x 10^6 | 1.1 | 8.5 |
| Untreated Control | 1.0 x 10^7 | N/A | 12.1 |
Optimizing protocols for complex microbial samples is not merely a procedural step but a critical factor in generating accurate, reliable, and meaningful viability data. By understanding the specific challenges posed by biofilms, environmental swabs, and probiotic formulations, researchers can adapt gold standard methods to these contexts. The integration of high-throughput screening for lyoprotectant development, effective biofilm dispersal techniques, and robust swab elution methods ensures that the viability measurements reflect the true state of the microbial population under study. Adhering to these optimized protocols will enhance the quality of research in drug development, microbial ecology, and public health monitoring, ultimately leading to more effective therapies and safety assurances.
Validating and Selecting the Right Viability Assay: A Critical Comparative Analysis
The colony-forming unit (CFU) assay remains the undisputed gold standard for enumerating viable microbes across diverse fields including clinical diagnostics, pharmaceutical development, and probiotic manufacturing [7] [13] [83]. This method's enduring primacy stems from its direct measurement of a fundamental biological property—a cell's capacity to proliferate and form visible colonies on culture media. Despite its widespread adoption and historical significance, the CFU assay faces significant limitations, including lengthy incubation times (typically 24-48 hours), inability to detect viable but non-culturable (VBNC) cells, and methodological constraints with fastidious microorganisms or complex multi-strain products [13] [83].
The growing recognition of these limitations has accelerated development of emerging technologies that offer faster, potentially more robust viability assessment. However, establishing the validity of these novel methods requires rigorous correlation studies against the established CFU benchmark. This technical guide examines the current landscape of these correlation methodologies, providing researchers with experimental frameworks and quality metrics for benchmarking emerging technologies against the CFU gold standard.
Limitations of the CFU Gold Standard
While the CFU assay provides a direct measure of replicative capacity, it operates under the fundamental constraint that it can only quantify microorganisms capable of division under the specific cultivation conditions provided. This limitation manifests in several critical aspects:
- Viable But Non-Culturable (VBNC) State: Numerous bacterial species can enter a VBNC state under stress, maintaining metabolic activity while losing culturalility on standard laboratory media. This creates a significant discrepancy where viability is underestimated by CFU counts [13] [83].
- Fastidious Growth Requirements: Many microorganisms, including emerging next-generation probiotics like Akkermansia muciniphila, have complex nutritional or atmospheric requirements that are difficult to replicate consistently in laboratory settings, leading to unreliable CFU data [83].
- Time-to-Result Constraints: The mandatory incubation period (often 24-72 hours) delays critical decisions in clinical diagnostics and manufacturing quality control [7] [84].
- Limited Throughput and Automation: Traditional plating methods remain labor-intensive and challenging to automate fully, creating bottlenecks in high-throughput applications such as drug discovery and functional genomics [7].
These limitations have driven the investigation of alternative viability markers, including membrane integrity, metabolic activity, and cellular respiration, each requiring careful validation against the replication-based CFU benchmark.
Emerging Enumeration Technologies and Correlation Approaches
Flow Cytometry-Based Methods
Flow cytometry has emerged as a powerful alternative, differentiating cell populations based on fluorescent staining patterns that report on cellular integrity and function. This approach typically categorizes cells into active fluorescent units (AFU), non-active fluorescent units (n-AFU), and total fluorescent units (TFU) [83].
Correlation Methodology: Studies typically involve parallel analysis of identical samples by both flow cytometry and CFU assays across a dilution series covering the method's dynamic range. The resulting data is analyzed through linear regression, with key metrics including correlation coefficients (R²) and bias factors [83].
Performance Data: A recent ring test demonstrated that fluorescence flow cytometry (FCC) showed robust reproducibility across different laboratories, instruments, and operators. When compared with impedance flow cytometry (IFC), both methods showed good agreement after initial per-strain optimization [13]. However, consistent observations indicate that AFU counts typically exceed CFU counts from the same sample, with the difference attributed to the VBNC population [83].
Table 1: Flow Cytometry Correlation with CFU Assays
| Metric | Typical Performance | Interpretation |
|---|---|---|
| Correlation Coefficient (R²) | 0.85-0.98 | Strong linear relationship with CFU |
| Bias Factor (AFU:CFU) | Often >1 | Suggests VBNC population present |
| Time-to-Result | 2-4 hours | Significant reduction vs. CFU (24-72 hours) |
| Repeatability | High (robust across operators and equipment) | Suitable for quality control environments |
Molecular-Based Viability Enumeration
Molecular methods, particularly propidium monoazide quantitative PCR (PMA-qPCR) and related techniques, have gained traction for species-specific viability enumeration in complex samples like multi-strain probiotics.
Principle: DNA-binding dyes like PMA selectively penetrate compromised membranes of dead cells. Upon photoactivation, dye-DNA adducts form, inhibiting PCR amplification. Thus, only DNA from intact (potentially viable) cells is amplified [13].
Correlation Methodology: Researchers validate PMA-qPCR methods by creating samples with known ratios of live and heat-killed target cells, followed by parallel analysis with CFU assays. Method specificity is demonstrated against non-target species to ensure no cross-reactivity [13].
Performance Data: Developed methods for Lactobacillus acidophilus, Bifidobacterium bifidum, Lacticaseibacillus rhamnosus, and other probiotics demonstrate efficient suppression of dead cell signals (>99%) and high specificity to target species. These methods successfully quantify viability in various matrices, including finished products during storage stability studies [13].
Innovative Phenotypic Methods
Geometric Viability Assay (GVA)
The Geometric Viability Assay (GVA) represents a novel approach that combines phenotypic growth detection with mathematical modeling to overcome throughput limitations of traditional CFU counting [7].
Principle: GVA leverages the predictable probability distribution of colonies forming within a conical volume (typically a pipette tip). The probability of colony formation at any point along the cone's axis is proportional to the cross-sectional area at that point, described by the probability density function: PDF(x) = 3x²/h³, where x is the distance from the tip and h is the total cone length [7].
Correlation Methodology: In validation studies, stationary-phase E. coli cultures are serially diluted and each dilution is analyzed by both GVA and traditional drop CFU method in technical quadruplicate. Colony distributions in the cone are compared against theoretical predictions across >6 orders of magnitude [7].
Performance Data: GVA demonstrated remarkable correlation (Pearson r = 0.98, p = 4 × 10⁻¹⁶) with drop CFU across 6 orders of magnitude. Bland-Altman analysis showed an average bias factor of 1.6 between methods. The method reduces time and consumable requirements by over 10-fold compared to traditional CFU, enabling a throughput of up to 1,200 viability measurements per researcher per day [7].
Table 2: Performance Comparison of Emerging Enumeration Technologies
| Technology | Principle | Dynamic Range | Time-to-Result | Key Advantage |
|---|---|---|---|---|
| Flow Cytometry | Membrane integrity via fluorescent staining | 10³-10⁸ cells/mL | 2-4 hours | Distinguishes live/dead populations |
| PMA-qPCR | Amplification from intact cells only | 10²-10⁷ cells/mL | 3-6 hours | Species-specific in complex mixtures |
| Geometric Viability Assay (GVA) | Colony distribution in conical volume | 1-10⁶ cells/sample | 18-24 hours (incubation) | Extreme throughput, minimal waste |
| Impedance Flow Cytometry | Electrical property changes | 10⁴-10⁸ cells/mL | 2-4 hours | Label-free, agrees with FCC after optimization |
Experimental Design for Robust Correlation Studies
Modified ISO 20391-2:2019 Framework for Microbial Cells
A modified ISO 20391-2:2019 standard provides a rigorous framework for designing correlation studies between emerging technologies and CFU assays. This approach was specifically adapted for microbial cell counting to accommodate the wide concentration ranges typical in microbiology [85].
Experimental Design: A stock microbial suspension (e.g., Escherichia coli NIST0056) is diluted across a log-scale range of concentrations (~5 × 10⁵ cells/mL to 2 × 10⁷ cells/mL). These blinded samples are then quantified using both reference (CFU) and test methods with fixed acquisition conditions and constant operators [85].
Quality Metrics: Key metrics modified from the ISO standard include:
- Proportionality: The degree to which measured values decrease proportionally with dilution factors
- Linearity: Assessment of the R² value from linear regression of expected vs. measured concentrations
- Variability: Calculation of coefficient of variation across replicate measurements
- Repeatability: Consistency of quality metrics across multiple experimental dates [85]
Application Example: In a recent study applying this framework, total cell count methods (Coulter principle, fluorescence flow cytometry, impedance flow cytometry) showed good agreement, while viable cell count methods exhibited greater variability. The quality metrics effectively illustrated differences in proportionality and variability across methods, guiding fit-for-purpose method selection [85].
Sample Preparation and Handling Considerations
Proper sample preparation is critical for meaningful correlation studies:
- Biological Replicates: Experiments should be performed on multiple separate dates with newly prepared cell stocks to account for day-to-day variability [85].
- Sample Replicates: Multiple replicate samples (typically 3) should be prepared at each dilution factor to assess measurement precision [85].
- Blinding: Operators should be blinded to sample identities and concentrations to prevent conscious or subconscious bias during measurement and analysis [85].
- Reference Materials: When available, standardized reference materials (e.g., polymeric beads) should be included to control for instrument performance [85].
Analytical Frameworks for Method Comparison
Statistical Approaches for Technology Benchmarking
Robust statistical analysis is essential for meaningful method comparison:
- Regression Analysis: Linear regression of test method results against CFU counts provides R² values indicating strength of correlation and regression parameters revealing proportional and constant biases [7].
- Bland-Altman Analysis: This method plots the difference between paired measurements against their mean, visualizing bias across the measurement range and establishing limits of agreement [7].
- Hierarchical Bayesian Modeling: Advanced modeling techniques can quantify different sources of experimental variability (instrument, operator, day-to-day) to identify primary contributors to measurement uncertainty [85].
Determining "Sub-Ranges" of Proportionality
Few methods perform equally well across their entire theoretical dynamic range. Correlation studies should specifically assess whether proportional "sub-ranges" exist where method performance is optimal [85]. This involves:
- Segmented regression analysis to identify breakpoints in linearity
- Calculation of quality metrics within defined concentration windows
- Practical determination of the usable range for each method based on predetermined acceptability criteria (e.g., R² > 0.95, CV < 15%)
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Viability Enumeration Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Propidium Monoazide (PMA) | DNA intercalator for dead cell discrimination | PMA-qPCR viability assessment |
| Fluorescent Viability Stains | Membrane integrity assessment (SYBR Green, propidium iodide) | Flow cytometry viability panels |
| Triphenyl Tetrazolium Chloride (TTC) | Redox indicator for metabolic activity | Colony visualization in GVA |
| Selective Culture Media | Optimization for fastidious organisms | CFU enumeration of specialized strains |
| Lyophilized Microbial Reference Materials | Standardized samples for method validation | Inter-laboratory comparison studies |
Correlation studies against the CFU gold standard remain essential for validating emerging viability enumeration technologies. The experimental frameworks and quality metrics described provide researchers with robust methodologies for these critical assessments. As the field advances, key areas for continued development include:
- Standardized reference materials certified for multiple measurands (CFU, total cell count, membrane integrity)
- Multi-laboratory ring trials to establish method reproducibility across sites and operators
- Expanded clinical validation connecting method results to patient outcomes in diagnostic applications
- Integrated approaches combining multiple technologies to address the limitations of any single method
The ongoing evolution of viability assessment methodologies promises enhanced accuracy, speed, and practicality while maintaining the essential connection to the biological relevance embodied by the CFU gold standard.
Experimental Workflow Visualization
Diagram 1: Technology Benchmarking Workflow. This workflow outlines the key stages in correlating emerging technologies against the CFU gold standard, from sample preparation through final validation.
Diagram 2: Viability Assessment Correlation Framework. This diagram illustrates the relationship between the CFU gold standard and alternative viability markers assessed through correlation studies, ultimately connecting to practical application outcomes.
Microbial viability testing serves as a cornerstone of microbiology research and development, with direct implications for pharmaceutical safety, drug discovery, and clinical diagnostics. The "gold standard" method for quantifying viable microorganisms has historically been the colony-forming unit (CFU) assay, which relies on the ability of viable cells to proliferate and form visible colonies on solid growth media. Despite its widespread adoption and reputation for reliability, the CFU assay possesses significant limitations, including extended time-to-results, labor-intensive processes, and limited dynamic range, which have driven the development of alternative methodologies.
This technical guide provides an in-depth comparative analysis of established and emerging microbial viability testing methods, focusing on their dynamic range, throughput, cost-effectiveness, and specific limitations. By synthesizing current research and technical specifications, this whitepaper aims to equip researchers, scientists, and drug development professionals with the data necessary to select optimal viability assessment strategies for their specific applications, ultimately enhancing research efficiency and diagnostic accuracy within the framework of gold standard methodology validation.
Established Gold Standard: CFU Assay and Its Evolution
Traditional CFU Methodology
The colony-forming unit (CFU) assay remains the universally recognized reference method for enumerating viable cells in microbiology laboratories worldwide. This method combines procedural simplicity with readily available reagents to achieve an enormous dynamic range, commonly measuring between 1 and 100,000,000 viable cells in a sample [7]. The fundamental principle involves serial dilution of a bacterial suspension, plating onto solid nutrient media, incubation under growth-permitting conditions, and counting discrete colonies that develop. Each colony theoretically originates from a single viable progenitor cell, allowing for backward calculation of the original viable cell concentration.
The CFU assay plays critical roles across diverse fields including food safety, functional genomics, and drug discovery against persistent cells [7]. However, this method presents significant limitations that impact its utility in modern high-throughput research environments. The most pronounced constraints include the extensive time investment (typically 1-7 days for incubation), substantial consumption of resources (plates, media, dilution tubes), and generation of considerable plastic waste [7] [86]. Furthermore, the method inherently detects only those microorganisms capable of forming colonies under the specific culture conditions provided, potentially underestimating viability for damaged, stressed, or unculturable cells [87] [88].
Enhanced CFU Approaches
Recent innovations have sought to address limitations of traditional plating methods while maintaining the fundamental viability principle of colony formation. The geometric viability assay (GVA) represents a significant advancement, replicating CFU measurements over 6 orders of magnitude while reducing time and consumable requirements by over 10-fold compared to traditional methods [7]. This approach computes viable cell counts based on the distribution of embedded colonies growing inside a pipette tip, leveraging probability functions related to the cone's geometry. The method has demonstrated compatibility with Gram-positive bacteria, Gram-negative bacteria, biofilms, and fungi, with a throughput of up to 1,200 viability measurements per researcher per day [7].
The GVA method has recently been adapted for anaerobic viability testing using Clostridium species as model organisms, demonstrating dynamic range extending over 5 orders of magnitude while maintaining compatibility with economical anaerobic jar systems [53]. This adaptation simplifies laborious procedures such as checkerboard assays, treatment time-courses, and drug screens against slow-growing cells, expanding the utility of plating-based viability assessment in specialized applications.
Emerging and Alternative Viability Assessment Methods
Flow Cytometry
Flow cytometry has emerged as a powerful alternative for viability assessment, particularly for complex microbial mixtures where traditional plating encounters limitations. This technique enables rapid, multi-parametric single-cell analysis based on light scattering and fluorescence characteristics [89] [88]. When coupled with viability dyes such as SYTOX Green or propidium iodide, flow cytometry can discriminate between live and dead cells based on membrane integrity, providing results in minutes rather than days [89].
Studies comparing flow cytometry with traditional CFU counts have demonstrated that flow cytometry typically detects higher viable cell counts, particularly in multi-species probiotic products [89]. This discrepancy is attributed to the ability of flow cytometry to detect viable but non-culturable (VBNC) cells that remain metabolically active and possess membrane integrity but cannot form colonies on conventional media [89] [88]. Additional advantages include high analysis speed (thousands of cells per second), ability to process hundreds of samples daily using automated samplers, and detection of subpopulations with intermediate characteristics [88].
The International Organization for Standardization and International Dairy Federation have recognized flow cytometry as a standard method for enumerating active lactic acid bacteria in dairy products, signaling growing acceptance despite requiring specialized instrumentation and expertise [89].
Metabolic and Staining Assays
Various staining methods have been developed as proxies for microbial viability and metabolic activity. Tetrazolium salts represent a large family of compounds used to measure redox activity in metabolically active cells [44]. Colorless tetrazolium salts penetrate bacterial cells and are reduced to colored formazan derivatives by NADH or NADPH-dependent oxidoreductases and dehydrogenases in metabolically active cells [44]. Different tetrazolium salts vary in solubility, reduction requirements, and the properties of their formazan products, enabling selection based on specific application requirements.
While these assays provide rapid results and are generally cost-effective, they measure metabolic activity rather than replication competence, potentially leading to discrepancies with CFU counts [44]. The assays may fail to detect slowly metabolizing cells, and results can be influenced by abiotic reduction or the presence of superoxide, necessitating appropriate controls [44].
Molecular and Spectroscopic Methods
Molecular methods such as precision metagenomics enable culture-independent identification and quantification of microorganisms by sequencing microbial DNA directly from samples. This approach has demonstrated superior diagnostic yield by detecting pathogens missed by both microbial culture and PCR, particularly in complex infections [90]. While not a direct viability assay, metagenomics can provide insights into microbial community composition without cultivation biases.
Surface-enhanced Raman spectroscopy (SERS) has emerged as a rapid, label-free technique for identifying pathogenic bacteria based on vibrational spectroscopy enhanced by noble metal nanoparticles [91]. The technique detects biochemical differences primarily associated with bacterial cell walls, allowing differentiation between species with high sensitivity. However, detection efficiency varies between Gram-positive and Gram-negative bacteria due to structural differences in their cell walls [91].
Comparative Analysis of Method Performance
Table 1: Dynamic Range, Throughput, Cost, and Limitations of Microbial Viability Methods
| Method | Dynamic Range | Throughput | Relative Cost | Key Limitations |
|---|---|---|---|---|
| CFU Assay | 1-10⁸ CFU/mL [7] | Low: 10s of samples/day; incubation 1-7 days [88] | Low (consumables only) [88] | Labor-intensive; only detects culturable cells; slow results; difficult to automate [7] [87] |
| Geometric Viability Assay (GVA) | 1-10⁶ CFU/mL [7] | High: 1,200 measurements/researcher/day [7] | Very Low (pipette tips, agarose) [7] [53] | Requires custom imaging setup; limited to compatible growth media [7] |
| Flow Cytometry | 10³-10⁸ cells/mL [87] | Very High: 100s of samples/day; immediate results [89] [88] | High (instrument, reagents) [88] | Requires expensive equipment; specialized training; viability dyes may not correlate perfectly with culturability [89] [88] |
| Tetrazolium Reduction | Varies by assay format | Medium-High: 100s of samples/day [44] | Low-Medium (reagents, plates) [44] | Measures metabolic activity, not necessarily viability; potential abiotic reduction [44] |
| Optical Density | 10⁶-10⁹ cells/mL [87] | Very High: 100s of samples/day; real-time [87] | Very Low (cuvettes/plates only) [87] | Cannot distinguish live/dead cells; interference from particles/nanomaterials [87] |
| SERS | 10⁴-10⁷ CFU/mL [91] | Medium: limited by sample preparation [91] | Medium-High (nanoparticles, instrument) [91] | Sensitivity varies between Gram-positive and Gram-negative bacteria; complex data interpretation [91] |
Table 2: Research Reagent Solutions for Microbial Viability Assessment
| Reagent/Kit | Function | Application Examples |
|---|---|---|
| Triphenyl Tetrazolium Chloride (TTC) | Tetrazolium salt reduced to red formazan in metabolically active cells [7] [44] | Visual colony contrast in GVA; metabolic activity assays [7] |
| SYTOX Green | Membrane-impermeant nucleic acid stain indicating compromised membranes [89] | Flow cytometry viability staining; distinguishes intact vs. compromised cells [89] |
| BacLight LIVE/DEAD Kit | Dual staining system with membrane-permeant and impermeant nucleic acid stains [87] | Flow cytometry differentiation of live vs. dead bacterial populations [87] |
| Gold Nanoparticles | SERS substrate enhancing Raman signals of proximate molecules [91] | Label-free bacterial detection and identification via SERS [91] |
| Bromocresol Green (BG) | pH-sensitive dye staining bacterial colonies against colorless background [53] | Visual enhancement in anaerobic GVA; qualitative MBC determination [53] |
| Double-ended Barcoded Primers | Unique identifier sequences for multiplexed sample analysis [92] | High-throughput 16S rDNA sequencing for species identification in biobanks [92] |
Experimental Protocols for Key Methods
Geometric Viability Assay (GVA) Protocol
The GVA method enables high-throughput viability testing with minimal consumables using the following detailed protocol [7] [53]:
Embedding Solution Preparation: Prepare embedding solution by melting agarose in appropriate culture medium (e.g., LB for bacteria, YEPD for yeast) to a final concentration of 0.5-0.66% (wt/vol). Cool the solution to ≤55°C before use. For enhanced colony contrast, add triphenyl tetrazolium chloride (TTC) to a final concentration of 0.05-0.1 mg/mL for aerobic bacteria, or Bromocresol Green for anaerobic applications [7] [53].
Sample Preparation and Dilution: Dilute bacterial samples in a 96-well plate to achieve expected maximum CFU/mL below 10⁷. For anaerobic applications, perform dilutions in an anaerobic chamber or using anaerobic dilution buffers [53].
Mixing and Solidification: Mix diluted samples 1:1 with embedding solution and aspirate into standard P200 pipette tips. Allow agarose to solidify completely at room temperature or 4°C for 10-15 minutes [7].
Incubation: Eject solidified tips into empty tip racks and incubate under appropriate conditions (temperature, atmosphere) for 12-48 hours depending on microbial growth characteristics. For anaerobic cultures, place tip racks in anaerobic jars with appropriate atmosphere generation systems [7] [53].
Imaging and Analysis: Image tips using a custom optical setup with consistent lighting and background. Manually identify colony positions along the tip axis or develop automated image analysis algorithms. Calculate original viable cell concentration using the probability density function: PDF(x) = 3x²/h³, where x is the perpendicular distance from the tip and h is the total cone length [7].
Flow Cytometry Viability Protocol
This protocol describes viability assessment for complex microbial mixtures using SYTOX Green staining [89]:
Sample Preparation: Dilute probiotic or microbial samples in appropriate buffer (e.g., PBS) to achieve approximately 10⁶-10⁷ cells/mL. For products containing particulate matter, preliminary filtration or centrifugation may be required [89].
Staining Solution Preparation: Prepare SYTOX Green working solution in buffer according to manufacturer recommendations. Typical final staining concentrations range from 0.5-1.0 μM [89].
Staining Incubation: Mix 100 μL of sample with 1-5 μL of SYTOX Green working solution. Incubate in darkness for 10-15 minutes at room temperature. Include controls with heat-killed cells (dead control) and unstained cells (autofluorescence control) [89].
Instrument Setup: Configure flow cytometer using forward scatter (FSC) and side scatter (SSC) parameters to identify bacterial populations. Establish fluorescence detection channels appropriate for SYTOX Green (excitation ~504 nm, emission ~523 nm). Adjust photomultiplier voltages using stained controls to optimally distinguish positive and negative populations [89].
Acquisition and Analysis: Acquire a minimum of 10,000 events per sample at low flow rate. Set threshold on FSC or fluorescence to ignore small debris. Analyze data using density plots of FSC vs. fluorescence, establishing gates for SYTOX Green-negative (viable) and positive (compromised) populations [89].
High-Throughput Species Identification Protocol
This protocol enables cost-effective species identification for large bacterial biobanks using Nanopore sequencing [92]:
Bacterial Culturing and Lysis: Culture bacterial isolates in 96-well plates with appropriate growth media. Perform cell lysis using thermal or chemical methods (e.g., 95°C for 10 minutes in alkaline lysis buffer) [92].
16S rRNA Gene Amplification: Perform PCR amplification of full-length 16S rDNA genes using double-ended barcoded primers (27F/1492R) with a high-fidelity DNA polymerase. Use optimized PCR conditions: initial denaturation 95°C for 2 min; 30 cycles of 95°C for 20s, 55°C for 30s, 72°C for 90s; final extension 72°C for 5 min [92].
PCR Product Pooling and Cleanup: Quantify PCR products using fluorometric methods. Pool equal masses of each barcoded amplicon into a single library. Clean pooled library using solid-phase reversible immobilization (SPRI) beads to remove primers and contaminants [92].
Nanopore Library Preparation and Sequencing: Prepare sequencing library using Nanopore Native Barcoding Expansion kits according to manufacturer instructions. Load library onto MinION or PromethION flow cells and sequence for 24-48 hours using standard operating procedures [92].
Bioinformatic Analysis: Demultiplex sequences based on barcodes using Guppy or similar tools. Perform taxonomic classification using reference databases (SILVA, Greengenes) with minimap2 or BLAST. Apply quality filtering to exclude reads with
[92].<="" p="" quality="" score="">
Visualized Workflows and Signaling Pathways
Diagram 1: Workflow comparison of major microbial viability assessment methods
Diagram 2: Decision framework for microbial viability method selection
The landscape of microbial viability assessment continues to evolve, with traditional gold standard methods being complemented and in some cases superseded by innovative approaches that address limitations in throughput, dynamic range, and applicability to complex samples. The CFU assay remains the reference method for viability determination based on reproductive capacity, but techniques such as geometric viability assay, flow cytometry, and advanced spectroscopic methods offer compelling advantages for specific applications.
Method selection should be guided by application requirements, with traditional plating optimal for regulatory applications requiring proven methodology, GVA excellent for high-throughput screening with maintained principle of viability, flow cytometry superior for rapid analysis of mixed populations, and molecular methods providing culture-independent community insights. As technology advances, the integration of these methods through orthogonal approaches provides the most comprehensive understanding of microbial viability, ultimately enhancing research efficiency and product safety across pharmaceutical, clinical, and industrial microbiology domains.
Method validation is the cornerstone of reliable and reproducible scientific research, ensuring that analytical techniques are fit for their intended purpose. In the critical fields of pathogen detection, probiotic potency, and biotherapeutic development, robust validation is not merely a regulatory formality but a fundamental requirement for ensuring product safety, efficacy, and public health. The core principle of validation applies universally: a method must be scientifically sound and consistently produce results that accurately reflect the biological reality, whether quantifying a deadly pathogen or the viable dose of a therapeutic microbe. This guide synthesizes current standards and emerging methodologies, providing a technical framework for researchers and drug development professionals to navigate the complex landscape of microbial viability research.
The "gold standard" for microbial viability has traditionally been the colony-forming unit (CFU) assay, which measures the ability of a microorganism to proliferate and form visible colonies [7]. While this method provides a direct measure of cultivability, the field is rapidly evolving to incorporate more sophisticated, mechanism-based potency assays and high-throughput technologies that offer deeper insights into biological function. This document details the specific validation strategies and experimental protocols required across three distinct applications, providing a comprehensive resource for advancing both research and regulatory-compliant product development.
Pathogen Detection: Validation during a Public Health Emergency
The rapid and accurate detection of emerging pathogens is a critical component of public health response. The U.S. Food and Drug Administration (FDA) provides specific guidance for the validation of in vitro diagnostic devices (IVDs) during a declared public health emergency under section 564 of the FD&C Act [93] [94]. The primary regulatory pathways for such devices are the Emergency Use Authorization (EUA) and pre-EUA submissions.
Core Validation Principles and Regulatory Framework
The goal of validation in this context is to ensure that IVDs for emerging pathogens are accurate, reliable, and clinically meaningful. The FDA's guidance outlines general recommendations for test data and information submitted for regulatory review [93]. This framework is designed to help manufacturers prepare for future outbreaks by clarifying regulatory expectations. The guidance was developed in response to independent assessments of the FDA's COVID-19 response, which recommended creating a standardized framework for test validation during a declared public health emergency (PHE) [93].
Key Analytical Performance Parameters
Validation of pathogen detection tests requires a thorough assessment of several key performance parameters. The following table summarizes the critical validation parameters and their general objectives for qualitative IVDs.
Table 1: Key Analytical Validation Parameters for Qualitative Pathogen Detection Tests
| Parameter | Validation Objective |
|---|---|
| Analytical Sensitivity (LoD) | Establish the lowest concentration of the pathogen that can be reliably detected. Determined using methods such as probit analysis. |
| Analytical Specificity | Evaluate interference from cross-reacting organisms and endogenous/exogenous substances to ensure the test detects only the target pathogen. |
| Inclusivity | Demonstrate that the test reliably detects the diverse genetic variants of the target pathogen that are circulating. |
| Exclusivity | Confirm that the test does not cross-react with near-neighbor organisms or other pathogens that could cause false positives. |
| Precision | Assess the degree of reproducibility under defined conditions (e.g., within-run, between-run, between-operator, between-lot). |
| Robustness | Determine the test's reliability when minor, deliberate changes are made to operational or environmental conditions. |
Experimental Protocol: Determining Limit of Detection (LoD)
A standard approach for establishing the LoD for a qualitative pathogen test is as follows:
- Sample Preparation: Serial dilutions of the target pathogen (e.g., cultured virus, bacterial cells, or synthetic genetic material) are prepared in a relevant matrix (e.g., universal transport medium, synthetic saliva, or nasal swab matrix).
- Testing: A minimum of 20 replicates per dilution level are tested with the IVD. The dilution levels should bracket the anticipated LoD.
- Data Analysis: The results are analyzed using a statistical model, such as probit regression, to determine the concentration at which the test detects the analyte with ≥95% probability.
- Verification: The calculated LoD is then verified by testing a minimum of 20 replicates at the determined concentration, requiring 100% detection or a statistically acceptable hit rate (e.g., ≥19/20).
This validation pathway and its key components can be visualized in the following workflow.
Probiotic Potency: Ensuring Viability and Efficacy
For probiotic products, "potency" is intrinsically linked to the viability (live microorganism count) and the stability of the product throughout its shelf life. Governing bodies, such as the Australian Therapeutic Goods Administration (TGA), define quality as encompassing "composition, strength, potency, stability, sterility, purity, [and] bioburden" [95]. The efficacy of a probiotic medicine is dependent on the presence of specific, live microorganisms in sufficient quantities to confer a health benefit.
Core Validation Principles and Regulatory Framework
The validation of probiotic potency methods focuses on controlling quality parameters to ensure safety and efficacy. A primary requirement is precise strain-level identification, for which Whole-Genome Sequencing (WGS) is increasingly considered the gold standard [96]. Furthermore, potency must be maintained through full-process quality control from manufacturing to the end of the shelf life, often requiring adherence to Good Manufacturing Practices (GMP) [96] [97]. The stability of the product must be validated to ensure the viable count at the end of shelf life meets the labeled claim, a critical factor for efficacy [95].
Enumeration Methods and Validation Parameters
The two primary methodologies for enumerating viable probiotics are traditional plating and flow cytometry. Each has distinct advantages and validation requirements.
Table 2: Comparison of Primary Methods for Probiotic Enumeration
| Parameter | Plate-Based Enumeration | Flow Cytometry |
|---|---|---|
| Principle | Culture-based growth and colony counting. | Laser-based detection and staining of individual cells. |
| Measure | Colony Forming Units (CFU). | Total viable cell count. |
| Throughput | Low (time and labor-intensive). | High (automated and rapid). |
| Key Validation Checks | Strain-specific growth conditions, media suitability, dilution linearity. | Stain specificity, instrument calibration, gating strategy robustness. |
| Regulatory Status | Traditional gold standard. | ISO 17025 accredited methods available (e.g., ISO 19344). |
Experimental Protocol: Plate-Based Enumeration (CFU Assay)
This is the foundational method for determining viable counts in probiotics [97].
- Sample Preparation: Homogenize the probiotic product (powder, capsule, gummy) in an appropriate diluent. For difficult matrices like gummies, additional steps may be needed to liberate the microbes.
- Serial Dilution: Perform a serial dilution (e.g., 1:10) in a neutral broth to obtain a countable range (typically 25-250 colonies per plate).
- Plating: Plate aliquots of the chosen dilutions onto sterile Petri dishes and pour with strain-specific growth medium tempered to ≤45°C. Alternatively, spread the sample on pre-poured agar plates.
- Incubation: Incubate plates under optimal conditions (specific temperature, atmosphere, and duration) for the target strain(s).
- Counting and Calculation: Count the number of colonies on a plate and multiply by the dilution factor to calculate the CFU per gram or per serving:
CFU/g = (Number of colonies) / (Dilution factor * Volume plated).
The critical quality attributes and the process for ensuring probiotic potency are multi-stage, as shown below.
Biotherapeutic Development: Functional Potency Assays
For advanced biotherapeutics, including Live Biotherapeutic Products (LBPs) and cell therapies, potency is defined as the "specific ability or capacity of the product to affect a given result" [98]. A potency assay must quantify the biological activity of the product and reflect its proposed Mechanism of Action (MoA). Regulatory agencies like the FDA and EMA consider potency a Critical Quality Attribute (CQA) that must be measured for each product lot [99] [98].
Core Validation Principles and Regulatory Framework
The development of potency assays for biotherapeutics is guided by several key principles. The assay must be mechanism-based, directly linked to the product's biological effect, rather than a mere surrogate like cell count [98]. For complex products, a potency assay matrix—a combination of multiple assays—may be necessary to fully capture the product's biological activity [98]. Finally, the strategy must be phase-appropriate, with development and validation increasing in rigor as the product moves from research to commercial stages [98].
Advanced Potency Assay Formats
Potency assays for biotherapeutics are diverse and tailored to the product's MoA.
Table 3: Common Potency Assay Formats for Biotherapeutics
| Assay Format | Principle | Example Application |
|---|---|---|
| Cell-Based | Measures a functional response in a live cell system (e.g., cytokine release, cell killing). | CAR-T cell potency via IFN-γ release upon target cell recognition [98]. |
| Binding Assay | Quantifies the ability of a therapeutic to bind to its target (e.g., antigen, receptor). | Monoclonal antibody binding to a specific antigen [100]. |
| Flow Cytometry | Multi-parameter analysis of cell surface markers or functional probes. | Potency determination for ATMPs by measuring surface antigen expression [99]. |
| High-Throughput Viability | Uses growth kinetics (e.g., lag time) as a proxy for viability in a micro-scale format. | Screening lyoprotectants for LBPs [37]. |
Experimental Protocol: High-Throughput Viability Screening for LBPs
This protocol, adapted from a 2025 study, describes a method to screen lyoprotectants for a Live Biotherapeutic Product (LBP) using growth lag time as a viability indicator [37].
- Formulation: Mix the LBP (e.g., a genetically modified yeast) with various lyoprotectant candidates (e.g., trehalose, sucrose, other alternatives) in a 96-well plate.
- Lyophilization: Lyophilize the formulations directly on the metal 96-well plate to simulate the drying stress of the manufacturing process.
- Reconstitution and Inoculation: Wash the lyophilized cakes to remove lyoprotectants, dilute, and inoculate into a nutritional growth medium in a new plate.
- Growth Monitoring: Continuously monitor the optical density (OD) or another growth metric of the cultures.
- Data Analysis: Calculate the lag time for each formulation. A significant inverse correlation (
Pearson's coefficient ≈ -0.99) has been demonstrated betweenlog10(cell viability)and growth lag time, meaning a shorter lag time indicates higher post-lyophilization viability [37].
The strategic development of a robust potency assay is critical for navigating the biotherapeutic development pathway.
The Scientist's Toolkit: Essential Reagents and Materials
Successful method validation relies on the use of high-quality, well-characterized reagents and materials. The following table details key solutions used in the experimental fields covered in this guide.
Table 4: Key Research Reagent Solutions for Microbial Viability and Potency Research
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Strain-Specific Culture Media | Supports the growth and enumeration of target microorganisms. | LB broth for E. coli; MRS broth for Lactobacillus; YEPD for yeast [7]. |
| Lyoprotectants (e.g., Trehalose, Sucrose) | Protect live microorganisms from damage during freeze-drying (lyophilization). | Screening protective excipients for Live Biotherapeutic Products (LBPs) [37]. |
| Viability Stains (for Flow Cytometry) | Fluorescent dyes that distinguish live from dead cells based on membrane integrity or enzymatic activity. | Probiotic enumeration via flow cytometry for accurate, high-throughput cell counts [97]. |
| Whole Genome Sequencing (WGS) Kits | Provide reagents for precise genetic identification of microbial strains to the species and strain level. | Confirm strain identity for probiotics and LBPs as a regulatory requirement [96]. |
| Reference Materials & Cell Mimics | Engineered cells providing stable, reproducible controls for functional assays. | TruCytes for standardizing CAR-T cell potency assays (e.g., IFN-γ release) [98]. |
| Triphenyl Tetrazolium Chloride (TTC) | Colorimetric indicator used in growth media; reduced by metabolically active cells to a visible red pigment. | Enhancing colony contrast in viability assays like GVA and plate counting [7]. |
Validation of methods in pathogen detection, probiotic potency, and biotherapeutic development is a demanding but essential discipline that bridges scientific innovation, manufacturing consistency, and regulatory compliance. The gold standards are evolving, from the foundational CFU assay towards more functional, mechanism-based potency measures and high-throughput technologies like flow cytometry and geometric viability assays. A successful validation strategy is iterative, phase-appropriate, and grounded in a deep understanding of the product's biological function. By adhering to the frameworks and employing the tools outlined in this guide, researchers and developers can ensure their methods are robust, reliable, and capable of generating data that protects public health and brings effective microbial-based therapies to patients.
The accurate assessment of microbial viability stands as a cornerstone of microbiology research, pharmaceutical development, and clinical diagnostics. For decades, the colony forming unit (CFU) assay has remained the gold standard for enumerating viable cells across diverse disciplines, combining simplicity with readily available reagents to achieve an enormous dynamic range of 1 to 100,000,000 viable cells per sample [101]. However, this conventional approach is increasingly revealing limitations in our modern research environment—it is notoriously time-intensive, resource-consuming, and generates significant plastic waste [101]. The fundamental definition of microbial viability itself is expanding beyond mere cultivability to encompass metabolic activity, membrane integrity, and genomic potential, necessitating more sophisticated assessment methodologies.
The future of viability assessment is rapidly evolving toward integrated, multi-parameter approaches that capture the complex physiological states of microorganisms while embracing standardized data management practices to ensure reproducibility and interoperability across studies. This transformation is being driven by concurrent advancements in three key areas: novel assay technologies that transcend traditional growth-based measurements, computational tools that extract maximal information from experimental data, and community-wide standardization initiatives that promote data quality and reuse. Within this framework, techniques such as the Geometric Viability Assay (GVA) are demonstrating how geometric probability can accelerate traditional CFU measurements by reducing time and consumables requirements over 10-fold while maintaining a dynamic range of 6 orders of magnitude [101]. Simultaneously, the field is recognizing that technological innovation must be paired with robust data standards, as evidenced by recent initiatives like the National Microbiome Data Collaborative (NMDC) Ambassador Program, which works to address the critical challenges of data comparability, reproducibility, and reusability in microbiome research [102].
Current Gold Standards and Their Limitations in Microbial Research
Established Methods and Emerging Challenges
Traditional viability assessment methods each offer distinct advantages and limitations, making them suitable for different experimental contexts while highlighting the need for complementary multi-parameter approaches. The table below summarizes the key characteristics of current fundamental methods:
Table 1: Comparison of Fundamental Microbial Viability Assessment Methods
| Method | Principle | Throughput | Time Required | Key Limitations |
|---|---|---|---|---|
| CFU Assay | Growth-based colony formation on solid media | Low | 18-24 hours to several days | Only detects culturable cells; lengthy incubation [101] |
| Live/Dead Staining | Membrane integrity discrimination using fluorescent dyes | Medium | Minutes to hours | Cannot distinguish between metabolically active and dormant cells with intact membranes |
| ATP Assay | Measurement of cellular ATP levels as indicator of metabolic activity | High | Minutes to hours | Does not indicate replicative capacity; sensitive to environmental conditions |
| Geometric Viability Assay (GVA) | Probability-based calculation from colony distribution in cones | High (1,200 measurements/day) | 18-24 hours with reduced hands-on time | Requires specialized imaging and analysis; newer method with evolving applications [101] |
The limitations of these established methods become particularly evident when investigating complex microbial states such as persistence, viability-but-non-culturability (VBNC), and biofilm-associated cells. The CFU assay, while considered the gold standard for replicative capacity, fundamentally fails to detect VBNC cells that remain metabolically active but have entered a dormant state where they cannot form colonies on conventional media [101]. Similarly, membrane integrity stains often misclassify dormant cells with intact membranes as "viable" despite their inability to replicate, while ATP assays may overestimate viability in stressed cells with transient metabolic activity but compromised reproductive capacity.
The Economic Context: Market Drivers for Innovation
The viability assays market reflects both the dominance of established methods and the growing investment in innovative approaches. The cell viability assays market size was over USD 2.68 billion in 2025 and is projected to exceed USD 6.46 billion by 2035, growing at a compound annual growth rate (CAGR) of over 9.2% [103]. This robust growth is fueled by several key factors:
- Rising prevalence of chronic diseases necessitating extensive drug discovery programs
- Expansion in pharmaceutical R&D spending, particularly in cell-based therapies
- Growing demand for stem cell research and regenerative medicine applications
- Increased adoption of high-throughput screening technologies in toxicology studies [104] [105]
The market is simultaneously experiencing a shift toward automated, multiplexed solutions that align with the multi-parameter approach advocated in this whitepaper. Key trends include the development of high-content screening methods, integration of AI and machine learning algorithms, creation of real-time monitoring systems, and adoption of automated liquid handling systems that collectively enable more comprehensive viability assessment [104].
Emerging Multi-Parameter Approaches in Viability Science
Technological Foundations for Integrated Assessment
Multi-parameter viability assessment represents a paradigm shift from single-endpoint measurements to integrated profiling of cellular states. This approach leverages complementary techniques to overcome the limitations of individual methods, providing a more nuanced understanding of microbial physiology. The core principle involves simultaneous measurement of multiple viability parameters—typically combining membrane integrity, metabolic activity, enzymatic function, and replicative capacity—to generate a composite viability profile that accurately distinguishes between different physiological states.
The experimental workflow for a comprehensive multi-parameter assessment typically integrates several complementary techniques:
Table 2: Multi-Parameter Viability Assessment Workflow Components
| Step | Technique | Parameters Measured | Implementation Considerations |
|---|---|---|---|
| Primary Sorting | Live/Dead staining with fluorescent dyes (e.g., SYTO 9/propidium iodide) | Membrane integrity | Rapid assessment; distinguishes clearly compromised cells [106] |
| Metabolic Profiling | ATP assays, resazurin reduction, tetrazolium salts (MTT, XTT) | Metabolic activity | Correlates with enzymatic activity; indicates functional metabolism |
| Growth Potential | Modified CFU assays, GVA, growth revival techniques | Replicative capacity | Ultimate test of reproductive ability; includes culturable and potentially revivable cells |
| Functional Analysis | Enzyme activity probes, membrane potential dyes, redox-sensitive fluorophores | Specific cellular functions | Reveals subpopulations with different metabolic capabilities |
The power of multi-parameter approaches is particularly evident when investigating complex biological scenarios such as antibiotic efficacy testing, where distinguishing between bactericidal, bacteriostatic, and persister-inducing effects requires complementary methods. For instance, combining live/dead staining with ATP assays can reveal subpopulations of membrane-intact but metabolically dormant persister cells that would be misclassified by either method alone.
Advanced Implementation: Geometric Viability Assay (GVA)
The Geometric Viability Assay represents a significant innovation in growth-based viability assessment that maintains the fundamental principle of the CFU assay while dramatically improving its efficiency. GVA computes a sample's viable cell count based on the distribution of embedded colonies growing inside a pipette tip, leveraging the predictable probability of colony formation along the cone's axis [101].
The probability density function (PDF) governing colony distribution in a cone is defined as:
Where x is the perpendicular distance from the tip along the x-axis and h is the total length of the cone. The total CFU concentration can then be estimated using:
Where (x₁, x₂) are the positions of the first and last colony in the counted sub-volume and V is the volume of the cone [101].
Experimental Protocol: Geometric Viability Assay
- Sample Preparation: Serially dilute microbial cells in appropriate buffer or growth medium.
- Agarose Embedding: Mix sample with melted LB agarose (cooled to ≤50°C) to a final agarose concentration of 0.5%.
- Loading: Transfer agarose-cell mixture into standard pipette tips using sterile technique.
- Solidification: Allow agarose to solidify completely at room temperature or 4°C.
- Incubation: Eject solidified agarose tips into sterile tip rack and incubate overnight at appropriate temperature.
- Imaging: Capture high-resolution images of colonies within tips using customized optical setup with mirrorless camera or smartphone macro lens attachment.
- Analysis: Measure position of colonies along tip axis and compute viable concentration using probability equations.
GVA has demonstrated compatibility with gram-positive and gram-negative planktonic bacteria, biofilms, and eukaryotic yeast cells, effectively replicating CFU measurements over 6 orders of magnitude while reducing time and consumable requirements by over 10-fold [101]. For pathogenic strains, the enclosed tip format facilitates safe handling through external decontamination procedures without affecting internal colony growth.
Research Reagent Solutions for Multi-Parameter Assessment
Implementing robust multi-parameter viability assessment requires carefully selected reagents and tools. The following table details essential research solutions:
Table 3: Essential Research Reagent Solutions for Multi-Parameter Viability Assessment
| Reagent/Tool | Function | Application Context |
|---|---|---|
| Fluorescent Viability Stains (SYTO 9/PI, FUN-1, CFDA-AM) | Differential staining based on membrane integrity and enzymatic activity | Rapid initial classification of cell populations; often used with fluorescence microscopy or flow cytometry |
| Metabolic Indicators (Resazurin, AlamarBlue, MTT, XTT) | Measure metabolic activity via reduction reactions | Assessment of functional metabolism; useful for high-throughput screening |
| ATP Detection Reagents (Luciferin-luciferase) | Quantify ATP levels as indicator of metabolic activity | Sensitive measurement of metabolically active cells; frequently used in bioluminescence assays |
| GVA Agarose Matrix (Low-melt agarose with TTC) | Provides solid support and contrast enhancement for colony formation | Enables probability-based viability assessment in pipette tips; tetrazolium red enhances colony visibility [101] |
| Microplate Assay Kits (CCK-8, PrestoBlue) | All-in-one formulations for high-throughput screening | Compatible with automated systems; designed for 96-well and 384-well formats |
The consumables segment dominates the product landscape in viability assessment, accounting for approximately 55% of the market share, reflecting the critical role of specialized reagents and kits in modern microbiology research [103].
Standardization and Data Management in Viability Research
Implementing FAIR Principles for Microbial Data
Standardization represents the essential corollary to technological advancement in viability assessment, ensuring that data generated through sophisticated multi-parameter approaches remains comparable, reproducible, and reusable across studies and laboratories. The FAIR (Findable, Accessible, Interoperable, and Reusable) principles have emerged as a foundational framework for addressing the critical data management challenges in microbial research, where inconsistent implementation of standards has historically limited data utility [102].
Community-led initiatives like the National Microbiome Data Collaborative (NMDC) Ambassador Program have demonstrated the effectiveness of "train-the-trainer" models for propagating best practices in data stewardship. Assessments following NMDC workshops revealed that 86% of participants reported increased familiarity with FAIR data principles, while 82% showed improved understanding of metadata standards and templates [102]. Perhaps most significantly, 99% of workshop attendees indicated plans to incorporate FAIR data concepts and data reuse practices into their future work, suggesting substantial potential for community-wide improvement in data quality and interoperability.
The implementation of standardized data management practices yields particularly valuable benefits in viability research:
- Enhanced cross-study comparisons through consistent metadata annotation
- Improved analytical reproducibility via standardized processing workflows
- Facilitated meta-analyses through interoperable data formats
- Long-term data preservation that maximizes research investment
Emerging Standards and Regulatory Frameworks
The standardization landscape for microbial research is evolving rapidly, with recent developments including the October 2025 release of two significant Chinese national standards: "Microbial Resource Institution Data Management and Release Specification" (GB/T 46408-2025) and "Metagenomic Data Processing and Analysis Requirements" (GB/T 46205-2025) [107]. These standards establish critical frameworks for data management in microbial resource centers and specify technical requirements for metagenomic data processing, directly impacting how viability data should be documented, processed, and shared.
The "Microbial Resource Institution Data Management and Release Specification" provides a standardized set of data fields for publication, introducing unique identifiers and unified data formats to improve accuracy and interoperability in data exchange between microbial resource centers [107]. Simultaneously, the "Metagenomic Data Processing and Analysis Requirements" addresses the crucial need for standardized analytical workflows in complex microbial community studies, where variations in processing methods directly impact the reliability of community composition and functional assessments [107].
For researchers implementing viability assessment protocols, several key standardization practices are recommended:
- Adopt community metadata standards such as the Minimum Information about a Microbial Culture (MIMaC) guideline
- Implement unique, persistent identifiers for microbial strains and datasets
- Document analytical workflows with version control for computational methods
- Participate in community proficiency testing to ensure methodological consistency
- Contribute to public repositories with comprehensively annotated data
Integrated Workflows and Visualizing Multi-Parameter Assessment
The power of contemporary viability assessment lies in the strategic integration of complementary methods into cohesive workflows that provide comprehensive insight into microbial physiological states. The following diagram illustrates a recommended integrated workflow for comprehensive viability assessment:
Diagram 1: Integrated Multi-Parameter Viability Assessment Workflow
This integrated approach enables researchers to overcome the limitations of individual methods by providing complementary data streams that collectively generate a comprehensive viability profile. The workflow emphasizes parallel assessment of membrane integrity, metabolic activity, and replicative capacity, with subsequent computational integration to resolve complex microbial populations including viable but non-culturable (VBNC) cells, persisters, and actively replicating populations.
For the implementation of the Geometric Viability Assay component within this broader workflow, the following experimental procedure visualization provides specific methodological details:
Diagram 2: Geometric Viability Assay (GVA) Experimental Workflow
The mathematical foundation of GVA enables its distinctive efficiency advantage over traditional methods. By leveraging the probability distribution of colony formation within a conical geometry, GVA achieves accurate quantification without requiring exhaustive enumeration of all colonies. Experimental validation has demonstrated that the CFU estimate derived from GVA deviates by less than a factor of 2 from the correct value in 97% of simulations based on the positions of just the first 10 colonies, even when the tip contains over 10,000 colonies [101].
Future Perspectives and Implementation Recommendations
Emerging Technologies and Methodological Convergence
The trajectory of viability assessment points toward increasingly integrated, automated, and information-rich approaches that leverage advances in multiple complementary fields. Several key technological developments are poised to further transform viability assessment in the near future:
- AI-enhanced image analysis for automated colony identification and classification in assays like GVA, improving accuracy and throughput while reducing subjective interpretation [104]
- Microfluidic single-cell analysis platforms that enable tracking of individual cell viability parameters across heterogeneous populations
- High-content multiparameter screening that combines viability assessment with simultaneous measurement of morphological and functional cellular markers
- Biosensor-integrated approaches incorporating genetically encoded viability reporters for continuous monitoring of microbial states
- Nanomaterial-based detection systems offering enhanced sensitivity for rare cell analysis and early viability changes
The convergence of these technological developments with standardized data practices will enable previously impractical large-scale viability studies, such as comprehensive antibiotic combination screening against persistent populations, longitudinal monitoring of microbial community dynamics, and high-throughput toxicity assessment in complex environmental samples.
Strategic Implementation Framework
For research organizations seeking to implement advanced viability assessment capabilities, a phased approach balancing technological investment with staff development is recommended:
- Technology Selection: Prioritize flexible platforms supporting multiple detection modalities rather than single-function instruments
- Workflow Integration: Develop standardized operating procedures that incorporate both experimental methods and data management practices
- Personnel Training: Invest in technical specialization while fostering cross-functional understanding of integrated data interpretation
- Quality Systems: Implement regular proficiency testing and reference standards to ensure methodological consistency
- Data Infrastructure: Establish structured data repositories with appropriate metadata standards to maximize research value
The remarkable convergence of novel analytical methods like GVA, which reduces traditional CFU assay time and consumables by over 10-fold, with robust data standardization frameworks creates unprecedented potential for advancing microbial viability research [101]. By embracing both technological innovation and methodological rigor, the field is positioned to transform how we define, measure, and interpret microbial viability across diverse applications from clinical diagnostics to environmental monitoring and biomanufacturing.
Conclusion
The landscape of microbial viability assessment is evolving from a sole reliance on the CFU assay toward a multi-faceted approach that incorporates metabolic and membrane integrity assays to capture the full spectrum of viable cells, including VBNC states. The choice of a 'gold standard' is now context-dependent, dictated by the specific research question, sample type, and required throughput. For researchers in drug development, embracing validated, high-throughput methods like GVA and flow cytometry is crucial for accelerating screening processes, while molecular techniques like PMA-qPCR offer essential strain-specificity for complex products. The future lies in integrated, multi-parameter validation frameworks that combine the strengths of different techniques to provide a more robust and comprehensive understanding of microbial viability, ultimately ensuring greater accuracy in public health risk assessment, therapeutic efficacy, and product quality control.
References
- 1. Recent Methods for the Viability Assessment of Bacterial Pathogens... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9500772/]
- 2. Emerging Technologies for Viability Enumeration of Live ... [https://www.frontiersin.org/research-topics/52945/emerging-technologies-for-viability-enumeration-of-live-microorganisms/magazine]
- 3. Frontiers | A Multi-Purpose Approach to the Mechanisms of Action of... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.842414/full]
- 4. Cell Viability Assays - Assay Guidance Manual - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK144065/]
- 5. WST-1 Assay: principles, protocol & best practices for cell ... [https://www.abcam.com/en-us/knowledge-center/cell-biology/wst-1-assay-principles-protocols-and-best-practices-for-cell-viability]
- 6. Assessment of Oral Microbial Viability by 2,6- ... [https://www.mdpi.com/2079-6382/14/6/590]
- 7. A high-throughput and low-waste viability assay for microbes | Nature Microbiology [https://www.nature.com/articles/s41564-023-01513-9]
- 8. A high-throughput and low-waste viability assay for microbes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10686820/]
- 9. CFU-S assay: a historical single-cell assay that offers ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9045698/]
- 10. CFU-S assay: a historical single-cell assay that offers ... [https://escholarship.org/uc/item/0kp3c9c7]
- 11. Culture and Analysis in Hematopoietic CFU Assays [https://www.stemcell.com/culture-and-analysis-of-hematopoietic-progenitor-cells-in-cfu-assays.html]
- 12. New method could replace microbiology CFU assay [https://www.europeanpharmaceuticalreview.com/news/188568/new-method-could-replace-microbiology-cfu-assay/]
- 13. Editorial: Emerging technologies for viability enumeration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11780490/]
- 14. High Throughput Viability Assay for Microbiology - PubMed [https://pubmed.ncbi.nlm.nih.gov/36712102/]
- 15. Frontiers | The importance of the viable - but in... non culturable state [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2014.00258/full]
- 16. Understanding the transition to viable but non-culturable state ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11098925/]
- 17. How to Evaluate Non-Growing Cells—Current Strategies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7912045/]
- 18. The Significance and Detection of VBNC Microorganisms [https://www.americanpharmaceuticalreview.com/Featured-Articles/113051-The-Significance-and-Detection-of-VBNC-Microorganisms/]
- 19. Nonculturable ( Viable ) Bacteria but VBNC [https://www.sigmaaldrich.com/CA/fr/technical-documents/technical-article/microbiological-testing/microbial-culture-media-preparation/viable-but-nonculturable-bacteria]
- 20. Induction of Viable - but in Clinically Relevant... Non Culturable State [https://pmc.ncbi.nlm.nih.gov/articles/PMC9952024/]
- 21. Detection of Viable but Nonculturable E. coli Induced by ... [https://www.sciencedirect.com/science/article/pii/S0362028X2400214X]
- 22. Quantitative Detection of VBNC State Pseudomonas ... [https://www.mdpi.com/2073-4441/16/2/236]
- 23. Detection and Quantification Methods for Viable but Non- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7214806/]
- 24. An Overview of the Current State of Cell Viability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11719996/]
- 25. Viable but non-cultivable state in oral microbiota [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1533768/full]
- 26. From dye exclusion to high-throughput screening: A review ... [https://www.sciencedirect.com/science/article/pii/S0734975025002502]
- 27. BacTiter-Glo™ Microbial Cell Viability Assay Protocol [https://www.promega.com/resources/protocols/technical-bulletins/101/bactiter-glo-microbial-cell-viability-assay-protocol/]
- 28. A Review of Methods to Determine Viability, Vitality, and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.547458/full]
- 29. Validating Flow Cytometry as a Method for Quantifying ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8865432/]
- 30. Methods to Quantify Microbial Viability | NIST [https://www.nist.gov/programs-projects/methods-quantify-microbial-viability]
- 31. Cell Viability - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/cell-viability]
- 32. Comparison of the XTT and resazurin assays for ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701217301549]
- 33. Comparison of three different cell viability assays for ... [https://pubmed.ncbi.nlm.nih.gov/25107459/]
- 34. Evaluation of an Automated System for the Counting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10433998/]
- 35. 3.1 Culture-dependent and culture-independent methods [https://fiveable.me/microbiomes-health-and-the-environment/unit-3/culture-dependent-culture-independent-methods/study-guide/ZQjjDgf0rRjZpFnx]
- 36. What is the role of microbial biotechnology and genetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10982607/]
- 37. A High-Throughput Microbial Viability Assay to Evaluate ... [https://pubmed.ncbi.nlm.nih.gov/41123384/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1FEu_gUSObj3DNTLNJL99sNQjL7WB4DgbcRhwqTBmQ0fEQTgLY&fc=None&ff=20251026023037&v=2.18.0.post22+67771e2]
- 38. To culture or not to culture: a snapshot of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8418214/]
- 39. Comparison of Platelet Count by Automated and Manual ... [https://jhscr.org/index.php/JHSCR/article/view/63]
- 40. Reshape [https://www.reshapebiotech.com/applications/cfu-counting]
- 41. Comparison of an Automated Most-Probable-Number ... [https://www.sciencedirect.com/science/article/pii/S0362028X22119654]
- 42. How do we know if a microbe is dead? [https://isappscience.org/how-do-we-know-if-a-microbe-is-dead/]
- 43. Recent Methods for the Viability Assessment of Bacterial ... [https://www.mdpi.com/2076-0817/11/9/1057]
- 44. A Review of Methods to Determine Viability, Vitality, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7705206/]
- 45. The Comparison of MTT and CVS Assays for the Assessment ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0155772]
- 46. tetrazolium reduction assay: Topics by ... [https://www.science.gov/topicpages/t/tetrazolium+reduction+assay]
- 47. An Optimized LIVE/DEAD Assay Coupled with Flow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12453624/]
- 48. LIVE/DEAD BacLight Bacterial Viability Kit Protocol [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/live-dead-baclight-bacterial-viability-protocol.html]
- 49. Rapid Detection of Escherichia coli Antibiotic Susceptibility ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8147107/]
- 50. Propidium iodide staining underestimates viability of ... [https://www.nature.com/articles/s41598-019-42906-3]
- 51. Geometry meets Microbiology: A New Approach to Viable ... [https://5dhpg.com/news/geometry-meets-microbiology-a-new-approach-to-viable-counts]
- 52. Geometric Viability Assay (GVA) [https://www.colorado.edu/research/geometric-viability-assay/geometric-viability-assay-gva]
- 53. A high-throughput anaerobic method for viability assays [https://pmc.ncbi.nlm.nih.gov/articles/PMC11960071/]
- 54. A flow cytometry method for safe detection of bacterial ... [https://pubmed.ncbi.nlm.nih.gov/37786349/]
- 55. Bacterial Viability and Vitality Assays for Flow Cytometry [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometry-assays-reagents/flow-cytometry-microbiology-assays/flow-cytometry-bacteria-viability-vitality-assays.html]
- 56. Proof of concept: real-time viability and metabolic profiling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11220157/]
- 57. Microcalorimetric assays for measuring cell growth and ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202314003260]
- 58. Practical application of PMA–qPCR assay for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11631528/]
- 59. Viability Quantitative PCR Utilizing Propidium Monoazide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6805072/]
- 60. Comparison of Droplet Digital PCR and qPCR for the ... [https://www.mdpi.com/2072-6651/8/5/157]
- 61. Viability PCR [https://biotium.com/technology/pma-for-viability-pcr/]
- 62. Development and validation of a PMA-qPCR method for ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1456274/full]
- 63. Comparison of Real-Time PCR and Droplet Digital PCR for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9105557/]
- 64. A Comparison between Droplet Digital and Quantitative PCR ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0110351]
- 65. Colony-forming unit (CFU) [https://examine.com/glossary/colony-forming-unit-cfu/?srsltid=AfmBOorta0UP4DGZAzUM3XT28H20VW1eThSbyLg9Rm6ddJzy7GxlgoTJ]
- 66. Probiotic Viability Reconsidered: Integrating VBNC ... [https://www.mdpi.com/2076-2607/13/11/2479]
- 67. Challenging the “gold standard” of colony-forming units [https://www.sciencedirect.com/science/article/pii/S0168160521003767]
- 68. State-of-the-Art Metabolic Toxicity Screening and Pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4862375/]
- 69. Cell viability assays [https://www.abcam.com/en-us/technical-resources/guides/cell-health-guide/cell-viability-assays]
- 70. Optimization of cell viability assays to improve replicability ... [https://www.nature.com/articles/s41598-020-62848-5]
- 71. A Twisted-Integrated Multifunctional Fiber Sensor for Real ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12635969/]
- 72. Advancing microbial risk assessment: perspectives from ... [https://www.nature.com/articles/s41538-025-00527-3]
- 73. Results [https://2025.igem.wiki/munich/results]
- 74. Viability Dye Selection Guide [https://fluorofinder.com/guide-to-viability-dyes/]
- 75. Fixable Viability Dyes for Flow Cytometry [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometry-assays-reagents/cell-viability-assays-flow-cytometry/fixable-viability-dyes-flow-cytometry.html]
- 76. Flow Cytometry: An Overview - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5939936/]
- 77. Fluorescence Microscopy Methods for Determining the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3814296/]
- 78. BactoView™ Viability Kits [https://biotium.com/product/bactoview-viability-kits/]
- 79. A High-Throughput Microbial Assay to Evaluate... Viability [https://pubmed.ncbi.nlm.nih.gov/41123384/]
- 80. Mimicking biofilm formation and development - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC8113887/]
- 81. Frontiers | A vacuum lyophilization and bacterial tablet-based method ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1493947/full]
- 82. ESKAPEE Pathogen Biofilm Control on Surfaces with ... [https://www.mdpi.com/2079-6382/12/5/871]
- 83. Probiotic enumeration for Akkermansia muciniphila products [https://www.nutraingredients.com/News/Promotional-features/probiotic-enumeration-for-akkermansia-muciniphila-products/]
- 84. Next-generation rapid phenotypic antimicrobial ... [https://www.nature.com/articles/s41467-024-53930-x]
- 85. Frontiers | Measurement quality metrics to improve absolute microbial ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1631377/full]
- 86. Challenges of growth-based microbiological methods in ... [https://link.springer.com/article/10.1007/s44395-025-00020-6]
- 87. A comparison of conventional methods for the quantification of ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-014-0222-6]
- 88. Life, Death, and In-Between: Meanings and Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3165249/]
- 89. High-Throughput Viability Testing of Microbial ... [https://www.mdpi.com/2673-8007/3/3/74]
- 90. Dataset for comparative analysis of precision ... [https://www.sciencedirect.com/science/article/pii/S235234092500071X]
- 91. Comparative analysis of the application efficiency of gold ... [https://link.springer.com/article/10.1007/s42452-024-05751-2]
- 92. High throughput construction of species characterized ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1545877/full]
- 93. Validation of Certain In Vitro Diagnostic Devices for ... [https://www.federalregister.gov/documents/2025/01/07/2024-31522/validation-of-certain-in-vitro-diagnostic-devices-for-emerging-pathogens-during-a-section-564]
- 94. Validation of Certain In Vitro Diagnostic Devices for ... [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/validation-certain-in-vitro-diagnostic-devices-emerging-pathogens-during-section-564-declared-emergency]
- 95. Demonstrating the quality of listed probiotic medicines [https://www.tga.gov.au/resources/guidance/demonstrating-quality-listed-probiotic-medicines]
- 96. Shaping the future of probiotics, live biotherapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12527994/]
- 97. Probiotic Enumerations: The Basics [https://www.eurofinsus.com/food-testing/resources/probiotic-enumerations-the-basics/]
- 98. How Potency Assay Strategy Can Accelerate Cell Therapy ... [https://slingshotbio.com/how-potency-assay-strategy-can-accelerate-cell-therapy-development/]
- 99. Flow Cytometry as a Tool for Potency Determination in ... [https://www.bioagilytix.com/resources/posters/flow-cytometry-as-a-tool-for-potency-determination-in-atmp-development-wcbp-2025/]
- 100. What is Biopharma Potency Assays? Uses, How It Works & ... [https://www.linkedin.com/pulse/what-biopharma-potency-assays-uses-how-works-top-companies-mveae/]
- 101. High Throughput Viability Assay for Microbiology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9881960/]
- 102. 微生物组数据标准与管理工作坊成效显著:推动数据规范,助力科研发展 - 生物通 [https://www.ebiotrade.com/newsf/2025-3/20250323061139017.htm]
- 103. Cell Viability Assays Market Size, Share & Growth Industry ... [https://www.researchnester.com/reports/cell-viability-assays-market/4785]
- 104. 2025 Cell Viability Assays Industry Trends Report [https://www.openpr.com/news/4131064/2025-cell-viability-assays-industry-trends-report-long-term]
- 105. Drivers of Change in Cell Viability Assay Kits Market 2025- ... [https://www.marketreportanalytics.com/reports/cell-viability-assay-kits-289091]
- 106. Live & Dead Cell Viability Assay Kit Analysis Report 2025 [https://www.archivemarketresearch.com/reports/live-dead-cell-viability-assay-kit-340908]
- 107. 微生物领域两项国家标准正式发布----中国科学院微生物研究所 [https://www.im.cas.cn/xwzx2018/jqyw/202510/t20251022_7995055.html]