Liposomal CRISPR-Cas9 Formulations: A Novel Strategy to Eradicate Pseudomonas aeruginosa Biofilms
Pseudomonas aeruginosa biofilms are a major cause of persistent, antibiotic-resistant infections, particularly in cystic fibrosis and immunocompromised patients.
Liposomal CRISPR-Cas9 Formulations: A Novel Strategy to Eradicate Pseudomonas aeruginosa Biofilms
Abstract
Pseudomonas aeruginosa biofilms are a major cause of persistent, antibiotic-resistant infections, particularly in cystic fibrosis and immunocompromised patients. This article explores the cutting-edge combination of CRISPR-Cas9 gene-editing technology with liposomal nanoparticle delivery systems as a precision antimicrobial strategy. We review the foundational science of biofilm-mediated resistance and the mechanisms by which liposomal Cas9 formulations disrupt key genetic pathways. The scope extends from methodological advances in gRNA design and liposome engineering to troubleshooting delivery efficiency and minimizing off-target effects. Furthermore, we validate this approach through comparative analysis with traditional antibiotics and emerging alternatives, synthesizing in vitro efficacy data that demonstrates over 90% biofilm reduction. This resource provides researchers and drug development professionals with a comprehensive overview of the potential and challenges of translating CRISPR-nanoparticle hybrids into clinical therapeutics.
The Biofilm Conundrum and the Rise of Precision Antimicrobials
Pseudomonas aeruginosa stands as a preeminent nosocomial pathogen, renowned for its formidable antibiotic resistance and capacity to cause life-threatening infections in immunocompromised hosts. This Gram-negative bacterium is particularly notorious for its ability to form resilient biofilms—structured communities of microorganisms encapsulated within a self-produced extracellular polymeric substance (EPS) that confers up to 1,000-fold increased tolerance to antimicrobial agents compared to their planktonic counterparts [1] [2]. The biofilm matrix functions as both a physical diffusion barrier and a protective niche, enabling persistent colonization on both biological tissues and abiotic surfaces such as medical implants, catheters, and respiratory equipment [1]. This biofilm-mediated resistance represents a principal factor driving the high mortality rates associated with P. aeruginosa nosocomial infections, necessitating innovative therapeutic approaches that extend beyond conventional antibiotic regimens.
P. aeruginosaBiofilm Composition and Development
The structural integrity and resistance properties of P. aeruginosa biofilms derive from their complex extracellular matrix, primarily composed of exopolysaccharides, extracellular DNA (eDNA), proteins, and lipids [1]. Three key exopolysaccharides perform distinct functional roles:
- Psl: A neutral pentasaccharide that facilitates initial surface attachment and cell-to-cell interactions during biofilm initiation, while also providing structural stability in mature biofilms and conferring protection against neutrophil phagocytosis [1].
- Pel: A cationic polysaccharide that maintains biofilm integrity and promotes tolerance to aminoglycoside antibiotics and colistin [1].
- Alginate: A negatively charged acetylated polymer predominantly produced by mucoid strains isolated from cystic fibrosis patients, which impedes antibiotic penetration and provides protection against phagocytosis [1] [2].
Additionally, extracellular DNA (eDNA) released through cell lysis contributes to biofilm architecture through cation chelation, facilitates nutrient acquisition, and creates an acidic environment that further limits antimicrobial penetration [1].
Biofilm Development Cycle
The formation of P. aeruginosa biofilms follows a sequential, cyclical process comprising distinct developmental stages [2] [3]:
- Initial Attachment: Planktonic cells reversibly adhere to surfaces through interactions mediated by flagella, pili, and electrostatic forces.
- Irreversible Attachment: Adhered cells transition to permanent attachment and begin synthesizing EPS components.
- Maturation: Microcolonies develop into complex three-dimensional structures characterized by water channels that facilitate nutrient distribution.
- Dispersion: Active detachment of biofilm cells enables dissemination to new colonization sites.
Table 1: Key Components of P. aeruginosa Biofilm Matrix and Their Functions
| Matrix Component | Chemical Composition | Primary Functions |
|---|---|---|
| Psl | D-glucose, D-mannose, L-rhamnose | Surface attachment, structural stability, protection from phagocytosis |
| Pel | N-acetylglucosamine, N-acetylgalactosamine | Biofilm integrity, aminoglycoside tolerance |
| Alginate | Mannuronic acid, guluronic acid | Antibiotic diffusion barrier, protection from immune recognition |
| Extracellular DNA | DNA fragments | Cation chelation, structural support, nutrient source |
| Proteins | Adhesins, lectins, appendages | Initial attachment, structural reinforcement |
CRISPR/Cas9 System: A Precision Armament Against Biofilm Resistance
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system represents a revolutionary gene-editing technology that enables precise targeting and modification of specific genetic sequences. This system functions through two fundamental components: the Cas9 nuclease, which introduces double-strand breaks in DNA, and a guide RNA (gRNA) that directs Cas9 to complementary genomic sequences [4]. For antimicrobial applications, CRISPR/Cas9 can be programmed to disrupt critical genetic determinants of biofilm formation and antibiotic resistance in P. aeruginosa.
Key Genetic Targets for Biofilm Disruption
Therapeutic strategies employing CRISPR/Cas9 against P. aeruginosa biofilms primarily focus on several high-value targets:
- Quorum Sensing Systems: The las, rhl, pqs, and iqs systems regulate biofilm maturation and virulence factor expression in a cell-density-dependent manner [2] [3]. Disruption of these signaling pathways can attenuate biofilm development without exerting direct bactericidal pressure.
- Biofilm Matrix Genes: Targeting exopolysaccharide biosynthesis operons (psl, pel, alg) can compromise structural integrity and enhance antibiotic penetration [1].
- Antibiotic Resistance Genes: Specific enzymatic resistance determinants (e.g., β-lactamases, aminoglycoside-modifying enzymes) can be selectively inactivated to restore antibiotic susceptibility [4].
- Regulatory Networks: Components of the cyclic-di-GMP signaling system that govern the transition between planktonic and biofilm lifestyles present attractive targets for intervention [5].
Liposomal Cas9 Formulations: Protocol for Enhanced Biofilm Penetration
Despite its considerable potential, the clinical translation of CRISPR/Cas9 antibacterials faces substantial delivery challenges, particularly regarding efficient penetration through the biofilm matrix and bacterial envelope barriers. Nanoparticle-based delivery systems, especially liposomal formulations, have demonstrated remarkable efficacy in overcoming these limitations by improving cellular uptake, protecting genetic payloads from degradation, and enabling controlled release within biofilm microenvironments [4].
Protocol: Preparation of Cationic Liposomal Cas9 Formulations
Principle: Cationic liposomes facilitate complexation with negatively charged nucleic acids and enhance interaction with bacterial membranes, thereby improving CRISPR/Cas9 component delivery into biofilm-embedded P. aeruginosa.
Materials:
- DOTAP (1,2-dioleoyl-3-trimethylammonium-propane)
- DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine)
- Cholesterol
- Cas9 protein purified from E. coli expression system
- in vitro-transcribed gRNA targeting P. aeruginosa lasR gene
- HEPES-buffered saline (pH 7.4)
- Extrusion apparatus with 100 nm polycarbonate membranes
- Dialysis tubing (MWCO 300 kDa)
Methodology:
Lipid Film Preparation:
- Combine DOTAP, DOPE, and cholesterol in a molar ratio of 50:45:5 in chloroform.
- Evaporate organic solvent under nitrogen stream to form a thin lipid film.
- Desiccate under vacuum for 12 hours to remove residual solvent.
Hydration and Extrusion:
- Hydrate lipid film with HEPES-buffered saline to a final lipid concentration of 10 mM.
- Vortex vigorously for 5 minutes to form multilamellar vesicles.
- Subject to five freeze-thaw cycles (liquid nitrogen/40°C water bath).
- Extrude through polycarbonate membranes with sequential pore sizes (400 nm, 200 nm, 100 nm).
Cas9/gRNA Complex Loading:
- Pre-complex Cas9 protein with gRNA at 1:2 molar ratio in assembly buffer (30 mM HEPES, 100 mM KCl, pH 7.5) for 15 minutes at 25°C.
- Add ribonucleoprotein complexes to pre-formed liposomes at 1:10 weight ratio.
- Incubate for 30 minutes at room temperature with gentle agitation.
Purification and Characterization:
- Remove unencapsulated material by dialysis against HEPES-buffered saline for 4 hours.
- Determine particle size and zeta potential by dynamic light scattering (expected size: 110-130 nm; zeta potential: +25 to +35 mV).
- Quantify encapsulation efficiency using fluorescence-labeled gRNA and standard curves.
Protocol: Assessment of Anti-Biofilm Efficacy
Biofilm Cultivation:
- Culture P. aeruginosa PAO1 in tryptic soy broth supplemented with 1% glucose at 37°C with shaking.
- Inoculate 96-well polystyrene plates with 1 × 10^6 CFU/mL and incubate statically for 48 hours at 37°C to establish mature biofilms.
Treatment and Analysis:
- Treat established biofilms with liposomal Cas9 formulations (50 μL/well) targeting lasR gene.
- Incubate for 24 hours at 37°C.
- Assess biofilm biomass using crystal violet staining.
- Quantify viable bacteria through colony counting after biofilm disruption.
- Evaluate lasR gene editing efficiency by DNA sequencing of recovered isolates.
Table 2: Expected Experimental Outcomes for Liposomal Cas9 Anti-Biofilm Activity
| Parameter | Control (Untreated) | Liposomal Cas9 (lasR-targeting) | Empty Liposomes |
|---|---|---|---|
| Biofilm Biomass | 100% | 10-15% [4] | 95-105% |
| Viable Bacteria | 1 × 10^8 CFU/mL | 1 × 10^5 CFU/mL | 1 × 10^8 CFU/mL |
| lasR Mutation Frequency | <0.1% | >90% | <0.1% |
| Pyocyanin Production | 100% | 15-20% | 95-105% |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Pseudomonas aeruginosa Biofilm and CRISPR/Cas9 Studies
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| CRISPR/Cas9 Components | Cas9 nuclease, guide RNA (gRNA) | Targeted gene disruption in biofilm-related genes | Program gRNAs to target lasR, rhlI, pslA, or pelA genes |
| Liposomal Transfection Reagents | DOTAP, DOPE, cholesterol | Nanoparticle formulation for enhanced delivery | Optimal lipid:nucleic acid ratio critical for efficiency |
| Biofilm Matrix Stains | Crystal violet, Congo red, FITC-conjugated lectins | Visualization and quantification of biofilm biomass | Use confocal microscopy for 3D architecture analysis |
| P. aeruginosa Strains | PAO1, PA14, clinical isolates | Biofilm formation studies | Mucoid strains essential for cystic fibrosis research |
| Quorum Sensing Inhibitors | Furanones, azithromycin, patulin | Control of virulence and biofilm formation | Use alongside CRISPR for combinatorial approaches |
| Gene Expression Analysis | RT-qPCR primers for lasR, rhlR, psl | Quantification of quorum sensing and biofilm gene expression | Normalize to proC or rpoD reference genes |
Workflow and Signaling Pathways
Experimental Workflow for Liposomal Cas9 Development
Diagram Title: Liposomal Cas9 Development Workflow
Quorum Sensing Regulation of Biofilm Formation
Diagram Title: Quorum Sensing Network in Biofilm Formation
The integration of CRISPR/Cas9 gene-editing technology with advanced liposomal delivery systems represents a paradigm shift in our approach to combating biofilm-associated P. aeruginosa infections. By enabling precise targeting of the genetic underpinnings of biofilm formation and antibiotic resistance, this strategy addresses the fundamental limitations of conventional antimicrobial therapies. Recent advances demonstrating that liposomal Cas9 formulations can reduce P. aeruginosa biofilm biomass by over 90% in vitro underscore the transformative potential of this approach [4]. As research progresses toward in vivo validation and clinical translation, the optimization of gRNA design, liposomal composition, and administration regimens will be critical to realizing the full therapeutic potential of this innovative technology. The application notes and protocols detailed herein provide a foundational framework for researchers pursuing these promising anti-biofilm strategies.
The extracellular polymeric substance (EPS) matrix is a foundational element of bacterial biofilms, constituting over 90% of their dry mass and forming a three-dimensional, protective architecture that shields embedded communities from antimicrobial agents [6] [7]. This matrix is not a mere physical barrier; it is a dynamic, functional component that confers a state of recalcitrance—encompassing both tolerance and resistance—to multidrug-resistant (MDR) pathogens like Pseudomonas aeruginosa [8] [9]. Within the context of developing liposomal Cas9 formulations to combat biofilm infections, a detailed understanding of the EPS matrix is paramount. Disrupting this protective shell is often a prerequisite for enabling therapeutic agents to access and eliminate the underlying bacterial cells. This Application Note details the composition, mechanisms of resistance, and key experimental protocols for characterizing the EPS matrix, providing a methodological foundation for advancing novel anti-biofilm therapeutics.
Deconstructing the EPS Matrix: Components and Resistance Functions
The EPS matrix is a complex amalgamation of biopolymers, primarily polysaccharides, proteins, extracellular DNA (eDNA), and lipids [6] [10]. The composition and abundance of these components are highly variable, influenced by the bacterial species, environmental conditions, and nutrient availability [6]. Each component contributes uniquely to the structural integrity and defensive capabilities of the biofilm. The following table summarizes the primary EPS constituents and their specific roles in fostering multi-drug resistance.
Table 1: Key Components of the EPS Matrix and Their Roles in Antimicrobial Resistance
| EPS Component | Chemical Nature | Primary Function in Resistance | Specific Examples & Mechanisms |
|---|---|---|---|
| Polysaccharides | Polymers of sugars (e.g., PNAG, cellulose, alginate) | Forms a dense diffusion barrier; mediates cohesion and adhesion [6] [7]. | • PNAG: A major polysaccharide in S. epidermidis and E. coli biofilms; degraded by Dispersin B [6].• Alginate: Produced by P. aeruginosa, contributes to mucoid phenotype and impedes antibiotic penetration [11]. |
| Proteins | Enzymes, structural proteins, amyloid fibers, S-layer proteins | Provides structural stability; can inactivate antimicrobials via enzymatic degradation [10] [12]. | • S-layer protein: In anammox biofilms, acts as a public-good exopolymer, organizing community structure and enhancing robustness [12].• Proteases: Can degrade antimicrobial peptides and inactivate certain antibiotics [8]. |
| Extracellular DNA (eDNA) | Double-stranded DNA released from lysed cells | Contributes to matrix stability via cation bridging; chelates cationic antimicrobials [7] [10]. | • Binds positively charged aminoglycosides (e.g., tobramycin), preventing their penetration into the biofilm [7]. Matrix stability is sensitive to DNase treatment [6]. |
| Lipids | Hydrophobic molecules | Influences hydrophobicity and potentially limits penetration of hydrophilic compounds [10]. | The specific role in resistance is less defined but contributes to the overall physicochemical properties of the matrix [10]. |
The synergistic interactions between these components create a robust, viscoelastic structure. For instance, divalent cations like Ca²⁺ and Mg²⁺ strengthen the matrix by forming ionic bridges between negatively charged functional groups on eDNA and polysaccharide chains [6]. This cross-linked network is fundamental to the matrix's mechanical stability and its function as a protective barrier.
Quantitative Analysis of EPS Composition
Understanding the relative proportions of EPS components is critical for evaluating the efficacy of matrix-disrupting agents, including potential liposomal formulations. Fourier Transform Infrared (FT-IR) Spectroscopy is a powerful, non-destructive technique for achieving this.
Table 2: Quantitative Analysis of Biofilm EPS by ATR/FT-IR Spectroscopy [10]
| IR Spectral Window (cm⁻¹) | Primary EPS Assignment | Functional Groups Detected | Interpretation and Application |
|---|---|---|---|
| 2800–3000 | Lipids | C-H, CH₂, CH₃ stretches | Indicates relative lipid content in the matrix. |
| 1500–1800 | Proteins | C=O, N-H, C-N (Amide I, II bands) | The Amide II band is a key biomarker for quantifying sessile cell biomass. |
| 900–1250 | Polysaccharides & Nucleic Acids | C-O, C-O-C, P=O, C-N stretches | Tracking the Amide II/Polysaccharide ratio reveals shifts in EPS production (e.g., preferential polysaccharide synthesis decreases the ratio) [10]. |
Core Mechanisms of EPS-Mediated Multidrug Resistance
The EPS matrix facilitates antimicrobial failure through a multi-faceted strategy that can be visualized as a series of layered defense mechanisms.
Diagram: Multifaceted Defense of the EPS Matrix. The EPS matrix employs sequential and synergistic mechanisms to protect bacterial cells from antimicrobials, leading to the development of resistant populations.
The pathways outlined in the diagram represent the core physiological strategies:
- Diffusion Limitation: The dense, hydrogel-like nature of the EPS physically hinders the penetration of antimicrobial molecules, causing a delayed and reduced accumulation at the core of the biofilm [8] [7]. This often results in sublethal doses that promote microbial selection and adaptation [8].
- Direct Interaction and Inactivation: Specific components of the matrix can directly bind and neutralize antimicrobials. For example, negatively charged eDNA chelates cationic aminoglycosides, while enzymes within the matrix can degrade certain drugs [7].
- Altered Microenvironment and Metabolic Heterogeneity: Consumption of nutrients and oxygen by peripheral cells creates gradients, leading to slow growth or metabolic dormancy in the biofilm interior. Since many antibiotics target active cellular processes, these dormant cells (including persisters) exhibit profound tolerance [7] [9].
Essential Protocols for EPS Matrix Disruption and Analysis
Targeting the EPS matrix is a key therapeutic strategy. The following protocols provide methodologies for assessing the efficacy of matrix-disrupting agents, which is essential for validating new anti-biofilm formulations like liposomal Cas9.
Protocol 1: Enzymatic Disruption of Biofilm Matrix
Principle: Specific enzymes degrade key structural components of the EPS, leading to a loss of mechanical integrity and biofilm detachment [6] [10].
Applications:
- Screening for potential matrix-disrupting compounds.
- Determining the structural contribution of specific EPS components (e.g., polysaccharides vs. proteins).
- Potentiating the activity of conventional antibiotics [10].
Workflow:
Diagram: Enzymatic Disruption of Biofilms. A standardized workflow for treating established biofilms with EPS-degrading enzymes and quantifying the resulting disruption.
Key Reagents and Materials:
- CDC Biofilm Reactor: Provides controlled, shear-relevant conditions for growing standardized, reproducible biofilms, superior to static well-plate models [6].
- Dispersin B: A glycoside hydrolase that specifically degrades poly-N-acetylglucosamine (PNAG), a key polysaccharide in many biofilms [6].
- Proteinase K/Trypsin: Serine proteases that hydrolyze peptide bonds in matrix proteins, effectively degrading protein-based EPS [6] [10].
- DNase I: Breaks down phosphodiester bonds in extracellular DNA (eDNA), a crucial component for matrix stability in many species [6].
Protocol 2: Mechanical Strength Assessment via Atomic Force Microscopy (AFM)
Principle: AFM measures the nanoscale cohesive strength of a biofilm by quantifying the force required to indent or rupture the EPS matrix, providing a direct readout of its mechanical integrity [6].
Applications:
- Evaluating the mechanical impact of EPS-targeting treatments (enzymes, antibiotics, novel agents).
- Correlating changes in EPS composition with biomechanical properties.
Workflow:
- Sample Preparation: Grow biofilms on suitable substrates (e.g., glass coverslips). Treat with EPS-modifying agents or leave as untreated control.
- AFM Calibration: Calibrate the AFM cantilever using a clean, rigid surface to determine its spring constant.
- Force Measurement: Position the cantilever over the biofilm surface. Approach the surface until contact is made, applying a defined force. Retract the cantilever and record the force-distance curve.
- Data Analysis: Analyze the retraction curve. The cohesive strength is proportional to the adhesion force or the work of adhesion required to detach the tip from the biofilm surface. A significant reduction in adhesion force after treatment indicates successful matrix disruption [6].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for EPS and Biofilm Research
| Research Reagent | Function/Application | Specific Example & Rationale |
|---|---|---|
| EPS-Degrading Enzymes | Targeted disruption of specific EPS components to study function and potentiate antibiotics. | Dispersin B: Targets PNAG polysaccharide [6]. DNase I: Degrades eDNA, critical for biofilms reliant on DNA for stability [6] [7]. |
| Cation Chelators | Weaken ionic cross-linking within the EPS matrix, reducing stability. | EDTA (Ethylenediaminetetraacetic acid): Chelates divalent cations (Ca²⁺, Mg²⁺), disrupting cation bridges that reinforce the matrix [6]. |
| Quorum Sensing Inhibitors (QSIs) | Interfere with bacterial cell-to-cell communication, potentially suppressing EPS production. | Cinnamaldehyde, AHL analogs: Natural and synthetic molecules that block quorum sensing, downregulating virulence and matrix synthesis [8] [9]. |
| Liposomal CRISPR-Cas9 Formulations | Precision targeting of bacterial resistance genes within biofilms. | Liposomal Cas9 + sgRNA: Nanoparticles encapsulating Cas9/sgRNA complexes. Enable targeted disruption of antibiotic resistance genes, quorum-sensing pathways (e.g., lasI/rhlI), or biofilm-regulating factors in P. aeruginosa [13] [9]. |
The architecture of the EPS matrix represents a master strategy in bacterial survival, directly challenging conventional antimicrobial therapies. A systematic, component-level understanding of this matrix is no longer optional but essential for innovating effective treatments. The integration of enzymatic disruptors, mechanical analysis, and advanced delivery systems like liposomal Cas9 represents the frontier of anti-biofilm research. The protocols and data presented herein provide a roadmap for researchers to deconstruct the defensive fortress of the biofilm, paving the way for combination therapies that first dismantle the matrix to then deliver a lethal, precise strike to the pathogen within.
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen responsible for life-threatening acute infections and chronic, difficult-to-eradicate conditions, particularly in healthcare settings and in patients with cystic fibrosis (CF) [14]. A defining feature of its pathogenicity is the formation of biofilms—structured, surface-attached communities of cells encased in a self-produced extracellular matrix [15]. It is estimated that 65–80% of all bacterial infections are associated with this biofilm mode of growth [14]. The biofilm lifestyle confers a profound level of intrinsic tolerance and resistance to antimicrobial therapies, which is a major driver of chronic infections [14] [16].
This protective effect is multifactorial, arising from a complex interplay of physical, physiological, and genetic factors [14]. The extracellular matrix, composed of exopolysaccharides, proteins, extracellular DNA (eDNA), and lipids, acts as a physical barrier that can restrict the penetration of certain antibiotics [14]. Furthermore, the biofilm environment is characterized by gradients of nutrients and oxygen, creating heterogeneous subpopulations of cells with varying metabolic states [14]. This heterogeneity is critical because many classic antibiotics require active bacterial growth to be effective. The most metabolically inactive cells, often located in the deeper layers of the biofilm, are therefore protected [14]. Adding to this challenge is the extraordinary capacity of P. aeruginosa to develop stable, genetic resistance to nearly all available antibiotics through chromosomal mutations, a feature known as its vast "mutational resistome" [14].
Within the context of a broader thesis focused on liposomal Cas9 formulations, understanding these intrinsic mechanisms of biofilm resistance is paramount. They represent the fundamental barriers that any novel therapeutic, including one based on gene editing, must overcome to be effective. The following sections will detail the key mechanisms of resistance and tolerance, present experimental protocols for their study, and visualize the complex regulatory networks involved.
Core Mechanisms of Biofilm Resistance and Tolerance
The recalcitrance of P. aeruginosa biofilms to antimicrobial treatment can be categorized into three primary, interconnected mechanisms: genetic adaptation, the activity of efflux pumps, and the formation of dormant persister cells.
Genetic Adaptation and the Mutational Resistome
P. aeruginosa exhibits a remarkable ability to develop antibiotic resistance through the selection of chromosomal mutations. This "versatile mutational resistome" is dramatically amplified in chronic infections, often driven by the emergence of hypermutable strains (mutators) [14]. Key genetic adaptations include:
- AmpC β-Lactamase Induction: Exposure to β-lactam antibiotics like imipenem or ceftazidime induces the expression of the AmpC β-lactamase. In biofilms, this enzyme shows a distinct structural distribution, concentrated at the biofilm periphery, and can be partially excreted via membrane vesicles to confer protection [14].
- Differential Gene Expression: Biofilm cells exhibit a distinct transcriptional profile compared to their planktonic counterparts. The activation of genes like brlR, a Mer-like transcriptional regulator, stimulates the expression of efflux systems (e.g., MexAB-OprM, MexEF-OprN) and other resistance determinants, lowering susceptibility to multiple antibiotic classes [14]. Another key gene, ndvB, codes for a glycosyltransferase involved in forming cyclic glucans that can sequester antibiotics such as aminoglycosides, preventing them from reaching their cellular targets [14].
- Adaptive Tolerance: Sub-inhibitory concentrations of antibiotics can induce a transient tolerant phenotype. For instance, the presence of colistin upregulates the pmr two-component system, which modifies lipopolysaccharide (LPS) to reduce its negative charge, thereby decreasing binding of this cationic antimicrobial peptide [14].
Efflux Pump-Mediated Resistance
Efflux pumps are membrane transporters that expel a wide range of toxic compounds, including antibiotics, from the bacterial cell. They are a cornerstone of intrinsic and acquired multidrug resistance in P. aeruginosa [15]. The Resistance-Nodulation-Division (RND) family pumps are particularly significant in a clinical context [15] [17].
Table 1: Key RND Efflux Pumps in P. aeruginosa Biofilm Resistance
| Efflux Pump | Key Substrates (Antibiotics) | Role in Biofilm Resistance |
|---|---|---|
| MexAB-OprM | β-lactams, tetracycline, chloramphenicol, quinolones, macrolides [15] | Contributes to resistance against aztreonam, gentamicin, tetracycline, and tobramycin in biofilms; expression is highest in cells located at the substratum [15]. |
| MexCD-OprJ | Macrolides (e.g., azithromycin), chloramphenicol [15] | Involved in biofilm resistance to azithromycin; induced in active subpopulations under colistin exposure [15]. |
| MexEF-OprN | Quinolones, chloramphenicol, trimethoprim [15] | Its role in biofilm resistance is less clear than MexAB-OprM [15]. |
| MexXY-OprM | Aminoglycosides, tetracycline, erythromycin [15] | Upregulated in response to oxidative stress and membrane-damaging agents; contributes to aminoglycoside resistance [14]. |
| PA1874-1877 | Ciprofloxacin, gentamicin, tobramycin [15] | A novel pump with expression ~10x higher in biofilms than in planktonic cells [14] [15]. |
The expression and function of efflux pumps in biofilms are intricately linked with other bacterial processes. They can export quorum-sensing (QS) signal molecules, thereby influencing cell-to-cell communication and biofilm development [15] [17]. Conversely, the BrlR regulator, which is upregulated in biofilms, can directly stimulate the expression of several efflux pumps, creating a feedback loop that enhances the multidrug-resistant phenotype [14].
Persister Cell Dormancy and Formation
Persisters are a small subpopulation of metabolically quiescent bacterial cells that exhibit extreme, non-genetic tolerance to high concentrations of antibiotics [18] [19]. They are not mutants; upon antibiotic removal, persisters can resume growth and give rise to a susceptible population, which is a primary cause of chronic infections and post-treatment relapse [18] [20] [19]. In biofilms, the frequency of persisters can be up to 1% of the population, significantly higher than in exponential-phase planktonic cultures [18].
The formation of dormant persisters is primarily governed by two interconnected mechanisms:
- Toxin-Antitoxin (TA) Systems: TA systems are genetic modules consisting of a stable toxin that disrupts essential cellular processes (e.g., translation, DNA replication) and a labile antitoxin that neutralizes the toxin [18]. Under stress conditions, the antitoxin is degraded, allowing the toxin to induce a state of dormancy. Key TA systems linked to persistence in various bacteria include HipBA, MqsR/MqsA, and TisB/IstR-1 [18]. For example, the TisB toxin can decrease the proton motive force and ATP levels, rendering the cell dormant and tolerant [18].
- The Stringent Response and (p)ppGpp: In response to nutrient starvation and other stresses, bacteria produce the alarmone guanosine tetraphosphate (ppGpp) [18] [20]. This molecule triggers a global transcriptional reprogramming known as the stringent response, which slows down bacterial growth and metabolism. ppGpp also activates stress response pathways and can stimulate the activity of TA systems, thereby promoting the formation of dormant persister cells [18] [20].
Table 2: Key Mechanisms of Bacterial Persister Cell Formation
| Mechanism | Key Components/Genes | Mode of Action in Persistence |
|---|---|---|
| Toxin-Antitoxin Systems | HipA, MqsR, TisB, RelE [18] | Toxin activity inhibits essential processes (translation, replication), inducing a dormant, antibiotic-tolerant state [18]. |
| Stringent Response | RelA, SpoT, (p)ppGpp [18] [20] | Nutrient stress triggers ppGpp accumulation, slowing metabolism and growth, and activating TA systems [18]. |
| SOS Response | RecA, LexA [20] | DNA damage induces this stress response, leading to cell cycle arrest and reduced metabolic activity [20]. |
| Reduced ATP Levels | [20] | Lowered intracellular ATP is correlated with increased antibiotic tolerance, as many drugs require energetic processes for killing [20]. |
Signaling Pathways Governing Biofilm Resistance
The mechanisms described above are not isolated but are coordinated by sophisticated regulatory networks that sense environmental cues and dictate bacterial behavior. The following diagram synthesizes the key signaling pathways that underpin genetic adaptation, efflux pump expression, and persister cell formation in P. aeruginosa biofilms.
Diagram 1: Signaling pathways in P. aeruginosa biofilm resistance. Environmental stimuli activate regulatory systems like Quorum Sensing (QS), Two-Component Systems (TCS), and the Stringent Response, which coordinately control the expression of resistance and tolerance effectors.
Experimental Protocols for Investigating Biofilm Resistance
To systematically study the mechanisms outlined above, robust and reproducible experimental protocols are essential. The following section provides detailed methodologies for key assays relevant to biofilm research and therapeutic development.
Protocol: Standardized Biofilm Cultivation and Treatment
This protocol describes a common in vitro method for growing P. aeruginosa biofilms in a 96-well microtiter plate format, suitable for high-throughput screening of anti-biofilm agents [15].
Key Research Reagent Solutions:
- Culture Medium: Lysogeny Broth (LB) or synthetic cystic fibrosis sputum medium (SCFM2) to mimic in vivo conditions [21].
- Staining Solution: 0.1% (w/v) Crystal Violet (CV) in distilled water for biomass quantification.
- Destaining Solution: 30% Acetic Acid or 95% Ethanol to solubilize bound CV.
- Microtiter Plate: Polystyrene, non-tissue culture treated plates to enhance bacterial attachment.
Procedure:
- Inoculation: Dilute an overnight culture of P. aeruginosa to an optical density at 600 nm (OD₆₀₀) of ~0.05 in fresh, pre-warmed medium. Dispense 200 µL per well into a 96-well microtiter plate.
- Biofilm Growth: Incubate the plate statically for 24-48 hours at 37°C. Do not agitate.
- Treatment (Optional): After biofilm formation, carefully remove the planktonic culture and medium by inverting and flicking the plate. Wash the adherent biofilm once with 200 µL of phosphate-buffered saline (PBS). Add 200 µL of the test agent (e.g., antibiotic, liposomal formulation) diluted in fresh medium to the wells. Incubate for a further desired period (e.g., 24 h).
- Biofilm Quantification (Crystal Violet Staining): a. Remove the contents of the wells and wash twice with 200 µL PBS to remove non-adherent cells. b. Air-dry the plate for 30-45 minutes. c. Add 200 µL of 0.1% CV solution to each well and incubate for 15 minutes at room temperature. d. Remove the CV and rinse the plate thoroughly under running tap water until the runoff is clear. e. Air-dry the plate completely. f. Add 200 µL of destaining solution (e.g., 30% acetic acid) to each well and incubate for 15 minutes with gentle shaking to solubilize the dye. g. Transfer 125 µL of the solubilized CV from each well to a new plate and measure the absorbance at 550 nm. The absorbance is proportional to the total biofilm biomass.
Protocol: Assessing Anti-biofilm Efficacy of Liposomal CRISPR/Cas9
This protocol outlines a method to evaluate the efficacy of nanoparticle-encapsulated CRISPR/Cas9 systems against established P. aeruginosa biofilms, measuring reduction in viable cells and biofilm biomass [13].
Key Research Reagent Solutions:
- Liposomal CRISPR/Cas9 Formulation: Cas9 protein and guide RNA (gRNA) complexes encapsulated in liposomal nanoparticles. The gRNA should be designed to target essential resistance genes (e.g., ampC), efflux pump components (e.g., mexB), or persister-related genes (e.g., hipA).
- Viability Stain: Live/Dead BacLight Bacterial Viability Kit (e.g., containing SYTO9 and propidium iodide).
- Mature Biofilm: 24-48 hour old P. aeruginosa biofilm grown as described in Protocol 4.1.
Procedure:
- Biofilm Preparation: Grow mature biofilms in a 24-well plate or on relevant surfaces (e.g., catheter pieces) for 48 hours.
- Treatment: Gently wash the biofilms with PBS. Apply the liposomal CRISPR/Cas9 formulation at the desired concentration (e.g., 1 × 10⁷ PFU/mL equivalent for phage-derived systems or µg/mL for synthetic formulations) in an appropriate buffer or diluted medium [13] [21]. Include controls: untreated biofilm, biofilm treated with empty liposomes, and biofilm treated with a non-targeting gRNA formulation.
- Incubation: Incubate the treatment plate for 4-24 hours at 37°C.
- Efficacy Assessment (Post-treatment analysis): a. Viable Cell Count (CFU assay): Aspirate the treatment, wash the biofilm with PBS, and disrupt the biofilm by sonication or vigorous pipetting in a known volume of PBS. Serially dilute the suspension and plate on LB agar. Count the Colony Forming Units (CFU) after overnight incubation. Calculate the log reduction compared to the untreated control. b. Biomass Quantification (Crystal Violet): Perform CV staining as in Protocol 4.1 to assess the physical disruption of the biofilm matrix. A >90% reduction in biomass has been demonstrated with effective liposomal CRISPR-Cas9 formulations in vitro [13]. c. Confocal Microscopy (Live/Dead Staining): Wash the treated biofilm and stain according to the Live/Dead kit instructions. Image using a confocal laser scanning microscope. Live cells (intact membranes) stain green (SYTO9), while dead/damaged cells (compromised membranes) stain red (propidium iodide). This provides a visual and quantitative measure of cell viability within the biofilm architecture.
The following diagram illustrates this integrated experimental workflow.
Diagram 2: Experimental workflow for evaluating liposomal CRISPR/Cas9. The protocol involves growing a mature biofilm, treating it with the therapeutic formulation, and using multiple analytical methods to quantify eradication efficacy.
Table 3: Essential Research Reagent Solutions for Biofilm and Persister Studies
| Item/Category | Specific Examples | Function/Application |
|---|---|---|
| Specialized Growth Media | Synthetic Cystic Fibrosis Sputum Medium (SCFM2) [21] | Mimics the in vivo lung environment of CF patients, promoting clinically relevant biofilm phenotypes for testing. |
| Efflux Pump Inhibitors | Phe-Arg β-naphthylamide (PAβN) [17] | A broad-spectrum inhibitor used to investigate the contribution of RND-type efflux pumps to antibiotic resistance and biofilm formation. |
| Viability Staining Kits | LIVE/DEAD BacLight Bacterial Viability Kit [13] | Differentiates between live and dead cells in a biofilm via membrane integrity, used for confocal microscopy analysis. |
| Gene Editing Tools | Liposomal CRISPR-Cas9 Formulations [13] | Enables targeted disruption of specific bacterial genes (e.g., resistance genes, TA systems) to study their function and as a therapeutic. |
| Model Bacteriophages | Pbunavirus phage PE1 and its evolved variants (PE1-3, PE1-5) [21] | Used as anti-biofilm agents and delivery vehicles; evolved phages show enhanced efficacy through improved LPS recognition. |
| Matrix Dispersal Agents | Dispersin B, DNase I | Enzymes that degrade specific biofilm matrix components (exopolysaccharides and eDNA, respectively), used to study matrix function and for combination therapies. |
The resilience of P. aeruginosa biofilms is not attributable to a single mechanism but is the result of a powerful synergy between genetic adaptation, efflux pump activity, and persister cell dormancy. These processes, regulated by interconnected networks like quorum sensing and the stringent response, create a formidable barrier to conventional antibiotics. The failure of monotherapies against such a multifaceted defense system is predictable.
This understanding underscores the critical need for innovative, targeted therapeutic strategies. The integration of CRISPR/Cas9 gene-editing technology, capable of precisely disrupting key resistance and persistence genes, with sophisticated nanoparticle-based delivery systems designed to penetrate the biofilm matrix, represents a promising frontier. This approach, which directly targets the genetic underpinnings of biofilm resistance, offers a pathway to dismantle the core defenses of P. aeruginosa and potentially overcome the treatment challenges posed by chronic biofilm infections.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) systems constitute an adaptive immune mechanism in bacteria and archaea, providing sequence-specific protection against invading genetic elements such as viruses and plasmids [22]. Originally identified as a biological curiosity in Streptococcus thermophilus in 1987, these systems were later recognized in the early 2000s for their role in microbial immunity [22]. The landmark 2012 discovery that the Cas9 protein could be reprogrammed with synthetic guide RNA to cut DNA at virtually any desired location transformed CRISPR-Cas9 into a versatile genome-editing platform with revolutionary implications across biological sciences and medicine [22].
CRISPR-Cas systems operate through a coordinated, three-stage immune process: adaptation, expression, and interference [22]. During adaptation, short fragments of foreign DNA (protospacers) are integrated into the host's CRISPR array as new spacers. In the expression stage, the CRISPR array is transcribed and processed into mature CRISPR RNAs (crRNAs). Finally, in the interference stage, RNA-guided Cas proteins recognize and cleave complementary nucleic acid sequences, providing defense against future invasions [22]. These molecular mechanisms form the foundation for adapting CRISPR-Cas9 as a programmable gene-editing tool.
CRISPR-Cas systems are broadly classified into two major classes based on their effector complex architecture. Class 1 systems (Types I, III, and IV) employ multi-protein complexes for target recognition and cleavage, while Class 2 systems (Types II, V, and VI) utilize single effector proteins, making them more suitable for biotechnological applications [22]. The Type II CRISPR-Cas9 system from Streptococcus pyogenes has become the most widely adopted platform for genome engineering due to its simplicity and well-characterized mechanism [23].
Figure 1: CRISPR-Cas9 Molecular Mechanism. The diagram illustrates the three-stage process of CRISPR-Cas9 function: adaptation (foreign DNA capture and integration), expression (crRNA processing), and interference (target recognition and cleavage).
CRISPR-Cas9 as a Precision Antimicrobial Tool
The application of CRISPR-Cas9 technology represents a paradigm shift in combating antimicrobial resistance (AMR), offering unprecedented precision in selectively eliminating resistant bacterial strains while sparing susceptible or beneficial members of the microbiome [22]. This specificity stems from the programmable nature of CRISPR guide RNAs, which can be tailored to recognize unique genetic sequences associated with resistance determinants, thereby avoiding the collateral damage often caused by conventional antibiotics [22].
Two primary CRISPR-based strategies have emerged for targeting AMR: strain-specific killing and resistance gene disruption. Strain-specific killing involves targeting essential genes or resistance-conferring loci to selectively eliminate resistant bacteria from mixed populations [22]. Resistance gene disruption focuses on inactivating specific antimicrobial resistance genes without directly killing the bacterial cell, potentially resensitizing the bacterium to conventional antibiotics [13]. Both approaches can be deployed against Pseudomonas aeruginosa biofilms, which are structured microbial communities embedded in extracellular polymeric substances that confer high levels of antibiotic resistance [1] [3].
The extracellular polymeric substance (EPS) matrix of P. aeruginosa biofilms comprises polysaccharides (alginate, Pel, Psl), extracellular DNA, and proteins, creating a protective barrier that limits antibiotic penetration and enhances bacterial persistence [1] [3]. This matrix, along with reduced metabolic activity of biofilm-embedded cells and increased horizontal gene transfer, contributes to the up to 1000-fold greater antibiotic tolerance observed in biofilms compared to planktonic cells [24]. CRISPR-Cas9 systems can be engineered to target key biofilm regulatory genes, including those involved in quorum sensing (QS) pathways (lasR, rhlR), EPS production, and stress response systems [3] [25].
Table 1: CRISPR-Cas9 Antimicrobial Strategies for Biofilm Control
| Strategy | Molecular Target | Mechanism of Action | Expected Outcome |
|---|---|---|---|
| Strain-Specific Killing | Essential genes (e.g., rpsL), species-specific sequences | Cas9-mediated double-strand breaks in chromosomal DNA | Selective elimination of target pathogens from mixed communities |
| Resistance Gene Inactivation | Antibiotic resistance genes (e.g., β-lactamases, efflux pumps) | Disruption of resistance gene function | Resensitization to conventional antibiotics |
| Virulence Attenuation | Quorum sensing systems (lasR, rhlR), toxin genes | Targeting regulatory networks controlling pathogenicity | Reduced biofilm formation and host tissue damage |
| Biofilm Disruption | EPS synthesis genes (psl, pel, alg), adhesion factors | Impairment of matrix production and structural integrity | Enhanced antibiotic penetration and immune clearance |
Advanced Delivery Systems for Biofilm Penetration
The clinical application of CRISPR-based antibacterials faces significant challenges, particularly in achieving efficient delivery and stability within bacterial populations, especially in the context of established biofilms [13] [24]. The complex architecture of biofilms, characterized by microcolonies interspersed with water channels and encased in a dense extracellular matrix, creates a formidable barrier to conventional delivery methods [24]. Recent advances in nanocarrier systems have shown remarkable potential for overcoming these limitations.
Nanoparticles (NPs) present an innovative solution, serving as effective carriers for CRISPR-Cas9 components while exhibiting intrinsic antibacterial properties [13] [24]. Nanoparticles can enhance CRISPR delivery by improving cellular uptake, increasing target specificity, and ensuring controlled release within biofilm environments [24]. Various types of nanoparticles, including lipid-based nanoparticles, polymeric nanoparticles, and metallic nanoparticles, have been explored for this purpose, with each offering distinct advantages for biofilm applications [24].
Recent advances have demonstrated that liposomal CRISPR-Cas9 formulations can reduce P. aeruginosa biofilm biomass by over 90% in vitro, while gold nanoparticle carriers enhance editing efficiency up to 3.5-fold compared to non-carrier systems [24]. These hybrid platforms also enable co-delivery with antibiotics, producing synergistic antibacterial effects and superior biofilm disruption [24]. The combination of CRISPR-Cas9 with nanoparticle-based delivery represents a promising approach for addressing the technical challenges of biofilm penetration and bacterial uptake.
Table 2: Nanoparticle Systems for CRISPR-Cas9 Delivery Against Biofilms
| Nanocarrier Type | Key Advantages | Editing Efficiency | Biofilm Reduction | Synergy with Antibiotics |
|---|---|---|---|---|
| Liposomal Nanoparticles | Enhanced biofilm penetration, fusogenic properties | Moderate to high | >90% in P. aeruginosa | Yes, with tobramycin and colistin |
| Gold Nanoparticles | Conjugation with gRNA, photothermal properties | 3.5-fold enhancement over non-carrier | ~70-80% | Yes, enhanced with functionalization |
| Polymeric Nanoparticles | Sustained release, high payload capacity | Moderate | ~60-75% | Variable depending on polymer |
| Magnetic Nanoparticles | External guidance, hyperthermia effects | Moderate | ~65-70% | Enhanced with thermal activation |
Application Notes: pCasPA/pACRISPR System forPseudomonas aeruginosa
The development of specialized CRISPR-Cas9 systems for Pseudomonas aeruginosa has significantly advanced genetic manipulation in this clinically important pathogen. The pCasPA/pACRISPR system represents a optimized platform for efficient and scarless genetic manipulation in P. aeruginosa, harnessing both the CRISPR-Cas9 and phage λ-Red recombination systems [23].
System Architecture and Components
The pCasPA/pACRISPR system employs a two-plasmid design that separates functional elements to enhance efficiency and flexibility [23]. The pCasPA plasmid expresses the Streptococcus pyogenes Cas9 (spCas9) nuclease and the λ-Red system proteins (Exo, Gam, Bet) under the control of the L-arabinose-inducible promoter ParaB [23]. This plasmid also contains the counter-selectable sacB gene, which confers sucrose sensitivity and facilitates plasmid curing after editing [23]. The pACRISPR plasmid expresses the single-guide RNA (sgRNA) under the control of the strong trc promoter and contains two seamless cloning sites: BsaI sites for Golden Gate assembly of the 20-nt spacer sequence, and XbaI/XhoI sites for Gibson assembly of repair arms (approximately 1 kb each) [23].
Workflow and Protocol
The experimental workflow for using the pCasPA/pACRISPR system involves sequential steps of plasmid construction, transformation, induction of recombination and cleavage functions, and selection of successful editants [23]. First, the pACRISPR plasmid is constructed by cloning the specific 20-nt spacer sequence targeting the genomic locus of interest into the BsaI sites via Golden Gate assembly, followed by insertion of the homologous repair arms into the XbaI and XhoI sites via Gibson assembly [23]. The pCasPA plasmid is then transformed into the P. aeruginosa strain, and successful transformants are selected using appropriate antibiotics. For editing, cultures containing pCasPA are grown to mid-log phase, and the λ-Red and Cas9 functions are induced by adding L-arabinose (0.2-0.5%) for 2 hours [23]. The induced cells are made electrocompetent and transformed with the constructed pACRISPR plasmid. Following recovery, cells are plated on selective media containing sucrose to counter-select against the pCasPA plasmid, which contains the sacB marker [23]. Surviving colonies are screened for the desired mutation via colony PCR and sequencing.
Figure 2: pCasPA/pACRISPR Experimental Workflow. The diagram outlines the step-by-step protocol for genome editing in Pseudomonas aeruginosa using the pCasPA/pACRISPR system, from sgRNA design to mutant validation.
Advanced Base Editing Systems for Pseudomonas Species
Beyond conventional CRISPR-Cas9 approaches, base editing technologies offer additional capabilities for genetic manipulation in Pseudomonas species. The pnCasPA-BEC system enables highly efficient gene inactivation and point mutations through cytidine deamination without generating double-stranded DNA breaks [23]. This system was developed by engineering a fusion of the cytidine deaminase APOBEC1 and the Cas9 nickase (Cas9n), creating a base editor that mediates C→T (or G→A) conversions in various Pseudomonas species, including P. aeruginosa, Pseudomonas putida, Pseudomonas fluorescens, and Pseudomonas syringae [23].
The pnCasPA-BEC system operates through a mechanism distinct from standard CRISPR-Cas9. The Cas9 nickase is guided to the target genomic locus by the sgRNA, where it generates a single-strand break (nick) in the non-target strand [23]. The fused APOBEC1 deaminase then catalyzes the conversion of cytidine to uridine within a narrow editing window (typically positions 4-8 within the protospacer) in the single-stranded DNA bubble generated upon Cas9 binding [23]. The cellular DNA repair machinery subsequently processes the U:G mismatch, resulting in a C:G to T:A base pair conversion. By targeting CAA, CAG, CGA, or TGG codons, the cytidine base editor can efficiently introduce premature stop codons (TAA, TAG, or TGA) to inactivate target genes [23].
Base editing systems offer several advantages for biofilm research, including the ability to create precise point mutations without donor templates, reduced indel formation compared to Cas9 nuclease, and potentially higher editing efficiencies for certain applications [23]. These systems are particularly valuable for studying essential genes where complete knockout would be lethal, for introducing specific amino acid changes to study protein function, and for creating graded reductions in gene expression through targeted nonsense mutations.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for CRISPR-Cas9 Biofilm Research
| Reagent / Material | Function and Application | Specific Examples |
|---|---|---|
| pCasPA/pACRISPR System | Two-plasmid system for efficient genome editing in P. aeruginosa | pCasPA (Cas9 + λ-Red), pACRISPR (sgRNA + repair arms) [23] |
| pnCasPA-BEC System | Base editing without double-strand breaks for point mutations | APOBEC1-Cas9n fusion for C→T conversions [23] |
| Liposomal Nanoparticles | Enhanced delivery of CRISPR components through biofilm matrix | CRISPR-Cas9 liposomal formulations (>90% biofilm reduction) [24] |
| Gold Nanoparticle Carriers | Improved editing efficiency and potential for photothermal activation | AuNP-CRISPR conjugates (3.5× efficiency enhancement) [24] |
| λ-Red Recombinase System | Enhanced homologous recombination efficiency in Pseudomonas | Exo, Gam, Bet proteins under arabinose-inducible promoter [23] |
| Counter-Selectable Markers | Plasmid curing and selection of editants | sacB gene (sucrose sensitivity) [23] |
CRISPR-Cas9 technology has revolutionized our approach to studying and combating biofilm-associated infections, particularly those caused by recalcitrant pathogens like Pseudomonas aeruginosa. The development of specialized systems such as pCasPA/pACRISPR and pnCasPA-BEC has dramatically simplified genetic manipulation in Pseudomonas species, accelerating investigations into bacterial physiology, drug target exploration, and metabolic engineering [23]. When combined with advanced delivery platforms like liposomal and gold nanoparticles, CRISPR-based approaches achieve unprecedented precision in targeting antibiotic resistance genes, quorum sensing pathways, and biofilm-regulating factors [13] [24].
Future directions in this field will likely focus on optimizing delivery platforms to enhance biofilm penetration and bacterial uptake efficiency while minimizing off-target effects and potential immune responses [24]. The integration of CRISPR diagnostics with therapeutic approaches will enable real-time monitoring of biofilm composition and treatment response, facilitating personalized antimicrobial strategies [26]. Additionally, the exploration of novel Cas variants with altered PAM specificities, reduced sizes, and higher fidelity will expand the targeting range and safety profile of CRISPR-based antimicrobials [27] [22]. As these technologies mature, CRISPR-Cas systems are poised to become transformative tools in the ongoing battle against antimicrobial resistance, offering targeted, programmable solutions to one of modern medicine's most pressing challenges.
Pseudomonas aeruginosa is a formidable opportunistic pathogen, particularly notorious for forming antibiotic-recalcitrant biofilms in clinical settings such as cystic fibrosis lungs, chronic wounds, and on medical implants [1] [28]. These biofilms are structured communities of bacteria encased in a self-produced matrix of extracellular polymeric substances (EPS) that confers a remarkable level of protection [29]. The biofilm matrix, composed of polysaccharides (Pel, Psl, alginate), extracellular DNA (eDNA), proteins, and lipids, creates a formidable physical and functional barrier to antimicrobial penetration [1]. This matrix facilitates bacterial survival in hostile environments, including under antibiotic pressure, with biofilm-embedded bacteria exhibiting up to 1000-fold greater resistance to antimicrobial agents compared to their planktonic counterparts [29] [4].
The challenge of delivering therapeutic agents to bacterial cells within bioframes represents a critical bottleneck in antimicrobial development. Conventional antibiotics fail to penetrate the dense EPS matrix effectively, demanding administration of dangerously high doses that risk toxicity while still often failing to eradicate the infection [29]. This therapeutic impasse necessitates innovative drug delivery strategies capable of navigating the biofilm barrier to deliver antimicrobial payloads directly to bacterial targets. Liposomal nanocarriers have emerged as a promising solution to this challenge, offering a versatile platform for enhancing drug penetration, protecting therapeutic cargo, and improving targeting efficiency within the complex biofilm microenvironment [29] [30].
Liposomal Formulations: Engineering Solutions for Biofilm Penetration
Liposomes are spherical vesicles composed of concentric lipid bilayers with an aqueous core, structurally mimicking biological membranes. This biomimetic architecture provides unique advantages for anti-biofilm drug delivery, including the ability to encapsulate both hydrophilic drugs within their aqueous interior and hydrophobic compounds within their lipid membranes [29]. The versatility of liposomal systems enables strategic engineering to overcome specific biofilm-associated delivery challenges through various specialized formulations:
- Conventional Liposomes: Basic phospholipid vesicles that provide fundamental encapsulation and protection for antimicrobial agents, enhancing stability and deposition within biofilm structures [29].
- Stealth Liposomes (PEGylated): Incorporation of polyethylene glycol (PEG) creates a hydrophilic protective layer that reduces opsonization and recognition by the mononuclear phagocyte system, significantly extending circulation half-life and enhancing accumulation at biofilm-infected sites [30].
- Stimuli-Responsive Liposomes: Engineered to release their payload in response to specific environmental triggers unique to the biofilm microenvironment, such as decreased pH, specific enzymes, or temperature gradients [30].
- Targeted Liposomes: Surface-functionalized with ligands, antibodies, or peptides that recognize specific bacterial surface components or biofilm matrix elements, enabling active targeting to enhance local drug concentrations [30].
Table 1: Engineering Strategies for Liposomal Biofilm Penetration
| Liposome Type | Key Features | Mechanism of Enhanced Penetration | Therapeutic Advantages |
|---|---|---|---|
| Conventional | Neutral or charged phospholipid bilayers | Passive diffusion through matrix porosity | Improved drug stability, reduced systemic toxicity |
| Stealth (PEGylated) | Surface-grafted polyethylene glycol polymers | Reduced immune clearance, prolonged circulation | Enhanced accumulation via EPR effect, repeated dosing capability |
| Stimuli-Responsive | Environment-triggered release mechanisms | Activated drug release in response to biofilm-specific signals (pH, enzymes) | Targeted payload release, minimized premature leakage |
| Ligand-Targeted | Surface-conjugated targeting moieties | Specific binding to bacterial cells or matrix components | Increased local drug concentration, species-specific targeting |
The effectiveness of these engineered liposomes against biofilms has been demonstrated across numerous bacterial pathogens. For gram-negative species, liposomal formulations have shown efficacy against P. aeruginosa, Escherichia coli, Acinetobacter baumannii, and members of the genera Klebsiella, Salmonella, and Serratia [29]. Against gram-positive pathogens, liposomal systems have proven effective against Staphylococcal strains including Staphylococcus aureus and Staphylococcus epidermidis, as well as Streptococcal strains and Cutibacterium acnes [29].
Quantitative Efficacy of Liposomal Anti-Biofilm Formulations
Substantial experimental evidence supports the superior efficacy of liposome-encapsulated antimicrobials compared to free drug administration. The enhanced performance stems from the ability of nanoscale liposomes to navigate the structural complexity of biofilms and overcome the physiological barriers that limit conventional antibiotic penetration.
Table 2: Quantitative Efficacy of Liposomal Formulations Against Bacterial Biofilms
| Therapeutic Approach | Biofilm Model | Key Efficacy Metrics | Proposed Mechanism of Action |
|---|---|---|---|
| Liposomal CRISPR/Cas9 | P. aeruginosa in vitro | >90% reduction in biofilm biomass [4] | Targeted disruption of bacterial resistance genes and quorum sensing pathways |
| Liposomal Antibiotics | Various gram-negative and gram-positive species | Up to 1000-fold increased efficacy over free antibiotics [29] | Enhanced penetration through EPS matrix, improved cellular uptake |
| Gold Nanoparticle-CRISPR Hybrids | Bacterial biofilms | 3.5-fold increase in editing efficiency vs. non-carrier systems [4] | Improved cellular uptake and endosomal escape of CRISPR components |
The strategic advantage of liposomal delivery extends beyond simple encapsulation. The enhanced permeability and retention (EPR) effect, well-documented in tumor environments, similarly operates in biofilm-associated infections due to the leaky vasculature and impaired lymphatic drainage characteristic of chronic inflammatory sites [30]. Liposomes in the 60-150 nm size range optimally exploit this phenomenon, extravasating through permeable blood vessels into infected tissue and achieving higher local concentrations than freely administered drugs [30].
Diagram 1: Liposomal strategies to overcome biofilm barriers. The diagram illustrates how engineered liposomes address specific biofilm defense mechanisms through tailored functionalization approaches.
Liposomal Cas9 Formulations forP. aeruginosaBiofilm Eradication: Experimental Protocol
The integration of CRISPR/Cas9 gene editing technology with liposomal delivery represents a cutting-edge approach for precision targeting of antibiotic resistance mechanisms in P. aeruginosa biofilms. The following protocol details the methodology for formulating, characterizing, and evaluating liposomal Cas9/sgRNA complexes for enhanced anti-biofilm activity.
Liposomal Cas9/sgRNA Complex Preparation
Materials Required:
- DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine)
- Cholesterol
- DOTAP (1,2-dioleoyl-3-trimethylammonium-propane)
- PEG2000-DSPE (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000])
- Cas9 nuclease with nuclear localization signal
- sgRNA targeting P. aeruginosa resistance genes (e.g., ampC, oprD, mexAB)
- Thin-film hydration apparatus (rotary evaporator)
- Extrusion system with polycarbonate membranes (100 nm, 400 nm)
- Size exclusion chromatography columns
Procedure:
- Lipid Film Formation: Prepare lipid mixture with molar ratio 55:40:5 (DSPC:Cholesterol:DOTAP) with 0.5-5 mol% PEG2000-DSPE. Dissolve in chloroform:methanol (2:1 v/v) and evaporate under reduced pressure at 45°C using rotary evaporation to form thin lipid film.
- Hydration and Loading: Hydrate dried lipid film with CRISPR/Cas9 complex solution (Cas9:sgRNA pre-incubated at molar ratio 1:2.5 in nuclease-free buffer) to final lipid concentration of 10-20 mM. Subject to five freeze-thaw cycles (liquid nitrogen/45°C water bath).
- Size Reduction and Purification: Extrude hydrated liposomes sequentially through polycarbonate membranes (400 nm followed by 100 nm, 10-15 passes each). Purify using size exclusion chromatography (Sepharose CL-4B column) to remove unencapsulated CRISPR/Cas9 components.
- Sterilization and Storage: Filter sterilize through 0.22 μm PVDF membrane. Store under inert atmosphere at 4°C for immediate use or freeze at -80°C with cryoprotectant for long-term storage.
Liposome Characterization and Quality Control
Physicochemical Characterization:
- Size and Polydispersity: Determine by dynamic light scattering (DLS). Target size: 80-120 nm with PDI <0.2.
- Zeta Potential: Measure by laser Doppler electrophoresis. Expected range: +15 to +30 mV due to cationic DOTAP component.
- Encapsulation Efficiency: Quantify using fluorescence-based assay (RiboGreen for sgRNA, BCA assay for Cas9). Minimum acceptable efficiency: 70%.
- Morphology: Confirm by transmission electron microscopy (TEM) with negative staining.
Functional Characterization:
- Stability: Assess size stability in biological buffers (PBS, simulated lung fluid) over 72 hours at 37°C.
- Release Kinetics: Quantify payload release using dialysis membrane method in biofilm-relevant conditions (varying pH, enzyme presence).
- Nuclease Protection: Verify protection of encapsulated nucleic acids via gel electrophoresis after nuclease exposure.
Anti-Biofilm Efficacy Assessment
Biofilm Cultivation:
- Grow P. aeruginosa reference strains (PAO1, PA14) or clinical isolates in CDC biofilm reactor or 96-well peg plates using tryptic soy broth with 1% glucose.
- Incubate for 48-72 hours at 37°C with medium refreshment at 24-hour intervals to establish mature biofilms.
Treatment and Analysis:
- Dosing Regimen: Apply liposomal formulations at concentrations of 10-100 nM CRISPR/Cas9 content. Include controls: free CRISPR/Cas9, empty liposomes, untreated biofilm.
- Viability Assessment: Post-treatment (24-48 hours), quantify viable cells via colony forming units (CFU) after biofilm disruption (sonication/vortexing with glass beads).
- Biomass Quantification: Measure total biofilm biomass using crystal violet staining at 570 nm.
- Confocal Microscopy: Evaluate biofilm architecture and bacterial viability using LIVE/DEAD BacLight staining (SYTO9/propidium iodide) with z-stack imaging.
- Gene Editing Confirmation: Verify target gene modification via PCR amplification and sequencing of treated vs. untreated bacterial genomes.
Data Analysis:
- Calculate percentage reduction in CFU/mL and biofilm biomass compared to controls.
- Perform statistical analysis (one-way ANOVA with post-hoc tests, p<0.05 considered significant).
- Determine minimum biofilm eradication concentration (MBEC) for formulation comparisons.
The Scientist's Toolkit: Essential Research Reagents for Liposomal Biofilm Studies
Table 3: Essential Research Reagents for Liposomal Anti-Biofilm Formulation Development
| Reagent Category | Specific Examples | Function and Application | Key Considerations |
|---|---|---|---|
| Lipid Components | DSPC, DOPC, Cholesterol, DOTAP, PEG-DSPE | Vesicle formation, stability, charge modification, stealth properties | Purity grade (>99%), storage conditions (-20°C under argon) |
| CRISPR/Cas9 Components | Cas9 nuclease, sgRNA targeting resistance genes | Precision gene editing of bacterial resistance mechanisms | Off-target potential, editing efficiency verification |
| Biofilm Matrix Reagents | Alginate, Psl, Pel, DNAse I, dispersin B | Matrix modeling and degradation studies | Purity, biological activity validation |
| Characterization Tools | Dynamic light scattering, TEM, fluorescence spectroscopy | Size, morphology, and encapsulation efficiency determination | Method standardization, instrument calibration |
| Biological Assays | LIVE/DEAD BacLight, crystal violet, CFU enumeration | Anti-biofilm efficacy assessment | Stain stability, appropriate control inclusion |
Diagram 2: Liposomal Cas9 formulation workflow. The process outlines key stages from lipid selection through final product characterization for consistent preparation of anti-biofilm formulations.
Liposomal delivery systems represent a rationally engineered solution to the formidable challenge of biofilm penetration in P. aeruginosa infections. By leveraging nanoscale properties, tunable surface characteristics, and programmable drug release mechanisms, liposomal formulations overcome the physical, chemical, and biological barriers that render conventional antibiotics ineffective against biofilm-associated infections. The integration of CRISPR/Cas9 gene editing technology with advanced liposomal carriers represents a paradigm shift in precision antimicrobial therapy, enabling targeted disruption of bacterial resistance mechanisms while exploiting the inherent biofilm-penetrating capabilities of nanoscale lipid vesicles.
Future developments in this field will likely focus on multifunctional liposomal systems that combine biofilm matrix degradation enzymes with antimicrobial payloads, personalized approaches targeting patient-specific bacterial strains, and innovative triggering mechanisms responsive to unique features of the biofilm microenvironment. As these advanced formulations progress through preclinical development toward clinical application, they hold significant promise for addressing the growing crisis of antibiotic-resistant biofilm infections that increasingly defy conventional treatment approaches.
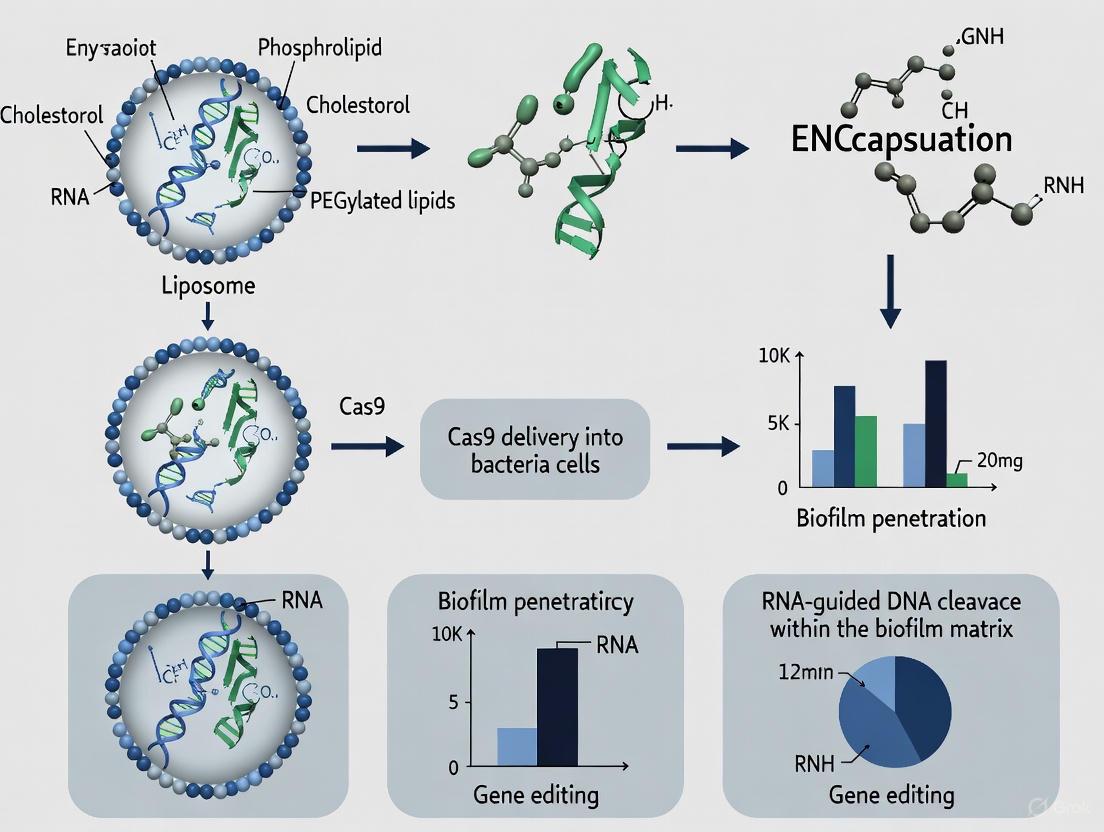
Engineering Liposomal Cas9 Formulations for Maximum Efficacy
The development of liposomal formulations for delivering Cas9 ribonucleoprotein (RNP) complexes represents a cutting-edge strategy in the fight against antibiotic-resistant biofilm infections caused by pathogens like Pseudomonas aeruginosa [4] [31]. Unlike plasmid DNA or mRNA formats, RNP delivery offers transient genome editing activity, which significantly reduces off-target effects and immune responses while enabling rapid editing upon cellular entry [32]. This application note details specialized protocols for designing liposomes that effectively encapsulate and deliver Cas9 RNP complexes to target biofilm-associated genes in P. aeruginosa, leveraging recent advances in lipid nanotechnology and gene editing [33].
The protective extracellular polymeric substance (EPS) matrix of biofilms reduces antibiotic penetration and creates microenvironments where bacteria exhibit up to 1000-fold greater tolerance to antibiotics compared to planktonic cells [4] [31]. By utilizing CRISPR/Cas9 to precisely target essential bacterial genes—such as those involved in antibiotic resistance, quorum sensing, and biofilm regulation—and delivering these molecular tools via engineered liposomes, researchers can achieve targeted disruption of biofilm integrity and resensitize resistant bacteria to conventional antibiotics [4].
Liposome Formulation Design
Lipid Composition Optimization
The lipid composition fundamentally determines the stability, encapsulation efficiency, and cellular interactions of liposomal Cas9 RNP carriers. The following table summarizes key lipid components and their functional roles based on recent research:
Table 1: Key Lipid Components for Cas9 RNP Liposomal Formulations
| Lipid Component | Molar Ratio | Function | Considerations for Biofilm Penetration |
|---|---|---|---|
| DOTAP | 40-50% | Cationic lipid providing positive surface charge for cell membrane interaction and sgRNA complexation [34]. | Enhances penetration through anionic EPS matrix of biofilms [4]. |
| Cholesterol | 30-40% | Modulates membrane fluidity and stability, prevents premature leakage [34]. | Improves liposome stability in hostile biofilm microenvironments [33]. |
| DOPE | 15-20% | Phospholipid promoting endosomal escape via transition to hexagonal phase [34]. | Crucial for intracellular delivery to bacteria within biofilm [33]. |
| DSPE-PEG2000 | 2-5% | Provides steric stabilization, reduces immune clearance, enhances circulation time [33] [34]. | PEG chain length can be optimized to balance stability and biofilm penetration [33]. |
Final optimized formulations should exhibit an average diameter of 200-230 nm, a polydispersity index (PDI) below 0.2, and a zeta potential of approximately +25 to +30 mV [34]. This size range facilitates effective biofilm penetration, while the positive surface charge enhances interaction with both anionic bacterial membranes and the biofilm matrix [4] [34].
Cargo Loading Strategies
Two primary approaches have been successfully demonstrated for loading Cas9 RNP complexes into liposomes:
- Standard Encapsulation: The Cas9 protein is encapsulated within the liposome's aqueous interior during the film hydration process. The single guide RNA (sgRNA) is then complexed electrostatically to the pre-formed cationic liposome's outer surface [34]. This method provides flexibility in sgRNA selection.
- Pre-complexed RNP Encapsulation: The Cas9 protein and sgRNA are first pre-assembled into a functional RNP complex, which is then encapsulated within the liposome during formulation [32]. This approach may enhance functional delivery but requires optimization of encapsulation conditions for the larger complex.
Table 2: Quantitative Performance of Liposomal Cas9 RNP Formulations
| Formulation Parameter | Performance Metric | Experimental Value | Reference |
|---|---|---|---|
| Encapsulation Efficiency | Cas9 protein encapsulation | 80.6% | [34] |
| Cellular Uptake | Efficiency in target cells | 45.6% | [34] |
| Biofilm Disruption | Reduction in P. aeruginosa biofilm biomass | >90% in vitro | [4] [13] |
| Gene Editing Efficiency | Knockdown of target gene (SRD5α2) | 29.7% reduction in mRNA expression | [34] |
Figure 1: Workflow for Liposome Formulation and Cargo Loading. The process begins with lipid film formation, followed by hydration with Cas9 protein, extrusion to create uniform vesicles, and final complexation with sgRNA.
Experimental Protocols
Liposome Preparation using Film Hydration Method
Principle: This method creates a thin lipid film that is subsequently hydrated with an aqueous buffer containing the Cas9 protein, forming multilamellar vesicles (MLVs) that are then downsized to uniform liposomes [34].
Materials:
- Lipids: DOTAP, Cholesterol, DOPE, DSPE-PEG2000 (Avanti Polar Lipids)
- Cas9 Protein: Commercially purified Cas9 nuclease
- Buffers: 200 mM HEPES buffer (pH 6.5, containing 1 M NaCl, 50 mM MgCl₂, 1 mM EDTA)
- Equipment: Rotary evaporator, extruder with polycarbonate membranes (0.8, 0.6, 0.22 μm)
Procedure:
- Lipid Film Formation: Weigh lipids according to the molar ratio Cholesterol:DOTAP:DOPE:DSPE-PEG2000 = 1:0.5:0.5:0.1 [34]. Dissolve in chloroform in a round-bottom flask. Remove organic solvent using a rotary evaporator at 45°C for 15 minutes to form a thin lipid film. Evaporate residual solvent under a stream of nitrogen gas.
- Hydration and Cas9 Encapsulation: Hydrate the dried lipid film with 200 mM HEPES buffer containing Cas9 protein (10 μM concentration). Gently agitate the mixture at 60°C for 1 hour to form multilamellar vesicles (MLVs) encapsulating Cas9.
- Size Reduction: Sequentially extrude the MLV suspension through polycarbonate membranes with decreasing pore sizes (0.8 μm, 0.6 μm, and finally 0.22 μm) using a liposome extruder. Perform 10 passes through each membrane to obtain small, unilamellar vesicles with uniform size distribution [34].
- sgRNA Complexation: Incubate the prepared cationic liposomes with sgRNA (targeting desired bacterial genes) at a liposome:sgRNA ratio of 1.5:1 (w/w) for 30 minutes at room temperature to allow electrostatic complex formation on the liposome surface [34].
Physicochemical Characterization
Dynamic Light Scattering (DLS) for measuring particle size, PDI, and zeta potential is essential. Protocols recommend three measurements per sample at 25°C [34]. Encapsulation efficiency is determined using a micro-BCA protein assay, comparing total protein against unencapsulated protein separated by ultracentrifugation [34].
Functional Assessment in Biofilm Models
Principle: Evaluate the efficacy of liposomal Cas9 RNP formulations against P. aeruginosa biofilms by measuring biofilm biomass reduction and gene editing efficiency [4].
Procedure:
- Biofilm Cultivation: Grow P. aeruginosa biofilms in flow cells or 96-well plates for 48-72 hours using appropriate media.
- Treatment: Apply liposomal Cas9 RNP formulations targeting specific antibiotic resistance genes (e.g., bla, mecA) or quorum-sensing genes (e.g., lasI, rhlI) [4]. Include appropriate controls.
- Biomass Quantification: Use crystal violet staining to quantify total biofilm biomass after treatment. Confocal laser scanning microscopy (CLSM) with live/dead staining can visualize biofilm architecture and cell viability [4].
- Gene Editing Analysis: Extract genomic DNA from treated biofilms and use T7E1 assay or sequencing to detect indels at the target locus. Quantify expression of target genes via qRT-PCR [34].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Liposomal Cas9 RNP Formulation
| Reagent/Category | Specific Examples | Function/Application | Notes for Bacterial Biofilm Research |
|---|---|---|---|
| Cationic Lipids | DOTAP, DC-Chol, DODAP | Imparts positive charge for nucleic acid binding and cellular uptake | DOTAP shows effective complexation with sgRNA [34] |
| Helper Lipids | DOPE, Cholesterol | Stabilizes bilayer structure and promotes endosomal escape | DOPE crucial for membrane fusion in bacterial systems [34] |
| PEGylated Lipids | DSPE-PEG2000, DMG-PEG | Provides stealth properties, reduces aggregation | Enhances stability in biofilm environments [33] |
| Cas9 Protein | S. pyogenes Cas9 | CRISPR nuclease for targeted gene disruption | Pre-complexing with sgRNA reduces off-target effects [32] |
| sgRNA | Custom-designed sequences | Guides Cas9 to specific genomic targets | Target biofilm-related genes (e.g., quorum sensing, resistance genes) [4] |
| Characterization Tools | DLS, Zeta Potential Analyzer | Measures particle size and surface charge | Essential for quality control of formulations [34] |
Figure 2: Mechanism of Liposomal Cas9 RNP Action Against Bacterial Biofilms. The diagram illustrates the journey of the liposome complex through the biofilm matrix, cellular uptake, intracellular release, and subsequent genetic disruption leading to biofilm elimination.
Troubleshooting and Optimization
Common challenges in liposomal Cas9 RNP formulation include low encapsulation efficiency, instability in biological fluids, and incomplete biofilm penetration. To address low encapsulation, ensure the hydration buffer is at the optimal pH (6.5) and osmolarity, and consider the charge balance between cationic lipids and Cas9 protein [34]. For stability issues, increase the cholesterol content to 40% and ensure proper PEGylation [33]. To enhance biofilm penetration, incorporate biofilm-penetrating peptides or use smaller liposome sizes (150-200 nm) while maintaining a slightly positive surface charge [4].
The global health crisis of antimicrobial resistance (AMR) demands innovative therapeutic strategies. Pseudomonas aeruginosa biofilm-associated infections represent a significant challenge in clinical settings, as biofilms can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [24]. The extracellular polymeric substance (EPS) matrix in biofilms limits antibiotic penetration while creating microenvironments conducive to horizontal gene transfer, facilitating the spread of resistance genes [24] [31].
CRISPR-Cas systems have emerged as powerful genetic tools to approach AMR, with liposomal Cas9 formulations demonstrating remarkable efficacy against P. aeruginosa biofilms, achieving over 90% reduction in biofilm biomass in vitro [13] [24]. These non-viral delivery vehicles protect genetic material and enhance cellular uptake through biofilm barriers, making them ideal for therapeutic applications [24]. The effectiveness of these formulations is critically dependent on precisely designed guide RNAs (gRNAs) that direct the Cas9 nuclease to specific genetic targets underlying resistance and virulence mechanisms.
This protocol details gRNA design strategies for targeting both antibiotic resistance genes and quorum-sensing (QS) pathways in P. aeruginosa, providing researchers with a methodological framework for developing effective liposomal Cas9 formulations against biofilm-associated infections.
gRNA Design Principles for Antibiotic Resistance Genes
Target Selection and Prioritization
Effective gRNA design begins with strategic target selection. For P. aeruginosa, priority should be given to genes conferring resistance to last-line antibiotics, particularly those located on mobile genetic elements which facilitate the spread of resistance across bacterial populations [35].
Table 1: High-Priority Antibiotic Resistance Gene Targets in P. aeruginosa
| Gene Target | Resistance Mechanism | Antibiotic Affected | Target Location |
|---|---|---|---|
| blaKPC | Carbapenemase production | Carbapenems | Plasmid/Chromosome |
| mecA | Altered penicillin-binding protein | β-lactams | Chromosome |
| NDM-1 | Metallo-β-lactamase production | β-lactams, Carbapenems | Plasmid |
| AmpC | β-lactamase production | β-lactams, Cephalosporins | Chromosome |
| gyrA/parC | Target site mutation | Fluoroquinolones | Chromosome |
gRNA Design Parameters
The design of gRNAs targeting these resistance genes should adhere to the following parameters to maximize editing efficiency:
- Sequence Specificity: gRNAs must target unique sequences within resistance genes to minimize off-target effects. For genes with multiple variants (e.g., blaKPC), identify conserved regions across variants.
- PAM Requirement: For Streptococcus pyogenes Cas9 (the most commonly used variant), the 5'-NGG-3' Protospacer Adjacent Motif (PAM) must be present immediately downstream of the target sequence.
- Target Site Positioning: Prioritize target sites in the 5' region of coding sequences to maximize disruption of gene function through frameshift mutations or premature stop codons.
- GC Content: Maintain GC content between 40-60% to balance stability and specificity.
- Avoidance of Secondary Structures: Ensure the gRNA spacer sequence does not form stable secondary structures that could impair Cas9 binding.
Experimental Protocol: gRNA Validation for Resistance Genes
Materials:
- P. aeruginosa target strains (e.g., PAO1, PA14)
- Liposomal formulation reagents (cationic lipids, cholesterol, PEG-lipids)
- Cloning vectors for gRNA expression (pACBSR, pCas9, or similar)
- Antibiotics for selection
- qPCR reagents for expression analysis
Methodology:
In Silico Design and Selection:
- Retrieve target gene sequences from NCBI GenBank.
- Use CRISPR design tools (CHOPCHOP, CRISPRscan, or Cas-Designer) to identify potential gRNA target sites with high on-target and low off-target scores [36].
- Select 3-5 candidate gRNAs per target gene to account for potential variability in efficiency.
gRNA Cloning and Vector Construction:
- Synthesize oligonucleotides corresponding to selected gRNA spacers with appropriate overhangs for cloning into your chosen Cas9 expression vector.
- Transform ligation products into competent E. coli, then isolate and sequence-confirm plasmid DNA.
Liposomal Formulation Preparation:
- Prepare liposomes using thin-film hydration method with cationic lipids (DOTAP, DOTMA), helper lipids, and PEG-lipids at molar ratios optimized for P. aeruginosa uptake.
- Incorporate CRISPR-Cas9 ribonucleoprotein complexes (RNPs) or plasmid DNA into liposomes using incubation or electroporation methods.
- Characterize liposome size (100-200 nm ideal) and zeta potential using dynamic light scattering.
Efficiency Validation:
- Treat 24-hour P. aeruginosa biofilms with liposomal formulations for 4-6 hours.
- Assess gene editing efficiency via T7E1 assay or tracking of indels by decomposition (TIDE) analysis.
- Evaluate phenotypic resistance changes through MIC determination against relevant antibiotics.
gRNA Design for Quorum-Sensing Pathways
Targeting the P. aeruginosa Quorum-Sensing Network
Quorum-sensing represents a promising target for anti-virulence approaches, as it coordinates the expression of numerous virulence factors and biofilm formation in P. aeruginosa without directly impacting bacterial viability, potentially reducing selective pressure for resistance [37].
Table 2: Key Quorum-Sensing System Targets in P. aeruginosa
| Target System | Key Regulatory Elements | Controlled Functions | gRNA Strategy |
|---|---|---|---|
| Las System | LasI, LasR, 3OC12-HSL | Early virulence factor production, biofilm maturation | Target lasI to prevent autoinducer synthesis |
| Rhl System | RhlI, RhlR, C4-HSL | Late virulence factors, rhamnolipid production | Target rhlI to disrupt secondary signal system |
| PQS System | PqsR, HHQ, PQS | Pyocyanin production, biofilm structuring | Target pqsA or pqsR to disrupt pseudomonas quinolone signal |
Strategic Considerations for QS Targeting
When designing gRNAs for QS pathways, consider the following strategic approaches:
- Hierarchical Targeting: The Las system sits atop the QS hierarchy in P. aeruginosa. Targeting lasI or lasR can produce cascading effects on downstream Rhl and PQS systems [37].
- Combination Approaches: Design gRNAs targeting both the Las system and specific virulence factors (e.g., pelA for Pel polysaccharide biosynthesis) for synergistic disruption of biofilm integrity.
- Temporal Considerations: Account for the sequential activation of QS systems during biofilm development, with Las activating first, followed by Rhl and PQS systems.
Experimental Protocol: Assessing QS Disruption
Materials:
- P. aeruginosa reporter strains (e.g., lasB-gfp, rhlA-gfp)
- Autoinducer standards (3OC12-HSL, C4-HSL)
- Confocal laser scanning microscope
- HPLC-MS for autoinducer quantification
Methodology:
gRNA Design and Cloning:
- Identify target sequences within key QS regulatory genes (lasI, lasR, rhlI, rhlR).
- Follow the same gRNA cloning procedure outlined in Section 2.3.
Liposomal Formulation with QS-Targeting gRNAs:
- Prepare liposomal Cas9-gRNA complexes as described previously, with modifications to optimize penetration through mature biofilms.
- Consider incorporating matrix-degrading enzymes (e.g., DNase I) to enhance liposomal penetration.
QS Disruption Assessment:
- Treat established P. aeruginosa biofilms (48-72 hours) with liposomal formulations.
- Quantify QS signal molecules using HPLC-MS or bioreporter assays.
- Assess virulence factor production (elastase, pyocyanin, rhamnolipids) using standardized protocols.
- Visualize biofilm architecture changes via confocal microscopy with appropriate stains (SYTO9, propidium iodide, concanavalin-A).
Phenotypic Characterization:
- Evaluate biofilm biomass reduction using crystal violet staining.
- Assess changes in biofilm mechanical stability through rheological measurements.
- Determine bacterial dispersal from biofilms by collecting and enumerating planktonic cells.
Liposomal Delivery Optimization for Biofilm Penetration
Formulation Strategies for Enhanced Efficacy
The effectiveness of gRNA-based approaches is critically dependent on efficient delivery through the protective biofilm matrix. Liposomal formulations must be optimized to address this challenge:
- Surface Functionalization: Modify liposome surfaces with biofilm-penetrating peptides (e.g., KSL-W) or P. aeruginosa-specific bacteriophage tail proteins to enhance target specificity and penetration.
- Stimuli-Responsive Release: Design liposomes that release their payload in response to biofilm-specific environmental cues such as decreased pH, increased reactive oxygen species, or specific enzymes.
- Co-delivery Strategies: Incorporate conventional antibiotics (e.g., tobramycin) or quorum-sensing inhibitors alongside CRISPR components to create synergistic effects [24].
Quantitative Assessment of Delivery Efficiency
Table 3: Key Parameters for Optimizing Liposomal Delivery
| Parameter | Optimal Range | Assessment Method | Impact on Efficacy |
|---|---|---|---|
| Particle Size | 100-200 nm | Dynamic Light Scattering | Determines biofilm penetration capability |
| Surface Charge | +20 to +40 mV | Zeta Potential Measurement | Influences bacterial membrane interaction |
| Payload Encapsulation | >85% | HPLC or Fluorescence Measurement | Affects delivered dose per particle |
| Stability in Biofilm | >4 hours | Fluorescence Resonance Energy Transfer | Determines release kinetics within biofilm |
Visualization of gRNA Design and Application Workflow
Diagram 1: Comprehensive workflow for gRNA design and application against P. aeruginosa biofilms, encompassing target identification, validation, liposomal formulation, and efficacy assessment.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for gRNA-Based Anti-Biofilm Studies
| Reagent Category | Specific Examples | Function/Application | Notes/Considerations |
|---|---|---|---|
| Cas9 Expression Systems | pCas9, pACBSR, pCasPA | Provides Cas9 nuclease for genome editing | Choose systems with appropriate bacterial selection markers |
| gRNA Cloning Vectors | pCRISPR, pTarget, pUC19-gRNA | gRNA expression and maintenance | Ensure compatibility with Cas9 system |
| Liposomal Components | DOTAP, DOPE, Cholesterol, DSPE-PEG | Formulation of delivery vehicles | PEGylation enhances stability but may reduce cellular uptake |
| P. aeruginosa Strains | PAO1, PA14, Clinical isolates | Biofilm models and efficacy testing | Include both laboratory and clinical strains |
| Biofilm Assessment Tools | Crystal violet, SYTO9, ConA | Quantification and visualization of biofilms | Combine multiple methods for comprehensive analysis |
| Gene Editing Validation | T7E1 assay, TIDE analysis, PCR | Confirmation of target gene modification | Include both molecular and phenotypic assessments |
| QS Monitoring Systems | lasB-gfp, rhlA-gfp reporters, HPLC-MS | Quantification of quorum-sensing activity | Reporters provide real-time monitoring capability |
The strategic design of gRNAs targeting both antibiotic resistance genes and quorum-sensing pathways represents a promising approach for combating P. aeruginosa biofilm-associated infections. When combined with optimized liposomal delivery systems, these molecular tools can achieve precise genetic manipulation that resensitizes bacteria to conventional antibiotics and disrupts virulence pathways.
Future developments in this field will likely focus on refining delivery systems for enhanced in vivo efficacy, developing multiplexed gRNA approaches to target multiple pathways simultaneously, and creating more sophisticated regulatory circuits that respond to bacterial population densities or specific environmental cues. As these technologies mature, they hold significant potential for addressing the growing threat of antimicrobial resistance in clinical settings.
Surface Functionalization of Liposomes for Enhanced Bacterial Targeting and Cellular Uptake
The efficacy of liposomal drug delivery systems is profoundly influenced by their surface characteristics. Within the context of a thesis focused on developing liposomal Cas9 formulations against Pseudomonas aeruginosa biofilms, surface functionalization transitions from a mere formulation step to a critical strategy for overcoming biofilm-associated resistance. Biofilms, with their complex extracellular polymeric substance (EPS) matrix, can exhibit up to 1000-fold greater tolerance to antimicrobials compared to their planktonic counterparts [4]. This application note details the protocols and key considerations for functionalizing liposomes to enhance their targeting and uptake in bacterial biofilms, specifically for delivering CRISPR/Cas9 components to eradicate P. aeruginosa biofilms.
The design of liposomes for biofilm targeting requires careful optimization of their physicochemical properties. The following tables summarize the key parameters and their impact on liposome performance.
Table 1: Impact of Liposome Physicochemical Properties on Biofilm Targeting [38]
| Physicochemical Property | Optimal Range for Biofilm Targeting | Impact on Biofilm Interaction and Uptake |
|---|---|---|
| Particle Size | Significantly below 500 nm | Smaller liposomes demonstrate superior penetration into the complex biofilm extracellular matrix. |
| Surface Charge | Positive (Cationic) | Promotes ionic interaction and fusion with negatively charged bacterial cell envelopes. |
| Membrane Rigidity | Relatively rigid bilayers | Composed of saturated phospholipids with higher cholesterol; enhances stability and target-site release. |
| Surface Functionalization | Antibodies, Lectins (e.g., Concanavalin A) | Enables specific (affinity-based) targeting to biofilm components, increasing retention. |
Table 2: Efficacy of Cationic and DBCO-Functionalized Liposomes
| Liposome Formulation | Target / Application | Key Experimental Findings | Reference |
|---|---|---|---|
| Cationic Liposomes | Negatively charged bacteria | Increased adsorption and effectiveness compared to anionic counterparts due to ionic interaction. | [38] |
| L-PEG2000-DBCO | Breast cancer cells (in vitro & in vivo) | Cellular uptake increased by 255% to 303% compared to non-targeted L-PEG2000; tumor uptake of ~54% vs. ~20% for conventional liposomes. | [39] |
| Liposomal Cas9 Formulations | Pseudomonas aeruginosa biofilm | Demonstrated reduction in biofilm biomass by over 90% in vitro. | [4] |
Experimental Protocols
Protocol: Preparation of Cationic Liposomes for Initial Bacterial Adhesion
This protocol outlines the formulation of basic cationic liposomes, which leverage electrostatic interactions for initial biofilm targeting [38].
Key Reagents:
- Cationic lipid (e.g., DOTAP, DC-Chol)
- Helper phospholipid (e.g., DOPC, DOPE)
- Cholesterol
- Chloroform or other organic solvent
- PBS (pH 7.4) or other aqueous hydration buffer
Procedure:
- Lipid Film Formation: Dissolve the lipid mixture (e.g., DOPC:Cholesterol:Cationic Lipid at a molar ratio optimized for positive charge and stability) in chloroform in a round-bottom flask.
- Solvent Evaporation: Rotate the flask under a stream of nitrogen gas to form a thin, uniform lipid film on the inner wall. Place the flask under vacuum for at least 2 hours to remove any residual organic solvent.
- Hydration: Hydrate the dried lipid film with PBS or the appropriate aqueous buffer (pre-warmed to a temperature above the phase transition of the lipids) to a final total lipid concentration of 1-10 mM. Rotate the flask gently for 1-2 hours until the film is fully suspended, forming multilamellar vesicles (MLVs).
- Size Reduction: Downsize the MLVs to form small, unilamellar vesicles (SUVs) by extruding the suspension through polycarbonate membranes (e.g., 100 nm pore size) using a liposome extruder for a minimum of 21 passes.
- Purification: Separate the formed liposomes from non-encapsulated material using dialysis or size-exclusion chromatography.
Quality Control:
- Measure particle size and polydispersity index (PDI) via Dynamic Light Scattering (DLS).
- Determine zeta potential using electrophoretic light scattering.
- Confirm desired positive surface charge.
Protocol: Surface Functionalization with Targeting Ligands (Post-Insertion Technique)
This method is ideal for attaching sensitive ligands, such as antibodies or peptides, to pre-formed liposomes without exposing them to harsh formulation conditions [40].
Key Reagents:
- Pre-formed liposomes (e.g., from Protocol 3.1)
- Lipid-PEG-Ligand conjugate (e.g., DSPE-PEG2000-Maleimide for thiol-coupled ligands)
- Micelles of the Lipid-PEG-Ligand conjugate
Procedure:
- Micelle Preparation: Dissolve the Lipid-PEG-Ligand conjugate (e.g., DSPE-PEG2000-DBCO) in an aqueous buffer by heating and vortexing to form micelles.
- Incubation: Incubate the pre-formed, sterile liposomes with the micellar Lipid-PEG-Ligand conjugate solution. A typical molar ratio of conjugate to total liposome lipid is 0.1-1.0%.
- Post-Insertion: Heat the mixture to a temperature above the phase transition temperature of the liposome lipids (e.g., 60°C for 30-60 minutes) with gentle agitation. This allows the Lipid-PEG-Ligand conjugates to insert their lipid anchors into the liposomal bilayer.
- Purification: Use dialysis or size-exclusion chromatography to remove unincorporated Lipid-PEG-Ligand conjugates.
Quality Control:
- Confirm ligand attachment and density using techniques like SDS-PAGE (for proteins), ELISA, or spectrophotometry.
- Re-measure size and zeta potential to confirm successful incorporation.
Protocol: Assessing Anti-Biofilm Efficacy of Liposomal Cas9 Formulations
This protocol describes an in vitro method to evaluate the effectiveness of functionalized liposomes carrying CRISPR/Cas9 components against P. aeruginosa biofilms [4].
Key Reagents:
- Functionalized liposomes encapsulating Cas9 ribonucleoprotein (RNP) or plasmid DNA.
- Pseudomonas aeruginosa bacterial strain.
- Culture media (e.g., LB, TSB).
- 96-well polystyrene microtiter plates.
- Crystal violet stain or fluorescent viability dyes (e.g., SYTO9/propidium iodide).
Procedure:
- Biofilm Formation: Grow P. aeruginosa in 96-well plates for 24-48 hours to allow for mature biofilm formation.
- Treatment: Treat the pre-formed biofilms with the following:
- Free Cas9 RNP/DNA
- Non-functionalized liposomal Cas9
- Functionalized liposomal Cas9 (e.g., cationic or ligand-targeted)
- Untreated control (media only)
- Incubation: Incubate the plates for a further 24 hours under appropriate conditions.
- Biomass Quantification (Crystal Violet Assay):
- Aspirate media and gently wash wells to remove non-adherent cells.
- Stain biofilms with 0.1% crystal violet for 15 minutes.
- Wash again to remove excess stain.
- Dissolve the bound crystal violet in 30% acetic acid.
- Measure the absorbance at 595 nm to quantify the total biofilm biomass.
- Viability Assessment (Live/Dead Staining):
- Use a fluorescent live/dead bacterial viability kit according to the manufacturer's instructions.
- Image the biofilms using confocal laser scanning microscopy (CLSM) to visualize the spatial distribution of live and dead cells within the biofilm structure.
Data Analysis:
- Calculate the percentage reduction in biofilm biomass and bacterial viability for each treatment group compared to the untreated control.
- A successful functionalized liposomal Cas9 formulation should demonstrate a significant reduction, potentially over 90% in biofilm biomass [4].
Workflow and Pathway Visualization
Workflow for Functionalized Liposomal Cas9 Delivery to Biofilms.
Mechanism of Liposomal Cas9 Action Against Biofilms.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Liposomal Formulation and Functionalization
| Reagent / Material | Function / Role | Specific Example(s) |
|---|---|---|
| Cationic Lipids | Imparts positive surface charge for electrostatic interaction with negatively charged bacterial cells and biofilm matrix. | DOTAP (1,2-dioleoyl-3-trimethylammonium-propane), DC-Chol (3β-[N-(N′,N′-Dimethylaminoethane)-carbamoyl]cholesterol) |
| PEGylated Lipids | Confers "stealth" properties by reducing opsonization and recognition by the mononuclear phagocyte system, prolonging circulation time. | DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]) |
| Functionalized PEG-Lipids | Serves as an anchor for conjugating targeting ligands (antibodies, peptides) to the liposome surface via chemical groups. | DSPE-PEG2000-Maleimide, DSPE-PEG2000-DBCO |
| Helper Phospholipids | Form the structural backbone of the liposomal bilayer. | DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) |
| Cholesterol | Modulates membrane fluidity and stability, reducing passive leakage of encapsulated contents. | Cholesterol (Pharmaceutical Grade) |
| CRISPR/Cas9 Components | The active therapeutic cargo for precise genetic disruption of antibiotic resistance or virulence genes within the biofilm. | Cas9 Protein (ribonucleoprotein, RNP), guide RNA (gRNA), plasmid DNA encoding Cas9/gRNA |
Application Notes
This document provides detailed protocols for the in vitro validation of a liposomal CRISPR-Cas9 formulation designed to combat Pseudomonas aeruginosa biofilms. The assays described herein quantitatively measure the formulation's dual efficacy: reducing established biofilm biomass and resensitizing antibiotic-resistant bacteria to conventional antibiotics. The integrated data from these methods is crucial for supporting the therapeutic potential of this novel strategy within a broader thesis research context.
Liposomal encapsulation of CRISPR-Cas9 components has been demonstrated to enhance delivery and efficacy, with one formulation reducing P. aeruginosa biofilm biomass by over 90% in vitro [4]. The protocols below are designed to confirm such anti-biofilm activity and quantify the subsequent restoration of antibiotic susceptibility.
Table 1: Summary of Key Quantitative Benchmarks for Anti-Biofilm and Resensitization Efficacy
| Parameter | Assay/Method | Exemplary Result from Literature | Potential Target for Liposomal Cas9 |
|---|---|---|---|
| Biofilm Biomass Reduction | Crystal Violet Assay [41] | >90% reduction with liposomal Cas9 [4] | >70% reduction vs. control |
| Bacterial Viability in Biofilm | Resazurin Staining / CFU Count [42] [43] | 3.5-fold increase in editing efficiency with nanoparticle carriers [4] | >2-log reduction in CFU |
| Matrix Alteration | Fluorescent Matrix Staining (e.g., WGA-Alexa Fluor 488) [43] | Matrix increase after penicillin G; no change after ciprofloxacin [43] | Quantifiable decrease in matrix components |
| Antibiotic Resensitization (Fold Change in MIC) | Checkerboard Microbroth Dilution [44] | Up to 30-fold enhanced efficacy with combinatory hydrophilic compounds [44] | ≥4-fold reduction in MIC of relevant antibiotics |
Experimental Protocols
Protocol 1: Biofilm Formation and Treatment Inhibition Assay
This protocol assesses the ability of the liposomal Cas9 formulation to inhibit biofilm formation [41].
Materials:
- Bacterial Strain: Pseudomonas aeruginosa clinical isolate (e.g., PAO1 or a cystic fibrosis-derived strain).
- Culture Media: Mueller-Hinton Broth (MHB) or Luria-Bertani Broth (LB).
- Treatment: Liposomal Cas9 formulation (targeting, for instance, quorum-sensing genes lasR or rhlR, or the alginate biosynthesis gene algD).
- Equipment: 96-well flat-bottom polystyrene plates, microplate reader, incubator.
Procedure:
- Culture Preparation: Grow P. aeruginosa overnight in MHB at 37°C with shaking. Dilute the culture in fresh MHB to an OD600 of 0.05, representing the start of the logarithmic growth phase (~10^7 CFU/mL) [41].
- Inoculation and Treatment: Dispense 180 µL of the diluted bacterial suspension into each well of a 96-well plate. Add 20 µL of the liposomal Cas9 formulation to achieve the desired final concentration. Include controls: medium-only (negative control), untreated bacteria (positive control), and empty liposomes (vehicle control).
- Incubation: Incubate the plate under static conditions at 37°C for 24-48 hours to allow for biofilm development and treatment.
- Biofilm Quantification: Proceed to the "Assessment of Biofilm Formation" detailed in Protocol 3.
Protocol 2: Biofilm Dispersal and Resensitization Assay
This protocol evaluates the ability of the formulation to disrupt pre-established biofilms and restore antibiotic sensitivity [41] [44].
Materials:
- Additional Reagents: Phosphate-buffered saline (PBS), antibiotics for resensitization testing (e.g., Tobramycin, Ciprofloxacin).
Procedure:
- Biofilm Formation: Prepare and incubate a 96-well plate as described in Protocol 1, steps 1-3, but without adding the treatment during this phase.
- Treatment of Mature Biofilms: After 24 hours, carefully remove the planktonic culture from each well by inverting the plate over a waste container. Gently rinse the adhered biofilms twice with PBS to remove non-adherent cells.
- Apply Formulation and Antibiotics: Add 180 µL of fresh MHB to each well. Add the liposomal Cas9 formulation alone and in combination with a sub-inhibitory concentration of an antibiotic. A critical control is biofilm treated with antibiotic alone to establish baseline resistance.
- Secondary Incubation: Incubate the plate again under static conditions at 37°C for an additional 24 hours.
- Assessment: Quantify the remaining biofilm biomass (Protocol 3) and determine the Minimum Biofilm Eradication Concentration (MBEC) of the antibiotic following treatment.
Protocol 3: Multi-Modal Assessment of Biofilm Formation
This sequential protocol allows for the quantification of total biomass, bacterial viability, and matrix composition within the same sample [43].
A. Total Biomass Quantification (Crystal Violet Staining) [41]
- Fixation and Staining: After treatment, remove the media from the plates and rinse gently with water. Air-dry the plates for 15 minutes. Add 125 µL of 0.1% crystal violet solution to each well and incubate for 10 minutes at room temperature.
- Destaining and Measurement: Carefully remove the crystal violet and rinse the wells thoroughly with water. Add 200 µL of a modified biofilm dissolving solution (e.g., 10% sodium dodecyl sulfate in 80% ethanol) to each well to solubilize the bound dye [41].
- Transfer 125 µL of the solubilized crystal violet solution to a new flat-bottom plate and measure the optical density at 570-600 nm using a plate reader.
B. Metabolic Activity Quantification (Resazurin Staining) [43]
- After biomass measurement, carefully remove the crystal violet solution from the wells.
- Add a dilute solution of resazurin (e.g., 0.0015% in PBS or media) to the same wells.
- Incubate the plate for 30-60 minutes at 37°C protected from light.
- Measure the fluorescence (Excitation: 560 nm, Emission: 590 nm). The fluorescent signal is proportional to the number of metabolically active cells in the biofilm.
C. Matrix Quantification (Fluorescent Staining)
- For parallel samples, stain the biofilm matrix with a specific fluorescent conjugate, such as Wheat Germ Agglutinin (WGA)-Alexa Fluor 488, which binds to extracellular polysaccharides [43].
- After staining and washing, measure the fluorescence to quantify relative matrix abundance.
Experimental Workflow and Mechanism Visualization
Experimental Workflow for Biofilm Validation
Liposomal Cas9 Mechanism in Biofilms
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for Biofilm Resensitization Studies
| Item | Function/Application | Exemplary Specifications |
|---|---|---|
| Liposomal Cas9 Formulation | Delivery of CRISPR-Cas9 machinery to bacterial cells within the biofilm. | Liposomes encapsulating Cas9 nuclease and target-specific gRNA [4]. |
| 96-well Polystyrene Plates | Substrate for standardized, high-throughput biofilm formation. | Clear, flat-bottom plates for optical density reading [41]. |
| Crystal Violet Solution | Total biofilm biomass staining. | 0.1% (w/v) in water [41]. |
| Resazurin Sodium Salt | Fluorometric/colorimetric indicator of metabolic viability in biofilms. | Ready-made solution or powder to prepare 0.0015%-0.015% solution [43]. |
| WGA-Alexa Fluor 488 | Fluorescent staining of biofilm matrix polysaccharides. | Conjugate for quantifying extracellular matrix components [43]. |
| Modified Biofilm Dissolving Solution (MBDS) | Solubilization of crystal violet stain for quantification. | 10% SDS in 80% Ethanol [41]. |
| Synthetic Cystic Fibrosis Sputum Medium (SCFM2) | Mimicking in vivo lung environment for more clinically relevant biofilm growth. | Medium for physiologically relevant assay conditions [21]. |
| Microplate Reader | Quantification of optical density (biomass) and fluorescence (viability/matrix). | Capable of reading OD 570-600 nm and fluorescence (Ex/Em ~560/590 nm) [41] [43]. |
{title page}
Synergistic Combination Therapies: Co-delivery of Cas9 and Conventional Antimicrobials
Application Notes and Protocols for Liposomal Formulations inPseudomonas aeruginosaBiofilm Research
The global health crisis of antimicrobial resistance is particularly acute in biofilm-associated infections, which exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [4]. These structured microbial communities, encased in a protective extracellular polymeric substance (EPS), are a hallmark of the opportunistic pathogen Pseudomonas aeruginosa, especially in chronic infections such as those found in cystic fibrosis airways [4] [45] [46]. Conventional monotherapies often fail to eradicate biofilms due to limited antibiotic penetration, reduced metabolic activity of persister cells, and enhanced horizontal gene transfer within the biofilm matrix [4]. The Clustered Regularly Interspaced Short Palindromic Repeats and associated Cas protein (CRISPR-Cas) system presents a revolutionary precision tool for targeting the genetic foundations of antibiotic resistance and biofilm integrity [4] [47]. Specifically, the CRISPR-Cas9 system can be programmed to disrupt chromosomal antibiotic resistance genes, plasmid-borne mobile resistance elements, and key genes regulating quorum sensing (QS) and biofilm formation [47].
The synergistic potential of co-delivering CRISPR-Cas9 with conventional antimicrobials lies in their complementary mechanisms of action. While antibiotics exert a physiological pressure, CRISPR-Cas9 applies a precise genetic pressure, such as by cleaving the bacterial chromosome at critical sites or eliminating resistance plasmids [4] [48]. This dual attack can resensitize resistant bacteria to traditional antibiotics and prevent the repair of Cas9-induced DNA breaks, leading to enhanced bacterial cell death [47]. However, the efficacy of this strategy is critically dependent on the development of advanced delivery systems that can co-encapsulate and protect these macromolecular complexes and ensure their simultaneous delivery to the target bacteria within the biofilm [4]. Liposomal nanoparticles have emerged as a leading platform for this purpose, offering improved cellular uptake, controlled release, and intrinsic biofilm-penetrating properties [4]. Recent advances demonstrate that liposomal Cas9 formulations can reduce P. aeruginosa biofilm biomass by over 90% in vitro, while hybrid systems incorporating antibiotics produce superior biofilm disruption [4]. These application notes and protocols detail the methodology for formulating, testing, and applying these synergistic combinations within the context of a research program focused on combating biofilm-associated infections.
Background and Key Concepts
The Challenge ofPseudomonas aeruginosaBiofilms
P. aeruginosa biofilms are structurally complex communities characterized by heterogeneous architecture with microcolonies interspersed with water channels [4]. Their development is a multi-stage process involving initial attachment, microcolony formation, maturation, and dispersion [45]. The biofilm's resilience is primarily attributed to its EPS matrix, which acts as a formidable physical and functional barrier against antimicrobial agents and host immune defenses [4] [45]. This matrix severely limits antibiotic penetration and creates heterogeneous microenvironments containing metabolically dormant persister cells, which are highly tolerant to conventional antibiotics that typically target active cellular processes [4]. Furthermore, biofilm formation and the expression of many virulence factors in P. aeruginosa are coordinated by a hierarchical quorum sensing (QS) system, comprising the las, rhl, and pqs circuits [49]. Targeting these regulatory networks and the structural genes essential for biofilm maintenance is a key strategic objective for effective eradication.
CRISPR-Cas9 as a Precision Antimicrobial Tool
The CRISPR-Cas9 system functions as an adaptive immune system in prokaryotes and has been repurposed for precise genome editing. The system comprises two core components: the Cas9 nuclease, which creates double-strand breaks in DNA, and a guide RNA (gRNA), which directs the nuclease to a specific genomic sequence complementary to its spacer region [4] [47]. In bacteria lacking efficient non-homologous end joining (NHEJ) pathways, such as E. coli and P. aeruginosa, these unrepaired breaks are lethal, providing the mechanistic basis for CRISPR-Cas9's use as an antimicrobial [48]. The system's programmability allows it to be targeted against essential bacterial genes, virulence factors, or, with extreme precision, antibiotic resistance genes themselves [47]. For instance, designing gRNAs to target and disrupt the blaNDM* or blaKPC* carbapenemase genes can resensitize resistant bacteria to carbapenem antibiotics [47]. A critical finding is that while the number of Cas9 cutting sites in the chromosome does not necessarily correlate with killing efficiency, it does reduce the emergence of chromosomal mutations conferring resistance [48]. However, the efficacy of killing is strongly linked to the intracellular expression level of Cas9, which must be high enough to overwhelm the bacterial RecA-mediated DNA repair machinery [48].
Application Notes: Quantitative Synergies and Material Solutions
The integration of CRISPR-Cas9 with nanoparticles and conventional antimicrobials has demonstrated significant synergistic effects in preclinical models. The table below summarizes key quantitative findings from recent research.
Table 1: Quantitative Efficacy of Combined Cas9 and Antimicrobial Therapies Against Biofilms
| Therapeutic Combination | Experimental Model | Key Efficacy Metric | Result | Citation |
|---|---|---|---|---|
| Liposomal Cas9 Formulation | P. aeruginosa biofilm ( in vitro ) | Reduction in biofilm biomass | >90% reduction | [4] |
| CRISPR-Gold Nanoparticle Hybrids + Antibiotics | Bacterial biofilms ( in vitro ) | Gene-editing efficiency & synergy | 3.5-fold increase in editing; superior biofilm disruption | [4] |
| Conjugative CRISPR/Cas9 vs mcr-1 | E. coli with MCR-1 plasmid | Plasmid elimination & re-sensitization | Successful plasmid elimination; restored colistin sensitivity | [47] |
| pCasCure System vs blaNDM/blaKPC | Carbapenem-resistant Enterobacteriaceae | Re-sensitization to carbapenems | Restored antibiotic sensitivity | [47] |
| N-Acetylcysteine (NAC) + Ciprofloxacin | P. aeruginosa biofilm (Cystic Fibrosis model) | Synergistic antibiofilm activity | Enhanced efficacy; NAC inhibits EPS matrix | [45] |
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs critical reagents and their functions for establishing a research pipeline for co-delivery therapies against P. aeruginosa biofilms.
Table 2: Essential Research Reagent Solutions for Co-delivery Studies
| Reagent / Material | Function and Application in Research | Key Considerations |
|---|---|---|
| Cationic Liposomes | Primary carrier for co-encapsulating Cas9-gRNA ribonucleoprotein (RNP) and antibiotics. Positively charged surface promotes interaction with negatively charged bacterial membranes and biofilm EPS. | Enable high loading efficiency for macromolecules; surface can be functionalized with PEG (pegylation) or targeting peptides for enhanced stability and specificity [4]. |
| Guide RNA (gRNA) | Confers target specificity to the Cas9 nuclease. Designed to target essential biofilm genes (e.g., pelA, pslG in EPS production) or antibiotic resistance genes (e.g., ampC, mex efflux pumps). | In silico design is critical to minimize off-target effects and maximize on-target activity. gRNA efficiency can be predicted using validated scoring models [48]. |
| S. pyogenes Cas9 (SpCas9) | The effector nuclease that induces double-strand DNA breaks at the site specified by the gRNA. | High-efficiency expression systems (e.g., strong, inducible promoters) are vital to ensure sufficient intracellular nuclease concentration to overwhelm bacterial DNA repair [48]. |
| Synthetic Cystic Fibrosis Sputum Medium (SCFM2) | A physiologically relevant growth medium for in vitro culturing of P. aeruginosa biofilms that mimics the nutritional environment of the CF lung. | Essential for generating biofilms with phenotypic resistance profiles comparable to clinical isolates, improving the translational value of in vitro data [46]. |
| Theophylline Riboswitch | An RNA-based regulatory element used to precisely control the expression of Cas9 in bacterial cells upon addition of theophylline. | Provides temporal control over Cas9 activity, allowing researchers to decouple bacterial growth from the induction of lethal DNA cleavage [48]. |
Detailed Experimental Protocols
Protocol 1: Formulation of Liposomal Cas9-Antibiotic Nanoparticles
This protocol describes a method for preparing co-encapsulated liposomal nanoparticles.
- Objective: To prepare and characterize cationic liposomal nanoparticles loaded with Cas9-gRNA ribonucleoprotein (RNP) and a conventional antibiotic (e.g., Tobramycin or Ciprofloxacin).
- Materials:
- Cationic lipid (e.g., DOTAP, DODAP), Helper lipid (e.g., DOPE), Cholesterol.
- Purified S. pyogenes Cas9 protein.
- In vitro-transcribed gRNA targeting a specific P. aeruginosa gene (e.g., lasR).
- Antibiotic (e.g., Tobramycin).
- PBS buffer (pH 7.4), Saline, Sucrose solution.
- Procedure:
- Lipid Film Formation: Dissolve lipid components (cationic lipid:helper lipid:cholesterol at a molar ratio of 50:25:25) in chloroform in a round-bottom flask. Remove the organic solvent by rotary evaporation to form a thin, uniform lipid film. Dry the film under vacuum overnight to remove trace solvent.
- Hydration and Co-Encapsulation: Hydrate the lipid film with an aqueous solution containing the pre-complexed Cas9-gRNA RNP and the antibiotic. Use a PBS or HEPES buffer at a temperature above the lipid transition temperature (T~c~). Vortex and allow the suspension to stand to form multilamellar vesicles (MLVs).
- Size Reduction and Homogenization: Extrude the MLV suspension through polycarbonate membranes of decreasing pore size (e.g., 400 nm, 200 nm, 100 nm) using a mini-extruder to form small, unilamellar vesicles (SUVs) with a uniform size distribution (~100-150 nm).
- Purification and Buffer Exchange: Purify the formed liposomes from non-encapsulated material using size-exclusion chromatography (e.g., Sephadex G-50 column) or dialysis against a suitable buffer like saline or PBS. This step removes free RNP and antibiotic.
- Characterization:
- Size and Zeta Potential: Determine the hydrodynamic diameter and polydispersity index (PDI) of the liposomes using Dynamic Light Scattering (DLS). Measure zeta potential to confirm a positive surface charge.
- Encapsulation Efficiency (EE): Quantify the EE for the antibiotic using HPLC. For Cas9 RNP, use a fluorescent nucleic acid stain (e.g., RiboGreen) after disrupting a liposome aliquot with 1% Triton X-100 and comparing to a standard curve.
Protocol 2: High-Throughput Biofilm Prevention and Eradication Assay
This protocol adapts a high-throughput method to evaluate the efficacy of formulations in preventing and eradicating P. aeruginosa biofilms in a physiologically relevant medium [46].
- Objective: To determine the Biofilm Preventing Concentration (BPC) and Biofilm Eradicating Concentration (BEC) of the liposomal co-delivery formulation.
- Materials:
- Synthetic Cystic Fibrosis Sputum Medium (SCFM2) [46].
- P. aeruginosa strain (e.g., PAO1 or a clinical isolate).
- 96-well flat-bottom polystyrene plates.
- Resazurin sodium salt solution (0.01% w/v).
- Microplate fluorometer.
- Procedure - Biofilm Prevention Assay (BPC):
- In a 96-well plate, serially dilute the liposomal formulation (and relevant controls) in SCFM2.
- Inoculate each well with a diluted overnight culture of P. aeruginosa to a final density of ~10^5^ CFU/mL.
- Incubate the plate statically for 24 hours at 37°C to allow biofilm formation in the presence of the treatment.
- After incubation, carefully aspirate the planktonic culture and wash the biofilms gently with PBS.
- Add a resazurin solution in PBS to each well and incubate for 30-60 minutes.
- Measure the fluorescence (Excitation: 560 nm, Emission: 590 nm). The loss of resazurin (a blue, non-fluorescent compound) to resorufin (a pink, highly fluorescent compound) is proportional to the number of metabolically active cells in the biofilm.
- The BPC is defined as the lowest concentration of the formulation that prevents any significant biofilm formation, indicated by fluorescence levels comparable to sterile medium controls.
- Procedure - Biofilm Eradication Assay (BEC):
- First, grow pre-established biofilms by inoculating a 96-well plate with bacteria in SCFM2 and incubating for 24 hours at 37°C without treatment.
- Gently wash the mature biofilms with PBS to remove non-adherent cells.
- Add fresh SCFM2 containing serially diluted liposomal formulations.
- Incubate for another 24 hours.
- Quantify the remaining viable biomass using the resazurin staining method described above.
- The BEC~50~/BEC~90~ is defined as the concentration that reduces the metabolic activity of the pre-established biofilm by 50% or 90%, respectively, compared to an untreated biofilm control.
Pathway and Workflow Visualizations
Mechanism of Synergistic Action
The following diagram illustrates the conceptual pathway and synergistic mechanism by which co-delivered liposomal Cas9 and antibiotics target and eradicate a P. aeruginosa biofilm.
Experimental Workflow for Therapeutic Evaluation
This workflow outlines the key steps for the development and testing of a synergistic co-delivery formulation, from design to final assessment.
Concluding Remarks and Future Perspectives
The co-delivery of CRISPR-Cas9 and conventional antimicrobials via advanced nanocarriers like liposomes represents a paradigm shift in the approach to treating resilient biofilm infections caused by pathogens like P. aeruginosa. The protocols and data outlined herein provide a foundational framework for researchers to explore and optimize these synergistic combinations. Future work must prioritize overcoming the challenges of in vivo delivery stability, potential immunogenicity, and the evolution of bacterial resistance to the CRISPR system itself, which can occur through mutations in the Cas9 gene or the target sequences [48]. The continued refinement of high-throughput screening assays, the exploration of new nanoparticle compositions, and the rational design of gRNAs based on evolving genomic data will be critical for translating this powerful therapeutic strategy from the laboratory to the clinic.
Navigating Technical Hurdles and Optimizing Delivery Platforms
The efficacy of liposomal Cas9 formulations against Pseudomonas aeruginosa biofilms is critically dependent on overcoming the dual challenges of maintaining liposomal stability and achieving controlled release of cargo within the complex biofilm microenvironment. Biofilms present numerous barriers to effective drug delivery, including a dense extracellular polymeric substance (EPS) matrix, varying chemical gradients, and heterogenous metabolic activity, which can lead to premature liposomal degradation or inadequate penetration [4] [29]. This Application Note details optimized protocols and characterization methods to ensure liposomal integrity during transit and precise Cas9 release upon reaching the target biofilm, thereby maximizing gene-editing efficiency.
Quantitative Performance Data of Liposomal Formulations
Table 1: Efficacy Metrics of Anti-Biofilm Liposomal Formulations
| Formulation Type | Target Pathogen | Biofilm Reduction (%) | Gene-Editing Efficiency Enhancement | Key Achievement |
|---|---|---|---|---|
| Liposomal Cas9 Formulation | P. aeruginosa | >90% in vitro [4] | Not Specified | Significant biofilm biomass disruption [4] [13] |
| CRISPR-Gold Nanoparticle Hybrid | Model Bacteria | Not Specified | 3.5-fold vs. non-carrier systems [4] | Superior cellular uptake and editing [4] |
| Cationic Liposomes (Stealth PEGylated) | S. aureus | 65-85% [29] | Not Applicable | Enhanced penetration and retention in biofilm matrix [29] |
| pH-Responsive Liposomes | P. aeruginosa | ~70% [50] | Not Applicable | Triggered release in acidic biofilm niches [50] |
Table 2: Liposomal Formulation Parameters for Biofilm Penetration
| Formulation Parameter | Optimized Characteristic | Impact on Biofilm Delivery |
|---|---|---|
| Size | 50 - 200 nm [29] [50] | Optimal penetration through EPS matrix and water channels [29] |
| Surface Charge (Zeta Potential) | Cationic (> +30 mV) [29] | Enhanced interaction with negatively charged EPS components [29] |
| Surface Functionalization | Polyethylene Glycol (PEG) Stealth Coating [50] | Improved stability, reduced immune clearance, and prolonged circulation [50] |
| Trigger Mechanism | pH-responsive lipids (e.g., DOPE, cholesteryl hemisuccinate) [50] | Controlled release in acidic microenvironment of mature biofilms [50] |
Experimental Protocols
Protocol: Formulation of pH-Responsive Liposomal Cas9
This protocol describes the preparation of liposomes designed for stable Cas9 RNP (ribonucleoprotein) encapsulation and triggered release in acidic biofilm regions [50].
Materials:
- Lipids: DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine), CHEMS (Cholesteryl hemisuccinate), DSPE-PEG2000 [50].
- Purified Cas9 protein and sgRNA (targeting, for instance, lasR quorum-sensing gene [4]).
- Chloroform, Methanol.
- HEPES Buffered Saline (HBS), pH 7.4.
- Acetate Buffered Saline (ABS), pH 5.5.
- Mini-extruder with 100 nm polycarbonate membranes.
- Dialysis tubing (MWCO 300 kDa).
Procedure:
- Lipid Film Formation: Dissolve DOPE, CHEMS, and DSPE-PEG2000 (molar ratio 5:4:1) in a chloroform:methanol mixture. Evaporate the solvent under a stream of nitrogen gas in a round-bottom flask to form a thin, uniform lipid film. Remove trace solvent under vacuum overnight.
- Hydration and Encapsulation: Hydrate the dried lipid film with HBS (pH 7.4) containing the pre-complexed Cas9:sgRNA RNP. Vortex vigorously and allow the suspension to stand for 1 hour above the lipid phase transition temperature. This results in the formation of multilamellar vesicles (MLVs) encapsulating the RNP.
- Downsizing and Purification: Pass the MLV suspension through a mini-extruder 21 times across two stacked 100 nm polycarbonate membranes to form small, unilamellar vesicles (SUVs). Separate the encapsulated liposomal Cas9 from free RNP using size-exclusion chromatography or dialysis against HBS (pH 7.4) for 4 hours.
- Characterization: Determine liposome size, polydispersity index (PDI), and zeta potential using dynamic light scattering. Quantify Cas9 encapsulation efficiency via a fluorescence-based assay or SDS-PAGE.
Protocol: In Vitro Assessment of Stability and Controlled Release
Materials:
- Formulated pH-responsive liposomal Cas9.
- Simulated Biofilm Fluid (SBF) containing lysozyme and DNase I.
- P. aeruginosa biofilm grown in a flow cell or 96-well plate.
- Fluorescence plate reader.
Procedure:
- Stability in Simulated Biofilm Conditions: Incubate liposomes in SBF at 37°C for up to 24 hours. Withdraw aliquots at 0, 2, 6, and 24 hours and measure particle size and PDI. A stable formulation will show less than a 15% increase in mean diameter over 24 hours [29].
- Triggered Release Kinetics: Use a fluorescent dye (e.g., calcein) co-encapsulated with Cas9 RNP. Dialyze the loaded liposomes against a large volume of HBS (pH 7.4) and ABS (pH 5.5) separately. Measure the fluorescence intensity in the dialysis medium over time. Calculate the percentage release, expecting a significantly higher release rate at the acidic pH [50].
- Biofilm Penetration Assay: Treat established P. aeruginosa biofilms (48-hour growth) with liposomes loaded with a lipophilic fluorescent dye (e.g., DiD). Analyze biofilm cross-sections using confocal laser scanning microscopy (CLSM) after 2 and 6 hours of incubation to visualize the depth and uniformity of liposomal penetration [29].
Pathway and Workflow Visualizations
Liposome-Biofilm Interaction Mechanism
Liposomal Cas9 Preparation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Liposomal Cas9 Formulation and Testing
| Reagent / Material | Function / Application | Specific Example / Note |
|---|---|---|
| Ionizable/Cationic Lipids | Confer positive surface charge for EPS interaction; enable pH-responsive release. | DOPE, CHEMS, DOTAP [29] [50]. |
| PEGylated Lipids | Create "stealth" liposomes to improve stability and reduce non-specific binding. | DSPE-PEG2000, DSG-PEG5000 [50]. |
| Cas9 Nuclease & sgRNA | The active gene-editing machinery targeted against bacterial resistance genes. | Target, for example, lasR or antibiotic resistance genes [4]. |
| Dynamic Light Scattering (DLS) Instrument | Critical for measuring liposome size, polydispersity index (PDI), and zeta potential. | Target size: 50-200 nm; Zeta potential: >+30 mV for cationic liposomes [29] [50]. |
| Confocal Laser Scanning Microscope (CLSM) | Visualize liposomal penetration and distribution within 3D biofilm structures. | Use with fluorescently tagged liposomes (e.g., DiD, Rhodamine-PE) [29]. |
| Simulated Biofilm Fluid (SBF) | In vitro testing of liposomal stability under biofilm-like conditions. | Contains enzymes like lysozyme and DNase I to challenge liposome integrity [29]. |
The application of CRISPR/Cas9 technology as a precision antimicrobial tool represents a paradigm shift in combating biofilm-driven infections, particularly those caused by Pseudomonas aeruginosa. A major obstacle in translating this technology into safe, effective therapies is the potential for off-target effects—unintended genetic modifications at sites other than the intended target. These effects raise significant safety concerns for clinical applications, as they could lead to the disruption of essential bacterial genes or unintended consequences in complex microbial communities [51]. The challenge is further amplified when using liposomal Cas9 formulations, as the lipid-based carriers must not only protect and deliver the CRISPR machinery efficiently but also ensure its highly specific activity within the heterogeneous and dense structure of bacterial biofilms. This document outlines evidence-based strategies and detailed protocols to quantify, minimize, and control for off-target effects in liposomal Cas9 anti-biofilm research.
Understanding the scale of the off-target problem and the performance of various detection methods is crucial for experimental planning. The tables below summarize key quantitative data and methodological characteristics.
Table 1: Performance Metrics of CRISPR-Cas9 Systems and Delivery Platforms
| System / Platform | Editing Efficiency (On-Target) | Reported Off-Target Reduction | Key Measurement |
|---|---|---|---|
| Gold Nanoparticle Carriers | Up to 3.5x increase | Not Specified | Editing efficiency vs. non-carrier systems [13] |
| Liposomal Cas9 Formulations | >90% biofilm reduction | Not Specified | P. aeruginosa biofilm biomass in vitro [13] |
| High-Fidelity Cas9 Variants | Varies by construct | Up to 10,000-fold | Off-target/on-target activity ratio [52] |
| RNP Delivery (vs. Plasmid) | High | Generally Lower | Reduced off-targets due to transient activity [51] |
Table 2: Comparison of Genome-Wide Off-Target Detection Methods
| Method | Principle | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|
| Digenome-seq [51] | In vitro digestion of purified genomic DNA with RNP, followed by WGS. | High (≤0.1% indel frequency) | High sensitivity; no predefined targets. | High sequencing depth/cost; omits chromatin effects. |
| CIRCLE-seq [51] | In vitro cleavage of circularized genomic DNA libraries. | Very High | Extremely sensitive; low background. | Cell-free; may not reflect intracellular chromatin state. |
| DIG-seq [51] | In vitro digestion of cell-free chromatin with RNP, followed by WGS. | High | Accounts for chromatin accessibility. | More complex than Digenome-seq. |
| SITE-Seq [51] | Selective enrichment and identification of biotinylated tagged ends. | High | Quantitative; provides cleavage frequency. | Requires biotinylated dsDNA break capture. |
Experimental Protocol: A Workflow for Assessing Specificity
This integrated protocol provides a step-by-step guide for designing, executing, and analyzing experiments aimed at minimizing off-target effects of liposomal Cas9 in biofilm studies.
Protocol 3.1: Specificity-Focused Design and Validation of Liposomal Cas9
Goal: To design a liposomal Cas9 formulation with maximal on-target activity and minimal off-target potential against P. aeruginosa biofilms.
Research Reagent Solutions:
| Reagent / Material | Function | Example & Notes |
|---|---|---|
| sgRNA Design Tools | Predicts on-target efficiency and nominates potential off-target sites. | Cas-OFFinder, CCTop. Use to screen for gRNAs with minimal off-target candidates [51]. |
| High-Fidelity Cas9 Protein | Reduces off-target cleavage while maintaining on-target activity. | eSpCas9(1.1), SpCas9-HF1 [51]. |
| Cationic Lipids | Forms stable, cationic liposomes for complexing anionic CRISPR cargo. | DOTAP, DODAP. Critical for encapsulating RNP complexes [13]. |
| PEGylated Lipids | Confers "stealth" properties, enhancing stability and biofilm penetration. | DSPE-PEG(2000). Improves delivery efficiency in biofilm matrix [13]. |
| Ribonucleoprotein (RNP) Complex | The active CRISPR machinery; pre-complexing Cas9 and sgRNA. | Purified Cas9 protein + in vitro transcribed sgRNA. Transient activity reduces off-target risk [51]. |
Procedure:
- sgRNA Selection: Design sgRNAs targeting essential biofilm-related genes (e.g., pelA, pslG, or quorum-sensing regulators like lasR). Use at least two in silico tools (e.g., Cas-OFFinder and CCTop) to cross-validate and select gRNAs with the lowest number of potential off-target sites across the P. aeruginosa genome, particularly those with ≤3 mismatches in the seed region [51].
- Liposome Formulation: Prepare liposomes using a thin-film hydration method. Use a lipid composition of, for example, DOTAP:Cholesterol:DSPE-PEG(2000) at a molar ratio of 50:45:5. The cationic DOTAP facilitates complexation with the negatively charged RNP.
- RNP Complexation and Encapsulation: Pre-complex purified High-Fidelity Cas9 protein with the selected sgRNA at a optimal molar ratio (e.g., 1:2) for 15 minutes at room temperature. Incubate the pre-formed RNP complexes with the pre-formed liposomes to allow for electrostatic encapsulation into liposomal nanoparticles (LNPs).
- Purification and Characterization: Purify the final Liposomal RNP (LRNP) formulation via size-exclusion chromatography. Characterize the LRNPs for particle size (target ~100 nm), zeta potential, and RNP encapsulation efficiency using standard assays.
Protocol 3.2: Genome-Wide Off-Target Assessment using Digenome-seq
Goal: To identify potential off-target sites in a cell-free system using purified genomic DNA from P. aeruginosa.
Procedure:
- Genomic DNA Isolation: Extract high-molecular-weight genomic DNA from a standard lab strain of P. aeruginosa (e.g., PAO1).
- In Vitro Cleavage: Divide the genomic DNA into two aliquots. Incubate the test aliquot with the LRNP formulation (or bare RNP for comparison). The control aliquot is incubated with nuclease-free water.
- Whole-Genome Sequencing (WGS): Subject both cleaved and control DNA to high-coverage whole-genome sequencing (≥100x coverage is recommended).
- Bioinformatic Analysis: Map the sequencing reads to the P. aeruginosa reference genome. Use the Digenome-seq analysis pipeline to identify sites with a significant concentration of sequence reads that start or end at the same genomic position, indicating a Cas9-induced double-strand break [51].
- Validation: The top-nominated off-target sites from Digenome-seq must be validated in a cellular context. Design specific PCR primers flanking these loci and perform targeted sequencing (e.g., Sanger sequencing or amplicon sequencing) of DNA extracted from biofilm cells treated with the LRNP.
Protocol 3.3: In-Biofilm Specificity Validation
Goal: To confirm the on-target activity and absence of off-target effects in mature P. aeruginosa biofilms.
Procedure:
- Biofilm Cultivation: Grow P. aeruginosa biofilms in a standardized reactor (e.g., CDC biofilm reactor or flow-cell system) for 48-72 hours.
- LRNP Treatment: Treat mature biofilms with the LRNP formulation, a naked RNP control, and a no-treatment control. Include a group treated with liposomes containing a non-targeting sgRNA as a critical control for non-specific effects.
- Assessment of Efficacy and Specificity:
- Biofilm Biomass: Quantify reduction in biofilm biomass using crystal violet staining [13].
- Bacterial Viability: Assess bacterial load via colony-forming unit (CFU) counts.
- On-Target Efficiency: Extract genomic DNA from treated and control biofilms. Use T7 Endonuclease I assay or, preferably, targeted amplicon sequencing to quantify indels at the intended on-target site.
- Off-Target Screening: Using the genomic DNA from step 3, perform targeted amplicon sequencing on the potential off-target loci identified in Protocol 3.2 to confirm the absence of mutations.
Strategic Pathways to Enhance Specificity
The following diagram and text outline the logical workflow and strategic interventions for ensuring specificity.
Diagram 1: A strategic workflow for minimizing off-target effects in liposomal Cas9 applications. Key interventions (linked via red dashed lines) are critical at each step to ensure the final therapeutic outcome is specific and effective.
Key Strategic Interventions
- Rational sgRNA Design: The foundation of specificity lies in sgRNA selection. Utilize computational tools that employ sophisticated algorithms (e.g., CFD score or MIT specificity score) to rank gRNAs not only by predicted on-target efficiency but also by a minimal number of potential off-target sites with low mismatch scores, especially avoiding mismatches in the PAM-distal seed region [51].
- High-Fidelity Cas9 Variants: Replace wild-type SpCas9 with engineered high-fidelity mutants such as eSpCas9(1.1) or SpCas9-HF1. These variants contain mutations that destabilize Cas9's interaction with the DNA duplex outside of the sgRNA-DNA hybridization, thereby requiring more perfect complementarity for cleavage and drastically reducing off-target activity without compromising on-target efficiency [51] [52].
- Ribonucleoprotein (RNP) Delivery: The delivery of pre-assembled Cas9 protein and sgRNA complexes (RNPs) is superior to plasmid DNA delivery for minimizing off-target effects. RNP delivery results in a rapid, but transient, burst of Cas9 activity that coincides with peak intracellular delivery, reducing the window of opportunity for off-target cleavage. This contrasts with plasmid-based expression, which can lead to prolonged Cas9 presence and accumulation, increasing off-target risks [51].
- Leveraging Biofilm Physiology for Specificity: The design of liposomal formulations can be informed by biofilm biology. Incorporating lipids with fusogenic properties or using ligands that target biofilm-specific markers could enhance local delivery and uptake specifically by biofilm-embedded bacteria, thereby reducing exposure of non-targeted bacteria and limiting potential off-target effects in complex communities.
Within the broader scope of a thesis investigating liposomal Cas9 formulations for combating Pseudomonas aeruginosa biofilms, this document details advanced optimization strategies employing gold and hybrid nanoparticles. The inherent resistance of biofilm-associated infections often limits the therapeutic efficacy of conventional antimicrobials and gene-editing systems. Nanoparticles, particularly those made from gold and silver-gold hybrids, offer a promising solution by enhancing the delivery and potency of CRISPR/Cas9 components. This note outlines specific application protocols and quantitative data supporting the integration of these nanocarriers to significantly boost gene-editing efficiency and biofilm disruption in P. aeruginosa research models.
The following tables summarize key quantitative findings from recent studies on nanoparticle-enhanced antimicrobial and gene-editing applications.
Table 1: Antibacterial and Anti-Biofilm Efficacy of Functionalized Gold Nanoparticles (Au NPs)
| Nanoparticle Type | Target Pathogen | Minimum Inhibitory Concentration (MIC) | Anti-Biofilm Activity | Key Findings | Source |
|---|---|---|---|---|---|
| DDM_Au NPs (Dichlorophen-functionalized) | Carbapenem-resistant Enterobacteriaceae (CRE) | 4 - 16 μg/mL | - Inhibited biofilm formation- Disrupted mature biofilms (2–6 log10 CFU/mL reduction) | Disruption of bacterial membrane integrity; Induction of ROS | [53] |
| Marine Sponge (A. compressa) AuNPs | Various bacteria | - | Significant anti-biofilm activity | Zone of inhibition: 26-31 mm; Spherical, 10-40 nm diameter | [54] |
| Photothermal AuNPs (Targeted) | S. epidermidis biofilms | - | Effective biofilm disruption upon NIR irradiation | Utilizes bacterial protein (autolysin) for targeted delivery | [55] |
Table 2: Efficacy of Hybrid and CRISPR-Loaded Nanoparticles against Biofilms
| Nanoparticle System | Cargo / Type | Target | Editing Efficiency / Biofilm Reduction | Key Findings | Source |
|---|---|---|---|---|---|
| Gold Nanoparticle Carrier | CRISPR/Cas9 | Bacterial Biofilms | Enhanced editing efficiency up to 3.5-fold vs. non-carrier systems | Enables co-delivery with antibiotics for synergistic effects | [4] [13] |
| Liposomal Formulation | CRISPR/Cas9 | Pseudomonas aeruginosa | Reduced biofilm biomass by >90% in vitro | Demonstrates high potency of nano-delivered CRISPR | [4] [13] |
| Silver-Gold Hybrid NPs (Ag-Au NPs) | N/A (Intrinsic activity) | Polymicrobial Biofilms | Suppressed formation & reduced intracellular infection by 70%-90% | Generates intracellular oxidative stress; 40±10 nm diameter | [56] |
Experimental Protocols
Protocol: Synthesis of Dichlorophen-Functionalized Gold Nanoparticles (DDM_Au NPs)
This protocol describes a one-pot synthesis for creating antimicrobial Au NPs, adapted from [53]. The resulting nanoparticles are effective against drug-resistant bacteria and their biofilms.
Reagents:
- Hydrogen tetrachloroaurate(III) trihydrate (HAuCl₄·3H₂O)
- Dichlorophen (DDM)
- Triethylamine
- Tween 80
- Sterile deionized water (ddH₂O)
Equipment:
- Magnetic stirrer with hot plate
- Sonicator
- Dialysis tubing (Molecular Weight Cut-Off: 7000 Da)
- 0.22 µm sterile syringe filters
Procedure:
- Mixture Preparation: In a clean vessel, combine 10 mL of sterile ddH₂O, 50 µL of triethylamine, 150 mg of Tween 80, and 0.1 mmol of DDM.
- Sonication: Sonicate the mixture for 15 minutes to ensure proper dissolution and mixing.
- Reduction Reaction: Place the mixture on a magnetic stirrer set to 500 rpm at room temperature. Using a dropper, add a solution of HAuCl₄·3H₂O (0.05 mmol in 200 µL) drop-wise to the stirring mixture.
- Reaction Completion: Continue stirring the reaction for 2 hours. Observe the color change from colorless to a stable purple-red, indicating the formation of DDM_Au NPs.
- Purification and Sterilization: Transfer the synthesized nanoparticle solution to dialysis tubing and dialyze against ddH₂O to remove unreacted impurities and reagents. After dialysis, sterilize the DDM_Au NP solution by passing it through a 0.22 µm syringe filter. Store at 4°C.
Characterization: The nanoparticles should be characterized using Dynamic Light Scattering (DLS) for size and dispersion, UV-Vis spectroscopy (absorption peak ~520-570 nm), and Transmission Electron Microscopy (TEM) for morphological analysis [53].
Protocol: Assessing Anti-Biofilm Efficacy Using a Catheter Model
This protocol evaluates the ability of synthesized nanoparticles to disrupt mature biofilms on a medical device surface, as described in [53].
Reagents:
- Prepared nanoparticle solution (e.g., DDM_Au NPs)
- Bacterial culture (e.g., CRE strain)
- Appropriate culture broth (e.g., LB, TSB)
- Phosphate Buffered Saline (PBS)
- Crystal violet stain (for biomass assessment, optional)
Equipment:
- Sterile urinary catheter segments
- 24-well cell culture plate
- Incubator
- Sonicating water bath
Procedure:
- Biofilm Formation: Cut the urinary catheter into small segments (~1 cm) and place them in the wells of a 24-well plate. Inoculate each segment with a standardized suspension of the test bacterium in culture broth. Incubate the plate under static conditions at 37°C for 24-48 hours to allow for mature biofilm formation.
- Treatment: After incubation, gently rinse the catheter segments with PBS to remove non-adherent (planktonic) cells. Add the nanoparticle solution at the desired test concentration (e.g., 1x-4x MIC) in fresh broth to the wells containing the biofilm-coated catheters. Include control wells with broth only.
- Incubation: Return the plate to the incubator for a specified treatment period (e.g., 4-24 hours).
- Biofilm Quantification (Viable Count):
- After treatment, transfer each catheter segment to a tube containing PBS.
- Sonicate the tubes in a water bath for 5-10 minutes to dislodge the biofilm bacteria.
- Serially dilute the resulting bacterial suspension and plate onto agar plates.
- Incubate the plates and count the resulting colonies to calculate the Log10 CFU (Colony Forming Units) per catheter segment.
- Data Analysis: Compare the bacterial load from nanoparticle-treated segments to untreated controls to determine the log reduction in biofilm viability.
Protocol: Enhancing CRISPR/Cas9 Delivery Using Gold Nanoparticles
This protocol outlines a strategy for complexing CRISPR/Cas9 components with gold nanoparticles (Au NPs) for improved delivery to bacterial cells, based on reports of 3.5-fold enhanced editing efficiency [4] [13] [57].
Reagents:
- Gold Nanoparticles (e.g., spherical, ~15-50 nm)
- CRISPR/Cas9 component (e.g., sgRNA, RNP complex)
- Polyethyleneimine (PEI) or other cationic polymer (for complexation)
- Appropriate buffer (e.g., HEPES)
Equipment:
- Microcentrifuge tubes
- Vortex mixer
- Incubator or water bath
Procedure:
- Preparation of Cargo: Depending on the experimental design, prepare the CRISPR/Cas9 cargo. The ribonucleoprotein (RNP) complex—pre-formed Cas9 protein and sgRNA—is often preferred for its rapid activity and reduced off-target effects [58] [57].
- Complexation: In a microcentrifuge tube, mix the Au NP solution with the CRISPR/Cas9 cargo (e.g., RNP) at a predetermined optimal mass ratio. This ratio must be determined empirically.
- Stabilization (Optional): To enhance complex stability and cellular uptake, a cationic polymer like PEI can be added. Incubate the mixture (Au NPs + Cargo + PEI) for 20-30 minutes at room temperature to allow for the formation of stable complexes via electrostatic interactions.
- Delivery: Add the completed complex directly to bacterial cultures containing pre-formed biofilms. For in vitro P. aeruginosa biofilms, this can be done in a 96-well plate or similar format.
- Assessment: Incubate the treated biofilms and assess editing efficiency (e.g., via sequencing of the target gene) or phenotypic outcomes (e.g., reduction in biofilm biomass or resensitization to antibiotics) after 24-48 hours.
Signaling Pathways and Workflows
Nanoparticle Antibiofilm Mechanism
CRISPR-Nanoparticle Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Nanoparticle-Enhanced CRISPR Delivery
| Reagent / Material | Function / Role | Specific Examples / Notes |
|---|---|---|
| Gold Chloride (HAuCl₄) | Gold ion precursor for nanoparticle synthesis. | Hydrogen tetrachloroaurate(III) trihydrate (HAuCl₄·3H₂O) is commonly used [53] [54]. |
| Functionalizing Agents | Impart targeting, stability, and enhanced activity to NPs. | Dichlorophen (DDM) [53]; Marine sponge extracts (e.g., A. compressa) [54]; Elastin-like polypeptides for targeting [55]. |
| CRISPR/Cas9 Components | The active gene-editing machinery. | Ribonucleoprotein (RNP) complexes are preferred for rapid action and reduced off-target effects [58] [57]. Plasmids or mRNA are alternatives. |
| Cationic Transfection Agents | Facilitate complexation and delivery of negatively charged CRISPR components. | Polyethyleneimine (PEI) [58]; Lipid nanoparticles (LNPs) can also be used for encapsulation [57]. |
| Biofilm Assay Kits | Quantify biofilm biomass and viability. | Crystal violet staining for total biomass; ATP-based assays or resazurin reduction for metabolic activity; CFU counting for viable cells [53]. |
For therapeutics designed for in vivo applications, particularly against resilient bacterial biofilms, overcoming host immune defenses is a critical barrier to success. Biofilms are structured communities of microorganisms embedded in a self-produced extracellular polymeric matrix that confer significant protection against both antimicrobial agents and host immune responses [59] [60]. Pseudomonas aeruginosa biofilms represent a formidable clinical challenge in healthcare settings, especially in immunocompromised patients or those with medical implants [61]. The biofilm matrix acts as a physical barrier that limits penetration of immune cells and therapeutic molecules while creating a microenvironment that actively suppresses immune function [59]. This application note explores strategic approaches to evade host immune recognition, specifically within the context of liposomal Cas9 formulations targeting P. aeruginosa biofilms, providing researchers with practical methodologies to enhance therapeutic delivery and efficacy.
Pathogen-Inspired Immune Evasion Strategies
Microbial pathogens have evolved sophisticated mechanisms to evade host immunity, providing valuable insights for therapeutic design. The table below summarizes key immune evasion strategies employed by pathogens that inform therapeutic delivery approaches.
Table 1: Pathogen-Inspired Immune Evasion Strategies with Therapeutic Applications
| Evasion Strategy | Mechanism of Action | Pathogen Examples | Therapeutic Application |
|---|---|---|---|
| Antigenic Variation | Alteration of surface antigens to avoid immune recognition [62] | Streptococcus pneumoniae, Influenza virus, African trypanosomes | Shield delivery vehicles with dynamically changing surface coatings |
| Surface Modification | Altering surface charge or composition to resist antimicrobial peptides [63] | E. coli, S. aureus, Salmonella | Optimize liposome surface charge to reduce clearance |
| Molecular Mimicry | Mimicking host molecules to avoid immune detection [63] | N. meningitidis, certain E. coli strains | Functionalize nanoparticles with host-derived CD47 markers |
| Biofilm Formation | Creating a physical barrier that impedes immune cell function [59] [61] | S. aureus, P. aeruginosa | Design matrix-penetrating delivery systems |
| Effector Proteins | Secreting proteins that directly inhibit immune cell function [63] | S. aureus Protein A, M. tuberculosis PtpA | Not directly applicable; target for therapeutic intervention |
| Latency/Low Metabolic State | Reducing metabolic activity to minimize immune detection [62] | Herpes viruses, M. tuberculosis | Not directly applicable |
Biofilm-Specific Immune Evasion Mechanisms
Biofilms employ multiple specialized strategies to evade host defenses, which must be considered when designing anti-biofilm therapeutics:
- Physical Barrier Function: The biofilm extracellular matrix, composed of polysaccharides, extracellular DNA (eDNA), and proteins, physically impedes immune cell infiltration and phagocytosis [59]. Macrophages show significantly reduced ability to penetrate and phagocytose bacteria within intact S. aureus biofilms [59].
- Immunosuppressive Metabolite Production: Biofilms actively reprogram the host immune response through metabolite secretion. S. aureus biofilms produce lactate that inhibits histone deacetylase 11, leading to increased IL-10 production and polarization of anti-inflammatory granulocytic myeloid-derived suppressor cells (G-MDSCs) [59].
- Toxin-Mediated Immune Cell Dysfunction: Biofilm-conditioned media containing toxins such as alpha-toxin (Hla) and leukocidin AB (LukAB) induce macrophage death and dysfunction, further protecting the bacterial community [59].
Integrated CRISPR-Nanoparticle Platforms for Biofilm Targeting
The integration of CRISPR/Cas9 gene editing with nanoparticle delivery systems represents a promising approach for precision targeting of biofilm-associated infections while overcoming immune barriers.
Liposomal Cas9 Formulations: Mechanisms and Efficacy
Liposomal delivery systems for CRISPR/Cas9 components offer significant advantages for anti-biofilm therapy by protecting the payload from immune recognition and degradation while enhancing biofilm penetration.
Table 2: Performance Metrics of Nanoparticle-Mediated CRISPR Delivery Against Biofilms
| Delivery Platform | Target Pathogen | Editing Efficiency | Biofilm Reduction | Key Advantages |
|---|---|---|---|---|
| Liposomal Cas9 Formulations | P. aeruginosa | Not specified | >90% in vitro [13] | Enhanced cellular uptake, biofilm matrix penetration |
| Gold Nanoparticle Carriers | Not specified | 3.5-fold increase vs. non-carrier systems [13] | Not specified | Improved editing efficiency, controlled release |
| Hybrid Nanoparticle Platforms | Not specified | Significant enhancement with antibiotic co-delivery [13] | Superior biofilm disruption [13] | Synergistic effects with conventional antibiotics |
The liposomal delivery system enhances CRISPR/Cas9 efficacy through multiple mechanisms: (1) protecting CRISPR components from serum nucleases and degradation in the host environment; (2) facilitating fusion with bacterial membranes for payload delivery; and (3) enabling co-delivery of Cas9 ribonucleoproteins with antibiotic adjuvants for synergistic effects [13].
Liposomal Cas9 Mechanism: This diagram illustrates the coordinated mechanism of liposomal Cas9 formulations, combining immune evasion with precise biofilm targeting.
Target Selection for CRISPR-Based Biofilm Disruption
Strategic selection of genetic targets is essential for effective CRISPR-based biofilm control. The most promising targets include:
- Antibiotic Resistance Genes: Direct disruption of resistance mechanisms restores antibiotic susceptibility [13].
- Quorum Sensing Pathways: Interference with bacterial communication reduces biofilm formation and virulence [13].
- Biofilm-Regulating Factors: Targeting structural components of the biofilm matrix enhances penetrability and susceptibility to host defenses [13].
- Essential Bacterial Genes: Disruption of core metabolic or structural genes induces bacterial cell death [13].
Experimental Protocols for Immune Evasion Assessment
Protocol: Macrophage Uptake and Phagocytosis Assay
Objective: Quantify the ability of liposomal formulations to evade uptake by immune cells, extending circulation time and enhancing biofilm targeting.
Materials:
- Primary macrophages (RAW 264.7 cell line or primary bone marrow-derived macrophages)
- Fluorescently labeled liposomal formulations (incorporate DiI or DiD membrane dyes)
- Flow cytometer or confocal microscopy system
- Tissue culture plates and complete cell culture media
- Phagocytosis inhibitors (cytochalasin D as control)
Procedure:
- Seed macrophages in 12-well plates at 5 × 10^5 cells/well and culture overnight.
- Incubate with fluorescently labeled liposomal formulations (100 μg/mL) for 1, 2, 4, and 6 hours at 37°C.
- For inhibition control, pre-treat cells with cytochalasin D (5 μM) for 30 minutes before adding liposomes.
- After incubation, thoroughly wash cells with cold PBS to remove non-internalized liposomes.
- Analyze cells using flow cytometry to quantify fluorescence intensity, or fix with paraformaldehyde (4%) for confocal microscopy visualization.
- Calculate phagocytosis index as (fluorescence of sample - fluorescence of inhibitor control) / total fluorescence added.
Expected Outcomes: Optimized liposomal formulations should show at least 40% reduction in macrophage uptake compared to unmodified nanoparticles, indicating improved stealth properties.
Protocol: Biofilm Penetration and Distribution Analysis
Objective: Evaluate the ability of liposomal Cas9 formulations to penetrate the biofilm matrix and distribute uniformly.
Materials:
- Established P. aeruginosa biofilms (48-hour growth in flow cells or on coupons)
- Fluorescently labeled liposomal Cas9 formulations
- Confocal laser scanning microscopy (CLSM) system
- Image analysis software (e.g., ImageJ, COMSTAT)
- Syto-9 and propidium iodide for biofilm staining
Procedure:
- Grow P. aeruginosa biofilms for 48 hours in optimal media under static or flow conditions.
- Treat mature biofilms with fluorescent liposomal formulations (200 μg/mL) for 4 hours.
- Gently wash to remove non-adherent particles.
- Stain biofilms with Syto-9 (5 μM) to visualize bacterial cells and propidium iodide (30 μM) to assess viability.
- Image using CLSM with z-stacking (1 μm steps through entire biofilm depth).
- Analyze fluorescence intensity distribution through biofilm depth using image analysis software.
- Calculate penetration efficiency as (fluorescence at base / fluorescence at surface) × 100%.
Expected Outcomes: Effective formulations should demonstrate uniform fluorescence distribution throughout the biofilm depth with penetration efficiency >60%, indicating successful matrix penetration.
Protocol: Immune Response Profiling in Biofilm-Infected Models
Objective: Characterize the host immune response to liposomal Cas9 treatment in biofilm-infected hosts.
Materials:
- Mouse model of implant-associated P. aeruginosa infection
- Liposomal Cas9 formulations targeting biofilm genes
- ELISA kits for cytokine analysis (TNF-α, IL-1β, IL-6, IL-10)
- Flow cytometry antibodies for immune cell markers (neutrophils, macrophages, G-MDSCs)
- Tissue processing equipment for homogenization
Procedure:
- Establish implant-associated P. aeruginosa biofilm infection in mice.
- Administer liposomal Cas9 formulations systemically or locally at infection site daily for 3 days.
- Collect serum and tissue samples at 24, 48, and 72 hours post-treatment.
- Process tissue samples for bacterial burden assessment (CFU counting) and immune cell profiling.
- Analyze cytokine levels in serum and tissue homogenates using ELISA.
- Characterize immune cell populations by flow cytometry using specific markers:
- Neutrophils: CD11b+ Ly6G+
- Inflammatory macrophages: CD11b+ F4/80+ MHC-II+
- G-MDSCs: CD11b+ Ly6G+ Ly6Cint
- Correlate immune parameters with bacterial clearance.
Expected Outcomes: Successful treatment should show reduced pro-inflammatory cytokines, decreased G-MDSC recruitment, and enhanced bacterial clearance compared to controls.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Liposomal Cas9 Immune Evasion Studies
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Liposomal Components | DOPC, DOTAP, cholesterol, PEG-lipids | Formulation of stealth liposomes with enhanced circulation time | PEG density affects macrophage uptake; charge influences biofilm interaction |
| CRISPR Components | Cas9 protein, sgRNAs targeting biofilm genes | Precision editing of bacterial virulence and resistance genes | sgRNAs must be designed for specific P. aeruginosa targets |
| Fluorescent Tags | DiI, DiD, FITC, Cy5 | Tracking nanoparticle distribution and uptake | Different wavelengths allow multiplexing in penetration studies |
| Biofilm Matrix Digesters | DNase I, dispersin B, proteinase K | Experimental disruption of matrix barriers | Used as controls to validate matrix role in immune evasion |
| Immune Cell Markers | Anti-CD11b, Ly6G, F4/80, MHC-II | Characterization of host immune responses | Panel design critical for comprehensive profiling |
| Cytokine Assays | TNF-α, IL-1β, IL-6, IL-10 ELISA | Quantification of inflammatory responses | Time course important for understanding kinetic response |
Strategic Implementation and Future Directions
The integration of immune-evasive liposomal Cas9 formulations represents a paradigm shift in approaching biofilm-associated infections. The strategic combination of pathogen-inspired stealth properties with precision gene editing enables targeted disruption of bacterial communities that have traditionally been refractory to conventional antibiotics. Future directions should focus on optimizing formulation parameters for clinical translation, including scale-up production, stability assessment, and comprehensive safety profiling. The development of triggered release mechanisms that respond to biofilm-specific environmental cues (e.g., low oxygen, specific enzymes) may further enhance specificity and reduce off-target effects. As antibiotic resistance continues to escalate, these integrated platforms offer a promising pathway for addressing the significant clinical challenge posed by biofilm-associated infections.
The escalating crisis of antibiotic-resistant bacterial infections poses a significant threat to global health, with Pseudomonas aeruginosa biofilm-associated infections representing a particularly challenging therapeutic target [4]. Biofilms, structured communities of microorganisms encapsulated in a self-produced extracellular polymeric substance (EPS), demonstrate up to 1000-fold greater tolerance to conventional antibiotics compared to their planktonic counterparts [4] [64]. This formidable resistance stems from multiple mechanisms, including reduced antibiotic penetration through the EPS matrix, altered metabolic states of embedded bacteria, and enhanced horizontal gene transfer [4] [3].
The emergence of CRISPR/Cas9 gene-editing technology has introduced a paradigm-shifting approach for precision targeting of bacterial resistance mechanisms. By specifically disrupting antibiotic resistance genes, quorum-sensing pathways, and biofilm-regulating factors, CRISPR/Cas9 offers a revolutionary strategy for combating biofilm-associated infections [4]. However, the clinical translation of this molecular technology faces significant delivery challenges, particularly in efficiently transporting CRISPR/Cas9 components through complex biofilm matrices and into bacterial cells [4] [65].
Liposomal formulations have emerged as a promising solution to these delivery challenges, serving as effective carriers for CRISPR/Cas9 components while exhibiting intrinsic antibacterial properties [4] [38]. These versatile nanocarriers protect genetic material from degradation, enhance cellular uptake, and can be engineered for controlled release within biofilm environments [66] [38]. This application note details standardized protocols for developing, optimizing, and scaling liposomal Cas9 formulations specifically designed to address the manufacturing challenges in translating laboratory success to clinically viable anti-biofilm therapeutics.
Quantitative Performance Assessment of Liposomal Formulations
Rigorous quantification of formulation efficacy is essential for benchmarking performance and guiding manufacturing optimization. The data below summarize key performance metrics for advanced anti-biofilm formulations.
Table 1: Quantitative Efficacy Metrics of Advanced Anti-Biofilm Formulations
| Formulation Type | Target Pathogen | Key Performance Metric | Result | Reference |
|---|---|---|---|---|
| Liposomal Cas9 Formulation | P. aeruginosa | Reduction in biofilm biomass (in vitro) | >90% | [4] |
| Cas9-Gold Nanoparticle Hybrid | P. aeruginosa | Gene-editing efficiency enhancement | 3.5-fold increase vs. non-carrier systems | [4] |
| Daphnetin + Tobramycin Combination | P. aeruginosa | Synergistic biofilm reduction (in vitro) | Significant reduction (P<0.05) in crystal violet staining & colony counts | [67] |
| Evolved Bacteriophage Cocktail | MDR P. aeruginosa | Bacterial suppression in biofilm (isothermal microcalorimetry) | ≥75% heat reduction (up to 91.5%) | [68] |
Table 2: Critical Quality Attributes (CQAs) for Liposomal Cas9 Formulations
| Critical Quality Attribute | Target Range | Impact on Efficacy & Manufacturing |
|---|---|---|
| Particle Size | <200 nm | Ensures effective penetration through biofilm EPS matrix [38] |
| Surface Charge (Zeta Potential) | Positive (cationic) | Promotes interaction with negatively charged bacterial membranes and EPS [38] |
| Polydispersity Index (PDI) | <0.2 | Indicates a homogeneous population, critical for batch-to-batch consistency and predictable drug release |
| Encapsulation Efficiency | >90% | Maximizes delivery of costly CRISPR/Cas9 components, reduces waste |
| Membrane Rigidity | High (controlled) | Reduces premature leakage; enables triggered release at target site [38] |
Experimental Protocols for Formulation Development & Assessment
Protocol: Thin-Film Hydration Method for Cationic Liposomal Cas9
This protocol describes the preparation of cationic liposomes for encapsulating CRISPR/Cas9 ribonucleoproteins (RNPs) or plasmid DNA.
Materials:
- Lipids: DOTAP, DOPE, Cholesterol (e.g., from Avanti Polar Lipids)
- CRISPR/Cas9 components: sgRNA, Cas9 protein (or plasmid encoding Cas9/sgRNA)
- Solvents: Chloroform, Ethanol (HPLC grade)
- Buffers: HEPES, Saline solutions
- Equipment: Rotary evaporator, Bath sonicator, Extruder apparatus
Procedure:
- Lipid Film Formation: Dissolve lipid components (DOTAP:DOPE:Cholesterol at 50:45:5 molar ratio) in chloroform in a round-bottom flask. Remove organic solvent using a rotary evaporator (≥ 40°C) to form a thin, homogeneous lipid film.
- Hydration: Hydrate the dried lipid film with sterile HEPES buffer (pH 7.4) containing the pre-complexed Cas9/sgRNA RNP. Gently agitate the mixture at 60°C for 1 hour to form multilamellar vesicles (MLVs).
- Size Reduction: Subject the MLV suspension to 5 cycles of freeze-thawing (liquid nitrogen/40°C water bath). Subsequently, extrude the suspension through polycarbonate membranes (starting with 400 nm, then 200 nm, and finally 100 nm) using a high-pressure extruder to form small, unilamellar vesicles (SUVs).
- Purification: Separate non-encapsulated CRISPR/Cas9 components using size-exclusion chromatography (e.g., Sephadex G-50 column) or dialysis against HEPES buffer for 24 hours.
- Sterilization & Storage: Filter-sterilize the final liposomal preparation through a 0.22 μm membrane. Store at 4°C under inert gas (e.g., Argon) for short-term use or lyophilize for long-term storage.
Protocol: In Vitro Assessment of Anti-Biofilm Efficacy
This standardized method evaluates the ability of liposomal Cas9 formulations to disrupt and eradicate pre-established P. aeruginosa biofilms.
Materials:
- Bacterial strain: P. aeruginosa PAO1 (or relevant clinical isolate)
- Culture media: Tryptic Soy Broth (TSB), Luria-Bertani (LB) Agar
- Stains: Crystal violet (1%), SYTO 9/propidium iodide (for live/dead staining)
- Equipment: Microplate reader, Confocal Laser Scanning Microscope (CLSM), Sonicator water bath
Procedure:
- Biofilm Formation: Inoculate a 96-well plate with P. aeruginosa PAO1 (1×10^8 CFU/mL in TSB) and incubate statically for 72 hours at 37°C to establish mature biofilms. Replace the medium every 24 hours to replenish nutrients.
- Treatment Application: Gently wash the 72-hour biofilms with PBS to remove non-adherent cells. Apply treatments to respective wells:
- Test Group: Liposomal Cas9 formulation (targeting, e.g., lasI quorum-sensing gene)
- Control Groups: Empty liposomes, free Cas9 RNP, conventional antibiotic (e.g., tobramycin), and untreated control (media only).
- Incubation: Incubate the treatment plate for 24 hours at 37°C.
- Biofilm Quantification (Crystal Violet Staining):
- Aspirate medium and gently wash wells with PBS.
- Fix biofilms with 200 μL of 99% methanol for 15 minutes, then air-dry.
- Stain with 1% crystal violet (200 μL/well) for 30 minutes.
- Wash thoroughly with water to remove unbound dye.
- Destain with 33% glacial acetic acid (200 μL/well) for 20 minutes with shaking.
- Transfer 100 μL of destain solution to a new plate and measure absorbance at 595 nm.
- Viability Assessment (Colony Counting):
- After treatment, wash separate biofilm wells with PBS.
- Add 100 μL of 0.1% Triton X-100 to wells and sonicate in a water bath for 5 minutes to disrupt the biofilm and disperse cells.
- Perform serial dilutions of the resulting bacterial suspension and plate on LB agar.
- Count Colony Forming Units (CFU) after 24 hours of incubation at 37°C.
- Confocal Microscopy Imaging: Use live/dead staining (SYTO 9/propidium iodide) on biofilms grown on coverslips, followed by imaging with CLSM to visualize biofilm architecture and bacterial viability in three dimensions.
Protocol: Scalability and GMP Considerations for Liposomal Cas9
Transitioning from laboratory-scale preparation to clinically viable batches requires careful process adaptation.
Key Considerations:
- Scaled-Up Manufacturing: Replace batch processes like thin-film hydration with continuous, scalable methods such as:
- Microfluidic Mixing: Provides superior control over particle size and PDI during hydration.
- Tangential Flow Filtration (TFF): Replaces extrusion and dialysis for more efficient concentration and buffer exchange at large volumes.
- Process Analytical Technology (PAT): Implement in-line monitoring (e.g., dynamic light scattering for particle size, UV for drug loading) to enable real-time quality control and ensure batch-to-batch consistency.
- Aseptic Processing: For sterile product manufacturing, the entire process from formulation to filling should be conducted in a Grade A environment. Terminal sterilization is often not feasible for liposomes; therefore, aseptic processing is mandatory.
- Lyophilization Development: Develop a robust lyophilization protocol to ensure long-term stability of the final drug product. This includes optimizing cryoprotectants (e.g., sucrose, trehalose) and freeze-drying cycles.
Pathway and Workflow Visualization
Diagram 1: Development workflow for a liposomal Cas9 product, from research to clinical batch production.
Diagram 2: Mechanism of action for liposomal Cas9 formulations targeting P. aeruginosa biofilms.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Liposomal Cas9 Anti-Biofilm Research
| Item | Function/Application | Example Specifications / Notes |
|---|---|---|
| Cationic Lipids | Forms positively charged liposome bilayer for bacterial cell interaction | DOTAP, DC-Chol; sourced from GMP-certified suppliers for translation |
| Helper Lipids | Enhances membrane fusion and stability | DOPE, Cholesterol; critical for forming fusogenic, non-lamellar structures |
| CRISPR/Cas9 Components | Active gene-editing machinery | High-purity Cas9 nuclease (e.g., SpyCas9) and sgRNA; use HPLC-purified sgRNA |
| Microfluidic Device | Scalable production of uniform liposomes | Nanoassembler or similar; enables continuous manufacturing with low PDI |
| Extrusion System | Laboratory-scale size reduction | Avanti Mini-Extruder with polycarbonate membranes (100-400 nm) |
| Particle Analyzer | Characterizes Critical Quality Attributes (CQAs) | DLS/Zetasizer for measuring size, PDI, and zeta potential |
| P. aeruginosa Strains | In vitro and in vivo biofilm models | PAO1 (reference), mucoid CF isolates (e.g., FRD1), clinical MDR strains |
| Biofilm Assessment Kit | Quantifies biofilm formation and eradication | Crystal violet staining, metabolic assays (resazurin), live/dead staining kits |
| Animal Model | Preclinical efficacy testing | Murine lung infection (for CF), implant-associated infection, burn wound models |
Benchmarking Liposomal Cas9 Against Established and Emerging Therapies
The Clustered Regularly Interspaced Short Palindromic Repeats associated protein 9 (CRISPR-Cas9) system represents a transformative tool for precision genome editing, with significant potential for combating antibiotic-resistant bacterial infections. However, its clinical application is hindered by challenges in delivery efficiency and stability, particularly against resilient bacterial structures like Pseudomonas aeruginosa biofilms [69] [4]. Non-viral delivery systems, especially lipid-based nanoparticles, have emerged as promising vectors to overcome these barriers, enhancing cellular uptake and protecting genetic material from degradation [69] [70]. This application note provides a direct quantitative comparison between liposomal Cas9 formulations and non-carrier delivery systems, framing the data within the context of anti-biofilm research against P. aeruginosa.
Quantitative Efficacy Metrics: Liposomal vs. Non-Carrier Delivery
The tables below summarize key efficacy metrics for liposomal and non-carrier Cas9 delivery systems, highlighting the enhanced performance of nano-formulations.
Table 1: Anti-Biofilm and Gene Editing Efficacy Metrics
| Efficacy Metric | Liposomal Cas9 Formulation | Non-Carrier Delivery System | Experimental Context |
|---|---|---|---|
| Biofilm Biomass Reduction | >90% reduction [4] | Not Specifically Reported | In vitro, against P. aeruginosa biofilm [4] |
| Gene Editing Efficiency | Up to 3.5-fold enhancement [4] | Baseline (1x) [4] | Gold nanoparticle data shown for comparison of carrier efficacy [4] |
| Cellular Uptake Efficiency | High (Dependent on lipid composition) [69] | Low (Due to electrostatic repulsion) [69] | Theoretical comparison based on nanoparticle properties |
Table 2: System Stability and Practical Handling
| Characteristic | Liposomal Cas9 Formulation | Non-Carrier Delivery System |
|---|---|---|
| Stability in Vivo | Enhanced stability; protection from nucleases [69] [70] | Vulnerable to degradation by nucleases and proteases [69] |
| Immunogenicity | Low immunogenicity [69] [70] | Can trigger immune responses [69] |
| Payload Packaging | High capacity for large RNP complexes [69] | Limited by complex size and charge [69] |
| Targeting Specificity | Can be engineered for targeting (e.g., with aptamers) [71] | Lacks inherent targeting capability |
Experimental Protocol: Assessing Anti-Biofilm Efficacy of Liposomal Cas9
This protocol details the methodology for evaluating the efficacy of liposomal Cas9 formulations against P. aeruginosa biofilms in vitro.
Materials and Reagents
- Bacterial Strain: Pseudomonas aeruginosa (e.g., strain collected from clinical isolates) [71].
- Culture Media: Luria-Bertani (LB) broth and LB agar plates [71].
- CRISPR-Cas9 Components: Cas9 protein, and sgRNA targeting specific bacterial resistance genes (e.g., ndm-1, mecA) or biofilm-regulating factors [4].
- Liposome Formulation: Cationic or ionizable lipid nanoparticles for RNP complex encapsulation [69] [4].
- Staining Solutions: Crystal Violet (CV) for total biofilm biomass, Calcein-AM/PI Double Stain Kit for bacterial viability/cytotoxicity within the biofilm [71].
- Equipment: Confocal Laser Scanning Microscope (CLSM), Scanning Electron Microscope (SEM) [71].
Procedure
Biofilm Formation:
Treatment with Liposomal Cas9:
- Prepare the liposomal Cas9-sgRNA RNP complex as described in Section 5.1.
- Carefully add the formulated liposomal Cas9 to the pre-formed biofilms. Include controls: untreated biofilm, biofilm treated with "empty" liposomes (without RNP), and biofilm treated with non-carrier Cas9-sgRNA RNP.
- Incubate the treated biofilms for a predetermined period (e.g., 24 hours) at 37°C [4].
Assessment of Biofilm Biomass (Crystal Violet Assay):
- Gently wash the biofilm to remove non-adherent cells.
- Fix the biofilm with methanol and stain with 0.1% Crystal Violet solution for 15-20 minutes.
- Wash again to remove excess stain.
- Elute the bound dye with acetic acid (33%) and measure the absorbance at 595 nm. The reduction in absorbance correlates with the reduction in biofilm biomass [71].
Assessment of Bacterial Viability (Live/Dead Staining):
- Stain the treated biofilms using the Calcein-AM/PI kit according to the manufacturer's instructions.
- Visualize under a Confocal Laser Scanning Microscope (CLSM). Calcein-AM stains live cells (green), and Propidium Iodide (PI) stains dead/damaged cells (red) [71].
- Quantify the ratio of red to green fluorescence to determine the percentage of bacterial cell death within the biofilm.
Visualization of Biofilm Structure (SEM):
- Fix the treated biofilms with glutaraldehyde, followed by dehydration in a graded ethanol series.
- Critical-point dry the samples and sputter-coat with gold/palladium.
- Observe the biofilm ultrastructure using Scanning Electron Microscopy (SEM) to visually confirm disruption of the biofilm matrix and bacterial cell damage [71].
Data Analysis
- Quantify the percentage of biofilm reduction from Crystal Violet data:
% Reduction = (1 - (Abs_treated / Abs_control)) * 100. - Perform statistical analysis (e.g., Student's t-test) to confirm the significance of the observed differences between liposomal Cas9 and control groups.
Visualization of Workflow and Mechanisms
The following diagrams illustrate the experimental workflow and the mechanism of action of liposomal Cas9.
Diagram 1: Experimental workflow for evaluating liposomal Cas9 anti-biofilm efficacy.
Diagram 2: Mechanism of liposomal Cas9 action against bacterial biofilms.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Liposomal Cas9 Anti-Biofilm Research
| Item | Function/Description | Example Application in Protocol |
|---|---|---|
| Cationic/Ionizable Lipids | Form the core of the nanoparticle, encapsulating and protecting Cas9 RNP; enhance cellular uptake [69]. | Formulation of the liposomal Cas9-sgRNA delivery vehicle. |
| Cas9 Ribonucleoprotein (RNP) | The active gene-editing complex; offers higher editing efficiency and reduced off-target effects compared to plasmid DNA [69] [70]. | Directly loaded into liposomes to target specific bacterial genes. |
| sgRNA Targeting Biofilm Genes | Guides the Cas9 protein to specific genomic sequences (e.g., quorum-sensing genes, antibiotic resistance genes) [4]. | Designed to disrupt genes critical for biofilm integrity and antibiotic resistance. |
| Clinical P. aeruginosa Isolate | A relevant, pathogenic bacterial strain expressing target genes and capable of robust biofilm formation [71]. | Source for in vitro biofilm models to ensure clinical relevance. |
| Crystal Violet Stain | A dye that binds to biomass, enabling quantitative assessment of total biofilm formation [71]. | Staining for the high-throughput biofilm biomass assay (Section 3.2, Step 3). |
| Calcein-AM/PI Viability Kit | Fluorescent stains for simultaneous quantification of live (green) and dead (red) bacterial cells [71]. | Determining bactericidal effects of the treatment within the biofilm via CLSM. |
| Confocal Laser Scanning Microscope (CLSM) | Enables high-resolution, 3D imaging of stained biofilms to assess structure and viability [71]. | Visualization and quantification of Live/Dead staining results. |
Antibiotic resistance represents one of the most urgent threats to global health, with biofilm-associated infections being particularly challenging to treat. Bacterial resistance manifests through two primary pathways: intrinsic resistance, which is an innate characteristic of bacterial species, and acquired resistance, which develops through genetic mutations or horizontal gene transfer [73] [74]. The structural differences between Gram-positive and Gram-negative bacteria play a significant role in intrinsic resistance, particularly due to the impermeable outer membrane of Gram-negative pathogens like Pseudomonas aeruginosa that limits antibiotic penetration [75]. Biofilms exacerbate this problem by creating structured communities embedded in an extracellular polymeric substance (EPS) that provides physical protection and enables bacterial survival in hostile environments [13] [24]. Understanding these mechanisms is crucial for developing effective strategies to overcome resistance.
Table 1: Fundamental Antibiotic Resistance Mechanisms
| Resistance Category | Key Mechanisms | Examples | Clinical Impact |
|---|---|---|---|
| Intrinsic Resistance | Impermeable outer membrane, natural efflux pumps, porin restrictions | Gram-negative resistance to vancomycin, anaerobic bacteria resistance to aminoglycosides | Limits initial treatment options, requires broader-spectrum antibiotics |
| Acquired Resistance | Enzymatic inactivation, target site modification, enhanced efflux pumps | β-lactamase production, ribosomal mutations, plasmid-encoded efflux systems | Renders previously effective antibiotics useless, spreads rapidly between pathogens |
| Biofilm-Mediated Resistance | Physical barrier, metabolic heterogeneity, persister cell formation, quorum sensing | Reduced antibiotic penetration in P. aeruginosa and A. baumannii biofilms | Causes chronic, recurrent infections that are extremely difficult to eradicate |
Quantitative Analysis of Traditional Antibiotics Versus Novel Approaches
Traditional antibiotics face significant limitations against resistant bacterial infections, particularly those involving biofilms. Conventional antimicrobial therapies often demonstrate up to 1000-fold reduced efficacy against biofilm-embedded bacteria compared to their planktonic counterparts [24]. The limitations of traditional approaches have prompted the development of innovative strategies that target resistance mechanisms more precisely.
Table 2: Efficacy Comparison Between Traditional and Novel Antimicrobial Approaches
| Therapeutic Approach | Efficacy Against Planktonic Cells | Efficacy Against Biofilms | Resistance Development Potential |
|---|---|---|---|
| Traditional β-lactams | High (MIC: 0.5-2 μg/mL for susceptible strains) | Low (>100-fold MIC increase required) | High (via β-lactamase acquisition) |
| Aminoglycosides | High (MIC: 1-4 μg/mL for susceptible strains) | Moderate to Low (limited penetration) | Moderate (via enzymatic modification) |
| Fluoroquinolones | High (MIC: 0.5-2 μg/mL for susceptible strains) | Moderate (affected by metabolic state) | High (via target site mutations) |
| Liposomal CRISPR/Cas9 Formulations | Variable (species-specific) | High (90% biomass reduction in P. aeruginosa) | Low (targets resistance genes directly) |
| Nanoparticle-Antibiotic Conjugates | Enhanced via improved uptake | Superior (3.5-fold efficiency increase) | Low (overcomes efflux/impermeability) |
Recent advances demonstrate that liposomal CRISPR-Cas9 formulations can reduce Pseudomonas aeruginosa biofilm biomass by over 90% in vitro, while gold nanoparticle carriers enhance editing efficiency up to 3.5-fold compared to non-carrier systems [13] [24]. These hybrid platforms enable co-delivery with antibiotics, producing synergistic antibacterial effects and superior biofilm disruption compared to traditional mono-therapies [24].
Experimental Protocols
Protocol for Assessing Liposomal Cas9 Efficacy Against Biofilms
Principle: This protocol quantifies the effectiveness of CRISPR/Cas9-loaded liposomes against established P. aeruginosa biofilms through biomass reduction and viability assessment.
Materials:
- P. aeruginosa strain PAO1 (or clinical isolate)
- Liposomal Cas9 formulation with gRNA targeting antibiotic resistance genes (e.g., β-lactamase)
- Tryptic Soy Broth (TSB)
- 96-well polystyrene plates
- Crystal violet solution (0.1%)
- Phosphate Buffered Saline (PBS)
- MTT solution (0.5 mg/mL)
- DMSO
- Microplate reader
Procedure:
- Biofilm Formation: Inoculate P. aeruginosa in TSB and adjust to 10⁶ CFU/mL. Dispense 200 μL per well into 96-well plate. Incubate statically for 48 hours at 37°C to establish mature biofilms.
- Treatment Application: Carefully remove planktonic cells and medium. Add 200 μL of liposomal Cas9 formulations at varying concentrations (50-500 μg/mL) to test wells. Include untreated controls and empty liposome controls.
- Incubation: Incubate plates for 24 hours at 37°C.
- Biomass Quantification (Crystal Violet Staining):
- Remove treatment and gently wash wells twice with PBS
- Fix biofilms with 200 μL methanol for 15 minutes
- Discard methanol and air dry plates
- Stain with 200 μL of 0.1% crystal violet for 15 minutes
- Wash extensively with distilled water until no residual stain appears
- Elute bound dye with 200 μL of 33% acetic acid
- Measure absorbance at 570 nm using microplate reader
- Viability Assessment (MTT Assay):
- After treatment incubation, add 50 μL MTT solution (0.5 mg/mL) to each well
- Incubate for 3 hours at 37°C
- Carefully remove solution and dissolve formed formazan crystals in 150 μL DMSO
- Measure absorbance at 570 nm with reference filter at 690 nm
- Data Analysis: Calculate percentage reduction in biofilm biomass and viability compared to untreated controls.
Diagram Title: Liposomal Cas9 Biofilm Efficacy Assessment Workflow
Protocol for Evaluating Outer Membrane Permeabilization
Principle: This assay assesses the ability of adjuvant compounds to disrupt the Gram-negative outer membrane, enhancing antibiotic penetration.
Materials:
- Polymyxin B nonapeptide (PMBN) or other permeabilizing agents
- N-phenyl-1-naphthylamine (NPN)
- HEPES buffer (5 mM, pH 7.2)
- P. aeruginosa cultures
- Fluorescence microplate reader
Procedure:
- Cell Preparation: Grow P. aeruginosa to mid-log phase (OD600 ≈ 0.5). Harvest cells by centrifugation and wash twice with HEPES buffer.
- NPN Addition: Resuspend cells to OD600 of 0.5 in HEPES buffer containing 10 μM NPN.
- Treatment: Dispense 200 μL of cell suspension per well in black 96-well plates. Add permeabilizing agents at varying concentrations.
- Measurement: Immediately measure fluorescence (excitation 350 nm, emission 420 nm) every 2 minutes for 30 minutes.
- Analysis: Calculate percentage increase in fluorescence compared to untreated controls, indicating outer membrane disruption.
Research Reagent Solutions
Table 3: Essential Research Reagents for Antibiotic Resistance Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| CRISPR/Cas9 Components | Cas9 nuclease, gRNA targeting resistance genes (e.g., blaSHV, mecA) | Precision editing of antibiotic resistance genes | Require efficient delivery systems; specificity verification essential |
| Nanoparticle Delivery Systems | Liposomal formulations, gold nanoparticles, polymeric NPs | Enhance cellular uptake and protect genetic material | Size, surface charge, and functionalization critical for biofilm penetration |
| Outer Membrane Disruptors | Polymyxin B nonapeptide (PMBN), deacylpolymyxin derivatives | Permeabilize Gram-negative outer membrane | Balance between efficacy and cytotoxicity; synergize with other antibiotics |
| Biofilm Matrix Disruptors | DNase I, dispersin B, EDTA | Degrade extracellular polymeric substance (EPS) | Improve antibiotic penetration; effectiveness varies by biofilm composition |
| Efflux Pump Inhibitors | Phe-Arg-β-naphthylamide (PAβN), berberine, reserpine | Block antibiotic extrusion from bacterial cells | Can have off-target effects in eukaryotic cells; specificity challenges |
| Traditional Antibiotics (Comparators) | Ciprofloxacin, meropenem, tobramycin, colistin | Efficacy benchmarks against novel approaches | Resistance profiles must be characterized for each bacterial strain |
Mechanistic Pathways for Overcoming Resistance
The integration of nanoparticle delivery systems with CRISPR/Cas9 technology represents a paradigm shift in addressing antibiotic resistance. This combinatorial approach simultaneously targets multiple resistance mechanisms through precision genetic editing and enhanced drug delivery.
Diagram Title: Integrated Strategy to Overcome Bacterial Resistance
The mechanistic basis for the superior efficacy of combined approaches lies in the simultaneous targeting of complementary resistance pathways. Liposomal Cas9 formulations directly address acquired resistance by precisely disrupting antibiotic resistance genes (e.g., β-lactamases), while the nanoparticle delivery component overcomes intrinsic resistance mechanisms by enhancing penetration through both bacterial membranes and biofilm matrices [13] [24]. This dual action creates a synergistic effect where traditional antibiotics regain their efficacy against previously resistant pathogens.
The combinatorial strategy is particularly effective against P. aeruginosa biofilms due to the ability of engineered nanoparticles to penetrate the EPS matrix and deliver functional CRISPR/Cas9 components to bacterial cells in deeper biofilm layers. This approach addresses the limitation of conventional antibiotics that predominantly target metabolically active cells in the biofilm periphery while leaving persistent cells in deeper regions unaffected.
Pseudomonas aeruginosa biofilms represent a critical therapeutic challenge, exhibiting resistance up to 1000-fold higher than their planktonic counterparts [76] [29]. This resilience is largely attributed to the biofilm's extracellular polymeric substance (EPS) matrix, which acts as a barrier to conventional antibiotics [38]. Nanotechnology offers promising strategies to overcome this barrier. This application note provides a quantitative comparison and detailed protocols for evaluating three major nanoparticle classes—liposomes, metallic, and polymeric nanoparticles—in the context of anti-biofilm research, with a specific focus on applications for advanced liposomal Cas9 delivery systems.
Comparative Performance of Nanotherapeutics
The table below summarizes the key characteristics and anti-biofilm efficacy of the three nanoparticle classes against P. aeruginosa.
Table 1: Head-to-Head Comparison of Anti-Biofilm Nanotherapeutics
| Parameter | Liposomes | Metallic Nanoparticles (e.g., AgNPs) | Polymeric Nanoparticles |
|---|---|---|---|
| Composition | Phospholipids (e.g., DOPC, DOPE), cholesterol [77] | Silver (Ag), Gold (Au), Copper (Cu), Metal Oxides (e.g., ZnO) [78] | Biodegradable polymers (e.g., PLGA, Chitosan) [76] |
| Key Anti-biofilm Mechanisms | Membrane fusion; enhanced drug penetration and retention; biofilm matrix disruption (when loaded with biosurfactants) [29] [79] | Reactive Oxygen Species (ROS) generation; degradation of the biofilm matrix; disruption of bacterial membranes [76] [78] | Controlled drug release; degradation of biofilm matrix (e.g., via enzyme delivery) [76] |
| Payload Capacity | High (Hydrophilic & hydrophobic cargo) [29] | Low (Intrinsic activity; can be functionalized) [78] | High (Hydrophilic & hydrophobic cargo) [76] |
| Synergy with Antibiotics | Demonstrated (e.g., liposomal meropenem) [77] | High (e.g., AgNPs with colistin) [80] | Demonstrated (Co-delivery systems) [76] |
| Reported Biofilm Inhibition | Up to 92% (Rhamnolipid-loaded, 100 nm) [77] | Up to 90% (Ag-Carth-NPs) [81] | Data varies by polymer and payload [76] |
| Key Design Parameter | Size, surface charge (Cationic > Anionic), PEG-modification [82] | Size, shape, surface coating [78] | Polymer composition, molecular weight, surface charge [76] |
| Toxicity Considerations | Generally biocompatible [38] | Potential cytotoxicity and environmental toxicity [76] [78] | Varies with polymer; generally favorable [76] |
Experimental Protocols for Anti-Biofilm Evaluation
Protocol: Preparation of Cationic Liposomes for Biofilm Penetration
This protocol details the synthesis of cationic liposomes, optimized for enhanced biofilm penetration and retention based on their surface properties [82].
Reagents:
- 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)
- 1,2-dioleoyl-sn-glycero-3-ethylphosphocholine (DOPEP) or DOTAP (cationic lipid)
- Cholesterol
- Chloroform
- PBS Buffer (pH 7.4)
- Payload (e.g., antibiotic, biosurfactant, or nucleic acids)
Procedure:
- Lipid Film Formation: Dissolve DOPC, cationic lipid, and cholesterol in a molar ratio (e.g., 50:45:5) in chloroform in a round-bottom flask. Evaporate the chloroform under reduced pressure using a rotary evaporator to form a thin, uniform lipid film.
- Hydration: Hydrate the dried lipid film with PBS buffer (or an aqueous solution of your hydrophilic payload) at a temperature above the lipid transition temperature (e.g., 50°C) for 30-60 minutes with gentle agitation.
- Size Reduction: To achieve a uniform size distribution critical for penetration, extrude the hydrated liposome suspension through polycarbonate membranes using a mini-extruder. Sequential extrusion through 400 nm, 200 nm, and finally 100 nm membranes is recommended [77].
- Purification: Separate unencapsulated payload via dialysis or gel filtration chromatography using a Sephadex G-50 column.
- Characterization: Determine the particle size, polydispersity index (PDI), and zeta potential using dynamic light scattering (DLS). Measure encapsulation efficiency via HPLC or spectrophotometry.
Protocol: Standardized Biofilm Inhibition Assay (TCP Method)
The Tissue Culture Plate (TCP) method is a widely accepted, quantitative assay for evaluating biofilm formation and inhibition [83] [81].
Reagents:
- Trypticase Soy Broth (TSB) with 1% glucose
- Phosphate Buffered Saline (PBS)
- Crystal Violet (0.1% w/v)
- Acetic Acid (33% v/v)
- P. aeruginosa culture (e.g., strain PAO1 or clinical isolate)
Procedure:
- Biofilm Formation: Adjust a fresh bacterial culture to 0.5 McFarland standard in TSB + 1% glucose. Dispense 200 µL per well into a sterile 96-well flat-bottom polystyrene plate. Incubate for 24-48 hours at 37°C.
- Treatment: Gently remove the planktonic cells and media. Wash the adhered biofilms twice with PBS. Add 200 µL of serial dilutions of the nanoformulation (e.g., liposomes, AgNPs) in fresh media to the wells.
- Incubation: Incubate the plate for another 24 hours at 37°C.
- Staining and Quantification:
- Wash the wells twice with PBS to remove non-adherent cells.
- Air-dry the plate and stain the biofilms with 200 µL of 0.1% crystal violet for 15 minutes.
- Wash extensively with water to remove excess stain.
- Elute the bound stain with 200 µL of 33% acetic acid.
- Measure the optical density (OD) of the eluent at 595 nm using a microplate reader.
- Analysis: Calculate the percentage of biofilm inhibition using the formula:
% Inhibition = [1 - (OD595 treated / OD595 control)] × 100
Visualization of Nano-Biofilm Interactions
The following diagram illustrates the sequential mechanisms by which different nanoparticles penetrate and disrupt bacterial biofilms.
Diagram Title: Nanoparticle Anti-Biofilm Mechanisms
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Liposomal Anti-Biofilm Research
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| DOPC / DOPE Lipids | Core structural phospholipids for liposome formation, imparting fluidity and fusogenic potential. | Avanti Polar Lipids. DOPE can enhance endosomal escape, crucial for intracellular delivery [77]. |
| Cationic Lipids | Imparts positive surface charge for enhanced interaction with negatively charged biofilm EPS and bacterial membranes. | DOTAP, DOPEP. Critical for increasing liposome retention within the biofilm [82]. |
| Rhamnolipid | A biosurfactant that disrupts biofilm matrix integrity; can be encapsulated in liposomes. | AGAE Technologies. Encapsulation shown to significantly boost eradication efficacy (up to 92%) [77]. |
| Silver Nitrate (AgNO₃) | Precursor for the synthesis of silver nanoparticles (AgNPs). | Sigma-Aldrich. Used in green synthesis with propolis or plant extracts for antimicrobial AgNPs [80] [81]. |
| Polycarbonate Membranes | For liposome extrusion and size control, a critical parameter for biofilm penetration. | Whatman. Use with Avanti Mini-Extruder for precise sizing (e.g., 100 nm, 200 nm) [77]. |
| 96-well Polystyrene Plates | Substrate for standardized in vitro biofilm cultivation and high-throughput inhibition assays. | Corning, Thermo Fisher. Essential for the Tissue Culture Plate (TCP) method [83] [81]. |
Discussion and Strategic Outlook
The comparative data indicates that liposomes hold a distinct advantage for the delivery of complex macromolecular payloads like Cas9 due to their high encapsulation efficiency and biocompatibility [29]. The demonstrated ability of cationic and PEG-modified liposomes to exhibit high retention and anti-biofilm effects [82] provides a clear design pathway. Furthermore, the success of liposome-encapsulated biosurfactants [77] [79] suggests a powerful combinatorial strategy: a liposomal Cas9 system could be co-delivered with a matrix-disrupting agent to first permeabilize the biofilm, thereby facilitating superior access for the gene-editing machinery.
In contrast, metallic nanoparticles like AgNPs, while highly effective at direct biofilm killing via ROS [78] [80], face challenges regarding payload capacity and potential toxicity, making them less suitable as primary carriers for Cas9. Their role may be more synergistic, as adjuvants to weaken the biofilm prior to targeted therapeutic intervention. The future of anti-biofilm nanotherapeutics lies in leveraging these complementary strengths through intelligent, multi-functional design.
The escalating crisis of antimicrobial resistance (AMR), particularly among ESKAPE pathogens such as Pseudomonas aeruginosa, necessitates the development of novel therapeutic strategies. Liposomal Cas9 formulations represent a promising approach for the precise targeting of biofilm-associated and antibiotic-resistance genes. However, to comprehensively evaluate their potential, it is crucial to contextualize their performance against other emerging antimicrobial modalities. This application note provides a comparative analysis of four key innovative strategies—Liposomal Cas9, Phage Therapy, Anti-Quorum Sensing Agents, and Antimicrobial Peptides—focusing on their efficacy, mechanisms, and practical application in combating P. aeruginosa biofilms. We summarize quantitative data into structured tables, provide detailed experimental protocols, and include visual workflows to serve researchers and drug development professionals.
The quantitative efficacy and key characteristics of the four novel antimicrobial modalities against P. aeruginosa are summarized in the table below.
Table 1: Comparative Analysis of Novel Antimicrobial Modalities for Pseudomonas aeruginosa Biofilms
| Modality | Reported Efficacy vs. Biofilms | Key Advantages | Primary Limitations | Ideal Use Case |
|---|---|---|---|---|
| Liposomal Cas9 Formulations | >90% reduction in biofilm biomass in vitro [4] | High precision for genetic targets; can resensitize bacteria to antibiotics [4] | Complex delivery system; potential for off-target effects; transient activity [4] [84] | Precision eradication of specific resistance genes in resistant infections |
| Phage Therapy | Effective biofilm penetration & disruption [85] | High specificity; self-replication at infection site; low environmental impact [85] [86] [87] | Narrow host range; potential for bacterial resistance; immunogenicity concerns [85] [86] | Targeted infection where a matching, specific phage is available |
| Anti-Quorum Sensing Agents | Dose-dependent reduction in virulence & biofilm formation [88] | Attenuates virulence without killing; lower selective pressure for resistance [88] [89] | Bacteriostatic (does not kill); efficacy can be species-specific [88] | Adjuvant therapy to disarm pathogens and enhance antibiotic efficacy |
| Antimicrobial Peptides (AMPs) | Eradication of MDR bacteria and biofilms (e.g., SAAP-148) [90] | Broad-spectrum activity; rapid membrane disruption; immunomodulatory effects [91] [90] | Potential cytotoxicity; susceptibility to proteolysis; high manufacturing cost [91] | Topical or localized treatment for multidrug-resistant infections |
Table 2: Quantitative Efficacy Metrics from Recent Studies (2022-2025)
| Modality | Specific Agent / Formulation | Target Pathogen | Key Metric | Result |
|---|---|---|---|---|
| Liposomal CRISPR/Cas9 | Liposomal Cas9-gRNA complex | P. aeruginosa | Biofilm Biomass Reduction | >90% [4] |
| Encapsulated Phage Therapy | Phage-loaded liposomes | P. aeruginosa | Loss of Viability during Nebulization | 1.08 ± 0.05 log (vs. 1.55 ± 0.04 log for free phage) [92] |
| Anti-Quorum Sensing | Coumaric Acid | Serratia marcescens | Minimum Inhibitory Concentration (MIC) | 700 µg/mL [88] |
| Antimicrobial Peptides | SAAP-148 | MDR Bacteria | Eradication of Biofilms & Persister Cells | Demonstrated, minimal resistance potential [90] |
Detailed Experimental Protocols
Protocol 1: Preparation and Evaluation of Liposomal Cas9 Formulations for Biofilm Eradication
This protocol details the formulation of liposomes for CRISPR/Cas9 delivery and the subsequent quantification of their anti-biofilm efficacy, adapted from recent hybrid study methodologies [4].
I. Liposomal Formulation via Thin-Film Hydration
- Lipid Film Preparation: Dissolve hydrogenated soybean phosphatidylcholine (HSPC), 1,2-distearoyl-sn-glycero-3-phosphoglycerol (DSPG-Na), DSPE-PEG, and cholesterol in a mass ratio of 3:2:0.5:1.7 in chloroform (5 mL) within a round-bottom flask [92].
- Solvent Evaporation: Use a rotary evaporator (e.g., Buchi Rotavapor) at 55°C to remove the organic solvent completely, forming a thin, uniform lipid film on the inner wall of the flask [92].
- Hydration and Encapsulation: Hydrate the dried lipid film with 3.75 mL of a CRISPR/Cas9 complex (e.g., Cas9 protein with guide RNA targeting a biofilm-specific gene) suspended in a sugar solution (80 mg/mL mannitol, 20 mg/mL sucrose). Maintain the suspension at 40°C with gentle agitation for 1 hour [92] [4].
- Downstream Processing: Size reduction is achieved via sonication or extrusion through polycarbonate membranes (e.g., 100 nm pore size). The final formulation is purified using dialysis or size-exclusion chromatography to remove non-encapsulated material [92].
II. Anti-Biofilm Efficacy Assay
- Biofilm Cultivation: Grow a static P. aeruginosa biofilm (e.g., strain PAO1 or a clinical isolate) for 24-48 hours in a 96-well polystyrene plate using a suitable growth medium like Tryptic Soy Broth (TSB).
- Treatment Application: Gently wash the mature biofilm and treat it with the liposomal Cas9 formulation, using empty liposomes and free CRISPR/Cas9 as controls. A typical treatment concentration is 100 µg/mL of total lipid, incubated for 24 hours at 37°C [4].
- Biomass Quantification: Assess the remaining biofilm biomass using a crystal violet (CV) assay. Fix the biofilm with methanol, stain with 0.1% CV, solubilize the bound dye with acetic acid, and measure the absorbance at 595 nm. Calculate the percentage of biofilm reduction relative to the untreated control [4].
Protocol 2: Assessing Anti-Quorum Sensing Activity of Phenolic Acids
This protocol outlines the in silico and in vitro methods for evaluating the anti-quorum sensing (QS) potential of plant-derived phenolic acids like coumaric acid, as recently described [88].
I. In Silico Molecular Docking and Dynamics
- Protein Preparation: If experimental structures are unavailable, generate 3D models of key QS-associated proteins (e.g., LuxS) via homology modeling using servers like I-TASSER. Validate model quality with Ramachandran plots [88].
- Ligand Preparation: Obtain the 3D structure data files (SDF) of coumaric acid (PubChem ID: 637542) and syringic acid (PubChem ID: 10742) from PubChem. Perform energy minimization using software like UCSF Chimera [88].
- Molecular Docking: Perform docking studies to predict the binding affinity and interaction profile between the ligands and target proteins. Analyze the results to identify the strongest binding complexes based on binding energy (e.g., ΔGbind for LuxS-coumaric acid was -21.74 ± 3.01 kcal/mol) [88].
- Molecular Dynamics (MD) Simulation: Run 100 ns MD simulations (e.g., using GROMACS) for the top protein-ligand complexes and the "naked" protein. Analyze root mean square fluctuation (RMSF), radius of gyration (Rg), and hydrogen bonding to assess complex stability [88].
II. In Vitro Validation: MIC and Time-Kill Assay
- Minimum Inhibitory Concentration (MIC): Determine the MIC using the broth microdilution method according to CLSI guidelines. The reported MIC for coumaric acid against S. marcescens is 700 µg/mL [88].
- Time-Kill Assay: Inoculate broth with bacteria and treat with the phenolic acid at concentrations like 0.5x, 1x, and 2x MIC. Take aliquots at 0, 2, 4, 6, 8, 10, 12, 16, 20, 24, and 48 hours, perform serial dilutions, and plate them to count Colony Forming Units (CFU/mL). Plot log CFU/mL versus time to determine if the effect is bactericidal or bacteriostatic [88].
Protocol 3: Evaluating Phage-Liposome Formulations for Pulmonary Delivery
This protocol describes the encapsulation of bacteriophages in liposomes for inhalable therapy and the evaluation of their stability and cellular uptake, based on a 2025 study [92].
I. Phage Propagation and Liposome Encapsulation
- Phage Propagation: Propagate P. aeruginosa bacteriophages using the double-layer agar plaque assay. Mix the host bacteria with molten soft agar and pour it onto a hard agar base. After overnight incubation, harvest the phages by scraping the top agar layer, followed by centrifugation and filtration (0.22 µm) to obtain a sterile phage lysate [92].
- Phage Encapsulation in Liposomes: Prepare liposomes using the thin-film hydration method as described in Protocol 3.1, Section I. Hydrate the lipid film with the phage stock. Using a low phage titer for hydration can achieve favorable encapsulation efficiency (e.g., 58 ± 6.02%) with minimal loss in viability [92].
- Nebulization Stability: Assess the viability of both free phages and phage-liposomes after nebulization using an air-jet nebulizer (e.g., PARI-LC Plus). The loss in phage viability for liposomal formulations (1.08 ± 0.05 log) is significantly lower than for phage suspensions (1.55 ± 0.04 log), demonstrating the protective effect of encapsulation [92].
II. Cellular Uptake Assay in Lung Epithelial Model
- Cell Culture: Grow H441 lung epithelial cells at the air-liquid interface (ALI) to form a differentiated, polarized monolayer that mimics the lung epithelium.
- Treatment and Tracking: Apply fluorescently labeled (e.g., with SYBR Gold) free phages or phage-liposomes to the apical side of the model.
- Quantification: After incubation, use fluorescence microscopy, flow cytometry, or a plaque assay to quantify the number of phages associated with or internalized by the cells. Encapsulation in liposomes has been shown to enable a two-fold reduction in phage cellular uptake, leading to longer extracellular phage retention for antibacterial action [92].
Visual Experimental Workflows and Signaling Pathways
Workflow for Comparative Modality Evaluation
The following diagram illustrates the core experimental workflow for evaluating the four antimicrobial modalities discussed in this note.
Mechanism of Action Against Bacterial Biofilms
This diagram summarizes the primary molecular and cellular mechanisms employed by each modality to combat bacterial biofilms.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Featured Experiments
| Reagent / Kit | Primary Function | Example Application in Protocols |
|---|---|---|
| Phage DNA Isolation Kit (e.g., Norgen Biotek #46800) | Purification of high-quality, sequencing-ready viral DNA from phage lysates. | Essential for genomic characterization and safety screening of therapeutic phages (Prop. 3.3.I.1) [87]. |
| Rotary Evaporator System | Gentle removal of organic solvents to form thin, uniform lipid films. | Critical step in the preparation of liposomes for encapsulating Cas9 or phages (Prop. 3.1.I.2 & 3.3.I.2) [92]. |
| Simulated Lung Fluid (with DPPC) | Mimics the composition and surface properties of human lung lining fluid. | Used for in vitro stability and release studies of inhalable formulations like phage-liposomes [92]. |
| I-TASSER Web Server | Protein structure prediction via homology modeling. | Generating 3D models of quorum-sensing proteins for in silico docking studies (Prop. 3.2.I.1) [88]. |
| GROMACS Software | Performing molecular dynamics simulations. | Assessing the stability and interaction dynamics of protein-ligand complexes over 100 ns (Prop. 3.2.I.4) [88]. |
The escalating crisis of antimicrobial resistance, particularly in biofilm-associated infections caused by pathogens like Pseudomonas aeruginosa, demands a paradigm shift in therapeutic development. Liposomal Cas9 formulations represent a frontier in precision medicine, offering the potential to target and disrupt the genetic underpinnings of antibiotic resistance and biofilm resilience. The path from laboratory research to clinical application for these novel biologicals is complex, requiring sophisticated pre-clinical models and innovative clinical trial designs. This document outlines detailed protocols and application notes to guide researchers and drug development professionals in this endeavor, framed within the context of advancing liposomal Cas9 therapies for P. aeruginosa biofilms.
The development of pre-clinical models must be informed by the performance metrics of existing and emerging technologies. The table below summarizes key quantitative data from recent studies on anti-biofilm strategies, providing benchmarks for evaluating novel liposomal Cas9 formulations.
Table 1: Quantitative Efficacy of Emerging Anti-Biofilm and Antimicrobial Strategies
| Strategy | Key Metric | Performance | Model System | Citation |
|---|---|---|---|---|
| Liposomal CRISPR-Cas9 | Reduction in P. aeruginosa biofilm biomass | >90% reduction in vitro | In vitro biofilm model | [4] |
| Gold Nanoparticle CRISPR Delivery | Gene-editing efficiency enhancement | 3.5-fold increase vs. non-carrier systems | In vitro bacterial culture | [4] |
| Glycoside Hydrolase Debridement | Biofilm dispersal (dose-dependent) | Comparable or superior to papain, bromelain, collagenase at ≥1% doses | In vitro and ex vivo P. aeruginosa biofilm models | [93] |
| CRISPR-engineered Bacteriophage (crPhage) | Target pathogen clearance | Significant clearance in animal models | Animal infection models | [22] |
| Conjugative Plasmid CRISPR Delivery | Selective eradication of resistant E. coli | Effective reduction in resistant populations within mixed communities | Mixed microbial communities in vitro | [22] |
The Scientist's Toolkit: Key Research Reagent Solutions
The following table catalogues essential materials and reagents critical for conducting research on liposomal Cas9 formulations against bacterial biofilms.
Table 2: Essential Research Reagents for Liposomal Cas9 Anti-Biofilm Research
| Reagent/Material | Function/Application | Examples & Notes |
|---|---|---|
| CRISPR-Cas9 System | Core gene-editing machinery for targeting resistance or virulence genes. | Cas9 nuclease, guide RNAs (gRNAs) designed against targets like β-lactamase genes, efflux pump regulators, or quorum-sensing systems. |
| Lipid Nanoparticles (LNPs) | Delivery vehicle for CRISPR components; offers affinity for specific tissues and potential for re-dosing. | Composed of ionizable lipids, phospholipids, cholesterol, and PEG-lipids; known to accumulate in the liver in systemic delivery. |
| Engineered Bacteriophages | Biological delivery vector for CRISPR payloads to specific bacterial pathogens. | Lytic or temperate phages engineered to be non-lytic and carry Cas9 and gRNA constructs; offer high specificity. |
| 3D Airway Organ Tissue Equivalents (OTEs) | Pre-clinical in vitro model that recapitulates human airway physiology, including for Cystic Fibrosis. | Fabricated from human primary cells from CF donors (CF-OTEs); supports robust P. aeruginosa biofilm formation for therapeutic screening. |
| Glycoside Hydrolase Cocktail | Biofilm dispersal agent used in combination studies to enhance penetration of antimicrobials. | A blend of alpha-amylase and cellulase; used as a pre-treatment to disrupt the extracellular polymeric substance (EPS) matrix. |
Experimental Protocols for Pre-clinical Development
Protocol: Assessment of Liposomal Cas9 Efficacy in a 3D CF Airway Model
Background: Traditional 2D cell cultures fail to capture the complexity of the in vivo airway environment, particularly in Cystic Fibrosis. The 3D Airway Organ Tissue Equivalent (OTE) model, which accurately recapitulates key traits of human CF airways, provides a robust platform for evaluating anti-biofilm therapies under physiologically relevant conditions [94].
Application Note: This protocol is designed to test the efficacy and initial safety of liposomal Cas9 formulations against P. aeruginosa biofilms established on a CF-relevant tissue model. It serves as a critical bridge between standard in vitro assays and in vivo animal studies.
Materials:
- CF-OTEs (commercially sourced or fabricated in-house from CF patient-derived cells).
- Mucoid or non-mucoid strain of P. aeruginosa (e.g., PAO1 or a clinical isolate).
- Liposomal Cas9 formulation (e.g., LNP encapsulating Cas9 mRNA and sgRNA targeting a predetermined resistance gene).
- Control formulations: Empty LNPs, scrambled sgRNA LNPs, and a standard-of-care antibiotic.
- Cell culture media specific for OTEs.
- Luminometer and supplies for ATP-based viability assays (e.g., CellTiter-Glo 3D).
- Materials for viable bacterial enumeration (homogenizer, serial dilution, agar plates).
- Histological staining kits (e.g., H&E, Alcian Blue/PAS for mucin).
Methodology:
- Biofilm Establishment:
- Culture P. aeruginosa to mid-log phase.
- Inoculate the apical surface of CF-OTEs with a standardized inoculum (e.g., 1x10^6 CFU) in a small volume of media.
- Allow for bacterial adhesion during a 2-hour incubation at 37°C, 5% CO2.
- Carefully wash the apical surface with sterile PBS to remove non-adherent bacteria.
- Add fresh media to the basolateral compartment and incubate for 24-48 hours to allow for mature biofilm formation.
Therapeutic Intervention:
- Randomize OTEs with established biofilms into treatment groups:
- Group 1: Liposomal Cas9 formulation (multiple dose levels recommended).
- Group 2: Empty LNP control (vehicle).
- Group 3: Scrambled sgRNA LNP control (editing control).
- Group 4: Standard-of-care antibiotic (e.g., tobramycin).
- Group 5: Untreated infected control.
- Apply treatments to the apical surface of OTEs in a small, defined volume. Incubate as per experimental design (e.g., single dose with 24-72 hour follow-up, or multiple doses).
- Randomize OTEs with established biofilms into treatment groups:
Efficacy and Safety Analysis:
- Bacterial Load Quantification: Post-treatment, homogenize the entire OTE. Perform serial dilutions and plate for viable bacterial enumeration (CFU/organoid).
- Biofilm Biomass Assessment: Use luminescence-based ATP assays on homogenates or specific biofilm staining (e.g., crystal violet) on the OTE surface to quantify total biofilm biomass.
- Host Cell Viability: Assess OTE cell viability using a 3D-optimized ATP assay (e.g., CellTiter-Glo 3D) to determine treatment-related toxicity.
- Histopathological Examination: Fix OTEs in formalin, process for histology, and section. Stain with H&E to assess tissue architecture and inflammation, and with Alcian Blue/PAS to visualize mucin content and distribution.
Diagram: Experimental Workflow for 3D OTE Model
Protocol: Combination Therapy with Biofilm Dispersal Agents
Background: The extracellular polymeric substance (EPS) of biofilms is a major barrier to the penetration of therapeutics. Combining liposomal Cas9 with a biofilm dispersal agent can synergistically enhance efficacy by improving access to the bacterial cells within the biofilm [93].
Application Note: This protocol outlines a method to pre-treat established biofilms with a glycoside hydrolase cocktail to disrupt the EPS matrix prior to the application of liposomal Cas9, mimicking a potential clinical debridement strategy.
Materials:
- In vitro biofilm model (e.g., 96-well plate assay or CDC biofilm reactor).
- Glycoside Hydrolase cocktail (alpha-amylase and cellulase, ≥1% final concentration) [93].
- Liposomal Cas9 formulation.
- PBS buffer.
- Microtiter plate reader or other relevant quantification equipment.
Methodology:
- Biofilm Formation: Grow P. aeruginosa biofilms in a standardized in vitro system for 24-48 hours.
- Dispersal Agent Pre-treatment: Carefully aspirate the planktonic culture. Add the glycoside hydrolase cocktail (in an appropriate buffer) to the established biofilms. Incubate for a predetermined time (e.g., 1-2 hours) at 37°C.
- Therapeutic Application: Remove the dispersal agent and wash gently with PBS. Immediately add the liposomal Cas9 formulation to the pre-treated biofilms.
- Analysis: After incubation with the liposomal Cas9, assess outcomes:
- Biofilm Disruption: Quantify remaining biofilm biomass via crystal violet staining or ATP measurement.
- Genetic Editing Efficiency: Extract genomic DNA from the biofilm and use PCR/digestion-based assays (e.g., T7E1) or next-generation sequencing to quantify indel frequencies at the target locus.
- Bacterial Killing: Perform CFU counts to determine the reduction in viable bacteria compared to controls (e.g., dispersal agent alone, Cas9 alone, untreated).
Signaling Pathways in Biofilm-Immune Interaction and Therapeutic Targeting
Understanding the host-pathogen interface is critical for designing effective therapies. The following diagram summarizes key immune evasion strategies of P. aeruginosa biofilms, highlighting potential intervention points for CRISPR-based strategies, such as targeting the quorum-sensing (QS) system that regulates rhamnolipid production [95].
Diagram: P. aeruginosa Biofilm Immune Evasion and CRISPR Targets
Considerations for Clinical Trial Design
Transitioning liposomal Cas9 from pre-clinical models to human trials requires a novel and adaptive design framework.
- Phase I Focus (Safety & Delivery): Initial human trials must prioritize safety. A key consideration is the delivery platform; lipid nanoparticles (LNPs) do not trigger the same immune responses as viral vectors, allowing for the possibility of re-dosing, which has been demonstrated in recent clinical trials [96]. Phase I trials should therefore explore both single and multiple ascending doses to establish a safety profile and define the pharmacokinetics/pharmacodynamics of the formulation.
- Biomarker-Driven Endpoints: Given the complexity of biofilm infections, trials should incorporate robust biomarkers. For liposomal Cas9, this includes:
- Pharmacodynamic Biomarkers: Reduction in specific bacterial resistance gene load in patient samples (e.g., sputum from CF patients) using qPCR or next-generation sequencing.
- Clinical Biomarkers: Reduction in inflammatory markers (e.g., C-reactive protein) and frequency of acute exacerbations (in CF or chronic wound settings).
- Patient Population Selection: Initial trials should focus on well-characterized patient populations with high, unmet need, such as CF patients with chronic, multidrug-resistant P. aeruginosa lung infections, or patients with complex, non-healing wound infections. These populations provide a clear context for evaluating targeted anti-biofilm therapy.
- Combination Therapy Arms: Reflecting the synergistic protocols from pre-clinical development, clinical trials should include arms where liposomal Cas9 is administered following standard-of-care debridement (surgical or enzymatic) or in conjunction with last-resort antibiotics, to assess combination efficacy in a clinical setting [93].
Conclusion
Liposomal CRISPR-Cas9 formulations represent a paradigm shift in combating biofilm-associated infections, moving from broad-spectrum cytotoxicity to precision genetic targeting. The synthesis of research confirms the potent ability of these hybrid systems to disrupt biofilm integrity and resensitize Pseudomonas aeruginosa to conventional antibiotics, demonstrating over 90% biomass reduction in vitro. However, the journey from bench to bedside requires overcoming significant challenges in delivery efficiency, specificity, and safety profiling. Future research must prioritize the development of advanced lipid nanoparticles with enhanced biofilm-penetrating capabilities, rigorous in vivo validation of efficacy and toxicity, and exploration of personalized gRNA cocktails targeting patient-specific bacterial isolates. Success in this field holds the promise of creating a powerful, next-generation arsenal against some of the most recalcitrant infections faced in modern medicine.
References
- 1. Pseudomonas aeruginosa Biofilms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7698413/]
- 2. Treatment of Pseudomonas aeruginosa infectious biofilms [https://pmc.ncbi.nlm.nih.gov/articles/PMC9459144/]
- 3. Treatment of Pseudomonas aeruginosa infectious biofilms [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.955286/full]
- 4. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12366227/]
- 5. Pseudomonas aeruginosa biofilm: treatment strategies to ... [https://pubmed.ncbi.nlm.nih.gov/40348909/]
- 6. Matrix matters: How extracellular substances shape biofilm ... [https://www.sciencedirect.com/science/article/abs/pii/S0927776524006003]
- 7. Mechanisms of antimicrobial resistance in biofilms [https://www.nature.com/articles/s44259-024-00046-3]
- 8. Multidrug-Resistant Biofilms (MDR): Main Mechanisms of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9784232/]
- 9. Biofilms and multidrug resistance: an emerging crisis and the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12237878/]
- 10. Extracellular polymeric substances, a key element in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6604936/]
- 11. Biofilm matrix and its regulation in Pseudomonas aeruginosa [https://pubmed.ncbi.nlm.nih.gov/24145749/]
- 12. Surface-layer protein is a public-good matrix exopolymer ... [https://www.nature.com/articles/s41396-023-01388-y]
- 13. Innovative approaches to combat antibiotic resistance [https://pubmed.ncbi.nlm.nih.gov/40830872/]
- 14. Mechanisms of antibiotic resistance in Pseudomonas ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10189392/]
- 15. Role of efflux pumps in the antibiotic resistance of bacteria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3711980/]
- 16. Biofilm formation and antibiotic resistance in Pseudomonas ... [https://www.sciencedirect.com/science/article/pii/S2950194624000451]
- 17. Efflux pumps as potential targets for biofilm inhibition [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1315238/full]
- 18. Bacterial Persister Cell Formation and Dormancy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3837759/]
- 19. Bacterial persisters: molecular mechanisms and ... [https://www.nature.com/articles/s41392-024-01866-5]
- 20. Are Bacterial Persisters Dormant Cells Only? [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.708580/full]
- 21. Directed evolution of phages in biofilms enhances ... [https://www.nature.com/articles/s41467-025-65014-5]
- 22. CRISPR-Cas Systems: Transformative Precision Tools to ... [https://www.genesispub.org/crispr-cas-systems-transformative-precision-tools-to-combat-antimicrobial-resistance-in-multidrug-resistant-gram-negative-pathogens]
- 23. CRISPR/Cas9-based Genome Editing in Pseudomonas ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6137401/]
- 24. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://bmcmedicine.biomedcentral.com/articles/10.1186/s12916-025-04323-4]
- 25. CRISPR control of virulence in Pseudomonas aeruginosa [https://www.nature.com/articles/cr20176]
- 26. CRISPR–Cas systems as emerging tools for precision ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925021416]
- 27. Development of the CRISPR-Cas12a system for editing ... [https://www.sciencedirect.com/science/article/pii/S2589004224014354]
- 28. Pseudomonas aeruginosa: pathogenesis, virulence ... [https://www.nature.com/articles/s41392-022-01056-1]
- 29. Liposomes-Based Drug Delivery Systems of Anti-Biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10215698/]
- 30. Advances in Liposomal Drug Delivery - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298344/]
- 31. Beyond antibiotics: CRISPR/Cas9 triumph over biofilm- ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2024.1408569/full]
- 32. Strategies in the delivery of Cas9 ribonucleoprotein for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7738854/]
- 33. Liposome-Based Carriers for CRISPR Genome Editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC10454197/]
- 34. Design of Liposome Formulations for CRISPR/Cas9 Enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10702188/]
- 35. Current Knowledge on CRISPR Strategies Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11672446/]
- 36. In silico sgRNA tool design for CRISPR control of quorum ... [https://www.sciencedirect.com/science/article/pii/S2352304218300564]
- 37. The effect of Quorum sensing inhibitors on the evolution of ... [https://www.nature.com/articles/s41396-021-00946-6]
- 38. Current Trends in Development of Liposomes for Targeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4932481/]
- 39. PEG 2000 -DBCO surface coating increases intracellular ... [https://www.nature.com/articles/s41598-022-14947-8]
- 40. Surface Functionalization and Targeting Strategies of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5796144/]
- 41. A Guideline for Assessment and Characterization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10632153/]
- 42. Quantitative and Qualitative Assessment Methods for Biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6133255/]
- 43. Combining biofilm matrix measurements with biomass and ... [https://www.nature.com/articles/ja201249]
- 44. Compounds and Methods to Resensitize Antibiotic- ... [https://mjima.org/articles/compounds-and-methods-to-resensitize-antibiotic-resistant-bacteria/mjima.galenos.2021.2021.46]
- 45. Combination drug strategies for biofilm eradication using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10149696/]
- 46. High throughput determination of the biofilm prevention ... [https://www.sciencedirect.com/science/article/pii/S2590207523000035]
- 47. The Application of the CRISPR-Cas System in Antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9356603/]
- 48. Bacterial resistance to CRISPR-Cas antimicrobials [https://www.nature.com/articles/s41598-021-96735-4]
- 49. High-Throughput Approaches for the Identification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8262948/]
- 50. Liposomal drug delivery strategies to eradicate bacterial ... [https://pubmed.ncbi.nlm.nih.gov/38554739/]
- 51. Frontiers | Off - target in CRISPR/ effects gene editing Cas 9 [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2023.1143157/full]
- 52. CRISPR/ Cas therapeutics: progress and prospects 9 [https://www.nature.com/articles/s41392-023-01309-7?error=cookies_not_supported]
- 53. Antimicrobial and Anti-Biofilm Activity of Dichlorophen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12396535/]
- 54. Eco‑friendly biosynthesis of gold nanoparticles from ... [https://www.nature.com/articles/s41598-025-11838-6]
- 55. Treatment of S. epidermidis biofilms with targeted ... [https://www.sciencedirect.com/science/article/pii/S0021979725026062]
- 56. silver- Hybrid suppress drug resistant... gold nanoparticles [https://pubmed.ncbi.nlm.nih.gov/32285904/]
- 57. Nanoparticle Delivery of CRISPR/Cas9 for Genome Editing [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.673286/full]
- 58. Nanoparticle based delivery of CRISPR/Cas9 genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6398936/]
- 59. Immune dysfunction during S. aureus biofilm-associated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12297603/]
- 60. Editorial: Immune Response to Biofilms [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.696356/full]
- 61. Implant infections: adhesion, biofilm formation and ... [https://www.nature.com/articles/s41579-018-0019-y]
- 62. Pathogens have evolved various means of evading or ... - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK27176/]
- 63. Microbial Ninja Warriors: Bacterial Immune Evasion [https://asm.org/articles/2018/december/microbial-ninja-warriors-bacterial-immune-evasion]
- 64. Pseudomonas aeruginosa in the Frontline of the Greatest ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11596597/]
- 65. Overcoming the Delivery Challenges in CRISPR/Cas9 Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12434829/]
- 66. Biomaterials-mediated CRISPR/Cas9 delivery: recent ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2023.1259435/full]
- 67. Effect of daphnetin combined with tobramycin on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12370681/]
- 68. Targeting Pseudomonas aeruginosa biofilm with an ... [https://www.nature.com/articles/s41467-024-52595-w]
- 69. CRISPR-Cas9 Gene Therapy: Non-Viral Delivery and Stimuli ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11820414/]
- 70. CRISPR/Cas9 gene editing strategy for cancer therapy [https://www.sciencedirect.com/science/article/abs/pii/S0141813025089469]
- 71. A targeted and synergetic nano-delivery system against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11772151/]
- 72. Optimization of an in vitro Pseudomonas aeruginosa Biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10907394/]
- 73. Mechanism of antibacterial resistance, strategies and next- ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1444781/full]
- 74. Novel Opportunity to Reverse Antibiotic Resistance [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.610070/full]
- 75. Overcoming Intrinsic Resistance in Gram-negative Bacteria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9885958/]
- 76. Nanoparticles Targeting Biofilms: A New Era in Combating ... [https://www.sciencedirect.com/science/article/pii/S2590097825000370]
- 77. Liposome-encapsulated antibiotics and biosurfactants [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02746-5]
- 78. Metallic and metal oxide nanoparticles in treating ... [https://www.sciencedirect.com/science/article/abs/pii/S1773224723011425]
- 79. Liposome encapsulated surfactant abetted copper ... [https://www.nature.com/articles/s41598-020-79976-7]
- 80. Biofilm inhibition of multidrug-resistant Pseudomonas ... [https://www.nature.com/articles/s41598-025-00005-6]
- 81. Impeding microbial biofilm formation and Pseudomonas ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02508-9]
- 82. The effects of surface properties of liposomes on their ... [https://www.sciencedirect.com/science/article/pii/S1773224720303646]
- 83. Inhibition of Pseudomonas aeruginosa Biofilm Formation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11847160/]
- 84. Antimicrobial resistance (AMR) management using ... [https://www.sciencedirect.com/science/article/pii/S2666388024000017]
- 85. Phage therapy as a revitalized weapon for treating clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12540056/]
- 86. Mini review advantages and limitations of lytic phages ... [https://www.sciencedirect.com/science/article/pii/S2405844024108808]
- 87. Phage Therapy: The Future of Fighting Antibiotic Resistance [https://norgenbiotek.com/blog/phage-therapy-future-fighting-antibiotic-resistance?srsltid=AfmBOorRqO7_EZVNzgjinwo3lJF2MxivZYwkf-6i9pDai_OYtrLPP1bn]
- 88. Mechanistic In-Silico Insights into the Anti-quorum Sensing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12594671/]
- 89. Natural product-based inhibitors of quorum sensing [https://www.sciencedirect.com/science/article/pii/S2405580825001980]
- 90. Antimicrobial peptides: structure, functions and ... [https://www.nature.com/articles/s41579-025-01200-y]
- 91. Multidrug-Resistant Microbial Therapy Using Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9379109/]
- 92. Development of Inhalable Bacteriophage Liposomes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12030023/]
- 93. Biofilm Dispersal and Wound Infection Clearance With ... [https://pubmed.ncbi.nlm.nih.gov/40069969/]
- 94. Farnesol emulsion for elimination of Pseudomonas ... [https://www.sciencedirect.com/science/article/pii/S2590207525000656]
- 95. Immune system dynamics in response to Pseudomonas ... [https://www.nature.com/articles/s41522-025-00738-2]
- 96. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]