Microbial Biofilm Formation: From Initial Attachment to Mature Structure and Therapeutic Targeting
This article provides a comprehensive analysis of the multi-stage process of microbial biofilm formation, a critical factor in chronic infections and antimicrobial resistance.
Microbial Biofilm Formation: From Initial Attachment to Mature Structure and Therapeutic Targeting
Abstract
This article provides a comprehensive analysis of the multi-stage process of microbial biofilm formation, a critical factor in chronic infections and antimicrobial resistance. Tailored for researchers, scientists, and drug development professionals, it synthesizes foundational knowledge on biofilm life cycles with advanced methodological approaches for study and disruption. The content explores the molecular mechanisms governing attachment, maturation, and dispersion; details cutting-edge analytical and computational models; evaluates challenges in biofilm eradication; and reviews innovative therapeutic strategies. By integrating foundational science with applied research, this review serves as a strategic resource for developing novel anti-biofilm interventions.
The Biofilm Lifecycle: Deconstructing the Stages of Microbial Community Development
Biofilms are complex, structured communities of microbial cells that are attached to a surface and embedded in a self-produced matrix of Extracellular Polymeric Substances (EPS) [1] [2]. This EPS matrix is a polymeric conglomeration typically composed of extracellular polysaccharides, proteins, lipids, and extracellular DNA (eDNA) [3] [2] [4]. Biofilms represent a fundamental mode of growth for microorganisms and are characterized by a unique, coordinated community lifestyle that is physiologically distinct from free-floating (planktonic) cells [5] [4].
The defining characteristics of biofilms include their three-dimensional architecture, the presence of an EPS matrix, and the emergence of community-level properties such as enhanced resistance to antimicrobial agents and environmental stresses [6] [7]. Biofilms are ubiquitous in nature and can form on both biotic (living) and abiotic (non-living) surfaces, making them significant in diverse environments from natural ecosystems to industrial settings and healthcare facilities [1] [4].
Table: Key Components of the Biofilm Extracellular Polymeric Substance (EPS) Matrix
| Component | Primary Functions | Clinical/Research Significance |
|---|---|---|
| Exopolysaccharides | Structural scaffold, adhesion, cohesion, water retention [1] [3] | Major barrier to antibiotic penetration [2] |
| Proteins | Structural support, enzymatic activities, adhesion [2] | Facilitate nutrient acquisition and matrix stability [2] |
| Extracellular DNA (eDNA) | Structural integrity, genetic information exchange [2] [4] | Contributes to antibiotic resistance spread; target for disruption (e.g., DNase) [2] |
| Lipids | Hydrophobicity, barrier functions [2] | Less characterized, but contributes to matrix complexity [2] |
Historical Context of Biofilm Research
The study of biofilms has undergone a significant evolution over the past several decades. The contemporary paradigm of biofilm microbiology began to take shape in the late 1970s, largely catalyzed by the observations of J. William Costerton, a Canadian microbiologist who played a pivotal role in changing the view that microorganisms exist primarily as individual planktonic cells [3]. Costerton's work emphasized that microbial aggregates, which he termed biofilms, represent a primary mode of bacterial growth in many environments [3].
Early biofilm research relied heavily on techniques such as microscopy and culturing to investigate the basic structure and composition of biofilms [1]. A significant milestone was reached in 2004 when biofilms were conceptually identified as the root cause of non-healing and long-term infections in the majority of chronic wounds [3]. This hypothesis was strengthened in 2008 by clinical evidence from James and colleagues, who demonstrated that 60% of chronic wounds contained biofilms [3]. Subsequent studies have further solidified this connection, with research in 2012 showing that more than 80% of surgical site infections (SSIs) involve biofilm development [3].
The field has progressively advanced with the integration of molecular biology techniques and sophisticated imaging technologies, such as Confocal Scanning Laser Microscopy (CSLM) and advanced Scanning Electron Microscopy (SEM), allowing researchers to study the complex architecture and behavior of biofilms in unprecedented detail [1] [6] [5]. This historical progression from observational studies to mechanistic and molecular investigations has fundamentally transformed our understanding of microbial biology and pathogenesis.
Clinical Significance of Biofilms
Biofilms are implicated in a substantial proportion of human microbial infections, particularly those that are chronic and recurrent in nature [2] [8]. It is estimated that 65-80% of all microbial infections are associated with biofilm formation [2] [8]. The clinical significance of biofilms stems primarily from their inherent tolerance and resistance to conventional antimicrobial therapies and host immune defenses [2]. Microorganisms within a biofilm can be up to 1,000 times more resistant to antibiotics than their planktonic counterparts [2] [7]. This recalcitrance makes biofilm-associated infections notoriously difficult to treat and eradicate, leading to persistent infections, increased morbidity, and significant healthcare costs [3] [2].
Table: Clinical Impact of Biofilms in Healthcare-Associated Infections (HAIs)
| Clinical Context | Key Biofilm-Related Challenges | Prevalence & Economic Impact |
|---|---|---|
| Chronic Wounds (e.g., Diabetic Foot Ulcers, Venous Leg Ulcers) | Delay healing, induce chronic inflammation, polymicrobial interactions [3] [2]. | ~80% of DFUs before amputation; U.S. cost >$50 billion annually for chronic wound management [3]. |
| Medical Device-Associated Infections (e.g., Catheters, Implants) | Formation on abiotic surfaces, source of recurrent bloodstream infections [2] [9]. | CAUTI is a common HAI; Biofilms contribute to ~65% of microbial infections [2] [9]. |
| Cystic Fibrosis Lung Infections | P. aeruginosa biofilms in lungs resist clearance and antibiotics [2] [5]. | Major cause of morbidity and mortality; resistance enabled by EPS matrix [2]. |
| Surgical Site Infections (SSIs) | Biofilms form on tissues or implanted medical devices [3]. | >80% of SSIs develop biofilms [3]. |
The economic burden of Healthcare-Associated Infections (HAIs), to which biofilms are a major contributor, is staggering. In the United States alone, HAIs account for over 88,000 fatalities annually, with an estimated economic burden of USD 4.5 billion [2]. The complex structure of the EPS matrix restricts the penetration of antimicrobial agents, while the heterogeneous microenvironment within biofilms leads to the presence of metabolically dormant "persister cells" that are highly tolerant to antibiotics [2]. Furthermore, the close proximity of cells within the biofilm facilitates horizontal gene transfer, accelerating the spread of antimicrobial resistance genes among bacterial populations [3] [2].
Mechanisms of Biofilm Formation
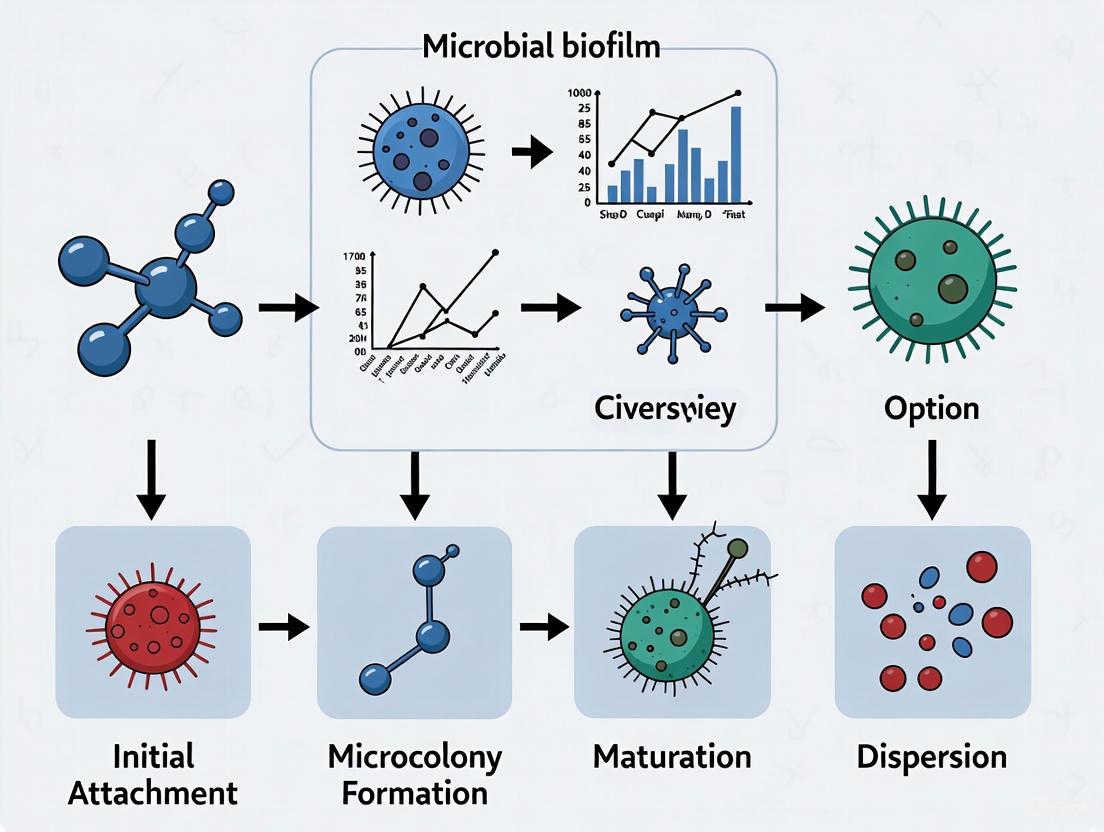
The development of a biofilm is a dynamic and cyclic process that occurs in several distinct, sequential stages [1] [3] [4]. Understanding these mechanisms is crucial for developing targeted anti-biofilm strategies.
Diagram: The Five Key Stages of Biofilm Development. The process begins with the attachment of free-floating planktonic cells and progresses through irreversible attachment, microcolony formation, maturation of a three-dimensional structure, and active dispersal, which can seed new biofilm sites [1] [3] [4].
Initial Attachment: The first stage involves the reversible attachment of planktonic microorganisms to a surface, mediated by weak physical forces such as van der Waals forces and hydrophobic interactions [4]. This attachment can be influenced by environmental cues such as nutrient availability and surface properties [8].
Irreversible Attachment and EPS Production: Once attached, cells begin to produce EPS, leading to a firm, irreversible attachment. Adhesive structures such as pili and fimbriae play a critical role in this stabilization [4]. The cells start to form a monolayer and begin coordinated cell-to-cell communication [1].
Early Development and Microcolony Formation: The attached cells proliferate and form microcolonies, which are the foundational units of the mature biofilm. The production of EPS increases significantly, creating a protective microenvironment [3].
Maturation: The biofilm develops a complex, three-dimensional architecture characterized by tower-like structures and mushroom-shaped microcolonies interspersed with a network of water channels that facilitate the distribution of nutrients, oxygen, and signaling molecules, while also removing waste products [1] [5]. This stage is regulated by a density-dependent communication system known as Quorum Sensing (QS), which allows bacterial cells to coordinate gene expression based on population density [5] [7].
Dispersal: This final stage is an active process where individual cells or small clusters detach from the mature biofilm to colonize new surfaces [1] [4]. Dispersal can be triggered by various factors including nutrient depletion, oxygen limitation, and signaling molecules such as nitric oxide or cis-2-decenoic acid [4]. Enzymes that degrade the EPS matrix, such as dispersin B and deoxyribonuclease, facilitate this process [4].
Research Methodologies and Experimental Protocols
The study of biofilms requires specialized methodologies for quantification and visualization, as traditional microbiological techniques designed for planktonic cells are often inadequate [6] [9]. A combination of quantitative and qualitative methods is typically employed to fully characterize biofilm formation, structure, and composition.
Quantitative Biofilm Assessment Methods
Various direct and indirect methods are used to quantify biomass and viable cells within a biofilm [6].
Table: Common Quantitative Methods for Biofilm Analysis [6]
| Method | Principle | Key Applications & Advantages | Limitations |
|---|---|---|---|
| Colony Forming Units (CFU) | Viable cells are dislodged, serially diluted, plated on agar, and counted after incubation [6]. | Gold standard for enumerating viable cells; differentiates live from dead [6]. | Time-consuming (24-72 hrs); labor-intensive; susceptible to cell clumping errors [6]. |
| Crystal Violet (CV) Staining | A dye that binds to proteins and polysaccharides is used to stain total biofilm biomass, which is then solubilized and measured spectrophotometrically [6]. | High-throughput; measures total adhered biomass (live and dead); inexpensive [6]. | Does not distinguish between live and dead cells; can be influenced by EPS composition [6]. |
| ATP Bioluminescence | Measures ATP from metabolically active cells using a luciferin-luciferase reaction that produces light [6]. | Very rapid (minutes); highly sensitive; correlates with viable cell count [6]. | Signal can be affected by environmental conditions and sample matrix [6]. |
| Quartz Crystal Microbalance (QCM) | Measures mass change (ng/cm²) per unit area by detecting the change in oscillation frequency of a crystal upon biofilm attachment [6]. | Real-time, label-free monitoring of initial attachment and growth [6]. | Requires specialized equipment; measures total mass including non-cellular material [6]. |
A Standardized Protocol for Biofilm Extraction from Medical Devices
The following detailed protocol, optimized for urinary catheters but adaptable to other devices, combines vortexing and sonication to achieve consistent and reproducible extraction of biofilm bacteria for subsequent quantification [9].
Objective: To effectively dislodge and homogenize biofilm bacteria from the surface of an indwelling medical device for accurate quantification via CFU or other methods [9].
Materials:
- Sterile phosphate-buffered saline (PBS)
- Sonicating water bath (with calibrated power output)
- Vortex mixer
- Sterile containers
- Equipment for serial dilution and plating
Diagram: Experimental Workflow for Biofilm Extraction. This standardized protocol using sequential vortexing and sonication steps ensures effective and reproducible recovery of biofilm bacteria from complex surfaces like catheters [9].
Procedure [9]:
- Sample Collection and Preparation: Aseptically remove the medical device (e.g., catheter). Cut into segments of appropriate size (e.g., 1 cm) using sterile instruments.
- Initial Rinse: Gently dip or rinse each segment in 5 ml of sterile PBS to remove non-adherent planktonic cells. Remove residual liquid by gently tapping the segment on sterile absorbent paper.
- Initial Vortexing: Place each segment in a tube containing a known volume of fresh PBS. Vortex the tube for 30 seconds. This step helps to dislodge loosely attached biofilm layers.
- Sonication: Subject the tube to sonication in a sonic water bath for 5-10 minutes at a frequency of 40-45 kHz. This application of sound energy disrupts the strong bonds between the deeply embedded cells and the surface and helps break apart the EPS matrix. Note: Optimization of sonication time and power may be necessary for different materials and biofilm densities to balance yield against potential cell lysis.
- Final Vortexing: Remove the tube from the sonicator and vortex again for 30 seconds. This final vortexing step helps to break down the dislodged biofilm material into a more homogenous suspension of individual cells or small clusters, which is critical for accurate quantification.
- Quantification: The resulting homogenized suspension can now be used for downstream analyses, such as performing serial dilutions for CFU enumeration, ATP bioluminescence, or total protein assays [6] [9].
Key Research Reagent Solutions
Table: Essential Materials and Reagents for Biofilm Research
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| Crystal Violet Stain | Total biofilm biomass quantification [6]. | Staining 96-well plate biofilms for high-throughput screening of anti-biofilm compounds [6]. |
| SYTO9/Propidium Iodide (PI) | Fluorescent live/dead cell viability staining [6]. | Confocal microscopy analysis to visualize spatial distribution of live/dead cells within a biofilm's 3D structure [6]. |
| Dispersin B & DNase I | Enzymatic biofilm disruption agents targeting polysaccharide and eDNA matrix components [2] [4]. | Evaluating biofilm dispersal efficacy; potential adjunctive therapeutic agents [2] [4]. |
| Calcein-AM / TMA-DPH | Alternative fluorescent viability stain and biomass probe [10]. | Assessing biofilm viability and residual biomass after antibacterial treatment [10]. |
| N-acyl Homoserine Lactones (AHLs) | Key signaling molecules in Gram-negative bacterial Quorum Sensing [5]. | Studying QS inhibition as an anti-virulence strategy; elucidating biofilm regulation pathways [5] [7]. |
| CLED / CHROMagar | Specialized culture media that prevent swarming and allow preliminary species identification [9]. | Culturing and identifying uropathogens from polymicrobial biofilm samples, such as from catheters [9]. |
Within the meticulously coordinated process of microbial biofilm formation, the initial stage of reversible attachment is a critical determinant for subsequent community development. This phase represents a transient yet essential period where planktonic microorganisms first engage with a surface, a process mediated by weak physical forces and profoundly influenced by the surface's physicochemical properties [11]. Framed within the broader thesis of biofilm research, understanding this inaugural stage is paramount, as it presents a strategic window for intervention to prevent the establishment of resilient, antibiotic-resistant communities [10] [11]. This guide provides an in-depth technical examination of the mechanisms governing reversible attachment, equipping researchers with the quantitative data and methodologies necessary to advance therapeutic and industrial applications in biofilm management.
Reversible attachment constitutes the first step in the multi-stage biofilm formation process, which progresses through attachment, microcolony formation, maturation, and dispersion [11]. During this initial phase, planktonic cells make initial contact with a surface but have not yet committed to a sessile lifestyle. The attachment is termed "reversible" because the cells can readily detach and return to their planktonic state, as the forces tethering them to the surface are weak and non-specific [12].
The primary forces responsible for this initial tethering are weak physicochemical interactions, including:
- Hydrophobic interactions
- Van der Waals forces
- Electrostatic interactions [12]
These forces collectively enable a provisional, low-affinity association between the microbial cell surface and the substrate. The success of this attachment is not solely dictated by microbial factors but is equally governed by surface preconditioning. This phenomenon involves the spontaneous formation of a conditioning film composed of organic molecules (e.g., proteins, polysaccharides) present in the surrounding environment, which effectively modifies the surface chemistry and topography before microbial arrival [11].
Concurrently, at the intracellular level, a crucial surface-sensing mechanism is activated. The Pil-Chp surface-sensing system detects contact with a surface and triggers an increase in the intracellular concentration of the key secondary messenger, bis-(3'-5')-cyclic dimeric guanosine monophosphate (c-di-GMP) [11]. This molecular switch promotes the transition from motility to adhesion by repressing flagella synthesis and stimulating the production of biofilm matrix components, thereby setting the stage for the transition to irreversible attachment [11].
Diagram: The molecular and physical progression from planktonic cells to irreversible attachment. The process initiates with weak forces and surface preconditioning, leading to surface sensing and increased c-di-GMP, which promotes irreversible attachment.
Quantitative Data and Surface Properties
The outcome of reversible attachment is highly dependent on the physicochemical properties of the surface. Research has demonstrated that parameters such as hydrophobicity, surface energy, and functional groups can dramatically alter attachment kinetics and microbial behavior.
Table 1: Impact of Surface Chemistry on Bacterial Attachment
| Surface Treatment | Chemical Character | Water Contact Angle (°) | Observed Biofilm Phenotype (Pantoea sp. YR343) | Key Experimental Findings |
|---|---|---|---|---|
| PFOTS | Hydrophobic | High (Not specified) | "Honeycomb" morphology | Robust biofilm propagation with logarithmic growth over time [13] |
| OTS | Hydrophobic | High (Not specified) | Conducive to biofilm formation | Used in vapor deposition for creating hydrophobic surfaces [13] |
| APTMS | Hydrophilic | Low (Not specified) | Minimal attachment | Pantoea sp. YR343 does not readily attach to hydrophilic surfaces [13] |
| MTMS | Hydrophilic | Low (Not specified) | Minimal attachment | Used in vapor deposition for creating hydrophilic surfaces [13] |
Table 2: Quantifiable Methodologies for Studying Reversible Attachment
| Methodology | Key Measurable Parameters | Technical Resolution / Throughput | Application in Reversible Attachment Studies |
|---|---|---|---|
| Droplet Microfluidics with AI Analysis [14] | Biofilm/aggregate area; Growth dynamics | High-throughput (1000s of droplets); Enables single-droplet analysis | Tracks early attachment and aggregation in controlled microenvironments; Quantifies kinetic parameters |
| Functionalized Silanes with Quantitative Image Processing [13] | Surface coverage; Morphological evolution (e.g., honeycomb structure) | Medium throughput (10s of images/chip); Quantifies 2D propagation | Directly correlates surface chemistry with attachment efficiency and early biofilm morphology |
| Quartz Crystal Microbalance with Dissipation (QCM-D) [13] | Absorbed mass; Viscoelastic properties | Sensitive to nanogram mass changes; Limited surface variety | Quantifies initial cell attachment and the role of appendages like flagella in real-time |
Experimental Protocols
To investigate the stage of reversible attachment, reproducible and controlled experimental protocols are essential. The following sections detail key methodologies for surface preparation and quantitative analysis.
Surface Functionalization via Silane Vapor Deposition
This protocol creates surfaces with defined chemical properties to study their effect on initial bacterial attachment [13].
Materials:
- Substrates: Silicon wafers with silicon dioxide coating (e.g., from Silicon Quest), diced into 20 mm x 20 mm squares.
- Silane Reagents:
- Trichloro(1H,1H,2H,2H-perfluorooctyl) silane (PFOTS) for hydrophobic surfaces.
- n-octadecyl(trimethoxy)silane (OTS) for hydrophobic surfaces.
- 3-aminopropyl trimethoxysilane (APTMS) for hydrophilic surfaces.
- Methoxytriethyleneoxypropyl-trimethoxysilane (MTMS) for hydrophilic surfaces.
- Equipment: Plasma cleaner (e.g., Harrick Plasma PDC-001), hot plate, enclosed glass dish.
Procedure:
- Surface Cleaning: Clean the silicon chips with filtered pressurized air (0.2 μm filter), followed by treatment in an air plasma cleaner for a minimum of 5 minutes.
- Vapor Deposition:
- For PFOTS: Place 20 μL per 80 cm² in an enclosed glass dish on a hot plate at 85°C for 4 hours.
- For APTMS: Place 40 μL per 80 cm² in an enclosed glass dish on a hot plate at 150°C for 2 hours.
- For OTS: Place 40 μL per 80 cm² in an enclosed glass dish on a hot plate at 150°C for 2 hours, followed by 2 hours with no heat.
- For MTMS: Place the sample in an enclosed glass dish for 4 hours at 65°C, followed by 1 hour at 115°C.
- Characterization: Validate surface properties by measuring the water contact angle using a goniometer (e.g., KRÜSS DSA 30). Perform measurements in triplicate with 1 μL droplets of distilled water.
Quantifying Early Attachment with Semi-Automated Image Analysis
This methodology quantifies bacterial attachment and early biofilm propagation on functionalized surfaces [13].
Materials:
- Bacterial Strain: Fluorescently tagged strains (e.g., Pantoea sp. YR343 expressing EGFP).
- Culture Media: Appropriate liquid medium (e.g., R2A for Pantoea).
- Equipment: Epifluorescence microscope (e.g., Olympus IX51) with FITC filter cube, image analysis software (e.g., ImageJ, MATLAB, or Python with appropriate libraries).
Procedure:
- Sample Inoculation:
- Grow bacteria to stationary phase overnight in liquid medium.
- Dilute the culture 1:100 in fresh medium and grow to early exponential phase (OD₆₀₀ ≈ 0.1).
- Place each functionalized substrate in a concave dish and add 3 mL of the bacterial culture.
- Incubate covered for specified time-points (e.g., 2, 4, 6, 8 hours).
- Sample Harvesting:
- Gently remove the substrate from the culture using tweezers, holding it by the corners.
- Rinse gently with 10 mL of DI water to remove loosely attached cells.
- Dry the sample using pressurized air blown through a 0.2 μm filter.
- Image Acquisition:
- Using an epifluorescence microscope, acquire a minimum of 10 images from random positions across each substrate.
- Ensure consistent microscope settings (exposure, gain) across all samples.
- Image Processing and Quantification:
- Develop or use a script in image analysis software (e.g., ImageJ) to perform the following:
- Apply background subtraction and thresholding to distinguish bacterial cells from the background.
- Use particle analysis functions to quantify parameters such as percent surface coverage, number of attached cells per unit area, and morphological descriptors (e.g., cluster size distribution).
- Develop or use a script in image analysis software (e.g., ImageJ) to perform the following:
Diagram: The experimental workflow for studying reversible attachment, from surface preparation to quantitative image analysis.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Reversible Attachment Studies
| Reagent / Material | Function / Role in Research | Example Use Case |
|---|---|---|
| Functionalized Silanes (PFOTS, OTS, APTMS, MTMS) [13] | Modulate surface hydrophobicity/hydrophilicity to study surface property effects on attachment. | Creating defined hydrophobic/hydrophilic surfaces on silicon/silica substrates for controlled attachment assays. |
| HFE-7500 Perfluorinated Oil with Fluorosurf Surfactant [14] | Forms the continuous phase in droplet microfluidics; surfactant stabilizes droplets and prevents coalescence. | High-throughput generation of monodisperse microdroplets serving as isolated microenvironments for biofilm studies. |
| c-di-GMP Assay Kits (e.g., ELISA, LC-MS/MS) | Quantify intracellular c-di-GMP levels, a key secondary messenger regulating the motile-to-sessile switch. | Correlating c-di-GMP concentration with the transition from reversible to irreversible attachment in surface-sentient cells. |
| Fluorescent Protein Plasmids (e.g., pBBR1-MCS5 with EGFP) [13] | Genetically tag bacterial strains for visualization and quantification via fluorescence microscopy. | Enabling tracking of bacterial attachment dynamics on functionalized surfaces over time. |
| Dispersin B (Glycoside Hydrolase) [12] | Degrades poly-N-acetylglucosamine (PNAG) in the biofilm matrix; used as a probe for matrix function. | Testing the role of PNAG in stabilizing early microcolonies after initial attachment; an anti-biofilm agent. |
| Microfluidic Chips & Pressure Pumps (OB1, Elveflow) [14] | Generate, trap, and manipulate picoliter-to-nanoliter droplets for high-throughput, in-situ analysis. | Studying the kinetics of reversible attachment and early aggregation in thousands of isolated microhabitats in parallel. |
Within the established research framework mapping the stages of microbial biofilm formation, the transition from reversible to irreversible attachment represents a critical commitment point for surface-associated bacteria [15]. This second stage, Irreversible Attachment, is characterized by a fundamental shift from transient, physical adsorption to permanent molecular anchoring [16]. This shift is mediated by the active production of bacterial adhesins such as pili and fimbriae, and the initial secretion of extracellular polymeric substances (EPS) [15]. For researchers and drug development professionals, understanding the precise mechanisms governing this phase is paramount, as it represents a key therapeutic target for preventing the establishment of resilient, chronic biofilm-based infections [16].
Core Mechanisms of Irreversible Attachment
The transition to a permanent, surface-anchored existence involves a coordinated change in bacterial behavior, gene expression, and polymer production. The following diagram illustrates the core mechanisms and their interrelationships during Stage 2.
Key Molecular Players and Genetic Regulation
The commitment to a biofilm lifestyle is underpinned by significant genetic reprogramming. Bacteria undergo a genetic shift, downregulating genes associated with motility and upregulating those responsible for the production of adhesins and EPS components [16]. The primary molecular effectors facilitating irreversible attachment include:
- Bacterial Adhesins: Hair-like appendages such as Type I and Type IV pili act as molecular grappling hooks, forming strong, specific interactions with the substrate surface [16]. Other surface structures, including lipopolysaccharides (LPS) in Gram-negative bacteria, also contribute to firm anchorage [15].
- Extracellular Polymeric Substances (EPS): The initial secretion of a matrix composed of exopolysaccharides, secreted proteins, lipids, and extracellular DNA (eDNA) creates a hydrated polymer network that physically enmeshes the cells and glues them to the surface [15] [16]. This matrix not only strengthens adhesion but also begins to form a protective barrier.
Quantitative Analysis of Irreversible Attachment
Quantifying the dynamics and properties of irreversible attachment is crucial for phenotyping bacterial strains and evaluating the efficacy of anti-biofilm strategies. The table below summarizes key parameters that can be measured experimentally.
Table 1: Key Quantitative Parameters for Analyzing Irreversible Attachment
| Parameter Category | Specific Parameter | Measurement Technique | Typical Observation in Stage 2 | Research Significance |
|---|---|---|---|---|
| Adhesion Strength | Cell Count Post-Wash | Microscopy with viability staining [17] | >80% retention of initially attached cells | Determines firmness of attachment; distinguishes from reversible phase. |
| Shear Force Resistance | Flow-cell systems with calibrated shear stress [18] | Stable adhesion under flow rates >0.5 dyn/cm² | Models biofilm resilience in physiological/industrial flow conditions. | |
| EPS Production | Polysaccharide/Protein Content | Colorimetric assays (e.g., phenol-sulfuric acid, Lowry) | Significant increase over planktonic cells & Stage 1 | Direct measure of matrix production initiation. |
| eDNA Quantification | Fluorescence staining (e.g., PicoGreen) & quantification [17] | Detectable signal in nascent matrix | Identifies a key structural and functional EPS component. | |
| Genetic Expression | Adhesin Gene (e.g., pilA) Expression | qRT-PCR or GFP transcriptional reporters [17] | Upregulation (e.g., 5-50 fold increase) | Links phenotypic attachment to genetic regulation. |
| Motility Gene (e.g., fliC) Expression | qRT-PCR | Downregulation (e.g., >10 fold decrease) | Marks the switch from motile to sessile lifestyle. | |
| Morphological | Microcolony Size & Distribution | 3D image analysis (e.g., BiofilmQ) [17] | Formation of small, structured clusters | Quantifies the transition from single cells to a community. |
Advanced image analysis software like BiofilmQ enables high-throughput, 3D quantification of these parameters directly within living biofilms, providing spatially resolved data on biofilm-internal properties [17]. The workflow for such an analysis is depicted below.
Experimental Protocols for Investigating Stage 2
Protocol: Quantifying Irreversible Attachment Using Static Adhesion Assay and Image Analysis
This protocol is designed to quantitatively assess the strength and extent of irreversible attachment under controlled laboratory conditions.
1. Principle: This assay differentiates reversibly and irreversibly attached cells by subjecting surface-associated bacteria to a gentle washing step. Cells that remain attached are considered irreversibly adhered and are quantified.
2. Materials:
- Bacterial culture (late logarithmic phase)
- Appropriate growth medium
- Sterile, tissue-culture treated multi-well plates (e.g., 24-well or 96-well)
- Inverted phase-contrast or fluorescence microscope
- Phosphate Buffered Saline (PBS), pH 7.4
- Fixative (e.g., 4% paraformaldehyde) if endpoint assay is used.
- Fluorescent stains (e.g., SYTO 9 for total cells, propidium iodide for dead cells) if using fluorescence microscopy.
3. Procedure: 1. Inoculation: Dilute the bacterial culture to an appropriate optical density (e.g., OD600 = 0.1) in fresh medium. Add equal volumes of this suspension to the wells of the multi-well plate. Incubate the plate under optimal growth conditions for a defined period (e.g., 2-4 hours) to allow attachment. 2. Washing: Carefully aspirate the planktonic (non-attached) culture from the wells. Gently add pre-warmed PBS to each well without disturbing the adherent layer. Tilt the plate and carefully aspirate the PBS. Repeat this washing step twice. 3. Fixation (Optional): For endpoint analysis, add a fixative like 4% paraformaldehyde to the wells for 15-30 minutes. Aspirate the fixative and wash twice with PBS. 4. Staining (Optional): If using fluorescent stains, apply the stain according to the manufacturer's protocol. For a simple total cell count, a DNA stain like SYTO 9 is sufficient. 5. Microscopy and Image Acquisition: For each well, acquire multiple random images using a 20x or 40x objective. If using a flow-cell system for shear force resistance, set the pump to a defined flow rate and duration after the initial attachment phase before imaging [18]. 6. Image Analysis: * Software: Load acquired images into analysis software such as BiofilmQ [17], ImageJ, or commercial alternatives. * Segmentation: Use the software's thresholding tools (e.g., Otsu, Maximum Correlation Thresholding) to accurately distinguish bacterial cells from the background [17]. * Quantification: Analyze the segmented images to count the number of adhered cells per field of view. Calculate the average number of cells per mm². In tools like BiofilmQ, you can also quantify the biovolume (µm³) and the spatial distribution of the microcolonies.
4. Data Analysis: Compare the post-wash cell counts or biovolume between different bacterial strains, growth conditions, or treatment groups (e.g., with/ without anti-adhesion compounds). Statistical analysis (e.g., Student's t-test, ANOVA) should be performed to determine significance.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 2: Essential Research Reagents for Studying Irreversible Attachment
| Item Name | Function/Application | Specific Example(s) |
|---|---|---|
| Tissue-Culture Treated Plates | Provides a uniform, sterile surface for biofilm growth in static assays. | 24-well or 96-well polystyrene plates. |
| Flow-Cell Systems | Creates controlled hydrodynamic conditions to study adhesion under shear stress. | Small-scale flow cells with syringe or peristaltic pumps [18]. |
| Fluorescent Dyes | Staining for viability, total cells, and specific EPS components. | SYTO 9 (nucleic acids), Concanavalin A-Tetramethylrhodamine (polysaccharides), PicoGreen (eDNA). |
| qRT-PCR Reagents | Quantifying gene expression changes during the reversible to irreversible transition. | Primers for adhesin (e.g., pilA) and EPS-related genes, reverse transcriptase, SYBR Green mix. |
| Transcriptional Reporters | Visualizing and quantifying promoter activity of target genes in real-time. | GFP, mCherry, or other fluorescent proteins fused to promoters of genes like psl or pel (EPS in Pseudomonas). |
| Image Analysis Software | High-throughput, 3D quantification of attachment and biofilm structure. | BiofilmQ [17], COMSTAT, Ilastik, ImageJ/FIJI. |
| Specific Antibodies | Detecting and localizing specific adhesin or EPS proteins. | Anti-Psl/Pel antibodies (Pseudomonas), Anti-RbmA antibodies (Vibrio cholerae) [17]. |
Stage 2, Irreversible Attachment, is a decisively commitment to the biofilm lifestyle, moving beyond transient physical forces to active, molecular-based permanence [15] [16]. The interplay of bacterial appendages and the nascent EPS matrix creates a robust foundation upon which the complex, 3D structure of a mature biofilm is built [19]. The quantitative methods and experimental frameworks detailed here provide researchers and drug developers with the tools to dissect this critical phase, offering actionable insights and specific molecular targets for innovative strategies aimed at biofilm prevention and eradication.
Microcolony formation represents a critical juncture in the biofilm life cycle, marking the transition from scattered, surface-attached cells to a structured, three-dimensional community. This stage, often designated as Stage 3 in the biofilm formation process, is characterized by bacterial aggregation and the initiation of a complex architecture that defines the mature biofilm [20] [21]. The shift from reversible to irreversible attachment triggers a rapid change in gene expression, driving the proliferating bacteria to form these initial, clustered aggregates known as microcolonies [22] [20]. This development is not merely a morphological change; it is a strategic survival adaptation. Life within a microcolony offers numerous advantages, including enhanced resistance to antimicrobial agents, protection from host immune responses and predation, and the facilitation of intercellular communication and genetic exchange [23] [24]. Understanding the mechanisms underpinning microcolony formation is therefore essential for developing strategies to combat persistent, biofilm-associated infections.
Structural and Compositional Foundations of the Microcolony
The architecture of a microcolony is fundamentally defined by the production of Extracellular Polymeric Substances (EPS). This matrix acts as a scaffold, encasing the microbial cells and forming the foundational structure of the biofilm [25] [21]. The composition of the EPS is complex and dynamic, primarily consisting of polysaccharides, proteins, nucleic acids (extracellular DNA, or eDNA), and lipids [22] [20] [21]. In the early stages of microcolony formation, eDNA plays a particularly critical role in stabilizing the initial aggregate, while polysaccharides and proteins become more dominant as the structure matures [21].
The EPS matrix is far from an inert scaffold. It confers critical physical and functional properties to the microcolony:
- Mechanical Stability: The dense, mesh-like structure of intertwined polysaccharide chains, often cross-linked by divalent cations (e.g., Ca²⁺ or Mg²⁺), provides significant mechanical strength, allowing the biofilm to withstand shear forces in fluid environments [20].
- Protective Barrier: The matrix serves as a biological barrier, impeding the penetration of antimicrobial agents and shielding internal cells from components of the host immune system [25] [26].
- Nutrient Reservoir: It acts as a resource-retaining system, trapping water, nutrients, and enzymes, thereby creating a unique microenvironment for the embedded cells [26].
The resulting structure is a highly viscoelastic, rubbery community that is predominantly composed of the EPS matrix, with microbial cells making up only about 10% of the total volume [20].
Table 1: Key Components of the Extracellular Polymeric Substance (EPS) Matrix in Microcolonies
| Component | Primary Function | Significance in Microcolony Formation |
|---|---|---|
| Polysaccharides | Structural scaffolding; mechanical stability | Forms a dense, cross-linked mesh that provides the 3D architecture and rubber-like properties. |
| Proteins | Structural support; enzymatic activity | Provides structural integrity and facilitates metabolic processes within the biofilm niche. |
| Extracellular DNA (eDNA) | Cell-to-cell adhesion; structural integrity | Critical for initial stabilization of the aggregate in early microcolony development [21]. |
| Lipids & Nucleic Acids | Unknown/Multiple | Additional structural and potentially functional roles within the complex matrix. |
| Divalent Cations (Ca²⁺, Mg²⁺) | Ionic cross-linking | Strengthens the EPS matrix by forming bridges between anionic polymer chains [20]. |
Molecular Mechanisms and Signaling in Microcolony Development
Genetic Regulation and Metabolic Adaptations
The transition from attached cells to a microcolony is driven by significant shifts in gene expression and metabolic physiology. Cells within the microcolony experience steep gradients of oxygen and nutrients, leading to a heterogeneous environment where different metabolic states coexist [25] [27]. Research on Pseudomonas aeruginosa has revealed that microcolony formation is specifically associated with stressful, oxygen-limiting conditions [27]. In response, the bacteria activate anaerobic and fermentative pathways.
A key regulator identified in this process is the two-component system MifSR. The response regulator, MifR, is essential for microcolony formation; inactivation of mifR results in thin, unstructured biofilms lacking microcolonies, while its overexpression leads to hyper-microcolony formation [27]. This regulatory system is functionally linked to central carbon metabolism. Under the oxygen-limited conditions within a developing microcolony, pyruvate fermentation becomes a critical adaptive process. The enzyme lactate dehydrogenase (LdhA), which converts pyruvate to lactate, is required for microcolony formation [27]. Experimentally, depleting pyruvate from growth medium impairs microcolony formation, whereas its supplementation significantly enhances it [27].
Cell-to-Cell Communication via Quorum Sensing
As cell density increases within the microcolony, bacteria begin to coordinate their behavior through a process called Quorum Sensing (QS). QS is a cell-cell communication system that relies on the production, detection, and response to small, diffusible signaling molecules called autoinducers [22] [24]. When the concentration of these molecules reaches a threshold (a "quorum"), it triggers a coordinated change in gene expression across the population. In the context of microcolony formation, QS regulates the production of virulence factors and EPS components, thereby facilitating the transition from a loose aggregate to a structured, functional community [22] [23]. This sophisticated communication system ensures that energy-intensive processes like EPS synthesis are only initiated once a sufficiently large population has been established.
The following diagram illustrates the core regulatory and metabolic pathway driving microcolony formation in P. aeruginosa, integrating the MifSR regulatory system with the critical metabolic shift to pyruvate fermentation.
Experimental Models and Methodologies for Studying Microcolonies
In Vitro Models and Analytical Techniques
The study of microcolony formation relies on robust experimental models that allow for controlled observation and manipulation. Individual-Based Models (IBMs) are computational approaches that simulate how individual bacterial behaviors, such as movement, adhesion, and reproduction, give rise to the emergent pattern of microcolony formation [23] [28]. These models help link individual cell actions to group-level fitness outcomes, such as survival under stress [23].
In the laboratory, a variety of physical models are used:
- Flow Cell Systems: These devices allow for the continuous provision of fresh nutrients and the removal of waste products, enabling the real-time observation of biofilm development under conditions that mimic natural environments (e.g., flowing water) or physiological conditions (e.g., blood flow over a catheter) [25].
- Static Microtiter Plate Assays: A high-throughput workhorse for biofilm research, this model is used to screen for genetic mutants or chemical compounds that affect biofilm formation. Biofilm mass is typically quantified using crystal violet staining [27].
- Drip-Flow Reactors: These simulate low-shear, semi-batch conditions and are useful for growing biofilms that are thicker and more representative of certain natural settings [25].
Analytical techniques are critical for dissecting the structure and composition of microcolonies. Confocal Laser Scanning Microscopy (CLSM) is arguably the most powerful tool, as it allows for the non-invasive optical sectioning of live biofilms, generating high-resolution, three-dimensional images of the microcolony architecture [20]. These images can be analyzed to determine biovolume, thickness, and spatial distribution of cells and matrix components. For molecular and biochemical analysis, transcriptomics and proteomics are used to identify genes and proteins that are differentially expressed during microcolony formation, as demonstrated in the study of the MifR regulon [27].
A Representative Experimental Workflow
The following workflow, based on the seminal study of the MifR-pyruvate pathway, provides a template for investigating molecular mechanisms of microcolony formation [27]:
- Generate Genetic Mutants: Create targeted gene knockouts (e.g.,
mifR::Mar,ldhAmutant) and overexpression strains (e.g., strain carryingpMJT-mifRplasmid) in the wild-type background (e.g., P. aeruginosa PAO1 or PA14). - Cultivate Biofilms: Grow biofilms of wild-type and mutant strains in a controlled system like a flow cell or a static plate, using a defined or rich medium (e.g., with varying pyruvate concentrations).
- Visualize and Quantify Architecture: Use CLSM to capture 3D images of the biofilms. Quantify architectural parameters such as microcolony biovolume, average thickness, and substratum coverage using image analysis software (e.g., COMSTAT, ImageJ).
- Conduct Molecular Profiling: Perform transcriptomic (e.g., RNA-Seq) and/or proteomic analyses on harvested biofilm cells to identify global changes in gene or protein expression between wild-type and mutant strains.
- Functional Validation:
- Biochemical Assays: Measure relevant metabolites (e.g., pyruvate, lactate) and enzyme activities (e.g., LdhA activity, c-di-GMP levels).
- Genetic Complementation: Re-introduce the functional gene into the mutant strain (e.g.,
mifR::Mar/pMJT-mifR) and confirm restoration of the wild-type microcolony phenotype. - Nutrient Manipulation: Systematically add or remove key metabolites (e.g., pyruvate) from the growth medium to assess their direct impact on microcolony formation.
The Scientist's Toolkit: Key Research Reagents and Solutions
The following table details essential materials and reagents used in the experimental investigation of microcolony formation, with a specific focus on the study of the P. aeruginosa MifR-pyruvate pathway [27].
Table 2: Essential Research Reagents for Investigating Microcolony Formation
| Reagent / Material | Type | Primary Function in Experimentation |
|---|---|---|
| Defined/Growth Media | Culture Reagent | Supports biofilm growth; used to manipulate nutrient availability (e.g., pyruvate depletion/addition) [27]. |
| P. aeruginosa PAO1 / PA14 | Bacterial Strain | Wild-type model organisms for studying biofilm formation and pathogenesis [27]. |
| mifR, mifS, ldhA Mutants | Genetic Tool | Isogenic mutant strains to determine the specific role of a gene in microcolony formation [27]. |
| Complementation Plasmid (pMJT-mifR) | Genetic Tool | Plasmid vector carrying the wild-type mifR gene used to restore function in a mutant, confirming gene-specificity of the phenotype [27]. |
| Flow Cell System | Physical Model | Provides a controlled hydrodynamic environment for real-time, in-situ observation of biofilm development [25]. |
| Confocal Laser Scanning Microscope | Analytical Instrument | Enables non-destructive, high-resolution 3D imaging of live microcolony architecture and matrix components [20]. |
| RNA Sequencing Kits | Molecular Biology Reagent | For global transcriptomic profiling to identify genes differentially expressed during microcolony development [27]. |
| Pyruvate | Metabolic Substrate | Key metabolite used to test the hypothesis that pyruvate fermentation is essential for microcolony formation [27]. |
| c-di-GMP Assay Kits | Biochemical Assay | To quantify intracellular levels of cyclic di-GMP, a central secondary messenger linking metabolism to biofilm formation [27]. |
Microcolony formation is a decisive phase in the establishment of a bacterial biofilm, representing a shift from a solitary to a collective lifestyle. This process is underpinned by the production of a protective EPS matrix, sophisticated genetic regulation via systems like MifSR, and a critical metabolic adaptation to hypoxia through pathways such as pyruvate fermentation [22] [27]. The resulting structure is not a homogeneous clump of cells, but a differentiated and heterogeneous community that is inherently more resistant to antimicrobials and environmental stresses [25] [23]. A deep understanding of the molecular mechanisms and environmental cues that drive microcolony development provides a solid foundation for the subsequent stage of biofilm maturation and unveils potential therapeutic targets. Disrupting the signaling pathways or metabolic processes essential for this developmental stage offers a promising strategy for preventing the formation of resilient, chronic biofilm infections.
Within the broader context of microbial biofilm formation research, the maturation stage represents a critical transitional phase where surface-attached microcolonies evolve into complex, three-dimensional communities. This stage, termed "Maturation II," is characterized by the development of a sophisticated architectural framework and the formation of functional water channels. This in-depth technical guide examines the structural and regulatory mechanisms that underpin this process, with a specific focus on Pseudomonas aeruginosa as a model organism. A deep understanding of this stage is paramount for the development of therapeutic strategies aimed at disrupting biofilm-associated infections, which are notoriously resistant to conventional antibiotics [29] [30].
Structural Composition and Key Components
The maturation of a biofilm is marked by the significant production of extracellular polymeric substances (EPS), which form a scaffold-like matrix. This matrix accounts for 75-95% of the biofilm's total biomass, with microbial cells constituting only 5-25% [11] [29]. This self-produced environment is fundamental to the biofilm's physical stability, pathogenicity, and recalcitrance.
Core Constituents of the EPS Matrix
The EPS is a complex amalgam of biomolecules that determine the biofilm's mechanical and functional properties.
- Exopolysaccharides: These are the primary structural components of the matrix in many bacterial biofilms. In P. aeruginosa, three exopolysaccharides are of particular importance:
- Pel: A glucose-rich polysaccharide that provides critical cell-cell adhesion and structural integrity to the biofilm [29].
- Psl: A mannose-rich polysaccharide that plays a key role in initial surface attachment and in maintaining the three-dimensional structure of mature biofilms [29].
- Alginate: Often overproduced by mucoid strains, it is crucial for the stability of the biofilm and for protecting the embedded cells from host immune responses and antibiotics [29].
- Extracellular DNA (eDNA): eDNA is a major accelerant of early biofilm development and is integral to the stability of the mature structure. It interacts with Pel and Psl polysaccharides, and eDNA-deficient biofilms show markedly increased sensitivity to detergents [29].
- Proteins: The protein component includes secreted enzymes, cell surface adhesins, and protein subunits of cell appendages. A key adhesin is CdrA, which is regulated by c-di-GMP and helps to stabilize the biofilm structure by cross-linking with Pel and Psl exopolysaccharides [29].
Table 1: Key Exopolysaccharides in Pseudomonas aeruginosa Biofilm Maturation
| Polysaccharide | Composition | Primary Function in Maturation |
|---|---|---|
| Pel | Glucose-rich | Provides cell-cell adhesion and structural integrity |
| Psl | Mannose-rich | Maintains 3D architecture and surface attachment |
| Alginate | Acetylated polymannuronic/guluronic acid | Enhances stability and resistance to host defenses/antibiotics |
Architectural Features: Water Channels and Heterogeneity
A defining characteristic of a mature biofilm is its heterogeneous three-dimensional architecture. This structure is not a uniform monolayer but rather a constellation of towering "mushroom-shaped" or "column-like" microcolonies separated by a network of interstitial voids, known as water channels [11] [29].
These water channels are critical for the functionality of the biofilm, acting as a primitive circulatory system. They facilitate the passive diffusion of nutrients and oxygen into the deeper layers of the biofilm and allow for the efficient removal of metabolic waste products [11]. This creates a myriad of microenvironments within the biofilm, leading to metabolic and phenotypic heterogeneity among the constituent cells. This spatial organization is a key factor in the biofilm's enhanced tolerance to antimicrobial agents [30].
Molecular Regulation of Maturation
The transition from a microcolony to a complex 3D structure is tightly regulated by intracellular signaling molecules and sensory systems.
The Central Role of c-di-GMP
The secondary messenger bis-(3',5')-cyclic dimeric guanosine monophosphate (c-di-GMP) is a master regulator of the switch from motile, planktonic lifestyles to sessile, biofilm-forming ones. A high intracellular concentration of c-di-GMP promotes biofilm formation by:
- Enhancing the production of exopolysaccharides (Pel, Psl, alginate) and adhesins like CdrA [29].
- Simultaneously repressing the expression of flagellar genes, thereby inhibiting motility and promoting irreversible attachment [29].
The cellular level of c-di-GMP is itself regulated by various signal transduction pathways. One key system is the Wsp chemosensory pathway, which is activated by the surface-sensing protein WspA. This pathway leads to the activation of the response regulator WspR, which in turn stimulates the production of c-di-GMP. Key receptors for c-di-GMP, such as FleQ and PelD, then drive the expression of exopolysaccharide operons, cementing the biofilm state [29].
Signaling Pathway and Regulatory Network
The diagram below illustrates the core regulatory network that drives the maturation of biofilm architecture.
Regulatory Network in Biofilm Maturation
Experimental Analysis of Maturation Architecture
The quantitative analysis of a mature biofilm's complex architecture requires specialized methodologies that go beyond traditional microbiological techniques like colony-forming unit (CFU) counts.
Key Experimental Protocol: Confocal Microscopy and Viability Staining
Confocal Laser Scanning Microscopy (CLSM) combined with fluorescent staining is the gold standard for visualizing and quantifying the 3D structure and viability of biofilms [31].
Detailed Methodology:
- Sample Preparation: Biofilms are grown on relevant substrates (e.g., plastic, glass, or biomaterial surfaces) under controlled conditions.
- Viability Staining: The biofilm is stained using a viability kit, most commonly the FilmTracer LIVE/DEAD Biofilm Viability Kit.
- SYTO 9: This green-fluorescent stain labels all bacteria, both live and dead, by penetrating intact cell membranes.
- Propidium Iodide: This red-fluorescent stain penetrates only bacteria with damaged membranes, labeling dead or dying cells.
- Critical Consideration: Propidium iodide can also bind to extracellular DNA (eDNA) present in the matrix, which can lead to false positives if not analyzed correctly. It is therefore essential to analyze the red and green channels separately during image processing [31].
- Image Acquisition: The stained biofilm is imaged using a CLSM, which collects high-resolution optical sections at various depths (z-stacks). These z-stacks are then used to reconstruct a 3D model of the biofilm.
- Image Analysis:
- Automated Analysis: To avoid user subjectivity and improve throughput, automated image analysis software is employed. Tools like BiofilmQ can quantify a wide range of parameters, including total biomass, biovolume, surface coverage, thickness, and spatial distribution of live/dead signals [32].
- Validation: The accuracy of automated methods is often validated by comparing results with traditional CFU counts and performing sensitivity/specificity analyses to ensure the algorithm correctly identifies bacterial pixels versus background [31].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents and Tools for Analyzing Biofilm Maturation
| Research Tool / Reagent | Function / Application |
|---|---|
| FilmTracer LIVE/DEAD Kit | Fluorescent staining for simultaneous assessment of biofilm viability and overall structure via CLSM. |
| Confocal Laser Scanning Microscope (CLSM) | High-resolution 3D visualization and image acquisition of biofilm architecture. |
| BiofilmQ Software | Comprehensive, open-source image analysis software for quantifying architectural features and fluorescence from CLSM data. |
| SYTO 9 Stain | Green-fluorescent nucleic acid stain labeling all bacterial cells in a biofilm. |
| Propidium Iodide | Red-fluorescent stain labeling dead cells and extracellular DNA (eDNA) within the matrix. |
| 96-well Polystyrene Plates | Standard substrate for high-throughput biofilm cultivation and assessment. |
| Crystal Violet Stain | Basic dye for total biofilm biomass quantification via spectrophotometry. |
Implications for Drug Development
The unique properties of mature biofilms pose a significant challenge for drug development. The EPS matrix acts as a barrier, inhibiting antibiotic penetration, while the metabolic heterogeneity and presence of dormant persister cells within the structure contribute to multidrug tolerance [33] [30]. The current clinical approach often mirrors strategies from cancer treatment, involving aggressive physical debridement to remove the biofilm nidus, combined with local delivery of high-dose antimicrobials (e.g., antibiotic lock therapy) [30].
Future therapeutic strategies are focusing on targeting the maturation process itself. These prospective approaches include:
- Quorum Sensing Inhibitors: Disrupting the cell-to-cell communication that coordinates biofilm development.
- Matrix-Degrading Enzymes: Using enzymes such as DNases to degrade eDNA or glycosidases to target exopolysaccharides, thereby destabilizing the biofilm structure and making it more susceptible to antibiotics [29] [30].
- c-di-GMP Signaling Interference: Developing compounds that lower intracellular c-di-GMP levels to trigger biofilm dispersion [29].
Given the multifactorial nature of biofilm resistance, the most promising strategies are likely to be combinatorial therapies that simultaneously target the extracellular matrix, disrupt signaling, and potentiate the activity of conventional antimicrobials [30].
Dispersion represents the crucial final stage in the biofilm life cycle, during which sessile, matrix-encased biofilm cells actively separate from the biofilm community and transition to a planktonic mode of growth [34]. This actively regulated process serves as a dissemination mechanism, enabling bacteria to colonize new niches and initiate fresh biofilm development cycles [35] [36]. Unlike passive detachment caused by external shear forces, dispersion is a genetically programmed response to specific environmental and cellular cues [34] [36]. From a therapeutic perspective, induced dispersion is being investigated as a promising antibiofilm strategy because the transition from biofilm to planktonic state renders bacteria more susceptible to conventional antimicrobial agents and host immune responses [35] [34].
This technical guide examines the molecular mechanisms, regulatory systems, and experimental methodologies underlying the biofilm dispersion process, with particular emphasis on applications for research and therapeutic development.
Molecular Mechanisms of Biofilm Dispersion
Matrix Degradation Enzymes
The liberation of cells from the biofilm structure requires controlled degradation of the extracellular polymeric substance (EPS) matrix. Various bacteria produce specific enzymes that target structural components of the biofilm matrix [36].
Table 1: Matrix-Degrading Enzymes Involved in Biofilm Dispersion
| Enzyme | Molecular Weight (kDa) | Substrate | Producing Bacterium | Function in Dispersion |
|---|---|---|---|---|
| Dispersin B | 42 | Poly-β(1,6)-N-acetyl-D-glucosamine (PNAG) | Aggregatibacter actinomycetemcomitans | Hydrolyzes polysaccharide component of biofilm matrix [36] |
| Alginate Lyase | 43 | Alginate (polymer of mannuronic and guluronic acids) | Pseudomonas aeruginosa | Degrades alginates in P. aeruginosa biofilms [36] |
| Aureolysin | 33 | Unknown matrix proteins | Staphylococcus aureus | Protease that degrades proteinaceous matrix components [36] |
| Hyaluronidase | 117 | Hyaluronan | Streptococcus spp. | Breaks down hyaluronic acid in biofilm matrix [36] |
| Glycosidases & Deoxyribonucleases | Variable | Exopolysaccharides & eDNA | Various species | Target polysaccharide backbones and extracellular DNA networks [34] [11] |
Regulatory Pathways and Signaling Molecules
Dispersion is regulated by complex signaling networks that integrate environmental cues with intracellular second messenger systems.
Figure 1: Regulatory pathways controlling biofilm dispersion. The diagram illustrates how environmental cues and signaling molecules converge through the c-di-GMP regulatory network to activate the dispersion response.
The intracellular second messenger cyclic dimeric guanosine monophosphate (c-di-GMP) serves as a central regulator of the transition between sessile and motile lifestyles in bacteria [37]. High intracellular c-di-GMP levels promote biofilm formation through increased matrix production, while low c-di-GMP levels induce dispersion [37]. This reduction in c-di-GMP is triggered when environmental signals such as nutrient limitation, oxygen availability, or accumulated waste products activate phosphodiesterases that degrade c-di-GMP [34].
Diffusible Signal Factors (DSF), particularly cis-2-decenoic acid, induce dispersion by binding to receptor proteins like RpfR, which stimulates c-di-GMP phosphodiesterase activity [37]. This decreases intracellular c-di-GMP concentrations, triggering downstream dispersion responses including production of matrix-degrading enzymes and activation of motility mechanisms [37].
Research Methodologies for Studying Dispersion
Experimental Models and Assay Systems
Table 2: Model Organisms for Dispersion Research
| Organism | Relevance | Key Dispersion Mechanisms | Research Applications |
|---|---|---|---|
| Pseudomonas aeruginosa | Well-characterized model system for biofilm development | DSF-mediated c-di-GMP reduction, matrix enzyme production | Study of regulatory networks, therapeutic testing [34] [37] |
| Staphylococcus aureus | Clinical relevance in medical device infections | Protease-mediated matrix degradation (e.g., aureolysin) | Antimicrobial susceptibility testing [36] |
| Aggregatibacter actinomycetemcomitans | Oral biofilm former | Dispersin B production | Enzyme-based dispersion studies [36] |
| Mixed cultures from MBR sludge | Environmental and industrial biofouling | Community-level responses to DSF signals | Biofouling control research [37] |
Quantitative Dispersion Assessment Methods
Optical Density-Based Assay
Protocol:
- Grow biofilms in standard microtiter plates or flow cells for 24-48 hours until mature.
- Treat with experimental dispersion-inducing compounds (e.g., CDA at 10-100 μM concentrations).
- Gently rinse to remove non-adherent cells.
- Measure optical density (OD₆₀₀) of the planktonic phase after induced dispersion.
- Compare treatment groups to untreated controls [37].
Data Interpretation: Increased OD in treatment groups indicates enhanced cell liberation from biofilms. This simple assay provides quantitative comparison of dispersion efficacy across different conditions [37].
Confocal Laser Scanning Microscopy (CLSM) with Image Analysis
Protocol:
- Grow biofilms on appropriate surfaces compatible with microscopy.
- Treat with test compounds and incubate for specified durations.
- Stain with appropriate fluorescent dyes (e.g., SYTO9 for live cells, ConA for polysaccharides).
- Capture z-stack images using CLSM at multiple random positions.
- Analyze using image analysis software (e.g., COMSTAT, ImageJ) to quantify structural parameters [37].
Quantitative Parameters:
- Total Biomass: Reduction indicates dispersion effectiveness.
- Mean Thickness: Thinner biofilms suggest active dispersion.
- Surface-to-Biovolume Ratio: Higher values correlate with more dispersed structures.
- Roughness Coefficient: Increased roughness may indicate irregular dispersion patterns [37].
Dispersion Index (DI)
The Dispersion Index integrates multiple CLSM-derived parameters into a single quantitative measure:
Calculation: DI = f(Biomass reduction, Thickness decrease, Structural complexity changes)
This integrated approach enables standardized comparison of dispersion effects across different experimental conditions and treatment modalities [37].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Dispersion Research
| Reagent/Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| Dispersion-Inducing Signals | cis-2-Decenoic acid (CDA), Nitric oxide donors | Experimental induction of dispersion response | Dose-dependent effects; typical range 1-100 μM [37] |
| Matrix Degrading Enzymes | Dispersin B, Alginate lyase, DNase I | Direct degradation of EPS components; positive controls | Useful for validating dispersion assays [36] |
| Fluorescent Stains | SYTO9, Propidium iodide, Calcein AM, TMA-DPH | Viability assessment and biomass quantification | Differential staining for live/dead cells; TMA-DPH for biomass [10] [37] |
| c-di-GMP Modulators | Phosphodiesterase activators, Guanylate cyclase inhibitors | Manipulation of intracellular c-di-GMP levels | Tools for mechanistic studies of regulation [37] |
| Quorum Sensing Inhibitors | Furanones, RNAIII-inhibiting peptides (RIP) | Interference with cell-cell communication | Study connection between QS and dispersion [10] |
| EPS Composition Analysis Kits | Phenol-sulfuric acid (polysaccharides), BCA/Lowry (proteins) | Quantitative assessment of matrix components | Monitor matrix changes during dispersion [37] |
Therapeutic Applications and Translational Potential
The strategic induction of biofilm dispersion creates a therapeutic window of opportunity when newly liberated planktonic cells exhibit transient susceptibility to antimicrobial agents [35] [34]. Several therapeutic approaches leveraging dispersion mechanisms are under investigation:
Enzyme-Based Dispersion Therapies: Localized application of matrix-degrading enzymes such as dispersin B or alginate lyase to disrupt biofilm integrity [36]. Clinical applications include combination therapy with conventional antibiotics for device-related infections [10].
Signal-Mediated Dispersion: Therapeutic application of DSF molecules like CDA to trigger endogenous dispersion mechanisms [37]. This approach demonstrated efficacy in membrane bioreactor systems, reducing biofouling and maintaining treatment efficiency [37].
Anti-biofilm Peptides: Novel peptides such as CRAMP-34 that induce dispersion by enhancing bacterial motility without directly killing cells, potentially reducing selective pressure for resistance [10].
Nanoparticle Delivery Systems: Engineered nanoparticles for targeted delivery of dispersion-inducing compounds to biofilm communities, improving therapeutic precision and reducing off-target effects [2].
Figure 2: Therapeutic strategy for biofilm control through induced dispersion. The workflow illustrates how triggering dispersion creates a vulnerable transitional state where conventional antimicrobials become effective against previously resistant biofilm cells.
Dispersion represents a critical, actively regulated developmental transition in the biofilm life cycle that enables bacterial dissemination and colonization of new environments. The molecular mechanisms underlying this process—particularly the central regulatory role of c-di-GMP signaling and matrix degradation pathways—provide promising targets for novel antibiofilm strategies. Advanced assessment methods, including CLSM analysis and the Dispersion Index, enable quantitative evaluation of dispersion responses in research settings. Therapeutic approaches that exploit dispersion mechanisms hold significant potential for improving outcomes in biofilm-associated infections, particularly when used in combination with conventional antimicrobials. Further research is needed to fully elucidate the complex regulatory networks controlling dispersion and to translate these findings into effective clinical interventions.
The transition from planktonic existence to structured, surface-attached biofilms represents a critical phase in the bacterial life cycle, governed by sophisticated molecular signaling systems. Among these, the second messenger cyclic di-GMP (c-di-GMP) and quorum sensing (QS) mechanisms serve as master regulators that coordinate bacterial lifestyle switching, biofilm development, and virulence expression. This technical review examines the intricate interplay between these signaling pathways, highlighting their convergent roles in regulating biofilm initiation, maturation, and dispersal. Through comprehensive analysis of current research, we demonstrate how c-di-GMP and QS systems form integrated networks that allow bacterial communities to synchronize their behavior in response to population density and environmental cues. The mechanistic insights provided herein offer valuable perspectives for targeting biofilm-associated infections in clinical and industrial contexts.
Biofilm formation constitutes a developmental process encompassing distinct, sequential stages: initial attachment, microcolony formation, maturation, and active dispersal [38]. Each transition through these phenotypic stages is governed by precise regulatory checkpoints that ensure bacterial commitment to the biofilm lifestyle while maintaining capacity for dissemination [39]. The second messenger cyclic di-GMP (c-di-GMP) has emerged as a central regulator driving the lifestyle switch between motile and sessile states, with elevated intracellular c-di-GMP concentrations promoting biofilm formation through exopolysaccharide production, adhesion expression, and motility inhibition [39] [38]. Concurrently, quorum sensing (QS) enables bacterial populations to coordinate group behaviors, including biofilm development, in response to critical threshold concentrations of secreted autoinducer signals [40]. Rather than operating in isolation, these systems form interconnected networks that process environmental and cellular information, creating a sophisticated regulatory circuit for biofilm morphogenesis. This review delineates the molecular mechanisms of c-di-GMP and QS signaling, their documented intersections in various bacterial species, and the experimental approaches driving their characterization.
Molecular Mechanisms of c-di-GMP Signaling
c-di-GMP Metabolism and Effector Systems
The intracellular concentration of c-di-GMP is dynamically regulated through the opposing activities of diguanylate cyclases (DGCs) and phosphodiesterases (PDEs). DGCs, containing conserved GGDEF domains, synthesize c-di-GMP from two GTP molecules, while PDEs, featuring EAL or HD-GYP domains, degrade c-di-GMP to pGpG or GMP, respectively [39] [38]. Pseudomonas aeruginosa, a model organism for biofilm studies, exemplifies the complexity of this signaling network with one of the highest complements of these enzymes: 18 GGDEF domain proteins, 5 EAL domain proteins, 16 hybrid GGDEF-EAL proteins, and 3 HD-GYP domain proteins [39].
c-di-GMP exerts its physiological effects through interaction with diverse effector molecules including PilZ domain proteins, transcription factors, and riboswitches [39] [38]. Upon binding, c-di-GMP modulates their activity, thereby regulating cellular processes such as exopolysaccharide production, flagellar motility, and gene expression. In *P. aeruginosa, elevated c-di-GMP levels (75-110 pmol/mg in biofilms versus <30 pmol/mg in planktonic cells) transcriptionally activate matrix genes (pel, psl, cdrA) while repressing flagellar gene expression and function [39] [38].*
Table 1: Key Enzymes in c-di-GMP Metabolism and Their Functions
| Enzyme Type | Domain | Function | Example | Role in Biofilm |
|---|---|---|---|---|
| Diguanylate Cyclase (DGC) | GGDEF | Synthesizes c-di-GMP from 2 GTP molecules | WspR (P. aeruginosa) | Activates exopolysaccharide production [39] |
| Phosphodiesterase (PDE) | EAL | Hydrolyzes c-di-GMP to linear pGpG | RocR (P. aeruginosa) | Promotes transition to motile lifestyle [39] |
| Phosphodiesterase (PDE) | HD-GYP | Hydrolyzes c-di-GMP to two GMP molecules | PA4108 (P. aeruginosa) | Regulates virulence and motility [39] |
c-di-GMP in Biofilm Development Stages
The role of c-di-GMP extends throughout the biofilm lifecycle, with non-uniform, hierarchical production occurring at specific developmental checkpoints rather than as a uniform cellular response [38]. Surface attachment triggers rapid c-di-GMP production through at least two established sensory systems in P. aeruginosa:
- The Wsp system: Surface contact sensed through membrane perturbation activates the WspA chemoreceptor, leading to phosphorylation of the WspR DGC and subsequent c-di-GMP production that induces Pel and Psl exopolysaccharide synthesis [38].
- The Pil-Chp system: Mechanical tension on type IV pili during surface attachment activates a signaling cascade through PilJ and ChpA, ultimately leading to cAMP-Vfr mediated production of PilY1, which stimulates c-di-GMP synthesis by the SadC DGC [38].
During biofilm maturation, elevated c-di-GMP levels promote matrix production and repress motility, cementing the sessile lifestyle. Finally, during dispersal, activated PDEs degrade c-di-GMP, facilitating the return to planktonic existence [41] [38].
Quorum Sensing Systems in Bacterial Communities
QS Signaling Molecules and Regulatory Networks
Quorum sensing enables bacterial populations to synchronize gene expression in response to cell density through the production, secretion, and detection of extracellular autoinducer molecules. In Gram-negative bacteria, the most prevalent QS signals are N-acyl homoserine lactones (AHLs), which vary in acyl chain length and substitution, determining system specificity [42] [40]. AHLs diffuse across cell membranes and, at sufficient concentrations, bind to and activate transcriptional regulators (LuxR-type proteins), which then modulate expression of target genes governing collective behaviors including bioluminescence, virulence factor production, and biofilm development [40].
Beyond AHLs, additional QS signals include autoinducer-2 (AI-2), a furanosyl borate diester utilized by both Gram-negative and Gram-positive species for interspecies communication, and the putrescine system recently identified in Dickeya oryzae [43] [44]. In Sinorhizobium fredii, QS mechanisms regulate crucial symbiotic functions including exopolysaccharide production, motility, and competitive nodulation in legume hosts [42].
QS in Biofilm Formation and Virulence
QS systems exert pleiotropic effects on biofilm architecture and functionality. In Pseudomonas aeruginosa, the LasI/LasR and RhlI/RhlR systems hierarchically regulate the production of virulence factors and biofilm matrix components [40]. Similarly, in Vibrio cholerae, QS synchronizes the expression of biofilm matrix proteins (RbmA, RbmC, Bap1) and exopolysaccharides (VPS) through the LuxO-HapR regulatory cascade [44]. QS-deficient mutants typically exhibit architecturally compromised biofilms with reduced biomass and enhanced susceptibility to antimicrobial agents, underscoring the critical role of population density signaling in biofilm robustness [40].
Integration of c-di-GMP and Quorum Sensing Signaling
Documented Mechanisms of Cross-Talk
Emerging evidence reveals extensive integration between c-di-GMP and QS pathways, forming sophisticated regulatory networks that fine-tune biofilm development in response to both intracellular and extracellular cues:
- Receptor-Mediated Interaction: In Dickeya oryzae, the c-di-GMP receptor YcgR directly interacts with and enhances the activity of SpeA, the rate-limiting enzyme in putrescine biosynthesis, thereby increasing intracellular putrescine levels. This interaction forms a regulatory loop wherein c-di-GMP molecules inhibit YcgR-mediated SpeA activation, collectively modulating bacterial motility [43].
- QS Regulation of c-di-GMP Metabolism: In Vibrio cholerae, quorum sensing activates expression of nspS-mbaA genes, increasing production of the NspS and MbaA proteins that mediate polyamine-dependent c-di-GMP biosynthesis. This QS-mediated regulation connects population density to environmental polyamine sensing, synergistically enhancing biofilm biomass and cell density [44].
- c-di-GMP Modulation of QS Signal Production: In Sinorhizobium meliloti, elevated c-di-GMP suppresses expression of the AHL synthase gene sinI and reduces AHL accumulation, demonstrating c-di-GMP-mediated repression of QS signaling [42].
Table 2: Documented Cross-Talk Mechanisms Between c-di-GMP and QS Systems
| Bacterial Species | Cross-Talk Mechanism | Biological Outcome | Reference |
|---|---|---|---|
| Dickeya oryzae | c-di-GMP receptor YcgR interacts with putrescine biosynthesis enzyme SpeA | Modulates bacterial motility and biofilm formation [43] | |
| Vibrio cholerae | QS activates nspS-mbaA expression, enhancing norspermidine-stimulated c-di-GMP production | Synergistic increase in biofilm biomass and density [44] | |
| Sinorhizobium meliloti | Elevated c-di-GMP suppresses AHL synthase gene sinI and AHL accumulation | Alters QS-mediated behaviors [42] | |
| Sinorhizobium fredii | c-di-GMP influences AHL production and regulates T6SS activity in strain-specific manner | Impacts symbiotic performance and competitive fitness [42] |
Convergence on Common Physiological Outputs
The integrated c-di-GMP and QS signaling networks ultimately converge to regulate key biofilm determinants:
- Exopolysaccharide Production: Both signaling pathways transcriptionally activate exopolysaccharide biosynthesis genes. In P. aeruginosa, high c-di-GMP induces pel and psl operons, while QS regulates Pel production through the Rhl system [39] [40].
- Motility and Adhesion: c-di-GMP directly inhibits flagellar motility through interaction with motor proteins and chemoreceptors, while QS systems transcriptionally repress flagellar gene clusters in multiple species [39] [40].
- Biofilm Dispersal: Nitric oxide-induced c-di-GMP degradation activates dispersal in P. aeruginosa, while certain QS signals, such as AI-2 in V. cholerae, can repress biofilm formation, creating a bistable switch between collective and individual behaviors [41] [44].
Experimental Approaches and Methodologies
Key Research Models and Techniques
The molecular dissection of c-di-GMP and QS signaling employs sophisticated genetic, biochemical, and biophysical approaches:
- Genetic Manipulation: Construction of in-frame deletions in genes encoding DGCs, PDEs, and QS components, coupled with epistasis analysis, reveals pathway hierarchy and functional relationships. For example, studies in P. aeruginosa identified specific contributions of SadC and RoeA (DGCs) and BifA (PDE) to biofilm formation through comprehensive mutant analysis [39].
- Fluorescent Reporter Systems: Ratiometric fluorescent biosensors (e.g., pCdrA::gfp) enable real-time monitoring of c-di-GMP levels in individual cells within developing biofilms, revealing spatial and temporal heterogeneity in signaling activity [41] [45].
- Microfluidic Biofilm Culture: The "biofilm-dispersal-then-recolonization" (BDR) microfluidic platform enables high-resolution analysis of dispersal dynamics and subsequent colonization capacity under continuous flow conditions, mimicking natural environments [41].
- Biochemical Assays: Quantitative measurements of c-di-GMP (via LC-MS/MS) and autoinducers (via reporter strains or chemical analysis) provide crucial correlation between signaling molecule concentration and phenotypic outputs [43] [42].
Figure 1: Integrated Signaling Network of c-di-GMP and Quorum Sensing in Biofilm Regulation. The diagram illustrates the convergence of surface-derived (c-di-GMP) and population-derived (QS) signals on common biofilm phenotypic outputs.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Investigating c-di-GMP and QS Signaling
| Reagent/Category | Specific Examples | Function/Application | Experimental Use |
|---|---|---|---|
| Genetic Tools | pleD* overexpression construct | Artificially elevates c-di-GMP levels | Studying c-di-GMP effects on exopolysaccharide production [42] |
| bdlA mutant strains | Disrupts chemically-induced dispersal | Analyzing biofilm dispersal mechanisms [41] | |
| Biosensors | pCdrA::gfp reporter | Reports c-di-GMP-mediated transcription | Visualizing c-di-GMP signaling in biofilms [41] |
| AHL bioreporter strains (e.g., A. tumefaciens NT1) | Detects AHL production | Quantifying QS signal production [42] | |
| Chemical Modulators | Sodium nitroprusside (SNP) | Nitric oxide donor induces dispersal | Studying biofilm dispersal pathways [41] |
| Norspermidine | Polyamine modulating c-di-GMP levels | Investigating polyamine-c-di-GMP interactions [44] | |
| Enzymatic Tools | Psl glycosidase | Degrades Psl exopolysaccharide | Studying enzymatic disassembly of biofilms [41] |
| YhjH PDE | Ectopic c-di-GMP degradation | Manipulating intracellular c-di-GMP levels [41] | |
| Model Systems | BDR microfluidic device | "Biofilm-dispersal-then-recolonization" platform | Analyzing dispersal dynamics and recolonization [41] |
| Static biofilm system | Studies early attachment and persistence | Investigating adhesion-linked persister formation [45] |
Detailed Experimental Protocol: Microfluidic Biofilm Dispersal and Recolonization Assay
The BDR microfluidic platform enables precise analysis of biofilm dispersal dynamics and subsequent colonization capacity:
Device Fabrication: Create a polydimethylsiloxane (PDMS) microfluidic device featuring a primary chamber with approximately 300 tapered microwells (150 × 250 × 150 μm) for 3D biofilm cultivation and a secondary chamber for capturing dispersed cells [41].
Biofilm Cultivation: Inoculate the primary chamber with bacterial suspension (e.g., P. aeruginosa PAO1) and perfuse with growth medium at constant flow rate (0.5-2 μL/min) for 12-24 hours to establish mature biofilms [41].
Dispersal Induction: Introduce dispersal stimuli through the flow medium:
- Chemical-induced dispersal (CID): Supplement with 25-500 μM sodium nitroprusside (SNP) as nitric oxide donor or other dispersal signals.
- Enzymatic disassembly (EDA): Introduce 0.1-1 mg/mL Psl glycosidase to degrade exopolysaccharide matrix [41].
Velocity and Population Tracking: Monitor bacterial departure from biofilms using time-lapse confocal microscopy. Track cell velocities and morphological forms (single cells vs. aggregates) using automated tracking software [41].
Recolonization Assessment: Direct flow from primary chamber to secondary chamber containing fresh microwells. Quantify colonization efficiency by measuring biofilm biomass (via crystal violet staining or direct cell counting) after 12-24 hours [41].
Infection Modeling: Incorporate host models (e.g., lung spheroids or C. elegans) in secondary chamber to assess virulence of dispersed populations [41].
Figure 2: Experimental Workflow for Microfluidic Biofilm Dispersal and Recolonization Assay. The diagram outlines the key steps in assessing biofilm dispersal dynamics and subsequent colonization capacity using the BDR platform.
The integrated signaling network comprising c-di-GMP and quorum sensing represents a sophisticated regulatory framework that enables bacterial communities to successfully transition between planktonic and biofilm lifestyles. The documented cross-talk mechanisms reveal how bacteria coordinate intracellular second messenger signaling with intercellular communication to optimize biofilm development in response to both physical surfaces and population density. From a therapeutic perspective, targeting these interconnected pathways offers promising approaches for combating biofilm-associated infections, particularly through dispersal-based strategies that sensitize communities to conventional antibiotics. Future research will undoubtedly uncover additional layers of complexity in these signaling networks, including intersection with other nucleotide second messengers and the role of small non-coding RNAs, providing novel targets for biofilm control across medical and industrial applications.
Advanced Tools and Models for Analyzing Biofilm Architecture and Dynamics
The study of microbial biofilms is critical in areas ranging from medical device infections to industrial corrosion and water treatment. Biofilms are complex three-dimensional microbial communities that adhere to surfaces and interact with their surroundings through a self-produced matrix. Understanding their structure and development requires techniques capable of capturing their dynamic formation and metabolic activity. This technical guide examines established imaging techniques—Scanning Electron Microscopy (SEM) and Confocal Laser Scanning Microscopy (CLSM)—alongside an emerging electrochemical monitoring approach, framing them within the broader context of microbial biofilm formation research. These methodologies enable researchers to quantify biofilm volume, architecture, and viability, providing essential data for developing effective antibacterial strategies and novel biomaterials.
Established Imaging Techniques: SEM and CLSM
Scanning Electron Microscopy (SEM)
Methodology and Workflow: SEM provides high-resolution spatial imaging of biofilm topography. Samples require extensive preparation including fixation, dehydration, and coating with conductive material. Fixation preserves the biofilm structure using chemical agents like glutaraldehyde. Dehydration is typically performed using a graded series of ethanol or acetone to remove water content. Critical point drying is then employed to avoid structural collapse from surface tension. Finally, samples are sputter-coated with a thin layer of gold or platinum to make the non-conductive biological material electrically conductive for imaging. The prepared sample is placed in a high-vacuum chamber where an electron beam scans the surface, and detectors capture emitted secondary or backscattered electrons to generate a topological image.
Technical Considerations: A significant limitation of conventional SEM is the requirement for sample preparation that alters the biofilm's physiological state. The fixation, dehydration, and coating processes can introduce artifacts and do not allow for the observation of live, hydrated biofilms. However, environmental SEM (ESEM) can partially overcome this by allowing the examination of hydrated samples under low-pressure conditions, though with some resolution trade-offs. The strength of SEM lies in its ability to reveal intricate surface details and the ultrastructure of the biofilm matrix at high magnification, providing resolution down to nanometers.
Confocal Laser Scanning Microscopy (CLSM)
Methodology and Workflow: CLSM enables the non-destructive visualization of fully hydrated, living biofilms in three dimensions. The technique relies on fluorescent staining to differentiate cellular components and the extracellular matrix. Common stains include SYTO dyes for nucleic acids in live/dead assays, concanavalin A conjugated with a fluorophore for polysaccharide matrix components, and propidium iodide for compromised cell membranes. The core of CLSM is a pinhole aperture that eliminates out-of-focus light, allowing optical sectioning of the sample. A laser beam scans across sequential focal planes (Z-stacks), and the emitted fluorescence from each plane is captured to reconstruct a 3D representation using image analysis software. Parameters such as biofilm thickness, biovolume, and surface coverage can be quantified.
Technical Considerations: A primary challenge with CLSM is photobleaching, where fluorophores permanently lose their ability to fluoresce due to photon-induced damage. This can limit the duration of real-time observation. While CLSM is superior to SEM for observing living biofilms, its resolution is lower, typically around 200 nanometers laterally. Furthermore, image collection can suffer from user bias if images are taken from different parts of the biofilm rather than through a standardized, randomized approach. Despite these limitations, CLSM remains the gold standard for quantifying the 3D architecture of live biofilms.
Comparative Analysis: SEM vs. CLSM
Table 1: Technical Comparison of SEM and CLSM for Biofilm Research
| Parameter | Scanning Electron Microscopy (SEM) | Confocal Laser Scanning Microscopy (CLSM) |
|---|---|---|
| Resolution | High (nanometer scale) | Lower (approx. 200 nm lateral) |
| Sample State | Fixed, dehydrated, coated (non-viable) | Hydrated, living (viable) |
| Dimensional Data | 2D surface topography | 3D internal architecture |
| Key Artifacts | Shrinkage from dehydration and coating | Photobleaching of fluorophores |
| Quantitative Capabilities | Limited morphological data | Biovolume, thickness, surface coverage |
| Real-time Monitoring | Not suitable | Limited by photobleaching; not ideal for continuous monitoring over days |
Emerging Technique: Electrochemical Impedance Monitoring
Principles and Experimental Protocol
Electrochemical Impedance Spectroscopy (EIS) has emerged as a powerful, non-invasive method for real-time monitoring of biofilm growth on various substrates. The technique measures changes in electrical impedance at the interface between a sensor and the developing biofilm, correlating these changes to biofilm volume and metabolic activity.
Sensor Fabrication and Setup: The impedance sensor is fabricated from a Teflon-coated platinum wire. Approximately 1.5 mm of the Teflon coating is removed, and the exposed Pt wire is washed with absolute alcohol. A conductive polymer layer, typically poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT-PSS), is electrodeposited on the exposed Pt wire via cyclic voltammetry, scanning from -0.2 V to 1.0 V vs Ag/AgCl in a solution containing ~10 mM EDOT and 0.1 mM PSS [46].
Biofilm Growth and Impedance Measurement: Biofilms are grown in appropriate culture media (e.g., baseline mucin medium for dental plaque biofilms) on the substrate of interest, which can include glass, dental filling resin, or Ca²⁺-releasing composites. For real-time monitoring, single-frequency impedance (1 Hz) is recorded at 0.1 V with a sinusoidal amplitude of 5 mV in a two-electrode configuration, where the PEDOT-PSS-deposited sensor serves as both working and reference electrode. Measurements are taken automatically at intervals (e.g., every ~20 minutes) over several days [46].
Data Normalization:
The impedance data is normalized to account for initial sensor variations. The percentage change in impedance is calculated using the formula:
% change in Impedance = (Z_T,1Hz - Z_BMM,1Hz) / Z_PBS,1Hz * 100
where Z_T,1Hz is the real-time impedance during biofilm growth, Z_BMM,1Hz is the stable background impedance in the culture medium, and Z_PBS,1Hz is the stable impedance in phosphate-buffered saline (pH 7.2) used for normalization [46].
Quantitative Correlation and Metabolic Monitoring
This electrochemical method provides a direct quantitative relationship between impedance changes and biofilm volume. Research on multispecies oral biofilms has established the following correlations: impedance changes of 2.5%, 35%, 50%, and 65% correspond to biofilm volumes of 0.10 ± 0.01, 16.9 ± 2.2, 29.7 ± 2.3, and 38.6 ± 2.8 μm³/μm², respectively [46].
The technique also enables parallel monitoring of metabolic activity. When combined with potentiometric pH microsensors, local pH changes at the biofilm-substrate interface can be tracked, providing insights into acid production by bacteria, which varies with biofilm volume. This dual monitoring capability offers a comprehensive view of both physical growth and metabolic state.
Substrate-specific growth rates can also be determined. For example, studies show that the time required to achieve a 50% change in impedance varies by substrate: approximately 3.5 days on glass, 4.5 days on dental filling resin, and 6 days on Ca²⁺-releasing dental composites [46].
Comparative Analysis of Biofilm Quantification Methods
Performance Metrics Across Techniques
Beyond the imaging and monitoring techniques already discussed, several other methods are commonly employed for biofilm quantification, each with distinct advantages and limitations.
Table 2: Comprehensive Comparison of Biofilm Quantification Methods
| Method | Accuracy | Reproducibility | Time Efficiency | Cost | Key Applications |
|---|---|---|---|---|---|
| Electrochemical Impedance | High (+++) | High (++++) | High (+) | Moderate (++) | Real-time monitoring on various substrates |
| SEM | Moderate (++) | Moderate (++) | Low (++++) | High (+++) | High-resolution surface topology |
| CLSM | High (+++) | High (+++) | Moderate (+++) | High (+++) | 3D architecture of live biofilms |
| XTT Assay | High (+++) | High (++++) | High (+) | Moderate (++) | Metabolic activity of viable cells [47] |
| Crystal Violet | Low (+) | High (++++) | Moderate (++) | Low (+) | Total biofilm biomass [47] |
| Viable Colony Counts | Moderate (++) | High (++++) | Low (+++) | Moderate (++) | Enumeration of cultivable cells [47] |
| DNA Quantification | Low (-) | Moderate (+++) | Moderate (++) | High (+++) | Total biological material [47] |
| qPCR | Low (-) | Moderate (++) | Low (+++) | Very High (++++) | Species-specific quantification [47] |
The XTT assay, which measures metabolic activity via tetrazolium salt reduction, has been identified as particularly effective for Candida biofilms, providing reproducible, accurate, and efficient quantitative estimation [47]. Crystal violet staining, while reproducible, may not accurately detect differences in biofilm formation between strains, as it measures total biomass without distinguishing between live cells and matrix [47].
Technical Workflow Integration
The following diagram illustrates the decision pathway for selecting appropriate biofilm analysis techniques based on research objectives:
Essential Research Reagents and Materials
Successful biofilm research requires specific reagents and materials tailored to the chosen methodology. The following table details key solutions and their applications in experimental protocols.
Table 3: Essential Research Reagents for Biofilm Studies
| Reagent/Material | Composition/Type | Function in Biofilm Research |
|---|---|---|
| Baseline Mucin Medium (BMM) | Protease peptone, tryptone, yeast extract, carboxymethylcellulose, KCl | Culture medium for supporting multispecies oral biofilm growth [46] |
| PEDOT-PSS | Poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) | Conductive polymer for electrochemical impedance sensor fabrication [46] |
| XTT Reagent | 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide | Tetrazolium salt used in metabolic assay to quantify viable biofilm cells [47] |
| Crystal Violet | C₂₅H₃₀ClN₃ | Histological stain for quantifying total biofilm biomass [47] |
| SYTO Stains | Various nucleic acid binding dyes | Fluorescent staining for visualizing and quantifying live cells in CLSM [46] |
| RPMI-MOPS Medium | Roswell Park Memorial Institute medium with MOPS buffer | Standardized medium for growing Candida biofilms for antifungal susceptibility testing [47] |
| Glutaraldehyde | C₅H₈O₂ | Fixative agent for preserving biofilm structure for SEM imaging [46] |
The choice of imaging and monitoring technique for biofilm research fundamentally depends on the specific research questions and requirements. SEM provides exceptional surface detail but requires destructive sample preparation. CLSM enables 3D visualization of live biofilms but is limited by photobleaching for extended real-time studies. The emerging electrochemical impedance method offers a powerful alternative for non-invasive, real-time monitoring of biofilm growth across various substrates, with defined correlations between impedance changes and biofilm volume. For comprehensive analysis, researchers often combine multiple techniques—using impedance for continuous growth monitoring, CLSM for architectural analysis, and targeted assays like XTT for viability assessment—to develop a complete understanding of biofilm formation and development across its distinct stages.
Molecular and Genetic Tools for Studying EPS Composition and Gene Expression
The study of Extracellular Polymeric Substances (EPS) is fundamental to understanding the biofilm phenotype, a dominant microbial lifestyle responsible for 65-80% of microbial infections and critical in environmental and industrial contexts [11] [20]. EPS constitute 50-90% of the biofilm's organic matter, forming a protective, structural matrix that encompasses microbial cells and determines the physicochemical properties of the biofilm [48] [11]. This matrix is a complex mixture of primarily polysaccharides, proteins, extracellular DNA (eDNA), and lipids, which interact to provide cohesion, adhesion, and protection against antimicrobials and host immune responses [48] [49]. Within the broader thesis of microbial biofilm formation research, analyzing EPS composition and the genetic regulation of its production is essential for developing targeted anti-biofilm strategies. This guide details the cutting-edge molecular, genetic, and analytical tools enabling researchers to deconstruct the EPS matrix and its regulatory networks.
Analytical Techniques for EPS Composition and Spatial Distribution
A multifaceted approach is required to fully characterize the EPS matrix, given its chemical complexity and dynamic spatial organization. The following techniques can be used in concert to provide a comprehensive analysis.
Table 1: Key Analytical Techniques for EPS Composition
| Technique | Primary Application in EPS Analysis | Key Metrics | Advantages | Limitations |
|---|---|---|---|---|
| ATR/FT-IR Spectroscopy [49] | Chemical content analysis; relative proportions of main EPS classes (proteins, polysaccharides, nucleic acids). | Absorbance bands: Amide I/II (proteins, 1500-1800 cm⁻¹), Polysaccharides (C-O, C-O-C, 900-1250 cm⁻¹), Lipids (C-H, 2800-3000 cm⁻¹). | In-situ, non-destructive analysis of hydrated biofilms; monitors changes in real-time. | Limited penetration depth (~2 µm); semi-quantitative. |
| Confocal Laser Scanning Microscopy (CLSM) [50] | Spatial distribution and 3D architecture of EPS components; biofilm viability. | Fluorescence intensity; layer thickness; biovolume; co-localization coefficients. | Non-destructive, in-situ 3D visualization; can be combined with fluorescent probes. | Requires staining; potential for probe impermeability. |
| Enzymatic Treatment [49] | Functional role determination of specific EPS components in biofilm integrity. | Percentage reduction in biofilm biomass or thickness post-treatment. | Reveals functional importance of targeted EPS (e.g., proteases for proteins, amylases for polysaccharides). | Potential lack of enzyme specificity; may not fully degrade cross-linked components. |
| EPS Extraction & Biochemical Assays [48] [50] | Quantitative and compositional analysis of specific EPS fractions. | Concentration (e.g., mg/g dry weight); carbohydrate-to-protein ratio; monosaccharide composition. | Provides material for detailed chemical analysis. | Method-dependent; may alter native EPS structure or selectively isolate components. |
Detailed Protocol: EPS Disruption via Enzymatic Treatment
This protocol determines the functional contribution of specific EPS polymers to biofilm structural integrity [49].
- Biofilm Growth: Grow biofilms in a relevant in vitro system (e.g., flow cell, microtiter plate, on a coupon material) for a defined period to reach desired maturity.
- Enzyme Preparation: Prepare fresh solutions of hydrolytic enzymes in an appropriate buffer (e.g., PBS for saline compatibility). Common enzymes and their targets include:
- Serine Proteases (e.g., Savinase, Subtilisin A): Target protein moieties in the EPS. Use at concentrations of 10-100 U/mL [49].
- Alpha-Amylase: Targets α-linked polysaccharides. Use at concentrations reported to be effective for S. aureus biofilms [49].
- DNase I: Degrades extracellular DNA (eDNA). Use at a common working concentration of 100 µg/mL.
- Dispersin B: A glycosyl hydrolase that specifically degrades polysaccharides like poly-N-acetylglucosamine (PNAG) [11].
- Treatment: Gently wash the mature biofilm to remove non-adherent cells. Apply the enzyme solution to cover the biofilm completely. Include a buffer-only control.
- Incubation: Incubate under optimal conditions for the enzyme (e.g., 37°C for Savinase) for a defined period, typically 24 hours.
- Analysis: Quantify the remaining biofilm using a method such as:
- Crystal Violet Staining: For total biomass in microtiter plates.
- Viability Staining & CLSM: For biovolume and 3D structure assessment.
- Sessile Cell Counts: By homogenizing and plating biofilm material for colony-forming units (CFUs).
- Interpretation: A significant reduction (e.g., ≥70% biomass reduction with Savinase [49]) in the treated biofilm compared to the control indicates the targeted polymer is critical for biofilm integrity.
Detailed Protocol: In-situ Analysis via ATR/FT-IR Spectroscopy
This technique monitors chemical changes in the biofilm matrix in real-time [49].
- Substrate Preparation: Use a germanium or diamond Internal Reflection Element (IRE) crystal as the biofilm growth substrate.
- Baseline Collection: Collect a background spectrum of the clean, sterile crystal in the presence of the growth medium or water.
- Biofilm Growth and Monitoring: Inoculate the medium with the test organism and allow biofilm formation directly on the IRE crystal. The ATR/FT-IR instrument can collect spectra at regular intervals through the hydrated biofilm.
- Spectral Acquisition: Collect infrared spectra over the 900-3000 cm⁻¹ range. The evanescent wave penetrates approximately 2 µm into the biofilm, analyzing the basal layers.
- Data Analysis: Analyze the acquired spectra by focusing on key absorption bands:
- Monitor the amide II/PS band intensity ratio to track relative changes in protein vs. polysaccharide content (decrease indicates preferential polysaccharide production) [49].
- Track the PO/PS band intensity ratio to observe nucleic acid synthesis relative to polysaccharides [49].
- Observe overall band intensity changes to monitor biomass accumulation and detachment.
Figure 1: ATR/FT-IR spectroscopy workflow for in-situ biofilm analysis.
Genetic Tools for Profiling Gene Expression in Biofilms
Understanding the genetic regulation of EPS production and the biofilm lifestyle requires comparing gene expression between planktonic and sessile cells. Key technologies for transcriptome profiling are summarized below.
Table 2: Comparison of Gene Expression Analysis Methods
| Method | Principle | Throughput | Key Advantages for Biofilm Research | Considerations |
|---|---|---|---|---|
| RNA Sequencing (RNA-Seq) [51] [52] | cDNA synthesis from total RNA followed by high-throughput sequencing. | High | Detects known and novel transcripts; broad dynamic range; applicable to any species; identifies allele-specific expression and gene fusions. | Data-intensive; requires bioinformatics expertise; can be time-consuming for low target numbers. |
| Gene Expression Microarrays [51] [52] | Hybridization of labeled cDNA to pre-defined, gene-specific probes on a chip. | High | High sample throughput for known genes; familiar workflow. | Cannot detect novel transcripts; limited dynamic range due to background and signal saturation. |
| qRT-PCR [51] [52] | Reverse transcription followed by fluorescent-based quantitative PCR. | Low | Gold standard for sensitivity and accuracy for few (<10) targets; fast; uses common lab equipment. | Low scalability; only detects known sequences. |
| Single-Cell RNA-Seq (scRNA-Seq) [51] | Barcoding and sequencing of RNA from individual cells. | Very High | Reveals heterogeneity within biofilm subpopulations; identifies metabolic states and key drivers of differentiation. | Technically challenging; expensive; data analysis is complex. |
Detailed Protocol: Transcriptome Profiling via RNA-Seq
This protocol outlines the steps for comparing gene expression between planktonic and biofilm populations [51].
- Sample Preparation:
- Planktonic Culture: Grow bacteria to the desired growth phase (e.g., mid-exponential) and harvest cells by centrifugation.
- Biofilm Culture: Grow biofilms in a system that allows for easy harvesting (e.g., biofilm reactors with scraping, or direct lysis on growth surfaces). Multiple biological replicates are crucial.
- RNA Extraction & Quality Control: Lyse cells using a mechanical method (e.g., bead beating) effective against biofilm matrices. Extract total RNA using a commercial kit designed to efficiently recover RNA and remove genomic DNA. Assess RNA integrity and purity (e.g., RIN > 8.0 using Bioanalyzer).
- Library Preparation & Sequencing: Deplete ribosomal RNA from the total RNA to enrich for mRNA. Synthesize cDNA and attach sequencing adapters. Use a platform like Illumina for high-throughput sequencing.
- Bioinformatic Analysis:
- Quality Control & Trimming: Use tools like FastQC and Trimmomatic to assess read quality and remove adapter sequences.
- Alignment: Map cleaned reads to a reference genome using aligners like Bowtie2 or HISAT2.
- Quantification & Differential Expression: Count reads mapping to genes/transcripts with featureCounts. Use packages like DESeq2 or edgeR in R to identify statistically significant differentially expressed genes (DEGs) between planktonic and biofilm conditions.
- Validation: Confirm key DEGs using an independent method like qRT-PCR.
Figure 2: RNA-Seq workflow for biofilm transcriptome analysis.
Integrating Tools: Signaling Pathways in Biofilm Development
The formation of biofilms is a genetically regulated process. A key intracellular signaling molecule is bis-(3',5')-cyclic dimeric guanosine monophosphate (c-di-GMP). A high level of c-di-GMP promotes the transition from motility to sessility by repressing flagella synthesis and stimulating EPS matrix production [11]. Genetic tools like mutagenesis and transcriptomics have identified key genes involved in this pathway.
Figure 3: Simplified c-di-GMP signaling pathway in biofilm formation.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents for EPS and Gene Expression Studies
| Reagent / Material | Function / Application | Example Use-Case |
|---|---|---|
| Hydrolytic Enzymes [49] | Functional dissection of EPS matrix; biofilm disruption. | Savinase (protease) for digesting proteinaceous matrix components; Alpha-amylase for polysaccharide degradation. |
| Fluorescent Probes & Stains [50] | Visualization and quantification of EPS components and live/dead cells via CLSM. | ConA lectin conjugate for staining polysaccharides; SYTO dyes for nucleic acids (eDNA); FITC for proteins. |
| Diguanylate Cyclase / Phosphodiesterase Mutants [11] | Genetic manipulation of c-di-GMP levels to study its role in EPS regulation. | Overexpression of diguanylate cyclase to lock cells in a high c-di-GMP, biofilm-forming state. |
| RNA Stabilization & Extraction Kits | Preservation and isolation of high-quality RNA from robust biofilm structures. | Commercial kits with bead-beating lysis for efficient mechanical disruption of biofilm matrix and cells. |
| Next-Generation Sequencing Kits [51] | Preparation of RNA-Seq libraries for whole-transcriptome analysis. | Kits for rRNA depletion, cDNA synthesis, and adapter ligation compatible with Illumina platforms. |
| Quorum Sensing Inhibitors [11] | Interruption of cell-cell communication to study its impact on EPS production and biofilm maturation. | Use of furanones or other small molecules to block acyl-homoserine lactone (AHL) signaling in Gram-negative bacteria. |
In Vitro and In Vivo Models for Studying Biofilm-Associated Infections
Microbial biofilms are structured communities of cells enclosed in a self-produced extracellular polymeric substance (EPS) matrix that are attached to biotic or abiotic surfaces [53]. This mode of growth presents a substantial global public health challenge, particularly in healthcare settings where biofilm formation on medical devices and tissues significantly enhances antimicrobial resistance (AMR) [54]. The protective biofilm matrix contributes to bacterial persistence and adaptation, exacerbating multidrug resistance (MDR) in nosocomial infections and leading to elevated patient morbidity and mortality [54]. These infections also impose considerable economic burdens on healthcare systems due to increased costs and prolonged hospital stays [54]. Understanding the unique biology of biofilm-associated infections is essential for developing adequate detection, prevention, and therapeutic strategies.
The research journey to understand biofilms spans from fundamental investigations of the initial attachment of planktonic cells to surface through microcolony formation and maturation to eventual dispersal [55]. Each stage presents distinct research challenges that require specialized models. The transition of bacteria from individual planktonic cells to organized sessile communities represents a profound shift in microbial behavior that demands sophisticated experimental approaches to replicate in laboratory settings. The complex interactions between microbial cells and host tissues, medical implants, and antimicrobial agents cannot be fully understood using simplistic monolayer cultures alone. This technical guide provides researchers with a comprehensive overview of current in vitro and in vivo models for studying biofilm-associated infections, with particular emphasis on their applications, limitations, and appropriate contexts for use within the broader framework of microbial biofilm research.
Biofilm Formation Stages and Research Objectives
The biofilm lifecycle follows a consistent developmental pattern across bacterial species, characterized by distinct phases that present unique research opportunities [55]. The attachment phase involves initial reversible adhesion of planktonic cells to surfaces through transient interactions mediated by surface chemistry, morphology, and environmental conditions [55]. During early development, surface colonization occurs through bacterial division, recruitment of additional cells, and initial EPS production, leading to microcolony formation [55]. The maturation phase involves the development of complex three-dimensional structures with characteristic architectures that vary by bacterial species and environmental conditions [55]. Finally, the dispersion phase sees the controlled release of cells from the biofilm through either external mechanical forces (detachment) or internally programmed responses to stimuli such as nutrient depletion or oxygen variations [55].
Each stage of biofilm development requires specific research approaches and model systems. The table below outlines key research questions appropriate for each phase:
Table 1: Research Focus Areas Across Biofilm Developmental Stages
| Biofilm Stage | Key Research Questions | Preferred Model Systems |
|---|---|---|
| Attachment | Mechanisms of initial adhesion, surface properties affecting colonization, early transcriptional changes | Flow cells, microfluidic devices, modified MBEC assays [55] [56] |
| Early Development | Microcolony formation, initial matrix production, early community interactions | Microfluidic systems, confocal microscopy with time-lapse imaging [55] |
| Maturation | 3D architecture development, gradient formation, metabolic specialization, antibiotic tolerance | Advanced 3D in vitro models, organotypic models, confocal microscopy with image analysis [57] [58] |
| Dispersion | Regulatory triggers, enzymatic matrix degradation, transition to planktonic state | In vivo infection models, complex 3D co-culture systems [55] |
The complexity of biofilm structure presents particular challenges for researchers. Biofilms are heterogeneous with clusters of cells incorporated in EPS and interstitial voids that facilitate transport processes [55]. This spatial organization creates gradients of nutrients, oxygen, and metabolic waste products that lead to distinct microbial subpopulations with different metabolic activities and phenotypic states [55]. The presence of dormant "persister cells" in protected regions of biofilms contributes significantly to antibiotic tolerance and chronic infection recurrence [55]. Understanding these complexities requires models that can replicate key aspects of the biofilm microenvironment.
In Vitro Models for Biofilm Research
Traditional and Advanced 2D Models
Traditional two-dimensional (2D) in vitro models have served as fundamental tools in biofilm research for decades. These include static systems such as microtiter plate assays, where biofilms form on the walls and bottoms of wells, and the Calgary Biofilm Device (MBEC assay) which uses specialized lids with pegs to support high-throughput biofilm formation [56] [6]. Modifications of these systems now incorporate stainless steel magnetic pins technology to improve reproducibility and better simulate industrial or medical implant surfaces [56]. While these traditional 2D models offer advantages in throughput, cost-effectiveness, and simplicity, they significantly simplify the complexity of real biofilm environments by lacking physiological cell-to-cell contacts, three-dimensional architecture, and relevant substrate properties [57].
Dynamic 2D flow systems represent an advancement over static models by incorporating fluid shear forces that more closely mimic natural and clinical environments. These include flow cell reactors with controlled laminar flow across surfaces, which enable real-time microscopic observation of biofilm development [55]. Rotating disk systems create defined hydrodynamic conditions that influence biofilm thickness and structure, while drip flow reactors simulate low-shear environments similar to medical implant sites [55]. These dynamic models allow researchers to study biofilm development under conditions that more closely resemble in vivo environments, where flow conditions significantly impact biofilm structure, thickness, and resistance mechanisms.
Advanced 3D and Organotypic Models
Advanced three-dimensional (3D) in vitro models have emerged to bridge the gap between traditional 2D models and in vivo experiments [57]. These systems offer more physiological cell-to-cell contacts and better replicate the interactions between tissue cells, implant materials, and pathogenic microorganisms [57]. Scaffold-based 3D models use structures made from various materials (ranging from soft hydrogels to rigid ß-TCP) to guide tissue-like cellular growth [57]. Transwell systems with semipermeable membranes can separate different cell types while allowing interaction via soluble substances, creating more realistic tissue interfaces [57]. These advanced models can incorporate human cells, ECM components, or be integrated with other tissue models to better mimic host-pathogen interactions [55].
Organotypic 3D models have been developed for specific infection scenarios. For dental implants, researchers have created models incorporating fibroblasts and keratinocytes to replicate the gum tissue interface [57]. For orthopedic applications, models using stem cells and fibroblast-like cells on novel biomaterials help predict implant integration and infection risk [57]. More complex systems incorporate immune components, such as co-cultures of fibroblasts and THP-1 derived macrophages, to study host-pathogen interactions in biofilm-associated infections [57]. These advanced models allow investigation of biofilm development in contexts that closely resemble specific infection sites, providing more clinically relevant data before proceeding to animal studies.
Table 2: Comparison of Advanced 3D In Vitro Biofilm Models
| Model Type | Key Components | Applications | Limitations |
|---|---|---|---|
| Scaffold-based | Hydrogels, rigid scaffolds (e.g., ß-TCP), tissue cells | Biomaterial testing, biofilm-biomaterial interactions | Variable reproducibility, may lack host factors [57] |
| Organotypic Dental | Fibroblasts, keratinocytes, implant materials | Periodontal infections, peri-implantitis | Limited complexity compared to oral microenvironment [57] |
| Organotypic Orthopedic | Stem cells, fibroblast-like cells, implant materials | Prosthetic joint infections, biofilm formation on implants | Does not fully replicate bone physiology [57] |
| Immune-component | THP-1 derived macrophages, fibroblasts, bacteria | Host-pathogen interactions, immune response to biofilms | Simplified immune representation [57] |
Microfluidic and Microcosm Models
Microfluidic devices, often called "lab-on-a-chip" systems, represent a cutting-edge approach to biofilm research by enabling precise control over spatial and temporal environmental conditions [55]. These systems can create stable chemical gradients that mimic conditions in chronic infections, allow real-time imaging of biofilm development, and subject biofilms to controlled shear stresses [55]. Microfluidic platforms are particularly valuable for studying early attachment and microcolony development phases under conditions that closely simulate natural environments, including nutrient gradients, antimicrobial penetration, and bacterial responses to changing conditions.
Microcosm models represent the most advanced category of in vitro systems, incorporating multiple elements of the natural environment to create highly realistic experimental conditions [55]. These models may include relevant human cell types, flow conditions, and multiple bacterial species to better simulate the complex interactions occurring in clinical biofilm infections. By incorporating human cells alongside bacterial populations, microcosm models enable researchers to study the bidirectional interactions between host and pathogen, including immune responses, tissue damage, and bacterial evasion mechanisms [55]. While these systems offer unprecedented biological relevance, they also present challenges in standardization, reproducibility, and technical complexity that may limit their widespread adoption.
In Vivo Models for Biofilm Research
Animal Models of Device-Associated Infections
In vivo models remain essential for understanding biofilm pathogenesis and evaluating potential therapeutic interventions, as they replicate the complex host-pathogen interactions that cannot be fully captured in vitro [59]. For device-associated infections, researchers have developed various animal models using different implant types and anatomical locations. Central venous catheter models in rats or mice allow study of Candida albicans and bacterial biofilms on intravascular devices [59]. Urinary catheter infection models in rabbits or mice enable investigation of biofilm formation in the urinary tract, particularly with pathogens like Escherichia coli and Pseudomonas aeruginosa [59]. Orthopedic implant models use rats or other animals to study biofilm formation on fracture fixation devices or joint prostheses, primarily with Staphylococcus aureus and Staphylococcus epidermidis [59].
These device-associated infection models typically involve surgical implantation of the device, followed by direct inoculation with the pathogen. The infected implants can be harvested after predetermined time periods to quantify bacterial burden, examine biofilm structure, and assess host tissue response [59]. More sophisticated versions of these models incorporate non-invasive imaging techniques, such as bioluminescent imaging, to monitor infection progression in real time without sacrificing animals [59]. These approaches allow for longitudinal studies and reduce the number of animals required for research, aligning with the 3R principles (Replacement, Reduction, and Refinement) of animal research ethics.
Tissue-Specific Infection Models
Beyond device-related infections, researchers have developed specialized animal models to study biofilm formation in specific tissues and organs. Pulmonary infection models, particularly in mice, mimic chronic Pseudomonas aeruginosa lung infections in cystic fibrosis patients and allow evaluation of antibiotic efficacy against biofilm-associated respiratory infections [59]. Otitis media models in chinchillas or other animals recreate middle ear infections and enable study of biofilm formation on mucosal surfaces [59]. Chronic wound models typically use pigs or mice to investigate biofilm development in cutaneous wounds and evaluate topical antimicrobial treatments [54]. Endocarditis models in rats study biofilm formation on heart valves, providing insights into this life-threatening infection [59].
Each tissue-specific model presents unique advantages and limitations. Porcine wound models closely resemble human skin anatomy and healing processes, providing highly relevant data for topical anti-biofilm treatments [54]. Mouse pulmonary infection models allow researchers to utilize the extensive available reagents for immunological studies but may not fully replicate human respiratory physiology [59]. Chinchilla otitis media models enable direct visualization and sampling of middle ear biofilms but require specialized facilities and expertise [59]. Researchers must select animal models based on the specific research questions, considering the anatomical, physiological, and immunological similarities to human infections.
Quantitative Assessment Methodologies
Traditional Quantification Methods
Accurate quantification of biofilm formation is essential for evaluating antibacterial efficacy and understanding biofilm biology. Traditional methods include colony forming unit (CFU) counting, which determines the number of viable cells in a biofilm through serial dilution and plating on agar media [6]. While this method provides information about cultivable cells, it is time-consuming, labor-intensive, and may underestimate bacterial numbers due to clumping or the presence of viable but non-culturable cells [6]. Crystal violet staining represents another common approach that measures total biofilm biomass through binding to cells and matrix components [6]. This method offers good reproducibility and throughput but does not distinguish between live and dead cells or provide information about biofilm architecture.
More advanced traditional methods include ATP bioluminescence, which quantifies metabolically active cells through measurement of cellular ATP content [6]. This approach provides rapid results and high sensitivity but may be influenced by environmental factors and the metabolic state of cells. The quartz crystal microbalance technique measures mass accumulation on sensor surfaces with exceptional sensitivity, enabling real-time monitoring of biofilm development [6]. These traditional quantification methods each provide specific types of information about biofilms, from viable cell counts to total biomass, and selection of appropriate methods depends on the specific research questions and available resources.
Advanced Imaging and Analysis Techniques
Modern biofilm research increasingly relies on advanced imaging technologies that preserve the three-dimensional structure of biofilms while providing detailed compositional information. Confocal laser scanning microscopy (CLSM) enables optical sectioning of thick biofilms without physical disruption, allowing visualization of the spatial organization of cells and matrix components [57] [58]. When combined with fluorescent markers specific for different bacterial species, matrix components, or gene expression patterns, CLSM provides rich information about biofilm architecture and heterogeneity [58]. Scanning electron microscopy (SEM) offers ultra-high resolution images of biofilm surface structures but requires extensive sample preparation that may introduce artifacts [6].
The field of biofilm image analysis has been revolutionized by sophisticated software tools that extract quantitative data from complex three-dimensional image sets. BiofilmQ represents a comprehensive image cytometry software tool for automated, high-throughput quantification, analysis, and visualization of numerous biofilm-internal and whole-biofilm properties in three-dimensional space and time [58]. This platform can analyze images ranging from microcolonies to millimetric macrocolonies, quantifying structural parameters, fluorescence intensities, and spatial relationships within biofilms [58]. COMSTAT, an earlier but still valuable tool, provides quantitative analysis of biofilm thickness, roughness, and surface coverage from image stacks [58]. These computational approaches have become essential for objective, reproducible analysis of biofilm properties across different experimental conditions.
Experimental Protocols for Key Methodologies
Protocol for 3D Organotypic Co-culture Model of Implant Infection
This protocol describes the establishment of a sophisticated 3D organotypic model for studying biofilm formation on implant materials in the context of host cells, adapted from recent literature [57].
Materials:
- Appropriate human cell types (e.g., fibroblasts, keratinocytes for dental models; stem cells for orthopedic models)
- Bacterial strains of interest (e.g., Staphylococcus aureus for orthopedic applications)
- Implant material specimens (e.g., titanium disks, polymer surfaces)
- Cell culture medium appropriate for selected human cells
- Bacterial culture medium
- Transwell inserts or custom scaffold systems
- Fixation and staining reagents for downstream analysis
Method:
- Prepare the implant material specimens according to manufacturer specifications, including sterilization and surface conditioning if required.
- Seed human cells onto the implant materials or surrounding scaffold at appropriate densities and culture for 7-14 days to allow tissue maturation.
- For immune-component models, differentiate THP-1 monocytes into macrophages using phorbol esters and add to the system during the tissue maturation phase.
- Inoculate with bacterial suspension at defined multiplicities of infection (MOI), typically ranging from 1:1 to 100:1 (bacteria:human cells).
- Maintain co-cultures for desired infection periods (typically 24-72 hours) with appropriate culture conditions for both cell types.
- For analysis, carefully separate implant materials from surrounding tissue constructs for independent assessment.
- Process samples for viability assays, histological analysis, gene expression studies, or imaging according to experimental requirements.
Applications: This model is particularly valuable for studying early events in implant-associated infections, host-pathogen interactions, and efficacy of antimicrobial coatings or treatments. It enables researchers to investigate how host cells influence biofilm formation and how biofilms affect host cell viability and function, providing insights beyond monoculture biofilm models.
Protocol for In Vivo Assessment of Anti-biofilm Efficacy Using a Porcine Wound Model
This protocol outlines the evaluation of anti-biofilm treatments using a porcine wound model, adapted from studies investigating nitric oxide formulations against MRSA biofilms [54].
Materials:
- Specific pathogen-free pigs (appropriate age and size)
- Clinical bacterial isolate (e.g., MRSA from nasal colonization)
- Test articles (e.g., nitric oxide formulations, antibiotic comparators like Mupirocin 2%)
- Wound creation apparatus (e.g., dermatome for partial-thickness wounds)
- Anesthesia and analgesic agents
- Sampling equipment (biopsy punches, sterile swabs)
- Materials for bacterial quantification (homogenizer, culture media)
Method:
- Anesthetize animals according to approved ethical protocols and maintain anesthesia throughout procedure.
- Create standardized deep partial-thickness wounds on dorsal skin using dermatome set to appropriate depth.
- Inoculate wounds with bacterial suspension (typically 10^7-10^8 CFU) and cover with semi-occlusive dressing for 24-48 hours to establish infection.
- Confirm biofilm establishment by sampling before initiating treatment.
- Apply test articles to wounds according to treatment schedule (e.g., once or twice daily for 7 days).
- Monitor wounds daily for clinical signs of infection and document with photography.
- Collect tissue biopsies at predetermined endpoints using sterile punch biopsy instruments.
- Homogenize tissue samples in appropriate buffer and perform serial dilutions for CFU enumeration on selective media.
- Process additional tissue samples for histology to assess biofilm structure and host response.
Applications: This model is highly relevant for preclinical evaluation of topical anti-biofilm treatments, particularly for chronic wound infections. The porcine skin model closely replicates human skin anatomy and healing processes, providing translational data for clinical development. The model enables quantification of bacterial burden reduction and assessment of wound healing progression in the context of biofilm infection.
Visualization of Biofilm Research Workflows
The following diagram illustrates the integrated experimental workflow for developing and evaluating anti-biofilm strategies, highlighting the complementary roles of in vitro and in vivo models:
Biofilm Research Workflow: This diagram outlines the sequential and iterative process for developing anti-biofilm strategies, beginning with high-throughput in vitro screening and progressing through increasingly complex models to clinical translation.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Biofilm Studies
| Category | Specific Examples | Research Applications | Technical Notes |
|---|---|---|---|
| Bacterial Strains | Pseudomonas aeruginosa (PAO1), Staphylococcus aureus (USA300), ESKAPE pathogens | Pathogenesis studies, antimicrobial testing | Clinical isolates provide greater translational relevance [54] |
| Implant Materials | Medical-grade titanium, surgical steel, polymer specimens | Biomaterial-associated infection studies | Surface roughness and chemistry significantly impact bacterial adhesion [57] |
| Cell Lines | THP-1 (macrophages), primary fibroblasts, keratinocytes | Host-pathogen interaction studies | Primary cells often better maintain physiological responses [57] |
| Matrix Stains | Concanavalin-A, SYPRO Ruby, FilmTracer FM | EPS visualization and quantification | Multi-channel staining reveals matrix architecture [58] |
| Viability Assays | LIVE/DEAD BacLight, ATP bioluminescence, resazurin | Antimicrobial efficacy testing | Combine multiple assays for comprehensive assessment [6] |
| Analysis Software | BiofilmQ, COMSTAT, ImageJ plugins | Image analysis and quantification | BiofilmQ enables single-cell resolution in 3D space [58] |
The strategic selection of appropriate in vitro and in vivo models is paramount for advancing our understanding of biofilm-associated infections and developing effective countermeasures. Simple 2D models remain valuable for high-throughput screening, while advanced 3D organotypic systems and microfluidic devices better replicate the complexity of in vivo conditions. Animal models continue to provide essential insights into host-pathogen interactions and therapeutic efficacy. The integration of quantitative assessment tools, particularly advanced imaging and computational analysis, enables researchers to extract meaningful data from these complex systems. As biofilm research continues to evolve, the development of even more sophisticated models that better replicate human physiology and disease states will accelerate the translation of basic research findings into clinical applications that address the significant challenges posed by biofilm-associated infections.
Computational and Machine Learning Approaches for Anti-biofilm Peptide Discovery
Microbial biofilms represent a structured growth state of bacterial communities, encased within a self-produced extracellular matrix and adhered to biological or abiotic surfaces [60]. Within the broader context of microbial biofilm formation research, it is now understood that biofilms are responsible for 65-80% of all microbial infections and are particularly prevalent in chronic conditions and medical device-related infections [61]. The biofilm growth state provides significant protection against conventional antibiotics, with bacteria within biofilms exhibiting 10- to 1000-fold greater resistance compared to their planktonic counterparts [62]. This resistance, combined with the fact that no approved anti-biofilm agents currently exist for clinical use, has created an urgent need for novel therapeutic strategies [61].
The traditional antibiotic discovery pipeline has proven inadequate for addressing the unique challenges posed by biofilm-associated infections. Anti-biofilm peptides (ABPs) have emerged as promising therapeutic candidates due to their ability to target biofilm-specific mechanisms while potentially minimizing the development of resistance [63]. The application of computational and machine learning approaches has significantly accelerated the discovery and optimization of these peptides, enabling researchers to navigate the vast combinatorial space of peptide sequences and identify candidates with desired properties [64] [65].
Biofilm Formation Stages and Computational Targeting Opportunities
The development of bacterial biofilms progresses through well-characterized stages, each presenting distinct opportunities for computational targeting and therapeutic intervention.
Stage 1: Initial Attachment and Reversible Adhesion
The biofilm lifecycle begins when planktonic bacteria attach to surfaces using weak, reversible adhesins. Computational targeting: Molecular dynamics simulations can model peptide interactions with surface adhesins, while machine learning can predict sequences that disrupt initial attachment without lethal pressure [60].
Stage 2: Irreversible Attachment and Microcolony Formation
Bacteria transition to irreversible attachment, forming structured microcolonies and beginning extracellular polymeric substance (EPS) production. Computational targeting: QSAR modeling and complex network analysis identify peptides that inhibit EPS matrix components or interfere with cell-cell adhesion mechanisms [66] [62].
Stage 3: Biofilm Maturation and Quorum Sensing
The biofilm develops its complex 3D architecture with water channels, while quorum sensing (QS) systems coordinate population-level behaviors. Computational targeting: Neural networks can design QS inhibitors, while classification models identify peptides that disrupt LuxR-type or peptide-based QS systems without affecting growth [61] [63].
Stage 4: Dispersion and Dissemination
Mature biofilms release planktonic cells to colonize new niches, often through enzymatic matrix degradation or phenotypic differentiation. Computational targeting: Regression models trained on MBEC (minimum biofilm eradication concentration) data identify peptides that trigger dispersion mechanisms or target dispersion-specific cell types [63].
Table 1: Key Signaling Pathways in Biofilm Formation and Computational Targeting Approaches
| Regulatory System | Role in Biofilm Formation | Computational Targeting Strategy | Validated Peptide Examples |
|---|---|---|---|
| Stringent Response (ppGpp) | Stress response activation, biofilm matrix production | Natural language processing to identify peptides that bind (p)ppGpp | Cationic amphipathic peptides targeting (p)ppGpp degradation [61] |
| c-di-GMP Signaling | Regulation of biofilm formation, matrix production | Structure-based design of c-di-GMP sequestering peptides | CSP peptide derived from CheY-like protein in Caulobacter crescentus [61] |
| Quorum Sensing Systems | Coordination of population behaviors, virulence expression | SVM classifiers trained on QS peptide sequences to identify inhibitors | Synthetic derivatives of LL-37 and Indolicidin [61] |
| Two-Component Systems | Environmental sensing, regulation of attachment genes | Molecular descriptor analysis to find kinase domain inhibitors | Peptides targeting AgrC in S. aureus [60] |
Machine Learning Frameworks for ABP Discovery
Feature Representation and Descriptor Calculation
The accurate computational representation of peptide sequences is fundamental to machine learning success. Multiple feature representation approaches have been developed:
Alignment-free sequence descriptors capture physicochemical properties including hydrophobicity, charge, residue distribution, and structural propensity without requiring sequence alignment [66]. For each peptide sequence, feature vectors are calculated considering the primary structure, the order of amino acids, their physicochemical properties, and their distributions [67].
3D molecular descriptors derived from quantitative structure-activity relationship (QSAR) modeling serve as numerical representations of the chemical information within peptide three-dimensional structures [62]. These include:
- TDB02s: 2D autocorrelation descriptors
- RDF040v and RDF130s: Radial distribution function descriptors
- Mor09v, Mor02s, Mor06s: 3D-MoRSE descriptors
- HATS0s: Leverage-weighted autocorrelation descriptors [62]
Classification Algorithms for ABP Identification
Supervised machine learning algorithms have demonstrated remarkable accuracy in classifying peptides with antibiofilm activity:
Table 2: Performance Metrics of Machine Learning Classification Models for ABP Prediction
| Model Architecture | Accuracy | Precision | MCC | F1 Score | Dataset Characteristics |
|---|---|---|---|---|---|
| Integrated ML/Structural Analysis [67] | 99% | 99% | 0.97 | 0.99 | Feature space with high antibiofilm activity |
| Classification-Regression Pipeline [63] | >98% | N/R | >0.90 | >0.90 | 242 ABPs vs. 10x negative dataset |
| SVM with Physicochemical Features [63] | N/R | N/R | 0.84 | N/R | BaAMPs database peptides |
| Weka-based Models [63] | N/R | N/R | 0.91 | N/R | BaAMPs with quorum-sensing negative set |
Advanced ML Pipelines for Sequence Mining
Recent approaches have implemented sophisticated multi-stage pipelines following a coarse-to-fine design principle. Huang et al. developed a cascading pipeline consisting of multiple machine learning modules that gradually narrow down the search space from hundreds of billions of possible sequences [65]. This pipeline includes:
- Empirical selection modules that perform initial filtering of sequence space
- Classification modules that distinguish antimicrobial from non-antimicrobial sequences
- Ranking modules that prioritize sequences by predicted potency
- Regression modules that predict minimum inhibitory concentrations [65]
This approach identified hexapeptides with strong activity against multidrug-resistant pathogens and demonstrated efficacy in murine pneumonia models with negligible toxicity and low resistance propensity [65].
Experimental Protocols for ABP Validation
QSAR-Driven Peptide Discovery Protocol
Objective: To discover novel anti-biofilm peptides using quantitative structure-activity relationship modeling.
Methods:
- Template Selection: Begin with a known antibiofilm peptide template (e.g., IDR-1018: VRLIVAVRIWRR-NH2) [62]
- SPOT-Synthesis: Create a comprehensive substitution library (96 single amino acid variants) on cellulose arrays [62]
- Biofilm Inhibition Screening: Test variants against MRSA biofilms using high-throughput assays [62]
- Descriptor Calculation: Generate ~2500 molecular descriptors based on 3D peptide structures [62]
- Model Training: Apply Linear Discriminant Analysis with forward stepwise variable selection [62]
- Virtual Screening: Deploy optimized QSAR model to screen 100,000 virtual peptide sequences [62]
- Validation: Synthesize and test top candidates, achieving ~85% prediction accuracy [62]
Key Result: Identification of peptide 3002 (ILVRWIRWRIQW-NH2) with 8-fold increased antibiofilm potency compared to 1018 and significant abscess reduction in a chronic MRSA mouse model [62].
Complex Network Analysis Protocol
Objective: To identify privileged scaffolds and motifs for next-generation antimicrobials using complex networks.
Methods:
- Data Curation: Collect 174 non-redundant antibiofilm peptides from StarPepDB after removing duplicates at 98% similarity [66]
- Network Construction: Build half-space proximal networks (HSPNs) using alignment-free sequence descriptors and Euclidean distance metrics [66]
- Centrality Analysis: Calculate betweenness, closeness, and degree centrality to identify representative and atypical ABPs [66]
- Motif Discovery: Extract recurring structural motifs within network communities [66]
- Metadata Integration: Construct multilayer networks (METNs) to incorporate associated metadata including origin, additional activities, and targets [66]
Key Result: Identification of a reduced but informative set of 66 ABPs representing the original antibiofilm space, containing both central and atypical sequences with desired properties for developing next-generation antimicrobials [66].
Diagram 1: Machine Learning Pipeline for Anti-biofilm Peptide Discovery. This workflow illustrates the integrated computational and experimental approach for identifying novel ABPs, from data collection through experimental validation.
Integrative Computational Approaches
Multi-Omics Integration for Target Identification
Systems biology approaches integrate genomics, transcriptomics, proteomics, and metabolomics data to identify novel targets for anti-biofilm therapy. Transcriptomic analyses of biofilm-forming bacteria have revealed differential expression of genes involved in stress response, matrix production, and metabolic adaptation [61]. By mapping these expression changes onto protein-protein interaction networks, researchers can identify central hubs that represent promising targets for peptide-based intervention [60].
Hybrid Modeling Frameworks
The combination of mechanistic modeling with machine learning has emerged as a powerful approach for understanding biofilm dynamics and predicting peptide efficacy. Continuum-discrete models that simulate biofilm growth, substrate diffusion, and antimicrobial penetration can generate synthetic training data for machine learning algorithms [68]. These hybrid frameworks can predict how peptide properties (charge, hydrophobicity, structure) influence penetration through the biofilm matrix and interaction with bacterial targets [68].
Diagram 2: Biofilm Signaling Pathways and Computational Targeting Strategies. This diagram illustrates the major regulatory systems controlling biofilm formation and the corresponding anti-biofilm peptide targeting approaches.
Table 3: Computational Resources and Databases for Anti-biofilm Peptide Research
| Resource Name | Type | Primary Function | Key Features |
|---|---|---|---|
| StarPep Toolbox [66] | Software Platform | Complex network analysis of peptide space | Half-space proximal network construction, centrality analysis, motif discovery |
| BaAMPs Database [63] | Specialized Database | Repository of biofilm-active antimicrobial peptides | Curated antibiofilm peptides with activity annotations |
| APD (Antimicrobial Peptide Database) [63] | Comprehensive Database | General antimicrobial peptide repository | >4,700 AMPs with structural and functional information |
| BIPEP [63] | Web Server | Prediction of antibiofilm peptides | SVM-based classification with 0.89 MCC score |
| QSAR Modeling Software (e.g., DRAGON) [62] | Molecular Modeling | Calculation of molecular descriptors | ~2500 descriptor types for 3D QSAR modeling |
| Gephi [66] | Network Visualization | Complex network visualization and exploration | Interactive network mining and community detection |
The integration of computational and machine learning approaches has fundamentally transformed the landscape of anti-biofilm peptide discovery. The ability to navigate vast sequence spaces, predict activity profiles, and identify novel structural motifs has dramatically accelerated the development of promising therapeutic candidates. As these technologies continue to evolve, several emerging trends are likely to shape future research:
The integration of deep generative models with molecular dynamics simulations promises to enable de novo design of peptides with customized properties [64] [69]. The emerging field of "molecular de-extinction" – using machine learning to mine proteomes of extinct organisms – represents another innovative approach that has already yielded novel antimicrobial sequences [69]. As multi-omics datasets continue to expand and computational power increases, the integration of mechanistic modeling with artificial intelligence will likely enable increasingly accurate predictions of peptide behavior in complex biofilm environments.
While significant challenges remain in translating computational predictions into clinically effective therapeutics, the continued refinement of these approaches offers considerable promise for addressing the critical unmet need for anti-biofilm agents. The interdisciplinary integration of computational science, systems biology, and experimental microbiology will be essential for realizing this potential and combating the growing threat of biofilm-associated infections.
The formation of microbial biofilms presents a formidable challenge in the treatment of infectious diseases. These structured communities of microorganisms, enveloped in a self-produced extracellular polymeric substance (EPS) matrix, establish resilient microenvironments on biological and inert surfaces [26]. Biofilms are significant in a clinical context because they exhibit insidious affinity for medical devices like catheters, prosthetics, and surgical instruments, leading to compromised device functionality and significantly increased risk of persistent infections [26]. The biofilm lifestyle confers substantial advantages to pathogens, primarily by acting as a physical barrier that shields microbial cells from antimicrobial agents and host immune responses [22]. This protective effect, combined with facilitated horizontal gene transfer among resident microbes, accelerates the dissemination of antibiotic resistance genes, making biofilm-associated infections particularly difficult to eradicate [22] [26].
The economic and clinical burden imposed by biofilm-associated infections is staggering. Recent analyses indicate that biofilm-related infections cost the United States healthcare system approximately $94 billion annually, with over 500,000 deaths attributed to these infections each year [26]. Furthermore, chronic wound infections, often associated with biofilm formation, alone are estimated to cost the U.S. healthcare system over $25 billion annually [26]. The treatment complexity for these infections necessitates prolonged antibiotic therapies, additional surgical interventions, and often device replacement, further amplifying healthcare costs and compromising patient outcomes [26]. Within this context, the process of drug discovery—from target identification to compound screening—requires specialized approaches to effectively address the unique challenges posed by biofilm-mediated infections.
Target Identification Strategies for Biofilm-Associated Infections
Target identification represents the foundational stage of antimicrobial drug discovery, involving the characterization of specific molecules or pathways that can be modulated for the development of new therapeutic agents [70]. This process entails pinpointing the specific molecular target, such as a protein or nucleic acid, with which a small molecule interacts, with particular focus on identifying the primary target responsible for the effectiveness of a drug [70]. In the context of biofilm-associated infections, several innovative strategies have emerged that shift the paradigm from traditional antimicrobial approaches.
Genomic and Proteomic Approaches
Genetic and genomic approaches take advantage of the convenience of manipulating DNA and RNA for extensive modifications and measurements, often employing the concept of genetic interaction, where genetic modifiers are used to generate hypotheses about potential targets [70]. Proteomic analysis provides a complementary method for discovering drug targets by comparing the variations in cellular proteins before and after drug intervention, allowing the identification of elements that specifically impact protein expression [70]. This approach offers a more comprehensive analysis compared to transcriptomics alone, as it directly examines the functional molecules within the cell.
A representative example of these approaches is illustrated in a study targeting methicillin-resistant Staphylococcus aureus (MRSA), where researchers conducted a comprehensive proteome analysis to identify novel therapeutic targets [71]. The workflow involved:
- Paralogous protein identification using the CD-HIT tool to eliminate duplicate proteins with a threshold value of 80% [71].
- Non-homologous analysis via NCBI BLASTp against Homo sapiens proteins to identify non-homologous proteins [71].
- Physicochemical characterization using the Expasy ProtParam server to compute properties including molecular weight, isoelectric point, aliphatic index, instability index, and GRAVY [71].
- Protein localization prediction with PSORTb to classify proteins based on cellular location, focusing on cytoplasmic proteins due to their established role in bacterial survival and virulence [71].
- Druggability analysis utilizing the DrugBank database and Therapeutic Target Database to compare the druggability efficiency of proteins [71].
- Virulence factors and essentiality analysis to identify proteins that play a major role in destabilizing the activity of immune cells in the host body [71].
This systematic approach led to the identification of the heme response regulator R (HssR) as a novel protein that critically controls heme levels in MRSA infections, representing a promising therapeutic target [71].
Emerging Paradigms in Target Selection
Contemporary research has revealed several innovative strategies for target identification that move beyond conventional approaches:
- Targeting pathogenicity rather than microbial growth, which may reduce selective pressure for resistance development [72].
- Targeting the host or host-pathogen interface rather than the pathogen itself, potentially broadening therapeutic effectiveness [72].
- Facilitating pathogen-specific immune responses to enhance natural defense mechanisms [72].
- Utilizing systems-based approaches to identify new drug targets and validate drug efficacy through comprehensive network analyses [72].
These strategies may allow identification of drugs that disrupt pathogenesis and allow the immune system time to protect, but do not easily engender resistance, representing a significant advancement in antimicrobial drug discovery philosophy [72].
Table 1: Biofilm-Specific Drug Targets and Their Therapeutic Implications
| Target Category | Specific Target Examples | Therapeutic Implications | Advantages |
|---|---|---|---|
| Virulence Regulators | Heme response regulator R (HssR) in MRSA [71] | Modulates bacterial heme levels, affecting survival | Disrupts pathogenesis without directly killing bacteria, potentially reducing resistance selection |
| Quorum Sensing Systems | Autoinducer peptides, LuxS/AI-2 system [22] | Interferes with cell-to-cell communication and biofilm coordination | Targets biofilm-specific behavior rather than essential growth processes |
| EPS Matrix Synthesis | Polysaccharide intercellular adhesin (PIA) in staphylococci [22] | Prevents initial biofilm formation and structural integrity | Weakens biofilm architecture, enhancing susceptibility to conventional antibiotics |
| Two-Component Signal Systems | Sensor kinases and response regulators [22] | Disrupts environmental sensing and adaptive responses | Affects multiple virulence pathways simultaneously |
Experimental Protocols for Biofilm Assessment and Compound Screening
Robust and standardized experimental protocols are essential for evaluating biofilm formation, assessing the efficacy of potential therapeutic compounds, and validating identified targets. This section details key methodologies used in biofilm research and compound screening.
Biofilm Formation Inhibition and Dispersal Assays
The microtiter plate assay represents a widely used method for quantifying biofilm formation and assessing the effects of inhibitory compounds. The following protocol, adapted from studies using Campylobacter jejuni as a model organism, provides a guideline for consistent and reproducible analysis of biofilm formation in the presence of naturally occurring inhibitory compounds [73]:
Protocol 1: Biofilm Formation Inhibition Assay
- Step 1: Recover bacterial strains from storage and plate on appropriate agar media supplemented with necessary antibiotics. Incubate under optimal conditions (e.g., microaerobically at 42°C for C. jejuni) [73].
- Step 2: Harvest bacterial cells from agar plates into liquid broth medium and transfer to a larger broth volume. Incubate with shaking overnight under optimal conditions [73].
- Step 3: Dilute overnight culture in fresh medium to achieve a standardized cell density (e.g., OD₆₀₀ of 0.05, approximately 10⁷ CFU/mL) at the start of the logarithmic growth phase [73].
- Step 4: Dispense diluted bacterial suspension into wells of a 24-well or 96-well plate (e.g., 180 μL per well for 96-well plates). Include uninoculated medium as a negative control in separate wells [73].
- Step 5: Add chosen concentrations of the test compounds directly to the culture in the wells. For water-soluble compounds, phosphate-buffered saline (PBS) can be used as a solvent [73].
- Step 6: Cover the plates and incubate under optimal conditions without shaking (static culture) for 24-48 hours [73].
- Step 7: Quantify biofilm formation using crystal violet staining or other appropriate methods as described in the assessment section below [73].
Protocol 2: Biofilm Dispersal Assay
- Step 1: Follow Steps 1-4 from the Biofilm Formation Inhibition Assay, but do not add test compounds during the initial biofilm formation phase [73].
- Step 2: After biofilm formation, remove the media from the wells and gently rinse to remove planktonic cells [73].
- Step 3: Add PBS containing an appropriate concentration of the test compound to each well (PBS-only serves as a negative control) [73].
- Step 4: Incubate the plates under optimal conditions without shaking for the desired treatment duration [73].
- Step 5: Remove the supernatants and measure OD₆₀₀ for each well to assess dispersed cells [73].
- Step 6: Quantify remaining biofilm using the assessment method below [73].
Assessment of Biofilm Formation
- Step A: Remove media from plates by inverting over an absorbent paper towel and rinse gently with distilled water twice to remove planktonic cells [73].
- Step B: Dry plates by gently tapping on paper towel and air-dry for 15 minutes in a laminar flow cabinet [73].
- Step C: Stain attached biofilm material by adding 0.1% crystal violet solution (125 μL for a 96-well plate) to each well and incubate for 10 minutes at room temperature [73].
- Step D: Remove crystal violet solution and rinse out unbound dye with distilled water until wells are free of liquid crystal violet [73].
- Step E: Air-dry the plates for 15 minutes in a laminar flow cabinet or overnight at room temperature [73].
- Step F: Add modified biofilm dissolving solution (MBDS), such as 33% glacial acetic acid or 80% ethanol with SDS, to each well to solubilize the crystal violet and incubate for 10 minutes at room temperature [73].
- Step G: Mix the solution by pipetting up and down, then transfer to a corresponding well of a flat-bottomed 96-well plate for absorbance measurement [73].
- Step H: Quantify OD at the appropriate wavelength (570-600 nm) in a plate reader, subtracting the measurement for blank wells (MBDS only) from the OD of each sample well [73].
Recent standardization efforts emphasize the importance of measuring absorbance at the λmax of resolubilized crystal violet: 585 nm for 33% acetic acid and 580 nm for 94-100% ethanol to enable accurate comparison across studies [74].
Advanced Screening Methodologies
Beyond traditional biofilm assays, several advanced screening methodologies are employed in antimicrobial drug discovery:
- High-Throughput Screening (HTS): Involves the rapid testing of thousands to millions of samples for biological activity at various levels using automated equipment. While beyond the reach of most academic researchers due to high costs and labor requirements, HTS has become the standard method in industrial drug discovery for understanding interactions between molecules of interest and biological systems [70] [75].
- Computer-Aided Drug Design (CADD): Complements experimental approaches by using predictive computational techniques to screen entire compound databases, recommending sets of compounds more likely to bind to a given drug target than compounds selected at random [75].
- Machine Learning Techniques: Advanced nonparametric machine-learning techniques learn directly from crystallographic and assay data without requiring explicit programmatic instruction, often achieving accuracies not possible with more conventional approaches [75]. Artificial neural networks (ANNs) and decision trees have been successfully used to identify experimentally validated antibiotics [75].
Diagram 1: Biofilm assay workflow for inhibition and dispersal studies
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of biofilm studies and antimicrobial compound screening requires specific research reagents and materials. The following table details essential solutions and their functions based on established protocols [73].
Table 2: Essential Research Reagent Solutions for Biofilm Studies
| Reagent/Material | Composition/Specifications | Function in Experimental Protocol |
|---|---|---|
| Culture Media | Mueller-Hinton agar/broth (MHA/MHB); Luria-Bertani broth (LB) | Supports bacterial growth and biofilm formation under standardized conditions [73] |
| Antibiotic Supplements | Trimethoprim (2.5 μg/mL); Vancomycin (10 μg/mL) | Selective pressure for maintaining plasmids or studying specific resistant strains [73] |
| Test Compounds | Natural compounds (e.g., D-amino acids like D-Ser); Phytochemicals; Synthetic antimicrobials | Investigational agents tested for biofilm inhibition or dispersal activity [73] [76] |
| Crystal Violet Solution | 0.1% crystal violet in demineralized water | Stains attached biofilm material for quantitative assessment of biomass [73] [74] |
| Modified Biofilm Dissolving Solution (MBDS) | 10% SDS in 80% Ethanol; or 33% glacial acetic acid | Solubilizes crystal violet stain from biofilms for spectrophotometric quantification [73] [74] |
| Phosphate-Buffered Saline (PBS) | pH 7.4 | Washing buffer to remove non-adherent cells while preserving biofilm integrity [73] |
| Multi-Well Plates | 24- or 96-well clear flat-bottom plates | Provide surface for biofilm formation and enable high-throughput screening [73] |
| Fixative Solution | 5% formaldehyde in distilled H₂O | Fixes biofilm structure for microscopic analysis by preserving cellular architecture [73] |
In Silico Approaches and Future Directions
The growing challenges of antimicrobial resistance have accelerated the development and application of computational approaches in drug discovery. These methods are particularly valuable in academic settings where traditional high-throughput screening may be prohibitively expensive and labor-intensive [75].
Machine Learning in Antibiotic Discovery
Machine learning techniques represent a sophisticated approach for identifying new targets and uncovering innovative drugs within biological networks [70] [75]. These networks can robustly maintain and quantitatively assess the interactions between various components of cell systems associated with human diseases [70]. Two primary machine-learning techniques have shown particular promise in antibiotic discovery:
Artificial Neural Networks (ANNs): These attempt to mimic the cellular architecture of the brain using virtual "neurodes" and "connections" [75]. During training, the strengths of connections are systematically modified to optimize the network's ability to accurately predict experimentally measured activities when given corresponding vectors of molecular descriptors [75]. For example, a study by Murcia-Soler et al. used an ANN mapping 62 structure-based molecular descriptors to biological activity, achieving 91.4% accuracy on a testing set and successfully identifying four compounds with low micromolar potency against bacterial strains [75].
Decision Trees: These methods create hierarchical structures of decisions based on molecular descriptors to classify compounds by their predicted activity [75]. While not explicitly detailed in the search results for antibiotic discovery, decision trees represent an important category of machine learning approaches with applications in compound prioritization [75].
Integrative Approaches and Validation
The future of antimicrobial drug discovery lies in the integration of multiple approaches. Chemical proteomics enables the identification of protein targets at the proteomic level by creating chemical probes that specifically bind to desired proteins, followed by retrieving and identifying these proteins [70]. Advances in mass spectrometry techniques have further played a crucial role in biological target identification and validation [70].
Target validation is the critical process that confirms whether targeting a specific entity offers potential therapeutic advantages [70]. This process involves a series of experiments and analyses to confirm the relevance and feasibility of a specific molecular target in the context of drug development, typically including both in vitro and in vivo experiments to demonstrate that modulating the target leads to the desired therapeutic effect [70]. Robust target validation is essential, as insufficient validation of drug targets in early development has been linked to costly clinical trial failures and lower drug approval rates [70].
Diagram 2: Integrated drug discovery pipeline for anti-biofilm therapeutics
The journey from target identification to compound screening in the context of microbial biofilm research represents a specialized and evolving frontier in drug discovery. The unique challenges posed by biofilm-mediated infections—including enhanced antimicrobial resistance, physical barrier properties, and phenotypic heterogeneity—demand innovative approaches that differ from traditional antimicrobial strategies. Successful strategies increasingly involve targeting pathogenicity rather than microbial growth, exploiting vulnerabilities in the host-pathogen interface, and utilizing systems-based approaches to identify novel targets. The integration of advanced computational methods, including machine learning and chemical proteomics, with robust experimental validation provides a powerful framework for addressing the persistent challenge of biofilm-associated infections. As these technologies continue to mature and our understanding of biofilm biology deepens, the prospects for developing effective therapeutics against these resilient microbial communities continue to improve, offering hope for addressing one of modern medicine's most intractable challenges.
Overcoming Biofilm Resistance: Mechanisms and Intervention Challenges
The extracellular polymeric substance (EPS) matrix is a self-produced, hydrated biofilm component that confers remarkable resistance to antimicrobials and host immune defenses. This matrix forms a functional and structural scaffold for microbial communities, creating a protected microenvironment that is a cornerstone of chronic and recalcitrant infections [77] [11]. Within the context of microbial biofilm development, the EPS matrix represents a key evolutionary adaptation, allowing prokaryotes to survive hostile conditions [4]. Its role in intrinsic resistance is multifaceted, involving physical barrier functions, chemical interactions with antimicrobial agents, and the creation of heterogeneous physiological states within the biofilm population [77] [11]. Understanding these mechanisms is paramount for researchers and drug development professionals aiming to overcome the challenges posed by biofilm-associated infections, which account for at least 65% of all bacterial infections in humans [78]. This whitepaper provides an in-depth technical analysis of the EPS matrix's protective mechanisms, supported by quantitative data and experimental methodologies relevant to current biofilm research.
The EPS matrix establishes the structural integrity of biofilms and is the primary mediator of their unique physicochemical properties [48]. This matrix is a complex polymer network, often described as a hydrogel, that can contain up to 97% water and constitutes 50% to 90% of a biofilm's total organic matter [11] [4] [48]. The matrix encases microbial cells and facilitates critical community functions, including cell-to-cell communication and the exchange of genetic material [4].
- Key Components: The EPS is a polymeric conglomeration composed of a diverse array of biopolymers in various structural forms. The major components include exopolysaccharides, proteins, lipids, and extracellular DNA (eDNA) [77] [4]. The exact composition is highly dynamic and varies depending on the microbial species, environmental conditions, and biofilm age [77].
- Structural Analogy: The matrix has been metaphorically described as "cities for microbes" and more functionally as "the house of biofilm cells," providing a stable, protective habitat for the resident microorganisms [4] [79]. In many biofilms, the EPS components are organized into a distinct macromolecular "honeycomb" structure, which contributes to its mechanical stability and functional versatility [77].
Table 1: Major Components of the Biofilm EPS Matrix and Their Primary Functions
| Component | Percentage of EPS | Primary Functions |
|---|---|---|
| Exopolysaccharides | 1-2% of total biofilm mass [11] | Provides architectural scaffolding, mediates adhesion to surfaces, and acts as a molecular glue [11]. |
| Extracellular Proteins | <1-2% of total biofilm mass [11] | Contributes to matrix stability, enzymatic activity, and biofilm integrity; includes enzymes and surface adhesins [11]. |
| Extracellular DNA (eDNA) | <1-2% of total biofilm mass [11] | Provides structural stability and cell-to-cell connectivity; facilitates horizontal gene transfer [77] [11]. |
| Water | Up to 97% of total biofilm mass [11] | Hydrates the matrix, forming a hydrogel; facilitates the diffusion of nutrients and signaling molecules [11]. |
Mechanisms of Resistance Conferred by the EPS Matrix
The EPS matrix is a fundamental determinant of the enhanced antimicrobial resistance observed in biofilm-associated infections. This resistance is not attributed to a single mechanism but is the result of a complex, synergistic interplay of physical, chemical, and physiological factors.
Physical and Chemical Barrier Functions
The EPS matrix acts as a formidable, dynamic barrier that restricts the penetration of antimicrobial agents and protects cells from external stressors.
- Limited Diffusion and Inactivation: The gel-like consistency and anionic nature of the EPS create a diffusion barrier that can slow down or even prevent the penetration of antimicrobial molecules into the deeper layers of the biofilm [77]. Furthermore, antibiotics may bind to and become inactivated by polysaccharides, DNA, and proteins within the biofilm before they can reach their cellular targets at effective concentrations [77]. Notably, the extracellular DNA (e-DNA) in the matrix of Pseudomonas aeruginosa biofilms forms grid-like structures that contribute to this structural and protective role [77].
- Enzyme Sequestration and Neutralization: The EPS matrix retains a wide range of extracellular enzymes, creating an "external digestion system" [77]. This system can neutralize reactive oxygen species and degrade or modify incoming antimicrobial compounds, thus providing an enzymatic shield for the embedded cells [11].
Physiological and Phenotypic Adaptations
Beyond acting as a passive barrier, the EPS matrix actively contributes to creating microenvironments that induce phenotypic changes in the resident cells, further enhancing resistance.
- Metabolic Heterogeneity and Dormancy: The limited and uneven diffusion of nutrients and oxygen through the EPS matrix leads to the formation of metabolic gradients. This results in heterogeneous microenvironments within the biofilm where subpopulations of cells, particularly those in the inner core, enter a slow-growing or dormant state [11] [78]. Since most conventional antibiotics target active cellular processes, these metabolically inactive cells exhibit heightened tolerance to treatment [78].
- Quorum Sensing and Coordinated Defense: The EPS matrix traps quorum sensing (QS) autoinducers, preventing their diffusion and thereby facilitating high local concentrations that enable efficient cell-to-cell communication [4]. QS systems regulate the expression of virulence factors and EPS production itself, allowing the biofilm community to coordinate its defensive responses in a density-dependent manner [77] [11].
The following diagram illustrates the coordinated resistance mechanisms provided by the EPS matrix.
Quantitative Analysis of EPS in Biofilm Resistance
The volume and composition of the EPS matrix evolve as the biofilm matures, directly correlating with increased resilience. Quantitative studies demonstrate that mature biofilms possess a greater volume of EPS and exhibit stronger internal adhesion, which contributes to their enhanced resistance profile compared to younger biofilms.
Table 2: Quantitative Changes in EPS During Oral Biofilm Maturation
| Biofilm Characteristic | 1-Week-Old (Young) Biofilm | 3-Week-Old (Mature) Biofilm | Measurement Technique |
|---|---|---|---|
| EPS Matrix Volume | Lower Volume | Significantly Higher Volume [80] | Confocal Laser Scanning Microscopy (CLSM) with fluorescent probes [80] |
| Surface Roughness | Significantly Higher Roughness | Lower Roughness [80] | Atomic Force Microscopy (AFM) [80] |
| Cell-Cell Adhesion Force | Lower Adhesion Force | Significantly More Attractive Forces [80] | AFM force-distance curve measurements [80] |
The data in Table 2, derived from a model of oral multispecies biofilms, confirms that maturation leads to a more robust and integrated structure. The increase in EPS volume and the strengthening of cell-cell adhesion forces create a more cohesive and less permeable barrier, explaining why mature biofilms are "significantly more resilient to disinfection than young biofilms" [80].
Experimental Protocols for EPS Analysis
For researchers investigating the EPS matrix and its role in resistance, several well-established methodologies provide critical quantitative and qualitative data. Below are detailed protocols for two key techniques.
Protocol 1: Quantifying EPS and Live Bacteria Volume via CLSM
This protocol is designed to measure the three-dimensional volume of both the EPS matrix and live bacterial cells within intact biofilms, providing a quantitative assessment of biofilm architecture [80].
- Sample Preparation: Grow multispecies biofilms anaerobically on relevant substrates (e.g., collagen-coated hydroxyapatite discs) for varying periods to represent different maturation stages. Incorporate a fluorescent marker, such as 1 mM Alexa Fluor 647-labelled dextran, directly into the growth medium before and during biofilm formation. This allows for the visualization of EPS as it is synthesized [80].
- Staining Procedure: After the incubation period, stain the live bacteria in the biofilms using a green-fluorescent nucleic acid stain like SYTO 9. Following staining, rinse the specimens gently with 0.85% physiological saline for 1 minute to remove unbound dye [80].
- Imaging and Analysis: View the fluorescence using a Confocal Laser Scanning Microscope (CLSM). For each sample, acquire images from at least 10 random areas at a resolution of 512 x 512 pixels with a 5-μm step size from the top to the bottom of the biofilm. Reconstruct the 3D volume stacks using software such as Imaris, and use the software's capabilities to calculate the volume (in μm³) of both the EPS (red fluorescence) and live bacteria (green fluorescence) [80].
Protocol 2: Probing Biofilm Topography and Adhesion via AFM
Atomic Force Microscopy is a powerful tool for characterizing the surface topography and nanomechanical properties of biofilms, including adhesion forces [80].
- Sample Preparation and Fixation: Grow biofilms as described in Protocol 1. Following growth, fix the samples in a solution containing 2% glutaraldehyde at 4°C for 3 minutes. Rinse the fixed samples twice in phosphate-buffered saline (PBS) and subsequently dry them overnight in a desiccator [80].
- Surface Roughness Analysis: Use a commercial AFM system operating in contact mode with sharpened silicon nitride cantilevers. Capture images at a scan size of 8 x 8 μm. The surface roughness should be quantified in terms of the root mean square average of the height deviations from the mean plane, as calculated by the instrument's software [80].
- Adhesion Force Measurement: Perform force-distance measurements using the same AFM setup, maintaining a scanning rate in the z-direction of 15 Hz. Conduct force mapping over a 64 x 64 grid on the sample surface. Measure the vertical adhesion forces at two distinct locations: between the AFM tip and the bacterial cell surface (tip-cell interface), and at the interface between two adjacent cells (cell-cell interface). Repeat each adhesion force experiment a minimum of three times to ensure statistical significance [80].
The workflow for the combined application of these techniques is outlined below.
The Scientist's Toolkit: Key Research Reagents and Materials
Successful experimental investigation into the EPS matrix requires a specific set of reagents and materials. The following table details essential items used in the featured protocols, along with their functions.
Table 3: Essential Research Reagents and Materials for EPS Analysis
| Item Name | Function/Application | Example from Protocol |
|---|---|---|
| Alexa Fluor 647-labelled dextran | A fluorescent probe incorporated during growth to label and visualize the EPS matrix in live biofilms. | Used at 1 mM concentration to stain EPS for CLSM visualization [80]. |
| SYTO 9 green-fluorescent stain | A nucleic acid stain used to label and identify live bacterial cells within the biofilm consortium. | Applied to stain live bacteria for dual-channel CLSM imaging with EPS [80]. |
| Hydroxyapatite (HA) Discs | A common substrate that mimics tooth or bone mineral surfaces for growing relevant biofilm models. | Used as a growth surface for oral multispecies biofilms, often coated with collagen [80]. |
| Type I Collagen | A protein coating applied to substrates to facilitate initial bacterial attachment and biofilm formation. | Used to coat HA discs to create a more biologically relevant surface for adhesion [80]. |
| Silicon Nitride AFM Cantilevers | Sharp tips with defined spring constants used for probing surface topography and measuring adhesion forces. | Used in contact mode for AFM imaging and force-distance measurements on biofilm surfaces [80]. |
| Glutaraldehyde | A cross-linking fixative agent used to preserve biofilm structure for AFM analysis by hardening the EPS matrix. | Used in a 2% solution to fix biofilm specimens prior to AFM examination [80]. |
The EPS matrix is far more than inert slime; it is a dynamically organized, functional extension of the microbial community that is fundamental to the intrinsic resistance of biofilms. Its role as a combined physical barrier, chemical inactivator, and inducer of phenotypic heterogeneity presents a formidable challenge in clinical and industrial settings. The quantitative data and experimental protocols detailed in this whitepaper provide a framework for researchers to systematically deconstruct these resistance mechanisms. As the field moves beyond the classic 5-step model to embrace the complexity of in vivo biofilm aggregates [81], future strategies aimed at disrupting the EPS matrix—through enzymatic degradation, quorum sensing inhibition, or nanotechnology-based targeting—hold the greatest promise for overcoming the defensive bastion of the biofilm and effectively treating the persistent infections they cause.
Within the context of microbial biofilm formation research, physiological heterogeneity represents a critical adaptive strategy that underpins biofilm resilience and pathogenicity. Biofilms, structured communities of microorganisms encased in an extracellular polymeric matrix, exhibit remarkable complexity in their internal organization [82]. This review focuses on two fundamental contributors to physiological heterogeneity: metabolic gradients and persister cells.
Metabolic gradients arise from the constrained diffusion of nutrients, oxygen, and metabolic waste products through the biofilm matrix, creating spatially distinct microenvironments [83]. Simultaneously, a subpopulation of bacterial cells enters a transient, dormant state known as persistence, exhibiting multidrug tolerance without genetic resistance mechanisms [82]. Together, these phenomena enable biofilm populations to survive diverse environmental stresses, including antimicrobial treatments, and contribute significantly to chronic and recurrent infections [84] [82].
Understanding the interplay between gradient-driven heterogeneity and persister cell formation is essential for developing effective anti-biofilm strategies. This technical guide examines the mechanisms underlying these phenomena, presents experimental approaches for their study, and discusses implications for therapeutic development.
Metabolic Gradient Formation and Consequences
Mechanisms of Gradient Formation
The biofilm matrix, composed of exopolysaccharides, proteins, and extracellular DNA, acts as a diffusion barrier that profoundly influences the internal chemical environment [82]. This structural limitation, combined with the metabolic activity of embedded cells, generates steep physical and chemical gradients from the biofilm periphery to the core.
Oxygen Gradients: Oxygen consumption by peripheral cells creates hypoxic or anoxic conditions in deeper layers. In many biofilms, oxygen concentrations decrease sharply with depth from the interface with air or water [83]. This gradient mirrors patterns observed in other biological systems, including developing embryos and tumors, where oxygen availability influences cellular differentiation and metabolic programming [83].
Nutrient Gradients: Similar patterns occur with carbon sources and other nutrients. For example, in Escherichia coli biofilms growing on glucose, the lower anoxic regions ferment glucose to produce acetate, which then diffuses upward to be consumed by aerobic cells in oxygen-rich zones [83]. This cross-feeding represents a form of metabolic division of labor that optimizes resource utilization.
The table below summarizes key gradient types and their physiological consequences:
Table 1: Metabolic Gradients in Biofilms and Their Physiological Effects
| Gradient Type | Formation Mechanism | Physiological Consequences | Experimental Evidence |
|---|---|---|---|
| Oxygen | Consumption by peripheral cells; diffusion limitation | Hypoxic stress response; shift to anaerobic metabolism; altered antibiotic efficacy | Direct measurement with microsensors; GFP-based hypoxia reporters [83] |
| Carbon Sources | Differential utilization; diffusion limitation | Metabolic specialization; cross-feeding; substrate stratification | 13C metabolic flux analysis; transcriptional profiling of subpopulations [83] |
| pH | Accumulation of acidic fermentation products | Activation of acid stress response; altered antibiotic activity | pH-sensitive fluorophores; ratiometric imaging [82] |
| Metabolic Waste Products | Limited diffusion from deep layers | Stress response activation; toxicity in biofilm interior | Mass spectrometry imaging; transcriptional reporters [83] |
Physiological Responses to Gradients
The heterogeneous conditions created by metabolic gradients drive profound physiological differentiation within biofilm populations:
Metabolic Specialization: Subpopulations within different microenvironmental niches express distinct metabolic pathways. For instance, aerobic respiration dominates in oxygen-rich regions, while fermentation or anaerobic respiration prevails in hypoxic zones [83]. This specialization represents a form of division of labor that enhances overall community fitness.
Stress Response Activation: Nutrient limitation and waste product accumulation induce stress responses in subpopulations. The universal stress response pathway, stringent response, and oxidative stress defenses are differentially activated across biofilm regions [82] [85].
Altered Growth Rates: Gradients create conditions where growth rates vary dramatically. Peripheral cells often maintain active replication, while nutrient-limited or waste-burdened interior cells enter slow-growing or dormant states [82]. This growth heterogeneity contributes significantly to antibiotic tolerance, as most antibiotics target active cellular processes.
Persister Cells in Biofilms
Persister Cell Formation and Characteristics
Persister cells represent a phenotypically dormant subpopulation capable of surviving high concentrations of antimicrobial agents. Unlike genetically resistant mutants, persisters exhibit transient, non-heritable tolerance that reverts upon regrowth [82].
Key characteristics of persister cells include:
- Dormancy: Greatly reduced metabolic activity and growth cessation
- Multidrug Tolerance: Survival against multiple antibiotic classes simultaneously
- Transient Phenotype: Reversion to susceptible state upon resuscitation
- Stochastic Formation: Both stochastic induction and environmental triggering
The formation of persister cells is strongly promoted by biofilm growth conditions. Biofilms contain persister cell concentrations 100-10,000 times higher than planktonic cultures [82]. This enrichment results from the combined effects of gradient-induced stress and potentially from increased expression of persistence-related genes.
Molecular Mechanisms of Persistence
Several molecular mechanisms contribute to persister formation and maintenance:
Toxin-Antitoxin Systems: These genetic modules consist of a stable toxin and a labile antitoxin. Under stress conditions, antitoxin degradation allows toxins to disrupt essential cellular processes, inducing dormancy [82].
Stringent Response: Nutrient limitation triggers accumulation of (p)ppGpp, which redirects cellular resources from growth to maintenance and stress survival [82].
Reduced ATP Levels: Dormant persisters exhibit dramatically lowered ATP levels, limiting energy-dependent antimicrobial activity [82].
ROS Defense: Enhanced protection against reactive oxygen species contributes to survival against bactericidal antibiotics [82].
Experimental Methodologies
Culturing Biofilms with Controlled Gradients
Microfluidic devices enable precise control over chemical gradients during biofilm growth. The following protocol adapts methods from JoVE for studying biofilm development under defined nutrient gradients [86]:
Table 2: Essential Research Reagents for Gradient-Based Biofilm Studies
| Reagent/Category | Specific Example | Function/Application | Technical Notes |
|---|---|---|---|
| Microfluidic Device | Dual-inlet flow chamber [86] | Creates defined, smooth chemical gradients | PDMS material bonded to glass coverslip via oxygen plasma treatment |
| Growth Media | Modified FAB medium [86] | Defined minimal medium for controlled nutrient delivery | Carbon source (e.g., 0.6 μM glucose) introduced through one inlet |
| Fluorescent Tracers | Cy5 (NHS ester) [86] | Conservative tracer for visualizing solute transport | Light-sensitive; prepare fresh and store in dark (20 μg/mL in sterile water) |
| Bacterial Stains | SYTO 62 [86] | Cell-permeant nucleic acid stain for viability assessment | Dilute stock (1 mM) 1:100 in sterile water; stain in dark for 30 minutes |
| Fixation Agents | Paraformaldehyde [86] | Cross-linking fixative for structural preservation | Typically 2-4% in buffer; optimize concentration for specific biofilm |
Protocol: Biofilm Growth Under Nutrient Gradients
Flow Cell Setup: Prepare polydimethylsiloxane (PDMS) flow chambers (23 mm × 13 mm × 0.24 mm) bonded to glass coverslips using oxygen plasma treatment [86].
Medium Preparation: Prepare modified FAB growth medium with 0.6 μM glucose. Filter-sterilize (0.2 μm pore size) before use. For gradient studies, introduce glucose-containing medium through one inlet and carbon-free FAB medium through the second inlet [86].
System Sterilization: Autoclave the entire flow path except for pre-sterilized disposable components (e.g., plastic three-way valves). Cover all openings with aluminum foil during assembly to prevent contamination [86].
Inoculation: Dilute overnight cultures to OD600 = 0.01 in sterile water. For mixed-species biofilms, combine species at 1:1 ratio with equivalent OD600 = 0.01 for each. Inject 1 mL inoculum through the three-way valve, then pause flow for 1 hour to allow bacterial attachment [86].
Growth Phase: Resume flow at 0.03 mL/min per inlet for 3 days under constant flow conditions. Maintain temperature at appropriate level for studied species (e.g., 37°C for P. aeruginosa and E. coli) [86].
Visualizing and Quantifying Metabolic Heterogeneity
Confocal Microscopy with Fluorescent Reporters:
- Use constitutive fluorescent protein expression (e.g., GFP) for biomass quantification
- Employ environment-sensitive dyes for pH, oxygen, or membrane potential
- Implement fluorescent transcriptional reporters for metabolic activity
- For mixed-species biofilms, use differential staining (e.g., SYTO 62 for nucleic acids) [86]
Solute Transport Visualization:
- Prepare conservative tracer (Cy5 at 20 μg/mL in sterile water)
- Stop flow and inject tracer solution upstream via three-way valve
- Restart flow and image using XY-T mode in confocal microscopy
- Adjust laser intensity and gain to avoid signal saturation for quantification [86]
Microscale Particle Tracking Velocimetry:
- Use fluorescent microspheres (0.5-1.0 μm diameter) as flow tracers
- Introduce at low concentration to avoid biofilm disruption
- Capture time-lapse images at high frame rate
- Calculate flow velocities from particle displacement between frames [86]
Persister Cell Isolation and Quantification
Protocol: Antibiotic Tolerance Assay in Biofilms
This protocol, adapted from mycobacterial biofilm studies, can be modified for other bacterial species [87]:
Biofilm Establishment: Grow biofilms in 6-well plates containing appropriate medium (e.g., Sauton's medium with 2% glucose for mycobacteria). Incubate statically for 4-7 days until mature biofilm forms at air-medium interface [87].
Antibiotic Exposure: Determine MIC90 for planktonic cells. Add antibiotic at MIC90 concentration (e.g., 64 μg/mL rifampin for M. smegmatis) to biofilm cultures, carefully introducing from the side of wells to minimize disruption [87].
Treatment Incubation: Incubate for 24 hours with antibiotic. Include untreated biofilm controls.
Biofilm Dissociation: Add detergent (e.g., Tween-80 to 0.2% final concentration) to all biofilm-containing wells. Seal plates and incubate at 4°C with gentle shaking (100 rpm) for 2 hours to dissociate cells [87].
Viability Assessment: Plate serial dilutions on appropriate agar media. Count colonies after incubation. Persister concentration is calculated as CFU/mL remaining after antibiotic treatment.
Validation: Confirm persistence (rather than resistance) by re-culturing surviving cells and repeating antibiotic susceptibility testing.
Technical Visualization
Metabolic Gradient Formation and Physiological Consequences
The following DOT script visualizes the relationship between metabolic gradients and physiological heterogeneity:
Diagram 1: Metabolic Gradient Formation and Consequences in Biofilms. The biofilm matrix and microbial consumption create chemical gradients that drive zonal metabolic specialization and phenotypic heterogeneity.
Experimental Workflow for Biofilm Heterogeneity Analysis
The following DOT script outlines a comprehensive experimental approach for analyzing physiological heterogeneity in biofilms:
Diagram 2: Experimental Workflow for Biofilm Heterogeneity Analysis. Integrated approach combining controlled biofilm growth with multiple analytical techniques to characterize physiological heterogeneity.
Research Implications and Future Directions
The study of physiological heterogeneity in biofilms has profound implications for addressing antimicrobial resistance and developing novel therapeutic strategies. Metabolic gradients and persister cell formation represent key challenges in eradicating biofilm-associated infections [82].
Therapeutic Development: Strategies targeting gradient mechanisms include:
- Permeability Enhancers: Compounds that disrupt matrix integrity to improve antibiotic penetration
- Metabolic Disruptors: Agents that exploit metabolic dependencies across biofilm zones
- Wake-Up Agents: Compounds that reverse persister dormancy to sensitize cells to conventional antibiotics
Technical Innovations: Advanced methodologies are enabling more precise interrogation of biofilm heterogeneity:
- Multiscale Imaging: Correlative approaches combining microsensor data with spatial transcriptomics
- In Silico Modeling: Computational models predicting gradient formation and heterogeneity patterns
- High-Throughput Screening: Platforms for identifying compounds targeting heterogeneous populations
Future research directions should focus on mapping the complex interplay between metabolic gradients, persister cell dynamics, and biofilm resilience mechanisms. Understanding how localized microenvironments drive phenotypic diversification will provide critical insights for combating biofilm-related treatment failures in clinical, industrial, and environmental contexts.
Microbial biofilms represent a dominant mode of growth for bacteria and fungi, characterized by surface-attached communities encased within a self-produced extracellular polymeric substance (EPS) matrix [11]. This structured consortium confers remarkable protection against antimicrobial agents and host immune responses, making biofilm-associated infections notoriously difficult to eradicate [2] [88]. The treatment recalcitrance observed in biofilm-mediated infections stems from three fundamental barriers: limited antimicrobial penetration through the EPS matrix, subtherapeutic antibiotic concentrations within biofilm subzones due to physiological heterogeneity, and specific challenges associated with medical device-related infections where biofilms thrive on abiotic surfaces [89] [88]. Understanding these interconnected barriers is essential for developing effective therapeutic strategies against chronic infections such as those affecting cystic fibrosis patients, chronic wounds, and indwelling medical devices [2] [89]. This review examines the structural and physiological basis of these treatment barriers within the context of biofilm development stages, providing a technical foundation for researchers developing novel anti-biofilm interventions.
Structural and Physiological Barriers to Treatment Efficacy
The Extracellular Matrix as a Physical Diffusion Barrier
The extracellular polymeric substance matrix forms the architectural foundation of biofilms and constitutes the primary physical barrier to antimicrobial penetration [11]. This hydrogel-like substance is composed of exopolysaccharides, proteins, extracellular DNA (eDNA), and lipids, with microbial cells comprising only 10-25% of the biofilm volume while EPS constitutes 75-90% [11]. The matrix demonstrates selective permeability, allowing passage of nutrients and signaling molecules while impeding the penetration of antimicrobial compounds through mechanisms including molecular sieving, charge interactions, and sequestration [90].
Table 1: Major Components of the Biofilm Extracellular Matrix and Their Roles in Antimicrobial Resistance
| Matrix Component | Chemical Properties | Contribution to Treatment Barrier |
|---|---|---|
| Exopolysaccharides (Pel, Psl, alginate, cellulose) | High molecular weight polymers; anionic | Molecular sieving; hydrogel formation; cation sequestration |
| Extracellular DNA (eDNA) | Polyanionic; structural polymer | Matrix stability; antibiotic binding through electrostatic interactions |
| Proteins (adhesins, amyloids, enzymes) | Variable charge and size | Enzyme-mediated antibiotic degradation; structural integrity |
| Lipids and Surfactants | Hydrophobic compounds | Hydrophobic compound sequestration; matrix architecture |
The diffusion limitation through biofilms follows the relationship Db/D0 = exp(-α·rm·δ), where Db and D0 represent diffusion coefficients in biofilm and water, α is the exclusion coefficient, rm is solute radius, and δ is biofilm thickness [11]. This relationship demonstrates how increasing biofilm thickness and antimicrobial molecule size significantly reduces penetration efficiency. For example, positively charged aminoglycosides experience charge-based interactions with anionic matrix components, further reducing their effective diffusion coefficients [11].
Physiological Heterogeneity and Concentration Gradients
Biofilms develop complex three-dimensional architectures featuring metabolic and physiological heterogeneity, creating microenvironments with distinct antimicrobial susceptibility profiles [2] [88]. Nutrient and oxygen gradients establish stratified zones of cellular activity, ranging from actively growing cells at the biofilm periphery to dormant persister cells in deeper anoxic regions [88]. These persister cells exhibit metabolically dormant states with reduced antibiotic susceptibility, contributing to treatment failure and infection recurrence [2].
The concentration gradients of antimicrobial agents within biofilms follow Fick's laws of diffusion, with concentration declining exponentially from the biofilm surface toward the substrate [11]. This results in subinhibitory antibiotic concentrations in deeper biofilm regions, creating selective pressure for resistance development and enabling survival of resistant subpopulations [88]. Techniques including microelectrode measurements and fluorescence recovery after photobleaching (FRAP) have demonstrated antibiotic concentration reductions of 10- to 1000-fold between the biofilm surface and basal layers [2].
Table 2: Biofilm Microenvironments and Their Impact on Treatment Efficacy
| Biofilm Zone | Metabolic Status | Oxygen Availability | Antibiotic Susceptibility |
|---|---|---|---|
| Surface Layer | High metabolic activity | Normoxic | High susceptibility to most antibiotics |
| Middle Region | Reduced metabolism | Gradients of hypoxia | Moderate susceptibility; some tolerance |
| Deep/Substrate Region | Dormant/persister cells | Anoxic | High tolerance to conventional antibiotics |
Medical Device-Related Challenges
Indwelling medical devices including catheters, prosthetic joints, pacemakers, and ventilators provide ideal abiotic surfaces for biofilm formation, accounting for approximately 65% of nosocomial infections [89] [90]. The foreign body material itself creates an immunocompromised zone where phagocytic immune cells function suboptimally, while the surface facilitates rapid microbial colonization often originating from contamination during implantation or subsequent hematogenous spread [89].
Device-related biofilms exhibit exceptional antimicrobial resistance, with biofilm-embedded bacteria demonstrating up to 1000-fold increased resistance compared to their planktonic counterparts [2] [88]. This recalcitrance necessitates aggressive combination therapy and frequently requires device removal—procedures that incur substantial healthcare costs exceeding $1.62 billion annually for prosthetic joint infections alone in the United States [89]. The problem is particularly acute with orthopedic implants, where over 500,000 biofilm-related infections occur annually in the U.S. [89].
Experimental Models and Analytical Methods for Studying Treatment Barriers
Penetration and Diffusion Measurement Techniques
Quantifying antimicrobial penetration through biofilms requires specialized methodologies that account for matrix interactions and three-dimensional architecture. Key experimental approaches include:
Fluorescence Correlation Spectroscopy (FCS): This technique tracks the diffusion of fluorescently labeled antibiotic molecules through biofilm sections, generating precise diffusion coefficients and revealing localized accumulation or exclusion zones [11]. The protocol involves: (1) cultivating biofilms on glass-bottom dishes for microscopy, (2) staining with fluorescent antibiotic conjugates maintaining biological activity, (3) collecting fluorescence fluctuations via confocal microscopy, and (4) calculating diffusion coefficients from autocorrelation analysis [11].
Microelectrode Measurements: Custom-fabricated antibiotic-selective microelectrodes with tip diameters of 1-10 μm can be precisely positioned within biofilms using micromanipulators to measure spatial concentration profiles with minimal disruption [11]. The methodology requires (1) electrode calibration in standard antibiotic solutions, (2) controlled insertion into biofilm at predetermined depths, (3) potential measurement relative to reference electrode, and (4) conversion to concentration values using the Nernst equation [11].
Transwell Diffusion Systems: These multi-compartment models separate biofilms grown on permeable membranes from antibiotic-containing donor compartments, allowing sampling from receiver compartments to establish penetration kinetics [11]. The standard protocol includes (1) biofilm cultivation on transwell membranes (0.4-8.0 μm pore size), (2) application of antibiotic solution to donor compartment, (3) sequential sampling from receiver compartment, and (4) antibiotic quantification via HPLC or bioassay [11].
Metabolic Heterogeneity Mapping
The physiological gradients within biofilms significantly influence antimicrobial efficacy, as most antibiotics require bacterial metabolic activity for optimal killing. Advanced techniques for mapping this heterogeneity include:
Nanosensor-based Measurements: pH and oxygen nanosensors with tip diameters <100 nm enable mapping of metabolic gradients without significantly disrupting biofilm architecture [11]. The experimental workflow involves (1) biofilm cultivation under relevant conditions, (2) precise sensor positioning using piezoelectric manipulators, (3) multi-point measurements throughout biofilm depth, and (4) spatial gradient reconstruction [11].
Fluorescent Reporter Systems: Genetically encoded fluorescent reporters under control of metabolic promoters (e.g., rRNA, ATP) provide real-time visualization of bacterial metabolic activity [88]. The standard methodology includes (1) incorporation of reporter constructs into target strains, (2) biofilm cultivation under flow conditions, (3) confocal microscopy imaging, and (4) quantitative image analysis to determine spatial expression patterns [88].
RAMAN Spectroscopy: Label-free chemical imaging provides information on metabolic state distribution based on intrinsic molecular vibrations [88]. The protocol involves (1) biofilm cryosectioning to preserve native structure, (2) spectral acquisition with spatial resolution <1 μm, (3) multivariate analysis to identify metabolic fingerprints, and (4) correlation with antibiotic susceptibility profiles [88].
Emerging Strategies to Overcome Treatment Barriers
Matrix-Targeting Disruption Approaches
Novel therapeutic strategies focus on degrading or bypassing the EPS matrix to enhance antimicrobial penetration:
Enzyme-based Matrix Disruption: Targeted enzymatic degradation of key matrix components can significantly enhance antibiotic penetration [2] [88]. DNase I treatment degrades eDNA networks, while dispersin B hydrolyzes polysaccharide adhesion polymers [2]. Combination therapies using matrix-degrading enzymes with conventional antibiotics have demonstrated 10-100 fold improvements in biofilm eradication in preclinical models [2].
EPS Biosynthesis Inhibition: Small molecule inhibitors targeting EPS synthesis pathways prevent matrix production without direct bactericidal pressure, potentially reducing resistance selection [10]. For example, Pel-specific inhibitors have shown efficacy in enhancing antibiotic susceptibility in Pseudomonas aeruginosa biofilms [10].
Quorum Sensing Interruption: Quorum sensing inhibitors (QSIs) disrupt cell-to-cell communication systems that coordinate matrix production and biofilm maturation [91]. Natural and synthetic QSIs including halogenated furanones and cinnamoyl hydroxamates have demonstrated significant reductions in biofilm formation and enhanced susceptibility to antimicrobial agents [10] [91].
Advanced Drug Delivery Systems
Nanotechnology-based delivery approaches offer promising strategies to overcome penetration limitations and address physiological heterogeneity:
Ultrasound-Activated Nanoparticles: Researchers at the University of Oxford developed antibiotic-loaded nanoparticles activated by ultrasound to simultaneously disrupt biofilm structure and release drugs at the infection site [92]. This approach reduced antibiotic concentrations required for biofilm eradication by more than 40-fold compared to conventional treatment and effectively targeted persister cells, reducing needed drug concentrations by 25-fold [92].
Liposome and Micelle Encapsulation: Nano-encapsulation protects antibiotics from matrix interactions and enables enhanced penetration through biofilm architecture [2]. Cationic liposomes additionally exploit charge-based interactions with anionic matrix components to achieve higher local drug concentrations [2].
Stimuli-Responsive Release Systems: Smart nanoparticles that release antimicrobial payloads in response to biofilm-specific stimuli (e.g., low pH, enzymes) improve target specificity and reduce off-target effects [88]. These systems demonstrate particular promise for medical device coatings where controlled, localized drug delivery is essential [89] [88].
Surface Modification and Anti-Fouling Strategies
For medical device-associated infections, surface modifications that prevent initial bacterial adhesion represent a proactive approach:
Antimicrobial Coatings: Impregnation of device surfaces with antimicrobial agents (silver, antibiotics) or non-biocidal antifouling polymers (PEG, zwitterions) creates surfaces resistant to biofilm formation [89] [93]. These approaches have demonstrated 70-90% reduction in bacterial adhesion in experimental models [93].
Topographical Modifications: Surface patterning at micro- and nanoscales creates topographies that impede bacterial adhesion through reduced contact area [93]. Sharklet-inspired patterns and nanopillars have shown particular efficacy against various pathogenic species including Staphylococcus aureus and Escherichia coli [93].
Electrical and Physical Disruption: Low-intensity electric fields enhance antibiotic penetration through electrophoretic movement, while surface-acoustic waves physically disrupt early biofilm formation [90]. These physical approaches avoid chemical agents and may reduce resistance development [90].
Table 3: Research Reagent Solutions for Biofilm Penetration and Efficacy Studies
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Fluorescent Antibiotic Conjugates | Vancomycin-FITC, Tobramycin-TAMRA | Penetration kinetics visualization | Validate biological activity post-labeling |
| Matrix Degrading Enzymes | DNase I, Dispersin B, Alginate lyase | EPS barrier disruption studies | Enzyme stability in biofilm microenvironment |
| Metabolic Reporters | GFP-rRNA fusions, pH-sensitive dyes | Physiological heterogeneity mapping | Reporter burden on bacterial metabolism |
| Nanoparticle Systems | PLGA nanoparticles, Cationic liposomes | Enhanced delivery testing | Characterization of size, charge, and release kinetics |
| Surface Modification Reagents | PEG-silanes, Zwitterionic polymers | Anti-adhesion surface development | Stability and biocompatibility testing |
The multifaceted barriers to effective biofilm treatment—including limited antimicrobial penetration, subtherapeutic concentrations due to physiological heterogeneity, and device-related challenges—represent significant obstacles in clinical management of chronic infections [2] [88]. Overcoming these barriers requires integrated approaches that target both the structural components of biofilms and the physiological adaptations of constituent cells [10] [91]. Emerging strategies including matrix-degrading enzymes, quorum sensing inhibition, and advanced nanoparticle delivery systems show promising results in experimental models [92] [88]. Future research directions should focus on combinatorial approaches that simultaneously address multiple resistance mechanisms while accounting for the dynamic nature of biofilm development stages [10] [89]. Translation of these innovative strategies to clinical practice will require robust validation in physiologically relevant models that accurately recapitulate the complex microenvironment of biofilm-associated infections [2] [88].
Optimizing Antimicrobial Efficacy Against Mature Biofilm Structures
Mature biofilms represent a significant challenge in clinical and industrial settings due to their profound resistance to antimicrobial agents. These structured microbial communities, encapsulated in a self-produced matrix of extracellular polymeric substances (EPS), are a key virulence factor for a wide range of microorganisms that cause chronic infections [94]. The transition from planktonic cells to a mature biofilm community involves a complex developmental process resulting in a heterogeneous structure with gradient environments that confer multifactorial drug tolerance [25] [94]. This architectural and physiological complexity necessitates specialized strategies that move beyond conventional antibiotic treatments, which are often ineffective against biofilm-associated infections. The optimization of antimicrobial efficacy against these structures requires a fundamental understanding of their composition, architecture, and the distinct resistance mechanisms they employ [22]. This guide synthesizes current understanding and emerging strategies to address the unique therapeutic challenges posed by mature biofilm structures, with particular emphasis on ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), which are notably resistant to antimicrobial action [25].
Architectural and Functional Complexity of Mature Biofilms
Structural Composition and Organization
The architecture of a mature biofilm is characterized by its structural heterogeneity and the presence of a protective EPS matrix. This matrix is composed of polysaccharides, proteins, nucleic acids, and lipids, which form a hydrated polymer network that encapsulates the microbial cells [25] [26]. Architecturally, distinct microcolonies are formed within the biofilms with different compositions and sizes, thus creating a heterogeneous and diverse environment which allows the effective exploitation of niches [25]. This spatial organization generates gradients of nutrients, oxygen, pH, and metabolic waste products, leading to varied microenvironments within the biofilm structure [26]. The EPS matrix serves not only as a physical barrier but also as a functional component that contributes to biofilm stability, surface adhesion, and resource capture while impeding the penetration of antimicrobial agents and host immune factors [26] [94].
Table 1: Key Components of the Mature Biofilm Extracellular Polymeric Substance (EPS)
| Matrix Component | Primary Functions | Contribution to Resistance |
|---|---|---|
| Exopolysaccharides | Structural scaffolding, adhesion, water retention, nutrient trapping | Physical barrier to antimicrobial penetration; cation sequestration |
| Proteins (including enzymes) | Structural support, enzymatic activity, cell-to-cell interactions | Degradation of antimicrobial molecules (e.g., β-lactamases) |
| Extracellular DNA (eDNA) | Structural integrity, genetic information transfer, cation chelation | Nutrient source for persister cells; binding site for antimicrobials |
| Lipids | Hydrophobicity modulation, secondary structure formation | Reduction of hydrophilic antibiotic diffusion |
Mechanisms of Antimicrobial Resistance in Biofilms
Biofilm resistance is multifactorial, arising from an interplay of physical, physiological, and genetic adaptations [22] [94]. The following key mechanisms collectively contribute to the remarkable resilience of mature biofilms:
- Physical Barrier and Altered Chemical Microenvironment: The EPS matrix physically impedes the penetration of antimicrobial molecules [94]. This retardation is not merely a passive sieving effect but involves direct binding and neutralization of antimicrobials by matrix components. Furthermore, the metabolic activity of surface-layer cells creates chemical gradients (e.g., oxygen, pH) within the biofilm that can render antimicrobials less effective against cells in deeper layers [25].
- Metabolic Heterogeneity and Persister Cells: The aforementioned gradients lead to zones of nutrient limitation and slow bacterial growth within the biofilm interior. Since many conventional antibiotics target active cellular processes (e.g., cell wall synthesis, protein production), these slow-growing or dormant cells exhibit heightened tolerance [94]. A sub-population of these, known as persister cells, exhibit multi-drug tolerance without genetic resistance and can repopulate the biofilm after antibiotic treatment is ceased [94].
- Activation of Stress Responses: The biofilm microenvironment induces a generalized stress response in resident cells. This adaptive state enhances cellular repair mechanisms and upregulates efflux pumps, which can expel a broad range of toxic compounds, including antibiotics, from the cell interior [22].
- Horizontal Gene Transfer: The dense, protected environment of a biofilm facilitates the efficient exchange of genetic material, including plasmids carrying antibiotic resistance genes, through conjugation, transformation, and transduction [26]. This accelerates the dissemination of resistance traits among the community.
Quantitative Assessment of Biofilms: Methodologies and Protocols
Accurate quantification of biofilm formation, viability, and structure is fundamental to evaluating the efficacy of antimicrobial strategies. The following section details key methodologies, summarizing their applications and providing specific experimental protocols.
Table 2: Comparison of Quantitative Methods for Biofilm Analysis
| Method | What It Measures | Key Advantages | Key Limitations | Reproducibility (CV) | Protocol Duration |
|---|---|---|---|---|---|
| XTT Assay [47] | Metabolic activity of viable cells | High accuracy, reproducibility, cost-effective, efficient | May not differentiate modest differences in biofilm density | ++++ (CV <0.1) | + (Short) |
| Confocal Microscopy with Live/Dead Staining & Automated Analysis [31] | 3D structure, biovolume, and cell viability (membrane integrity) | Provides spatial resolution, reduces user bias, high-throughput capable | Requires specialized equipment and software | ++++ (CV 4.24-11.5%) | ++ (Medium) |
| Crystal Violet Staining [47] | Total adhered biomass (cells + matrix) | Inexpensive, simple, high-throughput | Does not distinguish between live and dead cells | ++++ (CV <0.1) | ++ (Medium) |
| Colony Forming Units (CFU) [47] | Number of viable, cultivable cells | Direct measure of cell viability | Laborious, prone to human error, misses non-cultivable cells | +++ (CV 0.1-0.15) | +++ (Long) |
| qPCR [47] | Gene copy number (e.g., via ACT1 primers for C. albicans) | Highly sensitive, specific | Does not distinguish live/dead cells, costly, time-consuming | ++ (CV 0.15-0.2) | ++++ (Very Long) |
Detailed Experimental Protocols
The XTT assay measures the metabolic activity of biofilm cells via the reduction of the tetrazolium salt XTT to a water-soluble orange formazan product by metabolically active cells.
Procedure:
- Biofilm Growth: Grow biofilms in a 6-well polystyrene plate for 48 hours at 37°C on an orbital shaker (50 rpm) after a 1-hour adhesion period.
- Reagent Preparation: Prepare the XTT-menadione solution fresh. XTT salt is dissolved in PBS to a final concentration of 1 mg/mL and filter-sterilized. Menadione is prepared as a stock solution in acetone.
- Assay Incubation: Add the XTT-menadione solution directly to wells containing washed, mature biofilms.
- Incubation: Incubate the plate in the dark at 37°C for a specified period (e.g., 2-3 hours).
- Measurement: Transfer aliquots (e.g., 100 µL) from each well to a new microtiter plate and measure the absorbance at a wavelength of 490-500 nm using a spectrophotometer. Higher absorbance correlates with greater metabolic activity and viable biofilm biomass.
This protocol uses a live/dead stain (e.g., SYTO 9 and propidium iodide) combined with Confocal Laser Scanning Microscopy (CLSM) and automated image analysis to quantify viable biomass and 3D structure.
Procedure:
- Staining: Incubate the biofilm with a commercial LIVE/DEAD stain (e.g., FilmTracer LIVE/DEAD Biofilm Viability Kit) according to manufacturer's instructions. SYTO 9 stains all cells (green), while propidium iodide penetrates only cells with damaged membranes (red).
- Imaging: Acquire 3D image stacks of the stained biofilm using a CLSM with appropriate laser lines and emission filters for the fluorescent dyes.
- Automated Image Analysis (Biofilm Viability Checker): Use an open-source tool like the "Biofilm Viability Checker" macro for Fiji/ImageJ.
- Pre-processing: Separate the red and green channel images to avoid subjective color superposition.
- Segmentation: Apply an automated thresholding algorithm (e.g., Otsu, Maximum Entropy) to each channel to distinguish foreground (bacterial signal) from background.
- Quantification: The macro calculates the percentage of viable (green) and dead (red) pixels within the total biomass, providing metrics for viable biovolume, dead biovolume, and surface coverage.
For a more comprehensive spatial analysis of biofilm internal properties, the software BiofilmQ can be used.
Procedure:
- Image Acquisition and Segmentation: Acquire 3D fluorescence images and import them into BiofilmQ. The software offers automatic, semi-manual, or imported segmentation options to define the biofilm biovolume.
- Image Cytometry: BiofilmQ dissects the segmented biovolume into a cubical grid. For each cube, it calculates 49+ structural, textural, and fluorescence properties.
- Data Analysis and Visualization: Analyze spatially resolved parameters (e.g., distance from substratum, local cell density, fluorescence intensity gradients). The software includes batch-processing capabilities and extensive data visualization tools to generate graphs and renderings of the quantified data.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for Biofilm Research
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| Live/Dead Viability Stains | Differentiates cells with intact vs. compromised membranes. Essential for confocal microscopy viability assessment. | FilmTracer LIVE/DEAD Biofilm Viability Kit (SYTO 9 & Proidium Iodide). Note: PI can stain eDNA, requiring careful analysis [31]. |
| XTT Assay Kit | Quantifies metabolic activity of biofilms as a proxy for viability. | Often used with an electron-coupling agent like menadione for enhanced signal in fungal and bacterial biofilms [47]. |
| Crystal Violet | Stains total adhered biomass (cells and matrix). A classic, low-cost screening tool. | 1% aqueous solution. Requires solubilization (e.g., with ethanol or acetic acid) after staining and washing for absorbance reading [47]. |
| Extracellular Polymeric Substance (EPS) Degrading Enzymes | Selective disruption of biofilm matrix components for mechanistic studies or combination therapies. | DNase I (targets eDNA), Dispersin B (targets polysaccharides), proteinase K (targets proteins) [94]. |
| Quorum Sensing Inhibitors (QSI) | Interfere with bacterial cell-to-cell communication, potentially inhibiting biofilm development and virulence. | Natural and synthetic molecules that block autoinducer signaling pathways (e.g., furanones, halogenated furanones) [95]. |
| Non-Ionic Surfactants | Weaken hydrophobic interactions and enhance penetration of antimicrobials into the biofilm matrix. | Polysorbate 80 (Tween 80). Often used in combination with antibiotics or enzymes in disruption assays [25]. |
Strategic Approaches to Enhance Antimicrobial Efficacy
Overcoming biofilm resistance requires multi-targeted strategies that address its multifactorial nature. The following approaches, particularly when used in combination, show significant promise.
Combination Therapies and Emerging Strategies
- EPS Matrix Disruption: This strategy aims to degrade the physical barrier, allowing antimicrobials to penetrate effectively. This can be achieved using:
- Quorum Sensing Inhibition (QSI): QSIs do not kill bacteria but interfere with their ability to coordinate behavior. This can prevent biofilm maturation and the expression of virulence factors, potentially rendering the biofilm more susceptible to antibiotics and host immune clearance [95] [22].
- Targeting Dormant and Persister Cells: Eradicating these tolerant sub-populations is critical. Strategies include:
- Metabolic Stimulation: Using metabolites like mannitol or fructose to re-activate metabolism before antibiotic application.
- Anti-persister Compounds: Certain antimicrobial peptides (AMPs) and newer classes of antibiotics show activity against persister cells [94].
- Enhanced Physical Disruption:
- Ultrasound: Low-frequency ultrasound can induce acoustic cavitation, generating microbubbles whose implosion creates localized shock waves and micro-jets that physically disrupt the biofilm architecture [26]. This method is particularly effective when used to synergistically enhance the activity of concurrently applied antibiotics, a phenomenon known as sonodynamic therapy [25] [26].
- Smart Release Systems: Utilizing nanoparticles or hydrogels that release antimicrobial payloads in response to biofilm-specific stimuli (e.g., low pH, enzymes, or low oxygen) ensures targeted delivery and higher local drug concentrations [94].
The following diagram illustrates the logical workflow for developing and testing a combination therapy strategy against a mature biofilm.
Figure 1: Combination Therapy Development Workflow
The optimization of antimicrobial efficacy against mature biofilms necessitates a paradigm shift from single-target monotherapies to integrated, multi-targeted approaches. Success depends on a fundamental understanding of the complex biofilm architecture and its associated resistance mechanisms. By leveraging advanced quantitative assessment tools and combining strategies that disrupt the EPS matrix, counteract physiological heterogeneity, and neutralize specific tolerance mechanisms, researchers can develop more effective therapeutic interventions. The future of biofilm control lies in combinatorial therapies that are precisely targeted, potentially triggered by the biofilm microenvironment itself, to overcome the formidable defenses of these structured microbial communities [94]. Validating these sophisticated approaches in clinically relevant models will be essential for translating promising strategies from the laboratory to the clinic, ultimately addressing a critical unmet need in the treatment of persistent infections.
Pseudomonas aeruginosa biofilms represent a significant challenge in clinical settings due to their extreme tolerance to antimicrobial agents and ability to persist on both biological and abiotic surfaces. This recalcitrance stems from a complex interplay of physical, physiological, and genetic mechanisms that protect embedded bacterial communities. Within the broader context of microbial biofilm research, P. aeruginosa serves as a model organism for studying structured microbial communities and their resistance mechanisms. This whitepaper examines the architectural and regulatory complexity of P. aeruginosa biofilms, analyzes the multifactorial nature of their antimicrobial tolerance, and evaluates both conventional and emerging therapeutic strategies. For researchers and drug development professionals, understanding these mechanisms is crucial for developing effective countermeasures against this persistent pathogen.
P. aeruginosa is a Gram-negative opportunistic pathogen responsible for severe nosocomial infections, particularly in immunocompromised individuals [96]. It is recognized by the World Health Organization as a priority pathogen for research and development of new antibiotics due to its formidable resistance profiles [96]. The bacterium's success as a pathogen is largely attributable to its ability to form structured biofilms—complex microbial communities encased in a self-produced extracellular matrix that provides protection from environmental stresses, host immune responses, and antimicrobial treatments [96] [97].
Biofilm-associated infections are particularly problematic in clinical contexts, accounting for an estimated 65-80% of pathogenic infections in hospitals [97]. In the United States alone, approximately 6.5 million patients are affected by chronic wound infections annually, with substantial healthcare costs exceeding $25 billion per year [96]. The transition from planktonic to biofilm growth represents a fundamental shift in bacterial behavior that has profound implications for treatment outcomes and infection control strategies.
Biofilm Composition and Architecture
The biofilm matrix of P. aeruginosa is a complex amalgamation of biopolymers that function as a protective scaffold. This extracellular polymeric substance (EPS) accounts for over 90% of the biofilm biomass and is composed primarily of exopolysaccharides, proteins, extracellular DNA (eDNA), and lipids [96] [98]. The specific composition varies between strains and is influenced by environmental conditions and biofilm age [98].
Table 1: Major Exopolysaccharides in P. aeruginosa Biofilm Matrix
| Polysaccharide | Chemical Composition | Primary Function | Strain Prevalence |
|---|---|---|---|
| Psl | Neutral pentasaccharide of D-glucose, D-mannose, and L-rhamnose | Initial surface attachment, cell-to-cell interactions, structural stability, protection from neutrophils and antibiotics | Non-mucoid and mucoid strains |
| Pel | Cationic polymer of partially deacetylated N-acetylgalactosamine and N-acetylglucosamine | Pellicle formation at air-liquid interface, biofilm integrity, aminoglycoside tolerance | Non-mucoid strains |
| Alginate | Anionic acetylated polymer of mannuronic and guluronic acids | Biofilm maturation, protection from phagocytosis, reduced antibiotic diffusion, viscoelastic properties | Mucoid strains (common in cystic fibrosis isolates) |
In addition to exopolysaccharides, extracellular DNA (eDNA) serves multiple crucial functions in biofilms. It originates from cell lysis mediated by endolysins encoded in pyocin gene clusters [96]. eDNA provides structural support, functions as a cation chelator that activates virulence secretion systems, serves as a nutrient source, creates an acidic environment that limits antimicrobial penetration, and influences neutrophil-mediated inflammatory processes [96]. The matrix also contains various proteins including type IV pili, Cup fimbriae, adhesins, lectins, and Fap amyloid fibers that contribute to structural integrity and cellular adhesion [98].
Biofilm Development Cycle
The formation of P. aeruginosa biofilms follows a defined developmental cycle consisting of distinct morphological stages [97]. Understanding this cycle is essential for identifying potential intervention points.
Stage 1: Initial Attachment
The biofilm lifecycle begins with the reversible attachment of planktonic cells to surfaces through acid-base, hydrophobic, and electrostatic interactions [97]. Flagella play an essential role in this initial attachment to abiotic surfaces [99]. At this stage, attachment is reversible, and bacteria can detach to resume planktonic growth.
Stage 2: Irreversible Attachment and Microcolony Formation
Attached cells undergo a transition to irreversible attachment and begin forming microcolonies [97]. This stage is characterized by the production of Psl exopolysaccharide, which forms a fibrous matrix that promotes cell-to-cell interactions and strengthens surface adhesion [97]. Type IV pili-mediated twitching motility becomes important for microcolony development, with pilus-deficient mutants forming distinct, regularly spaced microcolonies rather than continuous biofilms [99].
Stage 3: Maturation
During maturation, biofilms develop complex three-dimensional mushroom-like structures containing water channels that facilitate nutrient transport and waste removal [96] [97]. This architectural organization is coordinated by quorum sensing (QS) systems that regulate the production of matrix components and enzymes [97]. Mature biofilms exhibit significantly enhanced resistance to environmental stresses and antimicrobial agents compared to earlier developmental stages [97].
Stage 4: Dispersal
The final stage involves active dispersal of biofilm cells, which can occur through erosion, sloughing, or regulated dispersal mechanisms [97]. This process is triggered by environmental cues such as nutrient limitation, oxygen availability, or signaling molecules including nitric oxide [100]. Dispersed cells can then colonize new surfaces and initiate fresh biofilm formation, completing the lifecycle [97].
Molecular Regulation of Biofilm Formation
The development and maintenance of P. aeruginosa biofilms are tightly regulated by interconnected molecular signaling systems that respond to environmental conditions and population density.
Cyclic di-GMP Signaling
The secondary messenger bis-(3'-5')-cyclic dimeric guanosine monophosphate (c-di-GMP) serves as a central regulator of the biofilm lifecycle [98]. High intracellular c-di-GMP levels promote biofilm formation by stimulating the production of adhesins and extracellular matrix components, while low levels favor the transition to planktonic growth [98]. The cellular concentration of c-di-GMP is controlled by the opposing activities of diguanylate cyclases (DGCs), which synthesize c-di-GMP, and phosphodiesterases (PDEs), which degrade it [98]. Psl polysaccharide can further amplify c-di-GMP production, creating a positive feedback loop that promotes thicker, more robust biofilms [96].
Quorum Sensing Networks
P. aeruginosa employs multiple interlinked quorum sensing (QS) systems that coordinate gene expression in response to population density [97] [98]. The Las and Rhl systems use acyl homoserine lactones as signaling molecules, while the PQS system utilizes alkyl quinolones [97] [98]. These interconnected circuits regulate the expression of numerous virulence factors and biofilm matrix components. The Las system represses the pel locus, the Rhl system positively regulates rhamnolipid production important for biofilm architecture, and the PQS system enhances eDNA release into the matrix [97] [98]. QS-deficient mutants form flat, undifferentiated biofilms that lack the characteristic mushroom-shaped structures of wild-type biofilms [99].
Two-Component Regulatory Systems
Environmental sensing through two-component systems such as GacA/GacS influences biofilm formation by regulating the expression of genes involved in exopolysaccharide production [98]. These systems intersect with c-di-GMP signaling and help modulate the transition between planktonic and biofilm lifestyles in response to external conditions [98].
Antimicrobial Tolerance and Resistance Mechanisms
Biofilm-associated bacteria exhibit dramatically enhanced tolerance to antimicrobial agents, surviving antibiotic concentrations up to 1000 times higher than their planktonic counterparts [96]. This recalcitrance is multifactorial, involving both physical barrier functions and physiological adaptations.
Table 2: Mechanisms of Biofilm-Associated Antimicrobial Tolerance in P. aeruginosa
| Mechanism Category | Specific Components | Effect on Antimicrobial Efficacy |
|---|---|---|
| Physical Tolerance | Matrix polysaccharides (alginate, Psl, Pel), eDNA | Restricts antibiotic penetration through binding and sequestration |
| Physiological Heterogeneity | Gradients of nutrients, oxygen, metabolic activity | Creates dormant subpopulations with reduced antibiotic susceptibility |
| Genetic Adaptation | Mutations in antibiotic target genes, efflux pump overexpression | Conventional resistance mechanisms with elevated MIC values |
| Stress Responses | RpoS stationary phase sigma factor, SOS response | Enhanced survival under antibiotic-induced stress conditions |
| Persister Cells | Dormant subpopulation with minimal metabolic activity | Survives high-dose antibiotic exposure without genetic resistance |
Restricted Antimicrobial Penetration
The biofilm matrix acts as a diffusion barrier that can retard antibiotic penetration through binding or sequestration [98]. Anionic components such as alginate and eDNA can bind positively charged aminoglycoside antibiotics, effectively reducing their bioavailability to embedded cells [96] [98]. The matrix environment may also promote antibiotic degradation through pH alterations or enzymatic activity before compounds reach their cellular targets [98].
Metabolic Heterogeneity
Biofilms develop gradients of nutrients, oxygen, and metabolic waste products that create distinct physiological zones with varied microbial activity [98]. Cells in oxygen-limited or nutrient-deprived regions enter slow-growing or dormant states that reduce their susceptibility to antimicrobials that target active cellular processes [98]. This metabolic heterogeneity means that a single antibiotic regimen must effectively target multiple physiological states simultaneously.
Genetic Resistance Development
The structured environment of biofilms facilitates horizontal gene transfer and promotes the emergence of genetic resistance through mutation and selection [98]. High bacterial densities in close proximity enhance the exchange of resistance determinants, while the presence of sublethal antibiotic concentrations within biofilms selects for resistant mutants [98]. Biofilm-grown P. aeruginosa demonstrates increased mutation rates compared to planktonic cultures, accelerating the development of heritable resistance mechanisms [98].
Experimental Models and Assessment Methodologies
Research on P. aeruginosa biofilms employs various experimental models and quantification methods to study biofilm formation, architecture, and antimicrobial susceptibility.
Flow Cell Biofilm Models
The flow cell system represents a sophisticated approach for studying biofilm development under controlled hydrodynamic conditions [99]. This method involves growing biofilms in channels with precise dimensions (typically 1×4×40 mm) through which medium flows at constant rates (e.g., 3 ml/h) [99]. The system provides laminar flow conditions that mimic natural environments while allowing real-time observation of biofilm architecture development. Biofilms are typically grown in modified FAB medium supplemented with citrate as a carbon source, with inoculation procedures standardized to an optical density of 0.1 at 600 nm [99].
Microtiter Plate Biofilm Assay
The crystal violet microtiter plate assay provides a high-throughput method for quantifying biofilm formation [101]. This protocol involves:
- Diluting overnight bacterial cultures to a standard turbidity (1 McFarland standard) in tryptic soy broth supplemented with 1% glucose
- Transferring 200 μL aliquots to sterile 96-well polystyrene microplates
- Incubating for 24 hours at 37°C without agitation
- Gently washing wells with phosphate-buffered saline (PBS) to remove non-adherent cells
- Fixing adherent biofilms with 99% methanol for 15 minutes
- Staining with 0.1% crystal violet for 5 minutes
- Destaining with 95% ethanol and measuring optical density at 570 nm [101]
Biofilm-forming capacity is categorized based on optical density values relative to a negative control cutoff: non-biofilm producer (OD < ODc), weak (ODc < OD < 2×ODc), moderate (2×ODc < OD < 4×ODc), or strong (4×ODc < OD) producer [101].
Microscopy and Image Analysis
Confocal laser scanning microscopy (CLSM) of GFP-tagged strains enables detailed visualization of three-dimensional biofilm architecture [99]. Image analysis with software such as COMSTAT provides quantitative measurements of structural parameters including biomass, average thickness, and surface roughness [99]. Statistical analysis of variance models incorporating factors such as bacterial strain, experimental round, and time points allow objective comparison of biofilm structures and temporal development patterns [99].
Table 3: Essential Research Reagent Solutions for P. aeruginosa Biofilm Studies
| Reagent/Category | Specific Examples | Research Function |
|---|---|---|
| Growth Media | Modified FAB + citrate, TSB + 1% glucose, LB medium | Supports biofilm development under standardized nutritional conditions |
| Genetic Tools | GFP-tagged strains, pilHIJK mutants, lasI/rhl mutants, rpoS mutants | Enables tracking and functional analysis of specific genes in biofilm formation |
| Detection Reagents | Crystal violet, fluorescent dyes (SYTO9, propidium iodide), ATP bioluminescence kits | Quantification of biofilm biomass and viability assessment |
| Molecular Assays | PCR primers for algD, pslD, pelF genes, RNA extraction kits | Detection of biofilm-related gene expression and matrix composition |
| Antimicrobial Agents | Aminoglycosides, fluoroquinolones, β-lactams, colistin | Assessment of biofilm tolerance and resistance mechanisms |
Therapeutic Strategies and Future Directions
Conventional antibiotic therapies often fail to eradicate P. aeruginosa biofilms, necessitating innovative approaches that target specific biofilm characteristics.
Anti-Virulence Approaches
Quorum sensing inhibitors represent a promising anti-virulence strategy that aims to disrupt bacterial communication without exerting direct lethal pressure [102] [97]. Small molecules that block QS signal reception or synthesis can prevent the coordinated gene expression required for biofilm maturation and virulence factor production [102]. Similarly, exopolysaccharide-degrading enzymes that target Pel, Psl, or alginate can disrupt biofilm integrity and enhance antibiotic penetration [100].
Combination Therapies
Bacteriophage-antibiotic combinations leverage the ability of phages to penetrate biofilms and target bacterial structures, potentially synergizing with conventional antibiotics [97] [100]. Antimicrobial photodynamic therapy uses light-activated compounds to generate reactive oxygen species that can damage multiple cellular targets simultaneously, potentially overcoming conventional resistance mechanisms [97].
Biofilm Dispersal Agents
Compounds that trigger the natural dispersal response of biofilms offer an alternative approach by promoting the transition from protected biofilm communities to vulnerable planktonic cells [100]. Nitric oxide signaling has been shown to mediate phosphodiesterase activity, decrease c-di-GMP levels, and enhance biofilm dispersal in P. aeruginosa [100].
Future Research Priorities
Future investigations should prioritize several key areas: (1) structural biology of QS receptors to guide rational inhibitor design; (2) single-cell and organ-on-a-chip models to dissect biofilm heterogeneity; (3) dual-omics approaches to map host-pathogen signaling crosstalk; and (4) environmental modulators such as iron availability and shear stress that fine-tune virulence expression [102]. Such multidisciplinary efforts will underpin the development of next-generation anti-virulence therapies capable of overcoming the formidable defensive capabilities of P. aeruginosa biofilms.
Evaluating Anti-Biofilm Strategies: From Traditional Agents to Novel Therapies
Comparative Analysis of Biofilm vs. Planktonic Cell Susceptibility
Within the context of microbial biofilm formation research, a critical area of investigation lies in understanding the phenotypic differences between biofilm-associated and planktonic (free-swimming) cells. A key distinction, and the focus of this analysis, is their markedly different susceptibility to antimicrobial agents. This divergence has profound implications for treating persistent infections in clinical settings, controlling contamination in industry, and managing biofouling in environmental contexts [11]. While traditional microbiology has largely relied on planktonic cultures for antimicrobial testing, a paradigm shift is recognizing that the biofilm mode of growth is the predominant and natural state for many bacteria and fungi [103]. This guide provides an in-depth technical analysis of the comparative susceptibility of these two growth modes, detailing the underlying mechanisms, experimental approaches for assessment, and the resultant data that underscores the recalcitrance of biofilm-associated infections.
The Biofilm Life Cycle and Cellular Differentiation
The formation of a biofilm is a multi-stage, developmental process that begins with the transition of planktonic cells to a surface-associated, sessile lifestyle. Understanding this cycle is fundamental to appreciating the physiological changes that confer altered susceptibility.
The lifecycle can be summarized in distinct phases [11] [104] [103]:
- Initial Attachment: Planktonic cells reversibly adhere to a surface via weak interactions like van der Waals forces and hydrophobic interactions. The intracellular signaling molecule bis-(3'-5')-cyclic dimeric guanosine monophosphate (c-di-GMP) plays a crucial role, with its concentration increasing upon surface contact, inhibiting flagella-mediated motility and promoting matrix production.
- Irreversible Attachment and Microcolony Formation: Adhered cells become irreversibly attached and begin to multiply, forming microcolonies. Type IV pili-mediated motility is often involved in cell-cell aggregation.
- Maturation: Cells within the microcolony produce a robust extracellular polymeric substance (EPS) matrix, developing a complex three-dimensional structure. This mature biofilm exhibits heterogeneity with gradients of oxygen, nutrients, and metabolic activity.
- Dispersion: Active or passive dispersal mechanisms release planktonic cells from the biofilm to colonize new niches. Dispersed cells can exhibit properties distinct from both planktonic and sessile biofilm cells [105].
This transition from a planktonic to a biofilm mode of growth is regulated by complex signaling pathways, with quorum sensing (QS) playing a central role in coordinating population-level behavior.
- Gram-Negative Bacteria (e.g., P. aeruginosa): Utilize systems like the LuxI/LuxR homologs. LuxI-type synthases produce N-acyl homoserine lactone (AHL) autoinducers that diffuse and, at a threshold concentration, bind to LuxR-type receptors. The complex then activates transcription of biofilm-related genes [104].
- Gram-Positive Bacteria: Often use modified oligopeptides as autoinducers, which are detected by two-component system sensor kinases, leading to phosphorylation of a response regulator that alters gene expression [104].
QS promotes biofilm formation by regulating the release of extracellular DNA (eDNA), which is critical for adhesion, structural stability, and defense [104].
Key Mechanisms of Biofilm-Associated Antimicrobial Tolerance
The heightened tolerance of biofilms to antimicrobial agents is multifactorial, arising from a combination of physical, physiological, and genetic adaptations.
Table 1: Core Mechanisms of Biofilm Antimicrobial Tolerance
| Mechanism | Description | Impact on Susceptibility |
|---|---|---|
| Physical Barrier of EPS | The extracellular polymeric substance matrix (polysaccharides, proteins, eDNA) limits penetration of antimicrobial agents, acting as a molecular sieve and neutralizing some molecules [11]. | Hinders drug diffusion; first line of defense. |
| Altered Microenvironment | Metabolic gradients within the biofilm create zones of nutrient limitation, low metabolic activity, and acidosis, placing a subpopulation of cells in a slow-growing or dormant state [11] [93]. | Renders cells less susceptible to growth-dependent antibiotics. |
| Physiological Heterogeneity | Biofilms contain a spectrum of cell phenotypes, including metabolically active surface cells and dormant or persister cells in the core. Persister cells are a small, transiently tolerant subpopulation [93]. | Increases population resilience; persisters can re-establish biofilm after treatment. |
| Upregulation of Efflux Pumps | Increased expression and activity of efflux pumps that expel a wide range of toxic compounds, including antibiotics, from the cell interior [105]. | Actively reduces intracellular drug concentration. |
| Genetic Adaptation | The biofilm environment facilitates horizontal gene transfer via the EPS, which can disseminate antibiotic resistance genes. Biofilm-specific gene expression also contributes to tolerance [11]. | Can lead to both inherited and phenotypic resistance. |
Experimental Assessment: Methodologies and Protocols
Accurately comparing the susceptibility of biofilm and planktonic cells requires specialized experimental protocols that account for the fundamental differences in their growth and physiology.
Cultivation Models
- Planktonic Cultures: Grown in liquid broth with shaking to ensure aerobic, free-floating conditions. Cells are typically harvested during the mid- to late-exponential growth phase [106].
- Biofilm Cultures: Cultured using models that support surface attachment and biofilm development.
- Static Microtiter Plate Assay: A high-throughput method where biofilms form on the walls and bottom of polystyrene plate wells. After incubation, non-adherent cells are removed by washing, and adherent biofilms are analyzed [11].
- Flow-Cell Systems: Allow for continuous nutrient supply and waste removal, enabling the development of thick, structurally complex biofilms that can be monitored in real-time via microscopy [103].
- Biofilm Reactors & Alginate Bead Models: Systems like the Calgary Biofilm Device or alginate bead encapsulation (used to mimic the EPS matrix) generate standardized, high-density biofilms for susceptibility testing [105].
Susceptibility Testing Protocols
A critical technical consideration is that standard planktonic Minimum Inhibitory Concentration (MIC) tests are inadequate for biofilms. Instead, methods like the Minimum Biofilm Eradication Concentration (MBEC) are used.
Table 2: Key Susceptibility Testing Methods for Biofilm vs. Planktonic Cells
| Method | Principle | Application & Workflow |
|---|---|---|
| Bacterial Enumeration (Viable Count) | The gold standard for quantifying viable cells. It measures the concentration of colony-forming units (CFU) before and after antimicrobial exposure [107]. | Biofilms are disrupted (e.g., by sonication or scraping) to create a homogeneous suspension. Serial dilutions are plated on agar, and CFUs are counted after incubation. The log reduction in CFU/mL is calculated. |
| Colorimetric Viability Assays (e.g., XTT) | Measures metabolic activity. Metabolically active cells reduce tetrazolium salts like XTT to a water-soluble, colored formazan product, which is quantified spectrophotometrically [107]. | After antimicrobial treatment, XTT solution is added to biofilms or planktonic cells. Following incubation, the absorbance of the supernatant is measured. This method is rapid and correlates well with bacterial enumeration [107]. |
| Minimum Biofilm Eradication Concentration (MBEC) | Determines the lowest concentration of an antimicrobial that eradicates a biofilm, typically defined as a ≥3-log reduction in viability [93]. | Biofilms are grown on pegs (e.g., in the Calgary Biofilm Device) and then transferred to a plate containing serial dilutions of the antimicrobial. After exposure, pegs are rinsed and viability is assessed (e.g., by sonicating into media and plating). |
The following diagram outlines a generalized experimental workflow for a comparative susceptibility study.
Quantitative Data and Comparative Analysis
Empirical data consistently demonstrates that biofilm populations are significantly more tolerant to antimicrobials than their planktonic counterparts, though the degree of tolerance varies by antibiotic class and bacterial species.
Table 3: Comparative Susceptibility of Planktonic vs. Biofilm Cells to Antibiotics
| Antimicrobial Agent / Class | Target | Efficacy Against Planktonic Cells | Efficacy Against Biofilm Cells | Key Findings & Context |
|---|---|---|---|---|
| Cell Wall Synthesis Inhibitors(e.g., Dicloxacillin, Cefazolin, Vancomycin) | Penicillin-binding proteins (PBPs) and cell wall synthesis. | Highly effective, causing rapid cell lysis [107]. | Markedly reduced efficacy; often ineffective at clinically achievable concentrations [107] [93]. | In a study on coagulase-negative staphylococci, inhibitors of cell wall synthesis were "highly effective" against planktonic cells over 3 hours but showed poor activity against biofilms [107]. |
| Protein Synthesis Inhibitors(e.g., Tetracycline) | 30S ribosomal subunit. | Effective against growing cells. | Reduced efficacy, but generally more active than cell wall inhibitors against biofilms [107]. | Susceptibility is "affected by the biofilm phenotype to a lesser degree" than cell wall inhibitors [107]. |
| RNA Synthesis Inhibitors(e.g., Rifampicin) | RNA polymerase. | Effective against growing cells. | Often retains good activity and is used in combination therapy for biofilm infections [107]. | Considered one of the more effective classes; susceptibility is less affected in the biofilm state [107]. |
| DNA Synthesis Inhibitors(e.g., Ciprofloxacin) | DNA gyrase and topoisomerase IV. | Highly effective. | Reduced efficacy, but can impact dispersed cells effectively [105]. | Dispersed cells from biofilms show higher susceptibility to ciprofloxacin than their biofilm counterparts, though they may still be less susceptible than naive planktonic cells [105]. |
| Disinfectants(e.g., Benzalkonium Chloride - BAC) | Cell membrane integrity. | Susceptible, with efficacy dependent on concentration and contact time. | Biofilm-detached and sessile cells show greater resistance, linked to modified membrane fluidity and other physiological adaptations [106]. | Planktonic S. aureus are more susceptible to BAC than biofilm-detached cells. Growth temperature that increases membrane rigidity further enhances resistance [106]. |
The Scientist's Toolkit: Essential Research Reagents and Materials
To conduct robust comparative susceptibility studies, researchers require a suite of specialized reagents and materials.
Table 4: Key Research Reagent Solutions for Biofilm Susceptibility Studies
| Reagent / Material | Function & Application in Research |
|---|---|
| Tetrazolium Salts (XTT, MTT) | Viability Indicator: Used in colorimetric assays to measure cellular metabolic activity. The reduction of XTT to a colored formazan product provides a rapid, quantitative measure of viable cells in both planktonic and biofilm states [107]. |
| Cation-Adjusted Mueller Hinton Broth (CAMHB) | Standardized Susceptibility Testing Medium: The internationally recognized medium for antibiotic susceptibility testing, providing consistent ion concentrations for reliable and reproducible results in planktonic MIC determinations. |
| Microtiter Plates (Polystyrene, 96-well) | High-Throughput Biofilm Cultivation: The standard platform for static biofilm formation assays. The wells serve as surfaces for initial attachment and biofilm development, allowing for parallel testing of multiple conditions and replicates [11]. |
| Flow-Cell Systems | Advanced Biofilm Cultivation: Enables the growth of biofilms under controlled hydrodynamic conditions, which more closely mimic natural and clinical environments (e.g., flowing water, urinary catheters). Ideal for real-time, non-destructive microscopy analysis [103]. |
| Calgary Biofilm Device | Standardized Biofilm Generation: Also known as the MBEC device, it uses a lid with pegs to grow equivalent, high-density biofilms for susceptibility screening. The pegs are dipped into antibiotic solutions for MBEC assays. |
| Neutralizing Solution (e.g., Dey-Engley) | Disinfectant Inactivation: Crucial for accurately determining viability after treatment with chemical disinfectants like benzalkonium chloride. It immediately neutralizes the antimicrobial agent at the end of the contact time to prevent carry-over effects during plating [106]. |
| Extracellular Matrix Components (e.g., Alginate) | Biofilm Mimicry: Used to create artificial biofilm models (e.g., alginate beads) for studying dispersed cells and the protective role of the matrix without the complexity of a natural biofilm [105]. |
The comparative analysis unequivocally demonstrates that biofilm populations exhibit profoundly greater tolerance to antimicrobial agents compared to planktonic cells. This recalcitrance is not attributable to a single mechanism but is an emergent property of the biofilm's structured, matrix-encased, and physiologically heterogeneous community. The data shows that tolerance is most pronounced against antibiotics targeting cell wall synthesis, while inhibitors of RNA and protein synthesis often retain relatively better activity. From a research perspective, these findings mandate a departure from purely planktonic-based antimicrobial testing. Effectively combating biofilm-related challenges in medicine, industry, and the environment requires the development and use of standardized biofilm-specific susceptibility protocols, such as MBEC assays, and the pursuit of novel anti-biofilm strategies that target the unique pathways and physical structures of the biofilm lifestyle.
Evaluating Conventional Antimicrobials and Their Limitations Against Biofilms
Biofilms represent the predominant mode of life for microorganisms, constituting structured communities of microbes encased within a self-produced matrix of extracellular polymeric substances (EPS) and attached to biotic or abiotic surfaces [108] [11]. This sessile existence contrasts sharply with the planktonic (free-floating) state, conferring upon biofilm-dwelling bacteria显著增强的耐受性to antimicrobial agents and environmental stresses [20]. It is estimated that 65-80% of all microbial infections are biofilm-associated, implicating biofilms in a wide array of persistent and chronic conditions, including medical device-related infections, cystic fibrosis pneumonia, chronic wound infections, and otorhinolaryngologic infections [11] [91]. The annual global economic impact of biofilms is staggering, exceeding 5 trillion USD,
driven by costs associated with healthcare interventions, industrial biofouling, and product contamination [109]. Understanding the limitations of conventional antimicrobials against these resilient structures is therefore not merely an academic exercise but a pressing clinical and economic necessity. This review evaluates the multifaceted mechanisms behind biofilm-mediated resistance and the consequent failure of standard antimicrobial regimens, framed within the context of the distinct developmental stages of biofilm formation.
The Biofilm Life Cycle and Architecture
The formation of a biofilm is a dynamic, multi-stage process that transforms free-living planktonic cells into a complex, surface-associated community. This life cycle can be broadly categorized into several key phases, each characterized by distinct genetic and phenotypic profiles [108] [11] [20].
Table 1: Key Stages of Biofilm Development
| Stage | Key Processes | Physiological and Molecular Events |
|---|---|---|
| Initial Attachment | Reversible and irreversible adhesion of planktonic cells to a surface. | Mediated by weak physical forces (van der Waals, electrostatic), flagella, pili, and surface adhesins. Expression of attachment-specific genes is triggered [11] [20]. |
| Microcolony Formation | Cell proliferation and aggregation, forming clustered microcolonies. | Initiation of EPS production. Regulation by intracellular signaling molecules (e.g., c-di-GMP). Downregulation of motility genes [11]. |
| Maturation | Development of a complex three-dimensional structure. | Establishment of a mature EPS matrix with water channels. Activation of Quorum Sensing (QS) for cell-cell communication. Increased heterogeneity and metabolic differentiation [108] [103]. |
| Dispersion | Active detachment of cells from the biofilm. | Triggered by nutrient depletion, oxygen gradients, or QS. Upregulation of motility genes and production of matrix-degrading enzymes (e.g., DNases, dispersin B) [11] [109]. |
The resulting architecture is a critical determinant of biofilm recalcitrance. The biofilm matrix, often comprising 75-90% of the total biomass, is a complex amalgam of exopolysaccharides, extracellular DNA (eDNA), proteins, and lipids [11]. This matrix is not an inert scaffold but a dynamic, biologically active component that provides structural integrity and creates a heterogeneous microenvironment. Gradients of nutrients, oxygen, and metabolic waste products establish distinct physiological zones within the biofilm, leading to varied metabolic activity and growth rates of the embedded cells [108] [103]. This spatial organization is fundamental to the multiple mechanisms of resistance that biofilms exhibit.
Diagram 1: The biofilm life cycle and associated resistance mechanisms. The process begins with planktonic cells attaching to a surface and progresses through irreversible attachment, microcolony formation, and maturation, culminating in dispersion. Key resistance mechanisms emerge at critical stages, highlighted in red.
Mechanisms of Biofilm-Mediated Antimicrobial Resistance
The resilience of biofilms to antimicrobial挑战 is not attributable to a single mechanism but is rather an emergent property of its organized, multicellular structure. The following interconnected mechanisms collectively contribute to the high levels of tolerance and resistance observed.
The Extracellular Polymeric Substance (EPS) as a Physical and Chemical Barrier
The EPS matrix is the first and most significant line of defense for a biofilm community. It acts as a formidable barrier that restricts the penetration of antimicrobial agents [11] [109]. The anionic nature of many EPS components, such as alginate in Pseudomonas aeruginosa, allows them to bind cationic antimicrobials (e.g., aminoglycosides), effectively sequestering them and preventing their reach to underlying cells [11]. Furthermore, the dense, gel-like consistency of the matrix slows down the diffusion of antimicrobial molecules, allowing time for enzymes within the EPS, such as β-lactamases, to degrade the incoming agents before they can exert their effect [109]. This limited penetration is a primary reason why biofilms can survive antibiotic concentrations that are 10 to 1000 times higher than the minimum inhibitory concentration (MIC) for their planktonic counterparts [11].
Metabolic Heterogeneity and Persister Cells
Within the mature biofilm, environmental gradients create zones with varying nutrient and oxygen availability. This leads to a heterogeneous population of cells with divergent metabolic states [108]. Cells in the outer layers, with better access to nutrients, are often metabolically active and more susceptible to antimicrobials. In contrast, cells in the deeper, nutrient-deprived regions enter a slow-growing or dormant state [11] [109]. Most conventional antibiotics, such as β-lactams and fluoroquinolones, target active cellular processes like cell wall synthesis and DNA replication. Consequently, these dormant cells are tolerant to treatment. A sub-population of these, known as persister cells, exhibit extreme multidrug tolerance without genetic mutation. They are believed to be the primary cause of biofilm recurrence post-treatment, as they can repopulate the biofilm once antibiotic pressure is removed [11].
Altered Microenvironment and Expression of Resistance Genes
The localized biofilm microenvironment can differ significantly from the bulk environment. For instance, bacterial metabolism and accumulation of waste products can lead to areas of low pH, which can reduce the efficacy of certain antibiotics [108]. Moreover, the biofilm mode of growth triggers a distinct genetic program. The sessile cells undergo rapid changes in gene expression, including the upregulation of genes encoding for multidrug efflux pumps (e.g., MexAB-OprM in P. aeruginosa) and stress response regulons (e.g., RpoS) [11] [91]. The close proximity of cells within the biofilm also facilitates horizontal gene transfer, efficiently spreading resistance genes, such as those encoding for β-lactamases, across the community [11].
Quorum Sensing (QS) Mediated Regulation
Quorum Sensing is a cell-cell communication system that allows bacteria to coordinate gene expression in a cell-density-dependent manner [109]. In biofilms, QS plays a crucial role in regulating the production of virulence factors, EPS components, and potentially, traits associated with antimicrobial tolerance [109] [91]. By disrupting QS, it is possible to attenuate biofilm formation and virulence, making the biofilm more susceptible to host immune responses and antibiotics, although its direct role in intrinsic antibiotic resistance is still a subject of research [10].
Table 2: Summary of Key Biofilm Resistance Mechanisms and Impact on Antimicrobial Efficacy
| Resistance Mechanism | Impact on Conventional Antimicrobials | Example Pathogens |
|---|---|---|
| EPS-Mediated Penriction Restriction | Delayed or reduced diffusion; binding and neutralization of antimicrobial molecules. | Pseudomonas aeruginosa (alginate), Staphylococcus epidermidis (PIA) [11] [20]. |
| Metabolic Heterogeneity | Inefficacy of antibiotics that target active cellular processes against dormant sub-populations. | Most chronic biofilm infections, leading to persister cell formation [11] [109]. |
| Enhanced Efflux & Stress Response | Active expulsion of antibiotics; general stress adaptation increases survival. | P. aeruginosa, Staphylococcus aureus [11] [91]. |
| Horizontal Gene Transfer | Rapid dissemination of antibiotic resistance genes (e.g., ESBL, carbapenemases) within the biofilm. | Enterobacteriaceae, Acinetobacter baumannii [11]. |
Experimental Models for Evaluating Anti-Biofilm Strategies
The transition from in vitro findings to clinical success requires robust and biologically relevant models for evaluating anti-biofilm therapies. The choice of model system is critical, as it must recapitulate key aspects of the in vivo biofilm environment to generate predictive data [110] [111].
In Vitro Models
Static Models: These include the classic microtiter plate (crystal violet) assay, which is high-throughput and excellent for initial screening of biofilm formation or inhibition [111]. However, these models lack the fluid dynamics present in many natural environments and do not generate the complex 3D architecture of mature biofilms.
Flow-Cell Systems: These models allow for a continuous supply of nutrients and the application of shear stress, supporting the development of structurally complex, mature biofilms that more closely resemble in vivo conditions [103] [111]. They are ideal for real-time, microscopic analysis of biofilm architecture and the efficacy of anti-biofilm agents over time.
Advanced In Vivo and Ex Vivo Models
There is a growing recognition that in vitro models often fail to fully capture the host-pathogen interactions and microenvironment of a clinical biofilm infection [110] [111]. Consequently, more sophisticated models are being developed.
Animal Models: Models in rodents (e.g., foreign-body infection, chronic wound, otitis media, lung infection) provide a whole-organism context, including a functional immune system [111]. For instance, a mouse wound infection model can be used to study polymicrobial biofilm interactions and test the efficacy of topical agents [111].
Organoid-Based Models: A cutting-edge development is the use of human organoids (e.g., airway, gut) infected with biofilms [111]. These systems offer a human-derived, physiologically relevant microenvironment that can bridge the gap between traditional in vitro models and clinical trials, allowing for the study of host-pathogen interactions and antibiotic penetration in a human tissue context [111].
Diagram 2: A workflow for designing anti-biofilm efficacy studies, progressing from the research question through model selection to analysis. The framework emphasizes the parallel use of in vitro and more complex in vivo/organoid models to generate translatable data.
The Scientist's Toolkit: Key Reagents and Materials
Table 3: Essential Research Reagents and Materials for Biofilm Studies
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Microtiter Plates | High-throughput screening of biofilm formation and anti-biofilm compounds. | 96-well polystyrene plates for crystal violet staining [111]. |
| Flow-Cell Systems | Culturing biofilms under continuous flow for structural maturation and real-time analysis. | Commercial flow cells coupled with confocal laser scanning microscopy (CLSM) [103]. |
| Confocal Microscopy Dyes | Vital staining for visualizing live/dead cells and EPS components in 3D. | SYTO9 (green, live), Propidium Iodide (red, dead), Calcein AM (viability), TMA-DPH (biomass) [10]. |
| Enzymatic Dispersal Agents | Experimental tools to dismantle specific EPS components. | DNase I (targets eDNA), Dispersin B (targets polysaccharides), proteases [11] [109]. |
| Quorum Sensing Inhibitors (QSIs) | To investigate the role of QS in biofilm virulence and tolerance. | Synthetic cinnamoyl hydroxamates (target P. aeruginosa), furanones [10] [91]. |
| Synthetic Growth Media | To simulate specific environmental conditions (e.g., cystic fibrosis sputum). | Artificial sputum medium (ASM), Mueller Hinton broth [111]. |
The inherent limitations of conventional antimicrobials against biofilms represent a fundamental challenge in modern medicine. The failure of these treatments is not a simple case of acquired genetic resistance but a complex consequence of the biofilm's physical structure, physiological heterogeneity, and coordinated multicellular behavior. The EPS matrix, the presence of dormant persister cells, and an altered microenvironment create a formidable fortress that most current drugs cannot reliably breach. Overcoming this challenge requires a paradigm shift from simply trying to kill growing bacteria to developing strategies that specifically target the biofilm state itself. Future directions must include the rational design of anti-biofilm agents that disrupt the EPS matrix, inhibit quorum sensing, or actively wake persister cells to render them susceptible to traditional antibiotics. Furthermore, the development of more predictive and clinically relevant biofilm models is essential for translating promising in vitro findings into effective clinical therapies. By deepening our understanding of the biofilm life cycle and its associated resistance mechanisms, the scientific community can pave the way for a new arsenal of therapeutic strategies to combat these persistent and costly infections.
Microbial biofilms represent a significant hurdle in modern medicine, contributing to persistent infections, medical device failures, and antimicrobial resistance. These structured communities of microorganisms, encased in a self-produced extracellular polymeric substance (EPS) matrix, exhibit tolerance to antimicrobial agents that can be up to 1,000 times greater than their planktonic counterparts [16] [11]. This resilience stems from a complex, multi-stage developmental process that creates physical and physiological barriers to conventional treatments. Within healthcare settings, biofilms are implicated in approximately 65-80% of microbial infections, causing significant morbidity, mortality, and economic burden estimated at billions of dollars annually [12] [11].
The extracellular matrix, comprising polysaccharides, proteins, nucleic acids, and lipids, forms a protective scaffold that limits antibiotic penetration, houses metabolically heterogeneous cell subpopulations, and facilitates adaptive resistance mechanisms [11] [112]. This biological complexity necessitates innovative approaches that target specific biofilm vulnerabilities throughout their developmental timeline. The emerging paradigm in anti-biofilm therapeutic development involves strategically intervening at critical junctures in the biofilm lifecycle using highly specific biological and nano-engineered tools.
This technical review examines three prominent emerging strategies—enzymes, bacteriophages, and nanoparticles—framed within the context of biofilm developmental biology. We present a detailed analysis of their mechanisms of action, experimental validation, and potential for clinical translation, with particular emphasis on combination approaches that target multiple biofilm vulnerabilities simultaneously.
Biofilm Formation Stages: Targeting Strategic Intervention Points
Biofilm development follows a defined sequence of events, each presenting unique vulnerabilities for therapeutic intervention. Understanding these stages is fundamental to designing effective anti-biofilm strategies.
Table 1: Stages of Biofilm Formation and Potential Intervention Points
| Formation Stage | Key Characteristics | Vulnerabilities for Intervention |
|---|---|---|
| Initial Attachment | Planktonic cells reversibly adhere via van der Waals forces, electrostatic interactions, and bacterial appendages like pili and flagella [16] [113] | Surface modifications, anti-adhesion coatings, interference with surface sensing |
| Irreversible Attachment | Production of EPS anchors cells firmly; downregulation of motility genes; increased c-di-GMP signaling [11] [104] | EPS-degrading enzymes, disruption of c-di-GMP signaling, matrix-targeting agents |
| Microcolony Formation | Cellular replication and aggregation into structured communities; beginning of metabolic differentiation [113] [104] | Quorum sensing inhibitors, targeted antimicrobials against actively dividing cells |
| Maturation | Development of complex 3D architecture with water channels; pronounced metabolic heterogeneity; maximal EPS production [11] [113] | Matrix-penetrating nanoparticles, combination therapies, dispersal agents |
| Dispersion | Active and passive release of planktonic cells to colonize new surfaces; often mediated by environmental cues and enzymatic matrix degradation [16] [11] | Dispersal triggers, anti-motility agents, preventation of secondary colonization |
The intracellular signaling molecule bis-(3'-5')-cyclic dimeric guanosine monophosphate (c-di-GMP) plays a crucial regulatory role throughout early biofilm development, inhibiting flagella-mediated motility while promoting EPS production and surface adherence [11] [104]. The transition from reversible to irreversible attachment represents a critical commitment point in biofilm development, characterized by significant transcriptional reprogramming that prioritizes matrix production over motility functions [16] [113].
Figure 1: Biofilm Developmental Cycle and Key Regulatory Transitions. The process begins with planktonic cell attachment and progresses through defined stages characterized by distinct genetic and structural changes, culminating in dispersion that enables new colonization sites.
Enzymatic Strategies: Targeted Matrix Disruption
Enzyme-based approaches represent a promising anti-biofilm strategy with precise targeting capabilities against specific EPS components. These biological catalysts offer an environmentally friendly solution with inherent biodegradability and minimal toxicity profiles [12].
Major Enzyme Classes and Their Mechanisms
Glycoside Hydrolases target the polysaccharide components of the biofilm matrix, which serve as critical structural elements and mediate surface adhesion. Dispersin B, a well-characterized glycosidase produced by Aggregatibacter actinomycetemcomitans, specifically hydrolyzes poly-N-acetylglucosamine (PNAG), a polysaccharide that facilitates biofilm formation and biocide tolerance in numerous Gram-positive and Gram-negative pathogens [12]. This enzyme has demonstrated efficacy in inhibiting biofilm formation, detaching established biofilms, and increasing susceptibility to conventional antimicrobials. Another prominent example, cellulase, exhibits concentration- and pH-dependent activity against Pseudomonas aeruginosa biofilms, with superior performance observed at pH 5 compared to pH 7 [12]. Size exclusion chromatography studies confirm that cellulase reduces the apparent molecular weight of P. aeruginosa exopolysaccharides while increasing reducing sugar levels, directly demonstrating EPS degradation.
Proteases target proteinaceous components of the EPS matrix, including surface adhesins and structural proteins that maintain biofilm integrity. Specific proteases can degrade these protein elements, compromising structural stability and facilitating biofilm removal [12] [11].
Deoxyribonucleases (DNases) attack the extracellular DNA (eDNA) component of biofilms, which contributes to structural stability and facilitates horizontal gene transfer [12] [11]. DNases are particularly effective against biofilms from certain bacterial species where eDNA comprises a substantial matrix fraction.
Table 2: Enzymes with Demonstrated Anti-Biofilm Activity
| Enzyme Class | Specific Examples | Target Substrate | Microbial Source | Reported Efficacy |
|---|---|---|---|---|
| Glycoside Hydrolases | Dispersin B | Poly-N-acetylglucosamine (PNAG) | Aggregatibacter actinomycetemcomitans | Inhibits biofilm formation, detaches established biofilms, increases antimicrobial susceptibility [12] |
| Cellulase | Cellulose-based polysaccharides | Various microbial sources | Reduces P. aeruginosa biofilm biomass by 40-70% at 37.6-75.2 U/mL, pH-dependent efficacy [12] | |
| Levan hydrolase | Levan | Various bacterial sources | Degrades fructan-based biofilm matrices [12] | |
| Proteases | Various extracellular proteases | Matrix proteins, adhesins | Multiple bacterial and fungal species | Disrupts protein-mediated structural integrity, enhances detachment [12] [11] |
| Nucleases | DNase I | Extracellular DNA (eDNA) | Bovine pancreas, microbial recombinants | Reduces biofilm stability, prevents initial attachment in some species [12] [11] |
Experimental Protocol: Evaluating Enzymatic Anti-Biofilm Activity
Objective: Assess the efficacy of glycoside hydrolases in preventing biofilm formation and disrupting pre-formed biofilms of Pseudomonas aeruginosa.
Methodology:
- Biofilm Cultivation: Grow P. aeruginosa biofilms in parallel flow chambers or 96-well peg plates for 4 days using glucose-minimal medium as nutrient source [12].
- Enzyme Treatment:
- For prevention studies: Add cellulase at concentrations of 9.4, 37.6, and 75.2 U/mL during biofilm development.
- For disruption studies: Apply enzymes to mature (4-day) biofilms for 24-hour exposure.
- Include controls with heat-inactivated enzyme.
- Efficacy Assessment:
- Biomass Quantification: Use crystal violet staining to measure total attached biomass [12].
- Viability Assessment: Determine colony-forming units (CFU) after biofilm disruption.
- EPS Analysis: Assess exopolysaccharide molecular weight distribution via size exclusion chromatography and monitor reducing sugar production [12].
- Synergy Testing: Combine sub-effective concentrations of cellulase (9.4 U/mL) with ceftazidime at 0.5× MIC to evaluate antibiotic enhancement [12].
Bacteriophage-Based Strategies: Biological Precision Targeting
Bacteriophages (phages) offer a biologically evolved approach to biofilm control, utilizing natural bacterial viruses with precision targeting capabilities. Their inherent ability to infect and replicate within specific bacterial hosts makes them particularly attractive for targeting biofilm-embedded cells.
Phage-Biofilm Interactions and Anti-Biofilm Mechanisms
The interaction between phages and biofilms is complex, with outcomes ranging from complete biofilm destruction to stable coexistence. Key advantages of phages include their self-replication at infection sites, production of matrix-degrading enzymes, and ability to penetrate biofilm structures through enzymatic activity [16] [113].
Phages employ multiple mechanisms to disrupt biofilms:
- Direct Lytic Activity: Phages attach to bacterial surface receptors, inject genetic material, and hijack cellular machinery to produce progeny virions, ultimately causing cell lysis through endolysin and holin activity [16] [114].
- Matrix Degradation: Many phages encode polysaccharide-depolymerases that specifically degrade key EPS components, facilitating phage penetration and biofilm disintegration [16] [113].
- Penetration via Water Channels: The inherent porosity of mature biofilms, characterized by water channels, enables phage access to deeper cellular layers [113].
Despite these advantages, phage therapy faces challenges including narrow host range, potential for bacterial resistance development through CRISPR-Cas systems or receptor modification, and hindered diffusion through dense EPS matrices [16] [113].
Experimental Protocol: Bacteriophage-Mediated Biofilm Removal
Objective: Evaluate the efficacy of bacteriophage cocktails and phage-antibiotic combinations against established biofilms.
Methodology:
- Phage Propagation and Characterization:
- Isolate phages from environmental sources using bacterial host enrichment.
- Purify via plaque assay and characterize host range against clinically relevant strains.
- Confirm lytic lifecycle (absence of integrase genes) for therapeutic safety [16].
- Biofilm Disruption Assay:
- Grow 48-hour mature biofilms in 96-well plates or on relevant material coupons (e.g., catheter segments).
- Treat with: (i) monophage suspension (10⁸ PFU/mL), (ii) phage cocktail (multiple phages with complementary host ranges), (iii) phage-antibiotic combinations (e.g., phage with colistin at 0.5× MIC) [114].
- Incubate for 24 hours at host-optimal temperature.
- Efficacy Assessment:
- Resistance Monitoring: Passage surviving cells with phage pressure to evaluate resistance development frequency.
Figure 2: Bacteriophage Mechanisms for Biofilm Destructuration. Phages employ a dual strategy: direct bacterial cell lysis through the infectious cycle, and enzymatic degradation of the EPS matrix to enhance penetration and biofilm structural collapse.
Nanoparticle-Based Strategies: Enhanced Penetration and Delivery
Nanoparticles (NPs) represent a versatile platform for anti-biofilm strategies, offering enhanced penetration, targeted delivery, and multifunctional capabilities. Their small size (typically <500 nm) enables improved diffusion through biofilm matrices, while their surface functionality allows for precise targeting and controlled release of antimicrobial payloads [115] [112].
Nanoparticle Types and Anti-Biofilm Applications
Lipid-Based Nanoparticles including liposomes and nanoemulsions can encapsulate both hydrophilic and hydrophobic antimicrobials, facilitating fusion with bacterial membranes for efficient intracellular delivery. Liposomal CRISPR-Cas9 formulations have demonstrated remarkable efficacy, reducing P. aeruginosa biofilm biomass by over 90% in vitro [112].
Metallic Nanoparticles such as gold and silver NPs possess intrinsic antimicrobial properties and can be functionalized with targeting ligands. Gold nanoparticle-CRISPR hybrids have shown a 3.5-fold increase in gene-editing efficiency compared to non-carrier systems while promoting synergistic action with antibiotics [112].
Polymeric Nanoparticles including chitosan and PLGA-based systems offer controlled release kinetics and mucoadhesive properties that prolong residence time at infection sites [115].
Bacteriophage-Nanoparticle Hybrids represent an emerging frontier, combining the biological targeting of phages with the enhanced delivery capabilities of nanomaterials. These systems can be engineered to display specific peptides for targeted delivery while encapsulating therapeutic payloads [115] [116].
Experimental Protocol: Nanoparticle-Mediated Biofilm Penetration and Treatment
Objective: Evaluate the biofilm penetration efficiency and anti-biofilm efficacy of engineered nanoparticle formulations.
Methodology:
- Nanoparticle Synthesis and Characterization:
- Prepare lipid nanoparticles via thin-film hydration with Cas9/sgRNA complexes targeting bacterial resistance genes (e.g., ndm-1, mecA) [112].
- Synthesize gold-chitosan hybrids using ionotropic gelation, conjugating with quorum sensing inhibitors (e.g., furanones).
- Characterize size (100-200 nm optimal), zeta potential (>+30 mV for enhanced biofilm interaction), and drug loading efficiency [112].
- Penetration Assessment:
- Grow 5-day mature biofilms in flow cells simulating in vivo conditions.
- Treat with fluorescently labeled NPs (e.g., Cy5-tagged) for 24 hours.
- Analyze penetration depth using confocal laser scanning microscopy with z-stack imaging and quantify fluorescence intensity at different biofilm strata [112].
- Efficacy Evaluation:
- Treat mature biofilms with: (i) NPs alone, (ii) free antimicrobials, (iii) NP-antimicrobial conjugates, (iv) NP-CRISPR complexes.
- Assess biofilm viability using resazurin metabolism assay and quantify biomass removal via crystal violet staining.
- Evaluate synergistic effects with conventional antibiotics by calculating fractional inhibitory concentration indices [112].
- Mechanistic Studies: Assess quorum sensing inhibition using reporter strains (e.g., C. violaceum for AHL detection) and resistance gene editing efficiency via PCR and sequencing.
Integrated Approaches and Future Perspectives
The complexity of biofilm biology necessitates combinatorial approaches that target multiple vulnerabilities simultaneously. Integrated strategies leveraging enzymes, bacteriophages, and nanoparticles represent the most promising direction for effective biofilm control.
Combination Strategies with Enhanced Efficacy
Enzyme-Nanoparticle Conjugates combine the precise matrix degradation of enzymes with the enhanced penetration capabilities of nanoparticles. For instance, DNase-functionalized gold nanoparticles demonstrate improved diffusion through biofilms and simultaneous degradation of eDNA components [12] [112].
Phage-Enzyme Cocktails utilize bacteriophages for bacterial targeting alongside purified depolymerases for enhanced matrix penetration. This approach has shown efficacy against multi-species biofilms, including Pseudomonas aeruginosa-Candida albicans mixed communities [114].
CRISPR-Nanoparticle Hybrid Systems represent a cutting-edge approach for precision targeting of resistance genes. These systems enable selective disruption of antibiotic resistance genes (e.g., bla, mecA) or quorum sensing networks (e.g., lasI/rhlI) while minimizing collateral damage to commensal flora [112].
Table 3: Quantitative Efficacy of Combined Anti-Biofilm Approaches
| Combination Approach | Components | Target Biofilm | Efficacy Metrics |
|---|---|---|---|
| Enzyme-Antibiotic | Cellulase + Ceftazidime | P. aeruginosa | 4-log reduction in CFU vs. 1-log with antibiotic alone [12] |
| CRISPR-Nanoparticle | Liposomal Cas9/sgRNA-ndm1 + Meropenem | Carbapenem-resistant E. coli | 99.9% biomass reduction; restored antibiotic susceptibility [112] |
| Phage-Antibiotic | Phage endolysin + Colistin | Acinetobacter baumannii | 3.5-fold enhancement in bacterial killing vs. monotherapy [114] |
| Nanoparticle-Antibiotic | Gold NP + Ciprofloxacin | S. aureus biofilm | 99% eradication at 64× lower antibiotic dose [112] |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Anti-Biofilm Research
| Reagent Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| Biofilm Cultivation Systems | Calgary biofilm device (peg plates), Flow cells with glass surfaces [12] | Standardized biofilm growth and treatment | Reproducible biofilm formation under static or shear stress conditions |
| Matrix-Degrading Enzymes | Dispersin B (0.1-5 µg/mL), DNase I (100 U/mL), Cellulase (37.6-75.2 U/mL) [12] | EPS disruption studies | Targeted degradation of specific matrix components (PNAG, eDNA, cellulose) |
| Engineered Bacteriophages | Phages with depolymerase activity, CRISPR-phage hybrids [16] [114] | Precision antimicrobial studies | Species-specific bacterial lysis with enhanced matrix penetration |
| Functionalized Nanoparticles | Gold NPs (10-100 nm), Cationic liposomes, Chitosan nanocarriers [115] [112] | Enhanced drug delivery | Improved biofilm penetration and targeted antimicrobial delivery |
| Quorum Sensing Inhibitors | Furano nes, AHL analogs, Quorum quenching antibodies [11] [104] | Anti-virulence studies | Disruption of cell-to-cell communication and coordinated gene expression |
| CRISPR-Cas Components | Cas9/sgRNA complexes targeting blaNDM-1, mecA [112] | Genetic targeting studies | Precise disruption of antibiotic resistance genes and virulence determinants |
| Viability Assessment Tools | Resazurin metabolism assay, ATP bioluminescence, LIVE/DEAD staining with confocal microscopy [12] [112] | Biofilm viability quantification | Metabolic activity measurement and spatial distribution of live/dead cells |
The multifaceted challenge of biofilm-associated infections demands equally sophisticated solutions that target specific developmental stages and resistance mechanisms. Enzymatic strategies offer precise matrix degradation, bacteriophages provide biological precision targeting, and nanoparticle systems enable enhanced penetration and delivery. The future of anti-biofilm therapy lies in intelligent combinations of these approaches, potentially including CRISPR-based genetic editing for resistance reversal and quorum sensing disruption for virulence attenuation.
Successful clinical translation will require careful consideration of biofilm heterogeneity, pharmacokinetic optimization for biofilm penetration, and addressing potential resistance development against these novel modalities. As our understanding of biofilm biology evolves, so too will these emerging strategies, potentially leading to a new arsenal of effective treatments for some of the most persistent challenges in clinical microbiology.
Biofilms are complex, three-dimensional microbial communities that grow at interfaces and are embedded within a self-produced matrix of extracellular polymeric substances (EPS) [6] [25]. This EPS matrix, composed of polysaccharides, proteins, nucleic acids, and lipids, provides structural stability and protects the residing cells from environmental threats, including antimicrobial agents and host immune responses [25] [12]. The biofilm lifecycle is a structured process, beginning with the initial reversible attachment of planktonic cells to a surface, proceeding through irreversible attachment and the development of a mature, complex structure, and culminating in a dispersal phase where cells detach to colonize new surfaces [25] [117]. This lifecycle is a key survival mechanism for bacteria in nature [12].
The clinical significance of biofilms is profound, as they are implicated in over 60-80% of microbial infections and represent a core challenge in treating chronic diseases and infections associated with medical devices [6] [118]. A primary reason for this is biofilm-associated antimicrobial resistance, where cells within a biofilm can exhibit up to 1,000-fold greater resistance to antimicrobials compared to their planktonic counterparts [12]. This recalcitrance is multifactorial, arising from factors such as impaired penetration of antimicrobials through the EPS matrix, the presence of metabolically dormant persister cells, and adaptive stress responses [25] [119]. Given the failure of conventional monotherapies to effectively eradicate biofilms, combination strategies have emerged as a promising paradigm. By targeting different aspects of biofilm structure and microbial physiology simultaneously, these approaches aim to achieve synergistic efficacy, disrupting the physical and biological barriers that make biofilms so difficult to treat [118] [119].
Model System I: β-Lactam Combination against Carbapenem-ResistantKlebsiella pneumoniae
Background and Rationale
Klebsiella pneumoniae (KP) is a formidable opportunistic pathogen responsible for severe infections including liver abscesses, urinary tract infections, and pneumonia [120]. The global rise of carbapenem-resistant K. pneumoniae (CRKP) poses a critical public health threat, as these strains are resistant to last-resort antibiotics, leading to limited therapeutic options and high mortality rates [120]. A key resistance mechanism in CRKP is the production of carbapenemases, such as KPC-2 (a serine β-lactamase) and NDM-1 (a metallo-β-lactamase) [120]. While the drug ceftazidime-avibactam (CZA) is effective against strains producing KPC-2, it is ineffective against those producing NDM-1. Conversely, aztreonam (ATM), a monobactam, is stable against metallo-β-lactamases but can be hydrolyzed by other β-lactamases. The combination of CZA and ATM is therefore rationalized: avibactam in CZA protects both ceftazidime and aztreonam from serine β-lactamases, while aztreonam retains its activity against metallo-β-lactamases, creating a comprehensive antibacterial profile [120].
Detailed Experimental Protocol
1. Bacterial Strains and Culture Conditions:
- Collect 150 non-duplicate clinical isolates of CRKP from various sample types (sputum, blood, drainage fluid) [120].
- Identify all isolates using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) [120].
- Detect common carbapenemase genes (blaKPC-2, blaNDM-1, blaIMP, blaVIM, blaOXA-48) via polymerase chain reaction (PCR) with specific primers [120].
2. Checkerboard Synergy Assay:
- Prepare stock solutions of CZA and ATM and perform twofold serial dilutions in a 96-well microtiter plate to create a concentration matrix [120].
- Inoculate each well with a bacterial suspension adjusted to approximately 5 × 10^5 CFU/mL [120].
- Incubate the plate at 37°C for 18-24 hours [120].
- Determine the Minimum Inhibitory Concentration (MIC) of each drug alone and in combination. Calculate the Fractional Inhibitory Concentration Index (FICI) using the formulas:
- FICI = FICA + FICB
- FICA = MIC of drug A in combination / MIC of drug A alone
- FICB = MIC of drug B in combination / MIC of drug B alone
- Interpret the FICI: < 0.5 indicates synergy; 0.5-4.0 indicates no interaction (additive or indifferent); > 4.0 indicates antagonism [120].
3. Time-Kill Assay:
- Exponentially growing CRKP strains are diluted to approximately 1 × 10^5 CFU/mL in fresh Mueller-Hinton broth [120].
- Apply treatments: CZA alone, ATM alone, and the CZA+ATM combination at predetermined concentrations (e.g., 0.5x, 1x, 2x MIC) [120].
- Incubate the cultures at 35°C with constant shaking [120].
- Withdraw samples at 0, 2, 4, 6, 8, and 24 hours, perform serial dilutions, and plate them onto agar for viable colony count (CFU/mL) [120].
- Plot time-kill curves (log10 CFU/mL vs. time). Synergy is defined as a ≥2-log10 decrease in CFU/mL by the combination compared to the most active single agent at 24 hours [120].
4. Biofilm Inhibition and Disruption Assays:
- Crystal Violet Staining for Biofilm Biomass:
- Grow biofilms in 96-well plates for 24-48 hours [120].
- Treat biofilms with CZA, ATM, or the combination for a further 24 hours.
- Fix biofilms with methanol, stain with 0.1% crystal violet for 15 minutes, and dissolve the bound dye in acetic acid [120].
- Measure the absorbance at 570 nm to quantify total biofilm biomass [120].
- Bacterial Cell Permeability Assay:
5. Gene Expression Analysis via qRT-PCR:
- Extract total RNA from CRKP biofilms treated with CZA, ATM, or their combination [120].
- Synthesize complementary DNA (cDNA) and perform quantitative real-time PCR (qRT-PCR) with primers specific for biofilm-related genes (LuxS, mrkA, wbbM, pgaA, wzm) [120].
- Normalize gene expression levels to a housekeeping gene (e.g., rpoB) and calculate the relative fold-change using the 2^(-ΔΔCt) method [120].
Key Quantitative Findings
Table 1: Synergistic Efficacy of CZA and ATM against CRKP Isolates
| Strain / Parameter | CZA MIC alone (μg/mL) | ATM MIC alone (μg/mL) | FICI (CZA+ATM) | Interpretation | Reduction in Biofilm Genes (qRT-PCR) |
|---|---|---|---|---|---|
| CRKP (blaKPC-2) | 16 | 32 | 0.25 | Synergy | Significant downregulation of LuxS, mrkA, wbbM, pgaA, wzm |
| CRKP (blaNDM-1) | 64 | 16 | 0.31 | Synergy | Significant downregulation of LuxS, mrkA, wbbM, pgaA, wzm |
| CRKP (Mixed Enzymes) | Variable | Variable | < 0.5 | Synergy (p<0.01) | Data not shown in source |
Table 2: Time-Kill Assay Results for a Representative CRKP Strain
| Time Point | Control (log10 CFU/mL) | CZA alone (log10 CFU/mL) | ATM alone (log10 CFU/mL) | CZA+ATM (log10 CFU/mL) |
|---|---|---|---|---|
| 0 hours | 5.0 | 5.0 | 5.0 | 5.0 |
| 4 hours | 5.8 | 5.5 | 5.6 | 4.8 |
| 8 hours | 6.5 | 6.2 | 6.3 | 3.5 |
| 24 hours | 8.0 | 7.5 | 7.8 | 2.0 (99% reduction) |
Mechanism of Action and Pathway Diagram
The CZA+ATM combination disrupts both the physical structure of the biofilm and the cellular physiology of the bacteria. CZA targets cell wall synthesis (PBP3) while avibactam inhibits serine β-lactamases. ATM independently targets cell wall synthesis and remains stable against metallo-β-lactamases. The combination leads to severe cell wall damage, increased permeability, and ultimately, cell lysis. Furthermore, the treatment significantly downregulates key genes involved in biofilm formation and maintenance, such as those responsible for adhesion (mrkA) and exopolysaccharide synthesis (wbbM, pgaA, wzm), thereby compromising the biofilm's structural integrity [120].
Diagram 1: Mechanism of CZA and ATM Synergy. The combined action disrupts cell wall synthesis and protects aztreonam from degradation, leading to cellular damage and biofilm gene downregulation.
Model System II: Synthetic Polymers with Antibiotics against Resistant Pathogens
Background and Rationale
The rise of pan-resistant pathogens has spurred the development of novel antimicrobial agents beyond traditional antibiotics. Synthetic nano-engineered antimicrobial polymers (SNAPs) are engineered mimics of antimicrobial peptides (AMPs), which disrupt bacterial membranes and are less prone to inducing resistance [119]. However, like AMPs, SNAPs can face challenges such as cytotoxicity and reduced activity in physiological environments. Combining SNAPs with conventional antibiotics offers a strategy to enhance efficacy, reduce required dosages, and mitigate potential side effects [119]. This approach is particularly valuable against biofilm infections, where the heterogeneous nature of the community requires agents that target both actively growing and dormant cells.
Detailed Experimental Protocol
1. Polymer Synthesis and Characterization:
- Synthesize SNAPs via Reversible Addition-Fragmentation Chain Transfer (RAFT) polymerization to ensure low dispersity and controlled block structure [119].
- Use monomers like (guanidino-ethyl)acrylamide (GEAM) for a guanidinium-functionalized SNAP (e.g., g-D50) and N-(2-aminoethyl)acrylamide (AEAM) for an ammonium-functionalized SNAP (e.g., a-T50). Incorporate N-isopropylacrylamide (NIPAM) to modulate hydrophobicity [119].
- Confirm polymer structure and purity using 1H NMR spectroscopy and Size Exclusion Chromatography (SEC) [119].
- Assess hydrophobicity via Reverse Phase HPLC (RP-HPLC) and check for thermoresponsive behavior using UV-vis spectroscopy [119].
2. Checkerboard Assay for Synergy Screening:
- Perform checkerboard assays in a 96-well format using cation-adjusted Mueller-Hinton broth (caMHB) and, crucially, in media mimicking infection sites: Synthetic Wound Fluid (SWF) for S. aureus and Synthetic Cystic Fibrosis Sputum Medium (SCFM) for P. aeruginosa [119].
- Prepare serial dilutions of the SNAP and the antibiotic in the chosen medium [119].
- Inoculate wells with a standardized suspension (~5 × 10^5 CFU/mL) of the target pathogen (Staphylococcus aureus USA300 or Pseudomonas aeruginosa PA14) [119].
- Incubate at 37°C for 18-24 hours. Determine the MIC of each agent alone and in combination, and calculate the FICI as described in Section 2.2 [119].
3. Resazurin Viability Assay and Synergy Landscape Modeling:
- For synergistic combinations, perform a dose-response matrix assay using a resazurin metabolic dye to distinguish between bactericidal and bacteriostatic effects [119].
- Incubate bacteria with varying concentrations of the SNAP and antibiotic for a set period (e.g., 4-6 hours) [119].
- Add resazurin solution and measure fluorescence after further incubation. The loss of metabolic activity (no fluorescence increase) indicates a bactericidal effect [119].
- Input the dose-response data into software like SynergyFinder to generate 3D synergy landscapes and calculate synergy scores (e.g., Zero Interaction Potency - ZIP scores) [119].
4. Anti-Biofilm Efficacy Assessment:
- In Vitro Biofilm Models: Grow biofilms in flow cells or on peg lids in 96-well plates for 24-48 hours [119].
- Treat pre-formed biofilms with the SNAP, antibiotic, or combination for 24 hours [119].
- Quantify viable cells within the biofilm by sonicating/scrubbing the biofilm, serially diluting, and plating for CFU counts. Alternatively, use metabolic assays like XTT or crystal violet staining for biomass [119].
- Ex Vivo Biofilm Models: Use an ex vivo wounded human skin model for S. aureus or an ex vivo porcine lung model for P. aeruginosa to validate findings under more physiologically relevant conditions [119].
- Treat the biofilms and quantify bacterial load via CFU counting or confocal microscopy with live/dead staining [119].
Key Quantitative Findings
Table 3: Synergistic Interactions of SNAPs with Antibiotics/Biocides
| SNAP Polymer | Target Pathogen | Synergistic Partner | FICI (in caMHB) | FICI (in Infection-Mimicking Media) | Key Outcome |
|---|---|---|---|---|---|
| g-D50 (Guanidinium) | S. aureus USA300 | Silver Sulfadiazine | < 0.5 (Synergy) | < 0.5 in SWF (Synergy) | Potent synergistic antibiofilm activity in vitro and ex vivo |
| g-D50 (Guanidinium) | S. aureus USA300 | Penicillin | ≥ 0.5 (Additive) | < 0.5 in SWF (Synergy) | Synergy is media-dependent |
| a-T50 (Ammonium) | P. aeruginosa PA14 | Colistin | < 0.5 (Synergy) | < 0.5 in SCFM (Synergy) | Potent synergistic antibiofilm activity in vitro and ex vivo |
| a-T50 (Ammonium) | P. aeruginosa PA14 | Tobramycin | ≥ 0.5 (Additive) | ≥ 0.5 in SCFM (Additive) | No synergy observed |
Mechanism of Action and Pathway Diagram
SNAPs are designed to mimic host defense peptides, primarily targeting and disrupting the bacterial cytoplasmic membrane through electrostatic interactions and pore formation. This membrane damage increases permeability, facilitating the entry of co-administered antibiotics. For example, the synergy between a-T50 and colistin is potent because both agents target the membrane: a-T50 causes initial damage, which allows colistin to more effectively integrate into and disrupt the outer and inner membranes of Gram-negative bacteria, leading to potent bactericidal activity even against biofilm-embedded cells [119].
Diagram 2: Generalized Mechanism of SNAP-Antibiotic Synergy. SNAPs disrupt the bacterial membrane, which enhances the uptake and efficacy of co-administered antibiotics.
The Scientist's Toolkit: Essential Reagents and Methodologies
Table 4: Key Research Reagent Solutions for Biofilm Combination Therapy Research
| Reagent / Material | Function / Application | Example from Reviewed Studies |
|---|---|---|
| Ceaftazidime-Avibactam (CZA) | A β-lactam/β-lactamase inhibitor combination used to treat infections caused by multidrug-resistant Gram-negative bacteria. | Testing synergy with aztreonam against CRKP [120]. |
| Aztreonam (ATM) | A monobactam antibiotic stable against metallo-β-lactamases. | Used in combination with CZA to overcome diverse carbapenemase-mediated resistance [120]. |
| Synthetic Nano-engineered Antimicrobial Polymers (SNAPs) | Engineered polymer mimics of antimicrobial peptides that disrupt bacterial membranes. | g-D50 and a-T50 polymers combined with antibiotics against S. aureus and P. aeruginosa biofilms [119]. |
| Crystal Violet Stain | A dye that binds to proteins and polysaccharides, used for the quantitative assessment of total biofilm biomass. | Used to measure the impact of CZA+ATM on CRKP biofilm biomass [120]. |
| SYTOX Green / Propidium Iodide | Cell-impermeant fluorescent nucleic acid stains used to label dead bacteria or cells with compromised membranes. | Used in flow cytometry and fluorescence microscopy to assess cell viability and membrane integrity after treatment [121]. |
| Adenosine Triphosphate (ATP) Bioluminescence Assay | A method to quantify viable, metabolically active cells by measuring intracellular ATP levels using a luciferin-luciferase reaction. | Used as a rapid method for quantifying biofilm cells on pipe materials [121]. |
| Resazurin Sodium Salt | A blue dye that is reduced to pink, fluorescent resorufin by metabolically active cells; used as an indicator of cell viability. | Employed to determine the bactericidal effect of SNAP-antibiotic combinations [119]. |
| PCR Primers for Biofilm & Resistance Genes | Specific oligonucleotide sequences used to amplify and detect genes involved in biofilm formation and antibiotic resistance via PCR/qRT-PCR. | Used to detect carbapenemase genes (blaKPC, blaNDM) and quantify expression of biofilm genes (mrkA, luxS) [120]. |
| Synthetic Wound Fluid (SWF) & Synthetic Cystic Fibrosis Medium (SCFM) | Complex growth media designed to chemically mimic the physiological environments of chronic wounds and cystic fibrosis lungs, respectively. | Used in checkerboard assays to evaluate synergy under clinically relevant conditions [119]. |
Advanced Quantitative Analysis and Emerging Tools
Modern biofilm research has moved beyond simple biomass quantification to sophisticated 3D spatial and temporal analyses. Software tools like COMSTAT and its successor BiofilmQ enable high-throughput, quantitative analysis of 3D image stacks obtained from confocal laser scanning microscopy (CLSM) [58] [99]. These tools can measure hundreds of parameters, including total biovolume, average thickness, surface area, roughness coefficient, and spatial distribution of fluorescent reporters (e.g., for specific gene expression or matrix components) within the biofilm architecture [58]. This allows researchers to objectively compare the structural integrity of biofilms before and after combination treatments with a high degree of statistical confidence. For instance, Analysis of Variance (ANOVA) models can be applied to COMSTAT data to account for inherent variability in biofilm experiments and rigorously validate the structural impact of a treatment across independent experimental rounds [99].
Furthermore, the field is increasingly leveraging enzymatic disruption as a component of combination strategies. Enzymes such as Dispersin B (a glycoside hydrolase that degrades poly-N-acetylglucosamine), cellulase, proteases, and DNase target specific components of the EPS matrix—polysaccharides, proteins, and extracellular DNA, respectively [12]. By breaking down the biofilm's physical scaffold, these enzymes can enhance the penetration and efficacy of subsequently administered antibiotics, representing a potent synergistic partnership for biofilm eradication [12].
The fight against biofilm-related infections necessitates a shift from monotherapy to sophisticated, multi-targeted approaches. The synergistic strategies detailed in this review—combining antibiotics to overcome enzymatic resistance (CZA+ATM) or pairing novel membrane-disrupting agents with conventional antibiotics (SNAPs+antibiotics)—demonstrate a powerful principle: targeting multiple bacterial vulnerabilities simultaneously can overcome the formidable defenses erected by the biofilm lifestyle. The success of these approaches is highly dependent on the specific pathogen, resistance mechanisms, and infection microenvironment, as evidenced by the critical influence of specialized media (SWF, SCFM) on synergy outcomes. Future research will be guided by advanced analytical tools like BiofilmQ for precise phenotypic characterization and the integration of enzymatic matrix degradation, paving the way for highly effective, tailored combination therapies to combat some of the most challenging infections faced in clinical practice.
Within the broader context of microbial biofilm formation research, the development of robust validation models is paramount for translating basic science into effective clinical interventions. Biofilms, which are structured communities of microorganisms embedded in an extracellular polymeric substance (EPS), are implicated in approximately 65–80% of all microbial infections and demonstrate extreme tolerance to antimicrobial agents and host immune defenses [122] [123]. This tolerance, often quantified by tolerance factors (TF) that can exceed 1,000-fold compared to planktonic cells, presents a formidable challenge in both preclinical development and clinical practice [124]. The biofilm lifecycle—comprising attachment, maturation, and dispersal phases—creates distinct therapeutic targets and challenges that must be modeled accurately to assess anti-biofilm efficacy meaningfully [125] [117]. This guide provides an in-depth examination of the current models and methods for validating anti-biofilm strategies, integrating traditional microbiology with advanced computational and imaging technologies to bridge the gap between laboratory findings and clinical application.
The Biofilm Lifecycle as a Framework for Validation
Understanding the biofilm lifecycle is fundamental to developing effective validation models, as each stage presents unique characteristics and vulnerabilities. The lifecycle begins with the initial attachment of free-swimming bacteria to a surface, transitioning to irreversible attachment and subsequent production of EPS [122] [125]. During maturation, microcolonies form and develop complex three-dimensional structures, a process globally regulated by quorum sensing (QS) cell-to-cell communication [122]. The final dispersal phase sees a subpopulation of cells actively leaving the biofilm to colonize new surfaces, contributing to disease transmission within the host [122]. Validation models must therefore account for this dynamic progression, as an agent effective against early-stage attachment may prove useless against a mature biofilm, and vice versa. This temporal consideration is critical for both preclinical assessment and clinical trial design, as the optimal timing of intervention depends on the specific biofilm stage being targeted.
Preclinical Validation Models and Methodologies
Preclinical validation employs a hierarchy of models, from simple in vitro screens to complex in vivo infections, to establish the proof-of-concept for anti-biofilm candidates.
In Vitro Models and Quantitative Assessment
In vitro models provide a controlled, high-throughput platform for the initial screening of anti-biofilm agents.
Microtiter Plate-Based Assays: The crystal violet (CV) staining assay is a cornerstone method for quantifying total biofilm biomass [123]. Following biofilm growth and treatment in 96-well plates, biofilms are fixed, stained with crystal violet, solubilized, and quantified by measuring the absorbance. This method is particularly useful for assessing a compound's ability to prevent biofilm formation or disrupt pre-formed biofilms. Table 1 summarizes key quantitative methods used in biofilm assessment.
Viability and Metabolic Assays: The Colony Forming Unit (CFU) assay determines the number of viable bacteria within a biofilm. After treatment, biofilms are homogenized via scraping, vortexing, or sonication, serially diluted, and plated on agar. After incubation, colonies are counted to calculate CFU/mL [6]. This method differentiates live from dead cells but can be labor-intensive and susceptible to errors from bacterial clumping. Alternative methods like ATP bioluminescence offer faster, indirect measurements of viable cell presence based on metabolic activity [6].
Advanced Imaging and Cytometry: Confocal Scanning Laser Microscopy (CSLM) enables non-destructive, three-dimensional analysis of biofilm architecture and viability. When used in conjunction with fluorescent stains (e.g., LIVE/DEAD BacLight, which differentiates live and dead cells based on membrane integrity), CSLM can quantify parameters like biovolume, thickness, and live/dead cell ratios in situ [126] [6]. For more sophisticated, high-throughput quantification, specialized software like BiofilmQ provides an image cytometry toolbox capable of automatically quantifying hundreds of structural, textural, and fluorescence properties of 3D biofilms, dissecting them into cubical grids or analyzing imported single-cell segmentations [127].
Minimum Inhibitory/Biofilm Eradication Concentrations (MIC/MBEC): While the Minimum Inhibitory Concentration (MIC) determines the lowest concentration of an antimicrobial that prevents the growth of planktonic cells, the Minimum Biofilm Eradication Concentration (MBEC) is a more relevant metric for biofilms. The MBEC assay, often performed using specialized peg lids, measures the lowest concentration required to eradicate a established biofilm, which is typically orders of magnitude higher than the MIC [123].
Table 1: Quantitative Methods for Biofilm Assessment in Preclinical Models
| Method | Measured Parameter | Principle | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Crystal Violet Staining [123] [6] | Total Biofilm Biomass | Dye binding to cells and matrix | High-throughput, inexpensive, simple | Does not differentiate live/dead cells |
| Colony Forming Unit (CFU) [6] | Viable Bacterial Count | Culture and enumeration of live cells | Direct measure of cell viability | Time-consuming, prone to clumping errors |
| ATP Bioluminescence [6] | Metabolic Activity | Luciferase reaction with cellular ATP | Rapid, sensitive | Does not directly correlate with cell number |
| Confocal Microscopy + Viability Stains [126] [6] | 3D Architecture & Viability | Fluorescence-based membrane integrity | Spatially resolved, in-situ data | Requires expensive equipment, complex analysis |
| BiofilmQ Analysis [127] | 3D Morphometry & Cytometry | Automated image analysis of 3D stacks | High-content, hundreds of parameters | Computational resource-intensive |
Diagram 1: Integrated Workflow for Preclinical Anti-biofilm Validation. This workflow outlines the progression from sample preparation through quantitative, qualitative, and computational analysis, highlighting the integration of in vitro and in vivo models.
In Vivo and Complex Infection Models
While in vitro models are essential for screening, in vivo models are indispensable for evaluating anti-biofilm efficacy in the context of a living host. These models better replicate the complex host-pathogen interactions, immune responses, and pharmacokinetic/pharmacodynamic (PK/PD) parameters that influence treatment outcomes.
Implant-Associated Infection Models: These models involve the surgical implantation of a material (e.g., catheter segment, titanium pin) pre-colonized with biofilm-forming bacteria (e.g., Staphylococcus aureus, Pseudomonas aeruginosa) into an animal, typically a mouse or rat [10]. The efficacy of a systemic or local anti-biofilm treatment is assessed by CFU counts from explained implants or histological analysis.
Wound Infection Models: Chronic wound models (e.g., burn wound, diabetic ulcer) are used to study biofilm formation and treatment in a soft tissue context. These models have been used to evaluate the efficacy of novel treatments, such as the synthetic iminosugar PDIA, which showed promise in treating biofilm-associated skin infections caused by S. aureus and P. aeruginosa in a mouse model [10].
Localized Therapy Models: Specific anatomical sites require specialized models. For instance, the efficacy of low-dose 265 nm UVC light for managing bacterial corneal infections has been validated in preclinical models, demonstrating potent anti-biofilm activity against P. aeruginosa biofilms [126]. Similarly, intrapleural enzyme-based therapy combined with antibiotic washes has been explored for P. aeruginosa biofilms in post-surgical scenarios [10].
Computational and In Silico Models
The integration of computational models represents a paradigm shift in anti-biofilm drug discovery and validation. These in silico approaches can de-risk and prioritize candidates before costly experimental work.
Molecular Docking and Dynamics: As demonstrated in a 2025 integrated dataset, compounds can be computationally screened against key biofilm-related protein targets, such as quorum-sensing regulators (e.g., P. aeruginosa LasR) or biofilm-forming enzymes (e.g., S. aureus sortase A) [123]. Molecular docking predicts the binding pose and affinity of a small molecule to a target protein. Following docking, Molecular Dynamics (MD) simulations (e.g., 100 ns trajectories) assess the stability of the compound-target complex over time, providing insights into the mechanism of action [123].
ADMET Prediction: Early prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties is critical for repurposing existing drugs or developing new antibiofilm agents. In silico tools (e.g., QikProp) can compute these profiles, helping to identify lead compounds with acceptable pharmacological properties and accelerating translational potential [123].
Table 2: Key Computational Approaches for Anti-biofilm Validation
| Computational Method | Application in Anti-biofilm Validation | Output Metrics | Software Examples |
|---|---|---|---|
| Molecular Docking [123] | Predict binding of repurposed compounds to biofilm targets (e.g., LasR, Sortase A) | Docking Score (kcal/mol), Binding Pose, Interaction Map | Schrödinger Glide, AutoDock Vina |
| Molecular Dynamics (MD) [123] | Assess stability of compound-target complex; understand mechanism | Root Mean Square Deviation (RMSD), Hydrogen Bonds, Simulation Trajectories | Desmond, GROMACS, NAMD |
| ADMET Prediction [123] | Predict pharmacokinetic and safety profiles of lead compounds | Predicted Caco-2 permeability, LogP, HERG inhibition, %Human Oral Absorption | QikProp, admetSAR |
| Bibliometric Analysis [117] | Identify research trends, gaps, and collaborative networks in biofilm cycle research | Keyword co-occurrence, Leading organizations/countries | VOSviewer |
Clinical Validation and Translational Considerations
Transitioning from preclinical success to clinical efficacy is the most significant challenge in anti-biofilm therapy development.
Clinical Trial Design and Endpoints
Clinical trials for anti-biofilm agents must be carefully designed to demonstrate clear patient benefits. Key considerations include:
- Patient Stratification: Enrolling patients with confirmed biofilm-associated infections (e.g., chronic wounds, device-related infections) is crucial. Diagnostic methods that can reliably detect biofilms in clinical samples are an area of active development.
- Endpoint Selection: Beyond standard microbiological endpoints (e.g., pathogen eradication), clinical endpoints such as time to resolution, need for device removal or surgical debridement, and rate of infection recurrence are critically important and more reflective of anti-biofilm efficacy [122] [10].
- Combination Therapy Trials: Given that many anti-biofilm agents function as anti-virulence therapies or dispersal agents, they may not be directly bactericidal. Therefore, clinical trials often evaluate them as adjuvants to conventional antibiotics [10]. For example, the anti-virulence potential of ibuprofen to enhance antibiotic efficacy against S. aureus is a relevant clinical hypothesis [10].
Innovative Clinical Strategies
Recent research highlights several innovative approaches entering clinical evaluation:
- Localized Drug Delivery: Systems like antibiotic-impregnated blood clots or enzyme-based irrigation therapies aim to enhance drug penetration and efficacy at the biofilm site, as explored in preclinical and early clinical scenarios [10].
- Non-Antibiotic Modalities: The clinical exploration of phage therapy, quorum-sensing inhibitors, and antimicrobial peptides (e.g., CRAMP-34) is growing. These strategies aim to disperse or sensitize biofilms without directly killing bacteria, potentially reducing selective pressure for resistance [10].
- Physical and Device-Based Strategies: Technologies like low-dose UVC light are being validated for managing localized biofilm infections, such as in the eye, offering an antibiotic-free alternative [126].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful anti-biofilm research relies on a suite of specialized reagents, assays, and tools. The following table details key components of the experimental toolkit.
Table 3: Essential Research Reagents and Materials for Anti-biofilm Validation
| Reagent / Material | Function / Application | Specific Examples / Notes |
|---|---|---|
| 96-well Microtiter Plates [123] | High-throughput biofilm cultivation and chemical treatment | Polystyrene plates are standard for crystal violet assays |
| Crystal Violet Stain [123] [6] | Quantitative staining of total biofilm biomass | Requires solubilization (e.g., with acetic acid or ethanol) for absorbance reading |
| LIVE/DEAD BacLight Bacterial Viability Kit [126] [6] | Fluorescent differentiation of live (green) vs. dead (red) cells in biofilms | Used for confocal microscopy or fluorescence plate readers |
| MBEC Assay Plates (Peg Lids) | Standardized testing of minimum biofilm eradication concentrations | Allows simultaneous testing of multiple antibiotics/concentrations |
| Specific Protein Targets [123] | Computational and biochemical studies of anti-virulence mechanisms | P. aeruginosa LasR (PDB: 3IX3), S. aureus sortase A |
| BiofilmQ Software [127] | Comprehensive 3D image cytometry and analysis of biofilm properties | Quantifies 49+ structural, textural, and fluorescence properties per object |
| Homogenization Equipment [6] | Dispersing biofilm into single-cell suspensions for CFU counting | Vortex mixers, sonication water baths, commercial homogenizers |
The landscape of anti-biofilm validation is evolving from reliance on simple in vitro screens toward an integrated, multi-scale approach. Robust validation requires correlating data across quantitative biomass assays, 3D spatial viability analyses, predictive computational modeling, and physiologically relevant in vivo models. The ultimate goal is to build a compelling preclinical package that accurately predicts clinical success. Future directions will be shaped by the integration of big data and machine learning to predict biofilm behavior, the standardization of methods across laboratories, and the continued development of clinical trial endpoints that are uniquely sensitive to the eradication of biofilm-associated infections [117]. By systematically applying this hierarchical validation framework, researchers can more effectively advance novel anti-biofilm strategies from the bench to the bedside, addressing a critical unmet need in modern medicine.
Conclusion
The formation of microbial biofilms is a sophisticated, multi-stage process that progresses from initial reversible attachment to the development of complex, resistant communities, culminating in active dispersion. This intricate lifecycle, governed by specific molecular signals like c-di-GMP and quorum sensing, presents a formidable challenge in clinical and industrial settings due to the enhanced antimicrobial resistance it confers. Future research must prioritize the development of novel diagnostic tools for early biofilm detection and innovative therapeutic strategies that specifically target biofilm integrity, regulatory pathways, and dormant cell populations. Interdisciplinary approaches combining advanced imaging, computational modeling, and machine learning with traditional microbiology hold the greatest promise for disrupting these resilient structures. Successfully translating this knowledge into clinical applications is paramount for addressing the global crisis of chronic, biofilm-associated infections and antimicrobial resistance, ultimately improving patient outcomes and public health.
References
- 1. Unlocking Biofilm Secrets [https://www.numberanalytics.com/blog/ultimate-guide-biofilms-microbiology]
- 2. Biofilm Resilience: Molecular Mechanisms Driving Antibiotic Resistance in Clinical Contexts - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11852148/]
- 3. : Biofilms , Formation Models, Potential Targets, and... Research [https://pmc.ncbi.nlm.nih.gov/articles/PMC9561771/]
- 4. - Wikipedia Biofilm [https://en.wikipedia.org/wiki/Biofilm]
- 5. | PPTX Biofilms [https://www.slideshare.net/slideshow/biofilm-ex-34791777/34791777]
- 6. Quantitative and Qualitative Assessment Methods for Biofilm Growth: A Mini-review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6133255/]
- 7. Frontiers | Editorial: Understanding biofilms : recent trends and... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1566230/full]
- 8. Mechanisms of Biofilm Development and Antibiofilm Strategies [https://www.frontiersin.org/research-topics/30984/mechanisms-of-biofilm-development-and-antibiofilm-strategies/magazine]
- 9. Extraction and quantification of biofilm bacteria: Method optimized for urinary catheters | Scientific Reports [https://www.nature.com/articles/s41598-018-26342-3]
- 10. Frontiers | Editorial: Fighting microbial biofilms: novel therapeutics and antibiofilm strategies [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1680391/full]
- 11. : formation, architecture, antibiotic resistance, and... Microbial biofilm [https://pmc.ncbi.nlm.nih.gov/articles/PMC8578483/]
- 12. enzymes as powerful natural anti- Microbial candidates biofilm [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02610-y]
- 13. Quantifying biofilm propagation on chemically modified surfaces - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9594113/]
- 14. Latent Space-Driven Quantification of Biofilm using... Formation [https://arxiv.org/html/2507.07632v1]
- 15. Beyond Risk: Bacterial Biofilms and Their Regulating Approaches... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7253578/]
- 16. Bacteriophage-mediated approaches for biofilm control - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11491440/]
- 17. image analysis of microbial... | Nature Microbiology Quantitative [https://www.nature.com/articles/s41564-020-00817-4?error=cookies_not_supported]
- 18. Analysis of Quantitative | Frontiers Biofilms Topic Research [https://www.frontiersin.org/research-topics/6536/quantitative-analysis-of-biofilms/overview]
- 19. Prokaryotic Biofilms | Biology for Majors II [https://courses.lumenlearning.com/wm-biology2/chapter/prokaryotic-biofilms/]
- 20. : A Review on Microbial , Infection, Antibiotic... Biofilm Formation [https://pmc.ncbi.nlm.nih.gov/articles/PMC10305407/]
- 21. and their role in pathogenesis | British Society for Immunology Biofilms [https://www.immunology.org/public-information/bitesized-immunology/pathogens-disease/biofilms-and-their-role-pathogenesis]
- 22. : Microbial , architecture, antibiotic resistance, and... biofilm formation [https://pubmed.ncbi.nlm.nih.gov/34558029/]
- 23. Microcolony and biofilm formation as a survival strategy for bacteria - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0022519307005188]
- 24. Development of Microbial and Their Role in device... Biofilms [https://journals.umt.edu.pk/index.php/BSR/article/view/6632]
- 25. : Biofilms , resistance mechanism, emerging control... structure [https://pubs.rsc.org/en/content/articlehtml/2025/pm/d5pm00094g]
- 26. Frontiers | Ultrasonic strategies for mitigating microbial adhesion and... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1558035/full]
- 27. Microcolony formation by the opportunistic pathogen Pseudomonas aeruginosa requires pyruvate and pyruvate fermentation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3527662/]
- 28. Microcolony and biofilm formation as a survival strategy for bacteria - PubMed [https://pubmed.ncbi.nlm.nih.gov/18083198/]
- 29. : mechanistic insights and therapeutic targets Biofilm formation [https://link.springer.com/article/10.1186/s43556-023-00164-w]
- 30. Targeting microbial biofilms: current and prospective therapeutic strategies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5685531/]
- 31. Biofilm viability checker: An open-source tool for automated biofilm viability analysis from confocal microscopy images | npj Biofilms and Microbiomes [https://www.nature.com/articles/s41522-021-00214-7]
- 32. Image analysis of microbial communities with BiofilmQ [https://communities.springernature.com/posts/image-analysis-of-microbial-communities-with-biofilmq]
- 33. Applying insights from biofilm biology to drug development - can a new approach be developed? - PubMed [https://pubmed.ncbi.nlm.nih.gov/24080700/]
- 34. Biofilm dispersion - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8564779/]
- 35. | Nature Reviews Microbiology Biofilm dispersion [https://www.nature.com/articles/s41579-020-0385-0?error=cookies_not_supported]
- 36. Biofilm Dispersal: Mechanisms, Clinical Implications, and Potential Therapeutic Uses - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3318030/]
- 37. Frontiers | Dispersive from membrane bioreactor strains... biofilm [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2023.1211761/full]
- 38. Controlling biofilm development through cyclic di-GMP signaling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9891824/]
- 39. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling: Lessons from Pseudomonas aeruginosa and Other Bacteria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4933438/]
- 40. The role of RNA regulators, quorum sensing and c-di-GMP in bacterial biofilm formation - PubMed [https://pubmed.ncbi.nlm.nih.gov/35234364/]
- 41. Distinct bacterial population dynamics and disease dissemination after... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10356768/]
- 42. Cyclic di - GMP Modulation of Quorum and Its... | Preprints.org Sensing [https://www.preprints.org/manuscript/202507.2632/v1]
- 43. The cyclic di - GMP receptor YcgR links the second messenger with the... [https://pubmed.ncbi.nlm.nih.gov/40444987/]
- 44. Synergy between c-di-GMP and Quorum-Sensing Signaling in Vibrio cholerae Biofilm Morphogenesis - PubMed [https://pubmed.ncbi.nlm.nih.gov/36154360/]
- 45. Cyclic di - GMP as an antitoxin regulates bacterial genome stability and... [https://elifesciences.org/articles/99194v1]
- 46. Real-Time Monitoring of Biofilm Using a Noninvasive... Formation [https://pmc.ncbi.nlm.nih.gov/articles/PMC9853957/]
- 47. analysis of Candida Comparative quantitation assays - PMC biofilm [https://pmc.ncbi.nlm.nih.gov/articles/PMC3251704/]
- 48. Extracellular polymeric substance - Wikipedia [https://en.wikipedia.org/wiki/Extracellular_polymeric_substance]
- 49. Extracellular polymeric substances, a key element in understanding... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6604936/]
- 50. Detection techniques for extracellular polymeric... :: BioResources [https://bioresources.cnr.ncsu.edu/resources/detection-techniques-for-extracellular-polymeric-substances-in-biofilms-a-review/]
- 51. | Profiling Gene & how-tos expression analysis methods [https://www.illumina.com/techniques/multiomics/transcriptomics/gene-expression-analysis.html]
- 52. Techniques Gene Expression [https://www.news-medical.net/life-sciences/Gene-Expression-Techniques.aspx]
- 53. What We Still Don’t Know About Biofilms—Current Overview and Key Research Information [https://www.mdpi.com/2036-7481/16/2/46]
- 54. Frontiers | Editorial: Controlling biofilm -related infections in healthcare... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1679631/full]
- 55. of Bacterial In : Innovative Tools to Improve... Vitro Models Biofilms [https://pmc.ncbi.nlm.nih.gov/articles/PMC10004855/]
- 56. JoVE | Peer Reviewed Scientific Video Journal - Methods and Protocols [https://app.jove.com/methods-collections/370/models-for-analyzing-biofilms-in-infection?]
- 57. Frontiers | Better models, better treatment? a systematic review of current three dimensional (3D) in vitro models for implant-associated infections [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1569211/full]
- 58. Quantitative image analysis of microbial communities with BiofilmQ | Nature Microbiology [https://www.nature.com/articles/s41564-020-00817-4]
- 59. of In | SpringerLink Vivo Models Biofilm Infection [https://link.springer.com/chapter/10.1007/978-1-4419-6084-9_16]
- 60. Computational and Systems Biology Approaches in Biofilm Research | SpringerLink [https://link.springer.com/chapter/10.1007/978-3-031-91863-6_16]
- 61. Frontiers | An Overview of Biological and Computational Methods ... [https://www.frontiersin.org/articles/10.3389/fmicb.2021.640787/full]
- 62. -aided Computer of Discovery that Specifically Attack... Peptides [https://www.nature.com/articles/s41598-018-19669-4?error=cookies_not_supported&code=cca8e615-a723-43c2-9033-3a590c17f28b]
- 63. Frontiers | Identification of Distinct Characteristics of Antibiofilm ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.783284/full]
- 64. Machine learning for antimicrobial peptide identification and design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11756916/]
- 65. Identification of potent antimicrobial peptides via a machine-learning pipeline that mines the entire space of peptide sequences - PubMed [https://pubmed.ncbi.nlm.nih.gov/36635418/]
- 66. Complex Networks Analyses of Antibiofilm : An Emerging... Peptides [https://pmc.ncbi.nlm.nih.gov/articles/PMC10135022/]
- 67. Integration of Machine Learning and Structural Analysis for Predicting... [https://pubmed.ncbi.nlm.nih.gov/38450220/]
- 68. Review of mathematical models for biofilms - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0038109810000281]
- 69. for antimicrobial Machine identification and design learning peptide [https://www.nature.com/articles/s44222-024-00152-x?error=cookies_not_supported&code=79461692-907c-4300-83b1-63868e763e93]
- 70. Importance of Target & Validation in Identification Development Drug [https://lifesciences.danaher.com/us/en/library/target-identification-validation-drug-discovery.html]
- 71. Frontiers | Identification of novel drug and small molecule... targets [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1562596/full]
- 72. Aligning antimicrobial drug discovery with complex and redundant host-pathogen interactions - PubMed [https://pubmed.ncbi.nlm.nih.gov/19218083/]
- 73. A Guideline for Assessment and Characterization of Bacterial Biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10632153/]
- 74. Standardizing biofilm quantification: harmonizing crystal violet absorbance measurements through extinction coefficient ratio adjustment - PubMed [https://pubmed.ncbi.nlm.nih.gov/39951131/]
- 75. Machine-Learning Techniques Applied to Antibacterial Drug Discovery - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4273861/]
- 76. Methods for screening and evaluation of antimicrobial activity: A review of protocols, advantages, and limitations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11097785/]
- 77. Therapeutic Strategies against Biofilm Infections - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9866932/]
- 78. Frontiers | Multiple biological characteristics and functions of intestinal... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1445630/full]
- 79. (PDF) The EPS as an adaptive bastion for matrix ... biofilms [https://www.academia.edu/54286654/The_EPS_matrix_as_an_adaptive_bastion_for_biofilms_introduction_to_special_issue]
- 80. Evaluation of extracellular polymeric substances matrix volume... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10547949/]
- 81. The biofilm life cycle– expanding the conceptual model of biofilm formation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9841534/]
- 82. 细菌生物膜的特征及抗细菌生物膜策略 [http://html.rhhz.net/YXXB/html/20181211.htm]
- 83. Gradients and consequences of heterogeneity in biofilms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9590228/]
- 84. 慢 性 创面细菌 生 机制及其诊断的 物 - PMC 膜 形 成 研 究 进 展 [https://pmc.ncbi.nlm.nih.gov/articles/PMC11917344/]
- 85. Physiological heterogeneity in biofilms - PubMed [https://pubmed.ncbi.nlm.nih.gov/18264116/]
- 86. 方法表征的共同发展 生 和 物 境 膜 生 异 质 性 [https://www.jove.com/cn/t/52602/methods-for-characterizing-co-development-biofilm-habitat]
- 87. 长分枝杆菌 生 作为 生 抗菌素 物 的模型 膜 研 究 耐 药 性 [https://www.jove.com/cn/t/66607/growing-mycobacterial-biofilm-as-model-to-study-antimicrobial]
- 88. Biofilm Resilience: Molecular Mechanisms Driving Antibiotic Resistance in Clinical Contexts [https://www.mdpi.com/2079-7737/14/2/165]
- 89. Targeting Bacterial Biofilms on Medical Implants: Current and Emerging Approaches [https://www.mdpi.com/2079-6382/14/8/802]
- 90. Frontiers | Breaking the barrier : disruption of bacterial biofilms using... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1670237/full]
- 91. Pharmacological strategies for targeting biofilms in ... [https://www.spandidos-publications.com/10.3892/br.2025.1973]
- 92. New ultrasound drug delivery system found to be highly effective against bacterial biofilms | University of Oxford [https://www.ox.ac.uk/news/2025-04-22-new-ultrasound-drug-delivery-system-found-be-highly-effective-against-bacterial]
- 93. Hostile Environments: Modifying Surfaces to Block Microbial Adhesion and Biofilm Formation [https://www.mdpi.com/2218-273X/15/6/754]
- 94. Targeting microbial : current... | Nature Reviews Microbiology biofilms [https://www.nature.com/articles/nrmicro.2017.99?error=cookies_not_supported&code=fdb2cd7b-7015-4e17-b264-052bcd5bc54e]
- 95. of Antibiofilm Therapeutics Strategies to Overcome... Development [https://pubmed.ncbi.nlm.nih.gov/35208758/]
- 96. Pseudomonas aeruginosa Biofilms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7698413/]
- 97. Frontiers | Treatment of Pseudomonas aeruginosa infectious biofilms: Challenges and strategies [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.955286/full]
- 98. Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents—How P. aeruginosa Can Escape Antibiotics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6509751/]
- 99. Statistical Analysis of Pseudomonas aeruginosa Biofilm Development: Impact of Mutations in Genes Involved in Twitching Motility, Cell-to-Cell Signaling, and Stationary-Phase Sigma Factor Expression - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC123874/]
- 100. Recent perspectives on the molecular basis of biofilm by... formation [https://link.springer.com/article/10.1007/s12223-018-0585-4]
- 101. Evaluation of antimicrobial resistance, biofilm forming potential, and... [https://bmcresnotes.biomedcentral.com/articles/10.1186/s13104-020-4890-z]
- 102. Frontiers | A comprehensive review of the pathogenic mechanisms of... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1619626/full]
- 103. The biofilm life cycle: expanding the conceptual model of biofilm formation | Nature Reviews Microbiology [https://www.nature.com/articles/s41579-022-00767-0]
- 104. Frontiers | Marine microbial biofilms on diverse abiotic surfaces [https://www.frontiersin.org/journals/marine-science/articles/10.3389/fmars.2025.1482946/full]
- 105. formation and antibiotic Biofilm in dispersed susceptibility ... cells [https://pubmed.ncbi.nlm.nih.gov/28770446/]
- 106. study of growth temperature impact on the... Comparative [https://annalsmicrobiology.biomedcentral.com/articles/10.1007/s13213-018-1419-y]
- 107. assessment of antibiotic Comparative of... susceptibility [https://pubmed.ncbi.nlm.nih.gov/15980094/]
- 108. : Formation, drug resistance and alternatives to Biofilms ... conventional [https://pmc.ncbi.nlm.nih.gov/articles/PMC9576500/]
- 109. Biofilm battleground: Unveiling the hidden challenges, current approaches and future perspectives in combating biofilm associated bacterial infections - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0882401024006223]
- 110. The Limitations of In Vitro Experimentation in Understanding Biofilms and Chronic Infection - PubMed [https://pubmed.ncbi.nlm.nih.gov/26344834/]
- 111. Towards improved biofilm models | Nature Reviews Microbiology [https://www.nature.com/articles/s41579-024-01086-2]
- 112. Innovative approaches to combat antibiotic resistance: integrating... [https://bmcmedicine.biomedcentral.com/articles/10.1186/s12916-025-04323-4]
- 113. Frontiers | Bacteriophage -mediated approaches for biofilm control [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2024.1428637/full]
- 114. Current and future directions in bacteriophage research for developing therapeutic innovations | Scientific Reports [https://www.nature.com/articles/s41598-024-76427-5]
- 115. Bacteriophages as nanocarriers for targeted drug delivery and enhanced therapeutic effects - Materials Advances (RSC Publishing) DOI:10.1039/D3MA00817G [https://pubs.rsc.org/en/content/articlehtml/2024/ma/d3ma00817g]
- 116. Application of Bacteriophages in Nanotechnology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7601235/]
- 117. Recent Advances in Biofilm Cycle by Bibliography... | Preprints.org [https://www.preprints.org/manuscript/202501.0368/v1]
- 118. Strategies to Enhance the Combination of Antimicrobial... Efficacy [https://pubmed.ncbi.nlm.nih.gov/29375486/]
- 119. SNAPs with antibiotics shows enhanced Combining ... synergistic [https://www.nature.com/articles/s41522-023-00401-8?error=cookies_not_supported&code=a922fee4-6481-42f3-b52b-bdbd993929a6]
- 120. Frontiers | Study on the invitro synergistic susceptibility and biofilm ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1542029/full]
- 121. Optimization of Quantitative Analysis of Biofilm Cell from Pipe Materials [https://www.mdpi.com/2079-6412/11/11/1286]
- 122. and their role on diseases | BMC Microbiology | Full Text Biofilms [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-023-02954-2]
- 123. Integrative experimental and computational dataset for repurposed antibiofilm drug candidates targeting quorum sensing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12446609/]
- 124. Antimicrobial Tolerance in Biofilms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4507308/]
- 125. Infographic: Stages of Biofilm | The Scientist Formation [https://www.the-scientist.com/infographic-stages-of-biofilm-formation-71140]
- 126. In vitro anti - biofilm of therapeutic low dose 265 nm UVC efficacy [https://pubmed.ncbi.nlm.nih.gov/39787975/]
- 127. Quantitative image analysis of microbial communities with BiofilmQ - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7840502/]