From Surface to Structure: Unraveling the Mechanisms of Bacterial Adhesion and Biofilm Initiation
This article provides a comprehensive analysis of the sophisticated mechanisms governing bacterial adhesion and the subsequent initiation of biofilms, a major contributor to chronic infections and antimicrobial resistance.
From Surface to Structure: Unraveling the Mechanisms of Bacterial Adhesion and Biofilm Initiation
Abstract
This article provides a comprehensive analysis of the sophisticated mechanisms governing bacterial adhesion and the subsequent initiation of biofilms, a major contributor to chronic infections and antimicrobial resistance. Tailored for researchers, scientists, and drug development professionals, it synthesizes foundational knowledge with cutting-edge methodological approaches. We explore the biofilm lifecycle from initial surface attachment governed by physicochemical forces and adhesins to the development of a mature, matrix-encased community. The content delves into advanced techniques for studying these processes, evaluates current and emerging therapeutic strategies to disrupt biofilms and prevent their formation, and offers a comparative analysis of conventional versus novel anti-biofilm technologies. The goal is to bridge fundamental research with clinical and industrial applications, offering insights that can inform the development of next-generation anti-infective therapies and biomaterials.
The Blueprint of Biofilms: Foundational Mechanisms of Bacterial Adhesion and Community Initiation
For decades, the five-stage model of biofilm development has served as a foundational concept for understanding how bacteria transition from free-swimming, planktonic cells to complex, surface-associated communities. This developmental cycle begins with the initial attachment of bacteria to a surface and progresses through irreversible attachment, maturation, and eventual dispersal. Framed within the broader context of mechanisms of bacterial adhesion, this model provides a structured framework for investigating the genetic, physical, and chemical processes that underpin biofilm initiation and resilience [1] [2]. The classic representation of this model, often exemplified by the mushroom-shaped structures of Pseudomonas aeruginosa, has been instrumental in guiding research [1].
However, it has become increasingly evident that this model does not fully capture the diversity of biofilm physiology, especially in clinical and environmental settings where biofilms often exist as non-surface attached aggregates [1]. This review will detail the established five-stage model while also acknowledging its limitations and the expanded, inclusive conceptualizations driving contemporary biofilm research forward [1].
The Classic Five-Stage Biofilm Lifecycle
The transformation from a planktonic to a sessile, biofilm lifestyle is a dynamic and regulated process. The following stages represent the core of the traditional model, observed in foundational studies of species like P. aeruginosa, Staphylococcus aureus, and Bacillus subtilis [1].
Stage 1: Initial Reversible Attachment
The biofilm lifecycle commences with the transient association of planktonic cells with a biotic or abiotic surface. This initial contact is mediated by weak, physical forces such as van der Waals interactions and electrostatic forces [3] [2]. The nature of the surface is critical; for instance, rough surfaces are known to promote better microbial adhesion than smooth ones [3]. At this stage, the attachment is reversible, meaning cells can easily detach and return to their planktonic state [2]. Bacterial structures like pili can facilitate this passive attachment in some species, such as Pseudomonas fluorescens [3].
Stage 2: Irreversible Attachment
The transition from reversible to permanent attachment is a critical commitment to the biofilm lifestyle. This stage is characterized by the strong, anchorage of cells to the substrate, primarily through the active secretion of extracellular polymeric substances (EPS) [3] [2]. The EPS matrix, initially rich in extracellular DNA (eDNA), acts as a "cellular glue" [2]. This stage also involves a shift in bacterial physiology, often regulated by signaling molecules like cyclic diguanylate monophosphate (c-di-GMP), which promotes a sessile existence by reducing motility [4].
Stage 3: Microcolony Formation and Maturation I
Following irreversible attachment, the anchored cells begin to proliferate, forming distinct microcolonies [1] [4]. This stage involves significant microbial growth and expansion of the EPS matrix, leading to the development of a rudimentary three-dimensional structure [1] [2]. Cell-cell communication through quorum sensing becomes increasingly important for coordinating population-level behaviors and the expression of biofilm-specific genes [3] [5].
Stage 4: Biofilm Maturation II
The biofilm matures into a complex, heterogeneous community with a characteristic 3D architecture [1] [5]. In many classic models, this includes the formation of towering "mushroom-shaped" structures interspersed with fluid-filled channels, which facilitate nutrient transport and waste removal [1] [6]. The matrix is now a sophisticated hydrogel composed of polysaccharides, structural proteins, lipids, and eDNA, providing mechanical stability and protection [2] [5]. This environment fosters metabolic diversity and ecological niches, allowing different species to coexist and interact synergistically or antagonistically [5].
Stage 5: Dispersion
The final stage of the lifecycle is dispersion, where a subpopulation of cells actively detaches from the mature biofilm to colonize new surfaces [1] [4]. This is a biologically regulated process, often triggered by environmental cues such as nutrient depletion or oxygen gradients [4]. The dispersed cells, which return to a planktonic state, are essential for propagating the infection or colonization cycle [2]. Passive detachment mechanisms, including erosion (loss of small clusters) and sloughing (detachment of large biofilm fragments) due to external shear forces, also contribute to dispersion [1] [4].
The following diagram synthesizes the core logic and regulatory drivers of this five-stage lifecycle.
Expanded Conceptual Models and In Vivo Complexity
While the five-stage model is a valuable heuristic tool, contemporary research emphasizes that biofilm formation is not a monolithic process. The conceptual model is being expanded to include scenarios more representative of in vivo conditions [1].
- Non-Surface Associated Aggregates: In many clinical infections, such as those in the viscous airway mucus of cystic fibrosis patients or in persistent soft tissue wounds, bacteria form cohesive aggregates that are not attached to a substratum. These self-contained aggregates display a biofilm-like phenotype and are now recognized as a form of biofilm [1] [4].
- Seeding from Cell Clumps: Rather than always initiating from single cells, biofilms can be seeded by pre-formed clumps of cells that have detached from an existing aggregate or formed in a mucus layer [4].
- Host Protein-Mediated Attachment: In infections involving medical implants, bacteria often do not attach directly to the device surface. Instead, they bind to host proteins like fibrin and fibrinogen that coat the surface, using specific adhesins like the S. aureus clumping factor A (ClfA) [4].
Quantitative Analysis of Biofilm Development
Tracking changes in biofilm architecture and biomass over time is crucial for quantifying developmental progression. The following table summarizes biovolume data from a study investigating Mycoplasma fermentans biofilm formation on glass surfaces over a one-week period, demonstrating the increase in biomass during the maturation stages [6].
Table 1: Biovolume Quantification of M. fermentans Biofilms at Early and Late Growth Stages
| M. fermentans Strain | Median Biofilm Volume at 3 Days (µm³) × 10³ | Median Biofilm Volume at 7 Days (µm³) × 10³ |
|---|---|---|
| ATCC19989 (Replicate 1) | 76 | 97 |
| ATCC19989 (Replicate 2) | 27 | 106 |
| M67910 (Replicate 1) | 4.9 | 5.8 |
| M67910 (Replicate 2) | 1.9 | 46 |
| MF1 (Replicate 1) | 7.7 | 40 |
| MF1 (Replicate 2) | 40 | 21 |
| M67195 (Replicate 1) | 1.9 | 2.0 |
| M67195 (Replicate 2) | 2.0 | 3.9 |
The data shows clear variability between strains and replicates, but a general trend of increased biofilm volume from day 3 to day 7 is evident, correlating with architectural maturation observed via imaging [6].
Advanced Methodologies for Investigating the Biofilm Lifecycle
Experimental Protocol: Confocal Laser Scanning Microscopy (CLSM) for 3D Architectural Analysis
CLSM is a cornerstone technique for non-destructively visualizing and quantifying the 3D structure of living biofilms [6] [7].
- Biofilm Growth: Grow biofilms on sterile, suitable substrates (e.g., 22 mm² glass coverslips) placed vertically in culture tubes containing appropriate growth medium, inoculated with a diluted planktonic culture (e.g., 1:100 dilution) [6].
- Incubation: Incubate under required conditions (e.g., 37°C in 5% CO₂) without agitation for defined periods (e.g., 3-7 days) to capture early and late growth stages [6].
- Fixation and Staining: Gently wash the biofilm-bearing coverslips with phosphate-buffered saline (PBS) to remove non-adherent cells. Fix the biofilms with a 4% formaldehyde solution in PBS for 10 minutes at room temperature. Stain with a fluorescent nucleic acid dye, such as propidium iodide, for 15 minutes [6].
- Imaging: Mount the coverslips and image using an inverted confocal microscope with appropriate laser excitation and emission detection settings. Acquire z-stack image series with high axial resolution (e.g., 0.12 µm slice thickness) [6].
- 3D Reconstruction and Quantification: Process the z-stack image datasets using specialized software (e.g., Amira). Apply median filters to reduce noise, threshold the images to define microcolonies, and generate 3D iso-surface visualizations. The software can then be used to quantify key parameters like total biovolume and structural features [6].
Cutting-Edge Technique: Single-Cell Morphometry in 3D Biofilms
Conventional microscopy and segmentation techniques struggle to accurately resolve individual cells within dense, 3D biofilms. The Bacterial Cell Morphometry 3D (BCM3D) workflow overcomes this by combining deep learning with non-invasive light-sheet microscopy [7].
- Image Acquisition: Acquire high-resolution 3D images of live biofilms with low phototoxicity using lattice light-sheet microscopy (LLSM). Cells can be labeled via internal fluorescent protein expression or membrane staining [7].
- In Silico CNN Training: Train 3D U-Net convolutional neural networks (CNNs) using computationally simulated biofilm images that mimic experimental conditions, including realistic cell densities, signal-to-background ratios (SBRs), and labeling methods. These simulated images have perfectly known "ground truth" segmentation maps [7].
- Cell Segmentation and Classification: Apply the trained CNNs to experimental LLSM image data. BCM3D uses the CNN output, followed by mathematical post-processing, to accurately segment and classify single bacterial cells in 3D space, enabling precise cell tracking and morphometric analysis over time [7].
This workflow achieves high voxel-level segmentation accuracy (>80%) and cell counting accuracy (>90%), even at high cell densities where conventional methods fail [7].
The BCM3D workflow integrates advanced imaging and computational analysis to enable single-cell resolution in dense biofilms, as visualized below.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Biofilm Lifecycle Studies
| Reagent/Material | Function in Biofilm Research | Example Application |
|---|---|---|
| Glass Coverslips | Provides a transparent, abiotic surface for standardized biofilm growth and direct microscopic observation. | Substrate for growing mycoplasma biofilms for CLSM and SEM analysis [6]. |
| Propidium Iodide | A fluorescent dye that stains nucleic acids; used to visualize the spatial distribution of cells and matrix components containing DNA (eDNA) within the biofilm. | Fluorescent staining of fixed M. fermentans biofilms to determine biovolume and structure via CLSM [6]. |
| Extracellular DNA (eDNA) | A key component of the biofilm matrix that contributes to structural integrity, adhesion, and antimicrobial tolerance. | Study of the role of eDNA in initial matrix formation and its ability to chelate cationic antibiotics like aminoglycosides [2] [4]. |
| Formaldehyde (4% in PBS) | A cross-linking fixative agent used to preserve the delicate 3D structure of biofilms for subsequent staining and imaging without structural collapse. | Fixation of M. fermentans biofilms on coverslips prior to CLSM imaging [6]. |
| Convolutional Neural Networks (CNNs) | Deep learning models for automated, accurate segmentation and classification of individual bacterial cells within 3D image stacks of dense biofilms. | Core of the BCM3D workflow for single-cell morphometry and tracking in living biofilms [7]. |
The five-stage biofilm lifecycle remains an essential framework for understanding the fundamental shift from planktonic to sessile bacterial existence, providing a critical context for research into bacterial adhesion mechanisms. However, the field is rapidly evolving beyond this classic model. The recognition of non-surface attached aggregates and the influence of the host environment demands a more flexible and inclusive conceptual model of biofilms [1]. The development of sophisticated tools like CLSM, LLSM, and deep learning-based analytics such as BCM3D is empowering researchers to dissect biofilm architecture and single-cell behaviors with unprecedented detail [6] [7]. This refined understanding, bridging traditional models with modern complexities, is vital for developing novel strategies to combat persistent biofilm-associated infections and for leveraging biofilms in industrial and environmental applications.
The initial adhesion of bacteria to surfaces is a critical, multi-stage process governed by the complex interplay of non-covalent physicochemical forces. Within the broader context of biofilm initiation research, understanding these forces—van der Waals, electrostatic, and hydrophobic interactions—is paramount for developing anti-fouling strategies and combating device-related infections. This in-depth technical guide synthesizes current knowledge on the individual and collective roles of these forces, providing researchers and drug development professionals with structured quantitative data, detailed experimental methodologies, and visual frameworks to advance the study of bacterial adhesion mechanisms.
Bacterial adhesion represents the critical first step in the establishment of biofilms, which are structured microbial communities embedded in an extracellular polymeric matrix and a significant contributor to persistent infections and global health problems [8]. This adhesion process is not random but is a sophisticated sequence of events directed by the aggregate of physicochemical interactions between the bacterial cell surface and the substrate. These initial interactions occur before any irreversible, biologically-mediated attachment and are largely governed by the physical chemistry of the interacting surfaces [9] [10]. The transition of planktonic bacteria to a sessile, biofilm-associated state is a multi-step process, beginning with the reversible adhesion phase controlled by these non-covalent forces [8] [9]. In both natural environments and complex medical settings, such as on the surface of implantable devices, the outcome of this first contact determines the success of microbial colonization [8]. Consequently, a rigorous understanding of van der Waals forces, electrostatic interactions, and hydrophobic attraction is essential for researchers aiming to disrupt the biofilm lifecycle at its earliest and most vulnerable stage.
Deconstructing the Key Physicochemical Forces
The initial attachment of bacteria to a surface is mediated by a balance of attractive and repulsive physical forces. The interplay between these forces determines the efficiency of colonization and is influenced by the properties of the bacterial surface, the substrate, and the surrounding liquid medium.
Van der Waals Forces: The Universal Attractor
Van der Waals (vdW) forces are ubiquitous, nonspecific attractive forces arising from induced dipole interactions between atoms and molecules. They play a fundamental role in the initial stage of bacterial adhesion by bringing the cell into close proximity with the surface [9]. These forces are always attractive between identical materials in a liquid medium and are effective over a range of up to 10 nanometers. While generally weaker than chemical bonds, their collective contribution is significant over the entire bacterial cell surface. Recent single-molecule force spectroscopy studies on hydrophobic surfaces have quantified the contribution of vdW interactions to the total adhesion force, revealing that in certain systems, their contribution can be less than 9 pN, with hydrophobic attraction playing a more dominant role [11]. The strength of vdW interactions is highly dependent on the material properties and the geometry of the interacting surfaces, but they provide the initial, long-range attractive force that enables closer-range interactions to come into effect.
Electrostatic Interactions: The Double-Edged Sword
Most bacterial cells carry a net negative surface charge at physiological pH due to the presence of ionizable functional groups in their cell wall components, such as teichoic acids in Gram-positive bacteria and lipopolysaccharides in Gram-negatives [8]. When a negatively charged bacterium approaches a typically negatively charged surface in an aqueous environment, a repulsive energy barrier is created. This electrostatic double-layer repulsion must be overcome by stronger attractive forces for adhesion to proceed [9]. However, the nature of electrostatic interactions can be complex. The surface charge of both the bacterium and the substrate is influenced by the environmental pH, ionic strength, and specific ion composition [8]. A positively charged surface will facilitate the attachment of planktonic bacteria by eliminating this repulsive barrier [8]. Furthermore, molecular dynamics simulations have demonstrated that introducing discrete charges or dipoles on a hydrophobic surface can significantly reduce its binding affinity for hydrophobic particles, effectively competing with and diminishing the hydrophobic attraction [12]. This highlights that electrostatic interactions are not merely repulsive but can be finely tuned to alter adhesion outcomes.
Hydrophobic Interactions: The Dominant Driver
Hydrophobic interactions represent a powerful attractive force that drives the adhesion of non-polar surfaces in an aqueous environment. This interaction is primarily an entropy-driven process where water molecules reorganize to minimize their contact with hydrophobic patches, effectively pushing these surfaces together [10]. The hydrophobicity of both the bacterial cell envelope and the substrate material is a critical determinant of adhesion efficiency. Hydrophobic bacterial strains, such as Staphylococcus aureus, demonstrate stronger native adhesion to surfaces compared to less hydrophobic species [10]. Similarly, hydrophobic materials like polystyrene are more prone to bacterial colonization than hydrophilic materials like stainless steel [9]. The dominance of hydrophobic attraction is quantified at the molecular level; on a hydrophobic MoS₂ surface, hydrophobic attraction can contribute up to 89% of the total single-molecule adhesion force, far surpassing the contribution from vdW interactions [11]. This makes the modulation of surface hydrophobicity a key strategy for controlling biofilm formation.
Table 1: Key Characteristics of Physicochemical Forces in Bacterial Adhesion
| Force Type | Nature | Effective Range | Role in Adhesion | Key Influencing Factors |
|---|---|---|---|---|
| Van der Waals | Always attractive (between identical materials) | Long-range (up to 10 nm) | Initial approach and reversible attachment | Material composition, surface geometry |
| Electrostatic | Typically repulsive | Medium-range (up to 100 nm) | Creates an energy barrier to overcome | pH, ionic strength, surface charge density [8] |
| Hydrophobic | Strongly attractive | Short-range (1-2 nm) | Dominant driver for firm attachment | Surface hydrophobicity, water structure [10] [11] |
Table 2: Quantitative Contributions of Forces in a Model System (Oligo Ethylene Glycol Copolymer on MoS₂) [11]
| Interaction Type | Contribution to Total Adhesion Force | Experimental Method | Condition |
|---|---|---|---|
| Van der Waals Interactions | < 9 pN (minor contribution) | Single-Molecule Force Spectroscopy (SMFS) | In water, excluding electrostatic effects |
| Hydrophobic Attraction | Up to 89% of total force | Single-Molecule Force Spectroscopy (SMFS) | In water, on hydrophobic basal surface |
| Electrostatic Interaction | Quantitatively partitioned | SMFS on anisotropic MoS₂ | Varied water chemistry |
Experimental Methodologies for Force Analysis
A comprehensive understanding of adhesion forces requires a multidisciplinary approach, combining macroscopic observations with sophisticated nanoscale measurements and computational models.
Assessing Bacterial Surface Physicochemistry (MATS Protocol)
The Microbial Adhesion To Solvents (MATS) assay is a standard method for characterizing the physicochemical properties of bacterial cell surfaces, which directly influence their adhesive behavior.
Protocol Overview: This method quantifies the affinity of a bacterial cell suspension for a panel of polar and non-polar solvents, which serve as proxies for different types of surface interactions [10].
- Sample Preparation: Bacterial cells are harvested in their mid- to late-exponential growth phase, washed, and resuspended in a buffer or saline solution to an optical density (OD) of approximately 0.4 at 400 nm (OD₄₀₀).
- Solvent Affinity Test: A volume of the bacterial suspension (typically 1.2 mL) is mixed with an equal volume of each test solvent (e.g., chloroform for electron-acceptor character, ethyl acetate for electron-donor character, and decane for hydrophobicity). The mixture is vortexed for 60 seconds to ensure adequate contact.
- Phase Separation & Measurement: The aqueous phase is allowed to separate, and the OD₄₀₀ is measured after 15 minutes. The percentage of cells adhering to or partitioning into the solvent is calculated as
[1 - (OD₄₀₀ after / OD₄₀₀ initial)] * 100%. - Data Interpretation: A high affinity for apolar solvents like decane indicates cell surface hydrophobicity. Affinity for chloroform (an acidic solvent) reflects the bacterium's electron-acceptor (Lewis acid) character, while affinity for ethyl acetate (a basic solvent) reflects its electron-donor (Lewis base) character [10]. This profile helps predict adhesion tendencies to variously charged and hydrophobic surfaces.
Single-Molecule Force Spectroscopy (SMFS)
SMFS, typically performed using atomic force microscopy (AFM), allows for the quantitative measurement of the unbinding forces between a single molecule or a functionalized tip and a surface.
Protocol Overview: This technique partitions the individual contributions of vdW, electrostatic, and hydrophobic interactions to the total adhesion force [11].
- Probe Functionalization: An AFM cantilever tip is functionalized with specific molecules or polymer chains (e.g., an oligo ethylene glycol methacrylate copolymer) relevant to the study.
- Surface Preparation: A substrate with well-defined properties, such as the anisotropic MoS₂ crystal which has a hydrophobic basal plane, is prepared and mounted.
- Force Curve Acquisition: The functionalized tip is brought into contact with the surface and then retracted in a controlled manner, thousands of times, in the desired aqueous environment. The deflection of the cantilever is recorded as a function of distance, generating a force-distance curve for each cycle.
- Data Analysis: The adhesion force is determined from the retraction curve's pull-off event. By systematically varying the environmental conditions (e.g., ionic strength to screen electrostatic interactions) and using surfaces with known properties, the individual contributions of vdW, electrostatic, and hydrophobic forces can be partitioned and quantified, as demonstrated in [11].
Computational Molecular Dynamics (MD) Simulations
MD simulations provide atomic-level insights into the dynamic interplay of forces that are challenging to observe experimentally.
Protocol Overview: This computational method models the time-dependent behavior of a molecular system under defined conditions, allowing for the calculation of binding free energies [12].
- System Setup: A model system is constructed, such as a hydrophobic particle (e.g., a united-atom methane) and two hydrophobic plates with defined charge distributions, solvated in a water box (e.g., using the SPC water model).
- Force Field Parameterization: The interactions between all atoms are defined by a classical force field, which includes parameters for bond stretching, angle bending, and non-bonded interactions (vdW and electrostatic).
- Free Energy Perturbation (FEP): The binding affinity is calculated using the FEP method. The interaction between the particle and the plates is gradually "turned on" over a series of simulation windows (defined by a coupling parameter λ). In each window, the system is equilibrated, and a production run is performed to collect energy data.
- Free Energy Calculation: The Bennett Acceptance Ratio (or similar method) is applied to the energy differences between neighboring λ windows to compute the total free energy change for the binding process. This allows researchers to observe how introducing charges on the plates (electrostatic perturbation) reduces the binding affinity of the hydrophobic particle, demonstrating the competition between hydrophobic and electrostatic interactions [12].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for Bacterial Adhesion Force Research
| Item / Reagent | Function / Application | Technical Notes |
|---|---|---|
| Microbial Adhesion to Solvents (MATS) Kit | Characterizes hydrophobicity and Lewis acid-base properties of bacterial surfaces. | Includes solvents like chloroform (electron-acceptor), ethyl acetate (electron-donor), and decane (hydrophobicity) [10]. |
| Atomic Force Microscope (AFM) | Measures single-molecule adhesion forces via Force Spectroscopy. | Requires functionalizable cantilevers and controlled liquid cells for in-situ measurements in relevant buffers [11]. |
| Anisotropic Crystals (e.g., MoS₂) | Serves as a model substrate to partition vdW and hydrophobic interactions. | The hydrophobic basal plane vs. hydrophilic edges allow for controlled experiments [11]. |
| Molecular Dynamics (MD) Software (e.g., DESMOND) | Simulates atomistic interactions and calculates binding free energies. | Utilizes force fields (e.g., OPLS) and explicit solvent models (e.g., SPC water) for accuracy [12]. |
| 3D Reconstructed Human Skin Model | Provides a physiologically relevant biotic surface for adhesion studies. | Standardized, sterile model with a differentiated epidermal barrier for testing anti-adhesion compounds [10]. |
| Rhamnolipids | Used as bioactive compounds to modulate bacterial surface hydrophobicity. | Demonstrates efficacy in significantly inhibiting S. aureus adhesion by altering its surface to strongly hydrophilic [10]. |
Integrated View: Force Interplay in Adhesion Pathways
The initial adhesion of bacteria is not a sequential process but a concurrent interplay of all physicochemical forces, whose net result determines the outcome of the first contact. Van der Waals forces provide the initial, long-range attraction that draws the cell toward the surface. As the bacterium approaches, the typically repulsive electrostatic double-layer force must be overcome. This is often achieved through the powerful, short-range hydrophobic attraction, which is strongly dependent on the physicochemical properties of both surfaces [10]. The balance of these forces can be strategically disrupted. For instance, introducing charges on a hydrophobic surface can make it "hydrophilic-like," ejecting hydrophobic particles and reducing binding affinity [12]. Similarly, compounds like Rhamnolipids can drastically reduce bacterial adhesion by increasing the hydrophilicity of the bacterial surface, thereby shifting the balance of forces away from attachment [10]. This integrated view underscores that the adhesive potential is a modifiable property, offering a roadmap for therapeutic intervention.
Bacterial adhesion represents the critical initial step in the pathogenesis of numerous infectious diseases and the establishment of beneficial host-microbe relationships. This process is mediated by a diverse array of specialized bacterial surface structures collectively termed adhesins, which function as molecular bridges facilitating attachment to host tissues and abiotic surfaces [13] [14]. The strategic importance of adhesins extends beyond mere attachment; they initiate complex signaling cascades in host cells, trigger biofilm formation, and ultimately determine tissue tropism and species specificity of microorganisms [15] [14]. For researchers and drug development professionals, understanding the molecular architecture and function of these adhesins provides a foundation for novel anti-infective strategies that target the earliest stages of microbial colonization [13] [15]. The escalating challenge of antimicrobial resistance (AMR), particularly among biofilm-forming pathogens, has further intensified research into adhesins as promising targets for prophylactic and therapeutic interventions [3] [4].
Structural Classification of Bacterial Adhesins
Bacterial adhesins can be broadly categorized based on their structural organization and assembly pathways. These sophisticated molecular machines have evolved distinct mechanisms to present receptor-binding domains at the bacterial surface, enabling specific recognition of host cell determinants.
Fimbrial Adhesins
Fimbrial adhesins are associated with hair-like appendages extending from the bacterial surface, known as pili or fimbriae. These multimeric protein complexes are assembled through several conserved pathways [16].
- Chaperone-Usher Pathway: This pathway produces composite fibers like P pili and type 1 pili in uropathogenic Escherichia coli (UPEC). P pili are heteropolymeric structures consisting of a rigid rod composed of repeating PapA subunits and a thin tip fibrillum containing the PapG adhesin, which specifically binds to Galα(1-4)Gal moieties in glycolipids [16]. Type 1 pili feature a FimA subunit rod and the FimH adhesin at the tip, which mediates D-mannose sensitive adhesion [16] [15].
- Extracellular Nucleation-Precipitation Pathway: This system assembles thin, aggregative fibers called curli, expressed by E. coli and Salmonella enteritidis. Curli subunits are secreted as unstructured proteins that polymerize into amyloids on the cell surface [16].
- General Secretion Pathway: This pathway assembles type IV pili, found in pathogens like Neisseria gonorrhoeae, Pseudomonas aeruginosa, and Vibrio cholerae. These pili are involved in both adhesion and a unique form of motility known as twitching motility [16].
- Alternate Chaperone Pathway: This pathway assembles adhesins such as CS1 pili in enterotoxigenic E. coli, which are associated with diarrheal diseases [16].
Table 1: Major Fimbrial Adhesin Assembly Pathways and Representative Structures
| Assembly Pathway | Representative Structure | Key Organism | Associated Disease(s) | Adhesin and Receptor Specificity |
|---|---|---|---|---|
| Chaperone-Usher | P pili | Uropathogenic E. coli | Pyelonephritis | PapG / Galα(1-4)Gal glycolipids [16] |
| Chaperone-Usher | Type 1 pili | Uropathogenic E. coli | Cystitis | FimH / D-mannose residues [16] [15] |
| General Secretion | Type IV pili | Neisseria gonorrhoeae | Gonorrhea | Minor Pilin / CD46 and other receptors [16] |
| Extracellular Nucleation-Precipitation | Curli | E. coli, Salmonella enteritidis | Sepsis | CsgA (major subunit) / Host matrix proteins [16] |
| Alternate Chaperone | CS1 pili | Enterotoxigenic E. coli | Diarrhea | CooD (major subunit) / Unknown intestinal receptor [16] |
Afimbrial Adhesins
Afimbrial adhesins are directly associated with the bacterial cell surface without forming extended pilus structures. Key examples include:
- Trimeric Autotransporter Adhesins (TAAs): These proteins, such as YadA in Yersinia enterocolitica and NadA in Neisseria meningitidis, consist of a head domain, a stalk, and a membrane anchor. They often bind to host extracellular matrix proteins like collagen and fibronectin [14].
- Other Outer Membrane Proteins: Many gram-negative bacteria utilize integral outer membrane proteins as adhesins. The Neisseria Opa protein family, for instance, binds to human carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) [14].
- MSCRAMMs (Microbial Surface Components Recognizing Adhesive Matrix Molecules): Primarily found in gram-positive bacteria like Staphylococcus aureus, these surface-anchored proteins (e.g., clumping factor A) bind to fibrinogen and other plasma proteins [4].
Mechanisms of Action: From Adhesion to Signaling
The role of bacterial adhesins extends far beyond physical tethering. Their engagement with host receptors initiates a complex microscale communication that profoundly alters host cell physiology [14].
Host Receptor Engagement and Signaling Cascades
The multivalent binding of adhesin-covered bacteria leads to clustering of host receptors, triggering intracellular signaling pathways [14]. The specific pathway activated depends heavily on the identity of the engaged host receptor.
- Integrin Engagement: Afimbrial adhesins like Yersinia invasin bind with high affinity to β1 integrins, mimicking natural ligands. This clustering activates focal adhesion kinase (FAK) and phosphoinositide 3-kinase (PI3K), leading to cytoskeletal rearrangements that can promote bacterial internalization [14].
- Uroplakin Signaling: The FimH adhesin of UPEC type 1 pili binds to mannosylated uroplakins on bladder epithelial cells. This interaction triggers a phosphorylation cascade via casein kinase II, resulting in localized elevation of calcium and PIP3, activation of PI3K and FAK, and ultimately actin rearrangement that facilitates bacterial invasion [14].
- Receptor Tyrosine Kinase Activation: Some adhesins indirectly activate growth factor receptors. For example, Helicobacter pylori CagA interacts with c-Met, dysregulating epithelial cell proliferation and motility [14].
The following diagram illustrates the key signaling pathways triggered by specific bacterial adhesins upon engaging their cognate host receptors.
Role in Biofilm Initiation
Adhesins are the cornerstone of the biofilm lifecycle. The transition from planktonic to biofilm growth begins with the initial, often reversible, attachment of bacteria to a surface, a process mediated by adhesins like fimbriae and pili interacting with surface-bound host proteins or abiotic conditioned layers [3] [4]. This attachment is strengthened by the production of extracellular polymeric substances (EPS), leading to irreversible adhesion and the development of microcolonies [3] [5]. The EPS matrix, composed of exopolysaccharides, proteins, extracellular DNA (eDNA), and lipids, provides structural integrity and protects the embedded community [3] [5] [4]. As the biofilm matures, its complex 3D architecture creates gradients of nutrients and oxygen, fostering metabolic diversity and enabling sophisticated behaviors like quorum sensing [3] [17]. The final stage of the lifecycle is dispersion, where cells detach to colonize new niches, completing a cycle that underscores the remarkable adaptability of bacteria [4] [17].
Experimental Methodologies for Studying Adhesins
A robust toolkit of molecular, biochemical, and cell-based assays is essential for characterizing adhesin function, structure, and their role in pathogenesis.
Molecular and Cell-Based Assays
- Adhesion and Invasion Assays: These fundamental protocols quantify bacterial attachment to and internalization into host cells. Typically, cultured epithelial cells are infected with bacteria for a set period. For adhesion assays, extracellular bacteria are washed away, and cell-associated bacteria are quantified by lysing the host cells and plating serial dilutions. For invasion assays, gentamicin or another membrane-impermeant antibiotic is added to kill extracellular bacteria before cell lysis and plating [14]. Isogenic mutants lacking specific adhesins serve as critical negative controls.
- Receptor Identification and Binding Specificity: Glycan array screening can define the carbohydrate specificity of lectin-like adhesins like FimH [14]. Co-immunoprecipitation and cross-linking experiments, followed by mass spectrometry, can identify proteinaceous host receptors. Surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) provide quantitative data on binding affinity and kinetics.
- Signal Transduction Analysis: The activation of host signaling pathways (e.g., FAK, PI3K) upon bacterial adhesion can be detected by Western blotting using phospho-specific antibodies [14]. Immunofluorescence staining allows for the visualization of cytoskeletal rearrangements and the recruitment of signaling molecules to the site of bacterial attachment.
Table 2: Key Research Reagents and Methodologies for Adhesin Research
| Research Tool Category | Specific Examples | Function/Application in Research |
|---|---|---|
| Cell Culture Models | Immortalized epithelial cell lines (e.g., HEK-293, HeLa, T24 bladder cells) | In vitro models for adhesion/invasion assays and host signaling studies [14]. |
| Biochemical Reagents | Phospho-specific antibodies (e.g., anti-phospho-FAK, anti-phospho-PI3K), Protein A/G beads | Detection and purification of activated signaling components in host pathways [14]. |
| Genetic Tools | Isogenic bacterial mutant strains (e.g., ΔfimH, Δinvasin), Plasmid complementation systems | Essential for establishing the specific role of an adhesin by comparing mutant to wild-type phenotype [14]. |
| Analytical Techniques | Surface Plasmon Resonance (SPR), Isothermal Titration Calorimetry (ITC) | Quantitative analysis of adhesin-receptor binding affinity (KD), kinetics, and thermodynamics [14]. |
| Imaging & Visualization | Confocal Laser Scanning Microscopy (CLSM), Atomic Force Microscopy (AFM) | High-resolution imaging of biofilm 3D architecture and biophysical measurement of adhesion forces [15] [5]. |
Structural Characterization andIn VivoModels
- Structural Biology Techniques: X-ray crystallography and cryo-electron microscopy (cryo-EM) have been instrumental in determining the high-resolution structures of adhesins like FimH and RrgA, revealing their domain organization and ligand-binding pockets [16] [15]. This structural information is critical for structure-based inhibitor design.
- In Vivo Infection Models: Animal models, including murine models of urinary tract infection (UTI) and pneumonia, are indispensable for validating the role of adhesins in pathogenesis within a complex host environment. Comparison of bacterial loads and histopathology between animals infected with wild-type and adhesin-deficient strains demonstrates the contribution of the adhesin to colonization and disease [15].
The following diagram outlines a generalized experimental workflow that integrates these methodologies to characterize a novel bacterial adhesin from initial genetic identification to functional assessment in vivo.
Therapeutic Applications and Future Directions
Targeting bacterial adhesins offers a promising anti-virulence strategy, aimed at preventing colonization and subsequent infection without exerting direct lethal pressure that drives resistance.
- Anti-Adhesin Vaccines: Vaccines targeting adhesins have shown promise. The FimH adhesin is a leading vaccine candidate for preventing UTIs. Anti-FimH antibodies in animal models significantly reduce bladder colonization by UPEC [15]. The Bordetella pertussis adhesins FHA and pertactin are already components of acellular pertussis vaccines [15].
- Inhibitory Compounds: Small molecules and peptides that block adhesin-receptor binding are under active investigation. Cranberry juice proanthocyanidins, for example, have been shown to inhibit FimH-mediated adhesion, and orally administered FimH inhibitors can prevent bladder infection in mice [15].
- Matrix-Targeting Enzymes: For disrupting established biofilms, enzymes such as glycoside hydrolases that degrade the EPS matrix are being explored to induce biofilm dispersal and improve antibiotic efficacy [4].
The future of adhesin research lies in overcoming challenges such as the redundancy of adhesin systems in many pathogens and their antigenic variation. Advanced techniques like single-cell analysis within biofilms, coupled with structure-guided design of multivalent inhibitors, will be crucial for developing the next generation of anti-adhesive therapeutics.
The second messenger bis-(3',5')-cyclic di-guanosine monophosphate (c-di-GMP) represents a central signaling system that governs the fundamental lifestyle transition between motile planktonic cells and sessile biofilm communities in bacteria. This switch is critical for bacterial environmental adaptation, persistence, and pathogenicity. Through a complex network of synthesizing enzymes (diguanylate cyclases, DGCs), degrading enzymes (phosphodiesterases, PDEs), and effector molecules, c-di-GMP integrates environmental cues to precisely control cellular processes including motility, surface adhesion, exopolysaccharide production, and virulence. This technical review examines the molecular architecture of the c-di-GMP signaling network, its regulation across diverse bacterial species, and its integral role in mechanisms of bacterial adhesion and biofilm initiation. We provide structured quantitative data, experimental methodologies, and visualization of key pathways to support research efforts aimed at targeting this system for therapeutic intervention.
Cyclic di-GMP functions as a ubiquitous intracellular secondary messenger, representing environmental or cellular cues and connecting sensory input with regulatory output in bacterial cells [18]. The c-di-GMP signaling network exhibits remarkable physiological versatility and mechanistic diversity, controlling a myriad of cellular processes including motility, biofilm formation, virulence, and cell cycle progression [19] [18]. The fundamental principle governing c-di-GMP-mediated lifestyle transitions is straightforward: elevated cellular c-di-GMP concentrations promote the transition from a motile, planktonic existence to a sessile, biofilm-forming lifestyle, while low c-di-GMP levels favor motility and dispersal [20]. In the model organism Pseudomonas aeruginosa, for instance, planktonic cells typically harbor less than 30 pmol/mg c-di-GMP, whereas biofilm cells can contain close to 100 pmol/mg [20].
The cellular pool of c-di-GMP is dynamically regulated by the opposing activities of two classes of enzymes: diguanylate cyclases (DGCs) that synthesize c-di-GMP from two GTP molecules, and phosphodiesterases (PDEs) that degrade c-di-GMP. DGCs contain characteristic GGDEF domains, while PDEs feature either EAL or HD-GYP domains [20]. These enzymes often possess complex sensory domain architectures that allow multisignal integration, enabling bacteria to modulate intracellular c-di-GMP levels in response to diverse environmental stimuli [18]. The downstream effects of c-di-GMP are mediated through various effector molecules including proteins and riboswitches that bind c-di-GMP and subsequently interact with target components to generate phenotypic outputs [21].
Molecular Architecture of the c-di-GMP Network
Core Enzymes and Effectors
The c-di-GMP regulatory network comprises three fundamental components: enzymes responsible for synthesis, enzymes responsible for degradation, and effector molecules that interpret concentration changes. The table below summarizes the key proteins and their functions in the c-di-GMP signaling network.
Table 1: Core Components of the c-di-GMP Signaling Network
| Component Type | Key Domain/Motif | Function | Example Proteins |
|---|---|---|---|
| Diguanylate Cyclase (DGC) | GGDEF | Synthesizes c-di-GMP from 2 GTP molecules | WspR, SadC, YfiN, PdcA, VpvC |
| Phosphodiesterase (PDE) | EAL or HD-GYP | Degrades c-di-GMP to pGpG/2 GMP | DipA, MorA |
| Effector/Receptor | Varied (e.g., PilZ) | Binds c-di-GMP to enact downstream responses | YcgR, VpsT, VpsR, FimX |
Regulatory Mechanisms and Sensory Integration
The activity of c-di-GMP metabolizing enzymes is precisely controlled through multiple regulatory mechanisms. Many DGCs and PDEs contain sensory domains that perceive specific environmental signals, translating them into changes in enzymatic activity and cellular c-di-GMP levels [18]. For example, the inner membrane DGC YfiN in E. coli is directly repressed by the periplasmic protein YfiR, which is inactivated by redox stress [19]. Furthermore, auto-inhibitory mechanisms provide critical control; the GGDEF domains of many DGCs contain inhibitory sites (I-sites) that bind c-di-GMP to provide feedback inhibition [19]. YfiN notably lacks these autoinhibitory sites, which allows for "run-away" c-di-GMP synthesis under certain conditions, leading to cellular GTP depletion and growth arrest when bacteria are cultured on gluconeogenic carbon sources [19].
Beyond individual enzyme regulation, operon-based signaling cascades create sophisticated control systems. In Burkholderia thailandensis, the pdcABC operon encodes a regulatory cascade where the DGC PdcA's activity is inhibited by phosphorylated PdcC (a response regulator), and the phosphatase PdcB dephosphorylates PdcC to derepress PdcA activity [22]. Homologous operons are widespread among betaproteobacteria and gammaproteobacteria, suggesting this represents a general mechanism for coordinating bacterial behavior and virulence [22].
Signaling Pathways and System Visualizations
The following diagrams illustrate core regulatory principles and specific c-di-GMP signaling pathways that govern the motile-to-sessile transition.
Core Regulatory Principle
YfiN-Mediated Growth Arrest Pathway
PdcABC Operon Cascade
Surface Sensing Mechanisms in P. aeruginosa
Quantitative Data and Phenotypic Outcomes
The intracellular concentration of c-di-GMP directly controls specific phenotypic outputs. The table below summarizes key quantitative relationships between c-di-GMP levels and bacterial behaviors.
Table 2: c-di-GMP-Mediated Phenotypic Transitions and Associated Mechanisms
| c-di-GMP Level | Motility Status | Biofilm Formation | Molecular Mechanisms | Pathogenic Consequences |
|---|---|---|---|---|
| Low | Motile (flagellar, twitching) | Repressed | Expression of flagellar and pilus genes; YcgR not bound to flagellar motor | Dissemination, acute virulence |
| High | Non-motile | Activated | YcgR binds flagellar motor switch complex; Transcription of matrix genes (Pel, Psl, VPS) | Chronic persistence, antibiotic tolerance |
Experimental measurements in P. aeruginosa indicate that the transition from planktonic to biofilm growth coincides with an increase in cellular c-di-GMP from <30 pmol/mg to approximately 100 pmol/mg [20]. Elevated c-di-GMP promotes surface aggregation by affecting the flagellar motor's torque-speed curve across all load conditions, effectively shifting the curve downward and promoting bacterial aggregation on surfaces [23].
Experimental Protocols for c-di-GMP Research
Protocol: Measuring c-di-GMP in Biofilm and Planktonic Cells
This protocol outlines the methodology for quantifying intracellular c-di-GMP levels, adapted from established procedures referenced in the search results [20].
Cell Cultivation and Harvesting: Grow bacterial strains under investigation in appropriate media under both planktonic and biofilm conditions. For biofilm cultivation, the experimental evolution model involves serially passaging biofilms on bead substrata (e.g., glass, PVC, stainless steel) in 24-well cell culture plates for 48 hours at 30°C with shaking at 60 rpm [24].
Sample Normalization: Harvest cells and normalize samples based on cell mass (mg wet weight) or protein content to enable comparative quantification.
Nucleotide Extraction: Perform nucleotide extraction using chilled methanol, acetonitrile, or formic acid to ensure complete inactivation of c-di-GMP metabolizing enzymes and efficient extraction of intracellular c-di-GMP.
Quantitative Analysis: Analyze c-di-GMP content using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Employ stable isotope-labeled internal standards (e.g., 13C15N-c-di-GMP) for precise quantification.
Data Interpretation: Express results as pmol c-di-GMP per mg cellular protein or wet weight. Compare levels between planktonic and biofilm populations to assess c-di-GMP flux during lifestyle transitions.
Protocol: Experimental Evolution of Biofilm-Adapted Lineages
This protocol describes an experimental evolution approach to study adaptation to biofilm lifestyle, as implemented in P. aeruginosa [24].
Setup: Inoculate 24-well plates containing 5 mm substrate beads (PVC, glass, or stainless steel) with 1 ml bacterial culture in lysogeny broth. Include control wells without beads for planktonic propagation.
Serial Passage: Incubate plates shaking at 60 rpm for 48 hours at 30°C. For each passage:
- Transfer one colonized bead to a fresh well containing three sterile beads of a different color.
- Archive one bead in 20% glycerol for subsequent phenotypic and genotypic analysis.
- For planktonic controls, perform 1:100 dilution into fresh media.
Phenotypic Monitoring: At designated timepoints (e.g., passages 10, 20, 30), assess:
- Biofilm productivity via colony-forming unit (CFU) counts per unit area.
- Biomass production using crystal violet staining.
- Morphotypic diversification through visual inspection of colony morphology.
Genomic Analysis: Sequence genomes of evolved lineages to identify mutations associated with biofilm hyperproduction, particularly in c-di-GMP signaling genes (dipA, yfiBNR, morA).
Protocol: Assessing c-di-GMP Pathway Regulation
This protocol examines transcriptional regulation of c-di-GMP metabolizing enzymes, based on studies of the vpvABC operon in V. cholerae [21].
Reporter Construction: Clone the promoter region of the target operon (e.g., vpvABC) upstream of a luminescence reporter gene (e.g., luxCDABE) in an appropriate plasmid vector.
Strain Generation: Introduce the reporter construct into wild-type and mutant strains (e.g., ΔvpsT, ΔvpsR, ΔrpoS) to dissect regulatory contributions.
Conditional Assays: Measure promoter activity under varying conditions:
- Induce c-di-GMP production using inducible DGC expression systems with varying IPTG concentrations.
- Compare exponential versus stationary growth phases.
- Assess spatial expression patterns in mature biofilms using reporter imaging.
Data Analysis: Quantify luminescence as a measure of promoter activity. Normalize readings to cell density. Perform statistical analyses to determine significant regulatory effects.
The Scientist's Toolkit: Research Reagent Solutions
The table below details essential research tools and reagents for investigating c-di-GMP signaling pathways, derived from methodologies cited in the search results.
Table 3: Essential Research Reagents for c-di-GMP Studies
| Reagent/Tool | Function/Application | Example Use Case |
|---|---|---|
| Inducible DGC Systems (e.g., Ptac-VCA0956) | Controlled increase of intracellular c-di-GMP | Dose-response studies of c-di-GMP effects on promoter activity [21] |
| Luminescence Reporters (e.g., luxCDABE) | Quantifying promoter activity of c-di-GMP-related genes | Dissecting transcriptional regulation of operons like vpvABC [21] |
| Substrate Beads (glass, PVC, stainless steel) | Providing surfaces for biofilm growth | Experimental evolution studies of biofilm adaptation [24] |
| c-di-GMP Immunoassays | Quantifying intracellular c-di-GMP levels | Comparing c-di-GMP in planktonic vs. biofilm cells [20] |
| Mutant Libraries (Δdgc, Δpde strains) | Determining contributions of specific enzymes | Genetic dissection of c-di-GMP network functionality [22] |
Discussion and Research Implications
The c-di-GMP signaling system represents a master regulator of bacterial lifestyle decisions, with far-reaching implications for understanding microbial ecology and pathogenesis. Recent research has revealed unexpected complexities in this regulatory network, including non-uniform increases in c-di-GMP during biofilm development [20] and the emergence of parallel regulatory circuits that integrate c-di-GMP sensing with growth phase signals via alternative sigma factors like RpoS [21]. Furthermore, studies have uncovered novel roles for c-di-GMP in processes such as antibiotic tolerance through growth arrest mediated by GTP depletion [19].
From a therapeutic perspective, targeting c-di-GMP signaling offers promising avenues for combating persistent biofilm-associated infections. The evolutionary selection for mutations in c-di-GMP pathways (dipA, yfiBNR, morA) during biofilm adaptation [24] underscores the central role of this system in bacterial persistence. However, the complexity and redundancy of c-di-GMP networks present significant challenges for therapeutic intervention, necessitating continued research into the specific pathways most critical for virulence in pathogenic species.
Future research directions should focus on elucidating the structural basis of effector recognition, developing specific inhibitors of key DGCs, and understanding how local signaling pools are maintained within cells to allow parallel processing of information. As our knowledge of this sophisticated signaling network deepens, so too will our ability to manipulate bacterial behavior for clinical and biotechnological applications.
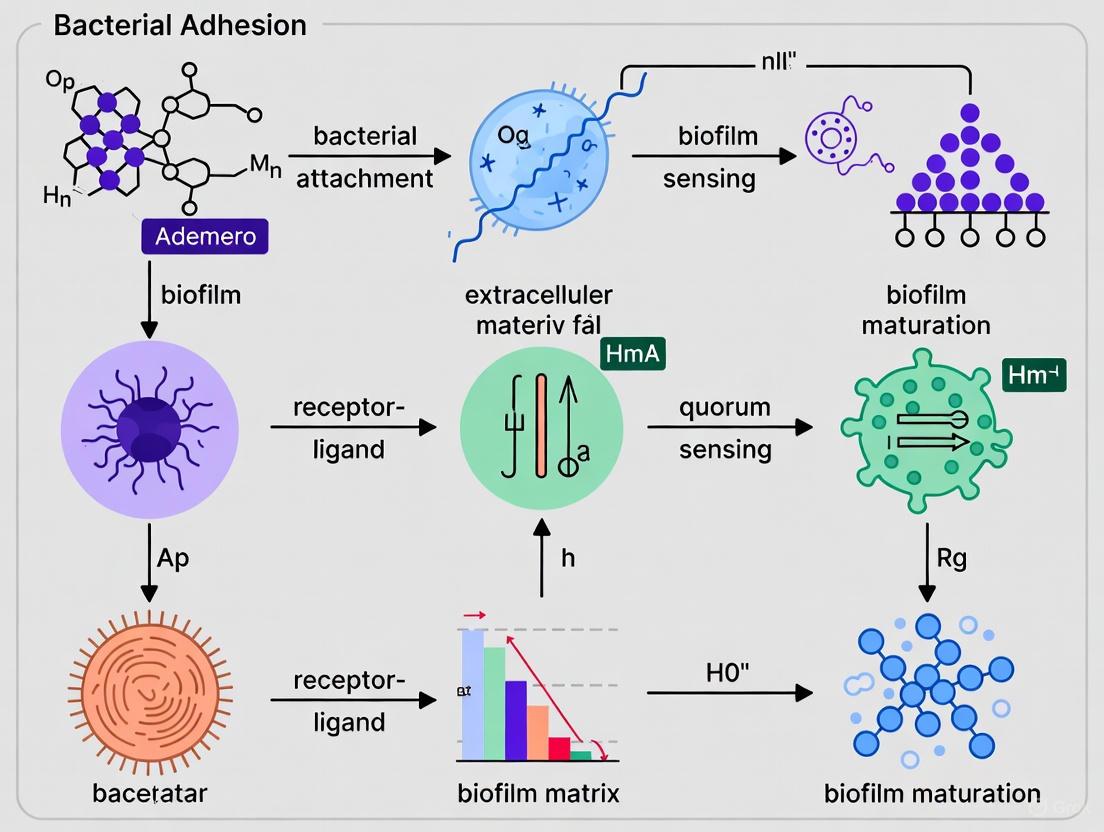
The extracellular matrix of bacterial biofilms, a complex amalgamation of extracellular polymeric substances (EPS), extracellular DNA (eDNA), and proteins, serves as the foundational scaffold that determines the structural integrity and functional dynamics of microbial communities. This matrix is not merely a passive barrier but an active component that facilitates bacterial adhesion, confers formidable resistance to antimicrobial agents, and enables adaptive responses to environmental stresses. Within the context of bacterial adhesion and biofilm initiation research, understanding the composition and interplay of these matrix components is paramount for developing novel anti-biofilm strategies. This whitepaper provides an in-depth technical analysis of the matrixome, detailing the quantitative composition, functional significance, and state-of-the-art methodologies for investigating its core constituents, with particular emphasis on their collective role in the initial stages of biofilm establishment.
Biofilms represent the predominant mode of bacterial growth in nature and clinical settings, characterized by surface-associated microbial communities encased in a self-produced matrix, often referred to as the "matrixome" [25]. This matrix is fundamentally composed of extracellular polymeric substances (EPS), which constitute 50% to 90% of a biofilm's total organic matter [26]. The EPS establishes the functional and structural integrity of biofilms and is considered the fundamental component that determines their physicochemical properties [26]. The matrixome is primarily composed of a sophisticated interplay of exopolysaccharides, proteins, lipids, and extracellular nucleic acids, which together create a protective, functional microenvironment for the embedded cells [3] [25].
The initial attachment of bacteria to surfaces marks a critical juncture in biofilm formation, triggering a shift from planktonic to sessile lifestyles that is energetically expensive but confers significant survival advantages [3] [17]. This shift is regulated by intracellular signaling pathways, including the accumulation of bis-(3ʹ-5ʹ)-cyclic dimeric guanosine monophosphate (c-di-GMP), which downregulates flagella-mediated motility and upregulates surface adhesins, facilitating firm attachment and subsequent matrix production [17]. The resulting matrix is far from a static scaffold; it is a dynamic, biologically active compartment that coordinates social behaviors, facilitates genetic exchange, and provides multimodal protection against environmental insults, host immune responses, and antimicrobial agents [3] [17]. The focus of this review is to dissect the core structural and functional components of this matrix—EPS, eDNA, and proteins—within the specific context of their roles in bacterial adhesion and biofilm initiation.
Core Components of the Biofilm Matrix
Extracellular Polymeric Substances (EPS): The Structural Backbone
EPS are natural polymers of high molecular weight secreted by microorganisms into their environment [26]. They are highly hydrated polymers that are mainly composed of polysaccharides (exopolysaccharides) and proteins, but also include other macromolecules such as DNA, lipids, and humic substances [26] [27].
Table 1: Major Exopolysaccharides in Bacterial Biofilms
| Exopolysaccharide | Producing Microorganism(s) | Key Characteristics |
|---|---|---|
| Alginate | Pseudomonas spp., Azotobacter vinelandii | Acetylated polymer of D-mannuronic and L-guluronic acids; contributes to mucoid phenotype and resistance [26]. |
| Cellulose | Acetobacter xylinum | Unbranched β-1,4-glucan; provides structural strength and resilience to mechanical stress [26]. |
| PNAG/PGA | Staphylococcus epidermidis, Escherichia coli | Poly-β-1,6-N-acetyl-D-glucosamine; crucial for cell-to-cell adhesion and biofilm accumulation [26]. |
| Xanthan | Xanthomonas campestris | Heteropolysaccharide with a cellulose backbone; excellent viscositying and stabilizing properties [26]. |
| Succinoglycan | Sinorhizobium meliloti, Alcaligenes faecalis | Acidic, octasaccharide repeating unit; essential for symbiosis and infection thread formation [26] [27]. |
The production of EPS is triggered by environmental signals and, despite being energetically costly, provides microorganisms with critical advantages including adhesion, cohesion, and protection [27]. The polysaccharides are particularly diverse, varying immensely in composition and structure, which dictates their specific functional properties [27]. Beyond polysaccharides, the structural proteins, enzymes, and extracellular DNA (eDNA) within EPS are now recognized as equally vital for the matrix's architecture and function [27].
Extracellular DNA (eDNA): A Multifunctional Matrix Polymer
eDNA is an ubiquitous component of the biofilm matrix, released through various mechanisms including autolysis, membrane vesicle-mediated release, phage-mediated release, active secretion, and Type VI secretion system (T6SS)-mediated release [25]. Quorum sensing (QS) plays a vital role in regulating eDNA release in a controlled manner by coordinating gene expression in response to cell density [25].
Table 2: Functional Roles of eDNA in Biofilm Development
| Function | Mechanism of Action | Representative Organisms |
|---|---|---|
| Initial Adhesion | Introduces favorable acid-base interactions, increases cell surface hydrophobicity [28]. | Staphylococcus epidermidis, Streptococcus mutans [28]. |
| Structural Cohesion | Acts as an adhesive, interacting with other EPS components to strengthen the biofilm architecture [28] [25]. | Bacillus subtilis, Pseudomonas aeruginosa [29] [25]. |
| Cation Sequestration | Negatively charged backbone attracts and sequesters cations (e.g., Ca²⁺, Mg²⁺), indirectly increasing antimicrobial resistance [29]. | P. aeruginosa, B. subtilis [29]. |
| Metabolic Reservoir | Serves as a source of phosphate and nucleotides, reclaimed by secreted nucleases later in biofilm development [29]. | B. subtilis [29]. |
Once released into the extracellular matrix, eDNA interacts with EPS components, enhancing matrix stability, structural cohesion, and integrity [25]. However, its role is not static. Recent research on Bacillus subtilis has revealed that eDNA is temporarily invested in the biofilm matrix before being globally degraded in a spatiotemporally coordinated pulse by secreted nucleases like YhcR, NucA, and NucB, highlighting a novel role for eDNA as a dynamic metabolic reservoir for phosphate [29].
Proteins: The Functional Workhorses
Proteins within the biofilm matrix include structural proteins, enzymes, and specialized adhesins, each playing distinct and critical roles.
Structural proteins contribute to the scaffold's integrity, while exoenzymes are secreted to break down large molecules in the environment into smaller, absorbable nutrients [26]. These include alkaline phosphatases, chitinases, β-d-glucosidases, and proteases, which can influence chemical signaling and biogeochemical cycling [26].
Surface adhesins, particularly the Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMMs) in Gram-positive bacteria, are critical for the initial attachment to host tissues and biomaterials [30]. These adhesins, such as fibronectin-binding proteins (FnBPs) and Serine-Aspartate Repeat proteins (Sdr), engage in sophisticated, force-dependent interactions with their ligands.
Table 3: Key Protein Adhesins in Bacterial Adhesion
| Adhesin | Bacterial Species | Ligand(s) | Binding Characteristics |
|---|---|---|---|
| Fibronectin-Binding Proteins (FnBPs) | Staphylococcus aureus | Fibronectin (Fn), fibrinogen (Fg) | Force-activated; forms cluster bonds of up to 80 proteins; unbinding force increases with loading rate [30]. |
| Serine-Aspartate Repeat Proteins (Sdr) | S. epidermidis, S. aureus | Fibrinogen (Fg) | "Dock, lock, and latch" mechanism; ultrastrong binding forces [30]. |
| Clumping Factor (Clf) | S. aureus | Fibrinogen (Fg) | Promotes cell clumping and adhesion to blood clots and implanted devices [30]. |
| SasG | S. aureus | Self (homophilic) | Zn²⁺-dependent; resists forces up to ~500 pN; unfolding under stress exposes cryptic domains [30]. |
A remarkable feature of these adhesins is their mechanosensitivity. Atomic force microscopy (AFM) studies have revealed that staphylococcal FnBPs, for instance, form catch bonds where the interaction strengthens under tensile load, a property ideal for resisting shear forces in the vasculature or on medical devices [30] [31]. The binding strength of a single FnBP-Fn bond is approximately 60 pN, but cluster bonds can be much stronger, and amino acid polymorphisms in clinical isolates can significantly alter bond lifetime and strength, impacting pathogenicity [30].
Experimental Protocols for Matrix Analysis
Quantifying eDNA in Adhesion and Aggregation
Protocol: The Role of eDNA in Initial Bacterial Adhesion (Based on [28])
Objective: To investigate the contribution of extracellular DNA (eDNA) to the initial adhesion and surface aggregation of Gram-positive bacteria.
Materials:
- Bacterial Strains: e.g., Staphylococcus epidermidis 1457 and its isogenic ΔatlE mutant (deficient in autolysin E and eDNA release).
- Substrata: Microscope slides coated with dimethyldichlorosilane (DDS) to create a hydrophobic surface, and clean glass for a hydrophilic surface.
- Enzyme: DNase I (with heat-inactivated DNase I as a control).
- Equipment: Parallel-plate flow chamber, microscope with camera for real-time monitoring, image analysis software.
Methodology:
- Culture Preparation: Grow bacterial cultures for 16 hours, wash in phosphate-buffered saline (PBS), and sonicate gently on ice to remove aggregates. Resuspend in PBS to a density of 3 × 10⁸ ml⁻¹.
- eDNA Removal: Treat a portion of the bacterial suspension with DNase I (in the presence of 10 mM MgCl₂) for 45 minutes at 37°C, followed by washing with PBS.
- Adhesion Assay: Place the substrata in the parallel-plate flow chamber. Perfuse the bacterial suspension through the chamber at a defined shear rate (e.g., 16 s⁻¹).
- Data Collection: Monitor and record bacterial adhesion for 60 minutes. Analyze images to calculate:
- The initial deposition rate (j₀), representing the number of cells attaching per unit area per second.
- The total number of bacteria adhered (N₆₀ min) after 60 minutes.
- The percentage of adhering bacteria involved in large aggregates (>5 cells).
- Surface Thermodynamic Analysis: Measure contact angles of bacterial lawns with various liquids before and after DNase I treatment. Use these to calculate the free energy of adhesion (ΔG) and aggregation, explaining the interaction from a physicochemical perspective.
Expected Outcome: Treatment with DNase I, or the use of the ΔatlE mutant, is expected to result in a statistically significant reduction in the initial deposition rate, total adhered cells, and the size of surface aggregates, particularly on hydrophilic surfaces, demonstrating the critical role of eDNA in early adhesion events [28].
Single-Molecule Force Spectroscopy of Bacterial Adhesins
Protocol: Measuring Binding Strength of Staphylococcal Adhesins (Based on [30])
Objective: To probe the single-molecule binding strength and kinetics of specific adhesin-ligand interactions under mechanical force.
Materials:
- AFM Setup: Atomic Force Microscope with cantilevers of known spring constant.
- Functionalized Probes: AFM tips chemically modified with the ligand of interest (e.g., fibronectin, fibrinogen).
- Sample Preparation: Living bacterial cells expressing the adhesin of interest, or a purified form of the adhesin immobilized on a solid substrate.
Methodology:
- Probe Functionalization: Covalently attach the purified ligand to the AFM tip using chemistry such as PEG-linkers.
- Force-Distance (FD) Curve Acquisition: Bring the functionalized tip into contact with the bacterial cell surface and then retract it at a constant velocity (pulling speed).
- Data Analysis:
- Adhesion Force: Identify the rupture event in the retraction curve and measure the force (in piconewtons, pN) at which the bond breaks.
- Binding Probability: Calculate the percentage of approach-retract cycles that result in a specific adhesion event.
- Dynamic Force Spectroscopy: Repeat the experiment at multiple pulling speeds (e.g., from 0.1 to 10,000 nm/s). Plot the most probable adhesion force versus the logarithm of the loading rate.
- Kinetic Parameter Extraction: Fit the dynamic force spectroscopy data with theoretical models (e.g., Bell-Evans model) to extract the zero-force dissociation rate (kᵒff) and the energy barrier width (xᵦ).
Expected Outcome: This protocol can reveal the mechanostability of adhesins, such as the ~125 pN homophilic bonds of FnBPA or the ultrastrong ~500 pN bonds of SasG [30]. It can also identify catch-bond behavior, where bond lifetime increases with applied force, providing deep insight into how pathogens sustain adhesion under physiological shear stress.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 4: Key Reagents for Biofilm Matrix Research
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| DNase I | Enzymatically degrades extracellular DNA (eDNA). | Used to dissect the structural and adhesive roles of eDNA in biofilms [28] [29]. |
| Proteinase K | Broad-spectrum serine protease that digests proteins. | Employed to determine the contribution of proteinaceous components to matrix integrity and adhesion [3]. |
| Atomic Force Microscope (AFM) | Measures piconewton-scale forces between single molecules or on living cells. | Essential for quantifying the binding strength of specific adhesin-ligand interactions (Single Molecule Force Spectroscopy) [30]. |
| Parallel-Plate Flow Chamber | Generates controlled, quantifiable fluid shear over a surface. | Used to study real-time bacterial adhesion and biofilm formation under hydrodynamic conditions mimicking in vivo environments [28]. |
| TOTO-1 / SYTO dyes | Fluorescent nucleic acid stains that are impermeant to intact cell membranes. | Allows for specific visualization and quantification of eDNA in biofilms via fluorescence microscopy or plate readers [29]. |
| Congo Red | A diazo dye that binds to polysaccharides and amyloid fibers. | Used to stain and assess the production of exopolysaccharides in biofilm matrices, e.g., in colony biofilms [29]. |
Visualizing Matrix Dynamics
The following diagrams, generated using Graphviz DOT language, illustrate key concepts and experimental workflows in biofilm matrix research.
Diagram 1: Biofilm Lifecycle and Matrix Composition
This diagram outlines the key stages of biofilm development and the associated changes in matrix composition, highlighting the dynamic role of eDNA.
Diagram 2: eDNA Dynamics and Experimental Workflow
This diagram illustrates the dual role of eDNA as a structural component and metabolic reservoir, alongside an experimental protocol for its study.
The biofilm matrixome, with EPS, eDNA, and proteins as its core components, is a masterfully engineered biological construct that is fundamental to the success of bacterial biofilms. Its composition is not static but is dynamically regulated in response to environmental cues and developmental stages. The initial adhesion of bacteria, a critical first step in biofilm formation, is mediated by a sophisticated synergy of specific protein adhesins capable of withstanding shear forces and a sticky web of eDNA and exopolysaccharides that consolidates attachment.
The detailed mechanistic insights provided by advanced techniques like single-molecule force spectroscopy and the discovery of metabolic reclamation pathways for eDNA underscore the complexity of the matrix. For researchers and drug development professionals, this evolving understanding opens new avenues for therapeutic intervention. Targeting matrix components—for instance, by disrupting key adhesins with anti-catch bond compounds, using DNases to dissolve the structural scaffold, or inhibiting the nucleases responsible for nutrient recycling—represents a promising strategy to prevent biofilm formation or sensitize established biofilms to conventional antibiotics. Future research will undoubtedly continue to unravel the intricate signaling networks that control matrix genesis, paving the way for next-generation anti-biofilm agents.
The paradigm of bacteria as solitary, free-floating organisms has been fundamentally overturned by the discovery that they predominantly exist in complex, surface-associated communities known as biofilms. Within these structured microbial societies, bacteria coordinate group behaviors through a sophisticated chemical communication process termed quorum sensing (QS). This cell-cell signaling enables bacterial populations to synchronously regulate gene expression in a cell-density-dependent manner, facilitating collective behaviors that would be futile if undertaken by individual cells [32] [33]. The formation of microcolonies—the critical transitional stage from single cells to mature biofilms—represents a pivotal point where quorum sensing mechanisms initiate profound changes in bacterial physiology and community organization [34].
The clinical implications of biofilm-associated infections are staggering. Approximately 65% of all bacterial infections and 80% of all chronic infections involve biofilm formation, presenting formidable challenges in healthcare settings due to their inherent resistance to antimicrobials and host immune responses [32]. Device-associated infections are particularly problematic, with biofilm formation occurring on implants, catheters, and other medical devices [32]. Understanding the intricate interplay between quorum sensing and microcolony formation is therefore not merely an academic pursuit but an urgent medical necessity for developing novel therapeutic strategies against persistent bacterial infections.
Molecular Mechanisms of Bacterial Communication
Fundamental Quorum Sensing Circuitry
Quorum sensing systems across bacterial species share a fundamental paradigm: the production, release, and population-wide detection of extracellular signaling molecules called autoinducers [33] [34]. This process enables bacteria to collectively regulate gene expression when a critical threshold signal concentration—corresponding to a sufficient population density or "quorum"—is reached [33].
The core molecular components typically include:
- Autoinducer synthases that produce signaling molecules
- Receptor proteins that detect these molecules
- Transcription factors that regulate target gene expression [33]
In Gram-negative bacteria, the most extensively studied QS systems utilize acyl-homoserine lactones (AHLs) as signaling molecules. These amphipathic molecules consist of a homoserine lactone ring attached to a fatty acid side chain, allowing them to freely diffuse through cell membranes and establish concentration gradients in the local environment [33]. The AHL-driven regulatory circuits are primarily governed by LuxI-type proteins (responsible for AHL synthesis) and LuxR-type proteins (involved in AHL-response regulation) that act as transcription factors regulating QS-related genes [34].
Pseudomonas aeruginosa: A Model for QS Studies
Pseudomonas aeruginosa serves as a paradigm for understanding quorum sensing mechanisms due to its well-characterized, multilayered signaling networks. This opportunistic human pathogen employs two primary interlinked AHL systems:
- LasI/LasR System: LasI produces N-3-oxododecanoyl-homoserine lactone, which binds to the transcriptional regulator LasR once a critical threshold is reached.
- RhlI/RhlR System: RhlI produces N-butanoyl-homoserine lactone, which activates RhlR [33].
These systems function hierarchically, with the Las system regulating the Rhl system, creating a sophisticated regulatory cascade that coordinates the expression of hundreds of genes [33]. A third system utilizing Pseudomonas quinolone signal (PQS), 2-heptyl-3-hydroxy-4-quinolone, further integrates into this regulatory network, adding another layer of control [33].
Quorum Sensing Thresholds and Sensitivity
Bacterial cells exhibit remarkable sensitivity to autoinducer concentrations, with response thresholds occurring at extremely low levels. Observed QS thresholds can be as low as 2.5-5 nM, corresponding to just a few signaling molecules per cell sufficient to trigger induction [34]. This exquisite sensitivity ensures rapid transition to communal behavior once a critical population density is achieved within microcolonies.
Quantitative Dynamics of Microcolony Induction
Spatial Heterogeneity in Induction Patterns
The transition from individual cells to coordinated microcolonies is not uniform across bacterial populations. Spatial distribution patterns significantly influence the timing and synchronization of QS activation. Computational models and experimental observations reveal two distinct induction scenarios:
- Homogeneous distributions lead to delayed but more concerted induction of the entire cell colony, exhibiting behavior close to mean-field dynamics where the population activates nearly simultaneously [34].
- Spatially heterogeneous distributions with higher local cell concentrations in clusters result in earlier but more localized induction events, where isolated "hot spots" activate well before the broader population [34].
This spatial heterogeneity creates a complex induction landscape where the local cell density variations, rather than the global population average, determine the timing and pattern of QS activation [34]. The emergence of higher-density clusters in an otherwise sparse colony can trigger premature induction events, fundamentally altering the developmental trajectory of biofilm formation.
Key Parameters in Microcolony Signaling
The dynamics of quorum sensing activation in developing microcolonies depend on several critical physical and biological parameters:
Table 1: Quantitative Parameters in Quorum Sensing and Microcolony Formation
| Parameter | Impact on QS Activation | Experimental Measurements |
|---|---|---|
| Cell Division Rate (γ) | Faster division reduces time to reach critical local density | Typically measured in generations per hour [34] |
| Cell Spreading Distance | Greater displacement delays local QS activation | Daughter cell displacement: fixed distance (d~new~) or Gaussian distribution (σ~str~) [34] |
| Autoinducer Signal Range | Longer range promotes earlier, more synchronized induction | Determined by diffusion coefficient and degradation rate [34] |
| Spatial Distribution | Clustered distributions cause localized early induction | Quantified by radial distribution functions and local density metrics [34] |
| Threshold Concentration | Lower thresholds enable earlier QS activation | As low as 2.5-5 nM AHL concentration [34] |
Burst Induction Statistics in Microcolonies
The transition to QS-controlled behavior in spatially heterogeneous colonies occurs through distinct "burst" events rather than synchronous population-wide activation. Computational models reveal that:
- Induction families emerge as groups of cells that become induced in rapid succession following initial activation of a founder cell [34].
- Burst sizes vary significantly based on spatial distribution, with highly clustered arrangements producing smaller, more frequent induction events compared to homogeneous distributions [34].
- The mean autoinducer concentration exhibits steep increases corresponding to these burst events, creating a positive feedback loop that accelerates the induction of remaining cells [34].
This burst-like induction pattern has profound implications for biofilm development, as heterogeneously induced microcolonies may exhibit different phenotypic characteristics and architectural features compared to synchronously induced populations.
Experimental Methodologies and Technical Approaches
Patterned Bacterial Systems for QS Studies
Advanced microfabrication and printing technologies have enabled precise spatial control of bacterial deposition for studying quorum sensing dynamics. Inkjet printing of bacterial cells provides a powerful platform for creating defined microcolony patterns with controlled initial conditions [35].
Table 2: Research Reagent Solutions for QS Experimentation
| Reagent/Technology | Function in QS Research | Specific Applications |
|---|---|---|
| Acyl-Homoserine Lactones (AHLs) | Native signaling molecules in Gram-negative bacteria | Direct activation of QS circuits; concentration gradient studies [33] [34] |
| Fluorescent Reporter Plasmids | Visualizing QS activation in real time | GFP/RFP constructs under control of QS-regulated promoters [35] |
| Inkjet Bioprinting Systems | Precise spatial patterning of bacterial colonies | Controlled microcolony formation with defined cell numbers and spacing [35] |
| LuxI/LuxR Pathway Components | Core molecular machinery for AHL signaling | Genetic manipulation of QS circuits; heterologous expression systems [33] [34] |
| Quorum Sensing Inhibitors (QSIs) | Blocking intercellular communication | Anti-virulence therapeutics; mechanistic studies [32] [36] |
Experimental Protocol: Inkjet Printing of Bacterial Patterns for QS Studies [35]
Bacterial Strain Preparation: Transform E. coli MG1655 with fluorescent reporter plasmids (GFP/RFP) under control of QS-responsive promoters. Culture overnight with appropriate antibiotic selection.
Printer Configuration: Utilize a commercial materials printer with 254 dpi (100 μm drop spacing) or 508 dpi (50 μm drop spacing) settings. Optimize droplet size to approximately 37 μm diameter.
Bioink Formulation: Suspend bacterial cells in solution containing 1% polyethylene glycol (PEG) to adjust viscosity and prevent nozzle clogging.
Pattern Design and Printing: Program specific geometric arrangements (circular arrays, grid patterns) with controlled inter-colony spacing (50-500 μm). Sequentially print different strains for co-culture experiments.
Incubation and Monitoring: Maintain printed patterns on agar surfaces with appropriate nutrients. Monitor microcolony growth and QS activation via fluorescence microscopy over 24-72 hours.
Signal Diffusion Analysis: Measure activation distances between sender and receiver colonies to quantify effective signaling range.
Computational Modeling Approaches
Mathematical modeling provides invaluable insights into QS dynamics that complement experimental approaches. Computational frameworks typically incorporate:
- Cell growth and division with appropriate generation times
- Spatial distribution of daughter cells following division (fixed displacement or Gaussian distribution)
- Autoinducer diffusion through the environment with characteristic diffusion coefficients
- Receptor binding kinetics and transcriptional activation thresholds [34]
These models enable researchers to systematically vary parameters that are difficult to control experimentally, revealing fundamental principles governing heterogenous induction patterns in developing microcolonies.
Therapeutic Implications and Intervention Strategies
Quorum Quenching Approaches
The critical role of quorum sensing in biofilm formation and virulence has inspired therapeutic strategies aimed at disrupting bacterial communication rather than killing pathogens outright. This "quorum quenching" approach includes:
- Enzymatic degradation of autoinducer signals (e.g., lactonases that inactivate AHLs)
- Small molecule inhibitors that competitively block signal receptor binding
- Signal analogs that interfere with native QS circuits [32]
These anti-virulence strategies offer potential advantages over conventional antibiotics by reducing selective pressure for resistance development, as they aim to disarm pathogens rather than eliminate them [32] [36].
Clinical Applications and Challenges
Biofilm-associated infections involving sophisticated QS systems present particularly formidable challenges in clinical settings. Pseudomonas aeruginosa lung infections in cystic fibrosis patients exemplify the critical role of QS in chronic infections, where biofilm formation significantly complicates treatment [32] [33]. Similarly, chronic wounds and indwelling medical device infections frequently involve QS-mediated biofilm communities that resist both antimicrobial therapy and host immune responses [32] [36].
The ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) represent particularly concerning biofilm-forming organisms that exploit QS mechanisms to enhance virulence and resistance profiles [3] [36].
Quorum sensing represents a fundamental mechanism governing the transition from individual bacterial cells to coordinated multicellular communities in microcolony formation. The spatial dynamics of bacterial populations critically influence the timing and pattern of QS activation, with heterogeneous distributions leading to localized induction bursts that shape subsequent biofilm architecture. The integration of advanced experimental techniques—including patterned bacterial systems via inkjet printing and sophisticated computational modeling—continues to reveal new insights into the complex interplay between spatial organization and collective behaviors in bacterial communities.
Future research directions will likely focus on manipulating QS dynamics for therapeutic benefit, understanding polymicrobial interactions in mixed-species biofilms, and exploiting emerging technologies to precisely control bacterial behaviors at the microcolony level. As our understanding of bacterial communication deepens, so too does our potential to develop novel interventions against biofilm-associated infections that pose serious challenges in clinical medicine.
Advanced Tools and Models: Methodologies for Probing Adhesion and Biofilm Architecture
Atomic Force Microscopy (AFM) has emerged as a powerful tool in microbiology, providing unprecedented capabilities for quantifying bacterial adhesion forces at the nanoscale. This technique enables researchers to probe the initial stages of bacterial attachment and biofilm formation—critical processes in both infectious disease and environmental microbiology. Unlike traditional microbiological assays that provide population-average data, AFM operates at the single-cell and single-molecule level, allowing for the direct measurement of interaction forces between bacterial cells and surfaces with piconewton sensitivity [37]. The ability to perform these measurements under physiological conditions makes AFM particularly valuable for studying fundamental microbial adhesion mechanisms, which often involve complex structures such as pili, flagella, and extracellular polymeric substances [38] [37].
The application of AFM to bacterial adhesion research represents a significant advancement in our understanding of how microorganisms interact with their environment. Bacterial adhesion to surfaces is a crucial first step in biofilm formation, which plays a pivotal role in chronic infections, antimicrobial resistance, and various industrial and environmental processes [8] [4]. By providing direct, quantitative measurements of the forces involved in bacterial attachment, AFM enables researchers to dissect the molecular mechanisms underlying these processes, potentially leading to novel strategies for controlling biofilm-related challenges in clinical and industrial settings [39].
Fundamentals of Bacterial Adhesion and Biofilm Initiation
The Biofilm Lifecycle
Bacterial biofilm formation is a multi-step process that begins with initial adhesion and progresses through distinct developmental stages. The lifecycle can be broadly described in five main steps [4]:
Initial Reversible Attachment: Planktonic (free-swimming) cells approach and loosely attach to surfaces through long-range physical forces including van der Waals forces, electrostatic interactions, and hydrophobic effects [8] [4]. This initial attachment is typically reversible, allowing cells to detach if conditions are unfavorable.
Irreversible Attachment: Following initial contact, bacteria strengthen their attachment through the production of adhesive surface structures such as pili and fimbriae, and begin secreting extracellular polymeric substances (EPS) that cement them to the surface [4].
Microcolony Formation: Attached cells proliferate and form clustered microcolonies, initiating cell-to-cell communication through quorum sensing systems.
Biofilm Maturation: The developing biofilm forms a complex three-dimensional architecture with characteristic features such as water channels that facilitate nutrient distribution and waste removal.
Dispersion: Cells detach from the biofilm, either individually or in clumps, to colonize new surfaces and initiate fresh biofilm formation elsewhere [4].
Key Bacterial Structures in Adhesion
Type IV pili are particularly important adhesive structures in many gram-negative bacteria such as Pseudomonas aeruginosa. These thin, hairlike appendages range from 0.5 to 7 μm in length and have diameters of 4-6 nm [38]. They function not only in adhesion but also in a form of surface motility called twitching motility. AFM studies have revealed that type IV pili can withstand rupture forces of approximately 95 pN and exhibit a persistence length of about 5 μm, reflecting their flexibility and strength [38].
Flagella also contribute significantly to initial surface interactions. Recent large-area AFM studies of Pantoea sp. YR343 have visualized flagellar structures measuring 20-50 nm in height and extending tens of micrometers across surfaces. These appendages appear to bridge gaps between cells during early attachment stages, suggesting a role in biofilm assembly beyond mere motility [40].
The following diagram illustrates the key stages of biofilm development and the corresponding structural changes at the bacterial cell surface:
AFM Methodologies for Quantifying Bacterial Adhesion
Core AFM Techniques
AFM offers multiple operational modes for studying bacterial adhesion, each providing unique insights into cell-surface interactions:
Topographic Imaging: Reveals the nanoscale surface morphology of bacterial cells and their appendages. This mode can visualize structures such as pili, flagella, and extracellular polymeric substances with resolution surpassing conventional optical microscopy [37] [40]. Imaging can be performed in both air and liquid environments, allowing researchers to observe bacterial surfaces under physiological conditions.
Single-Molecule Force Spectroscopy (SMFS): Utilizes functionalized AFM tips to probe specific molecular interactions. In this mode, the tip is modified with molecules of interest (e.g., adhesins, surface proteins, or carbohydrates), and force-distance curves are recorded to quantify binding strengths, mechanical properties, and interaction dynamics at the single-molecule level [37]. This approach has been used to measure the mechanical properties of peptidoglycan and to map the distribution of specific receptors on bacterial surfaces.
Single-Cell Force Spectroscopy (SCFS): Employs a complete bacterial cell attached to the AFM cantilever to measure adhesion forces at the cellular level [37]. This technique provides insights into the overall adhesive properties of bacteria, integrating contributions from various surface structures and molecules. SCFS has been used to quantify bacterial adhesion to diverse surfaces including medical implants, mineral particles, and host tissues.
Technical Considerations for Bacterial Adhesion Studies
Successful AFM measurements of bacterial adhesion require careful attention to several technical aspects. Cantilever selection is critical, with spring constants typically ranging from 8 ± 4 mN/m for imaging to even softer cantilevers for force spectroscopy to ensure sufficient sensitivity to measure weak interactions [38]. Sample preparation must preserve bacterial viability and surface structures while ensuring firm attachment to substrates. Common approaches include immobilizing bacteria on freshly cleaved mica or functionalized glass surfaces through physical adsorption or chemical cross-linking [38] [41].
Environmental control is another crucial consideration. Measurements performed in liquid environments better reflect physiological conditions but introduce challenges related to fluid dynamics and thermal drift. Buffer composition, ionic strength, and pH must be carefully controlled as these factors significantly influence electrostatic interactions and molecular binding affinities [41]. Recent advancements include the development of automated large-area AFM systems that combine high-resolution imaging with the ability to scan millimeter-scale areas, enabling researchers to link nanoscale adhesion events to larger biofilm structures [40].
Quantitative AFM Measurements of Bacterial Adhesion
AFM force spectroscopy provides direct quantification of bacterial adhesion forces across various biological systems. The following table summarizes key measurements reported in recent literature:
Table 1: Quantitative AFM Measurements of Bacterial Adhesion Forces
| Bacterial Species | Surface Type | Adhesion Force | Experimental Conditions | Reference |
|---|---|---|---|---|
| Pseudomonas aeruginosa | Mica | 95 pN (pili rupture) | PBS solution, retraction rate 3 μm/s | [38] |
| Escherichia coli | Goethite | 97 ± 34 pN (initial attraction) | Deionized water, pH 5.6-5.9 | [41] |
| Escherichia coli | Goethite | -3.0 ± 0.4 nN (maximum adhesion) | 4s contact time, bond strengthening | [41] |
| Sulfate-Reducing Bacteria | AFM tip | -3.9 to -4.3 nN (tip-cell interaction) | Seawater medium | [39] |
| Shewanella oneidensis | Goethite (010 face) | -0.80 ± 0.15 nN (anaerobic) | 30-45 min contact time | [41] |
| Shewanella oneidensis | Hematite (001 face) | -4.3 ± 0.04 nN (anaerobic) | Iron oxide surfaces | [41] |
The adhesion forces measured by AFM reflect the contribution of various bacterial surface components and their interaction with specific substrates. For example, the measured pili rupture force of 95 pN for Pseudomonas aeruginosa represents the strength of individual pilus filaments, while the stronger adhesion forces observed for Escherichia coli to goethite surfaces (ranging from 97 pN to 3.0 nN) likely involve multiple adhesins and surface structures [38] [41]. The variation in adhesion forces under different environmental conditions (e.g., anaerobic vs. aerobic) highlights how bacterial surface properties can change in response to their environment [41].
Time-dependent strengthening of bacterial adhesion has been observed in several AFM studies. For E. coli adhering to goethite, bond strengthening occurred within 4 seconds of contact, reaching maximum adhesion forces and energies of -3.0 ± 0.4 nN and -330 ± 43 aJ (10^-18 J), respectively [41]. This rapid strengthening likely reflects molecular rearrangements at the interface and the engagement of additional binding sites over time.
Experimental Protocols for AFM-Based Bacterial Adhesion Analysis
Bacterial Immobilization on AFM Cantilevers
The preparation of single-cell probes is a critical step for reliable SCFS measurements. One well-established protocol involves the following steps [38]:
Cantilever Functionalization: Incubate sharpened silicon nitride AFM cantilevers overnight in a 0.1% (wt/vol) aqueous poly-L-lysine solution. Poly-L-lysine provides a positively charged surface that promotes bacterial attachment through electrostatic interactions.
Drying and Preparation: Remove the cantilevers from the poly-L-lysine solution and allow them to dry for 2 hours by standing on edge on a paper towel. This ensures even coating and proper surface characteristics for bacterial attachment.
Bacterial Attachment: Place a drop of bacterial suspension in phosphate-buffered saline (PBS) at the tip of the poly-L-lysine-treated V-shaped AFM cantilever and allow to adsorb for 15 minutes. The bacterial concentration should be optimized to achieve single-cell attachment at the cantilever tip.
Rinsing and Storage: Gently rinse the cantilevers in PBS to remove loosely attached cells and use immediately for force measurements without drying. Verification of cell attachment can be performed using scanning electron microscopy after AFM measurements [38].
Force Spectroscopy Measurements
The following workflow outlines a standardized approach for collecting meaningful adhesion force data:
For consistent results, the following parameters should be carefully controlled during force spectroscopy measurements [38] [41]:
Approach and Retraction Rates: Standard retraction rates of 3 μm/s are commonly used, but varying this parameter can provide insights into the kinetic properties of adhesive bonds.
Contact Force: Maintain minimal contact forces (typically <200 pN) to avoid damaging bacterial surface structures while ensuring sufficient contact.
Dwell Time: Allow 1-4 seconds of contact between the bacterial cell and surface, as adhesion strength often increases with contact time due to molecular rearrangements and additional bond formation.
Environmental Conditions: Perform measurements in appropriate buffer solutions (e.g., PBS) at room temperature unless specific environmental conditions are being tested.
Statistical Rigor: Collect a minimum of 100-500 force curves from multiple cells and independent experiments to ensure statistical significance and account for biological variability.
Control experiments are essential to validate AFM adhesion measurements. These should include [38]:
- Using poly-L-lysine-coated AFM tips without bacteria to quantify non-specific interactions between the functionalized tip and the substrate.
- Testing non-adherent mutant strains (e.g., pilus-deficient mutants) to confirm the contribution of specific adhesion structures.
- Systematic variation of solution conditions (ionic strength, pH) to elucidate the physicochemical nature of the adhesive interactions.
Essential Research Reagents and Materials
Successful AFM studies of bacterial adhesion require carefully selected materials and reagents. The following table outlines key components and their functions in adhesion force measurements:
Table 2: Essential Research Reagents for AFM Bacterial Adhesion Studies
| Reagent/Material | Specification | Function in Experiment | Example Application |
|---|---|---|---|
| AFM Cantilevers | Silicon nitride, spring constant 8 ± 4 mN/m | Force sensing and bacterial attachment | Bacterial immobilization for SCFS [38] |
| Substrates | Freshly cleaved mica | Atomically flat surface for adhesion measurements | Standard substrate for initial adhesion studies [38] |
| Functionalization Reagent | Poly-L-lysine (0.1% aqueous solution) | Promotes bacterial attachment to cantilever | Creating bacterial probes for SCFS [38] |
| Buffer Systems | Phosphate-buffered saline (PBS), pH 7.4 | Maintains physiological conditions during measurement | Standard buffer for bacterial adhesion studies [38] |
| Clay Minerals | Kaolinite, montmorillonite | Representative soil mineral surfaces | Environmental adhesion studies [41] |
| Metal Oxides | Goethite, hematite | Model environmental surfaces | Studying bacterial adhesion to iron oxides [41] |
| Dental Materials | Porcelain, lithium disilicate, acrylics | Biomedical surfaces for oral biofilm studies | Dental adhesion research [42] |
The selection of appropriate substrates is particularly important, as surface properties significantly influence bacterial adhesion. Mica provides an atomically flat, negatively charged surface that is ideal for initial adhesion studies [38]. For environmental applications, clay minerals such as kaolinite and montmorillonite, as well as metal oxides like goethite, serve as relevant substrates that mimic natural surfaces [41]. In biomedical contexts, dental materials including porcelain and lithium disilicate enable studies of bacterial colonization on restorative materials [42].
Recent methodological advances include the development of automated large-area AFM approaches capable of capturing high-resolution images over millimeter-scale areas. When combined with machine learning algorithms for image stitching and analysis, these systems enable comprehensive characterization of biofilm heterogeneity and organization [40]. Additionally, the integration of AFM with complementary techniques such as fluorescence microscopy provides correlated structural and functional information, offering a more complete understanding of bacterial adhesion processes [37].
AFM has revolutionized our ability to quantify and understand bacterial adhesion forces at the nanoscale, providing critical insights into the initial stages of biofilm formation. The techniques and methodologies outlined in this guide enable researchers to directly measure interaction forces between bacterial cells and surfaces, unraveling the molecular mechanisms that underpin microbial colonization in diverse environments. As AFM technology continues to evolve, emerging approaches including automated large-area scanning, machine learning-assisted image analysis, and multi-modal integration with complementary techniques promise to further enhance our understanding of bacterial adhesion processes [40].
The quantitative data generated through AFM force spectroscopy provides a foundation for developing novel strategies to control bacterial adhesion in clinical, industrial, and environmental contexts. By understanding the fundamental forces that govern bacterial attachment, researchers can design surfaces that resist biofilm formation, develop more effective anti-fouling coatings, and create new therapeutic approaches to combat biofilm-associated infections. As these applications continue to expand, AFM will remain an indispensable tool in the ongoing effort to understand and manipulate bacterial adhesion at the nanoscale.
Within the broader study of bacterial adhesion and biofilm initiation, the transition from planktonic cells to a structured, surface-associated community represents a critical phase in bacterial life cycles. This process, fundamental to both environmental survival and clinical pathogenesis, begins with initial attachment and culminates in the formation of a complex, three-dimensional architecture. Confocal Laser Scanning Microscopy (CLSM) has emerged as a cornerstone technique for visualizing and quantifying this spatial organization in situ, allowing researchers to dissect the relationship between biofilm structure and function without causing significant damage to the delicate samples [43] [44]. The capability of CLSM to provide optical sectioning of fully hydrated, living biofilms makes it uniquely suited for the non-destructive analysis of the biofilm matrix's structural intricacies and the physiological heterogeneity of the encapsulated microorganisms [44] [45]. This technical guide details the application of CLSM for the spatial analysis of biofilms, providing a framework for investigating the mechanisms of adhesion and biofilm initiation central to microbial ecology and antimicrobial drug development.
The Biofilm Lifecycle: From Adhesion to Maturation
The formation of a biofilm is a dynamic, multi-stage process that begins with the adhesion of planktonic cells to a surface. Understanding this lifecycle is crucial for identifying potential targets for intervention.
Diagram 1: The biofilm lifecycle, from initial attachment to active dispersion.
The lifecycle initiates with the reversible attachment of free-living planktonic cells to a conditioned surface, mediated by weak interactions such as van der Waals forces and electrostatic interactions [3] [4]. This is followed by a transition to irreversible attachment, where microbial cells firmly anchor themselves using surface structures like pili and fimbriae, and begin secreting adhesive extracellular polymeric substances (EPS) [3] [46]. Subsequently, attached cells proliferate and form microcolonies, which then develop into a mature biofilm characterized by a complex 3D architecture with water channels and gradients of nutrients, oxygen, and signaling molecules [4] [46]. The final stage, active dispersion, allows cells to break free from the biofilm matrix to colonize new niches, completing the cycle [4].
Principles and Advantages of CLSM for Biofilm Analysis
CLSM operates on the principle of point illumination and a confocal pinhole aperture to eliminate out-of-focus light. A laser beam is focused onto a specific spot within the sample, and the resulting fluorescence emitted from that spot is detected through a pinhole. This configuration ensures that only light from the focal plane is detected, granting the microscope its optical sectioning capability [43]. By scanning the laser point-by-point across the x, y, and z planes, a series of high-contrast optical sections can be assembled into a precise three-dimensional reconstruction of the sample [43].
This principle offers several distinct advantages for biofilm research:
- Non-destructive, in situ imaging: Biofilms can be analyzed in a fully hydrated, living state, preserving their native architecture and enabling real-time observation of developmental processes [44] [45].
- Optical sectioning: The elimination of out-of-focus blur allows for the collection of clear images at various depths within the biofilm without physical sectioning [43].
- 3D reconstruction and quantification: Z-stack image series can be rendered into 3D models and analyzed to extract quantitative data on biofilm biovolume, thickness, roughness, and spatial distribution of components [47] [45].
- Spatial co-localization analysis: Multiple fluorescence channels can be used simultaneously to investigate the spatial relationships between different biofilm components, such as specific matrix polymers and bacterial populations [44] [47].
Methodological Workflow for CLSM Analysis of Biofilms
A standardized workflow is essential for obtaining reliable and reproducible data on biofilm architecture and composition. The following section outlines the key procedural stages.
Diagram 2: Core workflow for CLSM analysis of biofilms, from sample preparation to 3D quantification.
Sample Preparation and Staining Strategies
Proper sample preparation and staining are critical for meaningful visualization. Biofilms can be grown in various systems, including flow-cell reactors for continuous culture or on coupons and coverslips for static culture [45]. A key consideration is selecting appropriate fluorescent probes to target specific biofilm constituents.
Table 1: Common Fluorescent Probes for Biofilm Matrix Components
| Target Component | Probe Examples | Function and Application | Key Considerations |
|---|---|---|---|
| Exopolysaccharides (EPS) | Lectins (e.g., Con A, WGA) conjugated to fluorophores [44] [45] | Binds specifically to sugar residues; identifies spatial distribution of polysaccharides like Psl in P. aeruginosa [45]. | Specificity depends on lectin choice; may require validation with genetic mutants. |
| Extracellular DNA (eDNA) | SYTO dyes, TOTO, POPO [44] | Nucleic acid intercalating dyes that fluoresce upon binding DNA; visualizes the eDNA scaffold in the matrix. | Can penetrate live cells; may require cell-impermeant dyes (e.g., propidium iodide) to differentiate from intracellular DNA. |
| Proteins | FITC, Sypro Ruby [44] | Binds nonspecifically to proteins; provides a general overview of protein localization within the matrix. | Nonspecific staining; can be combined with immunofluorescence using antibodies for specific proteins. |
| Live/Dead Cells | SYTO 9 / Propidium Iodide (e.g., LIVE/DEAD BacLight) | SYTO 9 stains all cells; PI stains cells with compromised membranes; assesses biofilm viability. | Standard for viability assessment; results can be influenced by metabolic state. |
| Lipids | Nile Red [44] | Fluorescence emission shifts based on local hydrophobicity; identifies hydrophobic regions and lipid-rich components. | Useful for visualizing outer membrane vesicles (OMVs) in the matrix. |
Image Acquisition and Processing
For image acquisition, biofilm samples are typically imaged using a confocal microscope equipped with lasers corresponding to the excitation maxima of the chosen fluorophores [48]. A z-stack of sequential optical sections is collected with a step size (e.g., 0.5 - 1.0 µm) that satisfies the Nyquist sampling criterion to accurately reconstruct the 3D structure [47]. Key parameters such as laser power, gain, and pinhole size must be optimized to maximize signal while minimizing photobleaching and background noise [48].
Following acquisition, image processing is performed using software such as FIJI/ImageJ, ICY, or BiofilmQ [49] [47]. This involves:
- Deconvolution: A computational process that reassigns out-of-focus light to its point of origin, enhancing resolution and contrast [49].
- Segmentation: Differentiating the biofilm biovolume from the background is a critical step for quantitative analysis. BiofilmQ offers multiple segmentation options, including automatic thresholding and the import of pre-segmented images [47].
- 3D Reconstruction: The z-stack images are rendered into a 3D model for visualization and analysis of the overall biofilm architecture.
Quantitative Spatial Analysis
Quantitative analysis transforms image data into objective metrics for comparing biofilm phenotypes. Software tools like COMSTAT and BiofilmQ are specifically designed for this purpose [47] [45].
Table 2: Key Quantitative Parameters for Biofilm Architecture Analysis
| Parameter Category | Specific Metrics | Biological Significance | Analysis Tool |
|---|---|---|---|
| Global Architecture | • Biovolume (µm³)• Average Thickness (µm)• Surface Area to Biovolume Ratio• Roughness Coefficient | Describes the overall size, density, and topographical complexity of the biofilm. | COMSTAT [45], BiofilmQ [47] |
| Spatial Distribution | • Substrate Coverage (%)• Vertical Stratification• Distance to nearest neighbor | Quantifies how biomass is distributed relative to the attachment surface and within the 3D space. | BiofilmQ [47] |
| Fluorescence Intensity | • Mean/Median Intensity per channel• Correlation between channels (Manders', Pearson's) | Measures abundance and co-localization of specific components (e.g., matrix proteins with EPS). | BiofilmQ [47] |
| Textural Analysis | • Local Biovolume Density• Cluster Size Distribution | Reveals the heterogeneity and micro-scale organization within the biofilm. | BiofilmQ [47] |
The Scientist's Toolkit: Essential Reagents and Software
Table 3: Essential Research Reagents and Software for CLSM Biofilm Analysis
| Item Name / Category | Specific Examples | Function and Application in Biofilm Research |
|---|---|---|
| Fluorescent Stains & Dyes | • SYTO dyes (nucleic acids)• Fluorescently-conjugated Lectins (EPS)• FITC (proteins)• LIVE/DEAD BacLight viability kit | Targets and visualizes specific biochemical components of the biofilm matrix and assesses cell viability. |
| Biofilm Growth Systems | • Flow-cell reactors [45]• Static microtiter plates [45]• Drip-flow reactors | Provides controlled and reproducible environments for cultivating biofilms for experimental analysis. |
| Image Analysis Software | • BiofilmQ [47]• COMSTAT [45]• FIJI/ImageJ [49]• ICY [49] | Enables 3D quantification, visualization, and statistical analysis of biofilm architecture and composition. |
| Deconvolution Software | • Huygens Professional [49] | Improves image clarity and resolution by computationally removing out-of-focus light. |
| Model Organisms | • Pseudomonas aeruginosa [45]• Staphylococcus aureus [4]• Vibrio cholerae [47] | Commonly used bacterial species for studying biofilm formation, architecture, and resistance mechanisms. |
Application in Investigating Bacterial Adhesion and Biofilm Initiation
CLSM is instrumental in deciphering the mechanisms of bacterial adhesion and the subsequent initiation of biofilm formation. By applying the methodologies described above, researchers can:
- Visualize Initial Attachment: CLSM allows for the real-time observation of the transition from reversible to irreversible attachment, revealing the critical role of surface adhesins and conditioning films [46] [45].
- Define the Role of Matrix Components: Using isogenic mutants combined with specific staining, the function of individual EPS components (e.g., Psl, Pel, and Alginate in P. aeruginosa) in early microcolony development and structural stability can be directly visualized and quantified [45].
- Map Chemical Microenvironments: FLIM and the use of ratiometric dyes enable the measurement of pH, oxygen, and ion gradients that form within developing biofilms. These gradients are a direct consequence of limited diffusion through the matrix and create the heterogeneous metabolic landscape that influences phenotypic tolerance to antimicrobials [44].
- Track Resistance Dynamics: CLSM can be used to monitor the penetration and binding of fluorescently tagged antibiotics, revealing how the matrix acts as a barrier and how tolerance emerges in specific subpopulations within the biofilm structure [4].
Confocal Laser Scanning Microscopy provides an unparalleled window into the three-dimensional world of biofilms. By enabling the non-destructive, in situ analysis of biofilm architecture, composition, and physiology, CLSM has become an indispensable tool for advancing our understanding of the fundamental mechanisms of bacterial adhesion and biofilm initiation. The quantitative parameters derived from CLSM image analysis offer a robust framework for phenotyping biofilm mutants, evaluating the efficacy of anti-biofilm agents, and ultimately, for informing the development of novel strategies to combat biofilm-associated infections. As both imaging technologies and analytical software like BiofilmQ continue to evolve, their integration will undoubtedly yield deeper insights into the complex spatial ecology of microbial communities.
The study of bacterial adhesion and biofilm initiation is a critical area of research within microbiology and biomedical science. Biofilms, defined as structured communities of bacterial cells enclosed in a self-produced polymeric matrix and adherent to a surface, are implicated in over 65% of all microbial infections [50]. The development of these communities is a dynamic process, and their formation on biomedical implants and devices represents a significant healthcare burden, with estimated direct costs of 7 billion EUR annually in Europe alone [50].
The initial attachment of bacterial cells to a surface is a pivotal phase in biofilm development, and this process is profoundly influenced by hydrodynamic conditions. Hydrodynamics governs the rate at which planktonic (free-floating) cells are delivered to a surface, the residence time for attachment, and the shear forces that can either facilitate or hinder adhesion [51] [50]. While static models provide a preliminary understanding, they fail to replicate the fluid flow conditions present in many natural and clinical scenarios, such as the urinary tract, vascular system, or industrial pipelines [51]. Consequently, in vitro flow systems that accurately model these hydrodynamic conditions are indispensable tools for advancing both fundamental knowledge and therapeutic strategies in biofilm research.
Hydrodynamic Parameters in Bacterial Adhesion
Fundamental Concepts
Hydrodynamics affects initial bacterial adhesion through several key parameters. The shear rate (γ̇), defined as the derivative of fluid velocity in the direction perpendicular to the wall, quantifies the frequency at which cells contact the surface [50]. The shear stress (τ), representing the frictional force the fluid exerts on adhered cells, is the product of the shear rate and the fluid viscosity (μ) for Newtonian fluids (τ = μγ̇) [50]. These parameters, rather than flow rate alone, provide meaningful characterization of the forces acting on adhering cells, as they account for system geometry [50].
Impact on Adhesion Mechanisms
The process of initial adhesion begins with the reversible attachment of planktonic cells to a conditioned surface (a surface modified by the adsorption of environmental molecules) [52]. This "docking" phase is governed by physiochemical interactions including electrostatic forces, hydrophobic interactions, and van der Waals forces [52]. Hydrodynamics directly influences this stage by controlling the transport of cells to the surface and imposing shear forces that can disrupt early attachment. Research indicates that bacterial residence time and surface coverage tend to increase with shear stress up to an optimal point, reflecting a balance between the rate of cell delivery to the surface and the disruptive force acting on adhered bacteria [51] [50].
Table 1: Key Hydrodynamic Parameters and Their Influence on Initial Adhesion
| Parameter | Definition | Role in Initial Adhesion |
|---|---|---|
| Shear Rate (γ̇) | Derivative of fluid velocity in the perpendicular direction from the wall [50]. | Dictates the frequency at which planktonic cells contact the surface. |
| Shear Stress (τ) | Frictional force per unit area exerted by the fluid flowing over a surface (τ = μγ̇ for Newtonian fluids) [50]. | Determines the force attempting to detach reversibly adhered cells; influences biofilm structure. |
| Flow Velocity | Speed of the bulk fluid movement. | Influences the convective transport of cells and nutrients to the surface. |
| Pulsatility Index | Quotient of flow oscillation magnitude and mean flow rate [53]. | Affects endothelial cell activity and morphology in vascular systems; models physiological conditions. |
Platforms for In Vitro Flow Modeling
Several engineered platforms enable the study of bacterial adhesion under controlled hydrodynamic conditions. The selection of an appropriate system is crucial for generating reproducible and clinically relevant data.
Modified Robbins Device (MRD)
The Robbins device was originally developed to study biofilms in industrial water systems and has since been modified for biomedical applications [51]. The MRD typically consists of a pipe or channel with multiple sampling ports where material coupons can be mounted flush with the inner surface, allowing simultaneous testing of different surfaces under identical flow conditions [51] [50].
Key Applications: MRDs have been effectively used to mimic the hydrodynamic conditions in urinary catheters and stents (e.g., at a shear rate of 15/s) to study biofilm formation by pathogens such as Escherichia coli and Pseudomonas aeruginosa [51]. Their ability to allow periodic removal of coupons for analysis makes them suitable for time-course experiments [51].
Advantages and Limitations:
- Advantages: Generate large amounts of biomass; allow for long-term studies and periodic sampling; enable high-throughput screening of materials [51] [50].
- Disadvantages: Complex setup; limited potential for real-time, in-situ visualization of adhesion; coupon removal can disrupt the biofilm and flow dynamics [51] [50].
Flow Chambers
Flow chambers, particularly parallel-plate flow chambers (PPFCs), are designed to be mounted on microscope stages, enabling real-time, in-situ observation of microbial adhesion to transparent surfaces [51]. These systems typically include a recirculating flow loop with a pump, medium reservoir, and the flow chamber itself, allowing precise control over flow rate and temperature [51].
Key Applications: Custom-made flow chambers have been widely used to monitor the initial adhesion of uropathogens like E. coli and Enterococcus faecalis to polymeric surfaces such as polydimethylsiloxane (PDMS) over periods of 30 minutes to 4 hours, under shear conditions relevant to medical devices [51]. They are also used for longer-term biofilm studies (e.g., 24 hours) [51].
Experimental Protocol: Parallel-Plate Flow Chamber Assay
- Setup Assembly: Connect a parallel-plate flow chamber to a recirculating flow loop consisting of a medium reservoir, silicone tubing, and a peristaltic or centrifugal pump [51] [53].
- Surface Preparation: Place the test substrate (e.g., a functionalized glass slide or polymer coupon) into the flow chamber [53]. For adhesion studies, surfaces may be coated with relevant proteins to create a conditioned film [52].
- Hydrodynamic Calibration: Use Computational Fluid Dynamics (CFD) or established equations to determine the flow rate required to achieve the desired wall shear stress. For a PPFC, the wall shear stress (τw) is given by τw = (6μQ)/(wh²), where μ is fluid viscosity, Q is volumetric flow rate, w is chamber width, and h is half the chamber height [50].
- Inoculation and Flow: Introduce a bacterial suspension of known concentration (e.g., OD₆₀₀ = 0.01) into the system and initiate flow at the predetermined rate [51] [54].
- Real-Time Monitoring: Use phase-contrast or epifluorescence microscopy coupled with image analysis to quantify the number of adherent cells over time [51].
- Post-Assay Analysis: After the experiment, surfaces can be extracted for further analysis using techniques like confocal laser scanning microscopy (CLSM) to examine the distribution and viability of adhered cells [51].
Microfluidic Devices
Microfluidic platforms offer miniaturization, precise fluid control, and the ability to test multiple conditions in parallel [51] [50]. These devices are typically fabricated from PDMS and allow for rapid analysis under highly controlled flow conditions [51].
Key Applications: Microfluidic devices have been used to explore the initial bacterial adhesion on different materials, revealing that adhesion rates are higher in locations with sudden changes in shear forces [51]. They are particularly valuable for investigating the combined effects of surface topography and hydrodynamics on adhesion [51].
Advantages and Limitations:
- Advantages: High versatility; small reagent volumes; potential for high-throughput screening; precise control over local flow conditions [51] [50].
- Disadvantages: Often custom-designed for specific purposes; can be prone to channel clogging; may not be suitable for generating large biomass for subsequent molecular analysis [51] [50].
Table 2: Comparison of In Vitro Flow Platforms for Studying Initial Bacterial Adhesion
| Platform | Typical Shear Stress Range | Best Applications | Key Technical Considerations |
|---|---|---|---|
| Modified Robbins Device (MRD) | Wide dynamic range [50]. | Long-term biofilm studies, high-throughput material screening, mimicking conditions in catheters/stents [51]. | CFD modeling is recommended to characterize shear stress distribution and minimize entry effects [51]. |
| Flow Chambers (e.g., PPFC) | Precisely controlled, relevant to physiological flows (e.g., ~0.07 Pa in ureteral stents [51]). | Real-time visualization of adhesion, quantitative analysis of attachment kinetics, studies of single-species adhesion under well-defined flow [51]. | Requires transparent surfaces for microscopy. Flow must be fully developed; entrance length should be calculated [51]. |
| Microfluidic Devices | Highly controlled, from very low to high shear [51]. | Investigating adhesion in response to spatial shear variations, multi-parameter screening, studies requiring micro-scale resolution [51]. | Device design must be optimized via CFD to create relevant shear profiles. Fabrication requires cleanroom facilities [51]. |
Essential Methodologies and Workflows
Computational Fluid Dynamics (CFD) in System Design
Computational Fluid Dynamics is a critical tool for modeling flow systems, enabling the estimation of key parameters like shear stress and shear rate at a relatively low cost and with high speed compared to experimental techniques [50]. CFD simulations solve algebraic equations derived from the conservation of mass, momentum, and energy across a computational mesh of the flow geometry [50]. Its use is highly recommended for characterizing the hydrodynamic environment in any flow system before biological experiments, ensuring that the applied conditions accurately reflect the intended physiological or pathological scenario [51] [50].
Analytical Techniques for Quantifying Adhesion
Real-Time Microscopy: Flow chambers coupled with video capture systems allow for the direct observation and quantification of microbial adhesion. This can include tracking the number of adherent cells over time, analyzing their distribution, and monitoring early microcolony formation [51].
Confocal Laser Scanning Microscopy (CLSM): Following flow experiments, CLSM is used to obtain high-resolution images of adherent cells and early biofilms at various depths without disturbing their native structure. Subsequent image analysis with software like the COMSTAT ImageJ plugin or the PHLIP Matlab toolbox can provide quantitative metrics such as biomass, surface coverage, and biofilm thickness [51].
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Materials for In Vitro Flow Adhesion Studies
| Item | Function/Application | Examples/Notes |
|---|---|---|
| Parallel-Plate Flow Chamber | Creates a well-defined laminar flow field for adhesion studies under controlled shear stress [51] [53]. | Can be custom-made from acrylic or commercially sourced. |
| Peristaltic or Centrifugal Pump | Generates and controls the recirculating flow of bacterial suspension [51] [53]. | Must provide stable, pulse-free flow for accurate shear stress. |
| Polydimethylsiloxane (PDMS) | Material of choice for many microfluidic devices and for coating surfaces; transparent and biocompatible [51]. | Can be modified with antimicrobial peptides or carbon nanotubes [51]. |
| Polyether Ether Ketone (PEEK) | Medical-grade polymer used for implants; tested for bacterial adhesion propensity [54]. | Supports more biofilm formation than Polyamide 12 (PA12) [54]. |
| Artificial Urine Medium (AUM) | Mimics the chemical composition of human urine for studies relevant to urinary tract infections and device encrustation [51]. | Used in studies modeling adhesion to ureteral stents and catheters [51]. |
| Crystal Violet Stain | A common dye used to quantify total adhered biomass in endpoint assays [54]. | Stains cells and some matrix components; eluted dye is measured spectrophotometrically [54]. |
| Fibronectin Solution | Used to functionalize glass slides or membranes, creating a conditioned surface that mimics protein-coated biomedical implants [53]. | Typically applied at 25 μg/mL to coat surfaces before cell seeding [53]. |
In vitro flow systems are indispensable for elucidating the fundamental mechanisms of bacterial adhesion under conditions that mirror the dynamic environments found in the human body and on medical devices. Platforms such as the Modified Robbins Device, flow chambers, and microfluidic devices, when combined with rigorous hydrodynamic characterization via CFD, provide powerful and reproducible means to model the impact of hydrodynamics on initial attachment. The insights gained from these systems are critical for guiding the rational design of novel anti-fouling and antimicrobial surfaces, ultimately contributing to the development of strategies to mitigate biofilm-associated infections. Future research will likely focus on increasing the complexity of these models to better mimic in vivo conditions, such as incorporating multi-species communities, host cells, and more physiologically accurate fluid compositions and flow waveforms.
Diagram: Experimental Workflow for a Flow Chamber Adhesion Assay
The formation of bacterial biofilms represents a significant challenge in medical treatment, often leading to persistent infections and heightened antimicrobial resistance (AMR). Biofilms are complex, surface-delimited microbial communities encased within a matrix of extracellular polymeric substances (EPS) [3]. The initial, reversible attachment of free-floating (planktonic) microorganisms to a surface is a critical first step in establishing these structured communities [3] [17]. Consequently, anti-adhesion compounds that disrupt this initial attachment phase offer a promising therapeutic strategy to prevent biofilm-associated infections before they mature. This guide details the application of high-throughput screening (HTS) platforms to identify such compounds, a methodology positioned within the broader research objective of understanding and interrupting the mechanisms of bacterial adhesion and biofilm initiation.
Biological Foundation: Bacterial Adhesion and Biofilm Initiation
The Mechanism of Initial Attachment
The process of biofilm formation begins with the reversible attachment of microbial cells to a conditioned surface. This initial adhesion is governed by weak, non-specific physical forces such as van der Waals interactions, electrostatic forces, and hydrophobic effects [3] [17]. Surface characteristics, including roughness, play a key role, with rougher surfaces typically promoting better microbial adhesion [3]. At this stage, bacteria may employ passive attachment mechanisms or active approaches using surface structures like pili and fimbriae [3].
Transition to Irreversible Attachment
Following initial contact, bacteria undergo a phenotypic switch from a motile to a sessile lifestyle. This transition is often regulated by intracellular signaling pathways, such as those involving the secondary messenger bis-(3'-5')-cyclic dimeric guanosine monophosphate (c-di-GMP) [17]. The subsequent production of EPS, a sticky matrix of polysaccharides, proteins, and extracellular DNA, facilitates a strong, irreversible attachment and the development of microcolonies, paving the way for a mature biofilm [3] [17]. Anti-adhesion strategies aim to intervene before this irreversible attachment is established.
High-Throughput Screening (HTS) Platform Design
Core HTS Concepts and Assay Types
High-throughput screening enables the rapid testing of thousands of compounds for biological activity. A more advanced paradigm, quantitative HTS (qHTS), assays compounds across a range of concentrations directly in the primary screen. This approach generates concentration-response curves (CRCs) for each substance, providing data on both potency and efficacy while reducing false-positive and false-negative rates compared to traditional single-concentration HTS [55] [56].
Assays targeting anti-adhesion activity typically fall into two categories:
- Biochemical Assays: These target specific bacterial surface components (e.g., pili, fimbriae) or enzymes involved in their synthesis. They are highly reproducible but may not fully capture the complexity of bacterial-surface interactions.
- Cell-Based Phenotypic Assays: These measure the reduction in bacterial adherence to abiotic surfaces or host cell monolayers. They provide a more physiologically relevant context and can identify hits through various mechanisms.
Key Assay Development Parameters
To ensure a successful screening campaign, assay parameters must be rigorously optimized, as demonstrated in recent infectious disease research [57] [56].
Table 1: Key Assay Parameters for HTS Optimization
| Parameter | Description | Optimization Goal |
|---|---|---|
| Signal-to-Background (S/B) | Ratio of the assay signal in the presence of activity to the background signal without activity. | A ratio >3 is typically desirable for a robust assay window [57]. |
| Z'-factor (Z') | A statistical measure of assay quality and reproducibility that accounts for the dynamic range and data variation of both positive and negative controls. | Z' > 0.5 is generally acceptable for HTS; a score above 0.3 was used successfully in a macrophage-based infection screen [57]. |
| DMSO Tolerance | The maximum concentration of the compound solvent (DMSO) that does not interfere with the assay signal. | To ensure compound solubility without affecting biology (e.g., minimal effect at concentrations as high as 10%) [56]. |
Experimental Workflow and Protocols
A comprehensive screening pipeline involves multiple stages, from primary screening to mechanistic validation.
Workflow for Anti-Adhesion Compound Identification
The following diagram outlines the key stages of a qHTS pipeline for identifying anti-adhesion compounds.
Detailed Experimental Protocols
Protocol 1: Primary qHTS for Anti-Adhesion Activity Using a Luminescence Reporter
This protocol is adapted from a high-throughput screening campaign against Mycobacterium tuberculosis [57].
Bacterial Strain Preparation:
- Use a luminescence-based reporter strain (e.g., expressing lux operon) to quantify adhered bacteria rapidly.
- Grow bacteria to mid-log phase in appropriate medium. Centrifuge and resuspend in fresh assay medium to the desired optical density (e.g., OD~600~ = 0.05).
Assay Plate Preparation:
- Use 1536-well, solid-white assay plates.
- Pin-transfer nanoliter volumes of compounds from a library into assay plates. Include controls: negative control (DMSO vehicle only) and positive control (a known anti-adhesion agent or high-concentration antibiotic).
- Dispense bacterial suspension into all wells.
Incubation and Adhesion:
- Centrifuge plates briefly (e.g., 100 × g for 1 minute) to synchronize bacterial contact with the plate surface.
- Incubate plates under optimal growth conditions for a defined period (e.g., 2-4 hours) to allow for initial adhesion.
Signal Detection and Data Analysis:
- Gently wash plates with buffer to remove non-adherent planktonic cells.
- Add a luciferin substrate to wells and measure luminescence on a plate reader.
- Normalize raw luminescence values to positive and negative controls. Calculate a Z-score for each compound; a Z-score above 0.3 can be used as an initial threshold for hit selection [57]. In qHTS, fit data to the Hill equation to generate concentration-response curves and estimate AC~50~ (concentration for half-maximal response) and E~max~ (maximal response) values [55].
Protocol 2: Secondary Validation Using a Cell-Based Proteolytic Assay
This protocol, inspired by a CHIKV antiviral screen, validates hits in a more complex, cellular context [56].
Construct a Split-Protein Reporter System:
- Engineer a bacterial strain to express a fusion protein consisting of a surface adhesion protein (e.g., a key pilin subunit) linked to the C-terminal fragment of a reporter enzyme (e.g., Nanoluciferase).
- The complementary N-terminal fragment of the reporter is constitutively expressed and secreted.
Assay Execution:
- Seed the reporter bacteria into assay plates containing the qualified hits from the primary screen.
- Incubate to allow for adhesion and potential surface assembly of the adhesion protein.
- Functional assembly and adhesion bring the two fragments of the split reporter into proximity, facilitating complementation and generating a luminescent signal.
- A reduction in luminescence in treated wells indicates inhibition of the specific adhesion protein's function or surface presentation.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Anti-Adhesion HTS
| Research Reagent | Function in Anti-Adhesion HTS |
|---|---|
| Luminescent Reporter Strains | Genetically engineered bacteria (e.g., expressing lux operon) that enable rapid, high-sensitivity quantification of adhered biomass without washing steps [57]. |
| Fluorogenic Peptide Substrates | Short peptides labeled with a fluorophore/quencher pair; cleavage by surface-exposed or secreted proteases (a common virulence factor) generates a fluorescent signal to monitor bacterial presence and activity [56]. |
| Human iPSC-Derived Macrophages (hiPSC-Macs) | A physiologically relevant, scalable host cell model for studying bacterial adhesion and invasion in an intracellular infection context, overcoming the limitations of immortalized cell lines [57]. |
| Extracellular Polymeric Substance (EPS) Stains | Fluorescent dyes (e.g., concanavalin A, SYPRO Ruby) that bind to polysaccharides or proteins in the biofilm matrix, allowing for quantitative assessment of matrix inhibition [3]. |
| Quorum Sensing Inhibitors | Small molecules that disrupt bacterial cell-to-cell communication (e.g., by degrading autoinducers or blocking receptors), which can indirectly affect adhesion and biofilm maturation [17]. |
Data Analysis and Hit Prioritization in qHTS
In qHTS, the large number of concentration-response profiles necessitates robust statistical analysis. The Hill equation (also called the four-parameter logistic model) is widely used to fit the data and derive parameters for hit prioritization [55].
The model is defined as: ( Ri = E0 + \frac{(E{\infty} - E0)}{1 + \exp{-h[\log Ci - \log AC{50}]}} ) Where:
- ( Ri ) is the measured response at concentration ( Ci )
- ( E_0 ) is the baseline response
- ( E_{\infty} ) is the maximal response
- ( AC_{50} ) is the concentration for half-maximal response (potency)
- ( h ) is the Hill slope (shape parameter)
Table 3: Quantitative HTS (qHTS) Data Analysis and Hit Criteria
| Parameter | Interpretation in Anti-Adhesion Context | Application in Hit Prioritization |
|---|---|---|
| AC~50~ (Potency) | Concentration at which 50% reduction in adhesion is observed. | Lower AC~50~ indicates greater potency. Prioritize compounds with AC~50~ in the low micromolar to nanomolar range. |
| E~max~ (Efficacy) | Maximal percent inhibition of adhesion achieved by the compound. | Prioritize compounds with high efficacy (e.g., E~max~ >80% inhibition). A partial inhibitor may have a lower E~max~. |
| Hill Slope (h) | Steepness of the concentration-response curve. | Can indicate cooperativity in binding. Values significantly different from 1 may suggest a complex mechanism. |
| Curve Quality | Statistical goodness-of-fit of the data to the Hill model. | Ensure reliable parameter estimates by setting thresholds for fit confidence intervals or R-squared values [55]. |
It is critical to recognize that parameter estimates, particularly for AC~50~, can be highly variable if the tested concentration range fails to capture at least one of the asymptotes (E~0~ or E~∞~) of the curve [55]. Therefore, hit selection should rely on a combination of potency (AC~50~), efficacy (E~max~), and curve quality metrics.
Bacterial biofilms represent a predominant mode of life for microorganisms and are recognized as a critical factor in persistent infections and antimicrobial resistance. The transition from planktonic cells to structured, surface-attached communities involves dramatic reprogramming of bacterial physiology, governed by intricate regulatory networks. Within the broader context of bacterial adhesion and biofilm initiation research, multi-omics technologies have emerged as indispensable tools for deconstructing these complex biological systems. By integrating transcriptomics and proteomics, researchers can move beyond descriptive studies to uncover the mechanistic underpinnings of biofilm formation and maintenance [58]. This approach provides a systems-level view of the coordinated gene expression, protein synthesis, and metabolic rewiring required for the biofilm lifestyle, offering new avenues for therapeutic intervention against biofilm-associated infections.
Core Principles: Transcriptomic and Proteomic Profiling in Biofilm Research
Transcriptomics and proteomics provide complementary insights into biofilm biology. Transcriptomic analysis reveals how bacteria sense surfaces and alter gene expression to initiate adhesion, mapping the regulatory circuits that coordinate this transition. Proteomic profiling identifies the functional effectors that execute this developmental program, including structural components of the matrix, adhesion proteins, and enzymes for extracellular polymer synthesis [58]. Together, these approaches capture different levels of the central dogma of molecular biology as bacteria adopt the biofilm phenotype.
The power of omics approaches lies in their ability to generate hypothesis-free datasets that capture the global state of a cell population. When applied to biofilm research, these technologies have revealed that the planktonic-to-biofilm transition is not a simple, uniform program but rather a strain-specific and environment-dependent process [58]. For instance, transcriptomic studies of Pseudomonas aeruginosa have identified hundreds of differentially expressed genes during biofilm development, though inconsistencies between studies highlight the influence of experimental conditions on the resulting data [58].
Table 1: Core Omics Technologies in Biofilm Research
| Technology | Analytical Focus | Key Insights into Biofilm Biology | Common Platforms |
|---|---|---|---|
| Transcriptomics | RNA expression patterns | Identifies differentially expressed genes in adhesion, matrix production, and stress response | RNA-Seq, Microarrays |
| Proteomics | Protein abundance and modification | Reveals virulence factors, matrix enzymes, and metabolic enzymes critical for biofilm integrity | LC-MS/MS, Label-free quantification |
| Multi-omics Integration | Combined molecular layers | Uncovers regulatory networks connecting gene expression to functional protein outcomes | Bioinformatics pipelines |
Experimental Designs and Workflows
Establishing Biofilm Models and Controls
A critical first step in omics-based biofilm research is establishing appropriate biofilm culture systems that recapitulate key aspects of native biofilm environments. Common models include flow-cell systems, which allow for spatial and temporal analysis of biofilm development, and static biofilm models using microtiter plates or submerged surfaces for bulk analysis [58]. The choice of growth surface (e.g., plastic, glass, or biologically relevant surfaces) significantly influences biofilm architecture and gene expression, necessitating careful experimental design [58].
Proper control conditions are essential for meaningful data interpretation. Planktonic cultures harvested at similar growth phases typically serve as references for identifying biofilm-specific expression patterns. However, researchers must consider that differences between biofilm and planktonic cells extend beyond attachment status to include variations in nutrient availability, oxygen gradients, and cell density [58]. For time-series experiments, sampling at multiple stages of biofilm development (attachment, maturation, dispersion) provides dynamic resolution of the transcriptional and proteomic reprogramming.
Sample Preparation for Omics Analysis
Sample preparation represents a particularly challenging aspect of biofilm omics studies due to the extracellular polymeric matrix that encases biofilm cells. Effective lysis and nucleic acid or protein extraction require optimized protocols that account for matrix interference.
For transcriptomics of bacterial biofilms, RNA stabilization immediately upon sampling is critical due to the rapid turnover of bacterial mRNAs. Protocols typically include:
- Rapid harvesting of biofilm cells by scraping or enzymatic detachment
- Immediate stabilization using RNA preservatives such as RNAlater
- Efficient lysis using mechanical disruption (bead beating) combined with chemical lysis
- RNA purification with DNase treatment to remove genomic DNA contamination [58] [59]
Proteomic sample preparation requires:
- Comprehensive extraction of proteins from both cells and matrix components
- Protein digestion using trypsin or other proteases
- Peptide cleanup and concentration before mass spectrometry analysis [60] [61]
Table 2: Key Research Reagents and Solutions for Biofilm Omics
| Reagent/Solution | Function in Experimental Protocol | Specific Application Example |
|---|---|---|
| RNA stabilization buffers | Preserves RNA integrity immediately after sampling | RNAlater for biofilm transcriptomics |
| Crystal violet | Quantifies biofilm biomass in validation assays | Confirming biofilm formation phenotype [62] |
| Mass spectrometry-grade trypsin | Digests proteins into peptides for LC-MS/MS analysis | Proteomic profiling of biofilm matrix [60] |
| LC-MS/MS systems | Identifies and quantifies protein abundance | Quantitative proteomics of biofilm vs. planktonic cells [60] |
| Formaldehyde or glutaraldehyde | Fixes biofilm structure for electron microscopy | SEM visualization of biofilm architecture [59] |
Case Studies in Omics-Driven Biofilm Research
Multi-omics Analysis of Succinic Acid Inhibition onProteus mirabilisBiofilms
A comprehensive multi-omics study investigated the mechanism by which succinic acid inhibits biofilm formation in Proteus mirabilis, a major pathogen in catheter-associated urinary tract infections. Researchers employed transcriptomics, proteomics, and metabolomics to unravel the multimodal inhibitory action of succinic acid at 15 mM concentration, which reduced bacterial growth by ≥70% and biofilm formation by ≥50% [62].
The experimental protocol revealed that succinic acid induced widespread dysregulation across multiple cellular systems:
- Metabolomic profiling revealed disruptions in tryptophan and arginine metabolism, nucleotide biosynthesis, and the tricarboxylic acid cycle
- Transcriptomic analysis showed downregulation of ribosomal genes, oxidative phosphorylation pathways, and efflux pumps, alongside upregulated arginine transport
- Proteomic data demonstrated suppression of Type VI secretion system (T6SS) virulence factors and iron acquisition proteins [62]
This integrated approach identified a novel regulatory mechanism wherein succinic acid reduces K6 acetylation of the histone-like nucleoid structuring protein, enhancing its oligomerization to repress T6SS genes and inhibit biofilm formation [62]. The study exemplifies how multi-omics can uncover complex mode-of-action networks that would remain invisible in single-platform analyses.
Quantitative Proteomics of TetR Regulator inAeromonas hydrophilaBiofilm Formation
A label-free quantitative proteomics study investigated the role of a TetR family transcriptional regulator (UidR) in biofilm formation of Aeromonas hydrophila, an aquatic pathogen. The research demonstrated that deletion of the uidR gene significantly enhanced biofilm formation, prompting proteomic comparison between the ΔuidR mutant and wild-type strains [60].
The experimental workflow included:
- Biofilm cultivation of wild-type and ΔuidR strains under controlled conditions
- Protein extraction and digestion using filter-aided sample preparation
- LC-MS/MS analysis on a Q-Exactive mass spectrometer
- Bioinformatic analysis using MaxQuant and Perseus software
This proteomic approach identified 220 differentially expressed proteins (120 upregulated, 100 downregulated) in the mutant strain. Bioinformatics analysis indicated that UidR affects biofilm formation by regulating proteins in the glyoxylic and dicarboxylic acid metabolic pathways [60]. Follow-up genetic validation through deletion of four pathway-related genes (AHA_3063, AHA_3062, AHA_4140, and aceB) confirmed their importance in biofilm formation, demonstrating the power of proteomics for generating testable hypotheses in biofilm regulation.
Diagram 1: Multi-omics network of succinic acid inhibition on P. mirabilis biofilms
Transcriptomic Analysis of Adhesion Mechanisms inPseudomonasSpecies
Transcriptome analysis of Pseudomonas strains with varying adhesion capabilities to tilapia intestinal mucus revealed a complex regulatory network controlling initial attachment, a critical step in biofilm formation. Researchers used RNA sequencing to compare strains with naturally high and low adhesion capacity, as well as strains whose adhesion was modulated by NaCl exposure [63].
The study identified:
- 322 significantly differentially expressed genes in the low-adhesion strain under NaCl stress
- 1,550 differentially expressed genes in the high-adhesion strain under NaCl stress
- Key adhesion genes including flgC, fliC, and cheB that were differentially expressed between strains
- An adhesion regulatory network involving flagellar assembly, bacterial chemotaxis, quorum sensing, two-component systems, and bacterial secretion systems [63]
This transcriptomic approach identified ten key adhesion-related genes and demonstrated how environmental factors (NaCl concentration) can modulate adhesion capacity through transcriptomic reprogramming. The findings highlight how transcriptomics can decode the complex genetic basis of a critical phenotype in biofilm initiation.
Integrated Analysis of Multi-Omics Data
The true power of omics approaches emerges when datasets are integrated to build comprehensive regulatory networks. Bioinformatic integration of transcriptomic and proteomic data can reveal post-transcriptional regulatory mechanisms, metabolic bottlenecks, and functional validation of transcriptional changes [61]. For example, in a study of Bifidobacterium pseudocatenulatum biofilm formation, integrated transcriptomics and metabolomics identified how stress response, quorum sensing, and extracellular polysaccharide production are coordinated during biofilm development [61].
Several computational strategies facilitate this integration:
- Pathway enrichment analysis to identify biological processes consistently altered across omics layers
- Network correlation analysis to connect transcriptional regulators with downstream protein effectors
- Multi-omics factor analysis to identify latent factors that drive coordinated changes across molecular layers
When applied to Pseudomonas aeruginosa biofilm studies, integrated omics has revealed how this pathogen coordinates metabolic reprogramming, virulence factor expression, and matrix production during the transition from planktonic to biofilm growth [58]. These integrated models provide a more accurate representation of the biofilm state than any single omics approach could achieve.
Diagram 2: Integrated workflow for transcriptomic and proteomic analysis of biofilms
The application of transcriptomics and proteomics has fundamentally advanced our understanding of biofilm regulatory networks, moving beyond descriptive phenomenology toward mechanistic models of biofilm development. These technologies have revealed that biofilm formation is not governed by a single master regulator but rather emerges from interconnected networks of metabolic adaptation, stress response, and cell-cell signaling [61] [58]. The case studies highlighted in this review demonstrate how omics approaches can decode these networks, identifying key checkpoints in the transition from planktonic to biofilm growth.
Future directions in biofilm omics research will likely focus on single-cell analyses to resolve heterogeneity within biofilm subpopulations, spatial omics to map molecular gradients within biofilm structures, and temporal multi-omics to capture the dynamics of biofilm development and dispersal. As these technologies mature, they will provide an increasingly sophisticated understanding of biofilm biology, enabling targeted strategies to combat biofilm-associated infections and persistence across clinical and industrial settings.
Polymicrobial biofilms are structured communities of diverse microbial consortia, such as bacteria and fungi, enclosed in a self-produced extracellular polymeric substance (EPS) and adherent to a surface [64] [52]. These multi-species consortia represent a significant challenge in both clinical and industrial settings, exhibiting enhanced virulence and dramatically increased resistance to antimicrobials compared to their mono-species counterparts—often by orders of magnitude [64] [65]. Research into their formation and behavior is critical, as they are implicated in a vast majority of chronic infections, including over 80% of microbial infections in the human body and 60-80% of hospital-acquired device-related infections [64] [65]. Understanding the mechanisms of bacterial adhesion and the subsequent initiation of these complex biofilms provides the foundational knowledge required to develop effective anti-biofilm strategies.
The study of polymicrobial systems moves beyond simplistic monoculture models to capture the true ecological nature of microbial communities. In natural and clinical environments, bacteria and fungi rarely exist in isolation; instead, they form dynamic, interactive networks [64]. These interactions—which can be synergistic, additive, or antagonistic—profoundly influence the overall architecture, stability, and resilience of the biofilm [64]. For instance, the initial adhesion of a "helper bacterium" can condition a surface, facilitating the subsequent attachment and co-aggregation of secondary colonizers through specific molecular docking mechanisms [64] [52]. This cooperative colonization ultimately results in a heterogeneous structure that is notoriously difficult to eradicate. Therefore, research employing polymicrobial models is essential for deconstructing the multi-layered resistance mechanisms and devising therapeutic interventions that address this complexity.
Core Interkingdom Interactions in Polymicrobial Consortia
The pathogenesis of polymicrobial infections is driven by a complex web of chemical and physical interactions between different microorganisms. These interactions dictate the community's structure, function, and pathogenicity.
Bacterial–Bacterial Interactions
Co-aggregation and co-localization are key mechanisms. Co-aggregation involves specific adhesion between different bacterial genotypes via surface molecules [64]. In co-localization, a "helper bacterium" modifies the environment to support the growth and biofilm integration of other species [64]. A classic example found in cystic fibrosis lungs and chronic wounds is between Pseudomonas aeruginosa and Staphylococcus aureus, where P. aeruginosa can either enhance or suppress S. aureus [64]. Conversely, in catheter-associated urinary tract infections (CAUTI), Proteus mirabilis can inhibit the growth of Escherichia coli and Candida albicans, demonstrating antagonistic interactions [64].
Fungal–Bacterial Interactions
Interactions between Candida albicans and bacteria are among the most studied. In CAUTI, C. albicans and Staphylococcus epidermidis are predominant, with bacteria facilitating fungal attachment to bladder mucosa, and the fungus in turn enhancing bacterial growth and drug resistance [64]. In the oral cavity, adhesins such as SspB of Streptococcus gordonii and Als3 of C. albicans mediate tight binding [64]. The interplay between Aspergillus fumigatus and P. aeruginosa in CF lungs is another critical interaction; galactosaminogalactan from A. fumigatus enhances P. aeruginosa biofilm formation and antibiotic resistance, while bacterial phenazines inhibit fungal growth [64].
Fungal–Fungal Interactions
These are clinically significant in infections like vulvovaginal candidiasis, often caused by C. albicans and C. glabrata. In mixed-species biofilms, C. glabrata adheres to C. albicans hyphae, leveraging this physical association for enhanced colonization and deeper tissue invasion [64]. These dual-species biofilms show augmented resistance to antifungals like azoles and echinocandins, linked to a synergistic upregulation of efflux pump genes in both species [64].
Table 1: Types of Microbial Interactions in Polymicrobial Biofilms
| Interaction Type | Key Mechanisms | Representative Microorganisms | Outcome on Biofilm |
|---|---|---|---|
| Bacterial-Bacterial | Co-aggregation, Co-localization [64] | P. aeruginosa & S. aureus [64] | Enhanced stability & virulence; can be synergistic or antagonistic |
| Fungal-Bacterial | Specific adhesin-receptor binding, Metabolic cooperation, Toxin production [64] | C. albicans & S. epidermidis; A. fumigatus & P. aeruginosa [64] | Increased biomass, drug resistance, and severity of infection |
| Fungal-Fungal | Physical adhesion to hyphae, Metabolic cooperation [64] | C. albicans & C. glabrata [64] | Enhanced colonization, tissue invasion, and antifungal resistance |
Quantitative Assessment Techniques for Polymicrobial Biofilms
Accurately quantifying individual species within a polymicrobial biofilm is critical for understanding community dynamics and responses to stressors. No single method provides a complete picture; a combination of techniques is required for reliable data [66].
Culture-Dependent and Culture-Independent Methods
Traditional culture-based methods (plate counts) on specific and non-specific media allow for the isolation and enumeration of viable cells based on colony morphology [66]. However, these methods are constrained by their inability to detect viable but non-culturable (VBNC) cells, slow-growing organisms, and those requiring specific growth conditions, potentially missing up to 30-40% of co-pathogens in a sample [66] [65]. Molecular techniques overcome many of these limitations. Quantitative Real-Time PCR (q-PCR) provides rapid, sensitive, and specific quantification of target organisms based on their DNA, even those that are unculturable [66]. Peptide Nucleic Acid Fluorescence In Situ Hybridization (PNA-FISH) allows for the specific visualization, identification, and spatial localization of species within the intact biofilm architecture, providing insights into community organization [66].
Discrepancies and Method Validation
Discrepancies often arise between data from different quantification methods, highlighting the effect of biofilm heterogeneity on reliability [66]. For instance, q-PCR and PNA-FISH can yield bacterial counts up to 4 log₁₀ cells/cm² higher than culture-based methods, particularly in triple-species consortia and antibiotic-stressed biofilms, due to the detection of non-viable or VBNC cells [66]. Furthermore, technical limitations like primer specificity and efficiency in q-PCR can affect accurate quantification of all members, as seen with difficulties in assessing Dolosigranulum pigrum [66]. Therefore, validating each method with target species in both planktonic and biofilm states is essential before application to complex experimental consortia [66]. Relying on at least two, and preferably three, complementary quantification techniques is recommended for a comprehensive and reliable analysis of polymicrobial biofilm communities [66].
Table 2: Comparison of Biofilm Quantification Techniques
| Technique | Principle | Key Advantages | Key Limitations | Ideal for Quantifying |
|---|---|---|---|---|
| Plate Count (Culture) | Growth of viable cells on solid media [66] | Confirms cell viability; allows further analysis of isolates | Misses VBNC/fastidious organisms; slow (24-48h) [66] [65] | Total viable cells (CFUs) |
| q-PCR | Amplification and detection of species-specific DNA sequences [66] | High sensitivity and specificity; rapid; quantitative | Does not distinguish live/dead cells; requires DNA extraction [66] | Total cells of a specific target (viable and non-viable) |
| PNA-FISH | Fluorescent probes hybridize to species-specific rRNA [66] | Provides spatial distribution data; visual confirmation | Semi-quantitative at best; requires specialized microscopy [66] | Spatial localization and relative abundance in situ |
Detailed Experimental Protocol: Quantifying Species in a CF-Relevant Polymicrobial Biofilm
This protocol, adapted from a published study, details the steps for growing and quantifying a defined triple-species biofilm relevant to cystic fibrosis, using P. aeruginosa, I. limosus, and D. pigrum [66].
Phase 1: Preparation of Inoculum and Biofilm Growth
- Culture Pure Strains: Individually grow P. aeruginosa, I. limosus, and D. pigrum to mid-log phase in appropriate liquid media (e.g., Tryptic Soy Broth).
- Standardize Suspensions: Adjust the optical density of each culture to obtain standardized cell suspensions (e.g., ~10⁷ CFU/mL).
- Prepare Consortium: Combine equal volumes of each standardized suspension to create the polymicrobial inoculum.
- Grow Biofilms: Transfer sterile substrates (e.g., AMB Media carriers or peg lids) to a plate containing the mixed inoculum. Incubate under desired conditions (Aerobic, Microaerophilic, Anaerobic) for 24-48 hours at 37°C to allow biofilm formation [66].
Phase 2: Parallel Biofilm Quantification
- Harvest Biofilms: Remove substrates from the growth plate and gently rinse with sterile saline to remove non-adherent planktonic cells.
- Process for Analysis: Process the biofilm from multiple substrates for each analytical technique:
- For Plate Count: Dislodge biofilm from 3 substrates via sonication or vortexing into saline. Serially dilute the suspension and plate onto non-specific (TSA) and specific media (PIA for P. aeruginosa, supplemented BCSA for I. limosus). Incubate and count colonies after 24-48 hours. D. pigrum counts are estimated by difference between TSA and selective media counts [66].
- For q-PCR: Dislodge biofilm from 3 substrates into a DNAse/RNAse-free tube. Extract total genomic DNA using a commercial kit. Perform q-PCR with validated, species-specific primers (e.g., targeting the 16S rRNA gene) for each organism. Use standard curves of known cell concentrations for absolute quantification [66].
- For PNA-FISH: Fix biofilm on 3 substrates with 4% paraformaldehyde. Hybridize with species-specific PNA probes (e.g., Paer565 for P. aeruginosa, Ilim569 for I. limosus) conjugated to different fluorophores. Counterstain with DAPI. Image using epifluorescence or confocal microscopy. Quantify cells per unit area using image analysis software [66].
Phase 3: Data Analysis and Validation
- Calculate Loads: Express results as Log₁₀(CFU/cm²) for culture, Log₁₀(cells/cm²) for q-PCR, and cells/cm² for PNA-FISH.
- Compare and Contrast: Analyze data for discrepancies between methods, which can indicate the presence of VBNC states, DNA from dead cells, or methodological limitations [66].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Materials for Polymicrobial Biofilm Research
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| Specific Culture Media | Selective isolation and enumeration of target species from a consortium [66] | PIA for P. aeruginosa; supplemented BCSA for I. limosus [66] |
| Species-Specific PNA Probes | Fluorescently-labeled probes for precise identification and spatial localization in biofilms via FISH [66] | Paer565 probe for P. aeruginosa; Ilim569 probe for I. limosus [66] |
| Validated q-PCR Primers | Oligonucleotides for absolute quantification of target species DNA with high specificity [66] | Primers targeting 16S rRNA genes of P. aeruginosa, I. limosus, and D. pigrum [66] |
| Biofilm Growth Carriers | Provides high-surface-area, inert substrates for robust and reproducible biofilm growth [66] | AMB Media carriers used to grow triple-species CF biofilms [66] |
Future Perspectives and Concluding Remarks
The future of polymicrobial biofilm research lies in moving beyond descriptive studies to predictive modeling and precision targeting. Advanced 'omics' approaches (genomics, transcriptomics, proteomics, metabolomics) are crucial for elucidating the complex molecular dialogues that underpin interkingdom interactions and multi-layered resistance mechanisms [64]. The integration of artificial intelligence and machine learning (AI/ML) tools offers transformative potential, enabling the analysis of vast, multi-parametric datasets to predict community behavior and identify novel therapeutic vulnerabilities [64]. Furthermore, gene-editing technologies like CRISPR-Cas will allow for functional validation of specific genes within a mixed-species context, moving from correlation to causation in understanding polymicrobial dynamics [64].
The challenges of studying these complex communities are significant, but so is the payoff. A comprehensive understanding of polymicrobial biofilms, gained through the integrated use of validated quantitative techniques and advanced models, is the key to developing next-generation diagnostics and anti-biofilm therapies. By framing this research within the fundamental mechanisms of bacterial adhesion and biofilm initiation, scientists can deconstruct the very foundations of these resilient consortia, paving the way for breakthroughs in managing chronic infections and combating antimicrobial resistance.
Combating Colonization: Therapeutic Strategies to Inhibit Adhesion and Disrupt Biofilms
Surface-mediated pathogen transmission remains a critical vector for infectious disease, particularly in healthcare settings where biofilms on medical devices significantly enhance microbial tolerance to conventional antibiotics [67] [68]. This escalating challenge, compounded by the global emergence of antimicrobial resistance (AMR), has catalyzed the search for antibiotic-free antibacterial strategies that employ mechanisms distinct from traditional biocides [68]. Bioinspired nano- and micro-structured surfaces (NMSS) represent a revolutionary approach in this domain, offering non-leaching, physics-driven antifouling and mechano-bactericidal solutions [69]. These surfaces mitigate the risk of AMR by operating through physical principles that bacteria cannot develop resistance against, aiming to reduce reliance on toxic compounds that cause ecological harm [70]. Framed within broader research on bacterial adhesion and biofilm initiation, this technical guide examines the fundamental mechanisms, design parameters, and experimental methodologies underlying these advanced material solutions, providing researchers and drug development professionals with a comprehensive framework for developing next-generation antimicrobial surfaces.
Fundamentals of Bacterial Adhesion and Biofilm Initiation
The adhesion of contaminants to surfaces follows characteristic spatial and temporal sequences, beginning with the rapid conditioning of the interface by organic macromolecules which subsequently facilitates microbial attachment [70]. In marine biofouling, for instance, surfaces are initially coated by proteins and polysaccharides that create a favorable microenvironment for bacterial colonization [70]. These pioneer microorganisms then secrete abundant metabolic products that further alter surface morphology and chemistry, providing enhanced nutrient availability and adhesion sites for secondary colonizers [70].
The initial interaction between microbial cells and material interfaces is governed by a complex interplay of chemical, physical, and mechanical factors. Chemical interactions include covalent bonds, ionic bonds, and coordination chemistry, such as the stable binding of thiol-rich cysteine residues in proteins to gold surfaces through gold-thiol bonds [70]. Physical adsorption mechanisms encompass hydrogen bonding, van der Waals forces, and hydrophobic interactions, where lipids and organic compounds first displace interfacial water through hydrophobic effects before forming more permanent attachments via van der Waals forces [70]. Mechanical interlocking further contributes to adhesion, particularly in porous materials like filtration membranes where particulate matter becomes trapped within surface imperfections [70].
In medical contexts, thrombus formation on implanted devices exemplifies a sophisticated adhesion cascade. This process initiates with prothrombin activation by calcium ions, converting fibrinogen to insoluble fibrin polymers that form an interwoven network, increasing blood viscosity and promoting platelet adhesion through mediators like glycoprotein IB-IX-V, P-selectin, and von Willebrand factor (vWF) [70]. Understanding these dynamic interaction behaviors provides the fundamental basis for developing effective antifouling strategies that interrupt critical adhesion pathways.
Bioinspired Antifouling and Bactericidal Mechanisms
Primary Anti-Adhesion Strategies
Natural surfaces have evolved sophisticated mechanisms to resist fouling, providing valuable design principles for engineered solutions. Current bioinspired antifouling strategies primarily operate through four distinct mechanisms:
- Surface Energy Control: The relationship between surface free energy and contaminant adhesion is well-established, with the famous Baier curve demonstrating a defined correlation that guides optimal surface energy selection for minimal adhesion [70].
- Extreme Wettability: Creating superhydrophilic or superhydrophobic surfaces significantly reduces the available contact area for microbial attachment, with superhydrophobic surfaces mimicking the lotus leaf effect where air pockets prevent direct contact with contaminants [70] [71].
- Topographical Patterning: Introducing specific microstructures on surfaces creates physical barriers that either prevent adhesion or mechanically disrupt attached cells, taking inspiration from natural surfaces like shark skin that reduces microbial colonization through intricate ridge patterns [70] [68].
- Liquid-Infused Slippery Surfaces: Constructing superslippery surfaces through chemical modification or liquid infusion creates low-friction interfaces that facilitate the easy removal of attached contaminants before they form permanent bonds [70].
Mechano-Bactericidal Nanostructures
Bioinspired micro-/nanostructures that mimic natural surface patterns found on insects and plants can achieve bacterial inactivation through physical means alone [68]. These nanostructured surfaces impose mechanical stresses on bacterial membranes upon contact, leading to cell deformation and eventual lysis [69]. The bactericidal and antifouling efficacy depends critically on specific topographic parameters including pillar height, tip radius, spacing, substrate stiffness, and wettability [69].
Dense high-aspect-ratio nanostructures are particularly effective, as they minimize real contact area with approaching microorganisms while simultaneously increasing local shear forces and, upon sufficient penetration, inducing catastrophic membrane deformation [69]. For instance, nanostructures inspired by cicada wings exhibit remarkable bactericidal properties through this physical mechanism without releasing any biocidal chemicals [72] [68]. The mechano-bactericidal action occurs when bacterial cells attempt to adhere to the surface and their membranes stretch between nanofeatures, eventually reaching critical strain levels that cause rupture and cell death.
Dual-Functional and Hybrid Approaches
Recent innovations have focused on dual-functional surface coatings that integrate multiple antimicrobial and antibiofouling mechanisms to provide continuous protection against microbial contamination [67]. These advanced systems leverage combinations of physicochemical repulsion, contact-active biocides, controlled-release systems, and stimuli-responsive architectures to tackle both microbial adhesion and survival [67].
Hybrid strategies that combine topographic features with benign chemistries such as zwitterions, photocatalysts, and stimuli-responsive actuation demonstrate enhanced robustness under challenging conditions including protein conditioning and mixed-species biofilms [69]. Smart coatings that respond to environmental stimuli like pH changes, enzymatic activity, or radiation offer dynamic antibacterial properties that activate only when needed, reducing ecological impact while maintaining long-term efficacy [67].
Critical Design Parameters and Performance Data
Nanostructure Geometry Optimization
The antibacterial efficacy of nanostructured surfaces is governed by specific geometric parameters that must be carefully optimized for target microorganisms. Research has established clear correlations between structural dimensions and antimicrobial performance.
Table 1: Impact of Nanostructure Geometry on Bactericidal Efficacy
| Parameter | Effect on Antibacterial Activity | Optimal Range | Mechanistic Insight |
|---|---|---|---|
| Pillar Spacing | Determines contact area with bacterial membrane; influences strain distribution | 130-250 nm [69] | Must be smaller than target microorganisms to prevent full contact avoidance |
| Pillar Height | Affects penetration depth and mechanical stability | >500 nm [69] | Sufficient height prevents cells from contacting the substrate between features |
| Tip Radius | Influences local stress concentration on bacterial membrane | Sharp tips (<20 nm radius) [69] | Smaller radii create higher stress concentrations, promoting membrane penetration |
| Aspect Ratio | Balances mechanical robustness with bending capability | High (>5:1) [69] | Higher ratios increase flexibility but may compromise mechanical integrity |
Surface roughness, even at the nanoscale, exerts profound influence on microbial adhesion. Studies investigating bacterial adhesion patterns on hydrophobic surfaces with controlled roughness gradients (2-390 nm) revealed that contamination levels could vary by up to 75-fold depending solely on surface roughness [71]. Furthermore, research on the relationship between microorganism adhesion strength and nanopillar diameter demonstrates an inverse correlation, where adhesion strength decreases as nanopillar diameter increases [71].
Performance Comparison of Antimicrobial Strategies
Different antimicrobial approaches present distinct advantages and limitations, which must be considered when designing surfaces for specific applications.
Table 2: Comparative Analysis of Antimicrobial Surface Strategies
| Strategy Type | Mechanism of Action | Efficacy Duration | Advantages | Limitations |
|---|---|---|---|---|
| Nanotopography | Physical membrane disruption [69] [68] | Long-term (non-depleting) [69] | No chemical release; reduced AMR risk [68] | Geometry-specific; complex fabrication |
| Metal Ion Release | Chemical disruption of cellular processes [71] | Short-term (7 days to 6 weeks) [71] | Broad-spectrum activity | Limited duration; potential cytotoxicity |
| Cationic Polymers | Electrostatic membrane disruption [71] | Medium-term | Contact-based; non-release | Affected by environmental conditions |
| Stimuli-Responsive | Activated by pH, enzymes, or radiation [67] [71] | On-demand activation | Extended functional lifetime; smart response | Complex material design |
The integration of multiple mechanisms often yields superior performance. For example, hydrogel systems incorporating photothermal polydopamine and Mg²⁺ rapidly heat up (>20°C) under laser irradiation, causing simultaneous bacterial membrane rupture and protein denaturation [71]. Similarly, hydrogels with integrated black phosphorus nanosheets generate reactive oxygen species (ROS) upon light exposure, providing dual-mode antibacterial action [71].
Experimental Methodologies and Characterization Protocols
Nanostructured Surface Fabrication
Protocol 1: Bioinspired Nanostructure Replication via Soft Lithography
This protocol enables the faithful reproduction of natural nanotopographies (e.g., cicada wings, lotus leaves) onto polymer substrates:
- Template Preparation: Select natural templates (cicada wings, dragonfly wings, shark skin) or synthetic masters with desired nanotopographies. Clean templates sequentially with acetone, isopropanol, and deionized water in an ultrasonic bath for 10 minutes each [72] [68].
- Replica Molding: Prepare polydimethylsiloxane (PDMS) precursor (base:curing agent = 10:1 w/w). Degas under vacuum until bubbles dissipate. Pour over template and cure at 65°C for 4 hours [72].
- Negative Release: Carefully peel cured PDMS negative replica from template. Functionalize with trichloro(1H,1H,2H,2H-perfluorooctyl)silane via vapor deposition (100 µL, 30 minutes) to facilitate subsequent release [68].
- Positive Replication: Cast polyurethane or epoxy resin against PDMS negative. Cure according to manufacturer specifications (typically 24 hours at room temperature or 1 hour at 80°C) [72].
- Quality Control: Verify nanostructure fidelity using scanning electron microscopy (SEM) at 10-50 kV accelerating voltage after sputter-coating with 5-10 nm gold/palladium layer [68].
Protocol 2: Plasma-Enhanced Superhydrophobic Coating
A one-step surface modification technique for creating superhydrophobic coatings on biomedical implants:
- Substrate Preparation: Clean titanium substrates (10×10×1 mm) sequentially with acetone, ethanol, and deionized water in an ultrasonic bath (15 minutes each). Dry under nitrogen stream [72].
- Plasma Treatment: Place samples in low-pressure plasma chamber with fluorocarbon precursor (C₄F₈). Evacuate to 10-20 mTorr base pressure [72].
- Coating Deposition: Initiate plasma at 50-100 W for 5-30 minutes. Monitor coating thickness in situ with spectroscopic ellipsometry [72].
- Characterization: Measure water contact angles (>150°) and roll-off angles (<10°) using goniometry. Verify coating uniformity with atomic force microscopy (AFM) [72] [71].
Antibacterial Efficacy Assessment
Protocol 3: Quantitative Bacterial Adhesion and Viability Assay
Standardized methodology for evaluating antifouling and bactericidal performance:
- Bacterial Culture: Grow model organisms (Staphylococcus aureus ATCC 25923, Escherichia coli ATCC 25922) to mid-log phase (OD₆₀₀ = 0.4-0.6) in appropriate broth [68] [71].
- Surface Inoculation: Dilute bacterial suspension to 10⁶ CFU/mL in PBS or growth medium. Apply 100 µL aliquot to test surfaces (10×10 mm) in 24-well plate. Incubate at 37°C for 2 hours (adhesion phase) or 24 hours (biofilm formation) [68].
- Viability Quantification:
- Live/Dead Staining: Incubate with SYTO 9 (3.34 µM) and propidium iodide (20 µM) for 15 minutes in dark. Image with epifluorescence microscopy (5 random fields per sample) [68].
- CFU Enumeration: Gently rinse non-adhered cells with PBS. Detach adhered bacteria by sonication (5 minutes, 40 kHz) in 1 mL PBS. Serial dilute and plate on agar. Count colonies after 24-hour incubation [71].
- Morphological Analysis: Fix adhered bacteria with 2.5% glutaraldehyde for 2 hours. Dehydrate through ethanol series (30%, 50%, 70%, 90%, 100%), critical point dry, and image via SEM to assess membrane damage [68].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful development and evaluation of anti-fouling nanostructured materials requires specialized reagents and instrumentation. The following table compiles essential research tools referenced in experimental protocols.
Table 3: Research Reagent Solutions for Anti-fouling Material Development
| Category/Item | Function/Application | Representative Examples | Key Considerations |
|---|---|---|---|
| Natural Templates | Source of bioinspired nanotopographies | Cicada wings, dragonfly wings, lotus leaves [72] [68] | Structure fidelity preservation during replication |
| Polymer Substrates | Matrix for nanostructure fabrication | PDMS, polyurethane, epoxy resins [72] | Biocompatibility, mechanical properties, replication resolution |
| Surface Modifiers | Alter surface energy and chemistry | Trichloro(1H,1H,2H,2H-perfluorooctyl)silane [72] | Coating uniformity, durability, environmental stability |
| Antimicrobial Agents | Provide chemical bactericidal activity | Silver nanoparticles, chitosan, quaternary ammonium salts [71] | Release kinetics, cytotoxicity, spectrum of activity |
| Characterization Standards | Validate surface properties | Contact angle goniometry, SEM, AFM [72] [71] | Measurement standardization, instrument calibration |
| Biological Assay Components | Evaluate antibacterial efficacy | SYTO 9, propidium iodide, culture media [68] [71] | Stain stability, bacterial strain selection, incubation conditions |
Advanced material systems increasingly incorporate stimuli-responsive components such as polydopamine for photothermal activation, black phosphorus nanosheets as photosensitizers, and stanene nanosheets for ultrasound-triggered reactive oxygen species generation [71]. These smart materials represent the cutting edge of antimicrobial surface design, offering dynamic functionality that activates only in response to specific environmental triggers.
Future Perspectives and Translational Challenges
The continued advancement of anti-fouling nanostructured materials hinges on addressing several persistent challenges in balancing antimicrobial efficiency, surface stability, and ecological safety [67]. Future research directions should prioritize the development of multiscale models that link nanoscale membrane stresses to mesoscale transport phenomena and critical shear thresholds, enabling geometry selection specifically tailored to application-specific flow regimes and microbial types [69].
Machine-learning optimization represents a particularly promising pathway for accelerating the design of next-generation antifouling surfaces, allowing researchers to navigate the complex parameter space of topographic features, material properties, and environmental conditions more efficiently than traditional trial-and-error approaches [69]. Additionally, sensor-integrated maintenance systems coupled with bioinspired nanostructures can deliver scalable, lower-impact antifouling solutions capable of real-time performance monitoring and targeted intervention [69].
Translational pathways must address durability concerns under real-world conditions, where protein conditioning and mixed-species biofilms present significantly greater challenges than simplified laboratory models [69] [67]. Hybrid strategies that combine optimized nanotopography with benign chemistries such as zwitterions or photocatalysts demonstrate particular promise for enhanced robustness in these complex environments while reducing ecological load compared to conventional biocidal approaches [69]. As research in this field progresses, the establishment of standardized testing protocols and environmental impact assessments will be crucial for facilitating the transition from laboratory innovation to practical implementation across healthcare, industrial, and environmental applications.
The global rise of antibiotic resistance (AR) presents a critical threat to public health, prompting a paradigm shift in antimicrobial strategy development [73] [74]. Within this landscape, bacterial biofilms have emerged as a fundamental contributor to treatment failure, responsible for an estimated 65% of all bacterial infections and approximately 80% of all chronic infections [32]. Biofilms are complex, surface-associated microbial communities encased in an extracellular polymeric substance (EPS) matrix that confers intrinsic tolerance to antimicrobials and host immune responses [4] [3]. This review explores the strategic disruption of quorum sensing (QS)—the bacterial communication system governing virulence, biofilm formation, and resistance—as a transformative approach to combat biofilm-associated infections.
The QS system enables bacteria to coordinate population-wide behaviors through the synthesis, secretion, and detection of small signaling molecules called autoinducers [74]. When a critical threshold concentration of these molecules is reached, they trigger the expression of genes responsible for virulence factor production, biofilm maturation, and antibiotic tolerance [75]. Unlike conventional antibiotics that exert lethal selective pressure, quorum sensing inhibitors (QSIs) function by silencing bacterial communication rather than inhibiting growth, thereby attenuating pathogenicity without promoting resistance development [73] [74]. This non-lethal mode of action positions QS disruption as a promising therapeutic intervention within the broader context of bacterial adhesion and biofilm initiation research.
The Intricate Link Between Quorum Sensing and Biofilm Development
Molecular Mechanisms of Quorum Sensing
Bacteria employ sophisticated QS systems that vary between Gram-positive and Gram-negative species but follow a conserved principle: density-dependent gene regulation through diffusible signaling molecules [74]. In Gram-negative bacteria, such as Pseudomonas aeruginosa, QS is primarily mediated by acyl-homoserine lactones (AHLs) as autoinducers [74] [75]. These systems typically consist of signal synthases (LuxI homologs) that produce AHLs and cytoplasmic receptor-transcriptional activators (LuxR homologs) that detect them [74]. When AHLs accumulate to a threshold concentration, they bind their cognate LuxR-type receptors, forming complexes that activate transcription of QS-regulated genes [75].
P. aeruginosa possesses one of the most complex and well-studied QS systems, comprising three interconnected pathways—Las, Rhl, and Pqs—that establish a hierarchical regulatory network controlling more than 10% of its genome and approximately 20% of its proteome [75]. The Las system occupies the apex of this hierarchy, with LasI producing 3-oxo-C12-HSL that activates LasR to regulate genes associated with biofilm formation and virulence factors including hemolysins, proteases, elastase, and exotoxin-A [75]. The Rhl system, regulated by RhlI-produced C4-HSL binding to RhlR, activates expression of genes responsible for pyocyanin, hydrocyanic acid, siderophore, elastase, and alkaline protease production, while also regulating bacterial motility [75]. The Pqs system utilizes non-AHL signaling molecules called alkyl-4-quinolones, with 2-heptyl-3-hydroxy-4-quinolone (PQS) regulating genes involved in biofilm formation, swimming motility, and production of pyocyanin, proteases, elastases, rhamnolipids, and siderophores [75].
In Gram-positive bacteria, such as Staphylococcus aureus, QS is primarily mediated by autoinducing peptides (AIPs) [74]. These peptides are synthesized as precursor molecules and processed into active signaling molecules that are detected by two-component signal transduction systems consisting of membrane-bound histidine kinase receptors and response regulators that modulate gene expression [74]. Additionally, autoinducer-2 (AI-2), synthesized by the enzyme LuxS, serves as a universal QS signal facilitating interspecies communication among diverse bacterial species within microbial communities [74].
Quorum Sensing Directs Biofilm Formation
The biofilm lifecycle progresses through distinct stages—initial attachment, irreversible attachment, microcolony formation, maturation, and dispersion—with QS playing a critical regulatory role at multiple points in this process [32] [4]. During the maturation phase, QS systems coordinate the production of extracellular polymeric substances that form the protective biofilm matrix, including polysaccharides, lipids, proteins, and extracellular DNA (eDNA) [4]. In the final dispersion phase, QS triggers the release of bacterial cells from established biofilms to colonize new niches [4].
QS further enhances biofilm-mediated antibiotic resistance through multiple mechanisms. It regulates the expression of multidrug efflux pumps, such as MexAB-OprM in P. aeruginosa, which actively expel antibiotics from bacterial cells [74]. QS also triggers global stress response pathways, including oxidative stress and SOS responses, that help bacteria withstand hostile conditions and enter transiently tolerant states [74]. Additionally, QS facilitates horizontal gene transfer (HGT) of resistance determinants through conjugation and plasmid mobilization, especially within polymicrobial biofilm communities [74]. The biofilm matrix itself acts as a physical barrier that hinders antibiotic penetration through binding or enzymatic degradation of antimicrobial compounds [4].
Diagram Title: QS Regulatory Pathway in Gram-Negative Bacteria
Natural Product-Based Quorum Sensing Inhibitors
Natural products have emerged as a rich source of structurally diverse QSIs with potent anti-biofilm activities. These compounds can be broadly categorized based on their origins:
Plant-derived phytochemicals represent a major class of QSIs, with compounds such as flavonoids, phenolic acids, and alkaloids demonstrating significant efficacy against QS systems in pathogenic bacteria [73] [74]. For instance, galloylquinic acid compounds (GQAs) extracted from Copaifera lucens leaves have shown remarkable anti-QS and anti-biofilm activities against clinical isolates of multidrug-resistant P. aeruginosa [76] [77]. Other plant-derived QSIs include trans-cinnamaldehyde, salicylic acid, cinnamic acid, and 2′,4′-dihydroxy chalcone isolated from Oxytropis falcata [75].
Microbial secondary metabolites constitute another important category of QSIs, with various bacterial and fungal species producing compounds that interfere with QS pathways of competing microorganisms [73] [74]. For example, Mycoleptodiscus indicus PUTY1 has demonstrated QS inhibitory capability against P. aeruginosa [75].
Marine bioactive compounds represent an emerging source of novel QSIs, with marine organisms producing unique chemical structures not found in terrestrial ecosystems [73] [74] [78]. Marine invertebrates, including corals, sea anemones, and holothurians, have been found to harbor bacteria capable of producing QSIs or quorum quenching (QQ) molecules as defensive agents against pathogen colonization [78].
Mechanisms of Action
Natural QSIs disrupt bacterial communication through multiple molecular mechanisms, providing multi-faceted approaches to biofilm control:
Signal receptor antagonism: Many QSIs function as competitive inhibitors that bind to LuxR-type receptors without activating them, thereby blocking native AHL signaling [73] [74]. This molecular mimicry prevents the formation of functional AHL-receptor complexes necessary for transcription of QS-regulated genes.
Inhibition of signal synthesis: Certain compounds directly interfere with the enzymatic activity of AHL synthases (LuxI homologs), reducing the production of QS signaling molecules [73] [74].
Enzymatic degradation of signaling molecules: Known as quorum quenching (QQ), this approach utilizes enzymes such as lactonases, acylases, and oxidoreductases to degrade or modify AHLs, rendering them inactive [32] [78]. Both prokaryotic and eukaryotic organisms produce QQ enzymes as a natural defense mechanism against QS-dependent pathogens.
Suppression of QS-regulated gene expression: Some QSIs interfere with downstream signaling components or directly suppress the expression of QS-regulated genes without directly targeting signal synthesis or reception [73] [76].
Table 1: Representative Natural Quorum Sensing Inhibitors and Their Activities
| Compound/Source | Chemical Class | Target Bacteria | Mechanism of Action | Key Findings |
|---|---|---|---|---|
| Galloylquinic acid compounds (GQAs) from Copaifera lucens | Galloylquinic acids | P. aeruginosa | Downregulation of lasI, lasR, pqsA, pqsR genes; reduced pyocyanin and rhamnolipid production | 89% reduction in QS gene expression; 62.5% reduction in biofilm thickness at 128 µg/mL [76] [77] |
| Flavonoids from various plants | Flavonoids | P. aeruginosa | Inhibition of AHL-mediated virulence factors; competitive receptor binding | Reduced pyocyanin production, elastase activity, and biofilm formation [75] |
| Trans-cinnamaldehyde | Phenylpropanoid | P. aeruginosa | Interference with AI-2 based QS; reduced virulence gene expression | Inhibition of biofilm formation and virulence factor production [75] |
| Microbial secondary metabolites | Varied | Multiple species | Signal degradation; receptor antagonism | Species-specific QS inhibition through multiple mechanisms [73] [74] |
| Marine organism extracts | Varied | Marine and pathogenic bacteria | Signal interference; enzymatic degradation | Protection of hosts from QS-dependent pathogens [78] |
Experimental Approaches for Evaluating QSIs
Standardized Methodologies for Anti-Biofilm Assessment
Robust evaluation of potential QSIs requires integrated approaches assessing both anti-biofilm efficacy and QS disruption:
Antibacterial susceptibility testing establishes baseline antimicrobial activity using standard broth microdilution methods to determine minimum inhibitory concentrations (MIC) and minimum bactericidal concentrations (MBC) [76] [77]. For GQAs against MDR P. aeruginosa, MIC values ranged from 1-4 µg/mL while MBC values were 2-16 µg/mL [76] [77].
Anti-biofilm activity quantification employs specialized parameters including minimum biofilm inhibitory concentration (MBIC) and minimum biofilm eradication concentration (MBEC) [76]. For GQAs, MBIC80 and MBEC80 values were 64 µg/mL and 128 µg/mL respectively, indicating potent activity against pre-formed biofilms [76] [77].
Advanced imaging techniques provide visual confirmation of biofilm disruption. Confocal laser scanning microscopy (CLSM) with fluorescence staining enables quantification of biofilm thickness and viability, while scanning electron microscopy (SEM) reveals structural alterations in biofilm architecture [76]. GQA treatment at 128 µg/mL reduced biofilm thickness by 62.5% and severely compromised matrix integrity [76].
Virulence factor assays measure production of QS-regulated factors including pyocyanin, rhamnolipids, elastase, and proteases [76] [75]. GQAs at 128 µg/mL significantly reduced rhamnolipid and pyocyanin production while inhibiting bacterial motility [76].
Gene expression analysis using quantitative real-time PCR assesses downregulation of key QS genes (lasI, lasR, rhlI, rhlR, pqsA, pqsR) following QSI treatment [76]. GQAs achieved 89% downregulation of las and pqs system genes [76].
Diagram Title: QSI Bioactivity Assessment Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for QSI and Anti-Biofilm Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| Bacterial Strains | P. aeruginosa PAO1 (reference), clinical MDR isolates, S. aureus strains, AI-2 reporter strains (e.g., V. harveyi BB170) | Target organisms for QSI screening; interspecies QS studies | Clinical isolates should include MDR phenotypes; quality control strains essential for standardization [76] [77] |
| QS Signaling Molecules | Synthetic AHLs (3-oxo-C12-HSL, C4-HSL), PQS, AI-2 | Positive controls; competitive inhibition studies; QS activation assays | Commercially available; concentration-dependent responses must be established [74] [75] |
| Biofilm Growth Systems | Calgary biofilm device, flow-cell systems, microtiter plates with various surfaces (polystyrene, glass) | Biofilm cultivation under static or dynamic conditions | Surface material significantly influences biofilm architecture and QSI susceptibility [4] |
| Viability Stains | SYTO9/propidium iodide (LIVE/DEAD BacLight), resazurin, CTC-DAPI | Differentiation of live/dead cells; metabolic activity assessment in biofilms | Confocal microscopy with LIVE/DEAD staining enables 3D reconstruction of biofilm viability [76] |
| Molecular Biology Reagents | qPCR primers for QS genes (lasI, lasR, rhlI, rhlR, pqsA, pqsR), RNA extraction kits, reverse transcriptase | Gene expression analysis of QS pathways | Critical for confirming mechanism of action at transcriptional level [76] |
| Virulence Assay Components | Pyocyanin extraction solvents, elastase substrates (elastin-Congo red), rhamnolipid quantification reagents | Quantification of QS-regulated virulence factors | Standardized protocols essential for cross-study comparisons [76] [75] |
Synergistic Approaches and Clinical Translation
Enhancing Conventional Antibiotic Efficacy
QSIs demonstrate remarkable potential to enhance the efficacy of conventional antibiotics through synergistic combinations. This approach allows for significant reduction of antibiotic concentrations while maintaining or improving therapeutic outcomes, potentially overcoming biofilm-mediated resistance [73] [74]. The mechanisms underlying these synergistic effects include:
- Enhanced biofilm penetration: QSIs that disrupt matrix integrity can improve antibiotic access to embedded bacterial cells [74].
- Reduced virulence expression: By attenuating virulence factor production, QSIs may facilitate improved immune clearance of infections [75].
- Suppression of resistance mechanisms: QSIs can downregulate multidrug efflux pumps and other QS-controlled resistance elements [74].
- Prevention of persister cell formation: QS disruption may reduce the formation of dormant, antibiotic-tolerant persister cells within biofilms [4].
Natural QSIs like GQAs have demonstrated β-lactamase inhibitory activity, enhancing the efficacy of β-lactam antibiotics against resistant Escherichia coli strains expressing CTX-M-15 and KPC-2 β-lactamases [77]. The combination of GQAs with β-lactams resulted in a 64-512-fold reduction in MIC values, restoring susceptibility to these antibiotics [77].
Overcoming Translational Challenges
Despite promising preclinical results, several challenges impede the clinical translation of QSIs:
Bioavailability limitations plague many natural QSIs, necessitating advanced delivery strategies [73] [74]. Nanoparticle-based delivery systems offer promising solutions by improving solubility, stability, and targeted delivery of QSIs to infection sites [73] [74]. These platforms can be engineered for controlled release, enhancing therapeutic efficacy while reducing dosing frequency.
Bacterial adaptability remains a concern, as pathogens may evolve countermeasures against QS disruption [73]. While QSIs theoretically exert reduced selective pressure for resistance compared to bactericidal agents, compensatory mutations or efflux pump upregulation could potentially diminish efficacy over time [75]. Combination approaches utilizing multiple QSIs targeting different components of QS circuitry may mitigate this risk [75].
Regulatory barriers for anti-virulence agents present unique challenges, as traditional antibiotic efficacy endpoints may not fully capture the therapeutic benefits of QS disruption [73] [74]. Developing validated biomarkers and clinical endpoints specific to anti-virulence mechanisms will be essential for regulatory approval [73].
Quorum sensing inhibition represents a transformative approach in the ongoing battle against antibiotic resistance, particularly for biofilm-associated infections that evade conventional therapies. The strategic disruption of bacterial communication offers multiple advantages over traditional antibiotics, including reduced selective pressure for resistance and preservation of the host microbiome [73] [74]. Natural products serve as an invaluable source of structurally diverse QSIs with multi-target mechanisms capable of disrupting complex QS networks in pathogens like P. aeruginosa [73] [76] [75].
Future research directions should prioritize several key areas: First, the exploration of combination therapies integrating QSIs with conventional antibiotics or other anti-biofilm agents to enhance efficacy while minimizing resistance development [73] [75]. Second, the development of advanced delivery platforms, including nanoparticle-based systems and biomaterial coatings, to overcome pharmacokinetic limitations and enable targeted delivery to biofilm infection sites [73] [74]. Third, the investigation of microbiome-specific approaches that selectively target pathogenic QS systems while preserving beneficial commensal bacteria [74]. Finally, expanded clinical validation through well-designed trials establishing standardized efficacy endpoints for QSI-based therapies [73].
As our understanding of QS circuitry and its integration with biofilm development continues to evolve, so too will opportunities for therapeutic intervention. The strategic interception of bacterial communication signals represents a promising frontier in antimicrobial research, offering new hope for addressing the escalating crisis of antibiotic resistance, particularly in the context of biofilm-mediated infections that have long evaded conventional treatment approaches.
Bacterial biofilms are structured communities of microbial cells enclosed in a self-produced extracellular polymeric substance (EPS) that adhere to both biotic and abiotic surfaces [3]. This EPS matrix, a complex mixture of polysaccharides, proteins, and extracellular DNA (eDNA), constitutes over 90% of the biofilm's dry mass and serves as a primary defense mechanism, conferring remarkable resistance to antimicrobial agents and host immune responses [79]. The resilient nature of biofilms leads to persistent infections and complicates treatment across clinical and industrial settings, contributing significantly to the global challenge of antimicrobial resistance [3].
Matrix-degrading enzymes represent a promising therapeutic strategy that directly targets the structural integrity of the EPS rather than the bacterial cells themselves [80]. By hydrolyzing specific components within the biofilm matrix, these enzymes can disrupt biofilm architecture, release embedded cells, and potentially restore susceptibility to conventional antimicrobials [79]. This enzymatic approach offers several advantages, including broad-spectrum activity against diverse bacterial pathogens, reduced selective pressure for antimicrobial resistance, and compatibility with existing treatment regimens when used in combination therapies [81] [82]. This technical guide comprehensively examines three principal classes of matrix-degrading enzymes—DNases, glycoside hydrolases, and Dispersin B—detailing their mechanisms, efficacy, and practical applications in biofilm control strategies relevant to drug development and clinical translation.
Enzyme Classes and Mechanisms of Action
DNases: Targeting Extracellular DNA (eDNA)
Deoxyribonuclease I (DNase I) enzymes specifically degrade the extracellular DNA (eDNA) component within biofilm matrices. eDNA serves as a critical structural scaffold in many bacterial biofilms, facilitating initial cell attachment, maintaining structural integrity, and contributing to antimicrobial resistance [83]. DNase I hydrolyzes the phosphodiester bonds in DNA backbones, destabilizing the biofilm architecture and increasing permeability to antimicrobial agents.
Research demonstrates that DNase I coating on titanium surfaces significantly prevents bacterial adhesion and biofilm formation by Streptococcus mutans and Staphylococcus aureus over 24 hours [83]. The immobilized enzyme creates a biofilm-resistant surface that degrades eDNA as it is secreted by attaching bacteria, interrupting the initial stages of biofilm development. This approach has shown promise for medical implants, where preventing biofilm formation is clinically more feasible than eradicating established biofilms [83].
Glycoside Hydrolases: Degrading Polysaccharide Networks
Glycoside hydrolases (GHs) comprise a diverse family of enzymes that catalyze the hydrolysis of glycosidic bonds between carbohydrate moieties in expolysaccharides. Their substrate specificity depends on the specific glycosidic linkage they target:
- α-Amylase cleaves α-1,4 glycosidic linkages in starch-like polymers [79]
- Cellulase hydrolyzes β-1,4 linkages in cellulose and related polysaccharides [79]
- Dispersin B specifically targets β-1,6-glycosidic bonds in poly-β(1,6)-N-acetyl-D-glucosamine (PNAG) [80] [84]
These enzymes disrupt the polysaccharide backbone of biofilms, leading to structural collapse and release of embedded cells. Studies demonstrate that glycoside hydrolases effectively degrade biofilms formed by diverse pathogens including Staphylococcus aureus, Pseudomonas aeruginosa, Escherichia coli O157:H7, and Listeria monocytogenes [81] [79]. The engineered glycoside hydrolase CAase has shown particularly broad activity against multiple pathogens, disrupting biofilms on various surfaces including glass, plastic, and leafy greens [82].
Dispersin B: A Specialized Glycoside Hydrolase
Dispersin B is a 40-kDa glycoside hydrolase produced by the periodontal pathogen Aggregatibacter actinomycetemcomitans [84]. This enzyme specifically hydrolyzes β-1,6-glycosidic linkages in poly-N-acetylglucosamine (PNAG/PGA), a key polysaccharide component in biofilms of many Gram-positive and Gram-negative pathogens including Staphylococcus species and Escherichia coli [80] [84].
Structurally, Dispersin B features a TIM barrel fold with a large active site cavity containing three conserved catalytic residues: Asp183, Glu184, and Glu332 [84]. The enzyme employs a substrate-assisted catalytic mechanism where the N-acetyl group of the substrate participates in cleavage, resulting in hydrolysis with retention of anomeric configuration [84]. Dispersin B has demonstrated remarkable broad-spectrum activity, inhibiting biofilm formation, detaching preformed biofilms, disaggregating bacterial flocs, and sensitizing biofilms to various antimicrobial agents and host defenses [80].
Table 1: Key Characteristics of Major Matrix-Degrading Enzyme Classes
| Enzyme Class | Target Substrate | Mechanism of Action | Primary Bacterial Targets |
|---|---|---|---|
| DNases | Extracellular DNA (eDNA) | Hydrolysis of phosphodiester bonds in DNA backbone | S. aureus, S. mutans, P. aeruginosa |
| Glycoside Hydrolases | Exopolysaccharides | Cleavage of specific glycosidic linkages | Broad-spectrum activity |
| ∟ α-Amylase | α-1,4 glycosidic linkages | Hydrolysis of starch-like polymers | S. aureus, P. aeruginosa, V. cholerae |
| ∟ Cellulase | β-1,4 glycosidic linkages | Hydrolysis of cellulose polymers | B. cepacia, P. aeruginosa |
| ∟ Dispersin B | β-1,6-N-acetylglucosamine | Hydrolysis of PNAG/PGA polysaccharide | Staphylococcus spp., E. coli, A. actinomycetemcomitans |
Quantitative Efficacy Data
Enzyme Monotherapy Efficacy
Matrix-degrading enzymes demonstrate significant efficacy in biofilm disruption across multiple experimental models. Glycoside hydrolase treatment with α-amylase and cellulase (0.25% solution, 30 minutes) resulted in substantial biomass reduction in S. aureus and P. aeruginosa monoculture and coculture biofilms grown on coverslips, as quantified by crystal violet staining [79]. Importantly, efficacy was observed at concentrations as low as 0.0025% with treatment times of just 2 minutes, demonstrating potent activity at minimal concentrations.
In Burkholderia cepacia biofilms on stainless steel coupons, α-amylase treatment alone significantly reduced biofilm populations from 8.4 ± 0.2 to 6.03 ± 0.2 log₁₀ CFU/coupon, illustrating its utility against resilient environmental biofilms [85]. Similarly, the engineered glycoside hydrolase CAase effectively disrupted mature Listeria monocytogenes biofilms in laboratory assays, with visual assessment confirming extensive removal compared to untreated controls [82].
Combination Therapy Enhancement
The combination of matrix-degrading enzymes with conventional antimicrobials demonstrates enhanced efficacy through synergistic or additive interactions. In Burkholderia cepacia biofilms, enzyme-antimicrobial combinations significantly reduced the minimum inhibitory concentration against preformed biofilms (MIC-Bio):
- α-Amylase-ciprofloxacin exhibited synergistic effect (FICI = 0.50), reducing MIC-Bio to 4.0 μg/mL [85]
- Proteinase K-ciprofloxacin showed additive effect (FICI = 0.625) [85]
- α-Amylase-meropenem demonstrated additive effect (FICI = 0.750) [85]
When applied to preformed biofilms on stainless-steel coupons, the α-amylase-meropenem combination decreased biofilm populations from 7.5 ± 0.5 to 3.8 ± 1.0 log₁₀ CFU/coupon, representing a substantially greater reduction than either component alone [85]. Similar enhancement has been observed with Dispersin B, which sensitizes preformed biofilms to killing by antibiotics, antiseptics, bacteriophages, and predatory bacteria [80].
Table 2: Quantitative Efficacy of Matrix-Degrading Enzymes Against Bacterial Biofilms
| Enzyme Treatment | Pathogen | Biofilm Surface | Efficacy Measurement | Result |
|---|---|---|---|---|
| α-Amylase (monotherapy) | B. cepacia | Stainless steel coupons | Log reduction (CFU/coupon) | 8.4 ± 0.2 to 6.03 ± 0.2 log₁₀ |
| α-Amylase + Ciprofloxacin | B. cepacia | Stainless steel coupons | Fractional Inhibitory Concentration Index (FICI) | 0.50 (synergistic) |
| α-Amylase + Meropenem | B. cepacia | Stainless steel coupons | Log reduction (CFU/coupon) | 7.5 ± 0.5 to 3.8 ± 1.0 log₁₀ |
| Glycoside Hydrolases (α-amylase + cellulase) | S. aureus & P. aeruginosa | Plastic coverslips | Biomass reduction (crystal violet) | Significant reduction vs. controls |
| CAase | L. monocytogenes | Glass, plastic, leafy greens | Visual biofilm disruption | Effective removal of mature biofilms |
Experimental Protocols and Methodologies
Biofilm Cultivation and Treatment
Protocol 1: Microtiter Plate Biofilm Assay for Enzymatic Susceptibility Testing
Inoculum Preparation: Grow test strain (e.g., B. cepacia ATCC 25416) in appropriate broth (modified yeast extract dextrose calcium carbonate broth) under aerobic conditions for 24 hours at 37°C with shaking at 180 rpm [85]. Dilute overnight culture to approximately 10³-10⁴ CFU/mL in fresh medium.
Biofilm Formation: Dispense 200 μL aliquots of diluted culture into sterile 96-well polystyrene plates. Incubate under static conditions for 24-48 hours at optimal growth temperature to allow biofilm development [85].
Enzyme Treatment: Prepare serial dilutions of matrix-degrading enzymes in appropriate buffers. α-Amylase, DNase I, and Proteinase K are commonly tested at concentrations ranging from 0.0025% to 5% [85] [79]. Remove planktonic cells by gentle washing with phosphate-buffered saline (PBS) before adding enzyme solutions to preformed biofilms.
Incubation and Assessment: Incubate enzyme-treated biofilms for predetermined times (30 minutes to 24 hours). Assess biofilm disruption using crystal violet staining for total biomass, resazurin reduction for metabolic activity, or colony counting for viable cells [79].
Protocol 2: Biofilm Formation on Stainless Steel Coupons
Surface Preparation: Prepare stainless steel coupons (typically 1 × 1 cm) by cleaning with detergent, rinsing with distilled water, and sterilizing by autoclaving [85].
Biofilm Development: Immerse coupons in bacterial suspension and incubate with gentle shaking (50-100 rpm) for 24-48 hours to allow biofilm formation. Nutrient-limited media often support more robust biofilm development than nutrient-rich broths [85].
Enzyme Treatment: Transfer coupons to sterile containers containing enzyme solutions. For combination therapy, include appropriate antimicrobials at sub-MIC concentrations.
Post-Treatment Analysis: After treatment, gently rinse coupons to remove non-adherent cells and disrupted biofilm. Process for enumeration (sonication and viable counting), scanning electron microscopy, or confocal laser scanning microscopy [85].
Efficacy Assessment Methods
Crystal Violet Biofilm Staining This widely used method quantifies total biofilm biomass. After enzyme treatment and fixation, stain biofilms with 0.1% crystal violet for 15 minutes. Gently wash to remove unbound dye, then solubilize bound dye with acetic acid or ethanol. Measure absorbance at 570-600 nm, with higher values indicating greater biofilm biomass [79].
Viable Cell Enumeration For quantitative assessment of biofilm disruption, viable counts provide direct measurement of antibacterial efficacy. After enzyme treatment, dislodge biofilm-associated cells by sonication or scraping. Serially dilute and plate on appropriate agar media. Incubate and enumerate colonies to calculate log₁₀ CFU/cm² or CFU/coupon [85].
Scanning Electron Microscopy (SEM) SEM provides high-resolution visualization of biofilm architecture and enzyme-induced structural alterations [86]. Fix samples with glutaraldehyde and paraformaldehyde, followed by secondary fixation with osmium tetroxide. Dehydrate through ethanol series, critical point dry, sputter coat with conductive material, and image using SEM [85] [86].
Diagram 1: Experimental workflow for evaluating matrix-degrading enzymes
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Matrix-Degrading Enzyme Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| Matrix-Degrading Enzymes | DNase I, α-Amylase, Cellulase, Proteinase K, Dispersin B | Target specific EPS components (eDNA, polysaccharides, proteins) | Test concentration range: 0.0025% - 5%; assess synergy with antimicrobials [85] [79] |
| Growth Media | Tryptone Yeast Dextrose (TYD) broth, modified Yeast Dextrose Calcium Carbonate (mYDC) broth | Support robust biofilm formation; nutrient-limited media often enhance biofilm development | TYD: nutrient-rich for planktonic growth; mYDC: nutrient-limited for robust biofilms [85] |
| Assessment Reagents | Crystal violet, Resazurin, Live/Dead staining kits | Quantify total biomass, metabolic activity, and cell viability | Crystal violet measures total biomass; resazurin indicates metabolic activity [79] |
| Surface Materials | Polystyrene 96-well plates, Stainless steel coupons, Titanium disks | Substrates for biofilm formation; relevant to clinical/industrial applications | Surface roughness affects attachment; stainless steel mimics food processing surfaces [85] [83] |
Mechanisms and Pathways in Bacterial Adhesion
The enzymatic disruption of biofilms directly interferes with well-established mechanisms of bacterial adhesion and biofilm initiation. The process begins with reversible attachment of planktonic cells to conditioned surfaces through weak interactions (van der Waals forces, electrostatic interactions) [3]. This transitions to irreversible attachment through the production of EPS components that anchor cells firmly to surfaces [3].
Matrix-degrading enzymes target key polymers during this critical transition phase and in mature biofilms. DNases degrade the eDNA scaffold that facilitates initial adhesion and structural stability [83]. Glycoside hydrolases disrupt the expolysaccharide networks that provide architectural integrity and protection [79]. Dispersin B specifically cleaves the PNAG polysaccharide that mediates cell-to-cell adhesion in numerous bacterial species [80] [84].
This enzymatic approach capitalizes on the fundamental understanding that biofilm-associated tolerance stems primarily from physical and physiological adaptations enabled by the EPS matrix rather than genetic resistance [3]. By dismantling this protective barrier, matrix-degrading enzymes potentially revert bacterial cells to a planktonic-like state with restored antimicrobial susceptibility [79].
Diagram 2: Biofilm lifecycle and enzyme intervention points
Matrix-degrading enzymes represent a promising alternative or adjunct to conventional antimicrobials for controlling biofilm-associated infections and contaminations. Their targeted mechanism of action against EPS components offers advantages including broad-spectrum activity, potential synergy with antimicrobials, and reduced resistance selection compared to traditional biocides. The experimental data demonstrate significant efficacy both as monotherapies and in combination approaches across diverse bacterial species and surface types.
Future development should focus on optimizing enzyme stability and activity under application-relevant conditions, engineering novel enzymes with enhanced catalytic properties or broader substrate specificity, and developing effective delivery systems for clinical or industrial settings. The continued investigation of matrix-degrading enzymes will undoubtedly contribute valuable tools to address the persistent challenge of biofilm-related resistance in both clinical and industrial contexts.
Bacterial biofilms, structured communities of microorganisms embedded in a self-produced extracellular polymeric substance (EPS), represent a significant global health threat and a primary contributor to antimicrobial resistance (AMR) [3]. These complex biological barriers can exhibit up to a 1000-fold reduction in antibiotic susceptibility compared to their planktonic counterparts, rendering conventional treatments largely ineffective and complicating the treatment of chronic infections [87]. Of particular concern are biofilms formed by ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), which are responsible for numerous healthcare-associated diseases [3]. The biofilm lifecycle begins with initial reversible attachment to a surface, proceeds to irreversible attachment and maturation through EPS production, and culminates in dispersion, enabling bacteria to colonize new surfaces [87]. This cyclical process, combined with the physical protection offered by the EPS matrix and the phenotypic heterogeneity of biofilm inhabitants, creates a formidable defense against antimicrobial agents [3].
In the face of this challenge, and given the stalled pipeline for new conventional antibiotics, the scientific community has turned its attention to alternative therapeutic strategies [88]. Natural products, specifically plant extracts and antimicrobial peptides (AMPs), have emerged as promising candidates for novel anti-biofilm agents [88] [89]. These compounds, shaped by millennia of evolutionary pressure, often employ multi-targeted mechanisms of action, simultaneously disrupting multiple bacterial pathways and thereby reducing the likelihood of resistance development [88]. This in-depth technical guide evaluates the anti-biofilm properties of these natural arsenals, framing their efficacy within the fundamental context of bacterial adhesion and biofilm initiation mechanisms, and provides detailed methodologies for researchers and drug development professionals working to translate these findings into clinical applications.
Fundamentals of Bacterial Adhesion and Biofilm Initiation
The initial adhesion of bacteria to a surface is a critical physico-chemical process that sets the stage for subsequent biofilm formation. This process is governed by a complex interplay of factors, including the properties of the bacterial cell surface, the substratum surface, and the surrounding liquid medium [90].
Surface Properties Governing Initial Adhesion
- Surface Charge Density: Bacterial cells typically possess a net negative charge due to functional groups like carboxyl, amino, and phosphates on their cell walls. Consequently, positively charged surfaces often facilitate greater initial bacterial adhesion for many species, including Pseudomonas aeruginosa and Escherichia coli, through attractive electrostatic interactions [90].
- Surface Wettability and Hydrophobicity: Surface wettability, central to solid-liquid interactions, significantly impacts adhesion. The relationship is complex, but models stemming from Derjaguin-Landau-Verwey-Overbeek (DLVO) and extended-DLVO theories are often used to explain the interaction energies between bacteria and surfaces [90].
- Surface Roughness and Topography: Rough surfaces tend to promote better initial microbial adhesion compared to smooth surfaces, as surface irregularities provide an increased surface area and refuge from shear forces [3] [90].
- Surface Free Energy (SFE): Recent research suggests that the degree of bacterial adhesion is unambiguously mediated by the SFE difference between the bacterial cells and the solid substratum. A lower SFE difference correlates with a higher degree of bacterial adhesion [91].
The following diagram illustrates the sequential nature of biofilm development from initial adhesion to dispersion.
From Adhesion to Surface Sensing
Initial adhesion is not merely a physical event; it triggers profound biological responses in the bacterium. Adhesion forces can cause nanoscopic deformation of the bacterial cell wall, which is sensed by membrane-located sensor molecules [92]. This "surface sensing" activates signaling cascades that lead to phenotypic and genotypic changes, including the upregulation of genes responsible for EPS biosynthesis (e.g., the Psl matrix polymer in P. aeruginosa) and the activation of genes associated with antibiotic resistance (e.g., β-lactamase and efflux pumps) [92] [87]. This transition marks the shift from reversible to irreversible attachment and represents the first commitment to a biofilm lifestyle.
Anti-Biofilm Properties of Plant Extracts and Phytochemicals
Plant-derived compounds, or phytochemicals, represent a rich source of bioactive molecules with potent anti-biofilm activities. Their mechanisms are diverse and often multi-targeted, offering a significant advantage over single-target antibiotics [87].
Key Mechanisms of Action
Phytochemicals disrupt biofilm development through several key mechanisms:
- Inhibition of Initial Adhesion: Compounds like flavonoids and alkaloids can alter bacterial surface hydrophobicity or compete for binding sites, preventing the initial attachment of planktonic cells to surfaces [87].
- Quorum Sensing (QS) Interference: Many phytochemicals, including polyphenols and terpenoids, function as quorum sensing inhibitors (QSIs). They block the production or detection of acyl-homoserine lactones (AHLs) and other signaling molecules, disrupting bacterial communication and preventing the coordinated gene expression required for biofilm maturation [87].
- Disruption of Extracellular Polymeric Substances (EPS): Enzymes such as tannases and other compounds can degrade the polysaccharide and protein components of the EPS matrix, compromising the biofilm's structural integrity and making embedded cells more susceptible to antimicrobials [3] [87].
- Targeting Metabolic Pathways and Persister Cells: Some phytochemicals can penetrate the biofilm and target metabolic processes or effectively kill dormant persister cells, which are typically highly tolerant to antibiotics [87].
Prominent Phytochemical Classes and Representative Compounds
Table 1: Key Phytochemical Classes and Their Anti-Biofilm Activities
| Phytochemical Class | Representative Compounds | Primary Anti-Biofilm Mechanisms | Example Target Pathogens |
|---|---|---|---|
| Alkaloids | Berberine, Piperine | QS inhibition, reduction of virulence factor production, EPS disruption | Staphylococcus aureus, Pseudomonas aeruginosa [87] |
| Flavonoids | Quercetin, Naringenin, Myricetin | Inhibition of initial adhesion, QS interference, inhibition of efflux pumps | ESKAPE pathogens [87] |
| Non-Flavonoid Polyphenols | Curcumin, Resveratrol, Gallic acid | EPS degradation, disruption of membrane integrity, QS inhibition | E. coli, P. aeruginosa [87] |
| Terpenes & Terpenoids | Carvacrol, Thymol, Geraniol | Permeabilization of cell membranes, inhibition of biofilm maturation | Candida albicans, S. aureus [87] |
| Quinones | Emodin, Plumbagin, Hypericin | Generation of reactive oxygen species (ROS), interference with electron transport chains | Bacillus subtilis, S. aureus [87] |
Antimicrobial Peptides (AMPs) as Anti-Biofilm Agents
AMPs are small, cationic, and amphipathic peptides that are part of the innate immune system of most organisms. They are gaining traction as next-generation therapeutics due to their broad-spectrum activity and lower propensity for resistance development compared to conventional antibiotics [89] [93].
AMPs are structurally diverse, falling into several classes: linear α-helical peptides, β-sheet peptides (stabilized by disulfide bridges), and peptides with extended or looped structures [89]. They can be sourced from a wide range of organisms:
- Marine Organisms: Marine AMPs, such as pleurocidin from winter flounder and clavanins from the leathery sea squirt, exhibit unique adaptations to extreme conditions, resulting in high stability, salt tolerance, and often low cytotoxicity [89].
- Insects and Animal-Derived Compounds: Insect-derived AMPs (e.g., defensins and cecropins) and those from bee products (e.g., melittin in bee venom and bee defensin-1 in honey) show potent activity against drug-resistant pathogens like MRSA [88].
- Plants and Microbes: Plants and bacteria themselves are also prolific producers of AMPs, contributing to the over 5,000 peptides cataloged in the Antimicrobial Peptide Database [93].
Anti-Biofilm Mechanisms of AMPs
The anti-biofilm activity of AMPs extends beyond their direct bactericidal action and includes several sophisticated strategies:
- Membrane Disruption: The primary mechanism of many AMPs involves electrostatic interactions with the negatively charged bacterial membrane, leading to pore formation (via barrel-stave, toroidal-pore, or carpet models) and cell lysis [89] [93]. This mechanism is particularly effective against metabolically dormant persister cells within biofilms.
- Inhibition of Biofilm Formation and Adhesion: Sub-inhibitory concentrations of some AMPs can effectively prevent bacterial attachment and subsequent biofilm formation without killing the planktonic cells [89].
- Disruption of Pre-Formed Biofilms: Certain AMPs can penetrate existing biofilms and disrupt the EPS matrix, facilitating the killing of embedded cells and enhancing the efficacy of co-administered antibiotics [89].
- Immunomodulation: Some AMPs, such as epinecidin-1 from grouper, possess dual functionality, directly killing pathogens while also modulating the host's immune response to better clear the infection [89].
Table 2: Selected Marine Antimicrobial Peptides and Their Activity Against Resistant Pathogens
| Compound | Source | Mechanism of Action | Antibacterial Activity |
|---|---|---|---|
| Pleurocidin | Winter flounder (Pleuronectes americanus) | Membrane disruption, alteration of bacterial metabolic pathways, interference with QS | Active against multi-drug-resistant E. faecium, E. coli, P. aeruginosa, K. pneumoniae, and A. baumannii (MIC: 8–256 μg/mL) [89] |
| Clavanins | Leathery sea squirt (Styela clava) | Membrane disruption (enhanced with Zn²⁺); some translocate intracellularly without membrane damage | Active against MRSA ATCC 43300 (MIC: 16 μg/mL for clavanin C); active against MDR Enterobacter cloacae [89] |
| Epinecidin-1 | Grouper (Epinephelus coioides) | Membrane disruption and immunomodulation | Active against MRSA and other drug-resistant strains [89] |
The following diagram summarizes the multi-faceted anti-biofilm mechanisms shared by both phytochemicals and AMPs.
Experimental Protocols for Evaluating Anti-Biofilm Activity
Robust and standardized assays are crucial for quantifying the efficacy of potential anti-biofilm agents. The following section outlines key methodologies.
Phytochemical Extraction and Screening
- Extraction and Isolation: Conduct a systematic literature search to identify candidate plants. Active ingredients are typically extracted using solvents of varying polarities (e.g., methanol, ethanol, water). The crude extracts are then fractionated using techniques like liquid-liquid partitioning, column chromatography, and HPLC to isolate pure phytochemicals [87].
- Initial Antibacterial Screening: Determine the Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC) against target planktonic bacteria using standard broth microdilution methods according to CLSI/EUCAST guidelines [87].
Quantitative Anti-Biofilm Assays
- Minimum Biofilm Inhibitory Concentration (MBIC) Assay:
- Procedure: In a 96-well microtiter plate, add serial dilutions of the test compound to a suspension of log-phase bacteria in a suitable growth medium. Incubate under static conditions for 24-48 hours to allow biofilm formation.
- Analysis: Remove planktonic cells and wash the adhered biofilm gently. Quantify the biomass using crystal violet (CV) staining (absorbance at 570-600 nm) or metabolic activity assays using resazurin (AlamarBlue) or XTT [87]. The lowest concentration that shows a ≥50% reduction in biofilm biomass or activity compared to the untreated control is the MBIC.
- Minimum Biofilm Eradication Concentration (MBEC) Assay:
- Procedure: First, allow a mature biofilm to form on the surface of a specialized device (e.g., a Calgary Biofilm Device or a peg lid) for 48 hours. Then, transfer the biofilm-covered pegs to a new plate containing serial dilutions of the test compound and incubate for a further 24 hours.
- Analysis: The MBEC is defined as the lowest concentration of the compound that eradicates the pre-formed biofilm, demonstrated by no growth in the subsequent recovery medium [87]. This assay is critical for evaluating the ability of an agent to treat established infections.
Mechanistic Studies
- Quorum Sensing Inhibition (QSI) Assays: Utilize reporter strains that produce a detectable signal (e.g., bioluminescence, chromogenesis) in response to AHLs. A reduction in signal in the presence of the test compound indicates QS inhibition [87].
- EPS Disruption and Analysis: Treat pre-formed biofilms with the test compound and analyze the released EPS components. Quantify polysaccharides using the phenol-sulfuric acid method and proteins using the Lowry or Bradford assays [3] [87].
- Microscopic Visualization: Use Scanning Electron Microscopy (SEM) and Confocal Laser Scanning Microscopy (CLSM) to visually assess changes in biofilm architecture, thickness, and viability (using live/dead stains like SYTO9/propidium iodide) after treatment [87].
Advanced Tools for Biofilm Image Analysis
Powerful software tools like BiofilmQ enable automated, high-throughput quantification and visualization of 3D biofilm internal properties from microscopy images [94]. It can analyze hundreds of parameters, including biofilm volume, mean thickness, surface area, roughness, and spatially resolved fluorescence intensities, providing deep insights into the structural and functional impacts of anti-biofilm treatments [94].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Anti-Biofilm Research
| Reagent / Material | Function / Application | Examples & Notes |
|---|---|---|
| 96-well Microtiter Plates | Standard platform for high-throughput biofilm cultivation and screening assays. | Use flat-bottom plates for CV staining; specialized peg lids (e.g., MBEC Assay Kit) for MBEC determination. |
| Crystal Violet (CV) | A general stain for quantifying total biofilm biomass. | Simple and cost-effective; does not distinguish between live and dead cells. |
| Resazurin (AlamarBlue) / XTT | Metabolic dyes used to measure the metabolic activity of cells within a biofilm. | More reflective of viable cell count than CV; can be used sequentially after CV. |
| Live/Dead Staining Kits | Fluorescent viability assays for Confocal Laser Scanning Microscopy (CLSM). | Typically contain SYTO9 (green, stains all cells) and Propidium Iodide (red, stains dead cells). |
| Reporter Strains | Genetically engineered bacteria used to study specific mechanisms like Quorum Sensing. | e.g., C. violaceum CV026 for AHL detection; luminescent P. aeruginosa strains. |
| Biofilm Analysis Software | For quantitative image analysis of 3D biofilm structure and composition. | BiofilmQ [94], COMSTAT [94]. Essential for extracting robust data from microscopy. |
| Extraction & Chromatography | For isolating and purifying active phytochemicals from crude plant extracts. | Solvents (methanol, ethyl acetate), solid-phase extraction cartridges, HPLC/HPLC-MS systems. |
The escalating crisis of antimicrobial resistance, fueled in large part by the tenacity of bacterial biofilms, demands a paradigm shift in therapeutic development. Plant extracts and antimicrobial peptides represent a powerful "natural arsenal" with immense potential to meet this challenge. Their key strength lies in their multi-targeted mechanisms of action—simultaneously inhibiting adhesion, disrupting quorum sensing, degrading the EPS matrix, and killing persistent cells—which drastically reduces the probability of resistance evolution compared to conventional antibiotics [88] [89] [87].
While challenges in standardization, bioavailability, and scalable production remain, modern scientific approaches provide a clear path forward. The integration of omics technologies for discovery, rational nano-formulations to enhance stability and delivery, and sophisticated analytical tools like BiofilmQ for precise quantification, will be pivotal in translating these natural compounds from the laboratory to the clinic [88] [94] [87]. By systematically evaluating these agents within the fundamental context of bacterial adhesion and biofilm initiation, researchers and drug development professionals can unlock their full potential, paving the way for a new generation of effective anti-biofilm therapies.
Bacterial biofilms represent a formidable challenge in clinical settings, contributing significantly to the global crisis of antimicrobial resistance (AMR). These structured microbial communities, encased in a protective extracellular polymeric substance (EPS), exhibit intrinsic resistance to conventional antibiotics, making associated infections chronic and difficult to treat [3] [95]. The EPS matrix acts as a formidable biological barrier, impeding antibiotic penetration, while the heterogeneous microenvironment within biofilms fosters physiological states like dormancy, further enhancing tolerance [3] [96]. This is particularly concerning with biofilms formed by ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), which are frequently associated with healthcare-associated diseases and demonstrate multi-drug resistance [3].
The imperative to overcome these barriers has catalyzed a paradigm shift from monotherapy to innovative combination strategies. These approaches aim to disrupt the biofilm's physical integrity or regulatory networks, thereby sensitizing the embedded bacteria to subsequent antibiotic attack [96]. This technical guide synthesizes current research and emerging strategies for enhancing antibiotic efficacy against biofilms, providing a foundational resource for ongoing research into bacterial adhesion and biofilm initiation. By dissecting the mechanisms of combination therapies, this review aims to equip researchers and drug development professionals with the knowledge to design more effective interventions against these resilient microbial communities.
Biofilm Architecture and the Basis of Resistance
Structural and Physiological Barriers to Treatment
The resilience of biofilms is not attributable to a single mechanism but is instead a multifactorial consequence of their structured existence. The journey to a mature biofilm begins with the reversible attachment of planktonic cells to a surface, mediated by weak interactions such as van der Waals forces and electrostatic interactions [3]. This attachment becomes irreversible through the secretion of a sticky, three-dimensional matrix of EPS [3]. The EPS, composed of polysaccharides, proteins, nucleic acids, and lipids, forms a physical barrier that restricts the diffusion of antimicrobial agents and shields bacteria from host immune responses [3] [95].
Beyond the physical barrier, biofilms exhibit profound physiological heterogeneity. Gradients of nutrients, oxygen, and waste products create diverse microenvironments within the biofilm structure [3] [95]. This heterogeneity leads to varied metabolic activity, with subpopulations of bacteria, known as persister cells, entering a dormant state that renders them highly tolerant to antibiotics that typically target active cellular processes [96] [95]. Furthermore, the close proximity of cells within the dense biofilm structure accelerates horizontal gene transfer (HGT), facilitating the dissemination of resistance genes among the bacterial community and transforming biofilms into hotspots for the evolution of resistance [95].
Table 1: Key Mechanisms of Biofilm-Mediated Antimicrobial Resistance
| Resistance Mechanism | Description | Impact on Antibiotic Efficacy |
|---|---|---|
| EPS Barrier | Extracellular polymeric substances physically impede antibiotic penetration [3]. | Reduced drug accumulation at target sites within the biofilm. |
| Metabolic Heterogeneity | Gradients create zones of slow or non-growing cells [95]. | Tolerance to antibiotics effective only against metabolically active cells. |
| Persister Cells | Dormant, phenotypically variant cells that survive antimicrobial exposure [96] [95]. | Cause chronic, recurring infections post-treatment. |
| Horizontal Gene Transfer | Close cell proximity facilitates plasmid exchange [95]. | Spread of genetic resistance determinants across the community. |
| Upregulated Efflux Pumps | Increased expression of systems that expel toxins [46]. | Active removal of antibiotics from bacterial cells. |
Strategic Framework for Combination Therapies
Combination therapies are designed to target the specific resistance mechanisms outlined in Table 1. The overarching strategy involves a primary agent that disrupts the biofilm's defenses, followed by a secondary antibiotic that eradicates the now-vulnerable bacterial cells. This synergistic approach can significantly lower the required antibiotic concentration, mitigating toxicity and potentially overcoming resistance [96].
Quorum Sensing Inhibitors (QSIs) with Antibiotics
Quorum sensing (QS) is a cell-density-dependent communication system that coordinates biofilm development and virulence in many bacterial species. QSIs disrupt this signaling, leading to dysregulated biofilm formation and enhanced susceptibility.
- Mechanism of Action: QSIs function via multiple strategies, including inhibiting autoinducer synthesis, degrading signaling molecules, or interfering with signal receptor binding [96]. This disruption prevents the coordinated behavior necessary for maintaining a mature biofilm.
- Experimental Protocol: A standard protocol for evaluating QSI-antibiotic combinations against established biofilms involves several stages. First, grow a 24-hour biofilm in a suitable medium like Mueller-Hinton broth within a 96-well plate. Then, treat the mature biofilm with the QSI (e.g., hamamelitannin or baicalin hydrate) for a predetermined period (e.g., 2-4 hours). Next, add the antibiotic (e.g., tobramycin, vancomycin) at sub-inhibitory concentrations, either alone or in continuation with the QSI. Finally, quantify efficacy by assessing viable bacterial counts (CFU/mL) after disruption of the biofilm, or by using metabolic assays like resazurin reduction or crystal violet staining for biomass [96].
- Key Data: Studies have demonstrated that combining tobramycin with the QSI baicalin hydrate reduced pulmonary bacterial loads in a mouse model by 99.9%, a significantly greater effect than either component alone [96]. Similarly, hamamelitannin combined with antibiotics like cefazolin or tobramycin achieved ≥90% eradication of S. aureus biofilms in vitro [96].
Figure 1: Mechanism of Quorum Sensing Inhibition. QSIs disrupt bacterial communication by targeting autoinducer accumulation or receptor binding, preventing biofilm maturation.
Enzyme-Based Matrix Disruption
Glycoside Hydrolases (GHs) and other enzymes such as DNases target the structural integrity of the EPS matrix.
- Mechanism of Action: Enzymes like Dispersin B (targets polysaccharides) or DNase I (targets extracellular DNA) cleave specific structural components of the biofilm matrix, leading to its physical disintegration and enhanced antibiotic penetration [95].
- Critical Consideration: A crucial finding from recent research is that not all enzyme-antibiotic combinations are compatible. For instance, α-amylase from Aspergillus oryzae and Bacillus subtilis has been shown to degrade and sequester antibiotics like tetracycline and ciprofloxacin, potentially reducing treatment efficacy [97]. Therefore, compatibility screening via methods like Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) is essential before formulating such combinations [97].
- Experimental Protocol: To test enzyme-antibiotic synergy, first cultivate biofilms (e.g., 2-day P. aeruginosa biofilms). Then, incubate with a selected GH (e.g., α-amylase, Dispersin B) for a period to initiate matrix disruption. Subsequently, treat with an antibiotic and quantify the remaining viable cells. Parallel stability assays using LC-MS/MS should be conducted to monitor potential degradation of the antibiotic by the enzyme over time [97].
Phage-Antibiotic Synergistic (PAS) Therapy
Bacteriophages (phages), viruses that infect and lyse bacteria, offer a highly specific means of attacking biofilm-embedded cells.
- Mechanism of Action: Phages penetrate the biofilm matrix and lyse bacterial cells, which physically disrupts the biofilm structure and reduces bacterial density. This lysis can sensitize the remaining cells to antibiotics [98] [95]. This approach, known as Phage-Antibiotic Synergy (PAS), is particularly effective against dual-species communities, as demonstrated in 3D lung epithelial models [99].
- Key Data & Challenge: Preclinical models show that adding at least one phage to an antibiotic regimen enhances bacterial killing throughout the biofilm and prevents the emergence of antimicrobial resistance [98]. The primary challenge is access, as phage therapy is largely experimental, with timelines for obtaining specific phages for compassionate use often exceeding six months [98].
Table 2: Promising Combination Therapies Against Bacterial Biofilms
| Combination Type | Specific Agents | Target Pathogen | Reported Efficacy |
|---|---|---|---|
| QSI + Antibiotic | Hamamelitannin + Tobramycin | Staphylococcus aureus | ≥90% biofilm eradication in vitro [96] |
| QSI + Antibiotic | Baicalin hydrate + Tobramycin | Pseudomonas aeruginosa | 99.9% reduction in pulmonary bacterial load in vivo [96] |
| Phage + Antibiotic | Phage cocktail + Antibiotic | Methicillin-resistant S. aureus (MRSA) | Enhanced killing and prevention of resistance in vitro [98] |
| Nanoparticle + Antibiotic | mFeP-Cip NPs + Ultrasound | Pseudomonas aeruginosa | Effective biofilm eradication in a catheter implant mouse model [100] |
| Enzyme + Antibiotic | Dispersin B + Antibiotic | Various | Degrades polysaccharide matrix, enhancing antibiotic penetration [95] |
Advanced Delivery Systems for Enhanced Penetration
Even the most potent therapeutic agent requires effective delivery to its target. Recent innovations in nanomedicine have created sophisticated systems designed to physically disrupt biofilms and release antibiotics directly at the site of infection.
Ultrasound-Responsive Nanoparticles
These systems use nanoparticles that can be activated by external ultrasound to achieve targeted biofilm disruption.
- Mechanism of Action: Nanoparticles are loaded with a low-boiling-point perfluorocarbon (e.g., perfluoropentane, PFP) and an antibiotic. When stimulated by focused ultrasound, the PFP vaporizes, undergoing a phase change to gas (acoustic droplet vaporization). This process creates cavitation bubbles whose collapse generates microjets and shockwaves that physically disrupt the EPS matrix. This disruption simultaneously releases the antibiotic and enhances its penetration into the deeper layers of the biofilm [101] [100].
- Experimental Protocol: The multi-step process is visualized in Figure 2 and involves synthesizing and characterizing mesoporous nanoparticles (e.g., Fe₃O₄). These nanoparticles are co-loaded with PFP and an antibiotic like ciprofloxacin (Cip) to form the final construct (e.g., mFeP-Cip NPs) [100]. For in vitro testing, biofilms are grown on a substrate like a catheter piece and then incubated with the nanoparticles. A magnetic field (MF) can be applied first to guide and concentrate the nanoparticles at the biofilm site. Subsequently, ultrasound (e.g., 1.0 W/cm², 1 MHz) is applied to trigger vaporization and drug release. Efficacy is quantified via CFU counts, confocal microscopy of stained biofilms, and scanning electron microscopy (SEM) to visualize structural damage [101] [100].
- Key Data: This approach has demonstrated remarkable efficacy. One study reported that the combination of ultrasound-activated nanoparticles and antibiotics reduced the concentration of antibiotic required to eradicate biofilms by more than 40-fold compared to conventional treatment, achieving 100% killing of several clinical strains, including MRSA, at clinically feasible doses [101]. It was also highly effective against persister cells, reducing the required drug concentration by 25-fold [101].
Figure 2: Workflow for Ultrasound-Responsive Nanoparticle Therapy. Nanoparticles are targeted to the biofilm and activated by ultrasound, causing physical disruption and targeted drug release.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Anti-Biofilm Studies
| Reagent / Material | Function in Experimentation | Example Use Case |
|---|---|---|
| Hamamelitannin | Synthetic quorum sensing inhibitor (QSI) [96]. | Sensitizing S. aureus biofilms to β-lactam and aminoglycoside antibiotics [96]. |
| Dispersin B | Glycoside hydrolase that degrades polysaccharide matrix [95]. | Disrupting the EPS of staphylococcal biofilms to enable antibiotic penetration [95]. |
| Bacteriophages (Phage Cocktails) | Biological agents that lyse specific bacterial hosts [98] [99]. | Used in PAS (Phage-Antibiotic Synergy) to disrupt and eradicate dual-species biofilms [99]. |
| Mesoporous Fe₃O₄ Nanoparticles (mFe NPs) | Magnetic, porous core for drug and PFP loading [100]. | Serves as the core for constructing MF/US dual-responsive drug delivery systems [100]. |
| Perfluoropentane (PFP) | Low-boiling point phase-change agent [100]. | Loaded into nanoparticles; vaporizes upon US to disrupt biofilms via cavitation [100]. |
| α-Amylase (from various sources) | Glycoside hydrolase; can degrade matrix polysaccharides [97]. | Critical Note: Used to study EPS degradation, but requires compatibility checks with antibiotics due to potential for drug sequestration/degradation [97]. |
The fight against biofilm-mediated infections necessitates a move beyond conventional antibiotic monotherapies. The combination strategies detailed in this guide—ranging from signaling disruption with QSIs and matrix degradation with enzymes to the physical disruption achieved with phage therapy and ultrasound-activated nanoparticles—represent a promising frontier in antimicrobial research. The consistent theme across these approaches is synergy: using one agent to compromise the biofilm's formidable defenses, thereby allowing a second agent, typically an antibiotic, to effectively clear the infection. For researchers focused on the initial stages of bacterial adhesion and biofilm initiation, these therapeutic strategies offer a compelling link between foundational mechanisms and clinical application. The future of eradicating biofilm-related infections lies in the continued development and intelligent combination of these multifaceted treatment modalities.
Bacterial persisters are a subpopulation of growth-arrested, metabolically dormant cells that exhibit remarkable tolerance to high-dose antibiotic therapy without undergoing genetic resistance mutations [102] [103]. These phenotypic variants were first identified by Joseph Bigger in 1944 when he observed that a small subset of Staphylococcus cells survived penicillin exposure despite apparent antibiotic susceptibility [104] [102]. Unlike resistant bacteria, persisters do not possess specific resistance mechanisms but survive antibiotic challenge through their dormant state, which protects them from antibiotics that target active cellular processes [103]. When antibiotic pressure is removed, these cells can resuscitate and repopulate the environment, leading to chronic, recurrent infections that are notoriously difficult to treat [102] [103].
The significance of persister cells extends across clinical medicine, contributing to treatment failures in tuberculosis, recurrent urinary tract infections, Lyme disease, and biofilm-associated infections on medical devices [102] [103] [105]. Their role in persistent infections is particularly problematic in the context of the global antimicrobial resistance crisis, as surviving persister cells provide a reservoir from which genetically resistant mutants can emerge [103]. Understanding and targeting these dormant populations represents a critical frontier in infectious disease management and requires specialized approaches distinct from conventional antibiotic development.
Molecular Mechanisms of Persister Formation
Persister cells emerge through diverse molecular pathways that induce a transient, non-genetic dormant state. Research has identified several key mechanisms underlying this phenotype, with heterogeneity existing both in triggering stimuli and resulting physiological states.
Classification of Persister Types
Persisters are broadly classified into types based on their formation mechanisms and characteristics, as detailed in the table below.
Table 1: Classification of Persister Cell Types
| Type | Formation Trigger | Growth State | Key Characteristics |
|---|---|---|---|
| Type I (Triggered) | Stationary phase entry, environmental stress [104] | Non-growing [104] [102] | Pre-existing in population; associated with toxin-antitoxin systems [104] |
| Type II (Stochastic) | Spontaneous, stochastic switching during exponential phase [104] [102] | Slow-growing [104] [102] | Continuously generated at low rates; not growth-arrested [104] |
| Type III (Specialized) | Specific antibiotic-induced stress signals [104] | Variable (not necessarily slow-growing) [104] | Antibiotic-specific persistence mechanisms [104] |
Key Molecular Pathways
Multiple interconnected molecular systems contribute to persister formation and maintenance:
Toxin-Antitoxin (TA) Systems: These modular genetic elements consist of a stable toxin that disrupts essential cellular processes and a labile antitoxin that neutralizes the toxin. Under stress conditions, antitoxins are degraded, freeing toxins to induce dormancy. Key TA systems include HipBA, RelBE, and MazEF, which inhibit translation through mRNA cleavage, leading to growth arrest [104] [105]. In Pseudomonas aeruginosa, antibiotics like ciprofloxacin and colistin enhance expression of type II TA systems (relBE, vapBC), promoting persister formation in biofilms [105].
Stringent Response and Alarmone Signaling: Nutrient limitation and other stresses trigger the accumulation of the alarmone guanosine tetraphosphate (ppGpp), which dramatically reprogram cellular metabolism by downregulating energy-intensive processes and promoting survival mechanisms [104]. This signaling molecule redirects resources from growth to maintenance, facilitating the dormant state characteristic of persisters.
SOS Response: DNA damage activates the SOS pathway, inducing cell cycle arrest and DNA repair functions. This stress response can coordinate with other persistence mechanisms to promote multidrug tolerance [104].
The following diagram illustrates the core molecular pathways leading to persister cell formation:
Eradication Strategies: Overcoming Persister Tolerance
Traditional antibiotics typically fail against persisters because they target active cellular processes. Effective anti-persister approaches must therefore employ alternative strategies that either bypass dormancy mechanisms or reactivate cells to restore antibiotic susceptibility.
Direct Killing Approaches
Direct killing strategies target essential cellular structures that remain vulnerable even in dormant cells, with the bacterial membrane representing a particularly attractive target.
Membrane-Targeting Compounds: These agents disrupt membrane integrity, causing leakage of cellular contents and eventual cell lysis. Examples include synthetic cation transporters like SA-558, which disrupts bacterial homeostasis leading to autolysis [103], and compounds XF-70 and XF-73 that effectively kill non-dividing Staphylococcus aureus cells by membrane disruption [103]. Additionally, membrane-active compounds like the methylazanediyl bisacetamide derivative MB6 and synthetic retinoids (CD437, CD1530) embed in the MRSA lipid bilayer, disrupting membrane integrity and increasing antibiotic uptake [103].
Energy Disruption: Pyrazinamide, a frontline tuberculosis drug, targets persisters by disrupting membrane energetics. Its active form, pyrazinoic acid, binds to PanD (aspartate decarboxylase), triggering its degradation by the ClpC1-ClpP protease and disrupting coenzyme A biosynthesis [103].
Protein Degradation Activation: ADEP4, a semi-synthetic acyldepsipeptide, binds to and activates the ClpP protease, causing uncontrolled protein degradation in dormant cells. This results in the breakdown of hundreds of intracellular proteins, including metabolic enzymes essential for persister resuscitation [103].
Indirect and Synergistic Approaches
Indirect strategies focus on preventing persister formation or reactivating dormant cells to restore conventional antibiotic susceptibility.
Inhibition of Persister Formation: Targeting persistence at its developmental roots represents a promising preventive approach. The pheromone cCf10 reduces Enterococcus faecalis persister formation by limiting (p)ppGpp alarmone accumulation [103]. Similarly, inhibitors of hydrogen sulfide (H₂S) biogenesis, such as bacterial cystathionine γ-lyase (bCSE) inhibitors, reduce biofilm formation and persister numbers in S. aureus and P. aeruginosa [103].
Quorum Sensing Interference: Bacterial cell-cell communication systems regulate multicellular behaviors including persistence. Compounds with a benzamide-benzimidazole backbone inhibit the P. aeruginosa MvfR regulon, reducing persister formation without affecting growth [103].
Membrane Permeabilization for Antibiotic Sensitization: Increasing membrane permeability can restore antibiotic efficacy against persisters. Membrane-active compounds including bithionol, IMT-P8 (a cell-penetrating peptide), and polymyxin B nonapeptide (PMBN) disrupt membrane integrity, facilitating antibiotic entry into dormant cells [103].
Table 2: Quantitative Analysis of Persister Survival Across Bacterial Species and Antibiotics
| Bacterial Species | Antibiotic Class | Persister Survival Range | Key Observations |
|---|---|---|---|
| Escherichia coli [106] | Multiple classes (32 antibiotics) [106] | Variable across antibiotics [106] | Substantial variation between environmental isolates [107] |
| Staphylococcus aureus [106] | 18 different antibiotics [106] | MRSA: ~5% cell survival [106] | Membrane-active antibiotics admit fewest persisters [106] |
| Pseudomonas aeruginosa [106] | 16 different antibiotics [106] | Variable across antibiotics [106] | Higher persister numbers in stationary phase biofilms [105] |
| Acinetobacter baumannii [106] | Multiple classes [106] | Lowest persistence: 0.01% [106] | Species with lowest persistence levels [106] |
| Enterococcus faecium [106] | Multiple classes [106] | Highest persistence: up to 100% [106] | Species with highest persistence levels [106] |
Experimental Protocols for Persister Research
Isolation and Quantification of Persister Cells
Standardized methodologies are essential for reliable persister research. The following protocol describes persister isolation and quantification from biofilm populations:
Biofilm Cultivation: Grow bacterial cultures (e.g., P. aeruginosa) in appropriate media to exponential (OD₆₀₀ ≈ 0.5) or stationary (OD₆₀₀ ≈ 1.2) phase. Establish biofilms on suitable surfaces (e.g., peg lids, catheter segments) for 24-48 hours with medium refreshment [105].
Antibiotic Challenge: Expose biofilms to 5× MIC of selected antibiotics (e.g., ciprofloxacin, colistin) for 3.5 hours under optimal growth conditions [105].
Persister Collection and Enumeration: Remove antibiotic pressure by thorough washing with sterile saline. Disrupt biofilms by sonication (if necessary) and plate serial dilutions on antibiotic-free media. Count colony-forming units (CFUs) after 24-48 hours incubation [105].
Calculation of Persister Fractions: Express persister levels as the percentage of surviving cells relative to pre-treatment counts: (CFUpost-treatment / CFUpre-treatment) × 100% [105].
Advanced Computational Screening for Anti-Persister Compounds
Novel computational approaches enable rational design of anti-persister agents:
Compound Library Curation: Compile chemical libraries with known antimicrobial activity, such as the Asinex SL#013 Gram-Negative Antibacterial Library containing 80 iminosugar-scaffold molecules [108].
Molecular Descriptor Analysis: Extract structural and physicochemical parameters from reference anti-persister antibiotics (e.g., minocycline, rifamycin SV, eravacycline) using computational platforms (JOELib/ChemMine, Maestro). Key descriptors include logP (octanol-water partition), halogen content, hydroxyl groups, and globularity [108].
Chemoinformatic Clustering: Apply k-means clustering algorithms based on molecular descriptors to identify compounds with structural similarity to proven anti-persister agents [108].
Experimental Validation: Test computationally-selected compounds at standardized concentrations (e.g., 100 µg/mL) against high-persistence model strains (e.g., E. coli HM22 with hipA7 allele). Quantify killing efficacy against antibiotic-induced persisters [108].
The following workflow diagram illustrates the integrated experimental-computational pipeline for anti-persister drug discovery:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Persister Cell Research
| Reagent/Chemical | Function/Application | Specific Examples |
|---|---|---|
| High-Persistence Model Strains | Experimental models for persister studies | E. coli HM22 (hipA7 allele) [108]; Clinical P. aeruginosa isolates [105] |
| Membrane-Targeting Agents | Disrupt membrane integrity in dormant cells | XF-70, XF-73 [103]; SA-558 [103]; Synthetic retinoids (CD437, CD1530) [103] |
| Bacterial Signaling Inhibitors | Interfere with quorum sensing and persistence regulation | Benzamide-benzimidazole compounds (MvfR inhibitors) [103]; Brominated furanones [103] |
| Metabolic Modulators | Target persistence-related metabolic pathways | CSE inhibitors (H₂S biogenesis) [103]; cCf10 pheromone ((p)ppGpp reduction) [103] |
| Computational Screening Tools | Identify potential anti-persister compounds | JOELib/ChemMine [108]; Maestro software [108]; k-means clustering algorithms [108] |
The eradication of bacterial persister cells represents a critical challenge in overcoming chronic and recurrent infections. While significant progress has been made in understanding the molecular mechanisms underlying persistence and developing targeted strategies, the field continues to evolve rapidly. Future research directions should focus on several key areas: First, the clinical translation of anti-persister compounds requires optimization of therapeutic indices to minimize off-target toxicity while maintaining efficacy against dormant cells [103]. Second, the development of standardized methodologies for persister detection and quantification across laboratories will enhance reproducibility and comparability of findings [107]. Finally, innovative approaches such as nanoparticle-based delivery systems (e.g., red blood cell membrane-coated nanoparticles incorporating naftifine) and combination therapies that simultaneously target multiple persistence mechanisms show particular promise for clinical application [103].
The continuing global threat of antimicrobial resistance underscores the urgency of developing effective strategies against persister cells. By integrating mechanistic insights with innovative therapeutic approaches, the scientific community can address this persistent challenge in infection control and improve outcomes for patients suffering from chronic and recurrent bacterial infections.
Bench to Bedside: Validating and Comparing Anti-Biofilm Strategies and Technologies
Within the broader context of research on bacterial adhesion and biofilm initiation, the development of effective anti-biofilm strategies represents a critical frontier in combating persistent infections. Bacterial biofilms are structured communities of microbial cells enclosed in a self-produced extracellular polymeric substance (EPS) that adhere to biotic or abiotic surfaces [3] [4]. This biofilm lifestyle confers remarkable resistance to antimicrobial treatments and host immune responses, with biofilm-associated bacteria exhibiting up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [109] [110]. This review critically examines two principal approaches for combating biofilms: natural phytochemicals and synthetically engineered compounds, evaluating their respective efficacy, mechanisms of action, and limitations within the framework of modern anti-biofilm drug development.
The persistent nature of biofilm-associated infections is largely attributable to the complex architecture of the biofilm matrix and the heterogeneous metabolic states of embedded cells [4] [111]. The biofilm lifecycle progresses through systematic stages: initial reversible attachment, irreversible attachment, microcolony formation, maturation, and active dispersion [3] [4]. Each stage presents distinct molecular targets for therapeutic intervention, from initial adhesion mechanisms to the quorum sensing (QS) pathways that coordinate community behavior in mature biofilms [109] [110]. Understanding these fundamental mechanisms of biofilm development provides the necessary context for evaluating how natural and synthetic compounds disrupt this coordinated process.
Biofilm Formation and Resistance Mechanisms: Therapeutic Implications
The structured development of biofilms occurs in a multi-stage lifecycle that presents multiple intervention points for anti-biofilm agents. Figure 1 illustrates the key stages and the resistance mechanisms that emerge at each phase.
Figure 1. Biofilm lifecycle stages and emergence of resistance mechanisms. The diagram illustrates the progression from planktonic cells to mature biofilms and highlights the key resistance traits that develop during maturation. Based on content from [3] [4] [111].
The protective architecture of mature biofilms confers resistance through multiple simultaneous mechanisms. The EPS matrix, comprising polysaccharides, proteins, extracellular DNA (eDNA), and lipids, creates a physical barrier that restricts antibiotic penetration [4] [111]. This matrix can bind antimicrobial agents, particularly positively charged aminoglycosides that interact with negatively charged eDNA, significantly reducing their effective concentration within the biofilm interior [4]. Furthermore, metabolic heterogeneity within biofilms creates nutrient and oxygen gradients, resulting in subpopulations of dormant or slow-growing persister cells that exhibit exceptional tolerance to conventional antibiotics that typically target active cellular processes [4] [111]. The biofilm microenvironment also facilitates efficient horizontal gene transfer, accelerating the dissemination of antibiotic resistance genes among embedded cells [4].
Comparative Analysis of Natural and Synthetic Anti-Biofilm Agents
Natural Anti-Biofilm Compounds
Natural products, particularly phytochemicals derived from plants, represent a promising source of anti-biofilm agents with diverse mechanisms of action and potential synergies with conventional antibiotics [112] [110] [111]. These compounds typically target key virulence pathways rather than directly causing bacterial death, potentially reducing selective pressure for resistance development [110].
Table 1: Efficacy and Mechanisms of Selected Natural Anti-Biofilm Compounds
| Compound | Class | Primary Anti-Biofilm Mechanisms | Example Pathogens Tested | Key Efficacy Parameters |
|---|---|---|---|---|
| Quercetin | Flavonoid | QS inhibition; EPS disruption; reduces biofilm structural integrity [112] | P. aeruginosa [112] | Synergy with aminoglycosides; enhances antibiotic penetration [112] |
| Apigenin | Flavonoid | Antibacterial effects; biofilm reduction [112] | Not specified | Disrupts bacterial communication pathways [112] |
| Gallic Acid | Polyphenol | Antibacterial; inhibits biofilm formation & disrupts mature biofilms [112] | Not specified | Reduces free radicals and ROS at infection site [112] |
| Proanthocyanidins | Flavonoid | Antibacterial; inhibits biofilm formation & disrupts mature biofilms [112] | Not specified | Enhances accumulation in Gram-negative bacilli [112] |
| Arbutin | Glycoside | Anti-biofilm potential [112] | Associated with UTIs [112] | Found in bearberry leaf extract [112] |
| Rutin | Flavonoid | Anti-biofilm potential [112] | Not specified | Present in most plants; often with ascorbic acid [112] |
| ε-Viniferin | Stilbenoid | Inhibits biofilm formation [113] | E. coli O157:H7, P. aeruginosa [113] | Significant inhibition of both pathogens [113] |
| Berberine | Alkaloid | Inhibits biofilm formation and reduces bacterial activity within biofilms [110] | Not specified | Remarkable efficacy; multitargeted mechanisms [110] |
| Curcumin | Polyphenol | Inhibits biofilm formation and reduces bacterial activity within biofilms [110] | Not specified | Remarkable efficacy; multitargeted mechanisms [110] |
Natural compounds employ multiple strategic approaches to disrupt biofilm viability. Many phytochemicals, including quercetin and apigenin, effectively interfere with quorum sensing (QS) systems, bacterial communication networks that coordinate biofilm development and virulence factor production [112] [110]. By disrupting these signaling pathways, natural compounds can prevent the population-wide behavioral shifts necessary for biofilm maturation without imposing lethal pressure that drives resistance development [110]. Certain flavonoids demonstrate the ability to destabilize the structural integrity of the EPS matrix, thereby facilitating enhanced penetration of co-administered antibiotics [112]. Some phytochemicals can directly inhibit bacterial adhesion to surfaces, preventing the initial attachment phase that initiates biofilm formation [110] [111].
Despite their therapeutic potential, natural anti-biofilm agents face significant challenges in clinical translation. Many phytochemicals, including quercetin and curcumin, suffer from poor aqueous solubility, low bioavailability, and rapid systemic metabolism [112] [110]. Quercetin, for instance, demonstrates absorption rates as low as 2% for the aglycone form and 3-17% for glucoside forms after oral administration in humans [112]. The complex composition of plant extracts introduces additional variables that complicate standardization and dose optimization for clinical applications [110]. Furthermore, the multicomponent nature of many natural extracts makes it challenging to identify specific active constituents and their precise mechanisms of action [110].
Synthetic Anti-Biofilm Compounds and Strategies
Synthetic anti-biofilm approaches encompass rationally designed small molecules, repurposed pharmaceuticals, and engineered materials that target specific biofilm vulnerabilities. These compounds often emerge from high-throughput screening campaigns or structure-based drug design, allowing for precise optimization of pharmacokinetic properties and target engagement [114].
Table 2: Synthetic Anti-Biofilm Approaches and Characteristics
| Approach/Compound Type | Key Characteristics | Mechanisms of Action | Advantages | Limitations |
|---|---|---|---|---|
| Repurposed Drugs & Synthetic Analogs [114] | Screened from libraries of pharmacologically active compounds | Target biofilm-related proteins (e.g., QS regulators, biofilm-forming enzymes) [114] | Established safety profiles; known ADMET properties [114] | Often moderate efficacy; may require structural optimization [114] |
| 2-Aminoimidazoles [113] | Synthetic compounds inspired by natural marine products | Inhibit adhesion and biofilm formation in E. coli O157:H7 [113] | Specific molecular targets; designed bioavailability | Limited clinical validation; potential unknown long-term effects |
| Antimicrobial Peptides [109] | Short synthetic peptide sequences | Membrane disruption; EPS penetration; immune modulation [109] | Broad-spectrum activity; multiple mechanisms | Proteolytic instability; potential toxicity; high production costs |
| Engineered Nanomaterials [112] [110] | Precisely controlled nanostructures for drug delivery | Enhanced permeability and retention in biofilm matrix; responsive drug release [112] [110] | Improved drug solubility; targeted delivery; controlled release | Complex manufacturing; potential nanoparticle-specific toxicity |
| QS Inhibitors [109] | Synthetic analogs of autoinducer molecules | Competitive inhibition of QS receptor binding [109] | Attenuate virulence without growth pressure | Species-specific; may require combination therapies |
Synthetic compounds frequently exhibit superior pharmacokinetic profiles compared to natural products, with optimized solubility, stability, and tissue distribution characteristics [114]. Advanced delivery systems, including nanoparticles and functionalized materials, can be engineered to enhance biofilm penetration and provide sustained release of antimicrobial agents at the infection site [112] [110]. The defined chemical structures of synthetic compounds facilitate precise structure-activity relationship (SAR) studies, enabling methodical optimization of potency while minimizing off-target effects [114]. Computational approaches, including molecular docking and dynamics simulations, allow for rational design of compounds that selectively target biofilm-specific pathways such as c-di-GMP signaling systems [114].
However, synthetic anti-biofilm strategies are not without limitations. The high development costs and extensive timelines associated with novel compound development present significant barriers [109]. Synthetic molecules may exhibit unfavorable toxicity profiles or unexpected side effects that only become apparent in advanced preclinical testing [109]. The emergence of resistance to single-target agents remains a concern, particularly for compounds with specific molecular targets [110]. Additionally, regulatory hurdles for novel synthetic entities are typically more substantial than for natural products with established use histories.
Experimental Methodologies for Anti-Biofilm Evaluation
Standardized assays are essential for evaluating the efficacy of both natural and synthetic anti-biofilm compounds and facilitating direct comparison between different therapeutic approaches. The microtiter plate crystal violet assay represents the most widely employed method for quantifying biofilm formation inhibition or eradication [114]. This technique involves growing biofilms in 96-well plates, staining with crystal violet to visualize adhered biomass, and quantifying through spectrophotometric measurement [114]. Key parameters derived from these assays include:
- Minimum Biofilm Inhibitory Concentration (MBIC): The lowest concentration that prevents biofilm formation [109] [110]
- Minimum Biofilm Eradication Concentration (MBEC): The lowest concentration that eliminates established biofilms [110]
- Biofilm Inhibition Percentage: Quantitative measurement of reduction in biofilm biomass at various concentrations [114]
Advanced analytical methods provide deeper insights into anti-biofilm mechanisms. Confocal laser scanning microscopy (CLSM) with live/dead staining enables visualization of biofilm architecture and spatial distribution of viable cells following treatment [110]. Molecular docking studies predict interactions between candidate compounds and biofilm-related protein targets such as QS regulators and biofilm-forming enzymes [114]. Gene expression analysis through RT-qPCR assesses the impact of anti-biofilm agents on virulence gene transcription, particularly in QS pathways and matrix synthesis enzymes [110].
Figure 2 illustrates a comprehensive workflow that integrates these methods for systematic evaluation of anti-biofilm compounds.
Figure 2. Integrated experimental workflow for anti-biofilm compound evaluation. The diagram outlines key steps from initial screening to advanced characterization, highlighting the multi-faceted approach required for thorough assessment. Based on methodologies from [114] [110].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Experimental Materials for Anti-Biofilm Research
| Reagent/Material | Function/Application | Specific Examples & Notes |
|---|---|---|
| Crystal Violet Solution | Staining of adhered biofilm biomass for quantitative assessment [114] | Standardized concentration (typically 0.1-1%); ethanol-acetone for destaining [114] |
| 96-well Microtiter Plates | High-throughput screening of anti-biofilm compounds [114] | Polystyrene plates with non-treated surfaces for consistent biofilm formation [114] |
| QS Reporter Strains | Detection of quorum sensing inhibition potential | Engineered bacterial strains with luminescent or fluorescent reporters linked to QS promoters |
| Specific Protein Targets | Molecular docking and mechanism studies [114] | Quorum sensing regulators (e.g., LasR, LuxR); biofilm-forming enzymes [114] |
| Computational Tools | Prediction of compound-target interactions and ADMET properties [114] | Molecular docking software (AutoDock, Schrödinger); dynamics simulation packages (GROMACS) [114] |
| Nanocarrier Systems | Enhancement of compound delivery and penetration [112] [110] | Liposomes, polymeric nanoparticles; can be coated with phytochemicals [112] |
Molecular Mechanisms of Anti-Biofilm Action
The following diagram synthesizes current understanding of how natural and synthetic compounds target key pathways in biofilm formation and maintenance.
Figure 3. Molecular mechanisms of natural and synthetic anti-biofilm compounds. The diagram compares and contrasts the primary targets and modes of action for both compound classes, highlighting both distinct and shared pathways. Based on content from [112] [109] [110].
The critical comparison of natural and synthetic anti-biofilm compounds reveals complementary strengths that may be optimally leveraged through integrated approaches. Natural phytochemicals offer structurally diverse scaffolds with multi-target mechanisms that potentially reduce resistance development, while synthetic compounds provide precisely engineered properties for enhanced bioavailability and target specificity. The emerging strategy of combining natural product scaffolds with synthetic optimization represents a promising direction for future anti-biofilm drug development [112] [110].
Nanotechnology approaches are increasingly bridging the gap between natural and synthetic strategies by employing engineered delivery systems to overcome the pharmacokinetic limitations of natural compounds while maintaining their favorable bioactivity profiles [112] [110]. These advanced delivery platforms can enhance biofilm penetration through precise control of particle size, surface characteristics, and functionalization with targeting moieties [110]. Furthermore, the integration of computational methods with high-throughput screening enables more efficient identification and optimization of lead compounds from both natural and synthetic origins [114].
Future research priorities should include expanded investigation of combination therapies that pair sub-inhibitory concentrations of conventional antibiotics with natural anti-biofilm agents to achieve synergistic effects while minimizing toxicity [112]. Additionally, standardized protocols for evaluating anti-biofilm efficacy across research groups would facilitate more direct comparison of candidate compounds and accelerate translational progress. As understanding of biofilm biology continues to advance, particularly regarding interspecies interactions and immune evasion mechanisms, new therapeutic targets will undoubtedly emerge for both natural and synthetic intervention strategies.
The escalating global health threat of antimicrobial resistance (AMR) has necessitated a paradigm shift in how we combat bacterial infections, particularly those associated with medical devices and implants [115] [116]. The formation of bacterial biofilms on these surfaces is a primary concern, rendering infections notoriously difficult to treat as biofilms can exhibit tolerance to antibiotics up to 1,000 times greater than their free-floating counterparts [3] [117]. Traditional antibiotic-based strategies, which rely on biochemical mechanisms, increasingly face the challenge of AMR development. Within the protective extracellular polymeric substance (EPS) matrix of a biofilm, bacteria are shielded and can efficiently transfer resistance genes, accelerating the emergence of resistant strains [3] [117].
This crisis has spurred the exploration of antibiotic-free antibacterial strategies. Two principal approaches have emerged: chemical-based antimicrobial surfaces and mechanobactericidal nanostructured surfaces. Chemical strategies often involve the incorporation or leaching of biocidal agents such as antibiotics, silver nanoparticles, or polycations [118] [117]. In contrast, mechanobactericidal surfaces are inspired by natural structures like insect wings, which physically kill bacteria through nanoscale topography, mechanically rupturing cells upon contact [115] [116] [118]. This physical mode of action is a key differentiator from conventional antibiotics, which act upon bacteria by inhibiting cell membrane synthesis and interfering with the synthesis of vital proteins, DNA, and RNA, thus providing a promising approach to fight against antibiotic resistance [118].
Framed within the broader context of bacterial adhesion and biofilm initiation research, this review provides a critical assessment of the long-term performance of these two surface strategies. We delve into their fundamental mechanisms, evaluate their efficacy and durability through quantitative data, and outline advanced experimental protocols for their development and testing.
Mechanisms of Bacterial Adhesion and Biofilm Initiation
A comprehensive understanding of how bacteria colonize surfaces is fundamental to designing effective antibacterial strategies. Bacterial adhesion is a complex, multi-stage process governed by physicochemical and molecular interactions [119] [120].
The Two-Phase Adhesion Process
The initial interaction between a bacterial cell and a surface begins with a reversible attachment phase. This phase is primarily driven by weak, long-range physicochemical forces including van der Waals interactions, electrostatic forces, and hydrophobic interactions [119] [3]. The outcome of this phase is heavily influenced by the properties of both the bacterial cell and the material surface, such as surface charge, hydrophobicity, and roughness [119]. Rough surfaces, for instance, have been reported to provide more favorable sites for initial microbial attachment by increasing the surface area and protecting cells from shear forces [3].
Following initial attachment, bacteria undergo a transition to irreversible adhesion. This is a pivotal step where weak physical bonds are replaced by strong, permanent molecular interactions. Bacteria achieve this by producing adhesive structures like fimbriae and pili, and by secreting a sticky, self-produced matrix of EPS [119] [121]. The EPS matrix, composed of polysaccharides, proteins, nucleic acids, and lipids, acts as a biological glue, cementing the cells to the surface and forming the foundational scaffold of the biofilm [119] [3].
Biofilm Maturation and Resilience
Once irreversibly attached, bacteria proliferate and form structured microcolonies that evolve into a mature biofilm. This mature biofilm is characterized by a complex 3D architecture with heterogeneous conditions and enclosed bacterial communities [3]. A key feature of mature biofilms is quorum sensing (QS), a cell-to-cell communication process where bacteria secrete and detect signaling molecules called autoinducers. When a critical population density is reached, QS triggers the coordinated expression of virulence genes and enhances EPS production, further solidifying the biofilm's structural integrity and resistance [119] [117].
The resulting biofilm acts as a formidable biological barrier, significantly impeding the penetration of antimicrobial agents and providing a protected niche for the enclosed bacterial cells. This, combined with the metabolic heterogeneity of cells within the biofilm and the potential for horizontal gene transfer, makes biofilm-associated infections extremely challenging to eradicate [3] [117].
The following diagram illustrates the key stages and critical control points in the biofilm formation process:
Chemical Antimicrobial Surfaces: Mechanisms and Long-Term Challenges
Chemical antibacterial surfaces function by releasing or presenting biocidal agents that kill microbes upon contact. These can be broadly categorized into two release-based and contact-active mechanisms [118] [117].
Release-Based Mechanisms
This common strategy involves the incorporation of antimicrobial agents—such as antibiotics, silver ions, or zinc oxide nanoparticles—into a material matrix. These agents then leach out over time, creating a zone of inhibition around the material that kills approaching planktonic bacteria or those that have recently adhered [117]. While highly effective initially, a significant limitation is the depletion of the antimicrobial reservoir. The efficacy of these surfaces is inherently time-limited, as the active agent can be rapidly exhausted, after which the surface loses its protective function and becomes susceptible to colonization [118]. Furthermore, the sub-lethal release of antimicrobials can exert a selective pressure, potentially promoting the development of AMR [116] [117].
Contact-Active Mechanisms
Contact-active surfaces, on the other hand, do not leach biocides but are functionalized with immobilized antimicrobial molecules like polycations, antimicrobial peptides, or enzymes [118] [117]. Upon contact with the bacterial membrane, these molecules can disrupt membrane integrity, leading to cell death. While this approach avoids the issue of reservoir depletion, its long-term effectiveness can be compromised by the accumulation of dead bacteria and debris on the surface. This organic layer can mask the active sites, reducing bactericidal efficiency and potentially facilitating the attachment of new bacteria, leading to biofilm formation [118].
Table 1: Summary of Chemical Antimicrobial Surface Strategies and Their Long-Term Performance
| Strategy | Mechanism of Action | Key Agents | Long-Term Challenges |
|---|---|---|---|
| Release-Based | Diffusion of biocidal agents into the local environment. | Antibiotics, Silver nanoparticles, Metal oxides (e.g., ZnO) [117]. | Finite reservoir leads to transient efficacy; potential for AMR development due to selective pressure [118] [117]. |
| Contact-Active | Microbial membrane disruption upon contact with the surface. | Polycations, Antimicrobial peptides (AMPs), Enzymes [118] [117]. | Fouling and loss of activity due to accumulation of dead cells and debris; possible toxicity to host cells [118]. |
Mechanobactericidal Nanostructured Surfaces
Mechanobactericidal surfaces offer a fundamentally different, drug-free approach by leveraging physical forces to inactivate bacteria. This strategy is biomimetic, inspired by the naturally nanopatterned surfaces of insect wings, such as those of cicadas and dragonflies, which are lethal to bacterial cells [115] [116] [118].
The Mechano-bactericidal Mechanism
The bactericidal action is not chemical but mechanical. When a bacterial cell settles on a surface densely covered with high-aspect-ratio nanopillars, the nanopillars interact with the cell envelope. The primary mechanism is believed to be the adhesion-based killing [115]. As the bacterium attempts to adhere, its cell membrane tightly conforms to the nanopattern, leading to a massive increase in contact area. The differential adhesion between the basal membrane adhering to the nanopillar tips and the suspended membrane spanning the gaps between pillars generates substantial tensile stresses. If these stresses exceed the tensile strength of the cell membrane, it leads to localized rupture or critical deformation, causing cell death [115] [118]. This process is influenced by the geometric parameters of the nanostructures—such as diameter, height, and spacing—as well as the mechanical properties of the bacterial cell wall, explaining why Gram-negative bacteria with thinner cell walls are often more susceptible than Gram-positive species [115] [116].
Long-Term Performance and Self-Cleaning
A significant advantage of mechanobactericidal surfaces is their potential for long-term activity. Since the antibacterial effect is a direct function of the physical topography, the surface remains effective as long as the nanostructural integrity is maintained [115]. This makes them less susceptible to the issues of reservoir depletion that plague release-based chemical strategies.
However, a key challenge for long-term use is surface fouling by the corpses of killed bacteria, which can mask the nanostructures and nullify their bactericidal activity [118]. To address this, advanced smart surfaces are being developed. For instance, researchers have created a dual-functional surface combining cicada wing-inspired nanopatterns with salt-responsive polyzwitterionic brushes (polyDVBAPS) [118]. This design allows the surface to switch its function: in aqueous environments, the collapsed polymer brushes expose the nanopillars for bacterial killing, while upon exposure to a salt solution (e.g., 1 M NaCl), the polymer chains stretch, releasing the dead bacteria and regenerating a clean, active surface [118].
Table 2: Key Geometric Parameters and Their Influence on Mechanobactericidal Efficacy
| Geometric Parameter | Influence on Bactericidal Activity | Optimal Range (Approx.) | Remarks |
|---|---|---|---|
| Nanopillar Diameter | Critical for stress concentration; smaller diameters increase localized pressure on cell membrane [115]. | < 100 nm | Must be sufficiently sharp to penetrate or strain the cell envelope. |
| Nanopillar Spacing | Dictates the degree of membrane stretching; spacing smaller than the bacterial cell is essential [115] [118]. | ~ 50-200 nm | Spacing must allow for significant unsupported membrane area to be generated. |
| Nanopillar Height | Must be sufficient to prevent the cell from contacting the underlying substrate and "bottoming out" [115]. | > 200 nm | Ensures interaction is dominated by the nanostructures, not the base material. |
| Aspect Ratio | High aspect ratio (height/diameter) can enhance flexibility and adhesion-dependent killing [115]. | > 3:1 | Balances mechanical strength with the required deformation. |
Experimental Protocols for Development and Evaluation
Robust experimental methodologies are essential for the development and critical assessment of antibacterial surfaces. The following protocols outline key approaches for fabrication, efficacy testing, and mechanical characterization.
Fabrication of Biomimetic Nanopatterned Surfaces via Nanoimprinting
This protocol describes a method for creating biomimetic nanopatterns using anodic aluminum oxide (AAO) templates, a technique that allows for scalable production of highly ordered nanostructures [118].
- Template Preparation: Acquire a commercial porous AAO template with the desired pore diameter and spacing. Clean the template via sonication in acetone and ethanol, followed by oxygen plasma treatment to ensure a clean, hydrophilic surface.
- Polymer Substrate Preparation: Select a suitable polymer substrate (e.g., polycarbonate, epoxy resin). The substrate should be thoroughly cleaned and dried.
- Hot Embossing: Place the AAO template in contact with the polymer substrate. Apply a pressure of 0.5-2 MPa and heat the assembly to a temperature 10-20°C above the glass transition temperature (Tg) of the polymer. Maintain these conditions for 10-30 minutes to allow the polymer to flow and fill the nanopores of the AAO template.
- Demolding: Cool the system below the Tg of the polymer before carefully releasing the pressure and separating the AAO template from the polymer substrate. This results in a negative replica of the AAO pattern on the polymer surface.
- Surface Functionalization (Optional): To impart a bacterial-release capability, graft salt-responsive polyzwitterionic brushes (e.g., polyDVBAPS) onto the nanopatterned surface. This can be achieved via ultraviolet-induced surface-initiated polymerization. Immerse the substrate in a monomer solution and expose to UV light (e.g., 365 nm wavelength) for a predetermined time (e.g., 30-60 minutes) in an inert atmosphere [118].
Quantitative Assessment of Bactericidal Efficiency
A standardized assay is required to quantify the ability of a surface to kill bacteria upon contact [115] [118].
- Inoculum Preparation: Grow a bacterial strain of interest (e.g., Pseudomonas aeruginosa for Gram-negative, Staphylococcus aureus for Gram-positive) to the mid-logarithmic phase in a suitable broth (e.g., Lysogeny Broth). Centrifuge the culture, wash the cells, and resuspend them in a physiological buffer (e.g., phosphate-buffered saline, PBS) to a concentration of ~10^7 colony-forming units (CFU)/mL.
- Surface Inoculation and Incubation: Apply a small droplet (e.g., 20 µL) of the bacterial suspension onto the test surface. Cover with a sterile, oxygen-permeable membrane to prevent evaporation and ensure full contact. Incubate the inoculated surfaces for a set period (e.g., 2-4 hours) at 37°C and appropriate humidity.
- Cell Recovery and Enumeration: After incubation, transfer each sample into a tube containing 5 mL of PBS. Subject the tubes to vigorous vortexing (e.g., 2-5 minutes) followed by sonication in a water bath (e.g., 5-10 minutes) to detach the adhered bacteria. Serially dilute the resulting suspension and plate onto nutrient agar plates.
- Data Analysis: After incubating the agar plates for 18-24 hours at 37°C, count the resulting colonies. Calculate the bactericidal efficiency (BE) using the formula: BE (%) = [(CFU_control - CFU_test) / CFU_control] × 100 where CFU_control is the number of viable cells recovered from a reference material (e.g., a smooth, non-bactericidal surface), and CFU_test is the count from the nanostructured test surface.
Measuring Bacterial Adhesion Forces via Atomic Force Microscopy (AFM)
Atomic force microscopy (AFM) provides quantitative, nanoscale measurements of the forces involved in bacterial adhesion, a key parameter in understanding the initial stages of biofilm formation and the mechanism of mechanobactericidal surfaces [119].
- Probe Functionalization: A standard AFM cantilever is functionalized to represent a single bacterial cell. This can be done by chemically attaching a single bacterial cell to the tip-less cantilever using a bio-compatible epoxy glue. Alternatively, the cantilever can be coated with a layer of bacterial adhesins or extracellular polymeric substances to probe specific molecular interactions [119].
- Force Spectroscopy: The functionalized cantilever is brought into contact with the test surface in a liquid cell filled with PBS. Using a piezo-electric controller, a series of approach-retract cycles are performed at different locations on the sample surface.
- Data Collection: During each retraction cycle, a force-distance (F-D) curve is recorded. The adhesion force is determined from the "pull-off" force or the maximum negative force in the retraction curve. Hundreds of curves are collected to ensure statistical significance.
- Data Interpretation: Adhesion forces are reported in nanonewtons (nN). Studies have categorized bacterial adhesion into regimes: weak adherence (<1 nN), where bacteria may not initiate an adaptive response; intermediate adherence (1-10 nN); and strong adherence (>10 nN), which is often associated with irreversible attachment and the onset of biofilm formation [119]. This technique allows for direct comparison of how different surface chemistries and topographies influence the fundamental adhesion forces.
The following workflow diagram integrates these key experimental processes:
The Scientist's Toolkit: Essential Reagents and Materials
The research and development of advanced antibacterial surfaces rely on a suite of specialized reagents, materials, and instrumentation.
Table 3: Key Research Reagent Solutions for Antibacterial Surface Development
| Reagent / Material | Function / Application | Key Characteristics |
|---|---|---|
| Anodic Aluminum Oxide (AAO) Templates | Master template for nanoimprinting lithography; defines nanopattern geometry (diameter, spacing) [118]. | Highly ordered nanopores; tunable parameters; reusable. |
| Polycarbonate (PC) / Epoxy Resins | Substrate materials for hot embossing; allow for high-fidelity replication of nanoscale features [118]. | Thermoplastic properties; optical clarity; biocompatibility. |
| Salt-Responsive Polyzwitterions (e.g., polyDVBAPS) | Imparts "smart" bacterial-release functionality; grafts onto nanopatterns to create dual-action surfaces [118]. | Conformational change (collapse/stretch) in response to ionic strength. |
| Quorum Quenching Enzymes (Acylase, Lactonase) | Nano-formulated additives to disrupt bacterial communication (QS); inhibit virulence and biofilm formation without killing [117]. | Degrades AHL autoinducers; reduces selective pressure for resistance. |
| Atomic Force Microscopy (AFM) with Colloidal Probes | Quantifies nanoscale adhesion forces between bacteria and surfaces; maps mechanical properties [119]. | Piconewton (pN) to nanonewton (nN) force sensitivity; operates in liquid. |
The critical assessment of chemical and mechanobactericidal surfaces reveals a clear trade-off between initial potency and long-term, resistance-resistant performance. Chemical strategies, while potent, often face inherent limitations in longevity due to agent depletion and pose a tangible risk of fostering AMR. In contrast, mechanobactericidal nanostructured surfaces offer a durable, antibiotic-free alternative whose physical mode of action presents a formidable barrier to resistance development.
The future of antibacterial surface design lies in the convergence of these strategies to create intelligent, multi-functional systems. The integration of mechanobactericidal nanostructures with stimuli-responsive polymer brushes, as exemplified by the salt-responsive surface, represents a pioneering step toward regenerable, long-lasting antibacterial materials [118]. Further synergy can be achieved by combining nanopatterns with non-lethal anti-virulence agents like quorum-quenching enzymes, which disrupt biofilm formation without exerting lethal selective pressure [117]. As fabrication techniques advance, overcoming challenges in scalability and cost, these next-generation biomimetic surfaces are poised to play a transformative role in mitigating device-related infections and combating the global AMR crisis.
The transition from in vitro models to in vivo outcomes represents a critical pathway in biomedical research, particularly in the study of bacterial adhesion and biofilm initiation. This technical guide examines the strengths, limitations, and appropriate applications of both methodological approaches within the context of biofilm research. By comparing quantitative data, experimental protocols, and methodological frameworks, this review provides researchers with a structured approach for selecting appropriate models and interpreting translational data. The integration of both models offers the most robust strategy for advancing therapeutic interventions against biofilm-associated infections, which demonstrate significantly enhanced resistance to antimicrobial agents and host immune responses [3] [17].
Biofilm research presents unique challenges due to the complex, three-dimensional nature of microbial communities embedded in extracellular polymeric substances (EPS). These structures demonstrate emergent properties not observable in planktonic cultures, including enhanced resistance mechanisms and complex community interactions [3] [122]. Understanding the distinction between in vitro and in vivo approaches is fundamental to designing experiments that yield clinically relevant data.
In vitro (Latin for "in glass") refers to experiments conducted outside living organisms, typically in controlled laboratory environments such as test tubes, petri dishes, or microtiter plates [123] [124]. These systems allow for high precision and variable control but lack the complexity of whole-organism biology. Conversely, in vivo (Latin for "within the living") studies occur within living organisms, including animal models and human clinical trials, providing essential data on systemic interactions but introducing greater complexity and cost [123] [124].
For biofilm research specifically, this distinction is crucial as the biofilm lifecycle – from initial adhesion to maturation and dispersion – is influenced by environmental conditions that differ significantly between controlled laboratory settings and living hosts [3] [17]. The choice between these models should be guided by research phase, specific questions, and resources available.
Comparative Analysis: Methodological Approaches
Definition and Experimental Setup
Table 1: Fundamental Differences Between In Vitro and In Vivo Approaches
| Parameter | In Vitro | In Vivo |
|---|---|---|
| Definition | Experiments performed outside living organisms in artificial environments [123] | Experiments conducted within living organisms [123] |
| Environment | Controlled, simplified systems (test tubes, petri dishes) [123] [124] | Complex, natural systems (animal models, human trials) [123] [124] |
| Control Level | High precision, isolated variables [123] | Multiple interacting systems, limited control [123] |
| Cost & Duration | Lower cost, faster results [123] [124] | Higher cost, time-intensive [123] [124] |
| Biofilm Relevance | Ideal for studying initial adhesion mechanisms under controlled conditions [3] | Essential for understanding host-pathogen interactions and therapeutic efficacy [17] |
Applications in Biofilm Research
In vitro models are particularly valuable during early research phases investigating fundamental mechanisms of bacterial adhesion and biofilm initiation. These systems allow researchers to study the initial reversible attachment of microorganisms to surfaces, which is governed by weak physical forces including van der Waals interactions, electrostatic forces, and hydrophobic effects [3] [17]. The transition to irreversible attachment, facilitated by bacterial surface structures such as pili, fimbriae, and adhesins, can also be effectively studied in in vitro systems where surface properties can be carefully controlled [3].
In vivo models become essential when investigating how biofilms interact with host systems, including immune responses, nutrient availability, and multi-organism dynamics [17]. These models reveal critical aspects of biofilm pathogenesis that cannot be captured in vitro, such as the role of host inflammatory responses in chronic infections and the effect of biofilm structures on antibiotic penetration in living tissues [17].
Quantitative Assessment Methods for Biofilm Studies
Table 2: Biofilm Quantification and Characterization Methods
| Method Category | Specific Techniques | Key Applications in Biofilm Research | Advantages/Limitations |
|---|---|---|---|
| Direct Quantification | Colony Forming Units (CFU) [122] | Enumeration of viable cells in biofilm matrix | Advantages: Differentiates live vs. dead cells; Limitations: Time-intensive, requires culture viability [122] |
| Direct Quantification | Flow Cytometry [122] | Automated cell counting with fluorescence detection | Advantages: High-throughput, multi-parameter analysis; Limitations: Requires specialized equipment [122] |
| Indirect Quantification | Crystal Violet Staining [122] | Biomass quantification through EPS binding | Advantages: Simple, cost-effective; Limitations: Does not differentiate live/dead cells [122] |
| Indirect Quantification | ATP Bioluminescence [122] | Metabolic activity measurement via luciferase reaction | Advantages: Rapid results; Limitations: Indirect measure of cell viability [122] |
| Morphological Characterization | Scanning Electron Microscopy (SEM) [122] | High-resolution imaging of biofilm architecture | Advantages: Detailed structural information; Limitations: Requires extensive sample preparation [122] |
| Morphological Characterization | Confocal Scanning Laser Microscopy (CSLM) [122] | 3D visualization of living biofilms with fluorescent tags | Advantages: Real-time observation of biofilm dynamics; Limitations: Expensive equipment [122] |
Experimental Protocols for Biofilm Research
StandardizedIn VitroBiofilm Formation Assay
Purpose: To establish reproducible biofilms for studying initial adhesion and maturation under controlled conditions [122].
Materials:
- Sterile microtiter plates (96-well recommended)
- Appropriate bacterial strains (e.g., ESKAPE pathogens: Staphylococcus aureus, Pseudomonas aeruginosa)
- Culture media optimized for biofilm formation (e.g., Tryptic Soy Broth with 1% glucose)
- Incubator with controlled temperature and humidity
- Staining solutions (crystal violet, fluorescent dyes)
- Plate reader or microscope for quantification
Procedure:
- Prepare bacterial suspension from fresh overnight culture, adjusting to approximately 10^6 CFU/mL in appropriate media [122].
- Aliquot 200μL bacterial suspension into each well of sterile microtiter plate.
- Incubate under optimal growth conditions for 24-48 hours to allow biofilm formation.
- Carefully remove planktonic cells by washing wells with phosphate-buffered saline (PBS).
- Fix biofilm with 200μL of 99% methanol for 15 minutes.
- Stain with 200μL of 0.1% crystal violet solution for 5-15 minutes.
- Wash gently to remove unbound stain and air-dry.
- Dissolve bound stain in 200μL of 33% acetic acid.
- Quantify by measuring absorbance at 570-600nm using plate reader [122].
Validation: Include positive and negative controls with known biofilm-forming and non-forming strains respectively. Normalize results to control values.
2In VivoBiofilm Infection Model
Purpose: To evaluate biofilm formation, persistence, and treatment efficacy in a living system [17].
Materials:
- Appropriate animal model (typically murine)
- Bacterial strains with selectable markers
- Catheters or implant materials (if studying device-related infections)
- Anesthesia and surgical equipment
- Tissue processing equipment (homogenizers)
- Culture media for CFU enumeration
Procedure:
- Anesthetize animals according to approved ethical protocols.
- Introduce bacteria via appropriate route (intravenous, subcutaneous, or using implanted devices).
- Monitor animals for signs of infection and disease progression.
- Euthanize at predetermined time points post-infection.
- Harvest target tissues or devices under aseptic conditions.
- Homogenize tissues in sterile PBS using mechanical homogenizers.
- Prepare serial dilutions of homogenate and plate on selective media.
- Incubate plates for 24-48 hours and enumerate CFUs [122].
- Process additional tissue samples for histology or microscopy.
Validation: Confirm biofilm formation in tissues using histological staining (e.g., Gram stain, H&E) or microscopy techniques.
Integration Framework: From Laboratory to Clinical Applications
The most effective research strategy employs both in vitro and in vivo models sequentially, leveraging the strengths of each approach while mitigating their limitations [123] [124]. This integrated framework is particularly valuable for screening anti-biofilm compounds and therapeutic strategies.
The sequential approach begins with in vitro screening to identify promising candidates and elucidate basic mechanisms, followed by in vivo validation to assess efficacy in biologically relevant systems, and culminates in clinical trials to establish human applications [123] [124]. This framework maximizes resource efficiency by using low-cost, high-throughput in vitro methods for initial screening before committing to more resource-intensive in vivo studies.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents for Biofilm Studies
| Reagent/Material | Primary Function | Application Notes |
|---|---|---|
| Microtiter Plates | High-throughput biofilm cultivation [122] | Polystyrene surfaces most common; surface modification possible for specific adhesion studies |
| Crystal Violet | EPS and biomass staining [122] | Simple, cost-effective; binds to extracellular polymeric substances and cell surfaces |
| Fluorescent Dyes (SYTO9, PI) | Viability assessment and visualization [122] | Differentiate live/dead cells; compatible with confocal microscopy for 3D structure analysis |
| Cell Homogenizers | Biofilm dispersion for quantification [122] | Mechanical disruption required for accurate CFU enumeration from mature biofilms |
| Transwell Inserts | Co-culture and host-pathogen interaction studies | Permeable membranes allow molecular communication while maintaining separate compartments |
| Quorum Sensing Inhibitors | Interference with bacterial communication [3] | Target acyl-homoserine lactones (Gram-negative) or autoinducing peptides (Gram-positive) |
| Extracellular Matrix Components | Surface preconditioning for adhesion studies [3] | Fibronectin, collagen, or host fluid components to mimic in vivo surface conditions |
Bacterial Adhesion Signaling Pathway in Biofilm Initiation
The molecular pathway governing bacterial adhesion and biofilm initiation involves sophisticated signaling mechanisms. The process begins with environmental cues that trigger the transition from planktonic to sessile lifestyles through the key secondary messenger bis-(3'-5')-cyclic dimeric guanosine monophosphate (c-di-GMP) [17]. Elevated c-di-GMP levels downregulate flagellar motility while upregulating the production of bacterial surface adhesins and extracellular polymeric substances, establishing the structural foundation of biofilms [17]. As biofilms mature, quorum sensing mechanisms coordinate community behaviors through autoinducer signaling molecules, enabling population-density dependent gene regulation that optimizes biofilm architecture and function [3] [17].
The strategic integration of in vitro and in vivo models provides the most comprehensive approach for advancing biofilm research from basic mechanisms to clinical applications. While in vitro systems offer controlled environments for elucidating fundamental adhesion processes and initial screening, in vivo models deliver essential insights into host-pathogen interactions and therapeutic efficacy. The evolving landscape of biofilm research continues to benefit from technological advances in both methodological approaches, enabling more accurate modeling of the complex biofilm lifecycle and more effective translation of laboratory findings to clinical outcomes.
Comparative Analysis of Biofilm Resistance Mechanisms Across Key Pathogenic Species
Biofilms represent a protected mode of microbial growth that allows cells to survive in hostile environments. These structured microbial communities, encased in a self-produced extracellular polymeric substance (EPS), are a significant contributor to antimicrobial resistance (AMR) and a major cause of persistent infections [4] [3]. The complex relationship between biofilm formation and AMR presents a formidable challenge in clinical settings, particularly in device-related infections and chronic conditions such as those in cystic fibrosis patients [4]. Understanding the comparative biofilm resistance mechanisms across key pathogenic species is crucial for developing effective therapeutic interventions.
The biofilm lifestyle provides physical and physiological protection against antimicrobial agents and host immune responses. Bacteria within biofilms can exhibit 10–1000-fold greater antibiotic resistance than their planktonic counterparts [125]. This review systematically examines the biofilm resistance mechanisms of ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), which represent the most problematic clinical pathogens due to their ability to "escape" the biocidal action of antibiotics [3] [125]. We provide a comparative analysis of their resistance profiles, structural characteristics, and the molecular basis of biofilm-mediated resistance, supported by experimental data and methodological guidelines for researchers in the field.
Biofilm Architecture and Developmental Lifecycle
Structural Dynamics and Composition
Biofilms are complex, three-dimensional microbial communities embedded in a matrix of extracellular polymeric substances (EPS) that develop through several distinct stages [3]. This matrix typically comprises polysaccharides, lipids, proteins, and extracellular DNA (eDNA) [4]. The EPS composition varies significantly depending on microbial species, nutrient availability, and environmental conditions [4]. The matrix can make up over 90% of the biofilm mass, creating a structurally robust layer that acts as a protective barrier for embedded cells [4] [126].
Table 1: Key Components of Biofilm Extracellular Polymeric Substance Matrix
| Matrix Component | Primary Function | Representative Examples |
|---|---|---|
| Polysaccharides | Structural integrity, adhesion, barrier protection | PIA/PNAG in S. aureus and S. epidermidis, alginate in P. aeruginosa |
| Extracellular DNA (eDNA) | Structural support, cation chelation, genetic exchange | DNA from lysed cells, neutrophil extracellular traps (NETs) |
| Proteins | Adhesion, structural stability, enzymatic activity | Adhesins, amyloids, matrix-degrading enzymes |
| Lipids | Hydrophobicity, barrier function | Surfactants, membrane vesicles |
Developmental Lifecycle
Biofilm formation follows a conserved developmental sequence that can be divided into five key stages:
Initial Reversible Attachment: Free-living planktonic cells adhere to preconditioned surfaces through weak interactions such as van der Waals forces and electrostatic interactions [4] [3]. Surface properties including roughness significantly influence this attachment phase [3].
Irreversible Attachment: The reversibly attached cells transition to permanent attachment through the production of adhesive extracellular polymeric substances [4] [3]. In some species, this involves surface structures such as pili and microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) [126].
Microcolony Formation: Attached cells proliferate and form microcolonies, responding to signaling molecules such as cyclic diguanylate monophosphate (c-di-GMP) which promotes a sessile lifestyle [4].
Biofilm Maturation: Development of a complex three-dimensional architecture with defined water channels that facilitate nutrient distribution and waste removal [4] [3]. During this stage, metabolic heterogeneity emerges with gradients of oxygen, nutrients, and metabolic activity [127].
Dispersal: Active release of cells from the biofilm to colonize new niches [4]. This can occur through seeding, erosion, or sloughing mechanisms in response to environmental cues such as nutrient limitation [4].
The following diagram illustrates the key developmental stages and regulatory mechanisms in the biofilm lifecycle:
Diagram 1: Biofilm developmental lifecycle. The process begins with initial attachment of planktonic cells, progresses through irreversible attachment and maturation, and culminates in dispersal. Key regulatory mechanisms include c-di-GMP signaling and quorum sensing (QS). EPS production strengthens attachment and matrix formation.
Comparative Biofilm Resistance Mechanisms
Biofilms employ multiple concurrent strategies to achieve remarkable levels of antimicrobial resistance. These mechanisms can be categorized into physical barriers, physiological adaptations, and genetic determinants that operate synergistically to protect the microbial community.
Physical Barrier Mechanisms
The biofilm matrix acts as a formidable physical barrier that restricts antimicrobial penetration through several mechanisms:
Diffusion Limitation: The dense EPS matrix significantly retards the diffusion of antimicrobial molecules, particularly those with larger molecular weights [4]. This delayed penetration allows bacteria within the biofilm to deploy additional resistance mechanisms.
Binding and Inactivation: Matrix components can directly bind and neutralize antimicrobial agents. Positively charged aminoglycosides, for example, interact with negatively charged eDNA in the matrix, reducing their effective concentration [4]. Similarly, some antibiotics form complexes with matrix polysaccharides or are degraded by enzymes trapped within the EPS [4].
Host-Derived Protection: In chronic infections, host components can augment the protective barrier. In cystic fibrosis lungs, eDNA produced by P. aeruginosa combines with host eDNA to form a shield that protects the biofilm from tobramycin and immune cells [4]. Neutrophil extracellular traps (NETs) can similarly surround biofilms and limit antibiotic access [4].
Physiological and Metabolic Adaptations
Biofilms develop metabolic heterogeneity that significantly contributes to antibiotic tolerance:
Metabolic Gradients: The structured biofilm architecture creates gradients of oxygen, nutrients, and metabolic waste products [4] [127]. Cells in the biofilm interior often experience nutrient limitation and develop slow-growing or dormant phenotypes [127].
Persister Cell Formation: A subpopulation of metabolically dormant cells, known as persisters, exhibits exceptional tolerance to antimicrobials that target active cellular processes [4] [127]. These cells are not genetically resistant but can repopulate the biofilm after antibiotic treatment is discontinued.
Altered Microenvironment: The metabolic activity of biofilm cells creates localized microenvironments with distinct pH and oxygen tension that can compromise the activity of certain antibiotics [127].
Genetic Determinants and Horizontal Gene Transfer
Biofilms serve as hotspots for the exchange of genetic material, accelerating the dissemination of resistance genes:
Horizontal Gene Transfer: The dense, structured environment of biofilms facilitates efficient conjugation and transformation, enabling the spread of resistance genes, including those encoding extended-spectrum beta-lactamases (ESBLs) and carbapenemases [4] [128].
Resistance Gene Acquisition: Biofilm-associated bacteria demonstrate an enhanced capacity to acquire and maintain resistance genes. A 2025 study found significant correlations between biofilm formation and resistance to carbapenems, cephalosporins, and piperacillin/tazobactam in clinical isolates [125].
The following diagram illustrates the multifactorial nature of biofilm-associated antimicrobial resistance:
Diagram 2: Multifactorial mechanisms of biofilm-mediated antimicrobial resistance. Biofilms employ physical barrier functions, physiological adaptations, and genetic exchange mechanisms that operate synergistically to confer protection against antimicrobial agents.
Species-Specific Variations in Biofilm Resistance
While biofilms universally confer protection, significant differences exist in the resistance profiles and mechanisms employed by different pathogenic species. Understanding these species-specific variations is crucial for developing targeted anti-biofilm strategies.
Comparative Resistance Profiles of ESKAPE Pathogens
A comprehensive 2025 study of 165 clinical ESKAPE isolates revealed substantial differences in resistance profiles between species [125]:
Table 2: Comparative Biofilm Formation and Antimicrobial Resistance in ESKAPE Pathogens
| Pathogen | Biofilm Formation Capacity | Key Resistance Associations | Notable Resistance Markers |
|---|---|---|---|
| Acinetobacter baumannii | High (88.6% of isolates) | Carbapenems (74.29%), cephalosporins | Carbapenemase production (25.7% of isolates) |
| Klebsiella pneumoniae | High (91.4% of isolates) | Carbapenems (45.71%), colistin (42.86%) | Carbapenemase production (34.3%), MBL production |
| Pseudomonas aeruginosa | Moderate (82.9% of isolates) | Relatively lower resistance overall | Carbapenemase production (11.4%) |
| Staphylococcus aureus | Variable | Fluoroquinolones (53-66.67%) | MRSA (46.7% carrying mecA) |
| Enterococcus faecium | Variable | High MDR (90%), fluoroquinolones (86.67%) | Vancomycin resistance (20%, primarily vanB) |
The relationship between biofilm formation and antimicrobial resistance is species-dependent and antibiotic-specific. In gram-negative bacteria, specific relationships have been identified: gentamicin and ceftazidime resistance correlates with biofilm formation in Escherichia coli, piperacillin/tazobactam and colistin resistance in Klebsiella pneumoniae, and ciprofloxacin resistance in Pseudomonas aeruginosa [129]. Interestingly, multidrug-resistant isolates do not necessarily form more robust biofilms than non-multiresistant isolates, indicating that the relationship between overall resistance and biofilm formation is complex [129].
Molecular Basis of Species-Specific Variations
The variations in biofilm resistance mechanisms across species stem from differences in matrix composition, regulatory networks, and specific resistance determinants:
Matrix Composition Differences: The chemical nature of the EPS matrix varies significantly between species. P. aeruginosa produces alginate, Pel, and Psl polysaccharides; S. aureus and S. epidermidis generate poly-N-acetylglucosamine (PNAG); while K. pneumoniae produces capsular polysaccharides integrated into the biofilm matrix [4] [126]. These compositional differences influence the charge, porosity, and protective properties of the biofilm.
Regulatory Networks: Species-specific regulatory mechanisms control biofilm development and associated resistance. The ica locus regulates PNAG production in staphylococci [126], while complex quorum sensing systems involving Las, Rhl, and PQS circuits control biofilm maturation in P. aeruginosa [4].
Resistance Gene Prevalence: The distribution of specific resistance genes varies among species. Carbapenemase genes (OXA, NDM, KPC, VIM, IMP) are predominantly found in gram-negative pathogens like K. pneumoniae and A. baumannii [128] [125], while mecA mediates methicillin resistance in S. aureus and vancomycin resistance genes (vanA, vanB) are primarily associated with enterococci [125].
Experimental Methodologies for Biofilm Analysis
Robust and standardized methodologies are essential for comparative analysis of biofilm formation and associated resistance mechanisms. This section details key experimental protocols for quantitative and qualitative biofilm assessment.
Quantitative Biofilm Assessment Methods
Table 3: Quantitative Methods for Biofilm Characterization
| Method | Principle | Key Applications | Advantages/Limitations |
|---|---|---|---|
| Crystal Violet Staining | Dye binding to biomass | Total biofilm biomass quantification | High-throughput, but does not distinguish live/dead cells |
| Colony Forming Units (CFU) | Viable cell enumeration on agar plates | Quantification of cultivable cells | Gold standard for viability, but labor-intensive and misses VBNC cells |
| Microtiter Plate Assay | Biofilm formation in 96-well plates | High-throughput screening of biofilm formation | Standardized, scalable, but limited surface types |
| ATP Bioluminescence | Measurement of cellular ATP | Metabolic activity assessment | Rapid, sensitive, but correlates with metabolic activity rather than cell number |
| Quartz Crystal Microbalance | Mass-sensitive detection | Real-time biofilm growth monitoring | Label-free, real-time monitoring, but requires specialized equipment |
Advanced Imaging and Analysis Techniques
Modern biofilm research employs sophisticated imaging technologies that enable detailed structural and functional analysis:
Confocal Scanning Laser Microscopy (CSLM): This technique allows non-invasive optical sectioning of biofilms and three-dimensional reconstruction of biofilm architecture [122]. When combined with fluorescent viability stains or fluorescent protein reporters, CSLM can visualize spatial patterns of metabolic activity and gene expression within biofilms.
BiofilmQ Software Tool: BiofilmQ is a comprehensive image cytometry software designed for automated, high-throughput quantification of 3D biofilm properties [47]. It can analyze hundreds of parameters including biofilm volume, mean thickness, surface area, roughness coefficient, and spatially resolved fluorescence intensity distributions.
Scanning Electron Microscopy (SEM): Provides high-resolution images of biofilm surface topography and cell arrangements, though requires extensive sample preparation that may alter native biofilm structure [122].
The following workflow illustrates a standardized approach for biofilm assessment and analysis:
Diagram 3: Standardized workflow for biofilm assessment. The process begins with sample inoculation, progresses through biofilm maturation, and culminates in quantitative and structural analysis using complementary methodologies.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Materials for Biofilm Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| 96-Well Polystyrene Plates | Substrate for biofilm formation | High-throughput screening of biofilm formation [122] |
| Crystal Violet Solution | Biomass staining | Total biofilm quantification [122] |
| Live/Dead BacLight Viability Stains | Bacterial viability discrimination | CSLM analysis of spatial viability patterns [122] |
| DNase I | eDNA degradation | Matrix disruption studies [127] |
| Dispersin B | PNAG polysaccharide degradation | Staphylococcal biofilm disruption [127] |
| Cellulose Filters (0.45µm) | Bacterial lawn preparation | Cell surface hydrophobicity measurements [126] |
| TSB/Tryptic Soy Broth | Growth medium | Standardized biofilm growth conditions [126] [125] |
This comparative analysis reveals that while biofilm-mediated resistance mechanisms share common principles across pathogenic species, significant variations exist in their implementation and relative contributions to overall resistance. The ESKAPE pathogens employ distinct strategies rooted in their unique genetic backgrounds, matrix compositions, and regulatory networks. These species-specific adaptations present both challenges and opportunities for therapeutic development.
The relationship between biofilm formation and antimicrobial resistance is complex and context-dependent, influenced by specific pathogen-drug combinations rather than following a universal pattern. This understanding is crucial for guiding effective therapeutic decisions and research priorities. Future anti-biofilm strategies must account for these species-specific differences, potentially combining matrix-disrupting agents with conventional antibiotics tailored to the particular pathogen.
Advanced methodologies like BiofilmQ analysis and CSLM imaging provide powerful tools for deciphering the spatial and temporal dynamics of biofilm resistance mechanisms. As our understanding of these complex microbial communities deepens, so too will our ability to develop targeted interventions that overcome biofilm-mediated resistance, ultimately improving outcomes for patients suffering from persistent biofilm-associated infections.
The advent of implantable medical devices—from orthopedic prostheses and cardiac pacemakers to hernia meshes—has revolutionized modern medicine. However, this advancement has introduced a persistent clinical challenge: biofilm-associated infections (BAIs). Biofilms are structured communities of microorganisms encased in a self-produced extracellular polymeric substance (EPS) that adhere to biological or abiotic surfaces [130]. This biofilm mode of growth confers significant protection to pathogens; sessile bacteria within a biofilm can be 500–5,000 times more tolerant to antibiotics compared to their planktonic (free-floating) counterparts [131]. Consequently, biofilms are implicated in approximately 80% of all microbial infections in humans, presenting a formidable obstacle in clinical settings [131] [130].
The economic burden is substantial. In the United States alone, over 500,000 biofilm-related implant infections occur annually [130]. The cost of managing a single prosthetic joint infection often necessitates revision surgery, with annual costs for these procedures exceeding $500 million in the U.S.—a figure projected to rise to $1.62 billion by 2030 [130]. This financial impact, coupled with significant patient morbidity and mortality, underscores the urgent need for advanced preventive strategies, such as novel anti-biofilm coatings, which form the core of this cost-benefit evaluation within the broader context of bacterial adhesion and biofilm initiation research.
The Biofilm Problem on Medical Implants
Mechanisms of Biofilm Formation and Immune Evasion
Biofilm development on implant surfaces is a multi-stage, dynamic process initiated immediately upon device implantation. The process begins with the initial, reversible attachment of planktonic bacteria to the implant surface, governed by non-specific physical interactions such as electrostatic forces, hydrophobic interactions, and Lifshitz–van der Waals forces [130]. Bacterial surface structures like flagella and pili play a crucial role in this phase, enabling motility and initial contact [130].
This is followed by irreversible adhesion, mediated by short-range interactions including dipole-dipole forces, hydrogen bonds, and ionic bonding, facilitated by bacterial adhesins [130]. Subsequently, attached bacteria undergo multiplication and begin producing the extracellular polymeric substance (EPS) matrix, forming microcolonies. The intracellular secondary messenger cyclic di-guanosine monophosphate (c-di-GMP) upregulates EPS production, cementing the transition to a sessile lifestyle [130].
The biofilm then enters a maturation phase, developing a complex, three-dimensional architecture with water-filled channels that distribute nutrients and remove waste [130]. This stage is facilitated by quorum sensing (QS), a cell-density-dependent signaling system that coordinates population-wide gene expression, including the upregulation of biofilm-specific genes [131] [130]. The final stage is dispersal, where individual cells or clusters detach from the biofilm to colonize new surfaces, completing the lifecycle [130].
The resulting biofilm structure exhibits remarkable resilience to host defenses through multiple mechanisms. Biofilm-associated bacteria can reduce their metabolic activity, alter antigen expression, and secrete immunosuppressive molecules [130]. For instance, Staphylococcus aureus expresses Protein A (SpA), which enhances bacterial aggregation and binds to the Fc region of immunoglobulins, interfering with opsonophagocytosis [130].
Economic Impact of Biofilm-Related Implant Infections
The financial burden of BAIs stems from the complexity of treatment, which often involves prolonged antibiotic regimens, multiple surgical interventions, and extended hospitalization.
Table 1: Economic Burden of Biofilm-Associated Implant Infections
| Cost Component | Financial Impact | Notes |
|---|---|---|
| Prosthetic Joint Infection (PJI) Revision Surgeries | > USD 500 million annually (U.S.) | Projected to reach USD 1.62 billion by 2030 [130] |
| General Implant-Associated BAIs | > 500,000 annual cases (U.S.) | Contributes significantly to overall healthcare costs [130] |
| Treatment Cost per Case | Significantly higher than primary implantation | Costs include extended hospitalization, advanced diagnostics, high-dose antibiotics, and revision surgery [131] [130] |
Novel Anti-Biofilm Coating Technologies
Traditional systemic antibiotics often fail to eradicate biofilms due to poor penetration and bacterial tolerance. This has spurred the development of advanced anti-biofilm coatings designed to prevent initial bacterial adhesion or disrupt established biofilms directly at the implant surface.
Emerging Coating Strategies and Mechanisms of Action
Current research focuses on non-antibiotic, surface-modification strategies that mitigate the risk of fostering antimicrobial resistance (AMR) [130]. These approaches can be broadly categorized as anti-fouling (preventing adhesion) and bactericidal (killing microbes).
- Nanomaterial-based Coatings: Nanomaterials exploit their high surface-area-to-volume ratio and tunable chemistry for enhanced antimicrobial activity. Silver nanoparticles (AgNPs), for instance, are widely used for their broad-spectrum efficacy, disrupting bacterial membranes and enzyme activity [132] [133]. Other nanomaterials like zinc oxide, titanium dioxide, and graphene derivatives are also promising [133].
- Enzymes and Biosurfactants: These agents target the structural integrity of the biofilm matrix. Enzymes such as dispersin B degrade polysaccharides in the EPS, while proteases break down protein components, leading to biofilm disintegration [130].
- Quorum Sensing Inhibitors (QSIs): Also known as quorum quenching molecules, QSIs interfere with bacterial communication signals (e.g., AHLs), disrupting the coordinated behavior necessary for biofilm maturation without exerting a lethal pressure that drives resistance [130].
- Stimuli-Responsive Coatings: These "smart" coatings release antimicrobial agents (e.g., antibiotics, metal ions) in response to specific triggers from the infection site, such as a low pH, specific enzymes (e.g., lipases, proteases), or metabolites [132]. This provides a targeted, on-demand therapeutic approach.
- Chelating Agents: Compounds like EDTA disrupt biofilm stability by sequestering metal ions (e.g., Ca²⁺, Mg²⁺) that are essential for maintaining the integrity of the EPS matrix and bacterial cell walls [130].
The diagram below illustrates the multi-stage process of biofilm formation and the corresponding points of intervention for these novel coating strategies.
Diagram 1: Biofilm formation stages and coating intervention points. Anti-fouling coatings prevent initial attachment; nanomaterials disrupt adhesion and microcolonies; enzymes and quorum sensing inhibitors target maturation; stimuli-responsive coatings act against mature biofilms.
The Scientist's Toolkit: Key Reagents for Anti-Biofilm Coating Research
Table 2: Essential Research Reagents for Anti-Biofilm Coating Development
| Research Reagent / Material | Function & Utility in Experimental Protocols |
|---|---|
| Silver Nanoparticles (AgNPs) | Broad-spectrum antimicrobial agent; disrupts bacterial respiration and enzyme activity via ion release; commonly incorporated into polymer matrices [134] [133]. |
| Dispersin B | Enzyme that hydrolyzes poly-N-acetylglucosamine (PNAG), a key polysaccharide in the biofilm matrix of staphylococci; used for enzymatic biofilm disruption [130]. |
| Quorum Sensing Inhibitors (e.g., AHL analogs) | Interrupts bacterial cell-to-cell communication (quorum sensing), preventing the coordinated gene expression required for biofilm maturation and virulence [130]. |
| c-di-GMP | Key intracellular secondary messenger regulating the switch between planktonic and sessile lifestyles; modulating its levels is a strategy to control biofilm formation [130]. |
| Quaternary Ammonium Compounds (QACs) | Cationic surfactants that disrupt microbial cell membranes; frequently used in polymeric coatings for contact-killing surfaces [134]. |
| Cyclic Di-GMP Signaling Modulators | Used in research to induce biofilm dispersion by lowering intracellular c-di-GMP levels, a key trigger for the switch back to planktonic growth [130]. |
Methodologies for Evaluating Anti-Biofilm Coatings
Standardized Experimental Workflow for Coating Efficacy
A robust, multi-stage testing protocol is essential to transition anti-biofilm coatings from the laboratory to clinical application. The following workflow outlines key phases from initial material characterization to advanced in vivo modeling.
Diagram 2: Experimental workflow for evaluating anti-biofilm coatings, progressing from material analysis to advanced biological models.
Detailed Experimental Protocols
Protocol 1: Minimum Biofilm Eradication Concentration (MBEC) Assay [131] [135]
- Objective: To determine the minimum concentration of an antimicrobial agent required to eradicate a pre-established biofilm, which is crucial for evaluating coatings with incorporated active agents.
- Procedure:
- Biofilm Growth: Grow a standardized biofilm in a 96-peg lid (e.g., Calgary Biofilm Device) for 24-48 hours.
- Treatment: Transfer the lid with mature biofilm to a new plate containing serial dilutions of the antimicrobial agent (or place in contact with the coated material) and incubate for a further 24 hours.
- Recovery and Enumeration: Gently rinse the pegs to remove non-adherent cells and sonicate them in a recovery medium to dislodge viable biofilm-associated bacteria. Serially dilute the recovery medium and plate on agar to determine the Colony Forming Units (CFU/mL).
- Analysis: The MBEC is defined as the lowest concentration of antimicrobial that results in a ≥99.9% reduction (a 3-log kill) in viable counts compared to the untreated control.
Protocol 2: Assessment of Coating Biocompatibility with Mammalian Cells [130]
- Objective: To ensure that the anti-biofilm coating does not adversely affect the viability and function of host cells, a prerequisite for clinical translation.
- Procedure:
- Cell Seeding: Seed relevant mammalian cells (e.g., osteoblasts for orthopedic implants, fibroblasts for soft tissue integration) onto the coated surface or in extracts derived from the coating material.
- Viability Assay: After 24-72 hours of culture, assess cell viability using a standardized assay such as MTT or AlamarBlue, which measure metabolic activity. Compare results to cells grown on a control, uncoated material.
- Cytotoxicity and Morphology: Use Live/Dead staining (Calcein-AM for live cells, propidium iodide for dead cells) and visualize via fluorescence microscopy to assess membrane integrity and cell morphology directly on the coated surface.
Cost-Benefit Analysis: Weighing the Economic and Clinical Value
The adoption of novel anti-biofilm coatings represents a proactive investment. While these advanced coatings increase the initial cost of an implant, a comprehensive cost-benefit analysis must consider the substantial costs avoided by preventing BAIs.
Table 3: Cost-Benefit Analysis of Anti-Biofilm Coatings for Medical Implants
| Factor | Impact & Quantitative Consideration |
|---|---|
| Upfront Cost Increase | Coating Premium: Antimicrobial coatings can increase the device cost by 20-50% [134]. This includes R&D, advanced materials (e.g., silver nanoparticles), and specialized manufacturing. |
| Direct Cost Savings | Avoided Revision Surgery: A single revision for infection can cost 3-5 times more than the primary procedure. Preventing even a small percentage of infections yields massive savings, given the >$500M annual cost for PJI revisions [130]. |
| Indirect Cost Savings | Reduced Hospital Stays: Shorter lengths of stay and lower readmission rates justify the coating premium [134]. |
| Clinical & Patient Benefits | Improved Patient Outcomes: Mitigates physical and psychological trauma associated with recurrent infections and multiple surgeries. Reduces long-term antibiotic use, curbing AMR development [131]. |
| Market Dynamics | Growing Market: The anti-microbial coatings market is expected to reach USD 6.84 billion by 2030 (CAGR of 7.41%), reflecting strong industry confidence and adoption [134]. The biofilm treatment market is projected to grow from USD 2.38 Bn in 2025 to USD 4.13 Bn by 2032 (CAGR of 8.2%) [135]. |
The economic and clinical rationale for integrating novel anti-biofilm coatings into medical implants is compelling. The significant clinical burden and escalating costs associated with implant-related BAIs underscore the limitations of current treatment paradigms. Advanced coating strategies—including nanomaterial-based, enzyme-functionalized, and stimuli-responsive systems—offer a proactive, targeted, and multi-faceted approach to prevent and disrupt biofilms at their source.
Future research will be guided by several key trends. The integration of Artificial Intelligence (AI) is set to accelerate the discovery and design of novel anti-biofilm molecules and optimize material properties [132]. There is also a strong push towards multifunctional "smart" coatings that combine antimicrobial activity with other properties like osteoinduction (promoting bone growth) or self-reporting capabilities [133]. Furthermore, the field is increasingly prioritizing biodegradable and non-toxic nanomaterials to address long-term safety and environmental concerns [132] [133]. As these technologies mature and regulatory pathways become more defined, the next generation of anti-biofilm coatings will transition from being a premium option to a standard of care, ultimately reducing the global burden of medically related infections and improving patient outcomes.
Bacterial biofilm formation represents a fundamental survival strategy for microorganisms and a significant challenge in modern medicine. Biofilms are structured communities of bacterial cells enclosed in a self-produced polymeric matrix and adherent to an inert or living surface [52]. This matrix consists of extracellular polymeric substances (EPS), including exopolysaccharides, proteins, lipids, and extracellular DNA, which provides mechanical stability and protects resident bacteria from environmental threats [136]. The biofilm lifecycle progresses through distinct stages: reversible attachment, irreversible attachment, microcolony formation, maturation, and dispersal [136] [137].
The clinical significance of biofilms is profound. Bacteria within biofilms can be 100 to 1000 times more resistant to conventional antibiotic treatments compared to their planktonic counterparts [136] [137]. According to statistics, up to 80% of recurrent microbial and chronic infections in humans are related to bacterial biofilm formation [136]. This recalcitrance contributes significantly to the global antimicrobial resistance (AMR) crisis, which was associated with approximately 4.95 million deaths globally [138]. The protective nature of biofilms stems from multiple mechanisms including limited antibiotic diffusion through the EPS matrix, activation of antibiotic-degrading enzymes, horizontal gene transfer, and altered microbial physiology [137].
Table 1: Key Components of Bacterial Biofilms and Their Functions
| Component | Composition | Function in Biofilm |
|---|---|---|
| Extracellular Polysaccharides | Various polysaccharide polymers | Structural integrity, metal binding, adhesion |
| Proteins | Extracellular enzymes, structural proteins | Biofilm maintenance, inflammation, degradation |
| Lipids | Lipoproteins, hydrophobic compounds | Hydrophobicity, cellular integrity |
| Extracellular DNA (eDNA) | Bacterial DNA | Matrix stabilization, genetic exchange |
Phage Therapy: Mechanisms and Applications Against Biofilms
Bacteriophages (phages) are viruses that specifically infect and replicate within bacterial hosts, ultimately causing host cell lysis [139]. Discovered independently by Frederick Twort and Félix d'Hérelle in the early twentieth century, phages have re-emerged as promising alternatives to antibiotics amid the growing AMR crisis [136] [139]. Phages offer several unique advantages: high specificity toward target bacteria, self-amplification at infection sites, low inherent toxicity, and the ability to penetrate biofilm structures [139].
Mechanisms of Phage Action Against Biofilms
Phages employ multiple mechanisms to eradicate biofilms, with one of the most crucial being their ability to encode depolymerase enzymes that degrade the EPS matrix [136] [139]. These enzymes break down the defense barrier during host bacteria infection, allowing phages to access embedded bacterial cells [136]. Research has identified 160 putative depolymerases across 143 phages, categorized as either hydrolases or lyases [136]. Beyond enzymatic activity, phages can directly infect and lyse biofilm-resident bacteria through their replicative cycle and disrupt bacterial communication systems like quorum sensing [137].
The infection and replication of phages occur through either lytic or lysogenic cycles. Lytic phages follow five distinct stages: attachment, injection, replication and translation, assembly, and lysis [136]. This cycle rapidly destroys host cells, making lytic phages particularly valuable for therapeutic applications. In contrast, lysogenic phages integrate their genome into the host bacterium as prophages, replicating passively during cell division before potentially entering the lytic cycle under environmental stressors [139].
Experimental Protocols for Phage-Based Biofilm Control
Protocol 1: Phage Isolation and Characterization
- Sample Collection: Collect environmental samples from wastewater, soil, or clinical isolates where target bacteria are prevalent [139].
- Enrichment and Plaque Assay: Incubate samples with target bacterial strains in nutrient broth. Filter sterilize and perform dual-layer plaque assays to isolate individual phage variants [139].
- Genomic Sequencing: Extract DNA from purified phage plaques and perform whole-genome sequencing to identify virulence factors, depolymerase genes, and assess safety [139].
- Host Range Determination: Test phage susceptibility across multiple bacterial strains and species to determine host specificity using spot tests or efficiency of plating assays [137].
Protocol 2: Biofilm Prevention Assay
- Surface Selection: Choose relevant abiotic (catheters, grafts) or biotic surfaces based on intended application [140].
- Baseline Biofilm Formation: Allow target bacteria to adhere to surfaces for 2-4 hours, then establish mature biofilms over 24-48 hours [136].
- Phage Application: Apply purified phage preparations or cocktails at varying multiplicities of infection (MOI) during initial adhesion phase.
- Quantification: Assess biofilm biomass through crystal violet staining or viable cell counts after disruption [137].
Protocol 3: Phage-Antibiotic Synergy (PAS) Testing
- Subinhibitory Antibiotic Preparation: Prepare antibiotics at concentrations below the minimum inhibitory concentration (MIC) [139].
- Combination Treatment: Apply phages and sub-MIC antibiotics simultaneously or sequentially to biofilm cultures.
- Synergy Assessment: Compare biofilm reduction between monotherapies and combination therapy using metabolic assays (e.g., MTT, XTT) [139].
Table 2: Efficacy of Phage Therapy Against Biofilm-Forming Pathogens
| Pathogen | Biofilm-Related Infection | Phage Type/Name | Reported Efficacy |
|---|---|---|---|
| Staphylococcus aureus | Vascular graft infections [140] | Phage cocktail SniPha360 [140] | Clinical resolution in 70.3% of patients [140] |
| Mycobacterium abscessus | Cystic fibrosis lung infection [139] | Engineered phage cocktail (Muddy, ZoeJΔ45, BPsΔ33HTH-HRM10) [139] | Significant clinical improvement [139] |
| Escherichia coli | Gastrointestinal infections [141] | Not specified | 50-70% efficacy rate in MDR infections [139] |
| Pseudomonas aeruginosa | Respiratory and wound infections [139] | Phage PA5 in fibrin glue [140] | Improved antibacterial activity in sustained delivery [140] |
Nanoparticle-Based Delivery Systems: Engineering Solutions for Biofilm Eradication
Nanoparticle-based drug delivery systems represent a revolutionary approach to overcoming the limitations of conventional antimicrobial treatments. These systems are engineered materials ranging from 1 to 100 nm that can navigate biological barriers and improve therapeutic efficacy through targeted delivery and controlled release [142] [143]. Nanoparticles enhance drug bioavailability, reduce required dosages, minimize systemic toxicity, and can be functionalized with targeting ligands for precision therapy [144].
Nanocarrier Platforms for Anti-Biofilm Applications
Multiple nanocarrier platforms have been developed for biofilm eradication. Polymeric nanoparticles constructed from poly(lactic-co-glycolic acid) (PLGA), chitosan, or polyethylene glycol (PEG) provide controlled drug release and surface functionalization capabilities [144]. Lipid-based nanoparticles including liposomes and solid lipid nanoparticles excel at encapsulating both hydrophilic and hydrophobic agents while exhibiting high biocompatibility [142]. Inorganic nanoparticles such as silver, gold, zinc oxide, and copper nanoparticles possess intrinsic antimicrobial properties and can be combined with other therapeutic agents [138]. Dendrimers offer highly branched, monodisperse structures with multiple surface functional groups for conjugating antimicrobial agents [142].
The mechanism of nanoparticle-mediated biofilm disruption involves multiple approaches. Nanoparticles can penetrate the EPS matrix through their small size and surface modifications, disrupt quorum sensing signaling, generate reactive oxygen species (in the case of metal nanoparticles), and provide sustained release of encapsulated antimicrobials at the infection site [138].
Experimental Protocols for Nanoparticle-Based Biofilm Treatment
Protocol 1: Synthesis of Polymeric Nanoparticles for Drug Delivery
- Material Selection: Choose biodegradable polymers (PLGA, chitosan) based on drug compatibility and release requirements [144].
- Nanoprecipitation Method: Dissolve polymer and drug in water-miscible organic solvent (acetone, ethanol). Inject into aqueous phase under constant stirring [144].
- Purification and Characterization: Purify via centrifugation or dialysis. Characterize size (dynamic light scattering), zeta potential (laser Doppler anemometry), and morphology (SEM) [142].
- Drug Loading Efficiency: Determine encapsulated drug percentage via HPLC or UV-Vis spectroscopy after nanoparticle dissolution [144].
Protocol 2: Biofilm Penetration Assessment
- Biofilm Growth: Form mature biofilms on relevant substrates for 48-72 hours with medium refreshment [136].
- Fluorescent Labeling: Tag nanoparticles with fluorescent dyes (FITC, Rhodamine) using standard conjugation chemistry [143].
- Confocal Microscopy: Treat biofilms with labeled nanoparticles, section using cryostat, and image using confocal laser scanning microscopy [143].
- Penetration Quantification: Measure fluorescence intensity at different biofilm depths using image analysis software [143].
Protocol 3: Controlled Release Kinetics
- Dialyzer Setup: Place nanoparticle suspension in dialysis membrane with appropriate molecular weight cutoff [144].
- Sink Conditions: Immerse in release medium (PBS, pH 7.4) with mild agitation at 37°C [144].
- Sampling and Analysis: Withdraw samples at predetermined intervals, analyze drug content, and maintain sink conditions [144].
- Release Modeling: Fit data to mathematical models (Higuchi, Korsmeyer-Peppas) to determine release mechanisms [144].
Table 3: Nanoparticle Systems for Enhanced Anti-Biofilm Drug Delivery
| Nanoparticle Type | Materials | Loaded Agent | Key Advantages |
|---|---|---|---|
| Polymeric NPs | PLGA, Chitosan, PEG [144] | Antibiotics, Natural compounds [144] | Controlled release, biodegradability, functionalizable surface |
| Liposomes | Phospholipids, Cholesterol [142] | Antimicrobial peptides, Antibiotics [142] | High encapsulation efficiency, bilayer structure, biocompatibility |
| Metallic NPs | Silver, Zinc Oxide, Copper [138] | Intrinsic antimicrobial activity [138] | Reactive oxygen species generation, multiple mechanisms |
| Solid Lipid NPs | Triglycerides, Waxes [138] | Lipophilic drugs [138] | Enhanced stability, industrial scalability |
| Dendrimers | PAMAM, Polypropylene imine [142] | Antibiotics, Antimicrobial agents [142] | Multivalent surface, well-defined structure |
Integrated Approaches: Combination Therapies and Clinical Translation
The integration of phage therapy with nanoparticle delivery systems represents a promising frontier in combating biofilm-mediated infections. Combination approaches leverage the unique strengths of both technologies to overcome individual limitations. Phage-antibiotic synergy (PAS) demonstrates how certain antibiotics at subinhibitory concentrations can enhance phage replication and bacterial killing while reducing resistance development [139] [141]. Similarly, nanoparticle-mediated co-delivery of phages and antibiotics can protect therapeutic agents from degradation and facilitate targeted delivery to biofilm niches.
Advanced Experimental Protocols for Combination Therapies
Protocol 1: Phage-Loaded Nanoparticle Formulation
- Matrix Selection: Choose biocompatible polymers (alginate, chitosan, fibrin) compatible with phage viability [140].
- Encapsulation Method: Utilize double emulsion, ionic gelation, or spray-drying techniques to encapsulate phage particles [140].
- Viability Assessment: Determine phage titer before and after encapsulation via plaque assays to assess encapsulation efficiency [140].
- Release Kinetics: Characterize phage release profile under physiological conditions and assess infectivity of released phages [140].
Protocol 2: In Vivo Biofilm Model Evaluation
- Animal Model Selection: Utilize appropriate models (murine, porcine) based on infection site (cutaneous, systemic, implant-associated) [139].
- Biofilm Establishment: Allow 24-72 hours for biofilm formation post-bacterial inoculation before treatment initiation [139].
- Treatment Regimen: Administer monotherapies and combination therapies via relevant routes (topical, intravenous, local) [139].
- Outcome Assessment: Quantify bacterial burden (CFU/organ), inflammatory markers, histopathology, and biofilm imaging (SEM/confocal) [139].
Protocol 3: Resistance Development Monitoring
- Serial Passage Design: Expose bacteria to subtherapeutic concentrations of individual and combined therapies over multiple generations [141].
- Phenotypic Characterization: Assess MIC/MBC changes, biofilm-forming capacity, and colony morphology at regular intervals [141].
- Genomic Analysis: Perform whole-genome sequencing of resistant isolates to identify mutation patterns [141].
- Cross-Resistance Evaluation: Test resistance development to unrelated antimicrobial agents to assess collateral sensitivity [141].
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for Phage and Nanoparticle Biofilm Research
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Bacterial Strains | Biofilm formation studies | Reference strains: P. aeruginosa PAO1, S. aureus ATCC 25923; Clinical isolates with characterized biofilm phenotypes |
| Phage Libraries | Therapeutic agent discovery | Environmental phage banks; Commercially available phage collections; Characterized phage strains with sequenced genomes |
| Biocompatible Polymers | Nanoparticle synthesis | PLGA, PEG, Chitosan, Polyvinyl alcohol; Various molecular weights, block copolymers for tunable properties |
| Characterization Instruments | Material property analysis | Dynamic Light Scattering (size/zeta potential), SEM/TEM (morphology), HPLC (drug loading/release) |
| Biofilm Assay Kits | Biofilm quantification | Crystal violet staining kits, Metabolic activity assays (MTT/XTT), Extracellular DNA quantification kits |
| Animal Infection Models | In vivo efficacy assessment | Murine cutaneous abscess, Catheter-associated infection, Implant osteomyelitis models |
The convergence of phage therapy and nanoparticle-based delivery systems presents a transformative approach to addressing the persistent challenge of biofilm-associated infections. While both strategies demonstrate individual promise, their integration offers synergistic potential to overcome the limitations of conventional antibiotics. The specificity and self-amplifying nature of phages, combined with the enhanced bioavailability and targeted delivery capabilities of nanoparticles, creates a powerful platform for precision antimicrobial therapy.
Future research directions should focus on optimizing combination therapies, developing intelligent delivery systems responsive to biofilm microenvironments, and establishing standardized regulatory pathways for these innovative approaches. Additionally, the application of artificial intelligence in phage selection and nanoparticle design, along with advanced genetic engineering techniques to enhance phage potency and host range, will accelerate clinical translation. As these technologies mature, they hold significant potential to reshape our therapeutic arsenal against antimicrobial-resistant biofilm infections, ultimately addressing a critical unmet need in modern medicine.
Conclusion
The battle against biofilm-mediated infections hinges on a deep and integrated understanding of the initial adhesion and initiation phases. This synthesis of foundational science, advanced methodologies, therapeutic troubleshooting, and rigorous validation underscores that effective strategies must be multi-faceted, targeting not only the bacterial cells but also the physical and chemical forces that drive surface attachment, the signaling systems that coordinate community behavior, and the protective extracellular matrix. Future directions in biomedical research must prioritize the translation of laboratory insights into clinically viable solutions. This includes the rational design of next-generation smart biomaterials that resist fouling and kill on contact, the development of novel combination therapies that disrupt biofilm integrity while enhancing antibiotic susceptibility, and the refinement of in vivo models that accurately predict therapeutic success. By deconstructing the very foundations of biofilm formation, the field can move beyond conventional antibiotics and forge new paradigms for treating persistent and device-associated infections, ultimately alleviating a significant burden on global healthcare systems.
References
- 1. The biofilm life cycle– expanding the conceptual model of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9841534/]
- 2. Biofilms and their role in pathogenesis [https://www.immunology.org/public-information/bitesized-immunology/pathogens-disease/biofilms-and-their-role-pathogenesis]
- 3. Biofilms: structure, resistance mechanism, emerging control ... [https://pubs.rsc.org/en/content/articlehtml/2025/pm/d5pm00094g]
- 4. Mechanisms of antimicrobial resistance in biofilms [https://www.nature.com/articles/s44259-024-00046-3]
- 5. Formation, architecture, and persistence of oral biofilms [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1602962/full]
- 6. Visualisation and biovolume quantification in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8160185/]
- 7. Non-invasive single-cell morphometry in living bacterial biofilms [https://pmc.ncbi.nlm.nih.gov/articles/PMC7708432/]
- 8. Understanding bacterial biofilms: From definition to treatment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10117668/]
- 9. A Brief Recap of Microbial Adhesion and Biofilms [https://www.mdpi.com/2076-3417/9/14/2801]
- 10. Preventing bacterial adhesion to skin by altering their ... [https://www.nature.com/articles/s41522-024-00568-8]
- 11. Contributions of Van Der Waals Interactions and ... [https://pubmed.ncbi.nlm.nih.gov/30373365/]
- 12. Competition of electrostatic and hydrophobic interactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3040037/]
- 13. Bacterial adherence: adhesin-receptor interactions mediating ... [https://pubmed.ncbi.nlm.nih.gov/7014727/]
- 14. Microscale communication between bacterial pathogens ... [https://www.nature.com/articles/s41435-021-00149-1]
- 15. Bacterial adhesin [https://en.wikipedia.org/wiki/Bacterial_adhesin]
- 16. Bacterial Adhesins: Common Themes and Variations in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC93481/]
- 17. Biofilm formation by the host microbiota: a protective shield ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12093748/]
- 18. Recent advances and perspectives in nucleotide second ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10118264/]
- 19. Arresting Cell Growth by Altering Metabolic Flow - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/37036337/]
- 20. Controlling biofilm development through cyclic di-GMP signaling [https://pmc.ncbi.nlm.nih.gov/articles/PMC9891824/]
- 21. Parallel regulatory circuits orchestrate biofilm formation in ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1011870]
- 22. A c-di-GMP Signaling Cascade Controls Motility, Biofilm ... [https://pubmed.ncbi.nlm.nih.gov/35323023/]
- 23. The Second Messenger c-di-GMP Adjusts Motility and ... [https://pubmed.ncbi.nlm.nih.gov/30447993/]
- 24. Cyclic-di-GMP signalling mutants drive ecological succession ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12440571/]
- 25. Unraveling the multifaceted role of extracellular DNA ... [https://pubmed.ncbi.nlm.nih.gov/40322923/]
- 26. Extracellular polymeric substance [https://en.wikipedia.org/wiki/Extracellular_polymeric_substance]
- 27. Microbial Extracellular Polymeric Substances: Ecological ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2018.01636/full]
- 28. Role of Extracellular DNA in Initial Bacterial Adhesion and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2869138/]
- 29. Secreted nucleases reclaim extracellular DNA during ... [https://www.nature.com/articles/s41522-024-00575-9]
- 30. Binding Strength of Gram-Positive Bacterial Adhesins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7330015/]
- 31. High force catch bond mechanism of bacterial adhesion in ... [https://www.nature.com/articles/s41467-020-18063-x]
- 32. biofilms, quorum sensing, formation and prevention [https://pmc.ncbi.nlm.nih.gov/articles/PMC7086079/]
- 33. The contribution of cell-cell signaling and motility to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3204577/]
- 34. Burst statistics in an early biofilm quorum sensing model [https://www.nature.com/articles/s41598-019-48525-2]
- 35. Synthetic multicellular cell-to-cell communication in inkjet ... [https://www.sciencedirect.com/science/article/abs/pii/S0142961210015693]
- 36. Biofilm, resistance, and quorum sensing: The triple threat in ... [https://www.sciencedirect.com/science/article/pii/S2950194625003462]
- 37. Atomic Force Microscopy in Microbiology: New Structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4120197/]
- 38. Nanoscale Characterization and Determination of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1347306/]
- 39. Quantification of bacterial adhesion forces using atomic ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701299001372]
- 40. Analysis of biofilm assembly by large area automated AFM [https://www.nature.com/articles/s41522-025-00704-y]
- 41. Atomic force microscopy measurements of bacterial ... [https://www.nature.com/articles/srep16857]
- 42. Differential susceptibility of Streptococcus dentisani to ... [https://pubmed.ncbi.nlm.nih.gov/40760288/]
- 43. Confocal Laser Scanning Microscopy - an overview [https://www.sciencedirect.com/topics/chemistry/confocal-laser-scanning-microscopy]
- 44. Confocal microscopy imaging of the biofilm matrix [https://www.sciencedirect.com/science/article/abs/pii/S0167701216300367]
- 45. Confocal Laser Scanning Microscopy for Analysis of ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2019.00677/full]
- 46. Hostile Environments: Modifying Surfaces to Block ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190938/]
- 47. Quantitative image analysis of microbial communities with ... [https://www.nature.com/articles/s41564-020-00817-4]
- 48. Biofilm Analysis by Confocal Microscopy-Basics and ... [https://pubmed.ncbi.nlm.nih.gov/40927881/]
- 49. Innovative Biotechnological Approaches for Biofilm Control ... [https://scian.cl/scientific-image-analysis/innovative-biotechnological-apporaches-for-biofilm-control-and-characterization/]
- 50. A Selection of Platforms to Evaluate Surface Adhesion and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8468346/]
- 51. Bacterial Adhesion and Biofilm Formation: Hydrodynamics ... [https://link.springer.com/chapter/10.1007/978-3-031-04484-7_19]
- 52. Bacterial Adhesion: Seen Any Good Biofilms Lately? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC118072/]
- 53. In Vitro Model of Physiological and Pathological Blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4692682/]
- 54. In Vitro Biofilm Formation Kinetics of Pseudomonas ... [https://www.mdpi.com/2673-947X/5/3/32]
- 55. Quantitative high-throughput screening data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375054/]
- 56. A quantitative high-throughput screening pipeline to ... [https://www.nature.com/articles/s41598-025-14697-3]
- 57. High-throughput screening of small molecules targeting ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217486/]
- 58. A comprehensive review of genomics, transcriptomics ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813023054624]
- 59. Transcriptome analysis of the biofilm formation mechanism ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2023.1128166/full]
- 60. Quantitative proteomics analysis reveals an important role of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10995333/]
- 61. Multi-omics analysis reveals genes and metabolites involved ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10666050/]
- 62. Decoding the regulatory networks of Proteus mirabilis under ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12531150/]
- 63. Transcriptome Analysis Reveals the Molecular Mechanism of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11898514/]
- 64. Polymicrobial Biofilms: Interkingdom Interactions, Resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12373980/]
- 65. Polymicrobial Infections: A Comprehensive Review on ... [https://www.mdpi.com/2813-9054/70/4/39]
- 66. Quantitative assessment of individual populations within ... [https://www.nature.com/articles/s41598-018-27497-9]
- 67. Dual-functional surface coatings integrating antimicrobial and ... [https://pubs.rsc.org/en/content/articlehtml/2025/mh/d5mh01132a?page=search]
- 68. Bioinspired strategies for antibacterial surface design [https://www.sciencedirect.com/science/article/pii/S2950387625000054]
- 69. Bioinspired nanostructured surfaces for antimicrobial and ... [https://www.sciencedirect.com/science/article/pii/S2215038225000470]
- 70. Design and Application of Antifouling Bio-Coatings - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11945268/]
- 71. Design of Nanostructured Surfaces and Hydrogel Coatings ... [https://www.techscience.com/jpm/v42n3/64019/html]
- 72. Design of Nanostructured Surfaces and Hydrogel Coatings ... [https://www.semanticscholar.org/paper/Design-of-Nanostructured-Surfaces-and-Hydrogel-for-Cao-Luo/3d3209843d534c2cce94f1c680bd32d01c213e3d]
- 73. Natural product-based inhibitors of quorum sensing [https://www.sciencedirect.com/science/article/pii/S2405580825001980]
- 74. Natural product-based inhibitors of quorum sensing: A novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12242448/]
- 75. Multidrug-resistant bacteria quorum-sensing inhibitors [https://www.sciencedirect.com/science/article/abs/pii/S0223523424008900]
- 76. Anti-biofilm and anti-quorum sensing activities of ... [https://pubmed.ncbi.nlm.nih.gov/40205343/]
- 77. Anti-biofilm and anti-quorum sensing activities of galloylquinic ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-024-03712-8]
- 78. A review of quorum-sensing and its role in mediating ... [https://www.nature.com/articles/s42003-025-07608-9]
- 79. Glycoside Hydrolases Degrade Polymicrobial Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5278739/]
- 80. Aggregatibacter actinomycetemcomitans Dispersin B [https://pmc.ncbi.nlm.nih.gov/articles/PMC11357414/]
- 81. Disrupting Irreversible Bacterial Adhesion and Biofilm ... [https://pubmed.ncbi.nlm.nih.gov/33893112/]
- 82. Integrating Bacteriocins and Biofilm-Degrading Enzymes to ... [https://www.mdpi.com/1422-0067/26/1/399]
- 83. Effects of DNase I coating of titanium on bacteria adhesion ... [https://pubmed.ncbi.nlm.nih.gov/28576044/]
- 84. Dispersin B [https://en.wikipedia.org/wiki/Dispersin_B]
- 85. Enhancing the destruction of Burkholderia cepacia biofilm ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1662291/full]
- 86. Methods for the visualization and analysis of extracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6317720/]
- 87. Natural phytochemical-based strategies for antibiofilm ... [https://cmjournal.biomedcentral.com/articles/10.1186/s13020-025-01147-5]
- 88. Mechanism, Efficacy, and Safety of Natural Antibiotics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561696/]
- 89. Marine Antimicrobial Peptides: Emerging Strategies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383081/]
- 90. Implication of Surface Properties, Bacterial Motility, and ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.643722/full]
- 91. Quantitatively Predicting Bacterial Adhesion Using Surface ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4854535/]
- 92. Physico-chemistry from initial bacterial adhesion to surface ... [https://www.sciencedirect.com/science/article/pii/S000186861830229X]
- 93. A Comprehensive Overview of Antimicrobial Peptides [https://www.mdpi.com/2079-6382/14/11/1115]
- 94. Quantitative image analysis of microbial communities with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7840502/]
- 95. Biofilms and multidrug resistance: an emerging crisis and the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12237878/]
- 96. Combination Therapies for Biofilm Inhibition and Eradication [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2022.850030/full]
- 97. α-Amylase-Mediated Antibiotic Degradation and ... [https://pubmed.ncbi.nlm.nih.gov/41009919/]
- 98. Phage and Antibiotic Combination Therapy for Biofilm ... [https://www.contagionlive.com/view/phage-and-antibiotic-combination-therapy-for-biofilm-associated-bacterial-infections]
- 99. Combination of phages and antibiotics with enhanced ... [https://www.sciencedirect.com/science/article/pii/S2590207524000704]
- 100. Magnetic field-targeting and ultrasound-responsive ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925007643]
- 101. New ultrasound drug delivery system found to be highly ... [https://www.ox.ac.uk/news/2025-04-22-new-ultrasound-drug-delivery-system-found-be-highly-effective-against-bacterial]
- 102. Bacterial persisters: molecular mechanisms and ... [https://www.nature.com/articles/s41392-024-01866-5]
- 103. Mini review: Persister cell control strategies [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1706115/full]
- 104. From Dormancy to Eradication: Strategies for Controlling ... [https://www.mdpi.com/2304-8158/14/6/1075]
- 105. Isolation of persister cells within the biofilm and relative ... [https://www.sciencedirect.com/science/article/pii/S2213716521002630]
- 106. A Quantitative Survey of Bacterial Persistence in the Presence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7460088/]
- 107. Quantitative analysis of persister fractions suggests different ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/1471-2180-13-25]
- 108. A rational approach to discovering new persister control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406675/]
- 109. Strategies for combating bacterial biofilms: A focus on anti ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5955472/]
- 110. Natural phytochemical-based strategies for antibiofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12210829/]
- 111. Natural Anti-biofilm Agents: Strategies to Control ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.566325/full]
- 112. Potential Use of Selected Natural Compounds with Anti- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11765509/]
- 113. Natural and synthetic plant compounds as anti-biofilm ... [https://www.sciencedirect.com/science/article/pii/S1567134821003531]
- 114. Integrative Experimental and Computational Dataset for ... [https://www.sciencedirect.com/science/article/pii/S2352340925007474]
- 115. Mechano-bactericidal actions of nanostructured surfaces [https://pubmed.ncbi.nlm.nih.gov/32807981/]
- 116. Nanostructured antibacterial surfaces – What can be ... [https://www.sciencedirect.com/science/article/abs/pii/S1748013222000317]
- 117. Nanomaterials and Coatings for Managing Antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9952333/]
- 118. Biocompatible mechano-bactericidal nanopatterned ... [https://www.sciencedirect.com/science/article/abs/pii/S1742706122000472]
- 119. Bacterial adhesion to biomaterials: What regulates this ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8215285/]
- 120. Concise review of mechanisms of bacterial adhesion to ... [https://pubmed.ncbi.nlm.nih.gov/9730073/]
- 121. Dangerous bacterial biofilms have a natural enemy - UCR News [https://news.ucr.edu/articles/2025/01/10/dangerous-bacterial-biofilms-have-natural-enemy]
- 122. Quantitative and Qualitative Assessment Methods for Biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6133255/]
- 123. In Vivo vs. In Vitro Trials (and Why Combining Both Is Best) [https://science.howstuffworks.com/life/cellular-microscopic/in-vivo-vs-in-vitro.htm]
- 124. In Vitro vs. In Vivo: What's the Difference? [https://newlifefertility.com/blog/in-vitro-and-in-vivo/]
- 125. Comparative Analysis of Biofilm Formation and Antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382965/]
- 126. Quantitative analysis of adhesion and biofilm formation on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1356821/]
- 127. Biofilms and multidrug resistance: an emerging crisis ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1625356/full]
- 128. JCDR - Antimicrobial resistance , Biofilm formation, Bloodstream... [https://www.jcdr.net/article_fulltext.asp?issn=0973-709x&year=2025&month=June&volume=19&issue=6&page=DE01%20-%20DE06&id=21064]
- 129. Relationship Between Biofilm Formation and Antimicrobial Resistance ... [https://pubmed.ncbi.nlm.nih.gov/30142035/]
- 130. Targeting Bacterial Biofilms on Medical Implants [https://www.mdpi.com/2079-6382/14/8/802]
- 131. Bacterial biofilm formation on implantable devices and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6312881/]
- 132. Advanced Anti-Biofilm Technologies for Biomedical Devices [https://www.sciencedirect.com/science/article/pii/S3050837125000451]
- 133. Antimicrobial Nanocoatings Market Trends, 2025 to 2035 [https://www.futuremarketinsights.com/reports/anti-microbial-nanocoatings-market]
- 134. Anti-microbial Coatings Market Size & Share Analysis [https://www.mordorintelligence.com/industry-reports/anti-microbial-coatings-market]
- 135. Biofilm Treatment Market Trends, Share & Forecast, 2025- ... [https://www.coherentmarketinsights.com/industry-reports/biofilm-treatment-market]
- 136. Phages against Pathogenic Bacterial Biofilms and Biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8875263/]
- 137. Bacteriophage-Mediated Control of Biofilm: A Promising ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.825828/full]
- 138. A one health nanotechnology approach to address ... [https://pubs.rsc.org/en/content/articlehtml/2025/ma/d5ma00487j]
- 139. Phage therapy as a revitalized weapon for treating clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12540056/]
- 140. Bacteriophage Therapy as a Means of Resolving Biofilm ... [https://jmsgr.tamhsc.edu/bacteriophage-therapy-as-a-means-of-resolving-biofilm-formation-on-vascular-prostheses/]
- 141. Recent insights on challenges encountered with phage ... [https://gutpathogens.biomedcentral.com/articles/10.1186/s13099-025-00735-y]
- 142. Nanoparticles in Drug Delivery: From History to Therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9781272/]
- 143. Engineering precision nanoparticles for drug delivery [https://www.nature.com/articles/s41573-020-0090-8]
- 144. Nano based drug delivery systems: recent developments and ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-018-0392-8]