One-Pot RPA-CRISPR Diagnostics: A Revolutionary Approach for Rapid Biofilm Pathogen Detection
This article comprehensively reviews the emerging field of one-pot RPA-CRISPR/Cas diagnostics and its transformative application in detecting biofilm-forming pathogens.
One-Pot RPA-CRISPR Diagnostics: A Revolutionary Approach for Rapid Biofilm Pathogen Detection
Abstract
This article comprehensively reviews the emerging field of one-pot RPA-CRISPR/Cas diagnostics and its transformative application in detecting biofilm-forming pathogens. We explore the foundational principles of integrating recombinase polymerase amplification (RPA) with CRISPR/Cas systems like Cas12a into single-reaction assays, detailing the methodological advances that enable rapid, equipment-free detection. The content systematically addresses key optimization strategies to overcome sensitivity and specificity challenges, particularly for complex biofilm samples. By critically evaluating validation data and comparing performance against gold-standard culture methods and PCR, we demonstrate the significant potential of these platforms for point-of-care testing. This resource provides researchers, scientists, and drug development professionals with a state-of-the-art overview of a technology poised to revolutionize clinical microbiology and antimicrobial stewardship.
The Science Behind One-Pot RPA-CRISPR: Redefining Molecular Diagnostics for Biofilm Pathogens
The Urgent Need for Rapid Biofilm Pathogen Detection in Clinical and Industrial Settings
Microbial biofilms are complex, structured communities of bacteria encased in a self-produced extracellular polymeric substance (EPS) matrix that adhere to biological or inert surfaces [1]. These structures pose significant challenges in both clinical and industrial settings due to their inherent resistance to antibiotics and disinfectants, leading to persistent infections and biofouling [1] [2]. An estimated 65% of all human infections are associated with biofilms, affecting diverse areas including oral health (periodontal disease), respiratory systems (cystic fibrosis), chronic wounds, and urinary tracts [1]. The economic repercussions are substantial, with biofilms costing various industries an estimated exceeding $5 trillion USD annually due to prolonged medical treatments, equipment damage, and operational interruptions [2].
Traditional methods for detecting biofilm-forming pathogens, such as microbial culture and molecular techniques like quantitative PCR (qPCR), present significant limitations for rapid diagnostics. While culture remains the gold standard for viability determination, it is time-consuming, requiring 2-10 days for results, and demands specialized technical skills [3]. Molecular methods like qPCR, though faster, necessitate sophisticated equipment, trained personnel, and centralized laboratory facilities, making them unsuitable for point-of-care testing (POCT) during outbreak situations [3]. This diagnostic gap underscores the critical need for rapid, sensitive, and equipment-free detection methods that can be deployed at the point of need to enable timely intervention and biofilm control strategies.
Emerging Solution: One-Pot RPA-CRISPR Diagnostics
The integration of isothermal amplification techniques with CRISPR-Cas systems represents a paradigm shift in molecular diagnostics for biofilm-forming pathogens. Among these, one-pot assays combining Recombinase Polymerase Amplification (RPA) and CRISPR/Cas systems have emerged as particularly promising tools that meet the WHO's ASSURED criteria (Affordable, Sensitive, Specific, User-friendly, Rapid and robust, Equipment-free, and Deliverable to end-users) [4].
Recombinase Polymerase Amplification (RPA) is an isothermal amplification technique that operates at low temperatures (37-42°C) using recombinase enzymes, DNA polymerase, and single-strand binding proteins (SSBs) to amplify target DNA rapidly (within 30 minutes) without thermal cycling equipment [4] [3]. The CRISPR/Cas system, derived from bacterial adaptive immunity, utilizes Cas enzymes (such as Cas12, Cas13, and Cas14) that become activated upon recognition of a specific target sequence (guided by CRISPR RNA - crRNA) and exhibit promiscuous "trans-cleavage" activity, indiscriminately degrading nearby reporter molecules [3] [5]. This collateral cleavage generates a detectable signal, typically through fluorescence or lateral flow readouts.
The one-pot methodology consolidates nucleic acid amplification and CRISPR-based detection into a single tube, significantly simplifying the workflow, reducing contamination risks, and eliminating the need for specialized instrumentation [6]. Recent advances have further enhanced this system's practicality through extraction-free sample processing and two-temperature protocols that optimize both amplification and detection efficiency [6].
Table 1: Comparison of Biofilm Pathogen Detection Methods
| Method | Time Required | Limit of Detection | Equipment Needs | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Microbial Culture | 2-10 days [3] | Varies by pathogen | Incubators, biosafety facilities | Determines viability, gold standard [3] | Time-consuming, requires technical skill [3] |
| qPCR | 2-4 hours | High (copy number dependent) | Thermal cycler, trained personnel | High sensitivity and specificity [3] | Expensive equipment, central lab requirement [3] |
| One-Pot RPA-CRISPR | <1 hour [6] | 10 copies/test [6] | Minimal (water bath/heat block) | Rapid, equipment-free, suitable for POCT [6] | New technology, limited commercial availability |
Application Notes: One-Pot RPA-CRISPR/Cas12b for GBS Detection
A recently developed extraction-free, one-pot two-temperature CRISPR/Cas12b assay demonstrates the practical application of this technology for detecting Group B Streptococcus (GBS), a relevant biofilm-forming pathogen [6]. This integrated system combines RPA with CRISPR/Cas12b detection in a single reaction tube, achieving clinically relevant sensitivity and specificity without requiring nucleic acid extraction.
The optimized workflow begins with a simple sample pretreatment where DNA is released directly from swab samples through incubation in buffer or brief heat treatment (95°C for 5 minutes) [6]. The lysate is then added to a pre-mixed one-pot reaction containing all necessary RPA and CRISPR/Cas12b reagents. Amplification proceeds at 39°C for 40 minutes using RPA, after which the temperature is raised to 62°C for 5 minutes to activate AapCas12b-mediated trans-cleavage, generating a fluorescence signal detectable by the naked eye under blue or UV light [6]. The total assay time is under one hour, making it particularly suitable for resource-limited settings or scenarios requiring rapid diagnostics.
Validation using 60 vaginal-rectal swab samples demonstrated exceptional performance, with 96.7% concordance compared to culture methods and 98.3% concordance compared to qPCR methods [6]. The assay achieved a remarkable sensitivity of 10 copies/test (1 copy/μL), enabling detection even in low-copy samples [6].
Detailed Experimental Protocol
Primer and crRNA Design
- Target Selection: Identify a conserved region within the target pathogen's genome. For GBS, the cfb4 gene (GenBank: HQ148672.1) serves as an effective target [6].
- RPA Primer Design: Design primers using appropriate software (e.g., SnapGene). Optimal amplicon size typically ranges from 100-300 bp [6].
- crRNA Design: Design multiple single-guide RNAs (sgRNAs) targeting distinct regions of the same gene. Synthesize oligonucleotides and prepare high-quality sgRNA using commercial synthesis and purification kits (e.g., Cas12b High Yield sgRNA Synthesis and Purification Kit) [6].
One-Pot Reaction Setup
Prepare a 25 μL reaction mixture containing [6]:
- 2.5 μL 10× LAMP buffer (or appropriate reaction buffer)
- 1.0 μL dNTP mix (10 mM each)
- 2.5 μL 10× AapCas12b reaction buffer
- 1.0 μL AapCas12b nuclease (10 μM)
- 1.0 μL crRNA (10 μM)
- 1.0 μL ssDNA FQ reporter (10 μM)
- RPA primers (forward and reverse, 10 μM each)
- 2.5 μL MgCl₂ (20 mM)
- Nuclease-free water to volume
- 2-5 μL template DNA (extraction-free lysate)
Amplification and Detection
- RPA Amplification: Incubate the reaction mixture at 39°C for 40 minutes to allow isothermal amplification of the target DNA [6].
- CRISPR Detection: Increase the temperature to 62°C for 5 minutes to activate Cas12b trans-cleavage activity [6].
- Result Interpretation: Visualize under UV light (blue light ~485 nm). Positive samples emit green fluorescence, while negative samples show no signal [6].
Table 2: Key Research Reagent Solutions for One-Pot RPA-CRISPR
| Reagent/Component | Function | Specifications/Alternatives |
|---|---|---|
| AapCas12b nuclease | Target recognition and trans-cleavage | Thermostable Cas12b variant; Alternatives: LbCas12a, AsCas12a [6] |
| crRNA | Guides Cas protein to target sequence | Designed against conserved pathogen gene; requires synthesis [6] |
| ssDNA FQ Reporter | Signal generation | Fluorescently quenched ssDNA; FAM-TTATTATT-BHQ1 [6] |
| RPA Primers | Isothermal amplification | Target-specific; 30-35 nt length [6] |
| RPA Reaction Pellet | Isothermal amplification | Commercial RPA kits (TwistAmp) [6] |
| Maneval's Stain | Biofilm visualization | Differentiates cells (magenta-red) from matrix (blue) [7] |
Technical Considerations and Optimization Strategies
Implementing a Two-Temperature Protocol
The one-pot two-temperature approach represents a critical advancement for enhancing RPA-CRISPR/Cas12b detection sensitivity, particularly for low-copy targets [6]. While RPA operates optimally at lower temperatures (37-42°C), the thermostable AapCas12b enzyme functions most efficiently at higher temperatures (~62°C) [6]. Initial studies revealed that single-temperature protocols compromised either amplification efficiency or Cas12b activation. By systematically implementing a sequential temperature incubation (39°C for RPA amplification followed by 62°C for Cas12b activation), researchers significantly improved the signal-to-noise ratio and detection rate, achieving sensitivity as low as 10 copies/test even with challenging samples [6].
Signal Readout and Interpretation
The flexibility of one-pot RPA-CRISPR systems enables multiple readout modalities suitable for different settings:
- Fluorescence Visualization: Using FAM/BHQ1-quenched reporters, results can be visualized with a standard UV flashlight or blue light transilluminator, requiring no instrumentation [6].
- Lateral Flow Assays (LFA): For even simpler interpretation, reporters labeled with FAM/biotin can be detected on commercial lateral flow strips, providing a simple "line" readout familiar from pregnancy tests [3].
- Equipment-Based Quantification: For laboratory settings, fluorescence can be quantified using plate readers or microplate scanners for objective, quantitative results.
Adaptation for Biofilm Pathogen Detection
To specifically target biofilm-forming pathogens, the following protocol adaptations are recommended:
Sample Processing from Biofilms:
- Gently rinse the biofilm surface with sterile buffer to remove non-adherent cells.
- Resuspend biofilm cells in lysis buffer (e.g., TE buffer with lysozyme) [6].
- Incubate at 37°C for 15 minutes followed by heat inactivation at 95°C for 5 minutes [6].
- Use the supernatant directly as template in the one-pot reaction.
Biofilm Visualization and Validation:
- Implement the dual-staining method using Maneval's stain for parallel biofilm confirmation [7].
- Fix biofilm samples with 4% formaldehyde for 15-30 minutes at room temperature.
- Stain with 1% Congo red followed by Maneval's stain for 10 minutes [7].
- Visualize under light microscopy (100× oil immersion): bacterial cells appear magenta-red surrounded by a blue polysaccharide matrix [7].
The one-pot RPA-CRISPR diagnostic platform represents a transformative approach for rapid detection of biofilm-forming pathogens in both clinical and industrial settings. By combining the sensitivity of isothermal amplification with the exceptional specificity of CRISPR-based detection in a simplified, extraction-free format, this technology successfully addresses critical limitations of conventional diagnostic methods. The implementation of two-temperature protocols further enhances detection sensitivity while maintaining the equipment-free nature essential for point-of-care applications.
Future developments in CRISPR-based diagnostics for biofilm pathogens will likely focus on several key areas:
- Multiplexing Capabilities: Engineering systems to simultaneously detect multiple biofilm-forming pathogens or resistance markers using orthogonal CRISPR enzymes with distinct reporter specificities [4].
- Quantitative Detection: Incorporating digital droplet CRISPR or electrochemical sensors to enable quantification of pathogen load, which correlates with biofilm maturation stages [3].
- Direct Sample Integration: Developing advanced sample processing modules that can handle complex matrices like sputum, wound exudate, or industrial process fluids without purification [4].
- Lyophilized Reagent Formulations: Creating stable, room-temperature storage formats to enhance field-deployability and shelf-life [4].
As these advancements mature, one-pot RPA-CRISPR diagnostics are poised to become indispensable tools for combating biofilm-related challenges across healthcare, industrial, and environmental sectors, enabling rapid intervention and significantly improving patient outcomes and operational efficiency.
Recombinase Polymerase Amplification (RPA) is a single-tube, isothermal nucleic acid amplification technique that serves as a powerful alternative to the polymerase chain reaction (PCR) [8]. Developed in 2006, RPA utilizes a combination of enzymatic processes to exponentially amplify specific DNA target sequences at a constant low temperature of 37-42°C, without the need for thermal cycling [9] [10]. This core characteristic makes RPA particularly valuable for point-of-care testing (POCT), field applications, and resource-limited settings where access to sophisticated laboratory equipment is constrained. The technology has gained significant attention in molecular diagnostics for its rapid amplification capabilities, typically completing detectable amplification within 10-30 minutes [11] [10].
In the context of biofilm pathogen detection research, RPA presents unique advantages for developing one-pot RPA-CRISPR diagnostic systems. Its compatibility with low-temperature isothermal conditions aligns perfectly with the operational requirements of CRISPR-Cas systems, particularly Cas12a, which functions optimally at similar temperatures [11]. This synergy enables the development of integrated diagnostic platforms that can rapidly detect pathogenic microorganisms embedded in biofilms—structured microbial communities that pose persistent challenges in food safety, healthcare, and industrial environments [12].
Core Principles and Molecular Mechanism of RPA
Enzymatic Components and Reaction Mechanism
The RPA process employs three core enzymes that work synergistically to enable isothermal amplification: a recombinase, a single-stranded DNA-binding protein (SSB), and a strand-displacing DNA polymerase [9] [8]. The reaction begins when the recombinase protein (typically T4 bacteriophage UvsX) binds to oligonucleotide primers in the presence of ATP, forming recombinase-primer complexes [9] [10]. These complexes then interrogate double-stranded DNA, seeking homologous sequences, and facilitate strand invasion by the primers at cognate sites [10].
To prevent the ejection of inserted primers through branch migration, single-stranded binding proteins (SSB) immediately stabilize the displaced DNA strands [8]. Following recombinase disassembly, a strand-displacing DNA polymerase (such as the large fragment of Bacillus subtilis Pol I, Bsu) binds to the 3' end of the primer and initiates DNA synthesis in the presence of dNTPs [9]. The cyclic repetition of this process results in exponential amplification of the target DNA sequence [9]. When targeting RNA, a reverse transcriptase enzyme can be incorporated into the RPA reaction mixture, enabling direct detection of RNA without a separate cDNA synthesis step [9] [8].
RPA Reaction Workflow and Optimization
The typical RPA reaction workflow involves minimal sample preparation and can be completed within 20-40 minutes [9]. Optimal performance is achieved at temperatures between 37-42°C, though the reaction can proceed at temperatures ranging from 22-45°C [9]. This flexibility in temperature requirements allows RPA to be performed using simple heating sources including incubators, heating blocks, chemical heaters, or even body heat [9]. The reaction time depends on the initial copy number of the target nucleic acid, with detectable amplification possible in as little as 3-4 minutes for high-copy targets [9].
Several factors require careful optimization for robust RPA performance. Primer design typically employs 30-35 base oligonucleotides, though standard PCR primers can sometimes be effective [9]. The inclusion of a molecular crowding agent (typically polyethylene glycol) in the reaction mixture is essential for preventing spontaneous disassembly of the recombinase-primer complex [9]. However, this crowding agent can increase viscosity and impede reagent diffusion at low target concentrations, which can be mitigated through brief mixing after reaction initiation or reduced reaction volumes [9]. The development of lyophilized RPA reagents has significantly enhanced the technology's practicality for field applications by improving thermostability and eliminating cold-chain shipping requirements [13].
Comparative Analysis: RPA vs. PCR and Other Isothermal Methods
Performance Comparison with PCR
The following table provides a detailed comparison of the technical specifications and performance characteristics of RPA versus conventional PCR:
Table 1: Comprehensive Comparison of RPA and PCR Characteristics
| Parameter | Recombinase Polymerase Amplification (RPA) | Polymerase Chain Reaction (PCR) |
|---|---|---|
| Year Developed | 2006 [10] | 1980s [10] |
| Core Enzymes | Recombinase (UvsX), SSB, strand-displacing polymerase [9] [10] | Thermostable DNA polymerase (Taq) [13] [10] |
| Temperature Requirements | Constant low temperature (37-42°C) [9] [13] | Thermal cycling (50-95°C through denaturation, annealing, extension) [13] |
| Amplification Time | 5-20 minutes for detectable products [13] [10] | 1-2 hours typically [13] |
| Equipment Needs | Minimal; no thermal cycler required [13] | Requires thermal cycler [13] |
| Sensitivity | Capable of detecting 1-10 DNA target copies [9] | Highly sensitive, capable of detecting low copy numbers [13] |
| Primer Design | 30-35 bases recommended; standard PCR primers may work [9] | 18-25 bases typically; well-established design rules [10] |
| Specificity | High, but may tolerate certain mismatches [10] | High specificity with optimized conditions [10] |
| Resistance to Inhibitors | Generally resistant to inhibitors found in complex samples [10] | Often susceptible to inhibitors; may require sample purification [10] |
| Ease of Use | Simplified protocol; suitable for non-specialists [13] | Requires trained personnel and specialized equipment [13] |
| Commercial Kits | Limited availability primarily from specialized suppliers [8] | Widely available from multiple suppliers [8] |
| Cost Considerations | Higher reagent costs [10] | Lower reagent costs; established supply chains [10] |
| Quantitative Capability | Limited quantitative precision [10] | Excellent quantitative capabilities (qPCR) [10] |
Comparison with Other Isothermal Amplification Techniques
RPA occupies a distinct position among various isothermal amplification technologies, offering unique advantages in speed, temperature flexibility, and operational simplicity. The table below compares RPA with other commonly used isothermal amplification methods:
Table 2: Comparison of RPA with Other Isothermal Amplification Techniques
| Method | Template | Optimal Temperature | Time | Primers Required | Key Advantages |
|---|---|---|---|---|---|
| RPA | DNA/RNA [9] [10] | 37-42°C [9] | 5-20 min [10] | 2 [10] | Rapid; low temperature; simple primer design [9] |
| LAMP | DNA/RNA [10] | 60-65°C [9] [10] | 60 min [9] | 4-6 [9] [10] | High yield; resistant to inhibitors [10] |
| HDA | DNA/RNA [10] | 60-65°C [10] | 30-120 min [9] | 2 [9] | Simple reaction composition [10] |
| NASBA | RNA [9] [10] | 41°C [9] | 60-180 min [9] | 2 [9] | High selectivity for RNA [10] |
| RCA | DNA/RNA [10] | 30-65°C [9] | 60-240 min [9] | 1 [9] | Easy exponential amplification [10] |
| SDA | DNA [9] [10] | 30-55°C [9] | 60-120 min [9] | 4 [9] | Mild reaction conditions [10] |
Experimental Protocols and Methodologies
Basic RPA Protocol for DNA Detection
The following protocol outlines the standard procedure for conducting basic RPA reactions for DNA detection, adaptable for various applications including initial template amplification in biofilm pathogen detection:
Reaction Setup:
- Prepare RPA reaction mix according to manufacturer's specifications (typically 50 μL total volume)
- Include recommended concentrations of rehydration buffer, primers (typically 10 μM each), and dNTPs
- Add template DNA (1-10 target copies for optimal sensitivity [9])
- Initiate reaction by adding magnesium acetate (typically 14 mM final concentration)
Incubation Conditions:
- Incubate reaction at 37-42°C for 10-20 minutes
- Temperature control can be achieved using heating blocks, water baths, or portable thermal equipment
Product Detection:
- Analyze amplification products by agarose gel electrophoresis (basic RPA)
- Alternatively, use real-time fluorescence detection or lateral flow strips for endpoint analysis
For biofilm samples, preliminary processing may be required to liberate microbial DNA from the extracellular polymeric substance matrix. This can include enzymatic treatments (e.g., DNase, proteinase K) or mechanical disruption methods prior to RPA amplification [12].
Enhanced RPA (eRPA) for Improved Sensitivity
Recent advancements have led to the development of enhanced RPA (eRPA) protocols that significantly improve detection sensitivity. The key modifications include:
Reverse Transcriptase Selection: Use of engineered reverse transcriptases with minimal RNase H activity (e.g., SuperScript IV) for RNA targets [14]
RNase H Supplementation: Addition of exogenous RNase H to degrade RNA in RNA:DNA hybrids, reducing inhibition of RPA amplification [14]
Primer Screening Protocol: Systematic screening of multiple primer pairs using qPCR on diluted RPA products to identify candidates with high specific yield and minimal nonspecific amplification [14]
Reagent Concentration: Use of more concentrated reaction reagents than standard manufacturer protocols to allow increased sample input without sacrificing amplification efficiency [14]
This enhanced protocol has demonstrated detection limits as low as 5 viral copies in patient samples without requiring RNA purification, with results available in approximately 45 minutes from sample collection [14].
RPA-CRISPR Integrated Protocol for Pathogen Detection
The integration of RPA with CRISPR-Cas systems creates a powerful diagnostic platform particularly suited for detecting biofilm-associated pathogens. The following protocol outlines the single-pot RPA-CRISPR approach:
RPA Amplification Phase:
- Prepare RPA reaction mixture with target-specific primers
- Incubate at 37-42°C for 5-15 minutes to amplify target sequences
- For single-pot assays, use reduced RPA component concentrations to minimize interference with CRISPR components [15]
CRISPR Detection Phase:
- After RPA amplification, directly add CRISPR complex components to the same tube:
- Mix thoroughly by brief shaking or vortexing
- Incubate at 37°C for 5-15 minutes to allow CRISPR-mediated collateral cleavage and signal generation
Signal Detection:
- Visualize fluorescence under blue light or using portable readers
- Alternatively, use lateral flow strips for visual detection without instrumentation
This integrated approach has demonstrated exceptional sensitivity in detecting plant parasitic nematodes, with limits of detection reaching 10^-5 dilutions of genomic DNA, significantly surpassing both RPA-LFD and conventional PCR methods [15].
Research Reagent Solutions for RPA-Based Detection
The successful implementation of RPA and RPA-CRISPR platforms relies on specific reagent systems optimized for isothermal amplification and detection. The following table outlines essential research reagents and their functions:
Table 3: Essential Research Reagents for RPA and RPA-CRISPR Applications
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Recombinase Enzymes | T4 UvsX recombinase [9] [10] | Binds primers to form nucleoprotein filaments for strand invasion | Core RPA component; requires ATP cofactor |
| Single-Strand Binding Proteins | T4 gp32 SSB [9] | Stabilizes displaced DNA strands | Prevents primer displacement; essential for RPA efficiency |
| Strand-Displacing Polymerases | B. subtilis Pol I (Bsu) [9] | Extends primers from 3' end | Works at low temperatures; strong strand displacement activity |
| Reverse Transcriptases | SuperScript IV, Transcriptor [9] [14] | Converts RNA to cDNA for amplification | Selected for minimal RNase H activity; enables RT-RPA |
| CRISPR Components | Cas12a protein, crRNA [11] [15] | Specific target recognition and trans-cleavage | Enables highly specific detection of RPA amplicons |
| Lyophilized Reagent Formulations | TwistAmp kits [13] | Stable, room-temperature storage | Ideal for field applications; improved thermostability |
| Detection Probes | FQ reporters, exo probes, LF probes [11] [10] | Signal generation for readout | Fluorophore-quencher pairs for real-time or endpoint detection |
| Crowding Agents | Polyethylene glycol [9] | Molecular crowding to enhance interactions | Critical for recombinase-primer complex stability |
Integration of RPA with CRISPR Systems for Biofilm Pathogen Detection
The combination of RPA with CRISPR-Cas systems represents a transformative approach in the detection of biofilm-associated pathogens, addressing significant limitations of conventional methods. This integration leverages the rapid, isothermal amplification capability of RPA with the precise sequence recognition and collateral activity of CRISPR-Cas systems, particularly Cas12a and Cas13 [16] [11]. The operational compatibility of these technologies—both functioning optimally at 37-42°C—enables the development of streamlined one-pot reaction formats that minimize cross-contamination risks and simplify operational procedures [11] [15].
In the specific context of biofilm research, RPA-CRISPR platforms offer distinct advantages for detecting pathogens embedded within complex extracellular polymeric matrices. Traditional culture-based methods for biofilm analysis are time-consuming, requiring days to obtain results, while PCR-based approaches have stringent equipment requirements and are difficult to implement outside laboratory settings [11]. The RPA-CRISPR combination addresses these limitations by providing rapid (often under 30 minutes), highly sensitive detection with minimal equipment requirements [15]. Furthermore, the programmability of CRISPR systems allows for species-specific identification of pathogens through appropriate guide RNA design, targeting conserved genomic regions or antimicrobial resistance genes prevalent in biofilm communities [12].
Recent innovations have focused on overcoming the technical challenges associated with combining RPA and CRISPR components in single-reaction formats. Traditional approaches required sequential addition of reagents due to interference between RPA and CRISPR components, increasing contamination risks [15]. Advanced single-pot formulations now utilize optimized reagent concentrations and specialized delivery systems that maintain the functionality of both systems while enabling simplified workstreams [15]. These developments are particularly valuable for biofilm monitoring in food processing environments and clinical settings, where rapid, on-site detection can inform timely intervention strategies [12].
The application of RPA-CRISPR systems in biofilm research continues to evolve, with emerging innovations focusing on enhanced sensitivity, multiplexing capabilities, and integration with portable detection platforms. Current research directions include the development of lyophilized reagent formulations for improved field stability, incorporation of internal controls to ensure reaction validity, and implementation of quantitative readouts for assessing pathogen load in biofilm samples [13] [12]. As these technologies mature, RPA-CRISPR platforms are poised to become invaluable tools for precise biofilm monitoring and control across diverse applications from food safety to clinical diagnostics.
Originally identified as an adaptive immune system in bacteria and archaea, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) systems have revolutionized molecular biology. While their genome-editing capabilities are well-documented, this article focuses on their transformative application in molecular diagnostics, particularly for detecting biofilm-forming pathogens. The programmability of CRISPR/Cas systems, enabled by guide RNA sequences that direct Cas enzymes to specific nucleic acid targets, provides the foundation for both precise gene editing and highly specific pathogen detection [11] [16].
This technological expansion leverages the distinctive "collateral cleavage" activity of certain Cas proteins, such as Cas12 and Cas13. Upon recognition and cleavage of its target DNA, Cas12a exhibits promiscuous nuclease activity, indiscriminately degrading nearby single-stranded DNA (ssDNA) molecules [11] [16]. This phenomenon, known as trans-cleavage, provides a powerful signal amplification mechanism that can be harnessed for diagnostic purposes. When coupled with isothermal amplification techniques like Recombinase Polymerase Amplification (RPA), CRISPR/Cas systems form the basis for rapid, sensitive, and specific diagnostic platforms suitable for point-of-care testing (POCT) [11] [3]. This combination is particularly valuable for detecting challenging pathogens like Pseudomonas aeruginosa and Staphylococcus aureus, which form resilient biofilms and contribute significantly to healthcare-associated infections [17] [18] [19].
Molecular Mechanisms: From Target Recognition to Signal Generation
The diagnostic application of CRISPR/Cas systems relies on a two-step molecular mechanism: specific target recognition followed by activated nonspecific cleavage.
Core Mechanism of CRISPR/Cas12a Detection
The CRISPR/Cas12a detection mechanism begins with the formation of a ribonucleoprotein complex where the Cas12a enzyme is programmed with a CRISPR RNA (crRNA) complementary to the target DNA sequence. This complex scans DNA for a matching sequence adjacent to a protospacer adjacent motif (PAM), typically "TTTV" for Cas12a. Upon target binding, the Cas12a enzyme undergoes a conformational change that activates both its specific cis-cleavage activity (cutting the target DNA) and its nonspecific trans-cleavage activity. This collateral cleavage degrades nearby single-stranded DNA reporter molecules, generating a detectable signal [11] [16].
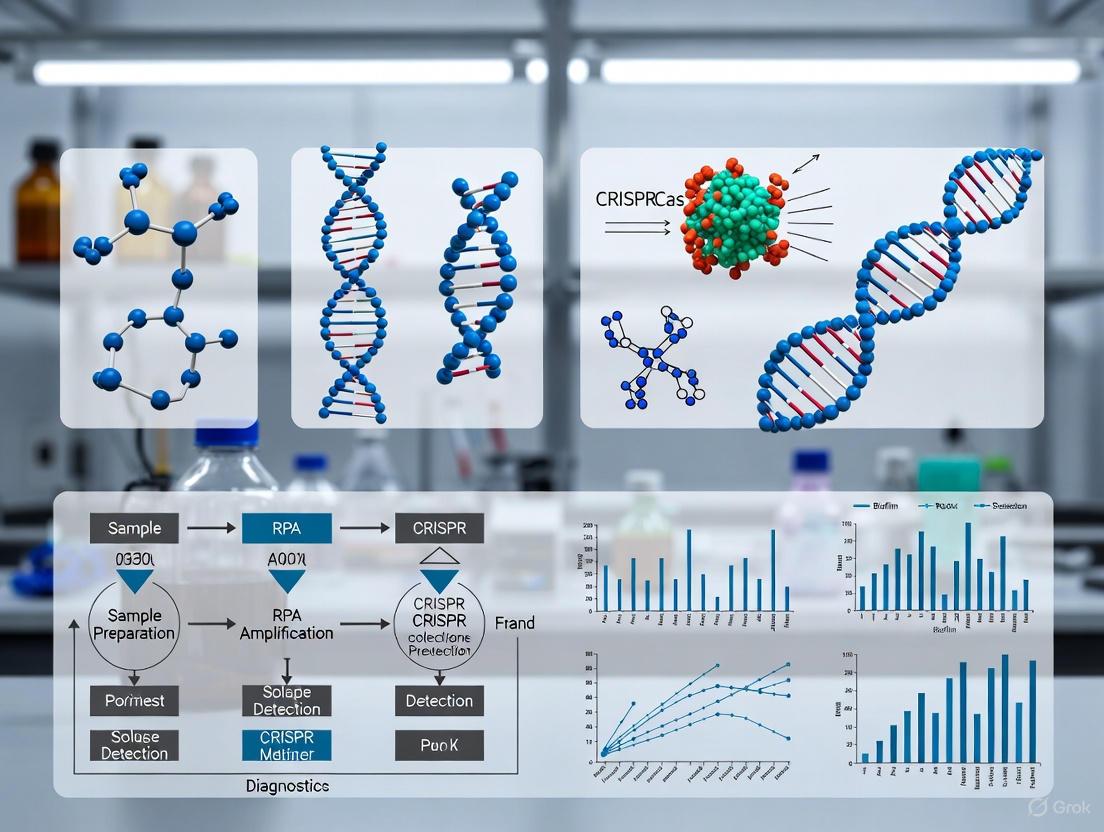
Figure 1: Core Mechanism of CRISPR/Cas12a Nucleic Acid Detection. The process begins with crRNA-guided target recognition, leading to Cas12a activation and subsequent trans-cleavage of fluorescently-quenched ssDNA reporters, generating a detectable signal.
CRISPR-Cas Protein Diversity and Their Diagnostic Applications
Different Cas proteins have unique properties that make them suitable for various diagnostic applications, as summarized in Table 1.
Table 1: Comparison of Key Cas Proteins Used in Diagnostic Applications
| Cas Protein | Nucleic Acid Target | PAM Sequence | Trans-Cleavage Substrate | Key Diagnostic Platforms | Primary Applications |
|---|---|---|---|---|---|
| Cas9 | dsDNA | 3'-NGG | None | CRISPRa, CRISPRi | Gene editing, regulation |
| Cas12a | dsDNA | 5'-TTTV | ssDNA | DETECTR, HOLMES | DNA virus, bacterial detection |
| Cas13a | ssRNA | Non-PAM specific | ssRNA | SHERLOCK | RNA virus detection |
| Cas14 | ssDNA | Non-PAM specific | ssDNA | -- | Single-nucleotide polymorphism |
Cas12a has emerged as particularly valuable for bacterial detection due to its DNA targeting capability and robust trans-cleavage activity. Its compatibility with isothermal amplification techniques like RPA, which operates at 37-42°C, enables the development of streamlined diagnostic workflows without requiring thermal cyclers [11] [3]. Furthermore, Cas12a's single guide RNA structure simplifies crRNA design compared to the dual-RNA system of Cas9, facilitating more straightforward assay development [11].
Application Note: One-Pot RPA-CRISPR/Cas12a for Biofilm Pathogen Detection
Workflow for Pseudomonas aeruginosa Detection
The one-pot RPA-CRISPR/Cas12a platform represents a significant advancement for detecting biofilm-forming pathogens like Pseudomonas aeruginosa. This integrated system consolidates nucleic acid amplification and CRISPR-based detection into a single reaction tube, simplifying the workflow and reducing contamination risks [18].
Figure 2: Integrated Workflow for One-Pot RPA-CRISPR/Cas12a Pathogen Detection. The process incorporates sample preparation, amplification, and detection in a single tube, significantly reducing hands-on time and contamination risk.
Performance Comparison of RPA-CRISPR/Cas12a Platforms
Recent studies have demonstrated the effectiveness of one-pot RPA-CRISPR/Cas12a systems for detecting various pathogens, with particular success against biofilm-forming bacteria, as shown in Table 2.
Table 2: Performance Metrics of RPA-CRISPR/Cas12a Detection Platforms for Pathogen Detection
| Target Pathogen | Target Gene | Assay Format | Detection Limit | Time | Clinical Concordance | Reference |
|---|---|---|---|---|---|---|
| Pseudomonas aeruginosa | lasB | One-tube RPA-CRISPR/Cas12a | 15.9 CFU/reaction | <40 min | 97.62% (n=84) | [18] |
| Pseudomonas aeruginosa | lasB | RPA-CRISPR/Cas12a | 100 copies/µL (fluorescence) 101 copies/µL (LFS) | ~30 min | Comparable to qPCR (n=150) | [17] |
| Staphylococcus aureus | nuc | Magnetic enrichment + single-step RPA-CRISPR/Cas12a | 10 CFU/mL | 40 min | Concordant with qPCR | [19] |
| Group B Streptococcus | cfb | One-pot two-temperature RPA-CRISPR/Cas12b | 10 copies/test | <60 min | 96.7% vs. culture 98.3% vs. qPCR (n=60) | [6] |
The one-pot RPA-CRISPR/Cas12a platform targeting the lasB gene of P. aeruginosa demonstrates particularly robust performance. This assay achieved 100% inclusivity for 21 P. aeruginosa isolates and 100% exclusivity against non-aeruginosa strains, confirming its specificity [18]. The clinical validation showing 97.62% concordance with traditional culture methods across diverse sample types highlights its diagnostic reliability [18].
Experimental Protocol: One-Pot RPA-CRISPR/Cas12a Assay for Pseudomonas aeruginosa Detection
Research Reagent Solutions
Table 3: Essential Reagents and Materials for One-Pot RPA-CRISPR/Cas12a Assay
| Reagent/Material | Function | Specifications/Alternative Products |
|---|---|---|
| LbaCas12a enzyme | CRISPR nuclease for target recognition and trans-cleavage | Commercial sources: New England Biolabs |
| RPA basic kit | Isothermal amplification of target DNA | Contains recombinase, single-stranded binding proteins, strand-displacing polymerase |
| crRNA | Guides Cas12a to specific target sequence | Designed against lasB gene; synthesized in vitro using T7 polymerase |
| ssDNA reporter | Signal generation upon trans-cleavage | Fluorescence: 5'-FAM-TTATT-BHQ1-3' Lateral flow: 5'-FITC-TTTTTTTTTT-Biotin-3' |
| RPA primers | Amplify target lasB gene | Forward and reverse primers targeting conserved lasB regions |
| Nuclease-free water | Reaction preparation | Ensure RNase-free and DNase-free conditions |
| Portable fluorescence detector | Signal detection | For quantitative readout; alternative: UV light for visual detection |
Step-by-Step Protocol
crRNA Design and Preparation (Day 1)
- Target Selection: Identify a conserved region within the P. aeruginosa lasB gene (elastase B) through sequence alignment.
- crRNA Design: Design crRNA sequences (approximately 20-24 nt) complementary to the target region with consideration of Cas12a PAM requirements.
- crRNA Synthesis:
- Anneal specific crRNA forward and reverse DNA oligonucleotides.
- Perform in vitro transcription using T7 RNA polymerase.
- Purify crRNA using RNA purification kits.
- Determine concentration and purity (A260/A280 ratio >1.8).
One-Pot Reaction Setup (Day 2)
Prepare Reaction Master Mix (25 µL total volume):
- 10 µL of 2× RPA rehydration buffer
- 2.1 µL of 10 µM forward primer
- 2.1 µL of 10 µM reverse primer
- 1.5 µL of 10 µM crRNA
- 1 µL of 10 µM ssDNA reporter (FAM-TTATT-BHQ1 for fluorescence)
- 0.5 µL of LbaCas12a enzyme (10 µM)
- 1 µL of template DNA (heat-lysed sample or extracted DNA)
- Nuclease-free water to 25 µL
Add Activation Buffer:
- Add 2.5 µL of 280 mM magnesium acetate (RPA activator) to the reaction tube lid.
- Centrifuge briefly to mix magnesium acetate with the reaction mixture.
Amplification and Detection
Incubate Reaction:
- Place tubes in a preheated block or dry bath at 39°C.
- Incubate for 15-30 minutes.
Signal Detection:
- Fluorescence Readout: Use a portable fluorescence detector to measure FAM signal at time intervals or endpoint.
- Visual Detection: Expose reaction tubes to UV light (blue light, 485 nm) – positive samples emit green fluorescence.
- Lateral Flow Readout: For FITC/biotin reporters, dip test strips and observe test line appearance within 5 minutes.
Troubleshooting and Optimization
- Low Signal Intensity: Extend RPA amplification time to 30 minutes or increase crRNA concentration to 1.5 µM.
- High Background: Titrate crRNA concentration; optimize Cas12a-to-crRNA ratio; verify primer specificity.
- Inconsistent Results: Include positive and negative controls in each run; ensure magnesium acetate is properly mixed.
- Sample Inhibition: Dilute sample template or implement a brief heat inactivation step (95°C for 5 minutes).
Discussion: Advantages and Implementation Considerations
The one-pot RPA-CRISPR/Cas12a system represents a significant advancement in molecular diagnostics for biofilm-forming pathogens. Its key advantages include rapid turnaround time (30-40 minutes versus 24-48 hours for culture), minimal equipment requirements (simple heat block), and excellent sensitivity and specificity comparable to qPCR but with greatly simplified workflow [17] [18].
For researchers implementing this technology, several factors require consideration. The one-pot format significantly reduces aerosol contamination compared to multi-step assays, but careful crRNA design remains critical for assay performance [18]. The lasB gene has proven particularly effective for P. aeruginosa detection due to its conservation and essential role in virulence [17] [18]. Furthermore, the system's flexibility allows integration with various signal detection methods, from portable fluorometers for quantitative results to UV lamps for visual readout in resource-limited settings [6] [18].
Recent innovations continue to enhance this platform's capabilities. The development of two-temperature protocols (RPA at 39°C followed by Cas12b activation at 62°C) has improved low-copy target detection [6]. Additionally, incorporating magnetic enrichment strategies for sample preparation, as demonstrated for S. aureus detection, can improve sensitivity by 100-fold, enabling detection as low as 10 CFU/mL [19].
These advancements position one-pot RPA-CRISPR/Cas systems as powerful tools for rapid detection of biofilm-forming pathogens in both clinical and point-of-care settings. Their simplicity, speed, and accuracy address critical needs in healthcare-associated infection control and antimicrobial stewardship, particularly for challenging pathogens like P. aeruginosa that exhibit extensive antibiotic resistance.
The CRISPR-associated proteins Cas12a and Cas12b belong to the Class 2, Type V CRISPR-Cas systems and possess a unique enzymatic property known as collateral cleavage or trans-cleavage activity [16] [20]. This activity is triggered when the Cas protein complex, guided by a CRISPR RNA (crRNA), binds to a specific target nucleic acid sequence. Upon target recognition and binding, the Cas protein undergoes a conformational change that activates its non-specific nuclease domain, enabling it to indiscriminately cleave surrounding single-stranded DNA (ssDNA) molecules [16] [21] [22]. This mechanism fundamentally differs from the targeted cis-cleavage activity used for gene editing, as the collateral cleavage is non-sequence-specific and operates in trans, making it exceptionally suitable for diagnostic applications.
The discovery of this collateral activity has paved the way for novel diagnostic platforms, such as DNA Endonuclease-Targeted CRISPR Trans Reporter (DETECTR) for Cas12a and specific high-sensitivity enzymatic reporter unlocking (SHERLOCK) for related systems [16] [5]. When combined with isothermal amplification methods like Recombinase Polymerase Amplification (RPA) or Loop-Mediated Isothermal Amplification (LAMP), these systems can detect attomolar to femtomolar concentrations of pathogen DNA, offering sensitivity comparable to or even surpassing traditional PCR-based methods, but with faster results and minimal equipment [23] [24] [25]. This combination is particularly powerful for detecting biofilm-forming pathogens in complex samples, as it allows for precise identification of specific nucleic acid sequences amidst background material.
Molecular Mechanisms of Cas12a and Cas12b
Cas12a Mechanism
Cas12a (formerly known as Cpf1) is a single RNA-guided endonuclease that requires only a CRISPR RNA (crRNA) for function, without the need for a trans-activating crRNA (tracrRNA) [20]. Its activity is directed by a crRNA containing a spacer sequence complementary to the target DNA. A critical requirement for Cas12a's recognition of double-stranded DNA (dsDNA) is the presence of a short Protospacer Adjacent Motif (PAM), typically a 5'-TTTV (where V is A, C, or G) sequence, located immediately adjacent to the target region [26] [20].
The mechanism unfolds in two stages:
- Target Recognition and cis-Cleavage: The Cas12a-crRNA complex scans DNA for the complementary protospacer sequence adjacent to a valid PAM site. Upon binding, the Cas12a protein cleaves the target DNA itself (cis-cleavage) [22] [20].
- Collateral trans-Cleavage: The successful binding and cis-cleavage activate a separate RuvC nuclease domain within Cas12a. This activated state triggers the indiscriminate cleavage of any nearby single-stranded DNA (ssDNA) molecules, which is the cornerstone of its diagnostic utility [26] [20].
It is noteworthy that Cas12a can also be activated by single-stranded DNA targets without requiring a PAM sequence, though with different catalytic efficiency [20].
Cas12b Mechanism
Cas12b (formerly known as C2c1) operates through a similar collateral cleavage mechanism but with distinct structural and functional characteristics. Unlike Cas12a, Cas12b is a dual RNA-guided system, requiring both a crRNA and a tracrRNA for target recognition and complex stability [24] [20].
A significant advantage of many naturally occurring Cas12b orthologs is their inherent thermostability. Wild-type Cas12b from species like Brevibacillus sp. (BrCas12b) functions optimally at higher temperatures (e.g., 55-65°C) than many Cas12a variants [24]. This property makes Cas12b exceptionally suitable for one-pot diagnostic assays, as its optimal temperature range overlaps perfectly with common isothermal amplification methods like RT-LAMP (60-65°C) [24]. Recent protein engineering efforts have further enhanced this thermostability. For instance, engineered BrCas12b (eBrCas12b) variants developed through computational design exhibit robust trans-cleavage activity at temperatures up to 67°C, enabling more flexible and efficient one-pot reactions [24].
Table 1: Comparative Analysis of Cas12a and Cas12b Properties
| Property | Cas12a | Cas12b |
|---|---|---|
| Guide RNA | Single crRNA | crRNA and tracrRNA |
| PAM Requirement | 5'-TTTV (for dsDNA) | Varies by ortholog |
| Key Domains | RuvC | RuvC |
| cis-cleavage | dsDNA or ssDNA | dsDNA |
| trans-cleavage | ssDNA | ssDNA |
| Thermostability | Moderate | High (enhanced via engineering) |
| Optimal Temperature | ~37-42°C [23] | ~60-67°C (eBrCas12b) [24] |
| Ideal for One-Pot | Typically two-pot | Yes, single-pot (e.g., SPLENDID) [24] |
Quantitative Performance of Collateral Cleavage
The sensitivity of CRISPR-Cas12a/b diagnostics is quantified by the Limit of Detection (LOD), which is significantly enhanced by coupling with a pre-amplification step. The following table summarizes performance data from various applications.
Table 2: Sensitivity and Performance of CRISPR-Cas12a/b Detection Systems
| Application Target | CRISPR System | Amplification Method | Limit of Detection (LOD) | Total Assay Time |
|---|---|---|---|---|
| Neospora caninum [23] | Cas12a | RPA | 1 parasite/mL (fluorescence)10 parasites/mL (LFS) | < 90 min |
| Hepatitis C Virus (HCV) [24] | Engineered Cas12b (eBrCas12b) | RT-LAMP (SPLENDID) | High clinical accuracy | ~60 min (incl. extraction) |
| SARS-CoV-2 [24] | Engineered Cas12b (eBrCas12b) | RT-LAMP (SPLENDID) | High clinical accuracy | ~20 min (detection only) |
| Human Adenovirus 3 & 7 [25] | Cas12b | MCDA | 5 fg/μL plasmid DNA | ~60 min |
| General Nucleic Acid Detection [21] | Cas12a (with engineered crRNA) | None (direct detection) | Femtomolar (fM) range | N/A |
The data demonstrates that pre-amplification is crucial for achieving high sensitivity in clinical or environmental samples. Furthermore, engineered proteins and optimized crRNAs can push the fundamental sensitivity of the CRISPR system itself into the femtomolar range even without amplification [21].
Experimental Protocols for One-Pot RPA-CRISPR Diagnostics
This section provides a detailed protocol for detecting biofilm-forming pathogens using a one-pot RPA-CRISPR/Cas12b assay, adapted from the SPLENDID (Single-pot LAMP-mediated engineered BrCas12b for nucleic acid detection of infectious diseases) methodology [24].
Protocol 1: One-Pot RPA-CRISPR/Cas12b Assay
Principle: This protocol leverages the thermostability of engineered Cas12b (eBrCas12b) to combine nucleic acid amplification and CRISPR detection in a single tube, simplifying the workflow, reducing contamination risk, and accelerating time-to-result [24].
Reagents and Materials:
- Template: Extracted DNA from biofilm samples or pure cultures.
- Enzymes: Engineered BrCas12b (eBrCas12b) nuclease [24], RPA dry pellet kit or equivalent isothermal amplification mix.
- Oligonucleotides: Target-specific RPA primers, crRNA designed for the pathogen of interest, ssDNA reporter probe (e.g., FAM-TTATT-BHQ for fluorescence or FAM-Biotin for LFA).
- Buffer: Appropriate reaction buffer (e.g., NEBuffer r2.1 or commercial CRISPR buffer).
- Equipment: Real-time fluorometer or thermal cycler capable of maintaining 60-65°C, or lateral flow strips (e.g., Milenia HybriDetect) for endpoint detection.
Procedure:
- Reaction Setup: In a single tube, prepare a master mix containing:
- 5 μL of template DNA.
- 29.5 μL of rehydration buffer from the RPA kit.
- Forward and reverse RPA primers (final concentration 420 nM each).
- crRNA (final concentration 60 nM).
- ssDNA reporter probe (final concentration 100 nM).
- eBrCas12b nuclease (final concentration 100 nM).
- Nuclease-free water to a final volume of 49.5 μL.
- Initiation: Add 0.5 μL of Magnesium Acetate (280 mM) from the RPA kit to the tube lid, briefly centrifuge to mix and initiate the reaction.
- Incubation: Incubate the reaction tube at 60-65°C for 40-60 minutes in a real-time fluorometer with fluorescence readings taken every minute, or in a heat block for endpoint analysis.
- Detection:
- Fluorometric Readout: Monitor the real-time fluorescence increase. A positive sample will show a significant increase in fluorescence signal over time.
- Lateral Flow Readout: For endpoint detection, pipette 5-10 μL of the reaction product onto the sample pad of a lateral flow strip and place the strip in a running buffer. Read the result after 5-10 minutes. The appearance of both test and control lines indicates a positive result; only the control line indicates a negative result.
Troubleshooting Notes:
- No Signal: Verify the activity of enzymes and the integrity of crRNA and reporter probes. Ensure the PAM site is correctly positioned in the amplicon.
- High Background: Titrate the crRNA and Cas protein concentrations to minimize non-specific activation. Ensure reagents are free of nucleases.
Protocol 2: crRNA Engineering for Enhanced Sensitivity
Principle: The sensitivity of Cas12a can be significantly improved by engineering the crRNA. Adding a short, single-stranded DNA extension to the 3'-end of the crRNA can augment the rate of collateral cleavage activity by up to 3.5-fold, a system known as ENHANCE [21].
Procedure:
- Design: Synthesize crRNA with a 7-mer deoxyadenosine (AAAAAAA) or other ssDNA extension on its 3'-end.
- Comparison: Test the modified crRNA (crRNA+3'DNA7) alongside the wild-type crRNA in a standard Cas12a fluorescent detection assay.
- Validation: The modified crRNA should demonstrate a significantly higher fluorescence slope and earlier time to positivity compared to the wild-type crRNA when detecting the same target concentration [21].
Visualization of Mechanisms and Workflows
Diagram 1: Cas12a Collateral Cleavage Mechanism.
Diagram 2: One-Pot RPA-CRISPR Assay Workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for RPA-CRISPR Diagnostic Development
| Reagent / Material | Function / Role | Example Specifications / Notes |
|---|---|---|
| Cas12a Enzyme (e.g., LbCas12a) | Target-specific binding and collateral ssDNA cleavage. | Requires TTTV PAM; operates at ~37°C [23] [26]. |
| Engineered Cas12b (e.g., eBrCas12b) | Thermostable variant for one-pot assays. | Optimal activity at 60-67°C; ideal for SPLENDID assays [24]. |
| crRNA | Guides Cas protein to the specific target DNA sequence. | Contains a scaffold (binds Cas) and a 20-24 nt spacer (complementary to target). Can be engineered with 3' DNA extensions (ENHANCE) for improved sensitivity [26] [21]. |
| ssDNA Reporter Probe | Signal generation upon collateral cleavage. | FQ Reporter: 5'-FAM, 3'-BHQ for fluorescence. FB Reporter: 5'-FAM, 3'-Biotin for Lateral Flow Assays [26] [22]. |
| RPA Kit | Isothermal amplification of target DNA. | Contains recombinase, polymerase, and proteins; amplifies DNA at 37-42°C in 15-20 min [23]. |
| Lateral Flow Strip (LFS) | Visual, equipment-free readout. | Typically contains a test line (anti-FAM antibody) and control line (streptavidin) [23] [26]. |
The collateral cleavage activity of Cas12a and Cas12b provides a powerful mechanism for translating specific nucleic acid recognition into an amplified, detectable signal. The development of thermostable, engineered Cas12b variants and optimized crRNAs has been instrumental in creating robust one-pot assays like SPLENDID, which merge amplification and detection into a single step [24]. These systems offer the speed, sensitivity, and portability required for point-of-care diagnostics. When applied to the challenge of biofilm pathogen detection, one-pot RPA-CRISPR protocols enable rapid and specific identification of pathogens directly on food-contact surfaces or in clinical samples, providing a powerful tool for ensuring safety and preventing the spread of infectious diseases.
The detection of biofilm-forming pathogens represents a significant challenge in clinical diagnostics and public health, often requiring complex, multi-step molecular assays that are impractical for point-of-care settings. The integration of Recombinase Polymerase Amplification (RPA) with CRISPR/Cas systems into a single reaction vessel—termed "one-pot" diagnostics—marks a revolutionary advancement in molecular detection technology. This integrated approach effectively addresses key limitations of conventional methods by eliminating aerosol contamination risks associated with amplicon transfer between tubes, significantly reducing total assay time, and simplifying operational procedures to make sophisticated diagnostics accessible in resource-limited environments [18] [15].
The fundamental innovation lies in the strategic coordination of two biologically distinct processes: isothermal nucleic acid amplification and CRISPR-mediated detection. RPA enables rapid DNA amplification at a constant temperature range of 37-42°C through the synergistic activity of recombinase enzymes, single-stranded DNA-binding proteins, and strand-displacing DNA polymerases [27] [11]. This isothermal characteristic makes RPA ideally suited for combination with CRISPR systems, which provide unparalleled sequence specificity through RNA-guided nucleic acid recognition [3]. The one-pot format strategically houses both systems in a single tube, where RPA first amplifies the target sequence, followed by CRISPR/Cas-mediated recognition and signal generation through trans-cleavage activity of reporter molecules [6] [18].
For biofilm pathogens, which often demonstrate heightened resistance to antibiotics and conventional treatments, this technology offers a rapid detection platform that can guide timely therapeutic interventions. The simplicity of the one-pot system, combined with its potential for visual readout without sophisticated instrumentation, positions it as an ideal solution for point-of-care testing where traditional laboratory infrastructure is unavailable [28] [18].
Technical Principles and System Design
Recombinase Polymerase Amplification (RPA) Fundamentals
RPA is an isothermal nucleic acid amplification technique that operates at low, constant temperatures (37-42°C) through a unique enzymatic mechanism. The core RPA reaction employs three essential enzyme components: a recombinase that forms complexes with oligonucleotide primers, single-stranded DNA-binding proteins (SSBs) that stabilize displaced DNA strands, and a strand-displacing DNA polymerase that extends primers from the recombination sites [27] [11]. This elegant biochemical process begins when recombinase-primer complexes scan double-stranded DNA for homologous sequences, facilitating strand invasion and displacement. The SSBs then stabilize the resulting displacement loops, preventing reannealing while the polymerase initiates synthesis using the opposing strand as a template [11].
A key advantage of RPA for one-pot diagnostics is its rapid reaction kinetics, typically completing amplification within 10-30 minutes—significantly faster than conventional PCR [27]. Additionally, RPA functions optimally at temperatures compatible with CRISPR enzyme activity (37-42°C), eliminating the need for thermal cycling equipment and enabling seamless integration with CRISPR detection systems in a single tube [6] [18]. Compared to other isothermal techniques like LAMP, RPA requires only two primers rather than four to six, simplifying assay design while maintaining high sensitivity and specificity [6] [27].
CRISPR/Cas12a Detection Mechanism
The CRISPR/Cas12a system (a Class 2, Type V CRISPR effector) provides the sequence-specific recognition and signal generation component of one-pot assays. Cas12a is guided by a short CRISPR RNA (crRNA) that directs the enzyme to complementary double-stranded DNA sequences adjacent to a protospacer adjacent motif (PAM) site [27] [11]. Upon recognizing its target, Cas12a exhibits two distinct cleavage activities: cis-cleavage (sequence-specific cutting of the target DNA) and trans-cleavage (non-specific degradation of single-stranded DNA molecules in the reaction environment) [11] [3].
The trans-cleavage activity forms the basis for detection in RPA-CRISPR assays. By including fluorescently-quenched single-stranded DNA reporters in the reaction mixture, Cas12a's collateral cleavage generates a detectable signal when the target pathogen DNA is present [18]. This mechanism provides exceptional specificity because signal generation depends entirely on precise crRNA-target recognition, effectively minimizing false-positive results that may occur from non-specific amplification in RPA [27] [18].
One-Pot Integration Strategy
Integrating RPA and CRISPR/Cas12a into a single reaction tube presents significant technical challenges due to potential interference between the two systems. The competing reactions can hinder overall efficiency if not properly coordinated [15]. Several strategic approaches have been developed to overcome these compatibility issues:
Two-Temperature Incubation: This method physically separates the amplification and detection phases through temperature manipulation. RPA amplification occurs first at 39°C for 40 minutes, followed by Cas12b activation at 62°C for 5 minutes to initiate detection [6]. The higher temperature for CRISPR activation takes advantage of the thermostable properties of certain Cas variants like Cas12b while simultaneously inactivating the RPA enzymes to prevent ongoing amplification during detection.
Sequential reagent activation: Some protocols initially physically separate RPA and CRISPR components within the same tube using barrier methods, then mix them after amplification through shaking or centrifugation [15]. This approach maintains the "one-pot" advantage of a single sealed container while minimizing interference during critical reaction phases.
Concentration optimization: Systematically balancing reagent concentrations through statistical design of experiments (DoE) can reduce mutual interference without requiring physical separation [29]. This method fine-tunes component ratios to ensure both systems function optimally in the shared environment.
The selection of appropriate Cas protein variants significantly impacts one-pot assay performance. While Cas12a operates efficiently at lower temperatures, thermostable variants like Cas12b (AapCas12b) offer advantages for two-temperature protocols by withstanding the higher temperatures needed to terminate RPA amplification before CRISPR detection [6].
Figure 1: Integrated workflow for one-pot RPA-CRISPR detection of biofilm pathogens, combining sample preparation, amplification, and detection in a single tube.
Application Notes: Detection of Biofilm-Forming Pathogens
Case Study: Pseudomonas aeruginosa Detection
Pseudomonas aeruginosa represents an ideal model for evaluating one-pot RPA-CRISPR diagnostics due to its clinical significance as a biofilm-forming pathogen with considerable antibiotic resistance. A recently developed platform targeting the lasB gene (encoding elastase B) demonstrates the practical application of this technology for clinical use [18].
The assay design employed comprehensive crRNA screening to identify optimal guide sequences for Cas12a recognition of the lasB target. Through systematic comparison of three candidate crRNAs, researchers identified the most efficient guide sequence by measuring real-time fluorescence accumulation rates, selecting the variant that produced the fastest signal generation with minimal background noise [18]. This optimization process highlights the critical importance of guide RNA design in achieving maximal assay sensitivity.
Clinical validation with 84 diverse patient samples (including sputum, wound secretions, and urinary specimens) demonstrated exceptional performance, with 97.62% concordance compared to traditional culture methods [18]. The assay achieved an impressive sensitivity of 15.9 CFU per reaction, enabling detection of clinically relevant bacterial loads without pre-amplification steps. The one-tube format effectively minimized aerosol contamination risks while maintaining robust performance across different sample matrices [18].
For biofilm-derived samples, which often contain inhibitory substances that compromise molecular assays, the direct detection capability without nucleic acid extraction represents a particular advantage. The simple heat-based sample preparation effectively lyzes bacterial cells while inactivating common inhibitors, making the platform suitable for complex clinical specimens where biofilm-forming pathogens are prevalent [18].
Comparative Performance Analysis
The performance characteristics of one-pot RPA-CRISPR platforms for pathogen detection are summarized in Table 1, highlighting their applicability for biofilm-forming organisms.
Table 1: Analytical Performance of One-Pot RPA-CRISPR Detection Platforms for Pathogen Detection
| Target Pathogen | Target Gene | Detection Limit | Assay Time | Clinical Concordance | Reference |
|---|---|---|---|---|---|
| Pseudomonas aeruginosa | lasB | 15.9 CFU/reaction | <60 minutes | 97.62% (vs. culture) | [18] |
| Group B Streptococcus | cfb4 | 10 copies/test | <60 minutes | 96.7% (vs. culture) | [6] |
| Genetically Modified Papaya | - | 20 copies | <60 minutes | - | [28] |
| Aphelenchoides besseyi | 18S rRNA | 10⁻⁵ dilution | <30 minutes | - | [15] |
The data demonstrate consistently excellent sensitivity across various applications, with detection limits sufficient for identifying clinically relevant pathogen concentrations. The rapid turnaround times (30-60 minutes) represent significant improvements over conventional culture methods that require 24-48 hours, particularly valuable for biofilm-associated infections where treatment timing critically impacts outcomes [6] [18].
Protocol: One-Pot RPA-CRISPR for Biofilm Pathogen Detection
Objective: To detect Pseudomonas aeruginosa and other biofilm-forming pathogens directly from clinical samples using an integrated one-pot RPA-CRISPR/Cas12a assay.
Principle: This protocol combines RPA-based isothermal amplification of pathogen-specific genetic markers with CRISPR/Cas12a-mediated detection in a single reaction tube, eliminating cross-contamination risks from amplicon transfer and enabling visual result interpretation under UV light [18].
Materials and Reagents
Table 2: Essential Research Reagent Solutions for One-Pot RPA-CRISPR Assays
| Reagent Component | Function/Principle | Recommended Concentration |
|---|---|---|
| RPA basic pellets (TwistAmp) | Isothermal amplification core components | 1 pellet per reaction |
| Cas12a protein (LbCas12a/EnGen) | Target-specific recognition and trans-cleavage | 10 μM [29] |
| crRNA (lasB-specific) | Guides Cas12a to target sequence | 2 μM [18] |
| ssDNA-FQ reporter (6-FAM/BHQ-1) | Fluorescence signal generation via collateral cleavage | 500 nM |
| RPA primers (lasB-specific) | Target amplification | 420 nM each |
- Equipment: Portable fluorescence detector or UV light source (365 nm), dry bath or heat block maintaining 39°C and 62°C, microcentrifuge tubes.
- Sample Preparation: Clinical samples (sputum, wound swabs, etc.) are processed using simple heat lysis (95°C for 5 minutes) or buffer incubation to release target DNA without extensive extraction [6] [18].
Experimental Procedure
Reaction Setup:
- Prepare master mix containing:
- 1× RPA rehydration buffer
- 420 nM each forward and reverse RPA primer (targeting lasB or pathogen-specific gene)
- 10 μM Cas12a protein
- 2 μM crRNA
- 500 nM FQ-reporter (5'-6-FAM-ZEN-IBFQ-3' or similar)
- Nuclease-free water to adjust volume
- Aliquot 47 μL master mix into 0.2 mL reaction tubes
- Add 2 μL template DNA (from heat-lysed sample) to reach 50 μL total reaction volume
- Mix thoroughly by pipetting and briefly centrifuge to collect liquid [18]
- Prepare master mix containing:
Amplification Phase:
Detection Phase:
Result Interpretation:
- Positive: Bright green fluorescence visible under UV light
- Negative: No fluorescence or minimal background signal
- Invalid: If no fluorescence develops in positive control reactions, repeat assay
Optimization Notes
- crRNA Design: Design crRNAs to target regions adjacent to appropriate PAM sequences (TTTV for LbCas12a). Screen multiple candidates to identify optimal guides with minimal background activity [18].
- Temperature Optimization: For two-temperature protocols, systematically test RPA and CRISPR phase temperatures to maximize sensitivity while minimizing non-specific signal [6].
- Clinical Validation: Establish assay performance using confirmed positive and negative clinical samples before diagnostic implementation. Compare results with reference culture methods to determine clinical sensitivity and specificity [18].
Technical Considerations and Optimization Strategies
Critical Implementation Factors
Successful implementation of one-pot RPA-CRISPR diagnostics requires careful attention to several technical factors that significantly impact assay performance:
Primer and crRNA Design: Effective primer design requires targeting conserved regions of pathogen genomes with balanced length (30-35 bp for RPA) and GC content to ensure efficient amplification. Similarly, crRNAs must be designed to target PAM-adjacent sequences with high specificity to the pathogen of interest. Computational tools should be employed to minimize off-target binding and secondary structure formation that could compromise assay efficiency [6] [18]. For biofilm-forming pathogens, target selection should focus on conserved virulence or species-specific genes rather than antibiotic resistance markers, which may be horizontally transferred.
Reagent Compatibility: The simultaneous presence of RPA and CRISPR components in a single tube creates a complex biochemical environment where enzymatic activities may interfere. Empirical optimization of component concentrations and reaction timing is essential to balance amplification efficiency with detection sensitivity [29] [15]. Statistical Design of Experiments (DoE) approaches efficiently identify optimal reagent ratios by systematically testing multiple variables simultaneously, significantly reducing optimization time compared to one-factor-at-a-time approaches [29].
Sample Processing: The direct use of minimally processed clinical samples represents both an advantage and challenge for one-pot assays. While eliminating DNA extraction saves time and resources, sample inhibitors can compromise reaction efficiency. Incorporation of sample dilution or simple heat treatment steps can mitigate inhibition while maintaining rapid processing times [6] [18]. For biofilm samples, which contain extracellular polymeric substances that may inhibit enzymatic reactions, additional optimization may be required.
Advanced System Configuration
Figure 2: Configuration options for one-pot RPA-CRISPR assays, showing different temperature protocols and detection modalities suitable for biofilm pathogen detection.
Troubleshooting Guide
Common challenges in one-pot RPA-CRISPR assay development and their solutions include:
- Low Signal Intensity: Increase crRNA concentration (up to 2 μM) or Cas protein concentration; extend CRISPR detection phase duration; verify RPA primer efficiency through gel electrophoresis; ensure reporter probe quality and concentration [18] [15].
- High Background Signal: Reduce crRNA concentration; implement hotter (higher temperature) CRISPR activation step; incorporate uracil DNA glycosylase (UDG) carryover contamination prevention; use truncated crRNA designs with improved specificity [6] [18].
- Inconsistent Results Between Replicates: Ensure thorough mixing of RPA pellets during master mix preparation; avoid repeated freeze-thaw cycles of enzyme reagents; include appropriate positive and negative controls in each run; verify consistent temperature distribution across reaction tubes [29].
- Reduced Sensitivity with Clinical Samples: Incorporate sample dilution to reduce inhibitors; increase RPA reaction time to 45-50 minutes; add bovine serum albumin (0.1-0.5 μg/μL) to mitigate inhibition; implement internal control targets to identify inhibition issues [18].
The integration of RPA and CRISPR technologies into single-reaction formats represents a transformative advancement in molecular detection for biofilm-forming pathogens. The one-pot platform successfully addresses critical limitations of conventional diagnostics by providing rapid results (under 60 minutes), exceptional sensitivity (down to single-digit copy numbers), and minimal technical requirements compatible with point-of-care settings [6] [18]. These characteristics make the technology particularly valuable for detecting challenging biofilm pathogens like Pseudomonas aeruginosa, where timely intervention significantly impacts clinical outcomes.
Future developments will likely focus on expanding multiplex detection capabilities for simultaneous identification of multiple pathogens or resistance markers, enhancing quantitative performance for treatment monitoring, and further simplifying sample processing to create truly sample-to-answer systems [3]. Additionally, the incorporation of novel Cas orthologs with improved properties—such as enhanced thermostability or different PAM requirements—will broaden the application scope and robustness of one-pot assays [6] [3].
The implementation of one-pot RPA-CRISPR diagnostics holds particular promise for resource-limited settings where biofilm-associated infections pose significant healthcare burdens. By providing accurate, rapid detection without sophisticated infrastructure, this technology can guide appropriate antibiotic use and infection control measures, ultimately improving patient outcomes while addressing the global challenge of antimicrobial resistance.
Key Biofilm-Forming Pathogens Amenable to RPA-CRISPR Detection
Biofilm-associated infections represent a significant challenge in clinical and industrial settings due to the inherent resistance of biofilms to conventional antibiotics and disinfectants. The complex extracellular polymeric substance (EPS) matrix limits pathogen detection and eradication, often leading to persistent and recurrent infections. The emergence of one-pot Recombinase Polymerase Amplification (RPA) coupled with CRISPR/Cas systems has revolutionized diagnostic approaches by enabling rapid, highly specific, and sensitive detection of pathogenic microorganisms without requiring sophisticated laboratory infrastructure. This integration leverages RPA's efficient isothermal amplification with CRISPR's precise sequence recognition, creating a powerful platform ideal for point-of-care testing (POCT) and resource-limited settings [11] [27]. This application note details the key biofilm-forming pathogens detectable by RPA-CRISPR platforms, provides quantitative detection performance data, and outlines standardized experimental protocols for research and diagnostic development.
Target Pathogens and Detection Performance
The following table summarizes primary biofilm-forming pathogens for which RPA-CRISPR detection systems have been successfully developed, along with their key genetic targets and analytical performance characteristics.
Table 1: Key Biofilm-Forming Pathogens and RPA-CRISPR Detection Performance
| Pathogen | Biofilm Association | Target Gene | CRISPR System | Sensitivity | Time-to-Result | Clinical Concordance |
|---|---|---|---|---|---|---|
| Pseudomonas aeruginosa | Healthcare-associated infections, ventilator-associated pneumonia, cystic fibrosis | lasB | Cas12a | 15.9 CFU/reaction | <1 hour | 97.62% (vs. culture) [18] |
| Acinetobacter baumannii | Medical device-related infections, ventilator-associated pneumonia | ompA | Multiple systems possible | Research phase | Research phase | Research phase [30] |
| Group B Streptococcus (GBS) | Neonatal infections, reproductive tract biofilms | cfb4 | Cas12b | 10 copies/test (1 copy/μL) | <1 hour | 96.7% (vs. culture), 98.3% (vs. qPCR) [6] |
| Staphylococcus aureus | Medical implant infections, chronic wounds | Multiple conserved genes | Cas12a, Cas9 | Research phase | Research phase | Research phase [11] [27] |
Detailed Experimental Protocol for P. aeruginosa Detection
The following protocol details the one-tube RPA-CRISPR/Cas12a method for detecting P. aeruginosa via the lasB gene, demonstrating the integration of amplification and detection in a single reaction vessel to minimize contamination and simplify workflow [18].
Materials and Reagents
Table 2: Essential Research Reagent Solutions
| Reagent Category | Specific Example | Function in Assay |
|---|---|---|
| Recombinase Enzyme | T4 UvsX recombinase or commercial RPA enzyme pellets | Binds primers and facilitates strand invasion of target DNA |
| DNA Polymerase | Bsu or Sau DNA polymerase | Extends primers from the 3' end following strand invasion |
| Single-Stranded Binding Protein (SSB) | T4 gp32 | Stabilizes displaced DNA strands during amplification |
| CRISPR Enzyme | LbCas12a or AapCas12b | Programmable nuclease providing specific target recognition and trans-cleavage activity |
| crRNA | lasB-specific crRNA (designed against target sequence) | Guides Cas protein to complementary target DNA sequence |
| Fluorescent Reporter | ssDNA-FQ reporter (e.g., 5'-FAM-TTATT-BHQ1-3') | Generates fluorescent signal upon Cas12 trans-cleavage |
| Primers | lasB-F: 5'-...3', lasB-R: 5'-...3' | Amplify specific target region during RPA reaction |
Step-by-Step Procedure
crRNA Design and Preparation
- Identify a conserved region within the target lasB gene (GenBank accession no. HQ148672.1) through multiple sequence alignment.
- Design multiple crRNA candidates (typically 3) targeting different regions of the conserved sequence.
- Synthesize crRNA through in vitro transcription using T7 RNA polymerase and purify using RNA purification kits.
- Validate crRNA efficiency through cleavage activity assays with target DNA [18].
RPA Primer Design
- Design RPA primers (forward and reverse, 30-35 bp length) flanking the crRNA target region.
- Avoid regions with secondary structure that may impede amplification efficiency.
- Screen multiple primer pairs using real-time fluorescence monitoring with intercalating dyes (e.g., Syto9/SYBR Green) or product analysis via agarose gel electrophoresis to select the highest efficiency pair [6].
One-Tube Reaction Setup
- Prepare a master mix containing:
- 2.5 μL 10× RPA buffer
- 1.0 μL dNTP mix (10 mM each)
- 1.2 μL forward primer (10 μM)
- 1.2 μL reverse primer (10 μM)
- 2.0 μL crRNA (10 μM)
- 1.0 μL Cas12a enzyme (10 μM)
- 1.0 μL ssDNA-FQ reporter (10 μM)
- 2.0 μL RPA enzyme pellets (commercial formulation)
- Nuclease-free water to 23 μL total volume
- Add 2 μL of extracted DNA or heat-lysed sample (95°C for 5 minutes) to reach a final reaction volume of 25 μL [18].
- Prepare a master mix containing:
Amplification and Detection
- Incubate the reaction tube at 39°C for 40 minutes in a portable fluorescence detector or dry bath.
- Monitor fluorescence in real-time or measure endpoint fluorescence using a portable fluorescence detector, UV light, or blue light transilluminator.
- For Cas12b-based systems, implement a two-temperature protocol: RPA at 39°C for 40 minutes followed by Cas12b activation at 62°C for 5 minutes to enhance detection sensitivity [6].
Result Interpretation
- Positive signal: Significant increase in fluorescence intensity compared to negative controls.
- Visual detection: Clear green fluorescence under blue light (∼470 nm) or UV light (∼365 nm) excitation.
- Quantitative analysis: Calculate target concentration based on standard curve from serial dilutions of known templates using fluorescence intensity values [11].
Molecular Mechanism of RPA-CRISPR/Cas12a Detection
The molecular mechanism of the integrated RPA-CRISPR/Cas12a system operates through two synergistic phases. During the RPA amplification phase, recombinase enzymes form complexes with primers and scan double-stranded DNA for homologous sequences. Upon locating target sequences, the recombinase-primer complexes facilitate strand invasion, while single-stranded DNA binding proteins stabilize the displaced strands. DNA polymerase then extends the primers, exponentially amplifying the target region at a constant temperature of 37-42°C within 10-30 minutes [11] [27].
The detection phase begins when CRISPR-Cas12a, guided by pathogen-specific crRNA, recognizes complementary sequences within the RPA amplicons. Target binding activates both the sequence-specific cis-cleavage activity and the non-specific trans-cleavage activity of Cas12a. The activated enzyme indiscriminately cleaves single-stranded DNA reporters in the reaction mixture, separating fluorophore-quencher pairs and generating a fluorescent signal proportional to the initial target concentration. This collateral cleavage activity enables ultrasensitive detection with single-base specificity, making it ideal for identifying specific biofilm-forming pathogens [11] [16].
Technical Considerations and Optimization
crRNA Design Optimization:
- Design 2-3 crRNAs targeting different regions of the same gene to identify the most efficient guide.
- Ensure target sequences contain appropriate PAM sites (TTTV for LbCas12a).
- Validate specificity through BLAST analysis against non-target genomes.
Temperature Optimization:
- For Cas12b systems, implement two-temperature protocols (RPA at 39°C, Cas12b activation at 62°C) to enhance sensitivity for low-copy targets [6].
- For one-tube reactions, balance temperatures to accommodate both RPA efficiency and Cas enzyme activity.
Sample Preparation:
- Implement extraction-free protocols using simple heat lysis (95°C for 5 minutes) for rapid sample processing.
- Validate sample inhibitors using internal controls in clinical specimens.
The integration of RPA with CRISPR/Cas systems presents a transformative approach for detecting key biofilm-forming pathogens with exceptional sensitivity, specificity, and speed. The one-tube format significantly reduces contamination risk and operational complexity while maintaining analytical performance comparable to traditional culture and PCR methods. This platform offers particular promise for point-of-care diagnostics in resource-limited settings where rapid identification of biofilm-associated infections can guide timely intervention and treatment decisions. Further development of multiplexed detection systems for simultaneous identification of multiple pathogens will enhance the clinical utility of this technology in managing complex biofilm-related infections.
Building a One-Pot RPA-CRISPR Assay: From Primer Design to Pathogen Detection
Step-by-Step Protocol for One-Pot RPA-CRISPR/Cas12a Assay Assembly
This application note provides a detailed protocol for assembling a one-pot Recombinase Polymerase Amplification (RPA) coupled with CRISPR/Cas12a for the detection of biofilm-forming pathogens. Traditional molecular diagnostics for biofilm-associated infections involve complex, multi-step processes that increase contamination risk and require specialized equipment. The one-pot RPA-CRISPR/Cas12a assay integrates nucleic acid amplification and detection in a single tube, enabling rapid, highly sensitive, and specific identification of pathogen DNA at the point of care [29] [31]. This methodology is particularly valuable for detecting pathogens with known genetic markers that are challenging to culture or require urgent clinical intervention.
The core innovation lies in leveraging the simultaneous activities of RPA enzymes and Cas12a/gRNA complexes under isothermal conditions. Following RPA amplification of the target DNA, the Cas12a/gRNA complex binds to specific amplicon sequences, activating its trans-cleavage activity that non-specifically degrades fluorescent reporter probes, generating a detectable signal [29] [32]. Strategic optimization using statistical design of experiments (DoE) has been shown to significantly enhance assay sensitivity, enabling detection of extremely low copy numbers of target nucleic acids [29].
Research Reagent Solutions
Table 1: Essential reagents and materials for one-pot RPA-CRISPR/Cas12a assay
| Reagent/Material | Function/Purpose | Example Specifications |
|---|---|---|
| RPA Basic Kit | Isothermal amplification of target DNA sequence | TwistAmp Basic (TwistDx) [29] |
| Cas12a Nuclease | Target-specific binding and trans-cleavage of reporter | EnGen Lba Cas12a (NEB) [29] [32] |
| crRNA/gRNA | Guides Cas12a to complementary target DNA sequence | Synthesized, e.g., from IDT [29] |
| ssDNA-FQ Reporter | Fluorescent signal generation upon Cas12a cleavage | 6-FAM-labeled ssDNA quenched with BHQ-1 [29] |
| Primers | Specific amplification of the target pathogen gene | Designed for target (e.g., P. sojae PHYSODRAFT_299276) [32] |
| NEBuffer 2.1 | Provides optimal ionic conditions for Cas12a activity | 1X final concentration [29] |
| Murine RNase Inhibitor | Protects gRNA from degradation in one-pot reactions | 40 U/μL (New England Biolabs) [29] |
Experimental Protocol
Reagent Preparation
- Cas12a/gRNA RNP Complex Pre-formation: Mix Cas12a nuclease, gRNA1, and gRNA2 (targeting two different regions on the template for enhanced signal) to a final concentration of 10 μM each in 1× NEBuffer 2.1 [29]. Incubate the mixture at room temperature for 15 minutes to form the ribonucleoprotein (RNP) complex. Aliquot and store at -20°C until use.
- Master Mix Preparation: Prepare the RPA master mix on ice according to the manufacturer's instructions (TwistAmp Basic). For a single 50 μL reaction, combine 29.5 μL of rehydration buffer, 2.4 μL of forward primer (10 μM), 2.4 μL of reverse primer (10 μM), and nuclease-free water to a final volume of 42.2 μL [29] [32].
- Additive Incorporation: To the master mix, add the pre-formed Cas12a/gRNA RNP complex (final concentration 400 nM), the ssDNA-FQ reporter (e.g., 200 nM), and critical additives identified via DoE optimization, such as a reverse transcription buffer and murine RNase inhibitor (40 U/μL) if detecting RNA targets [29].
One-Pot Assay Assembly and Execution
- Reaction Initiation: Distribute the complete master mix into individual reaction tubes. Add the template DNA (or RNA for RT-RPA) and manually mix the contents by pipetting. To initiate the RPA reaction, add 2.5 μL of 280 mM magnesium acetate (provided in the RPA kit) directly into the tube lid, then briefly centrifuge to mix [29].
- Isothermal Incubation: Immediately place the tubes in a pre-heated incubator or thermal block at 39°C for 30-40 minutes. Manual mixing during incubation, as recommended by some RPA kit suppliers, may be beneficial and should be tested [29].
- Signal Detection: Following incubation, results can be read using multiple methods:
- Fluorometer: Measure fluorescence intensity (e.g., FAM channel) in real-time or at end-point [29].
- Lateral Flow Strips: For visual detection, apply the reaction product to a lateral flow strip. The cleaved reporter is captured on the test line, producing a visible band [32].
- UV Light: Visualize fluorescence by exposing the tube to blue or UV light [29].
Critical Assay Performance Data
Table 2: Analytical performance of optimized one-pot RPA-CRISPR/Cas12a assays
| Target | Limit of Detection (LoD) | Assay Time | Key Optimized Parameters | Citation |
|---|---|---|---|---|
| Full-length SARS-CoV-2 RNA | 2 copies/μL | ~30-40 min | Addition of RT buffer & RNase inhibitor; DoE-optimized RT step [29] | [29] |
| Synthetic COVID-19 N gene (DNA) | 0.5 copies/μL | ~30-40 min | Template-specific optimization of reaction conditions [29] | [29] |
| Monkeypox Virus (MPXV) DNA | 2.5 copies/test | 40 min | Use of Cas12b and ssRNA blocker for thermal regulation (TRACE assay) [31] | [31] |
| Phytophthora sojae DNA | 10 pg/μL | < 1 hour | Specific crRNA design for target gene PHYSODRAFT_299276 [32] | [32] |
Troubleshooting and Optimization Guidelines
The one-pot nature of this assay introduces complexity, as multiple enzymatic reactions occur simultaneously. Successful implementation often requires careful optimization.
- Balancing Amplification and Detection: A key challenge is that Cas12a activity can deplete RPA amplicons, reducing amplification efficiency and overall sensitivity [31]. Strategies to mitigate this include:
- Thermal Regulation: Using Cas12b instead of Cas12a, as Cas12b has a higher temperature optimum. This allows for an initial lower temperature phase (37°C) favoring RPA, followed by a higher temperature phase (60°C) to activate Cas12b detection [31].
- gRNA Blocking: Adding a single-stranded RNA (ssRNA) blocker complementary to the gRNA spacer during the RPA phase. This inhibitor dissociates at higher temperatures, restoring Cas12 activity for the detection phase [31].
- Statistical Design of Experiments (DoE): Employ a Definitive Screening Design (DSD) to efficiently identify factors with a significant effect on performance (e.g., primer/probe concentrations, incubation time, temperature, additive concentrations). This approach accelerates optimization compared to traditional one-factor-at-a-time methods [29].
- Specificity and Internal Controls: Ensure crRNAs are designed to target unique, conserved regions of the pathogen genome to avoid cross-reactivity [32]. For clinical translation, incorporate a host gene or synthetic internal control in a duplex reaction to monitor sample quality and reaction validity, addressing a key limitation of many CRISPR assays [31].
Within the framework of developing one-pot RPA-CRISPR diagnostics, the precise selection of genetic targets is paramount. This document provides detailed application notes and protocols for identifying and validating highly conserved genetic targets in two major biofilm-forming pathogens, Pseudomonas aeruginosa and Staphylococcus aureus. The success of a diagnostic assay hinges on its ability to reliably detect diverse strains of these pathogens by targeting stable, essential genes involved in biofilm formation and pathogenicity. The protocols herein are designed for researchers and drug development professionals seeking to build robust, sequence-specific detection systems using RPA-CRISPR platforms.
Conserved Genetic Targets in Major Biofilm Pathogens
Target selection prioritizes genes that are universally present across strains, exhibit low sequence variability, and play fundamental roles in biofilm establishment and maintenance. The following tables summarize key conserved targets for each pathogen.
Table 1: Conserved Biofilm-Related Targets in Pseudomonas aeruginosa
| Gene/Element | Function/Description | Role in Biofilm Formation | Conservation & Notes |
|---|---|---|---|
cupA gene cluster [33] |
Encodes fimbriae via chaperone-usher pathway. | Critical for initial attachment to abiotic surfaces. [33] | Highly conserved; expression repressed by MvaT regulator. [33] |
oprF [34] |
Outer membrane porin. | Essential for biofilm stimulation by sub-inhibitory antibiotics; key in stress response. [34] | Highly conserved; homolog (OmpA) plays a similar role in E. coli. [34] |
sigX [34] |
Extracytoplasmic function (ECF) sigma factor. | Regulates response to cell envelope stress; required for antibiotic-induced biofilm stimulation. [34] | Part of a conserved regulatory pathway. |
pel and psl [34] |
Biosynthesis of Pel and Psl polysaccharides. | Major structural polysaccharides in biofilm matrix; act as adhesins. [34] | Core components of the extracellular polymeric substance (EPS). |
PA2146 [35] |
Small, previously uncharacterized hypothetical protein. | Contributes to biofilm architecture and antimicrobial tolerance; transcript abundance increases >70-fold in biofilms. [35] | Highly conserved in ɣ-proteobacteria; homologs in K. pneumoniae and E. coli complement P. aeruginosa mutant. [35] |
Table 2: Conserved Biofilm-Related Targets in Staphylococcus aureus
| Gene/Element | Function/Description | Role in Biofilm Formation | Conservation & Notes |
|---|---|---|---|
icaADBC operon [36] |
Encodes enzymes for synthesis of Poly-N-acetylglucosamine (PNAG/PIA). | Produces primary polysaccharide for cell-cell adhesion and biofilm matrix accumulation. [36] | The major mechanism for polysaccharide-based biofilm formation; subject to phase variation. [36] |
fnbA/B [36] |
Encodes fibronectin-binding proteins A and B. | Mediates initial attachment to host tissues and biomaterial surfaces. [36] | Classified as MSCRAMMs (Microbial Surface Components Recognizing Adhesive Matrix Molecules). |
bap (Biofilm-associated protein) [36] |
Large surface protein containing 2276 amino acids. | Promotes initial adhesion and intercellular adhesion via a polysaccharide-independent mechanism. [36] | Strong correlation exists between Bap presence and strong biofilm formation on abiotic surfaces. [36] |
cifA/B (Clumping factors) [36] |
Fibrinogen-binding proteins. | Mediate bacterial adhesion to natural tissues and biomaterial surfaces. [36] | Classified as MSCRAMMs. |
Pathway Diagram: Biofilm Stimulation by Sub-MIC Antibiotics inP. aeruginosa
The following diagram illustrates a key signaling pathway identified in P. aeruginosa that translates antibiotic stress into enhanced biofilm formation, integrating several of the conserved targets listed above.
Experimental Protocol: Validation of Conserved Targets for RPA-CRISPR Assay Development
This protocol outlines the key steps for validating the specificity and conservation of selected genetic targets prior to developing a one-pot RPA-CRISPR diagnostic assay.
Stage 1: In Silico Analysis and gRNA Design
Objective: To confirm target conservation and design highly specific guide RNAs (gRNAs) for the CRISPR detection system.
Materials:
- Bioinformatics Software: BLAST, Clustal Omega, Geneious Prime or equivalent.
- Genomic Databases: NCBI Nucleotide, PATRIC, Pseudomonas Genome Database, Staphylococcus aureus Typing.
- gRNA Design Tools: CHOPCHOP, CRISPRscan, or Cas-specific designer (e.g., for Cas12a/Cas13a).
Procedure:
- Target Sequence Retrieval: Download complete nucleotide sequences of the target gene (e.g.,
oprF,icaA) from a diverse panel of clinically relevant P. aeruginosa and S. aureus strains. - Multiple Sequence Alignment: Perform a multiple sequence alignment to identify conserved regions suitable for gRNA binding. The target region must be invariant across >99% of tested strains.
- gRNA Selection and Off-Target Screening:
- Design 3-5 candidate gRNAs (typically 20-24 nt) targeting the identified conserved region.
- The gRNA sequence must be specific to the target pathogen. Perform a whole-genome BLAST of the candidate gRNA sequences against the human genome and against genomes of common commensal flora to ensure no significant off-target matches exist.
- Select the final gRNA candidate based on high on-target efficiency scores and minimal predicted off-target effects.
Stage 2: Analytical Specificity and Sensitivity Testing
Objective: To experimentally verify that the RPA-CRISPR system specifically detects the intended target across various strains and exhibits high sensitivity.
Materials:
- Reagent Solutions: See "The Scientist's Toolkit" below.
- Strains: A collection of target pathogen strains (e.g., 10-20 different clinical isolates of P. aeruginosa and S. aureus) and non-target bacterial species for specificity testing.
- Equipment: Real-time PCR machine or fluorescence plate reader, heat block or water bath maintained at 37-42°C.
Procedure:
- Nucleic Acid Extraction: Purify genomic DNA from all bacterial strains to be tested using a commercial kit.
- One-Pot RPA-CRISPR Reaction Setup:
- Prepare a master mix for the one-pot reaction as described in the referenced literature [37]. A typical 50 µL reaction contains:
- 29.5 µL Nuclease-free water
- 2.5 µL each RPA forward and reverse primer (10 µM)
- 2.5 µL Cas13a/Cas12a protein (100-200 nM)
- 2.5 µL designed gRNA (100-200 nM)
- 2.5 µL fluorescent reporter (e.g., 500 nM FQ-ssRNA for Cas13a)
- 5.0 µL 10X RPA rehydration buffer
- Aliquot the master mix into reaction tubes. Add 1-5 µL of template DNA (or a synthetic gene fragment for LOD determination) to each tube.
- Initiate the reaction by adding 2.5 µL of 280 mM Magnesium Acetate, briefly centrifuging, and immediately placing it in the detector.
- Prepare a master mix for the one-pot reaction as described in the referenced literature [37]. A typical 50 µL reaction contains:
- Data Acquisition and Analysis:
- Incubate the reaction at 37-42°C for 30-60 minutes while monitoring fluorescence in real-time.
- For endpoint detection, use lateral flow strips after the incubation period [37].
- Specificity: The assay should produce a positive signal only for the target pathogen strains and show no cross-reactivity with non-target species.
- Limit of Detection (LOD): Perform a standard curve using a serially diluted target DNA template (e.g., from 10^6 to 1 copy/µL). The LOD is defined as the lowest concentration at which 95% of the replicates test positive. Well-optimized one-pot RPA-CRISPR assays can achieve LODs as low as 5-10 copies per reaction [37].
The Scientist's Toolkit
Table 3: Essential Research Reagents for RPA-CRISPR Assay Development
| Reagent / Material | Function / Description | Example / Notes |
|---|---|---|
| RPA Kit | Isothermal amplification of target DNA sequence. | TwistAmp Basic kit or equivalent. Provides enzymes and buffers for recombinase polymerase amplification. |
| Cas Protein | CRISPR effector; provides programmable detection. | Cas13a (for RNA detection) or Cas12a (for DNA detection). Catalyzes target-specific and collateral cleavage. |
| In Vitro Transcripted gRNA | Guides Cas protein to the specific target sequence. | Synthesized via in vitro transcription from a DNA template. Must be designed against conserved region. |
| Fluorescent Reporter | Signal generation for real-time detection. | For Cas13a: FQ-ssRNA (e.g., polyU- reporter with 5' fluorophore, 3' quencher). Signal increases upon collateral cleavage. |
| Lateral Flow Strips | Endpoint, visual readout of the assay. | Milenia HybriDetect strips or equivalent. Used with labeled reporters (e.g., FAM/Biotin) for yes/no results. |
| Synthetic Gene Fragments | Positive control and for determining LOD. | gBlock Gene Fragments containing the entire target region. Essential for quantitative validation without culture. |
crRNA Design Strategies for Maximum Specificity and Sensitivity
1. Introduction
Within the broader thesis on developing one-pot RPA-CRISPR diagnostics for biofilm pathogen detection, the design of the CRISPR RNA (crRNA) is the single most critical factor determining assay success. Biofilm-derived pathogens present unique challenges, including complex sample matrices and genetic heterogeneity. This application note details proven strategies for designing crRNAs that achieve maximum specificity to avoid off-target cleavage and maximum sensitivity for detecting low-abundance targets from difficult samples.
2. Key Design Parameters and Quantitative Guidelines
The following parameters must be optimized during the in silico design phase.
Table 1: crRNA Design Parameters for Specificity and Sensitivity
| Parameter | Objective | Optimal Value / Strategy | Rationale |
|---|---|---|---|
| Spacer Length | Sensitivity & Kinetics | 30-32 nt for Cas12a; 20 nt for Cas12b | Balances binding energy for rapid kinetics and sufficient specificity. |
| GC Content | Specificity & Secondary Structure | 40-60% | Prevents overly stable G-C pairs that promote off-target binding and minimizes spacer secondary structure. |
| Off-Target Screening | Specificity | BLASTn against host & microbiome genome; ≤3 nt mismatch in seed region (PAM-distal 5-8 nt) | The seed region is critical for initial recognition; mismatches here drastically reduce off-target effects. |
| Poly-T Sequences | Sensitivity | Avoid stretches of ≥4 T nucleotides | Can cause premature transcription termination in T7-based crRNA synthesis. |
| PAM Proximity | Sensitivity | Select spacer 1-5 nt downstream of PAM (e.g., TTTV for LbaCas12a) | Cas effector binding and activation is most efficient close to the PAM site. |
| Secondary Structure | Sensitivity | Minimize spacer self-complementarity & hairpins (ΔG > -5 kcal/mol) | Structured spacers are less accessible for guide:target hybridization, impairing cleavage. |
Table 2: Comparison of Cas Effectors for One-Pot RPA-CRISPR Diagnostics
| Feature | Cas12a (e.g., LbaCas12a) | Cas12b (e.g., AaCas12b) | Cas13a (e.g., LwaCas13a) |
|---|---|---|---|
| PAM Requirement | T-rich (TTTV) | T-rich (TTN) | None (targets RNA) |
| Pre-crRNA Processing | Self-processing | Requires mature crRNA | Self-processing |
| Trans-Cleavage Substrate | ssDNA (non-specific) | ssDNA (non-specific) | ssRNA (non-specific) |
| Optimal Temperature | ~37°C | ~48°C | ~37°C |
| Suitability for One-Pot | High (compatible with RPA ~37-42°C) | Medium (requires a two-step protocol) | High (compatible with RT-RPA ~37-42°C) |
3. Experimental Protocol: crRNA Design and Validation
This protocol outlines the steps from target selection to in vitro validation of crRNA performance.
Protocol 3.1: In Silico crRNA Design and Screening
Materials:
- Target genomic sequence (e.g., icaA gene for S. aureus biofilm)
- NCBI BLAST suite
- crRNA design software (e.g., CHOPCHOP, CRISPRscan) or custom scripts
- NUPACK or mFold for secondary structure prediction
Method:
- Identify Target Region: Select a ~200-300 bp unique and conserved region from the pathogen's genome, specific to the biofilm phenotype (e.g., a segment of the pslA gene for P. aeruginosa).
- PAM Scanning: Scan the target strand for all available PAM sites compatible with your chosen Cas effector (e.g., "TTTV" for LbaCas12a).
- Generate Spacer Candidates: For each PAM, extract the ~20-32 nt spacer sequence directly 5' to the PAM on the target strand.
- Filter by GC Content: Eliminate candidates with GC content <40% or >60%.
- Screen for Poly-T: Eliminate candidates containing 4 or more consecutive T nucleotides.
- Predict Secondary Structure: Analyze the remaining spacer sequences. Discard any with a predicted folding free energy (ΔG) more stable than -5 kcal/mol.
- Comprehensive Off-Target Analysis:
- Perform a BLASTn search of each spacer against the host genome (e.g., human) and a database of common commensal microbiome genomes.
- Manually inspect hits. Reject any spacer with a near-perfect match (≤3 mismatches, especially in the PAM-distal 5-8 nt seed region) to a non-target genome.
Protocol 3.2: In Vitro Validation of crRNA Efficacy
Materials:
- Synthesized candidate crRNAs (with direct T7 promoter sequence)
- Purified Cas effector protein (e.g., LbaCas12a)
- Synthetic target DNA amplicon (from RPA or PCR)
- Non-target DNA amplicon (for specificity testing)
- Fluorescent reporter (e.g., FQ-ssDNA reporter for Cas12a: 5'-/6-FAM/TTATT/3IABkFQ/-3')
- Real-time PCR instrument or plate reader
Method:
- Prepare Reaction Mix (20 µL):
- 1x NEBuffer 2.1 (or Cas-specific buffer)
- 50-100 nM purified Cas effector
- 50-100 nM candidate crRNA
- 100-200 nM FQ-ssDNA reporter
- Nuclease-free water to 19 µL
- Baseline Measurement: Incubate the mix at 37°C for 5-10 minutes in a real-time qPCR instrument with FAM channel acquisition every minute.
- Activation: Add 1 µL of target DNA amplicon (10-100 fM final concentration) to initiate the reaction.
- Data Collection: Continue fluorescence measurement for 60-90 minutes.
- Specificity Test: Repeat steps 1-4 using a non-target DNA amplicon (e.g., from a related non-biofilm forming strain).
- Data Analysis: Calculate the time-to-threshold (Tt) or the initial rate of fluorescence increase (slope). The crRNA with the fastest kinetics (lowest Tt, highest slope) with the target and no signal with the non-target is the lead candidate.
4. Research Reagent Solutions
Table 3: Essential Reagents for crRNA-Cas Diagnostic Development
| Reagent / Material | Function | Example Product / Note |
|---|---|---|
| LbaCas12a Protein | CRISPR effector for target recognition & trans-cleavage | Purified in-house or commercially sourced (e.g., from IDT). |
| T7 RNA Polymerase | In vitro transcription of crRNAs from DNA templates. | High-yield enzyme for robust crRNA production. |
| RPA Kit | Isothermal amplification of target DNA. | TwistAmp Basic kit for one-pot integration. |
| Fluorescent Reporter | Detection of trans-cleavage activity. | 5'-6-FAM/TTATT/3IABkFQ/-3' (ssDNA for Cas12). |
| Nucleic Acid Cofactor | Enhances Cas12a trans-cleavage kinetics. | Poly dT(30) or similar, used as a "activator DNA". |
| RNase Inhibitor | Protects synthesized crRNA from degradation. | Essential for maintaining assay sensitivity in complex buffers. |
5. Visualization of Workflows and Relationships
crRNA Design and Screening Workflow
One-Pot RPA-CRISPR Assay Workflow
The integration of Recombinase Polymerase Amplification (RPA) with CRISPR/Cas systems has revolutionized molecular diagnostics by enabling rapid, sensitive, and specific detection of pathogenic nucleic acids at constant temperatures. This combination is particularly valuable for detecting biofilm-forming pathogens in food processing environments and clinical settings, where timely identification is crucial for intervention [11] [12]. A fundamental decision in developing these assays lies in choosing between one-pot and two-step workflows, each presenting distinct trade-offs between convenience and performance.
In a two-step approach, the RPA amplification and CRISPR detection reactions are physically separated and performed sequentially. This method prevents interference between the enzymatic components but increases operational complexity and contamination risk [31]. In contrast, one-pot assays integrate both reactions within a single tube, simplifying the workflow and reducing contamination potential but potentially compromising sensitivity due to enzymatic competition [6] [38]. Understanding these trade-offs is essential for researchers and developers aiming to implement RPA-CRISPR diagnostics for precision biofilm control and pathogen detection.
This application note provides a systematic comparison of these workflows, detailing their mechanisms, performance characteristics, and optimized protocols to guide researchers in selecting the appropriate format for specific diagnostic applications.
Workflow Mechanisms and Comparative Analysis
Fundamental Principles of RPA-CRISPR Systems
RPA is an isothermal nucleic acid amplification technique that operates at 37-42°C, utilizing three core enzymes: a recombinase that facilitates primer binding to homologous sequences, single-stranded DNA-binding proteins that stabilize displaced strands, and a strand-displacing DNA polymerase that extends the primers [11]. This rapid amplification process (typically 15-30 minutes) generates target DNA sequences that subsequently activate CRISPR/Cas systems.
The CRISPR/Cas system, particularly Cas12a and Cas12b, provides sequence-specific detection through CRISPR RNA (crRNA)-guided recognition of target DNA. Upon binding to its target sequence, Cas12a exhibits both specific cis-cleavage of the target DNA and non-specific trans-cleavage activity, indiscriminately degrading single-stranded DNA reporters in the reaction mixture [11] [16]. This collateral cleavage enables signal generation through the release of fluorescent molecules or visualization on lateral flow strips [3].
Performance Comparison: One-Pot vs. Two-Step Workflows
The table below summarizes the key characteristics of one-pot and two-step RPA-CRISPR workflows based on current research findings:
Table 1: Performance Comparison of One-Pot vs. Two-Step RPA-CRISPR Workflows
| Parameter | One-Pot Workflow | Two-Step Workflow |
|---|---|---|
| Sensitivity | 10-100 copies/μL [6] [38]; Can be improved to 2.5 copies/test with optimization [31] | 2.5 copies/test (higher sensitivity) [31] |
| Assay Time | 20-60 minutes [38] [6] | 40-90 minutes (including transfer time) [31] |
| Contamination Risk | Lower (closed system) [39] | Higher (open tube transfer required) [31] |
| Operational Complexity | Simplified (minimal handling) [6] | Higher (manual transfer between steps) [31] |
| Equipment Needs | Compatible with simple incubators [38] | May require brief centrifugation [38] |
| Multiplexing Potential | Developing with temperature regulation [31] | Established with sequential reactions [3] |
| Key Challenges | Enzyme interference reduces sensitivity [31] | Aerosol contamination during transfer [38] |
Figure 1: Workflow comparison between one-pot and two-step RPA-CRISPR assays. The one-pot method maintains a closed-tube system, while the two-step approach requires manual transfer after amplification.
Optimized Protocols for Pathogen Detection
Two-Step RPA-CRISPR/Cas12a Protocol for MRSA Detection
This protocol demonstrates a specific application for detecting methicillin-resistant Staphylococcus aureus (MRSA), a biofilm-forming pathogen of significant clinical concern [38].
Reagent Preparation:
- RPA reaction mix (TwistAmp Liquid Basic kit)
- Primers targeting MRSA mecA gene (10 μM each)
- CRISPR/Cas12a system: LbaCas12a (1 μM), crRNA (2 μM) specific to mecA gene
- Fluorescent reporter: ssDNA FQ Reporter (500 nM)
- 10× NEBuffer 2.1
- RNA inhibitor
Procedure:
- RPA Amplification (Bottom of Tube):
- Prepare 25 μL RPA reaction containing:
- 10 μL of 2× Reaction Buffer
- 1 μL of dNTPs
- 2 μL of 10× E-mix
- 1 μL each of forward and reverse RPA primers
- 1 μL of 20× Core Reaction mix
- 2 μL of template DNA
- 2 μL of MgOAc (added last to initiate reaction)
- Incubate at 42°C for 10-15 minutes
- Prepare 25 μL RPA reaction containing:
CRISPR Detection (Tube Lid):
- During RPA incubation, prepare CRISPR mix in tube lid:
- 1 μL LbaCas12a (1 μM)
- 1 μL crRNA (2 μM)
- 0.25 μL RNA inhibitor
- 1 μL ssDNA FQ Reporter
- 2 μL 10× NEBuffer 2.1
- 4.75 μL nuclease-free water
- During RPA incubation, prepare CRISPR mix in tube lid:
Reaction Integration:
- After RPA amplification, briefly centrifuge tube to mix CRISPR reagents with RPA products
- Immediately incubate at 42°C for 10-15 minutes on a real-time PCR instrument
- Monitor fluorescence every minute or perform endpoint detection with UV illuminator
Performance Characteristics:
- Sensitivity: 10 copies/μL with fluorescence detection [38]
- Specificity: 100% concordance with qPCR for clinical MRSA isolates [38]
- Total assay time: 20-30 minutes
Thermally Programmed One-Pot RPA-CRISPR/Cas12b Protocol
This protocol employs temperature regulation to minimize interference between RPA and CRISPR reactions, enhancing one-pot assay sensitivity [6] [31].
Reagent Preparation:
- RPA reaction mix (TwistAmp Basic kit)
- AapCas12b enzyme (thermostable Cas12b variant)
- crRNA specific to target pathogen (e.g., Group B Streptococcus cfb gene)
- ssRNA blocker complementary to crRNA spacer region
- Poly-T fluorescent reporter (400 nM)
- 10× isothermal amplification buffer
Procedure:
- One-Pot Reaction Setup:
- Prepare 25 μL reaction containing:
- Complete RPA reaction components (as in section 3.1)
- 400 nM AapCas12b RNP (pre-complexed crRNA:Cas12b)
- 1.6 μM ssRNA blocker (1:4 ratio to gRNA)
- 400 nM poly-T fluorescent reporter
- Mix gently and avoid premature activation
- Prepare 25 μL reaction containing:
Thermally Programmed Incubation:
- Stage 1 (Amplification): Incubate at 39°C for 40 minutes
- ssRNA blocker inhibits Cas12b activity during RPA amplification
- Stage 2 (Detection): Increase temperature to 62°C for 5-10 minutes
- Higher temperature dissociates ssRNA blocker, activating Cas12b trans-cleavage
- Fluorescent signal generation indicates target detection
- Stage 1 (Amplification): Incubate at 39°C for 40 minutes
Signal Detection:
- Visualize under UV/blue light (qualitative)
- Use fluorometer for quantitative measurement
- Apply to lateral flow strips for point-of-care applications
Performance Characteristics:
- Sensitivity: 10 copies/test for Group B Streptococcus [6]
- Assay time: <60 minutes total [6]
- Clinical concordance: 96.7% vs. culture, 98.3% vs. qPCR [6]
Table 2: Optimization Strategies for Enhanced One-Pot Assay Performance
| Strategy | Mechanism | Performance Improvement |
|---|---|---|
| Two-Temperature Protocol | Spatial separation of RPA (39°C) and Cas12b activation (62°C) | Significantly improved low-copy detection [6] |
| ssRNA Blockers | Complementary RNA sequences inhibit Cas12b during amplification | 40x lower LOD (2.5 copies/test) [31] |
| RNP Concentration Optimization | Balancing amplification and detection efficiency | Improved signal-to-noise ratio [31] |
| Reporter Engineering | Poly-T reporters instead of mixed-sequence | Enhanced fluorescence signal [31] |
| crRNA Design | Targeting suboptimal PAM sites | Reduced interference with amplification [6] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for RPA-CRISPR Assay Development
| Reagent | Function | Examples & Specifications |
|---|---|---|
| RPA Kits | Isothermal amplification of target DNA | TwistAmp Liquid Basic Kit (TwistDx) [38] |
| Cas Enzymes | Target-specific recognition and trans-cleavage | LbaCas12a (NEB), AapCas12b (thermostable) [6] [38] |
| crRNA | Guides Cas enzyme to specific target sequence | In vitro transcribed with T7 promoter [38] |
| Fluorescent Reporters | Signal generation via trans-cleavage | ssDNA FQ Reporter (FAM-TTATT-BHQ1) [38] |
| ssRNA Blockers | Temporarily inhibit Cas activity in one-pot assays | Complementary to crRNA spacer region [31] |
| Lateral Flow Strips | Point-of-care visual detection | Milenia HybriDetect strips [38] |
Figure 2: Essential reagents and their roles in RPA-CRISPR assay systems. Dashed lines indicate which components are added at the reaction setup stage.
The choice between one-pot and two-step RPA-CRISPR workflows depends on the specific application requirements and available resources. Two-step protocols currently offer superior sensitivity (as low as 2.5 copies/test) and are recommended for applications requiring maximum detection capability, such as identifying low-abundance biofilm pathogens in food safety monitoring [31]. One-pot assays provide operational simplicity and reduced contamination risk, making them ideal for point-of-care testing and resource-limited settings, particularly when optimized with thermal programming and blocking agents [6] [31].
For biofilm pathogen detection specifically, where sensitivity is often critical for early intervention, researchers should consider implementing a two-step approach for initial assay development and validation, then transitioning to optimized one-pot formats with temperature regulation and ssRNA blockers once the target sensitivity requirements are met. The ongoing innovation in CRISPR enzyme engineering, reagent formulation, and instrumentation will likely continue to narrow the performance gap between these workflows, making one-pot assays increasingly capable for the precise detection and control of biofilm-forming pathogens in both clinical and industrial settings.
The detection and analysis of biofilm-forming pathogens are critical in clinical diagnostics and antimicrobial resistance surveillance. Signal readout methods form the bridge between complex molecular recognition events and interpretable results. Within the context of one-pot RPA-CRISPR diagnostics for biofilm pathogen detection, selecting an appropriate readout method directly impacts assay sensitivity, specificity, usability, and applicability in point-of-care settings. This document details three principal readout methodologies—fluorescence, lateral flow strips, and naked-eye visualization—providing application notes and experimental protocols tailored for researchers developing diagnostic platforms for biofilm pathogens.
Core Readout Methodologies
Lateral Flow Assays (LFAs)
Principle and Components: Lateral flow assays are paper-based platforms that detect analytes via capillary action without external forces [40]. A typical LFA strip consists of overlapping membranes mounted on a backing card: a sample pad, conjugate pad, porous nitrocellulose membrane (detection zone), and absorbent pad [40]. The sample pad is impregnated with buffer salts and surfactants to prepare the sample for optimal interaction with subsequent components [40]. The conjugate pad contains detection agents (e.g., antibody-conjugated nanoparticles), which resuspend as the sample migrates through. The detection membrane has immobilized capture molecules (e.g., antibodies or antigens) in test and control lines. The absorbent pad wicks excess fluid to maintain continuous flow [40].
Formats and Signaling: LFAs primarily operate in two formats: sandwich (for larger analytes with multiple epitopes) and competitive (for small molecules with single epitopes) [41]. In sandwich assays, the signal intensity at the test line increases proportionally with target concentration, making results intuitively interpretable [41]. In competitive formats, the target analyte competes with a labeled competitor for binding sites, resulting in decreased test line signal with increasing target concentration [41]. This inverse relationship can be less intuitive but is necessary for detecting small molecules.
Detection Labels and Signal Amplification: While colloidal gold is the most widely used label for colorimetric detection due to its intense color and stability [40], recent advancements have incorporated fluorescent microspheres (FMs) [42], quantum dots [40], and other nanomaterials to enhance sensitivity. Laser-induced signal amplification techniques that exploit the plasmonic properties of gold nanoparticles (GNPs), such as surface-enhanced Raman scattering (SERS), photothermal, and photoacoustic methods, can significantly lower detection limits without modifying the LFA architecture [43]. These approaches generate stronger signals than colorimetric readouts and enable precise quantification when paired with appropriate reader systems [43].
Fluorescence Detection
Principle and Applications: Fluorescence-based detection relies on emitting light at a specific wavelength after exciting a fluorophore at a lower wavelength. This method offers high sensitivity and is amenable to quantification and multiplexing. In biofilm research, fluorescence is widely employed for spatial organization analysis through techniques like fluorescence in situ hybridization (FISH) [44] and differential staining fluorescence microscopy (DSFM) [45]. FISH uses fluorescently labeled oligonucleotide probes to target specific microbial sequences, allowing identification, quantification, and spatial localization of pathogens within complex biofilm structures without disrupting their 3D architecture [44].
Advanced Fluorescence Imaging: DSFM is a powerful tool for tracking enteric pathogens within mixed-species biofilms [45]. This protocol involves staining pathogens with a cell-permeant dye (e.g., BacLight Red Stain) and counter-staining the entire biofilm with a general biofilm stain (e.g., FM-1-43) before visualization using fluorescence microscopy [45]. This differential staining allows researchers to distinguish specific pathogens from the background biofilm matrix and precisely determine their location within the biofilm structure, which is crucial for understanding pathogen persistence and designing effective interventions [45].
Naked-Eye Visualization
Direct Colorimetric Readout: The simplest readout method involves direct visual interpretation of color changes or line appearances without instrumentation. In LFA, this typically relies on the accumulation of colored nanoparticles, such as colloidal gold, at test and control lines [40]. The control line must always appear to validate the test; a positive result in a sandwich assay is indicated by the appearance of both test and control lines [40]. This method is highly user-friendly and low-cost but generally offers lower sensitivity compared to instrumental methods and is typically semi-quantitative at best.
Enhancement Strategies: To improve the visibility and sensitivity of naked-eye readouts, strategies include using high-contrast labels (e.g., carbon nanoparticles which appear black against the white nitrocellulose background) [40], enzymatic reactions that generate colored products [43], and metallic enhancement techniques that catalytically deposit silver onto gold nanoparticles, amplifying the signal [43].
Quantitative Comparison of Readout Methods
The table below summarizes the key performance characteristics and implementation requirements of the three signal readout methods.
Table 1: Quantitative Comparison of Signal Readout Methods
| Parameter | Lateral Flow (Colorimetric) | Lateral Flow (Fluorescent) | Fluorescence Microscopy | Naked-Eye (Direct) |
|---|---|---|---|---|
| Typical LOD | ~ng/mL (can reach pg/mL with enhancements) [43] | ~pg/mL [42] | Single cell [45] | ~ng/mL [40] |
| Quantification | Semi-quantitative (reader enables quantitative) [43] [40] | Yes, with reader [42] | Yes (e.g., biovolume measurement) [45] | Semi-quantitative/Qualitative [40] |
| Multiplexing Potential | Moderate (multiple test lines) [40] [41] | High (different fluorescent labels) [40] | High (multiple fluorophores) [44] | Low |
| Assay Time | 5-30 minutes [40] | 5-30 minutes (plus reader time) [42] | Minutes to hours (including staining) [45] | 5-30 minutes [40] |
| Instrumentation Required | No (for visual readout) [40] | Yes (fluorescence reader) [42] | Yes (microscope) [45] | No |
| Cost | Low | Moderate | High | Very Low |
| Key Applications | Point-of-care diagnostics, food safety [46] [40] | Sensitive POC, quantitative analysis [42] | Biofilm structure analysis, pathogen localization [45] [44] | Rapid screening, home testing [40] |
Experimental Protocols
Protocol: FluorescenceIn SituHybridization (FISH) for Biofilm Pathogens
This protocol enables the identification and spatial localization of specific pathogens within a multi-species biofilm using FISH [44].
Research Reagent Solutions:
- Hybridization Buffer: Contains formamide, salts, detergents, and blocking reagents to create optimal conditions for probe binding.
- Washing Buffer: Saline-sodium citrate (SSC) buffer to remove non-specifically bound probes.
- FISH Probes: Fluorescently labeled oligonucleotide probes targeting species-specific 16S rRNA sequences.
- Mounting Medium: A medium to preserve fluorescence and prepare the sample for microscopy.
Procedure:
- Biofilm Fixation: Grow biofilms on a suitable substrate (e.g., glass coverslip). Apply fixative (e.g., 4% paraformaldehyde) for 1-4 hours at 4°C to preserve the 3D structure and permeability cells. Wash with phosphate-buffered saline (PBS) [44].
- Hybridization: Apply a hybridization buffer containing the specific fluorescent FISH probe to the fixed biofilm sample. Incubate in a dark, humidified chamber at the appropriate temperature (e.g., 46°C for 90 minutes) to allow the probe to penetrate cells and bind to the target rRNA [44].
- Washing: Immerse the sample in a pre-warmed washing buffer to remove excess and unbound probes. Incubate for 10-30 minutes at 48°C [44].
- Drying and Mounting: Gently air-dry the sample in the dark. Apply a drop of antifading mounting medium and carefully place a coverslip over the sample [44].
- Imaging and Analysis: Visualize the biofilm using a confocal laser scanning microscope (CLSM). Acquire Z-stack images to reconstruct the 3D structure. Use image analysis software to quantify the biovolume and spatial distribution of the target pathogen [44].
Protocol: PCR-Lateral Flow Strip (PCR-LFS) Detection
This protocol couples nucleic acid amplification with LFA for highly specific detection of pathogens or resistance genes, such as blaCTX-M in E. coli and Salmonella [47].
Research Reagent Solutions:
- Primers: Forward primer labeled with FITC and reverse primer labeled with biotin.
- PCR Master Mix: Contains DNA polymerase, dNTPs, and buffer.
- Lateral Flow Strip: Strip with a test line coated with anti-FITC antibody and a control line coated with an antibody that captures the nanoparticle conjugate.
Procedure:
- Nucleic Acid Amplification: Perform a standard PCR reaction using the FITC- and biotin-labeled primers. The resulting amplicons will be dual-labeled [47].
- Strip Assay Assembly: Place the completed PCR reaction tube in a rack. Insert the lateral flow strip directly into the amplicon solution, ensuring the sample pad is fully immersed.
- Migration and Conjugation: Allow the solution to migrate up the strip via capillary action. The dual-labeled amplicons will bind to anti-FITC antibodies conjugated to colored nanoparticles (e.g., colloidal gold) in the conjugate pad [47].
- Capture and Detection: The complex continues to migrate until captured at the test line by immobilized anti-FITC antibodies, generating a visible band. Excess gold-conjugated antibodies are captured at the control line, validating the test [47].
- Result Interpretation: Read the results visually after 5-15 minutes. The appearance of both control and test lines indicates a positive result. The presence of only the control line indicates a negative result [47].
Workflow and Signaling Pathways
The following diagram illustrates the logical workflow for selecting an appropriate signal readout method based on the requirements of the diagnostic assay.
Signal Readout Method Selection Workflow
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Signal Readout
| Item | Function/Description | Example Application |
|---|---|---|
| Colloidal Gold Nanoparticles | Red-colored label for colorimetric detection; conjugated to antibodies or other bioreceptors. | Standard visual LFA [40]. |
| Fluorescent Microspheres (FMs) | Latex beads embedded with fluorescent dyes; excited at specific wavelengths for sensitive detection. | Quantitative fluorescent LFA [42]. |
| Nitrocellulose Membrane | Porous membrane that serves as the solid support for immobilizing capture molecules in test/control lines. | Core component of all lateral flow strips [40]. |
| FISH Probes | Fluorescently labeled oligonucleotides designed to bind complementary rRNA sequences inside fixed cells. | Identifying specific pathogens within biofilms [44]. |
| BacLight & FM-1-43 Stains | Differential fluorescent stains for labeling live/dead cells and general biofilm matrix. | DSFM for pathogen localization in biofilms [45]. |
| Bst DNA Polymerase | Recombinant polymerase for isothermal amplification (e.g., SEA, RPA), enabling constant temperature nucleic acid amplification. | Integrating nucleic acid amplification with LFA [42]. |
| Anti-FITC & Anti-Biotin | Antibodies used to capture labeled amplicons on the test and control lines of nucleic acid LFAs. | PCR-LFS for detecting AMR genes [47]. |
The detection of biofilm-associated pathogens is a critical challenge in clinical diagnostics. Biofilms, implicated in over 80% of human microbial infections, contribute significantly to treatment failures and chronic wound persistence [48]. Conventional nucleic acid testing for pathogen detection often relies on time-consuming and equipment-intensive nucleic acid extraction methods, which are unsuitable for point-of-care (POC) settings. Extraction-free protocols represent a transformative approach that eliminates this bottleneck by enabling direct amplification from clinical samples. When integrated with one-pot recombinase polymerase amplification (RPA) and CRISPR/Cas detection systems, these protocols facilitate rapid, sensitive, and instrument-free diagnostics ideal for resource-limited environments [6] [49]. This Application Note details optimized extraction-free protocols for detecting biofilm-forming pathogens such as Pseudomonas aeruginosa and Group B Streptococcus, providing researchers with standardized methodologies to accelerate development of POC diagnostic solutions.
Technical Principles and Mechanisms
Principles of Extraction-Free Nucleic Acid Release
Extraction-free protocols bypass conventional nucleic acid purification through simplified lysis conditions that liberate DNA while maintaining compatibility with downstream enzymatic amplification. The fundamental mechanisms include:
- Thermal Lysis: Brief incubation at elevated temperatures (e.g., 95°C for 5 minutes) disrupts cell membranes and inactivates nucleases without significantly damaging nucleic acid targets [6].
- Chemical Lysis: Incubation in specialized buffers at room temperature or with mild heating disrupts cell walls and viral envelopes, releasing DNA while neutralizing PCR inhibitors commonly found in clinical specimens [49].
- Dual-Function Buffers: Optimized formulations simultaneously lyse cells and create a chemical environment compatible with both RPA and CRISPR/Cas enzymes, eliminating the need for buffer exchange steps [6].
This approach significantly reduces processing time from hours to minutes while minimizing cross-contamination risks by eliminating multiple liquid transfer steps [49].
Integration with One-Pot RPA-CRISPR Systems
The successful integration of extraction-free samples with one-pot RPA-CRISPR detection relies on strategic reaction compartmentalization and temperature modulation:
- Reaction Compartmentalization: In single-tube formats, RPA primers, CRISPR/Cas components, and reporter molecules are physically separated from the sample lysate during the initial amplification phase, preventing premature Cas enzyme activation and reporter cleavage [18].
- Two-Temperature Protocol: A lower temperature (37-42°C) optimizes RPA amplification, while a subsequent higher temperature (55-62°C) activates Cas12a/Cas12b trans-cleavage activity, enhancing signal-to-noise ratio in complex samples [6].
- crRNA Engineering: Strategic crRNA design and chemical modifications enhance resistance to degradation in complex sample matrices and prevent interference from lysis reagents [6].
Table 1: Comparison of Extraction-Free Lysis Methods for Different Sample Types
| Lysis Method | Typical Conditions | Compatibility | Advantages | Limitations |
|---|---|---|---|---|
| Thermal Lysis | 95°C for 5 minutes | RPA, LAMP, CRISPR/Cas | Rapid, simple, cost-effective | May be less effective for Gram-positive bacteria |
| Chemical Lysis | Room temperature incubation in buffer | RPA, CRISPR/Cas | Preserves enzyme activity, effective for diverse pathogens | Requires buffer optimization |
| Combined Thermal-Chemical | Buffer incubation with mild heating (e.g., 65°C) | RPA-CRISPR one-pot systems | Enhanced lysis efficiency, broad compatibility | Potential for inhibitor carryover |
Experimental Protocols
Extraction-Free Protocol for Pseudomonas aeruginosa Detection from Wound Secretions
This protocol adapts the one-tube RPA-CRISPR/Cas12a platform targeting the lasB gene of P. aeruginosa, achieving sensitivity of 15.9 CFU/reaction and 97.62% concordance with culture methods [18].
Sample Pretreatment and Lysis
- Sample Collection: Using a sterile swab, collect wound secretions from chronic wounds suspected of P. aeruginosa biofilm infection.
- Direct Lysis: Transfer swab to 200 μL of lysis buffer (10 mM Tris-HCl, 0.1% Triton X-100, pH 8.0).
- Incubation: Vortex vigorously for 15 seconds, then incubate at 95°C for 5 minutes for thermal lysis.
- Cooling: Centrifuge briefly and cool to room temperature. Use 2 μL of supernatant directly in the RPA-CRISPR reaction.
One-Tube RPA-CRISPR/Cas12a Reaction
Reaction Setup: In a single tube, prepare a 25 μL reaction mixture containing:
- 2.5 μL 10× RPA buffer
- 1.0 μL lasB-specific forward primer (10 μM)
- 1.0 μL lasB-specific reverse primer (10 μM)
- 1.0 μL lasB-specific crRNA (1 μM)
- 0.5 μL Cas12a enzyme (10 μM)
- 0.5 μL fluorescent reporter (10 μM, FAM-ssDNA-BHQ1)
- 2.0 μL direct sample lysate
- Nuclease-free water to 25 μL
Amplification and Detection:
- Incubate at 39°C for 40 minutes for RPA amplification.
- Activate Cas12a by incubating at 62°C for 5 minutes.
- Visualize results under UV/blue light or using a portable fluorescence detector.
Extraction-Free Protocol for Group B Streptococcus Detection
This protocol utilizes a one-pot two-temperature RPA-CRISPR/Cas12b system targeting the cfb gene of Group B Streptococcus, achieving sensitivity of 10 copies/test with 98.3% concordance to qPCR [6].
Sample Preparation from Vaginal-Rectal Swabs
- Sample Collection: Collect vaginal-rectal swab samples using standard clinical procedures.
- Minimal Pretreatment: Place swab in 500 μL of sample release buffer (provided in commercial RPA kits).
- Incubation: Incubate at room temperature for 5 minutes with occasional vortexing.
- Clarification: Centrifuge at 2000 × g for 1 minute and use supernatant directly without further purification.
One-Pot Two-Temperature RPA-CRISPR/Cas12b Reaction
Reaction Assembly: Prepare a 25 μL reaction mixture containing:
- 2.5 μL 10× RPA buffer
- 1.0 μL cfb-specific RPA primers (10 μM each)
- 1.0 μL AapCas12b enzyme (10 μM)
- 1.0 μL cfb-specific crRNA (1 μM)
- 0.5 μL fluorescent reporter (10 μM)
- 2.0 μL prepared sample
- RPA rehydration buffer to 25 μL
Amplification and Detection:
- Incubate at 39°C for 40 minutes for RPA amplification.
- Transfer to 62°C for 5-10 minutes to activate Cas12b trans-cleavage.
- Visualize fluorescence under UV light (positive: bright green fluorescence; negative: no fluorescence).
Table 2: Performance Comparison of Extraction-Free RPA-CRISPR Protocols
| Pathogen | Target Gene | CRISPR System | Sensitivity | Assay Time | Clinical Concordance |
|---|---|---|---|---|---|
| Pseudomonas aeruginosa | lasB | Cas12a | 15.9 CFU/reaction | <50 minutes | 97.62% (n=84) |
| Group B Streptococcus | cfb | Cas12b | 10 copies/test | <60 minutes | 98.3% vs qPCR (n=60) |
| General Pathogen Detection | Varies | Cas12a/Cas12b | Varies by target | 45-90 minutes | 90-99% in validation studies |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Extraction-Free RPA-CRISPR Assay Development
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Sample Lysis Reagents | Triton X-100, Tris-HCl buffer, commercial sample release agents | Cell membrane disruption, nucleic acid release | Must be compatible with enzymatic amplification; avoid strong inhibitors |
| RPA Components | TwistAmp basic kits, recombinase enzymes, single-stranded DNA binding proteins | Isothermal amplification of target sequences | Primer design critical; typically 30-35 bp length |
| CRISPR Enzymes | AapCas12b, LbCas12a, AsCas12a | Sequence-specific recognition and trans-cleavage activity | Temperature optimization required; Cas12b more thermostable |
| crRNA Reagents | Target-specific crRNA, T7 transcription kits, RNA purification kits | Guides Cas enzyme to target sequence | Requires careful design to avoid off-target effects |
| Reporters | FAM-ssDNA-BHQ1, FQ-ssDNA-QSY, lateral flow reporters | Signal generation upon trans-cleavage | Fluorescent reporters enable real-time or endpoint detection |
| Portable Detection | Portable fluorescence detectors, UV lamps, blue light transilluminators | Result visualization | Enables true point-of-care application |
Troubleshooting and Optimization Guidelines
Common Challenges and Solutions
- Low Sensitivity: Optimize lysis conditions for specific pathogen types; Gram-positive bacteria may require enhanced lysis with lysozyme or proteinase K pretreatment.
- Inconsistent Results: Include positive and negative controls in each run; ensure consistent sample collection and lysis procedures.
- High Background Signal: Titrate crRNA concentration; optimize Cas enzyme to reporter ratio; implement hot-start protocols to prevent premature activation.
- Inhibition Issues: Dilute sample lysate 1:2-1:10; include amplification facilitators such as BSA or betaine in reaction mix.
Validation and Quality Control
- Analytical Specificity: Test against closely related species to verify minimal cross-reactivity.
- Limit of Detection: Determine using serial dilutions of quantified target pathogen or synthetic DNA standards.
- Clinical Validation: Perform method comparison with gold-standard culture and PCR methods across relevant sample matrices.
Extraction-free protocols represent a paradigm shift in molecular diagnostics for biofilm-associated pathogens, dramatically simplifying sample preparation while maintaining high sensitivity and specificity. The integration of these protocols with one-pot RPA-CRISPR systems creates a powerful platform for point-of-care detection that requires minimal instrumentation and technical expertise. The protocols detailed herein for P. aeruginosa and Group B Streptococcus detection provide researchers with validated methodologies that can be adapted for other biofilm-forming pathogens. As these technologies continue to evolve, they hold significant promise for revolutionizing infectious disease diagnostics in both clinical and resource-limited settings, ultimately enabling earlier intervention and improved patient outcomes in biofilm-related infections.
Biofilm-forming pathogens such as Methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa represent a significant threat in healthcare settings, contributing to persistent infections, antimicrobial resistance, and increased morbidity and mortality [50]. The complex extracellular polymeric substance (EPS) of biofilms confers inherent resistance to conventional antibiotics and host immune responses, making infections difficult to eradicate [50] [51]. Traditional diagnostic methods, including culture-based techniques and PCR, are often time-consuming, require specialized equipment, or lack the sensitivity for early detection [52] [53].
The integration of recombinase polymerase amplification (RPA) with CRISPR-Cas systems has emerged as a revolutionary approach for molecular diagnostics [16]. This combination enables rapid, highly specific, and sensitive detection of pathogens at the point-of-care, addressing critical limitations of conventional methods [12]. This application note details successful case studies implementing one-pot RPA-CRISPR platforms for detecting clinically relevant biofilm formers, providing validated protocols and analytical performance data to guide researchers and clinical scientists.
Core Principles and Advantages
RPA-CRISPR diagnostics combine isothermal nucleic acid amplification with the sequence-specific recognition and collateral cleavage activity of CRISPR-associated enzymes. The system leverages:
- RPA: Amplifies target DNA at a constant temperature (37-42°C) within 15-20 minutes, eliminating the need for thermal cycling [52] [53].
- CRISPR-Cas12a/Cas12b: Upon recognition of the target sequence guided by crRNA, the Cas enzyme exhibits trans-cleavage activity, indiscriminately degrading single-stranded DNA reporters [52] [16].
- Signal Detection: Cleavage of fluorescent- or lateral flow-based reporters generates detectable signals, enabling result quantification or visual interpretation [52] [54].
The one-pot configuration, where amplification and detection occur in a single tube, minimizes contamination risk, simplifies operational procedures, and reduces total assay time [6].
Molecular Mechanism of RPA-CRISPR Detection
The following diagram illustrates the molecular mechanism of one-pot RPA-CRISPR detection for biofilm-forming pathogens:
Case Study 1: MRSA Detection Targeting the mecA Gene
Experimental Protocol
Target Gene: mecA gene, encoding penicillin-binding protein 2a (PBP2a) responsible for methicillin resistance [52] [55].
Sample Preparation:
- Isolate bacterial DNA from clinical samples using commercial kits or heat lysis.
- For heat lysis, suspend colonies in nuclease-free water, incubate at 95°C for 5-10 minutes, and centrifuge at 12,000 × g for 1 minute. Use supernatant as template [54].
One-Tube RPA-CRISPR/Cas12a Reaction:
- Prepare a master mix containing:
- 10 μL of 2× Reaction Buffer
- 1 μL of dNTPs
- 2 μL of 10× E-mix
- 1 μL each of forward and reverse RPA primers (10 μM)
- 1 μL of 20× Core Reaction mix
- 2 μL of template DNA
- Nuclease-free water to 23 μL
- Add 2 μL of MgOAc (280 mM) to initiate RPA reaction.
- Simultaneously, prepare CRISPR detection mix on tube cap:
- 1 μL of LbaCas12a (1 μM)
- 1 μL of crRNA (2 μM)
- 0.25 μL RNA inhibitor
- 1 μL ssDNA FQ Reporter (FAM-BHQ1 or HEX-BHQ1, 10 μM)
- 2 μL of 10× NEBuffer 2.1
- Incubate tube at 42°C for 10 minutes for RPA amplification.
- Briefly centrifuge to mix RPA amplicons with CRISPR reagents.
- Incubate at 42°C for additional 10-15 minutes for Cas12a detection [52].
Signal Detection:
- Fluorescence: Monitor real-time fluorescence every minute or visualize under UV light [52].
- Lateral Flow: Use FAM-biotin labeled reporters. Apply 5 μL reaction mixture to strip and observe test and control lines within 3-5 minutes [52] [54].
Performance Metrics
Table 1: Analytical performance of one-pot RPA-CRISPR for MRSA detection
| Parameter | Fluorescence Detection | Lateral Flow Detection |
|---|---|---|
| Limit of Detection | 10 copies/reaction [52] | 10-100 copies/reaction [52] |
| Assay Time | 20 minutes [52] | 20 minutes [52] |
| Specificity | 100% (no cross-reactivity with other common bacteria) [55] | 95.7% concordance with qPCR [52] |
| Clinical Sensitivity | 100% vs. qPCR [52] | 95.7% vs. qPCR [52] |
Case Study 2: P. aeruginosa Detection Targeting Virulence Genes
Experimental Protocol
Target Genes: lasB (elastase) or oprL (outer membrane lipoprotein) genes, conserved virulence factors in P. aeruginosa [53] [56].
Sample Preparation:
- For swab samples, blister into sterile EP tube with 300 μL nuclease-free water.
- Heat at 100°C for 8 minutes, centrifuge at 11,000 × g for 1 minute.
- Use supernatant as template for RPA [54].
One-Pot RPA-CRISPR/Cas12b Reaction:
- Prepare a pre-mixed one-pot reaction containing all RPA and CRISPR/Cas12b reagents.
- Add extracted DNA or crude lysate to the reaction mixture.
- Perform RPA amplification at 39°C for 40 minutes.
- Increase temperature to 62°C for 5 minutes to activate AapCas12b trans-cleavage activity [6].
- Visualize results under UV light.
Alternative Two-Tube Method:
- Perform RPA amplification in a separate tube at 42°C for 20 minutes.
- Transfer 2 μL RPA product to CRISPR/Cas12a detection mix containing:
- 10.5 μL nuclease-free water
- 2 μL 10× NEBuffer 2.1
- 1 μL RNase inhibitor
- 2 μL LbCas12a (1 μM)
- 2 μL crRNA (2 μM)
- 0.5 μL FQ-labeled ssDNA reporter (10 μM)
- Incubate at 37°C for 10 minutes [54].
Detection Methods:
- Fluorescence: Use qPCR instrument with HEX channel or portable UV lamp [54].
- Lateral Flow: Replace FQ reporter with FAM-biotin labeled probe. Dip strip for 3 minutes [53] [54].
Performance Metrics
Table 2: Analytical performance of RPA-CRISPR for P. aeruginosa detection
| Parameter | lasB Gene Target | oprL Gene Target |
|---|---|---|
| Limit of Detection | 100 copies/μL (fluorescence), 101 copies/μL (LFTS) [53] | 60 fg/reaction (~8 copies) [56] |
| Assay Time | <30 minutes [53] | 30 minutes [56] |
| Specificity | 100% (no cross-reactivity with 15 non-Pseudomonas strains) [53] | 100% [56] |
| Clinical Concordance | Comparable to qPCR [53] | 100% vs. microfluidic chip [56] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents and materials for one-pot RPA-CRISPR assays
| Reagent Category | Specific Examples | Function | Manufacturer/Source |
|---|---|---|---|
| CRISPR Enzymes | LbaCas12a (Cpf1), AapCas12b | Target recognition and collateral cleavage | New England Biolabs [52] [54] |
| Amplification Kits | TwistAmp Basic RPA Kit, Isothermal DNA Amplification Basic Kit | Isothermal nucleic acid amplification | TwistDx [52], EZassay [53] |
| crRNA Synthesis | Custom crRNA designs targeting mecA, lasB, oprL | Sequence-specific guidance of Cas enzyme | Sangon Biotech [52] [53] |
| Reporters | ssDNA-FQ (FAM/HEX-BHQ1), ssDNA-FB (FAM-Biotin) | Fluorescent or lateral flow signal generation | Sangon Biotech [52] [53] |
| Lateral Flow Strips | Colloidal gold-based strips with anti-FAM and streptavidin lines | Visual result interpretation | Tiosbio, Lesunbio [53] [54] |
| Buffer Systems | NEBuffer 2.1, 10× LAMP buffer | Optimal enzyme activity and stability | New England Biolabs, Tolo Biotech [52] [6] |
Technical Considerations and Optimization Strategies
Workflow Design and Temperature Optimization
The experimental workflow for one-pot RPA-CRISPR assays requires careful temperature management, particularly when using thermostable Cas variants like Cas12b:
Critical Optimization Parameters
Successful implementation requires optimization of several key parameters:
- crRNA Design: Design crRNAs to target conserved regions with appropriate PAM sequences. Tools like CHOPCHOP facilitate design [54].
- Primer-CrRNA Compatibility: Ensure RPA primers amplify regions accessible to crRNA binding without interference [53].
- Magnesium Optimization: MgOAc concentration critically affects RPA efficiency. Titrate from 0-280 mM for optimal results [52].
- Reporter Concentration: Balance between signal intensity and background noise. Test ssDNA reporters at 0.5-5 μM [54].
One-pot RPA-CRISPR diagnostics represent a transformative approach for detecting clinically relevant biofilm-forming pathogens. The case studies presented demonstrate robust detection of MRSA and P. aeruginosa with sensitivity down to 10-100 copies, specificity exceeding 95%, and completion times under 30 minutes [52] [53] [54]. These characteristics make the technology particularly suitable for point-of-care testing in resource-limited settings.
Future development should focus on:
- Multiplexing Capabilities: Simultaneous detection of multiple pathogens or resistance genes [12].
- Extraction-Free Protocols: Simplified sample processing using heat lysis or direct specimen application [6].
- Quantitative Detection: Correlation between signal intensity and bacterial load for infection monitoring [16].
- Broad-Spectrum Applications: Adaptation to other biofilm-forming pathogens beyond MRSA and P. aeruginosa [12] [16].
The integration of artificial intelligence with CRISPR diagnostics shows particular promise for optimizing guide RNA design, interpreting complex results, and predicting resistance patterns [12]. As these technologies mature, one-pot RPA-CRISPR platforms are poised to become indispensable tools in clinical microbiology, enabling rapid intervention and improved management of biofilm-associated infections.
Optimizing One-Pot RPA-CRISPR Assays: Overcoming Sensitivity and Specificity Challenges
The integration of recombinase polymerase amplification (RPA) with CRISPR/Cas systems has revolutionized molecular diagnostics, particularly for detecting biofilm-forming pathogens. A critical advancement in this field is the implementation of dual-temperature protocols, which strategically separate the nucleic acid amplification phase from the CRISPR-mediated detection phase. This approach significantly enhances assay sensitivity and specificity by optimizing the enzymatic efficiency of each component. Research demonstrates that a brief high-temperature pre-incubation of the Cas12a protein at 65°C before its use at 37°C can dramatically improve its collateral activity, reducing the limit of detection by 33-fold without incurring false positives [57]. Furthermore, precise thermodynamic modeling of reaction components has been shown to account for up to 99.42% of variability in optimal assay conditions, underscoring the indispensable role of temperature optimization in assay performance [58]. This application note details the protocols and mechanistic basis for employing dual-temperature strategies to maximize the performance of one-pot RPA-CRISPR diagnostic platforms for challenging targets like biofilm-associated pathogens.
The Scientific Basis for Dual-Temperature Protocols
Fundamental Principles of RPA-CRISPR Reactions
The one-pot RPA-CRISPR system combines two powerful biochemical processes that have distinct, and often competing, optimal temperature requirements. Recombinase Polymerase Amplification (RPA) is an isothermal amplification technique that operates optimally between 37-42°C, leveraging a recombinase-primer complex to facilitate strand invasion and DNA synthesis [11] [54]. Concurrently, the CRISPR/Cas12a detection module functions through a crRNA-guided Cas12a enzyme that, upon recognizing its target DNA sequence, exhibits trans-cleavage activity, indiscriminately degrading single-stranded DNA reporters [3] [38].
The core challenge in one-pot assays lies in the fact that while RPA performs efficiently at 37-42°C, the Cas12a enzyme from commonly used orthologs like LbaCas12a demonstrates enhanced enzymatic activity and faster reaction kinetics at elevated temperatures. Research has revealed that heating the wild-type AsCas12a protein to 65°C accelerates nucleic acid detection by inducing a transient acceleration of Cas12a collateral activity and stabilizing the protein-nucleic acid complexes [57]. This thermal enhancement creates a thermodynamic dilemma when both reactions are forced to occur at a single compromise temperature.
Thermodynamic and Kinetic Rationale
The mathematical modeling of biochemical reactions provides a robust framework for understanding temperature effects on assay performance. Sophisticated predictive models incorporating multivariate Taylor series expansion and thermodynamic functions have demonstrated exceptional capability in forecasting optimal reaction conditions, achieving a coefficient of determination (R²) of 0.9942 for critical parameters like MgCl₂ concentration, which is intrinsically linked to temperature optimization [58].
The underlying mechanism for thermal enhancement involves:
- Accelerated reaction kinetics at higher temperatures, following Arrhenius equation principles
- Stabilization of enzyme-substrate complexes through optimized hydrogen bonding and van der Waals interactions
- Reduced secondary structures in target nucleic acids that might impede hybridization
- Enhanced collateral cleavage activity of Cas12a enzymes when temporarily exposed to supra-optimal temperatures
Table 1: Thermodynamic Parameters Influencing Temperature Optimization in RPA-CRISPR Assays
| Parameter | Impact on RPA | Impact on Cas12a | Optimization Approach |
|---|---|---|---|
| Primary Temperature Range | 37-42°C | 37-65°C (enhanced) | Sequential temperature phases |
| Activation Energy (Eₐ) | Lower, suitable for isothermal amplification | Higher, benefits from thermal activation | Brief high-temperature pulse for Cas12a |
| Melting Temperature (Tₘ) | Critical for primer binding | Critical for crRNA-target hybridization | Balanced through bioinformatic design |
| Mg²⁺ Dependency | High (cofactor for polymerase) | Moderate (influences cleavage efficiency) | Modeled concentration at 4-6 mM [58] |
| Time Constant | 15-20 minutes for amplification | 5-10 minutes for detection | Sequential timing optimization |
Essential Reagents and Materials
Research Reagent Solutions
The successful implementation of dual-temperature RPA-CRISPR protocols requires carefully selected reagents and materials. The table below details the essential components and their specific functions within the assay system.
Table 2: Essential Research Reagents for Dual-Temperature RPA-CRISPR Assays
| Reagent/Material | Function/Purpose | Specifications/Notes |
|---|---|---|
| TwistAmp Liquid Basic RPA Kit | Isothermal amplification of target DNA | Contains recombinase, single-stranded DNA-binding protein, strand-displacing polymerase [38] |
| LbaCas12a or AsCas12a Nuclease | Target-specific recognition and trans-cleavage | Wild-type proteins benefit from high-temperature pre-incubation [57] |
| Custom crRNA | Guides Cas12a to specific target sequences | Designed using CHOPCHOP webtool; synthesized in vitro with T7 polymerase [54] |
| Fluorescent Reporters | Signal generation via collateral cleavage | ssDNA labeled with FAM/BHQ-1 or HEX/BHQ-1 for real-time detection [59] [54] |
| Lateral Flow Reporters | Visual detection on strips | ssDNA labeled with FAM/Biotin for compatible lateral flow strips [38] |
| NEBuffer 2.1 | Reaction buffer for Cas12a activity | Provides optimal ionic conditions for enzyme function [38] [54] |
| MgOAc (280 mM) | Initiator for RPA reaction | Critical cofactor added last to start amplification [38] |
| RNase Inhibitor | Protects crRNA from degradation | Essential for maintaining assay integrity in one-pot formats [38] |
| Lateral Flow Strips | Point-of-care compatible detection | Integrated with colloidal gold probe, capture antibody, and streptavidin [54] |
Dual-Temperature Protocol for Biofilm Pathogen Detection
The following diagram illustrates the complete experimental workflow for the dual-temperature RPA-CRISPR assay, from sample preparation to result interpretation:
Step-by-Step Protocol
Pre-Assembly and Physical Separation of Reaction Components
The one-tube format is maintained through physical separation of RPA and CRISPR components prior to mixing:
RPA Master Mix Preparation (Bottom of Tube)
- Prepare a 25 μL RPA reaction mixture containing:
- 10 μL of 2× Reaction Buffer (TwistAmp Basic Kit)
- 1 μL of dNTPs (10 mM each)
- 2 μL of 10× E-mix
- 1 μL each of forward and reverse RPA primers (10 μM)
- 1 μL of 20× Core Reaction mix
- 2 μL of template DNA (from biofilm samples)
- Nuclease-free water to 23 μL
- Note: Do not add MgOAc at this stage [38]
- Prepare a 25 μL RPA reaction mixture containing:
CRISPR/Cas12a Master Mix Preparation (Tube Lid)
- Prepare CRISPR detection mixture containing:
- 1 μL of LbaCas12a (1 μM)
- 1 μL of crRNA (2 μM)
- 0.25 μL RNA inhibitor
- 1 μL ssDNA reporter (10 μM; FAM/BHQ for fluorescence or FAM/Biotin for LFS)
- 2 μL of 10× NEBuffer 2.1
- Nuclease-free water to 10 μL
- Carefully pipette this mixture onto the inner surface of the tube lid [38]
- Prepare CRISPR detection mixture containing:
Sequential Temperature Incubation Protocol
The critical dual-temperature implementation proceeds as follows:
Initiate RPA Amplification
High-Temperature Cas12a Activation
- Immediately following RPA completion, transfer tubes to a 65°C heat block
- Incubate for 5 minutes to activate Cas12a collateral cleavage activity
- This critical step enhances detection sensitivity by 33-fold [57]
Combine and Complete Detection
- Briefly centrifuge tubes (10-15 seconds) to mix RPA amplicons with CRISPR reagents
- Immediately transfer to 37°C incubation for 10 minutes
- During this phase, Cas12a recognizes targets and cleaves reporters [38]
Signal Detection and Interpretation
Real-Time Fluorescence Monitoring
Endpoint Fluorescence Visualization
- Illuminate reaction tubes with a blue light transilluminator (470 nm)
- Positive samples emit bright green fluorescence visible to naked eye
- Capture images with smartphone camera for documentation [38]
Lateral Flow Strip Detection
- Dilute 5 μL of final reaction mixture with 95 μL of lateral flow assay buffer
- Insert lateral flow strip for 3-5 minutes
- Interpret results: Control line only (negative); Control and Test lines (positive) [54]
Molecular Mechanism of Enhanced Detection
The following diagram illustrates the molecular mechanism by which the dual-temperature protocol enhances RPA-CRISPR detection sensitivity:
Key Performance Metrics and Validation
Implementation of the dual-temperature protocol should be validated using the following performance parameters:
Table 3: Performance Metrics for Dual-Temperature RPA-CRISPR Assays
| Performance Parameter | Expected Outcome | Validation Method |
|---|---|---|
| Limit of Detection (LoD) | 10-100 copies/reaction | Probit analysis with serial dilutions |
| Assay Time | 20-35 minutes total | From sample addition to result |
| Specificity | 100% for target pathogen | Testing against panel of related species |
| Analytical Sensitivity | 33-fold improvement vs single-temperature | Comparison of LoD with/without high-temperature step [57] |
| Clinical Sensitivity | 95.7-100% vs qPCR | Testing with clinical isolates [38] [54] |
| Interference Resistance | Robust in biofilm matrix | Spike-in recovery experiments |
Troubleshooting and Optimization Guidelines
Common Challenges and Solutions
- High Background Signal: Reduce crRNA concentration (1-2 μM optimal), ensure Cas12a purity, and verify reporter probe quality [59]
- Low Signal Intensity: Increase high-temperature activation time (up to 10 minutes), check RPA primer efficiency, and verify MgOAc concentration [57] [58]
- Inconsistent Lateral Flow Results: Optimize dilution factor (typically 1:5 to 1:10), ensure proper strip orientation, and check reporter labeling efficiency [54]
- Failed RPA Amplification: Verify template quality, use positive control primers, and ensure MgOAc is properly added last [38]
Advanced Optimization Strategies
For challenging applications like biofilm pathogen detection where inhibitor resistance is crucial:
- Buffer Optimization: Supplement with 0.2% BSA or 0.1% Tween-20 to mitigate biofilm-derived inhibitors
- crRNA Design: Target highly conserved regions with minimal secondary structure using RNAplex analysis [59]
- Temperature Calibration: Fine-tune high-temperature step (60-70°C range) for specific Cas12a orthologs
- Time-Resolved Activation: Implement real-time monitoring to determine optimal reaction termination point
The dual-temperature protocol represents a significant advancement in one-pot RPA-CRISPR diagnostics, enabling rapid, ultra-sensitive detection of biofilm-forming pathogens with minimal equipment requirements. This approach effectively resolves the thermodynamic compromise inherent in single-temperature assays, unlocking the full potential of CRISPR-based diagnostics for point-of-care applications.
Mitigating Primer-Dimer Formation and Non-Specific Amplification in RPA
Recombinase Polymerase Amplification (RPA) is a powerful isothermal nucleic acid amplification technique, but its application, particularly in complex one-pot RPA-CRISPR diagnostics for biofilm pathogens, is hampered by primer-dimer formation and non-specific amplification. These artifacts compete for reaction reagents, reduce target amplification efficiency, and can generate false-positive signals that compromise assay reliability [60] [61]. The fundamental mechanism of RPA, which relies on recombinase-primer filaments to scan and invade double-stranded DNA at low temperatures (37-42°C), inherently increases the risk of off-target priming and primer-dimer artifacts compared to higher-stringency PCR methods [60] [62]. In CRISPR-coupled systems, these non-specific products can inadvertently activate Cas protein trans-cleavage activity, leading to detectable signals even in the absence of the true target pathogen [6] [3]. Addressing these challenges is therefore essential for developing robust diagnostic assays for biofilm-based pathogens.
Understanding Amplification Artifacts
Types and Causes of Non-Specific Amplification
Non-specific amplification in RPA manifests in several distinct forms, each with different causes and consequences for diagnostic accuracy:
Primer-dimers are short, amplifiable duplexes formed by two primers, typically producing fragments of 20-60 bp visible as bright bands at the bottom of electrophoresis gels [63]. These artifacts form through direct hybridization of primers and can join with other primer-dimers to form larger multimers (100-200 bp) that create ladder-like patterns on gels and compete significantly with target amplification [63].
PCR smears appear as a continuous background of randomly amplified DNA fragments of varying lengths, often caused by highly fragmented DNA templates, excessive template concentration leading to self-priming, degraded primers, or suboptimal annealing temperatures [63]. These smears can obscure specific amplification bands and make subsequent CRISPR detection impossible.
Unexpected amplicon sizes occur when primers bind to non-target regions with sufficient complementarity to initiate elongation, resulting in discrete bands at sizes different from the expected product [63]. This frequently happens when primer sequences share homology with non-target regions in complex samples like biofilm DNA extracts.
The recombinase enzyme in RPA can tolerate a significant number of primer-template mismatches, with one study demonstrating that RecA-ssDNA filaments can recognize mismatches with a minimum of 18 (23%) bases in an 80-mer DNA sequence [61]. This permissiveness is particularly problematic in biofilm pathogen detection, where complex sample matrices and abundant background DNA from multiple microbial species further increase the likelihood of non-specific amplification events [61].
Strategic Approaches for Mitigation
Primer and Probe Design Optimization
The most effective approach to minimizing amplification artifacts begins with careful primer and probe design employing multiple complementary strategies:
Strategic base substitutions introduce deliberate mismatches at specific positions in primer and probe sequences to reduce formation of primer-probe complexes without significantly affecting target binding. One study targeting Salmonella enterica serotype Typhimurium successfully eliminated primer-dependent artifacts by incorporating such substitutions following specific design guidelines [60].
Probe incorporation in the RPA reaction provides an additional layer of specificity, as demonstrated in RPA-lateral flow strip (LFS) detection systems where probe-based assays eliminated false positives that occurred with primer-only configurations [60].
Analysis of primer-probe complex formation during the design phase helps identify and eliminate sequences prone to intermolecular interactions. This pre-emptive screening, combined with careful selection of primers targeting unique virulence genes, enables development of highly specific RPA assays [60].
Bioinformatic validation through comprehensive alignment checks against relevant genomic databases ensures minimal homology with non-target sequences present in biofilm samples, reducing the risk of off-target amplification.
Reaction Composition Enhancement
Optimizing the chemical environment of RPA reactions significantly reduces artifact formation:
Betaine enhancement at optimal concentrations (0.8 M) dramatically improves RPA specificity and efficiency simultaneously. This inexpensive additive addresses the fundamental challenge of non-specific amplification in samples with large amounts of background DNA, as demonstrated in clinical testing for hepatitis B virus where betaine-assisted RPA (B-RPA) achieved 95% agreement with clinically approved qPCR assays [61].
Crowding agents like polyethylene glycol (PEG) included in standard RPA formulations modulate biochemical efficiency but contribute to viscosity that can restrict reagent distribution. Understanding this balance is crucial for optimizing reaction conditions for specific biofilm pathogen targets [62].
Table 1: Quantitative Experimental Data on Mitigation Strategies
| Mitigation Approach | Experimental Findings | Impact on Specificity | Reference |
|---|---|---|---|
| Betaine supplementation (0.8 M) | Eliminated non-specific bands in hepatitis B virus detection; 95% agreement with qPCR | Significant improvement in complex samples | [61] |
| Reduced reaction volume (50 μL to 5 μL) | Achieved similar sensitivity with or without mixing step; improved low-copy detection | Reduced need for manual intervention | [62] |
| Solid-phase RPA on nanoplasmonic microarrays | Limit of detection: 4 copies/reaction; minimized primer-dimer formation | Enhanced specificity through spatial separation | [64] |
| Primer design with base substitutions | Eliminated false positives in S. Typhimurium detection | Enabled specific detection in 30 min at 42°C | [60] |
| Two-temperature RPA-CRISPR (39°C → 62°C) | Achieved sensitivity of 10 copies/test for Group B Streptococcus | Improved signal-to-noise ratio in low-template samples | [6] |
Physical and Workflow Parameters
Adjusting physical aspects of the RPA process can further reduce artifacts:
Reduced reaction volumes (5 μL vs. standard 50 μL) improve sensitivity without requiring the recommended mixing step, as demonstrated in HIV-1 detection assays where smaller volumes showed similar performance with and without mixing, potentially due to more efficient thermal transfer and reagent distribution [62].
Strategic mixing protocols counteract localized reagent depletion in standard 50 μL reactions. The manufacturer recommends vigorous mixing after 4-5 minutes of incubation, with studies showing that omitted mixing leads to longer amplification times and inferior detection signals, particularly for low-copy targets [62].
Temperature optimization plays a crucial role in coupled RPA-CRISPR systems. A two-temperature approach (39°C for RPA followed by 62°C for Cas12b activation) significantly improved detection sensitivity for Group B Streptococcus, achieving 10 copies/test by optimizing the trans-cleavage activity of the thermostable AapCas12b enzyme [6].
Solid-phase amplification spatially separates amplification events using surface-immobilized primers. Nanoplasmonic microarray-based solid-phase RPA demonstrated exceptional sensitivity (4 copies/reaction) for SARS-CoV-2 detection by minimizing primer-dimer formation through stringent washing processes that remove non-specifically bound primers [64].
Detailed Experimental Protocols
Betaine-Assisted RPA (B-RPA) Protocol
The following protocol details the implementation of betaine to enhance RPA specificity for detecting biofilm pathogens:
Reagents and Equipment:
- TwistAmp Basic RPA kit (lyophilized or liquid format)
- Betaine (molecular biology grade)
- Target-specific primers (optimized design)
- Template DNA (from biofilm samples)
- Nuclease-free water
- Magnesium acetate (280 mM)
- Heating block or water bath (39°C)
Procedure:
- Prepare a 5M betaine stock solution in nuclease-free water and filter sterilize.
- Reconstitute or prepare the RPA reaction mixture according to manufacturer instructions, adjusting the formulation to contain 0.8 M betaine in the final reaction volume.
- For a 50 μL total reaction volume, combine:
- 29.5 μL of rehydration buffer containing betaine
- 2.4 μL of forward primer (10 μM)
- 2.4 μL of reverse primer (10 μM)
- 5-10 μL of template DNA (optimized for biofilm samples)
- Nuclease-free water to 47.5 μL total
- Add the mixture to the RPA reaction pellet (if using lyophilized format) or add enzyme components according to kit instructions.
- Briefly centrifuge to mix components.
- Initiate amplification by adding 2.5 μL of 280 mM magnesium acetate.
- Mix immediately and incubate at 39°C for 30-40 minutes.
- Include appropriate controls (no-template, positive template) to validate performance.
Validation: In the hepatitis B virus detection study, this B-RPA protocol successfully eliminated non-specific amplification observed in conventional RPA and demonstrated 95% concordance with clinical qPCR results when testing clinical plasma samples [61].
Solid-Phase RPA on Nanoplasmonic Microarrays
This protocol adapts RPA for solid-phase implementation to minimize primer-dimer formation in multiplex detection scenarios:
Reagents and Equipment:
- Nanoplasmonic substrate (fabricated as described in [64])
- Biotin-labeled forward primers
- Cy5-labeled reverse primers
- Streptavidin solution (1.8 μM in PBS)
- TwistAmp Liquid Basic RPA kit
- PDMS frame and glass slides
- Microarray spotter
- Blocking solution (0.5% BSA)
Procedure:
- Microarray Fabrication:
- Mix streptavidin (100 nM final) with each biotin-modified forward primer (2.5 μM final) in DEPC-treated water.
- Spot 20 nL-100 nL volumes onto nanoplasmonic substrates using a microarrayer with 2 mm spacing between spots.
- Incubate at 4°C overnight for immobilization.
- Wash three times with DEPC-treated water to remove excess reagents.
- Block non-spotted areas with 0.5% BSA solution for 2 hours at 25°C.
- Wash again and dry at room temperature.
Solid-Phase RPA:
- Attach the prepared nanoplasmonic microarray to a glass slide using a PDMS frame to contain reagents.
- Prepare the RPA reaction mixture according to manufacturer instructions, including reverse primers and template.
- Apply the reaction solution to the microarray.
- Incubate at 39°C for 30-40 minutes for amplification.
- Perform stringent washing to remove non-specifically bound amplicons.
Detection:
- Visualize results using appropriate fluorescence detection.
- For CRISPR-coupled detection, add Cas12a-crRNA complex with reporter molecules after amplification.
Performance: This approach achieved exceptional sensitivity (4 copies/reaction) for SARS-CoV-2 detection while effectively minimizing primer-dimer formation through spatial separation and stringent washing [64].
Table 2: Research Reagent Solutions for RPA Artifact Mitigation
| Reagent/Material | Function in Mitigation | Application Example | Optimal Conditions |
|---|---|---|---|
| Betaine | Prevents non-specific amplification by modifying DNA duplex stability | Hepatitis B virus detection in clinical samples | 0.8 M final concentration in RPA reaction [61] |
| Polyethylene glycol (PEG) | Crowding agent that enhances enzyme activity and reaction efficiency | Standard component of RPA formulations | Included in manufacturer's formulation [62] |
| Biotin-labeled primers | Enable solid-phase immobilization for spatial separation of amplification | Nanoplasmonic microarray RPA for SARS-CoV-2 | 2.5 μM primer with 100 nM streptavidin for spotting [64] |
| Streptavidin-coated surfaces | Provide solid support for primer immobilization | Multiplexed solid-phase RPA-CRISPR detection | Pre-coated surfaces incubated with biotin-primers [64] [65] |
| Modified crRNAs | Enhance specificity in coupled RPA-CRISPR detection | One-pot GBS detection with Cas12b | Specific design to prevent early CRISPR activation [6] |
| TwistAmp RPA kits | Provide optimized core reagents for RPA reactions | HIV-1 detection in resource-limited settings | Stable at 25°C for 12 weeks, 45°C for 3 weeks [62] |
Integrated Workflow for RPA-CRISPR Diagnostics
The following integrated protocol combines multiple mitigation strategies for robust one-pot RPA-CRISPR detection of biofilm pathogens:
Procedure:
- Primer/Probe Design: Implement strategic base substitutions in RPA primers targeting biofilm pathogen genes. Design crRNAs with minimal off-target potential against the amplified region.
- Reaction Assembly: Prepare a one-pot reaction containing:
- Betaine-enhanced RPA reagents (0.8 M final betaine concentration)
- Target-specific primers with base substitutions
- crRNA targeting the amplified sequence
- AapCas12b enzyme
- Fluorescent reporter (e.g., FAM-TTATT-BHQ-1)
- Template DNA from biofilm samples
- Adjust to reduced volume (5-10 μL) if using non-mixed format
- Two-Temperature Incubation:
- Incubate at 39°C for 40 minutes for RPA amplification
- Without opening the tube, increase temperature to 62°C for 5-10 minutes to activate Cas12b trans-cleavage activity
- Detection:
- Visualize results using UV flashlight or blue LED excitation
- Positive samples show bright fluorescence due to reporter cleavage
Validation: This approach demonstrated 96.7% concordance with culture methods and 98.3% with qPCR when detecting Group B Streptococcus in clinical samples, achieving sensitivity of 10 copies/test [6].
Effective mitigation of primer-dimer formation and non-specific amplification in RPA requires a multifaceted approach combining strategic primer design, reaction enhancement with additives like betaine, optimization of physical parameters, and potentially solid-phase implementation. For one-pot RPA-CRISPR diagnostics targeting biofilm pathogens, these strategies collectively address the fundamental challenge of maintaining specificity in complex sample matrices while preserving the rapid, isothermal advantages of RPA technology. The protocols detailed herein provide researchers with validated methodologies to enhance assay robustness, enabling reliable detection of low-abundance pathogens in challenging diagnostic scenarios. As RPA-CRISPR platforms continue evolving toward point-of-care applications, these artifact mitigation approaches will remain essential for ensuring diagnostic accuracy and reliability.
Strategies to Minimize Aerosol Contamination in Single-Tube Formats
In the development of one-pot RPA-CRISPR diagnostics for biofilm pathogen detection, aerosol contamination presents a significant challenge to assay reliability. Aerosols, defined as suspensions of solid or liquid particles in a gas, form during many laboratory activities including pipetting with air-displacement pipettes [66]. These microscopic droplets can carry amplicons from previous reactions into subsequent tests, leading to false-positive results and compromising diagnostic accuracy. The extreme sensitivity of CRISPR-based detection systems, while advantageous for identifying low-abundance targets, simultaneously increases vulnerability to such contamination events.
Within single-tube diagnostic formats, the risk of contamination is particularly pronounced during reagent transfer steps. Traditional CRISPR detection workflows often require physical opening of reaction tubes after nucleic acid amplification to introduce CRISPR reagents, creating opportunities for aerosol release and cross-contamination between samples [52]. For researchers focusing on biofilm-derived pathogens, which may be present in low quantities in clinical samples, preventing such contamination is essential for obtaining clinically actionable results. This document outlines evidence-based strategies to minimize aerosol contamination while maintaining the analytical sensitivity required for effective biofilm pathogen detection.
Aerosol Contamination Mechanisms and Risks
Contamination Pathways in Molecular Diagnostics
Aerosol-mediated contamination in molecular diagnostics follows three primary pathways, each with distinct mechanisms and consequences for assay integrity. Understanding these pathways is fundamental to implementing effective contamination control strategies.
Sample-to-pipette contamination: This occurs when pipetted liquid or aerosol particles enter the pipette body during aspiration. This risk is heightened when pipetting techniques are too rapid or when pipettes are held at incorrect angles. Contaminated pipettes can then transfer foreign nucleic acids to reagent stocks or subsequent reactions [66]. The slow release of the pipette plunger is critical to minimize aerosol formation during dispensing.
Sample-to-sample contamination (carry-over): This pathway involves transfer of aerosol or liquid residue from one sample to another, typically when the same pipette tips are reused or when amplified products are handled near pre-amplification areas [66]. In RPA-CRISPR workflows, the high concentration of amplicons generated during amplification makes them particularly problematic contaminants.
Pipette-to-sample contamination: This occurs when contaminated pipettes or tips introduce foreign materials into clean reactions. Contaminants may include DNase, RNase, endotoxins, or trace nucleic acids from previous procedures [66]. For biofilm pathogen detection, where target organisms might be present in low abundance, these contaminants can inhibit amplification or detection reactions.
Contamination Risks in RPA-CRISPR Workflows
The integration of RPA amplification with CRISPR detection creates unique contamination challenges. Conventional two-step approaches require physical transfer of amplicons from amplification to detection reactions, generating significant aerosol contamination risk [52]. Each tube opening presents an opportunity for aerosol release, potentially contaminating laboratory surfaces, equipment, and subsequent reactions. The single-tube formats developed to address this vulnerability employ spatial or temporal separation of amplification and detection components, but still require careful implementation to maintain contamination-free conditions throughout the workflow.
Contamination Control Strategies for Single-Tube Formats
Single-Tube Platform Designs
Innovative single-tube designs represent the most effective strategy for minimizing aerosol contamination in RPA-CRISPR diagnostics. These platforms physically contain amplification products while allowing sequential execution of RPA and CRISPR reactions.
Physical segregation with cap-based systems: This approach involves placing the RPA reaction mixture at the bottom of a tube while depositing CRISPR reagents in the cap. Following amplification, a brief centrifugation step mixes the components without tube opening. Research demonstrates this method effectively contains amplicons while reducing hands-on time [52]. A study detecting methicillin-resistant Staphylococcus aureus (MRSA) achieved detection limits of 10 copies for fluorescence methods and 100 copies for lateral flow strips using this approach, with 100% concordance with qPCR for clinical samples [52].
Photocontrolled activation systems: This advanced method employs crRNA modified with photodegradable chemical groups (NPOM-dt) that temporarily silence the CRISPR system. Following RPA amplification, brief ultraviolet light exposure removes the modifying group, restoring CRISPR activity without physical reagent mixing [67]. This platform demonstrated a detection limit of 10 copies/μL for Acinetobacter baumannii with no cross-reactivity against six common pathogenic bacteria [67]. The complete closure of the reaction vessel throughout the process virtually eliminates aerosol escape.
Table 1: Comparison of Single-Tube Platform Performance Characteristics
| Platform Type | Detection Limit | Assay Time | Contamination Risk | Pathogen Validated |
|---|---|---|---|---|
| Cap-based system [52] | 10 copies (fluorescence)100 copies (LFS) | 20 minutes | Low | MRSA |
| Photocontrolled system [67] | 10 copies/μL | ~30 minutes | Very Low | Acinetobacter baumannii |
Laboratory Practice and Workflow Optimization
Effective contamination control extends beyond reaction design to encompass comprehensive laboratory practices. Implementing structured workflows and standardized procedures significantly reduces contamination risk.
Spatial segregation of laboratory workflows: Maintaining physically separated work areas for different procedural stages prevents cross-contamination. Dedicated spaces should be established for reagent preparation, sample processing, amplification, and detection [68]. Movement between areas should follow a unidirectional workflow, with personnel moving from clean to potentially contaminated areas without backtracking.
Pipetting techniques and equipment: Proper pipetting practices significantly reduce aerosol generation. These include: releasing the pipette plunger slowly to minimize aerosol formation; holding pipettes vertically during use to prevent liquid ingress into the pipette body; using filter tips as physical barriers against aerosol contamination; and regularly decontaminating pipette exteriors with appropriate disinfectants [66].
Reagent and consumable management: Strategic handling of reagents and consumables further mitigates contamination risk. Key practices include: aliquoting reagents into single-use volumes to prevent contamination of master stocks; using high-quality, nuclease-free tips certified for molecular biology applications; and regularly disinfecting work surfaces with freshly prepared sodium hypochlorite solution (5-10%) [68].
Table 2: Essential Research Reagent Solutions for Contamination Control
| Reagent/Consumable | Function | Application Specifics |
|---|---|---|
| Filter pipette tips [66] | Prevent aerosol entry into pipette shafts | Use during all reagent handling steps |
| Nuclease-free water [67] | Molecular biology reagent preparation | Certified free of DNase/RNase activity |
| Sodium hypochlorite [68] | Surface decontamination | 5-10% solution for work surfaces |
| High-quality polypropylene tips [66] | Sample handling | Certified free of leachables and human DNase |
| UV light source [67] | Activation of caged crRNA | 365nm for photochemical uncaging |
Experimental Protocols
One-Tube RPA-CRISPR Protocol with Physical Segregation
This protocol enables contamination-resistant detection of biofilm pathogens using a single-tube, physically segregated format adapted from established methodologies [52].
Reagents and Equipment:
- TwistAmp Liquid Basic RPA kit (TwistDx)
- EnGen LbaCas12a (New England Biolabs)
- crRNA targeting desired biofilm pathogen marker
- Fluorescent or lateral flow reporters
- Nuclease-free water
- Thermal cycler or heat block (37-42°C)
- Microcentrifuge
Procedure:
- RPA Master Mix Preparation (bottom of tube):
- Combine the following in a 0.2mL tube:
- 10μL 2× Reaction Buffer
- 1μL dNTPs
- 2μL 10× E-mix
- 1μL each forward and reverse primer (10μM)
- 1μL 20× Core Reaction mix
- 2μL template DNA
- Nuclease-free water to 23μL
- Briefly centrifuge to collect liquid at tube bottom
- Combine the following in a 0.2mL tube:
CRISPR Detection Mix (tube cap):
- Prepare the detection mix containing:
- 1μL LbaCas12a (1μM)
- 1μL crRNA (2μM)
- 0.25μL RNA inhibitor
- 1μL ssDNA reporter (for fluorescence) or 5μM biotin/FAM-modified reporter (for LFS)
- 2μL 10× NEBuffer 2.1
- Carefully pipet this mixture into the tube cap, avoiding contact with tube walls
- Prepare the detection mix containing:
Amplification and Detection:
- Add 2μL magnesium acetate to the RPA mix, close tube securely
- Incubate at 37-42°C for 10-15 minutes to complete RPA amplification
- Briefly centrifuge (10 seconds) to combine RPA amplicons with CRISPR reagents
- Incubate at 37-42°C for an additional 10-20 minutes for Cas12a detection
Signal Detection:
- For fluorescence: Measure in real-time or endpoint using appropriate excitation/emission
- For lateral flow: Apply reaction mixture to strip and interpret after 5-10 minutes
Photocontrolled One-Tube RPA-CRISPR Protocol
This protocol utilizes photodegradable crRNA modifications to temporally control CRISPR activation, eliminating contamination risk from tube opening [67].
Specialized Reagents:
- NPOM-dt modified crRNA (custom synthesis)
- UV light source (365nm)
- PUC19 plasmid with target sequence
- TIANamp Bacterial DNA Kit
Procedure:
- Reaction Assembly:
- Prepare a master mix containing:
- RPA reagents as in Protocol 4.1
- LbaCas12a protein
- NPOM-dt modified crRNA targeting desired pathogen sequence
- Appropriate fluorescent reporter
- Distribute 25μL aliquots into reaction tubes
- Prepare a master mix containing:
Amplification Phase:
- Incubate tubes at 37-42°C for 15 minutes to complete RPA amplification
- Maintain tube closure throughout this process
Photoactivation:
- Expose closed reaction tubes to 365nm UV light for 2-5 minutes
- This cleaves the NPOM-dt group, activating the crRNA for target binding
Detection Phase:
- Incubate tubes at 37°C for 15 minutes to allow CRISPR detection
- Monitor fluorescence in real-time or measure endpoint signal
Validation:
- Test against a panel of clinically relevant biofilm pathogens
- Compare with conventional PCR for concordance
- Assess sensitivity using serial dilutions of target DNA
Workflow Visualization
Effective minimization of aerosol contamination in single-tube RPA-CRISPR formats requires integrated approach combining innovative reaction design with rigorous laboratory practice. The single-tube platforms described herein—employing either physical segregation of reagents or photochemical control of CRISPR activation—provide robust solutions to contamination challenges while maintaining the sensitivity and specificity required for detecting biofilm-associated pathogens. Implementation of these strategies will enhance the reliability of one-pot diagnostic platforms in both research and potential clinical applications, ultimately supporting more accurate detection and management of biofilm-related infections. As these technologies evolve, continued attention to contamination control measures will be essential for realizing the full potential of RPA-CRISPR diagnostics in complex clinical matrices where biofilm pathogens pose significant diagnostic challenges.
crRNA and Cas Protein Ratio Optimization for Robust Collateral Activity
The integration of recombinase polymerase amplification (RPA) with CRISPR-Cas diagnostics in a one-pot format represents a transformative advancement for detecting biofilm-forming pathogens. This streamlined approach minimizes contamination risk and simplifies operational procedures, making it particularly valuable for point-of-care settings [31]. However, a significant technical challenge impeding its widespread adoption is the suboptimal sensitivity often observed in one-pot systems compared to traditional two-step assays, frequently resulting from premature collateral activity that depletes nucleic acid targets before amplification reaches completion [31].
The core of this challenge lies in the precise stoichiometric relationship between crRNA and Cas proteins. Achieving robust collateral activity—essential for generating strong detection signals—requires careful optimization of this ratio to prevent interference with the initial amplification phase while maintaining high detection sensitivity in the subsequent phase [69] [31]. This protocol details a systematic framework for optimizing crRNA:Cas protein ratios, specifically tailored for one-pot RPA-CRISPR assays targeting biofilm-associated pathogens, incorporating recent advances in thermally regulated systems and collaborative crRNA strategies.
Background and Significance
The Critical Role of Collateral Activity in Detection
CRISPR-based diagnostics leverage the trans-cleavage activity of certain Cas proteins, such as Cas12a and Cas13a, which non-specifically degrade reporter molecules (ssDNA or RNA) after recognizing a target sequence [16] [3]. This collateral activity serves as the primary signal generation mechanism in platforms like DETECTR (Cas12a-based) and SHERLOCK (Cas13a-based) [16]. The intensity and timing of this signal are directly influenced by the concentration and activation kinetics of the Cas ribonucleoprotein (RNP) complex [69].
The One-Pot Assay Challenge
In one-pot RPA-CRISPR systems, all reaction components coexist. Without proper regulation, Cas proteins activated by early-amplified targets can degrade primers and amplicons, severely inhibiting amplification efficiency and reducing overall sensitivity. Studies have shown that unoptimized one-pot assays can exhibit up to 40-fold lower sensitivity compared to their two-step counterparts [31]. Optimal crRNA:Cas ratios establish a delicate balance where the CRISPR system remains sufficiently suppressed during amplification yet achieves maximal activation afterward.
Quantitative Optimization Data
The table below summarizes key experimental parameters and their impact on assay performance, synthesized from recent studies.
Table 1: Quantitative Parameters for crRNA and Cas Protein Optimization
| Optimization Parameter | Optimal Range or Value | Impact on Collateral Activity & Assay Performance | Key Supporting Findings |
|---|---|---|---|
| crRNA:Cas12a Protein Molar Ratio | 1:1 to 3:1 (RNP complex formation) | Maximizes target cleavage and collateral activity; lower ratios risk incomplete complex formation; higher ratios may sequester Cas proteins [69]. | A 1.2:1 ratio demonstrated a 3.5-fold increase in gene-editing efficiency against Pseudomonas aeruginosa biofilms compared to non-carrier systems [70]. |
| Combinatorial crRNA Strategy | 4-6 crRNAs targeting a single gene | Synergistically enhances signal, achieving up to 85-fold improvement in sensitivity versus single-crRNA systems [69]. | A 6-crRNA cocktail reduced the limit of detection (LoD) for the hantavirus M gene to 0.086 pM, compared to 7.31 pM for a single crRNA [69]. |
| ssRNA Blocker-to-gRNA Ratio | 1:4 (molar ratio) | Suppresses premature Cas activity during RPA, enabling a 40x lower LoD in one-pot assays [31]. | The TRACE assay used this with Cas12b, achieving an LoD of 2.5 copies/test, matching two-step assay sensitivity [31]. |
| Final RNP Concentration in Reaction | 200 - 400 nM | Balances high signal output with reagent cost; critical for strong fluorescence in lateral flow detection [31]. | 400 nM RNP yielded the best signal-to-noise ratio for MPXV detection in the TRACE assay [31]. |
Detailed Experimental Protocols
Protocol 1: Determining Optimal crRNA:Cas Protein Ratio
This protocol establishes the foundational stoichiometry for robust RNP complex formation.
4.1.1 Research Reagent Solutions
Table 2: Essential Reagents for Ratio Optimization
| Reagent | Function/Explanation |
|---|---|
| Purified Cas Protein (e.g., LbCas12a) | The effector nuclease; its purity is critical for consistent activity and accurate molar quantification. |
| In Vitro-Transcribed or Synthetic crRNA | Guides Cas protein to the target sequence; requires precise concentration measurement. |
| Fluorescent Reporter Probe (e.g., FAM-TTATT-BHQ1 ssDNA) | A quenched reporter molecule that, when cleaved, produces a fluorescent signal to measure collateral activity. |
| Target DNA Template (Synthetic Amplicon) | A known positive control containing the target sequence to activate the CRISPR system. |
| Nuclease-Free Water | Ensures the reaction environment is free of contaminants that could degrade nucleic acids. |
4.1.2 Procedure
- crRNA Preparation: Dilute the stock crRNA to 1 µM in nuclease-free water.
- Cas Protein Preparation: Dilute the stock Cas protein to 1 µM in provided storage buffer.
- RNP Assembly: Set up a series of 10 µL RNP pre-assembly reactions with varying crRNA:Cas molar ratios (e.g., 0.5:1, 1:1, 1.2:1, 1.5:1, 2:1, 3:1). Incubate at 25°C for 10 minutes.
- Activity Reaction: To each pre-assembled RNP, add:
- 1 µL of 1 µM target DNA template
- 1 µL of 10 µM fluorescent reporter probe
- 3 µL of nuclease-free buffer
- Bring the total volume to 20 µL with nuclease-free water.
- Signal Measurement: Incubate the reactions at 37°C for 30 minutes in a real-time PCR machine or fluorescence reader. Monitor fluorescence every minute.
- Data Analysis: Calculate the maximum fluorescence slope (∆F/∆t) for each reaction. The ratio yielding the highest slope without a significant increase in background signal is optimal.
Protocol 2: Implementing a Combinatorial crRNA Strategy
This protocol enhances sensitivity and robustness by using multiple crRNAs against a single target.
4.2.1 Procedure
- crRNA Design: Using bioinformatics tools, design 4-6 crRNAs targeting highly conserved regions of your biofilm pathogen's gene of interest (e.g., a virulence or antibiotic resistance gene).
- Individual crRNA Testing: Test each crRNA individually using the optimal ratio from Protocol 1 to determine their baseline activities.
- Cocktail Assembly: Prepare a master crRNA cocktail containing equimolar concentrations of all individual crRNAs. The total concentration should match the optimal crRNA concentration determined in Protocol 1.
- Performance Validation: Assemble the RNP complex using the cocktail and compare its limit of detection (LoD) and signal intensity against the best-performing single crRNA using a serial dilution of the target DNA.
Protocol 3: Integrating a Thermo-Regulated Blocker for One-Pot Assays
This protocol, adapted from the TRACE assay, spatially and temporally separates RPA from CRISPR detection using temperature [31].
4.3.1 Procedure
- Blocker Design: Synthesize an ssRNA oligonucleotide that is fully complementary to the spacer region of your chosen crRNA.
- One-Pot Master Mix: Prepare a master mix on ice containing:
- RPA dry powder pellet
- RPA primers
- Target DNA template
- Pre-assembled RNP (at the optimal ratio from Protocol 1)
- Fluorescent reporter probe
- ssRNA blocker at a 4:1 molar ratio relative to the crRNA.
- Thermally Programmed Reaction:
- Amplification Phase: Incubate the reaction at 37°C for 10-15 minutes. The blocker inhibits the RNP, allowing unimpeded RPA amplification.
- Detection Phase: Shift the reaction to 55-60°C for 30 minutes. The higher temperature dissociates the blocker, activating the Cas RNP for target detection and robust collateral cleavage of the reporter.
- Result Interpretation: Analyze the fluorescence curve. A well-optimized assay will show minimal signal during the 37°C phase and a sharp, exponential increase in fluorescence after the temperature shift.
Diagram 1: Workflow of a thermally regulated one-pot RPA-CRISPR assay. The ssRNA blocker prevents premature Cas activity during the RPA phase. A temperature shift deactivates the blocker, enabling the detection phase.
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for crRNA and Cas Protein Studies
| Research Reagent Solution | Function & Application Context |
|---|---|
| LbCas12a (Cas12a from Lachnospiraceae bacterium) | A widely used Type V Cas protein with strong DNA target recognition and collateral ssDNA cleavage activity, ideal for DETECTR-style diagnostics [71]. |
| crRNA Library (Combinatorial Cocktail) | A pool of 4-6 crRNAs targeting a single pathogen gene; employed to enhance sensitivity and diagnostic robustness by synergistically activating Cas proteins [69]. |
| ssRNA Blocker Oligonucleotide | A short, synthetic RNA strand complementary to the crRNA spacer; used in thermally regulated one-pot assays to temporarily inhibit Cas activity during the RPA phase [31]. |
| Fluorescent Reporter Probe (FAM-TTATT-BHQ1) | A single-stranded DNA molecule with a fluorophore (FAM) and a quencher (BHQ1); cleavage by an activated Cas protein produces a measurable fluorescent signal [72]. |
| Nucleic Acid Amplification Kit (RPA) | An isothermal amplification kit used to amplify pathogen DNA directly at 37-42°C, making it compatible with CRISPR enzymes for one-pot diagnostic formats [31]. |
The meticulous optimization of crRNA to Cas protein ratios is not merely a procedural step but a critical determinant for the success of one-pot RPA-CRISPR diagnostics aimed at biofilm pathogens. The synergistic application of a stoichiometrically optimized RNP complex, a multi-crRNA strategy, and a thermo-regulated blocker system provides a comprehensive solution to the historical challenge of low sensitivity in integrated assays. By implementing these detailed protocols, researchers can develop robust, field-deployable diagnostic assays that meet the required sensitivity and specificity for detecting challenging targets like biofilm-forming pathogens in both clinical and environmental settings.
The detection of low-abundance nucleic acid targets in complex samples, such as those containing biofilm-derived pathogens, remains a significant challenge in molecular diagnostics. The presence of inhibitors, background debris, and the intrinsic heterogeneity of biofilms can severely impair assay sensitivity and reliability. For research focusing on one-pot RPA-CRISPR diagnostics, enhancing the detection limit is paramount for accurate pathogen identification and subsequent drug development. This Application Note outlines optimized approaches and detailed protocols to overcome these barriers, leveraging the synergistic combination of RPA pre-amplification with CRISPR/Cas12a's specific detection capabilities. The strategies discussed herein are designed to achieve sensitivity down to single-copy levels, enabling researchers to reliably detect minimal pathogen loads within intricate sample matrices.
Key Technical Approaches for Enhanced Sensitivity
Strategic Temperature Optimization
A primary strategy for enhancing detection limits involves the systematic optimization of reaction temperatures. The one-pot, two-temperature protocol represents a significant advancement over single-temperature incubations. This approach physically separates the amplification and detection phases, optimizing each under different conditions. Recombinase Polymerase Amplification (RPA) is first performed at 39°C for optimal enzyme activity and rapid primer binding. Subsequently, the temperature is raised to 62°C to activate the AapCas12b enzyme's trans-cleavage activity [6]. This elevated temperature enhances the signal-to-noise ratio by maximizing Cas12b's collateral cleavage efficiency while minimizing non-specific background signal, which is crucial for distinguishing true low-abundance targets from false positives. This method has been clinically validated, showing a 96.7% concordance with culture methods and 98.3% with qPCR when testing clinical swab samples [6].
Streamlined, Extraction-Free Sample Preparation
The requirement for nucleic acid extraction presents a major bottleneck where target loss frequently occurs, disproportionately affecting low-abundance targets. Implementing extraction-free protocols with minimal pretreatment can drastically improve the recovery of trace nucleic acids. Clinical samples, such as vaginal-rectal swabs, can be processed by incubating the swab in a simple buffer at room temperature or via a brief heat treatment (e.g., 95°C for 5 minutes) to lyse cells and release DNA directly into the solution [6]. This lysate can then be added directly to the pre-mixed one-pot reaction. This approach not only simplifies the workflow but also preserves the integrity of low-copy-number targets that might otherwise be lost during column-based or magnetic bead-based purification steps.
Research Reagent Solutions for RPA-CRISPR/Cas12a Assays
The following table details essential reagents and their critical functions in establishing a robust one-pot RPA-CRISPR detection system for challenging samples.
Table 1: Key Research Reagent Solutions for One-Pot RPA-CRISPR/Cas12a Assays
| Reagent / Component | Function & Importance in Low-Abundance Detection |
|---|---|
| AapCas12b / LbaCas12a | Thermostable Cas enzymes; AapCas12b is ideal for two-temperature protocols due to high-temperature activation (62°C), reducing background noise [6]. LbaCas12a is widely used for its efficient trans-cleavage at ~37°C [38]. |
| RPA Basic Kit (TwistAmp) | Provides core enzymes (recombinase, SSB, polymerase) for rapid isothermal amplification at 39-42°C, enabling quick target enrichment without thermal cycling [6] [38]. |
| Fluorescent ssDNA Reporter | (e.g., FAM-TTATT-BHQ1). The Cas enzyme's trans-cleavage substrate. Signal generation is directly proportional to target amplification; crucial for quantifying low-level signals [38] [73]. |
| Custom-designed crRNA | Guides Cas enzyme to the specific target sequence. High-quality, specific crRNA is vital for distinguishing homologous sequences in complex samples and minimizing off-target activation [18]. |
| Silicon Hydroxyl Magnetic Beads | Used for rapid, efficient genomic DNA extraction from lysed samples. Offers higher recovery rates of low-concentration DNA compared to some column-based methods [73]. |
| Portable Fluorescence Detector / UV Light | Enables endpoint or real-time fluorescence readout. Essential for Point-of-Care Testing (POCT) and resource-limited settings, allowing for visual or instrument-based detection of positive signals [18] [38]. |
Detailed Experimental Protocol for a One-Pot Two-Temperature RPA-CRISPR/Cas12b Assay
This protocol is designed for the detection of low-abundance pathogen DNA from complex samples, such as biofilm isolates or clinical swabs, with a total processing time of under 60 minutes.
Sample Pretreatment and DNA Release
- Sample Collection: Collect the sample (e.g., a swab from a biofilm surface or clinical specimen) and place it into a microcentrifuge tube containing 200 µL of a mild lysis buffer (e.g., 10 mM Tris-HCl, pH 8.0).
- DNA Release: Incubate the tube at room temperature for 5 minutes, with occasional vortexing. Alternatively, for more robust lysis, heat the tube at 95°C for 5 minutes.
- Clarification: Centrifuge the tube at 10,000 x g for 2 minutes to pellet debris. The supernatant, containing the crude nucleic acid lysate, is ready for use.
One-Pot Reaction Setup
- Prepare Master Mix: In a single 0.2 mL PCR tube, combine the following components to form a 25 µL reaction [6]:
- 10 µL of 2.5x RPA rehydration buffer (from TwistAmp Basic kit)
- 1.0 µL of each RPA primer (10 µM each)
- 1.0 µL of crRNA (2 µM)
- 1.0 µL of AapCas12b (1 µM)
- 1.0 µL of fluorescent ssDNA reporter (e.g., FAM-TTATT-BHQ1, 10 µM)
- Nuclease-free water to adjust the volume.
- Add Template: Add 2 µL of the prepared sample lysate (supernatant from step 4.1) directly to the master mix.
- Initiate Amplification: Add 2.5 µL of Magnesium Acetate (280 mM) to the tube lid, close the tube, and briefly centrifuge to mix. This step initiates the RPA reaction.
Two-Temperature Incubation and Detection
- RPA Amplification: Immediately place the tube in a preheated thermal block or water bath at 39°C. Incubate for 20-40 minutes to allow for isothermal amplification of the target DNA [6].
- CRISPR/Cas12b Detection: Without opening the tube, transfer the reaction to a second preheated block at 62°C. Incubate for 5-10 minutes to activate the trans-cleavage activity of AapCas12b.
- Result Visualization: After incubation, visualize the results under a blue light transilluminator. A positive reaction will emit bright green fluorescence, while a negative reaction will remain dark. For quantification, use a portable fluorescence reader to measure real-time or endpoint fluorescence intensity.
Quantitative Performance of Optimized RPA-CRISPR Assays
The following table summarizes the demonstrated sensitivity and performance of various optimized RPA-CRISPR platforms in detecting pathogens in complex matrices.
Table 2: Analytical Performance of RPA-CRISPR Platforms for Pathogen Detection
| Pathogen / Target | Assay Format | Sample Matrix | Limit of Detection (LoD) | Key Enhancement for Sensitivity |
|---|---|---|---|---|
| Group B Streptococcus (GBS) [6] | One-pot, two-temperature RPA-CRISPR/Cas12b | Vaginal-rectal swabs | 10 copies/test (1 copy/µL) | Two-temperature protocol (39°C → 62°C) |
| Candida albicans [73] | Affinity-AMS + RPA-CRISPR/Cas12a one-pot | Blood, BAL fluid | 30 CFU/mL | Affinity-magnetic separation (AMS) for target enrichment |
| Pseudomonas aeruginosa (lasB gene) [18] | One-tube RPA-CRISPR/Cas12a | Sputum, wound secretions | 15.9 CFU/reaction | Optimized crRNA design and one-tube format |
| Methicillin-Resistant Staphylococcus aureus (mecA gene) [38] | One-tube RPA-CRISPR/Cas12a | Clinical isolates | 10 copies (fluorescence) | Spatial separation of RPA and CRISPR in a single tube |
Workflow and Mechanism Visualization
The diagram below illustrates the integrated workflow and molecular mechanism of the one-pot two-temperature RPA-CRISPR/Cas12b assay.
Addressing Inhibitor Interference from Biofilm Matrices and Clinical Samples
The development of robust one-pot recombinase polymerase amplification (RPA)-CRISPR diagnostics for biofilm-forming pathogens represents a transformative advancement in clinical microbiology. However, the complex composition of biofilm matrices and clinical samples introduces significant inhibitor interference that can compromise assay sensitivity and specificity. Biofilms are structured microbial communities embedded in extracellular polymeric substances (EPS) that provide physical and chemical barriers to molecular diagnostics [12] [74]. These EPS matrices contain polysaccharides, proteins, lipids, and extracellular DNA that can inhibit nucleic acid amplification enzymes and CRISPR-Cas protein activity [2] [12]. Similarly, clinical specimens such as vaginal-rectal swabs contain hemoglobin, immunoglobulins, and other compounds that interfere with molecular assays [6]. This application note provides detailed protocols for addressing these challenges within the context of one-pot RPA-CRISPR diagnostics, enabling reliable detection of biofilm-associated pathogens in complex samples.
Composition and Interference Mechanisms
Biofilm matrices create multiple interference mechanisms that challenge molecular diagnostics. The extracellular polymeric substance (EPS) presents a physical barrier that limits reagent penetration and target accessibility [74]. Chemically, the matrix contains nucleases that degrade RNA and DNA targets, polysaccharides that inhibit polymerase activity, and proteins that sequester Cas nucleases [12]. Clinical samples introduce additional challenges, including heme compounds from blood that inhibit RPA amplification, proteases that degrade Cas proteins, and high viscosity that impedes reagent mixing [6].
The one-pot RPA-CRISPR/Cas12b system for Group B Streptococcus (GBS) detection demonstrates that inhibitor interference can reduce sensitivity by 1-2 orders of magnitude in unoptimized assays [6]. Without proper countermeasures, this interference leads to false-negative results, particularly in samples with low bacterial loads (<100 CFU/test). The integration of extraction-free processing further compounds these challenges, requiring robust inhibitor mitigation strategies within the simplified workflow.
Quantitative Impact of Inhibitors on Assay Performance
Table 1: Quantitative Effects of Common Inhibitors on RPA-CRISPR Components
| Inhibitor Type | Source | Effect on RPA Amplification | Effect on Cas12b Activity | Overall Signal Reduction |
|---|---|---|---|---|
| Polysaccharides | Biofilm EPS | 40-60% reduction in amplicon yield | 20-30% reduction in cleavage activity | 50-70% |
| Heme Compounds | Blood contamination | 70-80% inhibition at 10 µM concentration | Minimal direct effect | 60-75% |
| Mucopolysaccharides | Vaginal-rectal swabs | 30-50% reduction in amplification efficiency | 15-25% reduction in Cas activity | 35-55% |
| Proteases | Bacterial secretion/Inflammation | Minimal direct effect | 40-60% degradation of Cas proteins | 40-60% |
| EDTA | Sample collection media | Complete inhibition at >1 mM concentration | No significant effect | >90% |
Methodologies for Inhibitor Management
Extraction-Free Sample Processing Protocol
The following protocol enables effective inhibitor management while maintaining the rapid, simple workflow essential for point-of-care applications:
A. Sample Collection and Pre-treatment
- Collect vaginal-rectal swabs using standard clinical procedures
- Place swab in 1 mL of specialized pretreatment buffer (10 mM Tris-HCl, pH 8.0, 0.1% Triton X-100, 1% polyvinylpyrrolidone)
- Vortex vigorously for 30 seconds to dislodge biofilm aggregates
- Incubate at room temperature for 5 minutes [6]
B. Heat-Based DNA Release and Inhibitor Denaturation
- Transfer 200 µL of sample supernatant to a clean microcentrifuge tube
- Heat at 95°C for 5 minutes in a dry bath or heat block
- Centrifuge at 10,000 × g for 1 minute to pellet insoluble debris
- Retain supernatant for direct addition to RPA-CRISPR reactions [6]
C. One-Pot Reaction Assembly
- Prepare master mix containing:
- 10.2 µL nuclease-free water
- 2.5 µL 10× RPA buffer
- 1.0 µL RPA primer mix (10 µM each)
- 1.0 µL crRNA (2 µM)
- 0.5 µL AapCas12b (10 µM)
- 0.3 µL single-stranded DNA reporter (100 µM)
- 4.5 µL rehydration buffer
- Add 5 µL of processed sample supernatant to 20 µL of master mix
- Incubate at 39°C for 40 minutes for RPA amplification
- Increase temperature to 62°C for 5 minutes to activate Cas12b cleavage
- Visualize results under UV light (blue light: positive = green fluorescence) [6]
Optimization Strategies for Enhanced Resistance
A. Reaction Composition Optimization
- Include 0.1% bovine serum albumin (BSA) to sequester proteases and stabilize enzymes
- Add 2% polyvinylpyrrolidone (PVP) to adsorb polyphenolic compounds
- Incorporate 0.01% Tween-20 to reduce surface binding of reagents
- Use 50-100 mM betaine as a crowding agent to enhance enzyme stability [6]
B. Two-Temperature Incubation Protocol The sequential temperature optimization significantly improves signal-to-noise ratio in complex samples:
- Amplification Phase: 39°C for 40 minutes enables efficient RPA amplification while minimizing non-specific products
- Detection Phase: 62°C for 5 minutes optimally activates AapCas12b trans-cleavage activity while denaturing residual inhibitors [6]
Table 2: Optimization of Two-Temperature Protocol for Inhibitor Resistance
| Temperature Scheme | Sensitivity (Copies/Test) | Time to Positive Signal | Inhibitor Resistance Level |
|---|---|---|---|
| Single Temperature (39°C) | 100 | 25-35 minutes | Low-Moderate |
| Single Temperature (62°C) | 1000 | >60 minutes | High |
| Two-Temperature (39°C→62°C) | 10 | 40-45 minutes | High |
| Gradual Ramp (39°C→62°C over 30 min) | 50 | 50-60 minutes | Moderate-High |
Experimental Validation and Performance Metrics
Protocol for Inhibitor Spike-in Studies
To quantitatively evaluate inhibitor resistance, conduct controlled spike-in experiments:
A. Inhibitor Stock Preparation
- Prepare 10 mg/mL alginate solution in TE buffer to simulate biofilm EPS
- Create 10 mM hemin solution in DMSO to represent blood contamination
- Reconstitute mucin from porcine stomach at 20 mg/mL in PBS for clinical sample simulation
B. Dose-Response Analysis
- Add inhibitors to GBS-positive samples at clinically relevant concentrations:
- Alginate: 0.1, 0.5, 1.0 mg/mL
- Hemin: 1, 5, 10 µM
- Mucin: 0.5, 1.0, 2.0 mg/mL
- Process samples using the extraction-free protocol
- Compare results to uninhibited controls using fluorescence intensity and time-to-positive metrics [6]
C. Data Analysis
- Calculate percentage signal reduction relative to uninhibited controls
- Determine limit of detection shift for each inhibitor concentration
- Establish threshold values for acceptable performance degradation
Clinical Validation with Complex Samples
The extraction-free one-pot RPA-CRISPR/Cas12b assay was validated using 60 vaginal-rectal swab samples, demonstrating 96.7% concordance with culture methods and 98.3% concordance with qPCR despite the complex sample matrix [6]. The assay maintained sensitivity of 10 copies/test (1 copy/μL) in these challenging clinical samples, representing a significant improvement over unoptimized protocols.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Managing Inhibitor Interference
| Reagent | Function | Optimized Concentration | Mechanism of Action |
|---|---|---|---|
| AapCas12b | Thermostable Cas nuclease for detection | 0.5 µL of 10 µM stock | CRISPR-based specific DNA detection; thermostability resists inhibitor denaturation |
| Triton X-100 | Surfactant for sample processing | 0.1% in pretreatment buffer | Disrupts biofilm matrix and enhances cell lysis |
| Polyvinylpyrrolidone (PVP) | Polymer for inhibitor sequestration | 1-2% in reaction mix | Binds polyphenolic compounds and polysaccharides |
| Betaine | Osmoprotectant and crowding agent | 50-100 mM in reaction | Stabilizes enzyme structure and enhances amplification efficiency |
| BSA (Bovine Serum Albumin) | Protein stabilizer | 0.1% in reaction mix | Competes for protease activity and prevents reagent adsorption |
| Single-stranded DNA Reporter | Fluorescent signal generation | 0.3 µL of 100 µM stock | Cas12b collateral cleavage substrate for visual detection |
Workflow Visualization
Diagram 1: Complete workflow for addressing inhibitor interference in one-pot RPA-CRISPR diagnostics, showing sample processing, reaction assembly, and detection phases with critical optimization steps.
Diagram 2: Inhibitor interference mechanisms and mitigation strategy framework, showing relationships between inhibitor sources, their mechanisms of action, and corresponding countermeasures.
Benchmarking One-Pot RPA-CRISPR: Performance Validation Against Gold-Standard Methods
In the development of molecular diagnostics for biofilm-forming pathogens, analytical sensitivity defines the lowest concentration of a target that an assay can reliably detect. Establishing this for plasmid DNA and clinical isolates is a critical step in validating one-pot RPA-CRISPR assays, which combine recombinase polymerase amplification (RPA) with CRISPR-Cas detection in a single reaction tube. These assays are being engineered for precision detection of pathogens in complex biofilm matrices, where sensitivity and speed are paramount for timely intervention [12]. This protocol details the methodologies for determining the limits of detection (LOD) for both plasmid DNA standards and clinical isolates, providing a framework for validating one-pot RPA-CRISPR diagnostics within biofilm pathogen detection research.
Experimental Protocols
Plasmid DNA Standard Preparation and LOD Determination
This protocol describes the generation of a quantitative standard for precisely determining the analytical sensitivity of a one-pot RPA-CRISPR assay.
- Primer and crRNA Design: Identify a conserved, species-specific genomic region of the target pathogen (e.g., the cfb gene for Group B Streptococcus or the mecA gene for MRSA) via multiple sequence alignment [6] [38]. Design RPA primers using software such as SnapGene. Design crRNAs to target adjacent, non-overlapping regions within the amplified sequence [6].
- Plasmid Construction and Quantification: Clone the target amplicon sequence into a standard plasmid vector (e.g., pUC57). Transform into an appropriate E. coli strain. Isolate plasmid DNA using a commercial kit and quantify concentration spectrophotometrically [6] [38].
- Standard Curve Generation: Perform a 10-fold serial dilution of the purified plasmid in a background solution such as nuclease-free water or TE buffer, spanning a range from 10^6 to 10^0 copies/µL. Confirm dilution accuracy via digital PCR if available.
- LOD Determination Assay: Run the one-pot RPA-CRISPR assay in replicate (n ≥ 8-10) for each plasmid dilution. The LOD is defined as the lowest copy concentration at which ≥95% of the replicates return a positive result, as determined by fluorescence or lateral flow readout [6] [38].
Clinical Isolate Processing and LOD Determination
This protocol establishes the LOD using live bacterial cells, validating assay performance with biologically relevant samples.
- Bacterial Culture and Cell Counting: Streak the target clinical isolate (e.g., methicillin-resistant Staphylococcus aureus [MRSA] or biofilm-forming Escherichia coli) onto an agar plate and incubate overnight. Pick single colonies to inoculate liquid broth and grow to mid-log phase. Determine the cell density (CFU/mL) by measuring the optical density at 600 nm (OD₆₀₀) and correlating with a standardized growth curve [75].
- Sample Preparation (Extraction-Free): To maximize point-of-care applicability, employ a simple, extraction-free sample preparation. Pellet a known volume of bacterial culture, resuspend in nuclease-free water or a mild lysis buffer, and incubate at 95°C for 5 minutes to lyse cells and release genomic DNA. Centrifuge briefly, and use the supernatant directly as the template for the assay [6].
- Serial Dilution and Assay: Perform a 10-fold serial dilution of the heat-treated lysate in a solution matching the sample matrix. Quantify the number of CFU/mL in the initial culture to back-calculate the approximate genomic copy number per test in each dilution.
- LOD Determination: Test each dilution in multiple replicates (n ≥ 8-10) using the one-pot RPA-CRISPR platform. The LOD is reported as the lowest number of CFU per reaction that yields a positive signal in ≥95% of replicates [6].
Results and Data Presentation
The following tables consolidate typical LOD results for one-pot RPA-CRISPR assays using plasmid DNA and clinical isolates.
Table 1: Limits of Detection for Plasmid DNA and Clinical Isolates in One-Pot RPA-CRISPR Assays
| Pathogen Target | Assay Type | Template | Reported LOD | Reference |
|---|---|---|---|---|
| Group B Streptococcus | RPA-CRISPR/Cas12b | Plasmid DNA | 10 copies/test | [6] |
| Methicillin-Resistant S. aureus (MRSA) | RPA-CRISPR/Cas12a | Plasmid DNA | 10 copies | [38] |
| Group B Streptococcus | RPA-CRISPR/Cas12b | Clinical Vaginal-Rectal Swabs | Comparable to 10-copy plasmid standard | [6] |
| Methicillin-Resistant S. aureus (MRSA) | RPA-CRISPR/Cas12a | Clinical MRSA Isolates | Coincidence rate of 95.7-100% vs. qPCR | [38] |
Table 2: Key Parameters for One-Pot RPA-CRISPR/Cas12b Assay
| Parameter | Specification | Protocol Reference |
|---|---|---|
| RPA Amplification | 39°C for 40 min | [6] |
| CRISPR/Cas12b Detection | 62°C for 5-10 min | [6] |
| Total Assay Time | < 60 min | [6] [38] |
| Sample Input Volume | 2-5 µL | [6] |
| Readout Methods | Fluorescence (real-time/end-point), Lateral Flow Strips | [6] [38] |
Experimental Workflow Visualization
The following diagram illustrates the integrated workflow for determining the analytical sensitivity of a one-pot RPA-CRISPR assay.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for One-Pot RPA-CRISPR Assay Development
| Item | Function/Description | Example |
|---|---|---|
| RPA Basic Kit | Provides recombinase, polymerase, and proteins for isothermal amplification at 39-42°C. | TwistAmp Liquid Basic Kit [6] [38] |
| Cas Nuclease | CRISPR-associated nuclease that provides specific target binding and collateral cleavage activity. | AapCas12b (thermostable) or LbaCas12a [6] [38] |
| crRNA | CRISPR RNA; guides the Cas nuclease to the specific target DNA sequence. | Synthesized in vitro from a DNA template with a T7 promoter [6] [38] |
| Fluorescent Reporter | Single-stranded DNA oligonucleotide labeled with a fluorophore and quencher; cleaved collateraly for real-time signal generation. | ssDNA FQ Reporter (e.g., 5'-6-FAM/TTATT/3'-BHQ1) [38] |
| Lateral Flow Strips | For endpoint visual detection, utilizing labeled reporter molecules. | Commercial strips for biotin- and FAM-labeled products [38] |
| Plasmid Cloning System | For generating a quantifiable standard containing the target sequence. | pUC57 Vector, DH5α Competent Cells [38] |
The shift from traditional culture-based methods to molecular diagnostics represents a significant advancement in clinical microbiology. This application note details the clinical validation of one-pot Recombinase Polymerase Amplification (RPA) coupled with CRISPR/Cas systems for detecting biofilm-forming pathogens, focusing on concordance rates with traditional culture and quantitative PCR (qPCR) from patient samples. This integrated approach combines the rapid, isothermal amplification capability of RPA with the precise targeting of CRISPR/Cas systems, creating a powerful diagnostic tool particularly suitable for detecting pathogens in complex biofilm-associated infections [27].
Biofilm-forming pathogens present a substantial challenge in clinical settings due to their inherent resistance to conventional antibiotics and limitations of traditional diagnostic methods. Culture-based techniques, while considered the historical gold standard, are time-consuming, require viable pathogens, and often fail to accurately represent the polymicrobial nature of biofilms [76] [77]. Molecular methods like qPCR offer improved sensitivity and speed but still require sophisticated equipment and laboratory infrastructure, limiting their point-of-care utility [78]. The one-pot RPA-CRISPR platform addresses these limitations by enabling rapid, highly specific detection with minimal instrumentation, making it particularly valuable for diagnosing biofilm-related infections where timely, targeted treatment is critical [6] [27].
Comparative Performance of Diagnostic Methods
Concordance with Reference Standards
Table 1: Clinical Concordance Rates of RPA-CRISPR with Reference Methods
| Pathogen | Sample Type | Comparison Method | Concordance Rate | Key Performance Metrics | Source |
|---|---|---|---|---|---|
| Group B Streptococcus | Vaginal-rectal swabs | Culture | 96.7% | Sensitivity: 10 copies/test | [6] |
| Group B Streptococcus | Vaginal-rectal swabs | qPCR | 98.3% | Specificity: 100% | [6] |
| Klebsiella pneumoniae | Clinical isolates | PCR | 100% (30/30) | LOD: 4.072×10² copies/reaction | [79] |
| Common Respiratory Pathogens* | Sputum (COPD patients) | Culture | 75.6%-82.0% | Higher sensitivity with qPCR | [78] |
*Includes Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae.
The one-pot RPA-CRISPR platform demonstrates exceptional concordance with both culture and qPCR reference methods across multiple pathogens. For Group B Streptococcus detection, the platform achieved 96.7% concordance with traditional culture methods and 98.3% concordance with qPCR in a clinical validation study of 60 vaginal-rectal swab samples [6]. The assay demonstrated high sensitivity, detecting down to 10 copies per test, which is particularly valuable for identifying low-level colonization [6].
Similarly, for Klebsiella pneumoniae detection, a light-controlled one-pot RPA-CRISPR/Cas12a method showed 100% concordance with conventional PCR in clinical sample validation, perfectly aligning with PCR results across all 30 tested samples [79]. This method demonstrated a detection limit of 4.072×10² copies per reaction, highlighting the sensitivity achievable with this technology [79].
When compared to culture-based methods, molecular techniques consistently show advantages. A comprehensive analysis of respiratory pathogen detection in COPD patients revealed that real-time PCR had significantly higher positivity rates for fastidious organisms like Haemophilus influenzae (43.4% vs 26.2%) and Moraxella catarrhalis (12.9% vs 6.3%) compared to culture [78]. The overall agreement between culture and PCR for H. influenzae detection ranged from 75.6% to 82.0% across three clinical studies, with most discrepancies arising from culture-negative but PCR-positive samples, indicating the superior sensitivity of molecular methods [78].
Methodological Comparison
Table 2: Comparison of Diagnostic Method Capabilities
| Parameter | Traditional Culture | qPCR | One-pot RPA-CRISPR |
|---|---|---|---|
| Time to Result | 24-72 hours | 1-2 hours | <1 hour (<50 minutes) |
| Equipment Needs | Incubators, biosafety cabinets | Thermal cyclers, detection systems | Minimal (isothermal, visual readout) |
| Hands-on Time | High (processing, interpretation) | Moderate | Low (<5 minutes) |
| Sensitivity | Moderate (requires viable organisms) | High (1-100 copies/μL) | Very High (as low as 10 copies/test) |
| Specificity | Moderate (phenotypic) | High | Very High (sequence-specific) |
| Point-of-Care Suitability | Low | Low | High |
| Biofilm Pathogen Detection | Limited (planktonic bias) | Moderate | High (molecular detection) |
| Cost per Test | Low | High | Moderate |
The one-pot RPA-CRISPR system offers substantial advantages in detection speed, completing analysis in under 1 hour (as little as 50 minutes for KP detection) compared to 24-48 hours for culture and 1-2 hours for qPCR [6] [79]. This rapid turnaround is critical for initiating targeted therapy, especially in biofilm-associated infections where delayed treatment can lead to poor outcomes.
For biofilm-forming pathogens like Pseudomonas aeruginosa, conventional susceptibility testing often fails to predict clinical response because standard MIC determinations target planktonic cells, while biofilms exhibit dramatically increased resistance to antimicrobial agents [76]. One study demonstrated that biofilm inhibitory concentrations (BICs) for β-lactam antibiotics were 32-64 times higher than corresponding MICs for planktonic cells [76]. This highlights the critical need for detection methods that can identify pathogens regardless of their growth state, a key advantage of molecular approaches like RPA-CRISPR that detect genetic material rather than relying on viable, culturable organisms.
Experimental Protocols
One-Pot RPA-CRISPR/Cas12b Assay for GBS Detection
Protocol: Extraction-Free, One-Pot Two-Temperature RPA-CRISPR/Cas12b Assay
- Sample Preparation: Clinical swab samples undergo minimal pretreatment. DNA is released by incubating swabs in buffer at room temperature or via brief heat treatment (95°C for 5 minutes). The lysate is directly added to the reaction mixture without nucleic acid extraction [6].
- Reaction Setup: The one-pot reaction contains all necessary RPA and CRISPR/Cas12b reagents in a single tube:
- RPA rehydration buffer
- Forward and reverse primers (400 nM optimal concentration)
- crRNA (300 nM optimal concentration)
- AapCas12b enzyme (300 nM optimal concentration)
- Fluorescent reporter molecule
- Magnesium acetate (to initiate amplification)
- Pretreated sample lysate
- Amplification Phase: Incubate the reaction at 39°C for 40 minutes to allow RPA-mediated isothermal amplification of target DNA [6].
- Detection Phase: Increase temperature to 62°C for 5 minutes to activate AapCas12b-mediated trans-cleavage. At this optimal temperature, the Cas12b-crRNA complex binds to amplified target DNA, activating collateral cleavage that degrades the reporter molecule and generates a fluorescence signal [6].
- Result Interpretation: Detect fluorescence using a portable reader or visualize with the naked eye under blue or UV light. The entire assay, from sample to result, is completed in under 1 hour [6].
Light-Controlled RPA-CRISPR/Cas12a for KP Detection
Protocol: Single-Tube, Light-Controlled RPA-CRISPR/Cas12a Detection
- Primer and crRNA Design: Design RPA primers targeting the conserved rcsA gene of KP. Design crRNA with photocleavable group (NPOM-dt) incorporation to temporarily inhibit Cas12a activation [79].
- Reaction Assembly: Combine in a single tube:
- RPA reaction components (primers at 400 nM)
- Cas12a enzyme (300 nM)
- Photocaged crRNA (300 nM)
- Fluorescent-quenched reporter
- Sample DNA
- Amplification: Incubate at 37-42°C for 20 minutes for RPA amplification [79].
- Photoactivation: Expose the reaction tube to UV light for 30 seconds to cleave the photocleavable group and activate the crRNA [79].
- Detection: Continue incubation at 37°C for signal development (approximately 10-20 minutes). The activated CRISPR/Cas12a system recognizes amplified targets and exhibits trans-cleavage activity, generating a detectable fluorescence signal [79].
- Analysis: Read results fluorometrically or visually under UV light. The method achieves detection limits of 4.072×10² copies/reaction with 100% specificity for KP against seven common clinical strains [79].
Technological Workflow and Mechanisms
The following diagram illustrates the integrated workflow and molecular mechanism of the one-pot RPA-CRISPR detection system:
This integrated workflow demonstrates how the one-pot RPA-CRISPR system combines sample processing, nucleic acid amplification, and specific detection in a single tube, significantly simplifying the diagnostic process while maintaining high sensitivity and specificity. The molecular mechanism highlights the sequence-specific recognition through crRNA guidance followed by the collateral trans-cleavage activity that enables sensitive signal detection.
Research Reagent Solutions
Table 3: Essential Research Reagents for RPA-CRISPR Assay Development
| Reagent Category | Specific Examples | Function in Assay | Implementation Considerations |
|---|---|---|---|
| Recombinase Enzyme | T4 uvsX recombinase | Binds primers and invades dsDNA to initiate RPA | Requires single-stranded binding proteins for stability |
| DNA Polymerase | Bsu DNA polymerase | Extends primers after strand invasion | Must have strand-displacing activity |
| Cas Proteins | Cas12a, Cas12b | Target-specific binding and trans-cleavage | Cas12b offers higher thermostability; Cas12a has simpler PAM requirements |
| crRNA Components | Custom crRNA designs | Guides Cas protein to specific target sequence | Can be modified with photocleavable groups (NPOM) for temporal control |
| Signal Reporters | FQ-labeled ssDNA reporters | Fluorescent signal generation upon trans-cleavage | Various fluorophore-quencher pairs available for different detection platforms |
| Primer Sets | Species-specific RPA primers | Amplification of target pathogen DNA | Typically 30-35 bp; designed to conserved genomic regions |
| Reaction Buffers | Optimized buffer systems | Maintain optimal pH and reaction conditions | Often include crowding agents and ATP regeneration systems |
Discussion and Clinical Implications
The validation data presented demonstrate that one-pot RPA-CRISPR diagnostics achieve excellent concordance with both culture (96.7%) and qPCR (98.3%) methods while offering significant advantages in speed, simplicity, and potential for point-of-care use [6]. This technology is particularly suited for detecting biofilm-associated pathogens where traditional culture methods often fail due to the viability challenges and antimicrobial resistance of biofilm-embedded bacteria [76].
The ability of RPA-CRISPR platforms to detect pathogens without cultivation makes them invaluable for identifying uncultivable or fastidious organisms in biofilm communities. Studies have shown that molecular methods like qPCR can detect uncultivable periodontal pathogens such as Eubacterium brachy, E. saphenum, and Filifactor alocis with high sensitivity (82.2-100%) and specificity (100%), organisms that are frequently missed by conventional culture [80]. Similarly, in diabetic foot ulcer infections, next-generation sequencing revealed an average of 5.1 pathogens per sample compared to only 2.6 pathogens detected by standard culture, highlighting the limitations of culture in polymicrobial biofilm infections [77].
For clinical researchers and drug development professionals, one-pot RPA-CRISPR systems offer a rapid, precise tool for pathogen identification that can guide targeted antibiotic therapy, especially crucial for biofilm-related infections. The technology's capability to detect resistance genes, such as mecA/C (methicillin resistance), blaKPC (carbapenemase), and blaCTX-M (extended-spectrum beta-lactamase), further enhances its clinical utility for antimicrobial stewardship [81]. As these platforms continue to evolve with innovations like light-controlled activation and extraction-free protocols, they hold promise for transforming the diagnosis and management of complex biofilm-associated infections across diverse clinical settings.
The development of one-pot recombinase polymerase amplification (RPA)-CRISPR diagnostics for biofilm-forming pathogens represents a transformative advancement in molecular detection technologies. However, the complex ecological landscape of clinical and environmental samples presents a significant challenge: the potential for false-positive results due to cross-reactivity with commensal microorganisms or genetically related pathogenic strains. Specificity testing through rigorous cross-reactivity assessment is therefore a critical validation step to ensure diagnostic accuracy and clinical utility [4] [16].
The unique combination of RPA isothermal amplification with CRISPR-Cas12a detection creates a system with exceptional sensitivity, capable of detecting target nucleic acids at attomole (aM) levels [11] [82]. This very sensitivity, however, increases vulnerability to non-specific signal generation when closely related non-target organisms share genetic homology with the intended pathogen target. This challenge is particularly acute when detecting biofilm-associated pathogens, as these organisms frequently coexist within complex polymicrobial communities where commensal bacteria and related pathogens may be present in high abundance [83] [84].
This application note provides a comprehensive framework for evaluating the specificity of one-pot RPA-CRISPR assays through systematic cross-reactivity assessment. By establishing standardized protocols and validation criteria, we aim to enable researchers to develop robust diagnostic tests that maintain high sensitivity while minimizing false-positive results, thereby ensuring reliable performance in real-world applications targeting biofilm-forming pathogens.
Principles of RPA-CRISPR Specificity
Molecular Basis of Specificity in One-Pot RPA-CRISPR Assays
The exceptional specificity of RPA-CRISPR diagnostics arises from the multi-layered molecular recognition built into the system. The initial specificity layer occurs during RPA amplification, where primer binding specificity determines which nucleic acid sequences undergo exponential amplification. The recombinase-primer complexes scan double-stranded DNA and form primer-template junctions only when sufficient complementarity exists [11]. This process is facilitated by single-strand binding proteins that stabilize the displaced DNA strands, preventing primer displacement by branch migration [11].
The second, and more stringent, specificity layer is provided by the CRISPR-Cas12a system. Cas12a ribonucleoproteins programmed with CRISPR RNA (crRNA) recognize and bind to specific protospacer adjacent motif (PAM) sequences and subsequently unwind the adjacent DNA to enable crRNA-target DNA hybridization [11] [16]. Only upon precise target binding is the trans-cleavage activity of Cas12a activated, leading to cleavage of fluorescent reporter molecules and signal generation [16]. This dual-recognition mechanism – requiring both PAM recognition and crRNA-DNA complementarity – provides exceptional discrimination, potentially down to single-base differences [4].
The integration of these processes into a one-pot reaction introduces additional complexity for specificity optimization. Reaction conditions must simultaneously support efficient RPA amplification and highly specific Cas12a detection, requiring careful balancing of magnesium concentrations, temperature, and reaction pH to maximize both sensitivity and specificity [85].
Cross-Reactivity Mechanisms in Microbial Communities
In complex sample matrices such as biofilms, several mechanisms can contribute to cross-reactivity in molecular diagnostics. Genetic homology represents the most straightforward mechanism, where shared genomic regions between target and non-target organisms lead to non-specific amplification or detection [83]. Functional homology presents a more subtle challenge, where different genetic sequences may encode similar functional domains with sufficient similarity to permit crRNA binding under suboptimal conditions.
Biofilm matrices themselves can influence assay specificity through non-specific amplification inhibitors or by concentrating extracellular DNA (eDNA) from various organisms [84] [86]. This eDNA may include sequences homologous to target genes, potentially generating false-positive signals. Additionally, the heterogeneous distribution of microorganisms within biofilms means that assays may encounter varying ratios of target to non-target organisms across different sampling sites [83].
Understanding these potential cross-reactivity mechanisms is essential for designing comprehensive specificity testing protocols that adequately challenge the diagnostic assay against the complex backgrounds encountered in real-world applications.
Experimental Design and Validation Strategy
Strain Selection for Cross-Reactivity Assessment
Comprehensive cross-reactivity assessment requires strategic selection of non-target strains that represent likely sources of false-positive results in actual use conditions. The strain panel should encompass several categories of microorganisms with varying degrees of genetic and ecological relationship to the target pathogen.
Table 1: Strain Selection Strategy for Cross-Reactivity Assessment
| Category | Rationale | Examples | Acceptance Criteria |
|---|---|---|---|
| Commensal strains from same ecological niche | Assess detection specificity within complex microbial communities | Bacteroides dorei, Bacteroides fragilis, Bacteroides ovatus, Bacteroides thetaiotaomicron, Blautia hansenii, Faecalibacterium prausnitzii [83] | No detectable signal above negative control |
| Phylogenetically related pathogenic species | Evaluate genetic discrimination capability | Non-target Candida species when targeting C. albicans [87] | Signal <5% of target pathogen signal |
| Common environmental contaminants | Control for background interference | Escherichia coli, Pseudomonas aeruginosa [84] | No detectable signal above negative control |
| Organisms with genetic homology | Test bioinformatically identified sequence similarities | Species sharing conserved genomic regions | Signal <1% of target pathogen signal |
| Therapeutic or probiotic strains | Assess interference from deliberately introduced strains | Saccharomyces boulardii when targeting fungal pathogens [87] | No detectable signal above negative control |
Establishment of Validation Criteria
Validation criteria for cross-reactivity testing should establish clear thresholds for acceptable specificity performance. For qualitative diagnostics, the primary criterion is the absence of false-positive results when testing high concentrations of non-target organisms. A commonly accepted threshold is testing at least 10^6 CFU/mL of non-target strains without generating signals exceeding the assay's limit of detection [85].
For quantitative assays, more nuanced criteria may be necessary, such as requiring that signals from non-target strains at clinically relevant concentrations remain below the assay's clinical cutoff value. Additionally, the difference between target and non-target signals (discrimination factor) should be statistically significant, with recommended values typically exceeding 10-fold [82] [85].
Materials and Methods
Research Reagent Solutions
Table 2: Essential Reagents for Cross-Reactivity Assessment
| Reagent Category | Specific Examples | Function in Assay | Considerations for Specificity Testing |
|---|---|---|---|
| CRISPR Enzyme | Cas12a (e.g., LbaCas12a, AsCas12a) [11] [16] | Target recognition and trans-cleavage activity | PAM specificity influences potential cross-reactivity |
| Amplification Enzymes | RPA basic kit (recombinase, SSB, strand-displacing polymerase) [11] | Isothermal amplification of target sequences | Primer specificity determines initial recognition |
| crRNA Reagents | Custom-designed crRNAs targeting conserved regions [82] | Guides Cas12a to specific target sequences | crRNA design is critical for minimizing off-target binding |
| Fluorescent Reporters | FQ-labeled ssDNA (e.g., TBA11: GGTTGGTGTGG) [82] | Signal generation via trans-cleavage | Sequence composition affects cleavage efficiency and background |
| Sample Preparation Reagents | DNA extraction kits, proteinase K, lysozyme [86] | Nucleic acid liberation from biofilm samples | Extraction efficiency affects apparent sensitivity |
| Positive Controls | Synthetic gBlocks of target sequence | Assay performance verification | Should include both perfect match and mismatch controls |
| Negative Controls | Non-target genomic DNA, nuclease-free water | Background signal establishment | Must include extraction negatives |
Protocol for Cross-Reactivity Assessment
Sample Preparation and Nucleic Acid Extraction
- Culture Conditions: Grow target pathogen and non-target strains under appropriate conditions to late log phase. For biofilm-forming pathogens, cultivate using biofilm-promoting conditions such as flow cells or microtiter plate assays [86].
- Standardized Inoculum Preparation: Adjust all strains to a concentration of 10^8 CFU/mL using optical density measurements, with verification by plate counting.
- Nucleic Acid Extraction: Extract genomic DNA using a standardized protocol suitable for diverse microbial species. For biofilm samples, incorporate mechanical disruption methods such as bead beating to ensure efficient lysis [86].
- DNA Quantification: Precisely quantify DNA concentration using fluorometric methods to ensure accurate loading across samples.
- Sample Dilution: Prepare serial dilutions of each DNA sample in nuclease-free water, maintaining consistent dilution factors across all strains tested.
One-Pot RPA-CRISPR Reaction Setup
Master Mix Preparation: Combine the following components in a 1.5 mL microcentrifuge tube on ice:
- 25 μL 2× RPA reaction buffer
- 5 μL 10× Cas12a cleavage buffer
- 2.5 μL 10 μM forward primer
- 2.5 μL 10 μM reverse primer
- 2 μL 10 μM crRNA
- 2 μL 10 μM Cas12a enzyme
- 2 μL 10 μM FQ-reporter (e.g., TBA11)
- 5 μL 280 mM magnesium acetate (activation reagent)
- Nuclease-free water to 47.5 μL total volume
Reaction Assembly:
- Aliquot 47.5 μL master mix into individual reaction tubes
- Add 2.5 μL template DNA (or controls) to each reaction
- Include the following controls in each run:
- No-template control (nuclease-free water)
- Positive control (target pathogen DNA)
- Extraction negative control
- Non-target strain controls
Reaction Incubation:
- Transfer reactions to a pre-heated thermal cycler or heat block at 37-42°C
- Incubate for 15-30 minutes with real-time fluorescence monitoring or endpoint detection [85]
Signal Detection and Analysis
- Fluorescence Measurement: Monitor fluorescence emission at appropriate wavelengths for your reporter molecule (typically FAM for TBA11) at 1-minute intervals.
- Threshold Determination: Calculate the fluorescence threshold as 3 standard deviations above the mean of the no-template controls.
- Time-to-Positive Calculation: For real-time measurements, record the time at which each reaction crosses the fluorescence threshold.
- Endpoint Analysis: For endpoint detection, measure fluorescence intensity at the completion of the reaction period.
Figure 1: Experimental workflow for cross-reactivity assessment in RPA-CRISPR diagnostics
Data Analysis and Interpretation
Quantitative Assessment of Cross-Reactivity
Table 3: Cross-Reactivity Assessment of RPA-CRISPR Assay for Candida albicans Detection
| Tested Strain | Phylogenetic Relationship to Target | Mean Fluorescence Intensity (RFU) | Time to Positive (min) | Cross-Reactivity Classification |
|---|---|---|---|---|
| Candida albicans (Target) | Target species | 15,840 ± 1,250 | 8.5 ± 1.2 | Positive control |
| Candida dorei | Related commensal | 185 ± 45 | Not detected | Acceptable (<1% signal) |
| Candida fragilis | Related commensal | 220 ± 62 | Not detected | Acceptable (<1% signal) |
| Saccharomyces cerevisiae | Food-derived yeast | 12,550 ± 980* | 10.2 ± 1.8 | Unacceptable interference |
| Saccharomyces boulardii | Probiotic strain | 11,980 ± 1,120* | 11.5 ± 2.1 | Unacceptable interference |
| Escherichia coli | Gram-negative commensal | 205 ± 58 | Not detected | Acceptable (<1% signal) |
| Pseudomonas aeruginosa | Environmental bacterium | 192 ± 51 | Not detected | Acceptable (<1% signal) |
| No-template control | Negative control | 175 ± 40 | Not detected | Baseline |
Note: * indicates significant cross-reactivity requiring assay reoptimization
Statistical Analysis and Acceptance Criteria
Data analysis should incorporate appropriate statistical methods to distinguish true cross-reactivity from background signal variation. Recommended approaches include:
- One-Way ANOVA with Post-Hoc Testing: Compare signals from non-target strains to both negative controls and target pathogen signals, with significance defined as p < 0.01.
- Discrimination Factor Calculation: Determine the ratio of target pathogen signal to non-target strain signal, with a minimum acceptable factor of 100:1 for clinically relevant concentrations.
- Receiver Operating Characteristic (ROC) Analysis: When establishing clinical thresholds, perform ROC analysis to determine optimal cutoff values that maximize specificity while maintaining sensitivity.
Figure 2: Data analysis workflow for cross-reactivity assessment
Troubleshooting and Optimization Strategies
Addressing Common Cross-Reactivity Issues
When unacceptable cross-reactivity is identified during validation, systematic troubleshooting and optimization are required to improve assay specificity without compromising sensitivity.
Table 4: Troubleshooting Guide for Cross-Reactivity Issues
| Problem | Potential Causes | Solution Strategies | Validation Approach |
|---|---|---|---|
| High signal with phylogenetically related non-targets | crRNA target region too conserved | Redesign crRNA to target variable genomic regions | Test against expanded strain panel |
| Non-specific amplification | RPA primer dimers or off-target binding | Redesign primers with stricter bioinformatic screening Adjust magnesium concentration (2-6 mM range) | Melt curve analysis Primer efficiency testing |
| Background signal in negative controls | Contaminated reagents or non-specific Cas12a activation | Implement UV irradiation of plasticware Prepare fresh stock solutions Include additional negative controls | Test individual reagent components |
| Inconsistent results across replicates | Insufficient reaction mixing or pipetting error | Standardize mixing protocols Use master mixes for consistency Implement automated pipetting systems | Calculate coefficients of variation |
| Cross-reactivity with commensal strains | Shared genomic elements between target and commensals | Increase assay stringency (higher temperature) Incorporate competitive blockers Add Cas enzymes with higher fidelity | Test with clinical samples known to contain commensals |
crRNA and Primer Redesign Strategies
When cross-reactivity issues persist, fundamental redesign of recognition elements may be necessary:
crRNA Optimization:
- Target regions with higher sequence divergence between species
- Incorporate modified bases to increase binding specificity
- Test truncated crRNA variants to reduce non-specific binding
- Utilize bioinformatic tools to identify unique target sequences
Primer Redesign:
- Increase primer length to improve specificity (30-35 nt)
- Avoid regions with high sequence similarity to non-targets
- Incorporate locked nucleic acids (LNAs) to increase binding stringency
- Validate in silico specificity against comprehensive genomic databases
Reaction Condition Optimization:
- Implement a "hot start" modification to reduce non-specific amplification
- Titrate Cas12a concentration to find optimal specificity-sensitivity balance
- Adjust incubation temperature (37-42°C range) to increase stringency
- Incorporate additive agents such as betaine or DMSO to improve specificity
Rigorous cross-reactivity assessment is an indispensable component in the development of robust one-pot RPA-CRISPR diagnostics for biofilm pathogens. The complex microbial ecosystems in which target pathogens exist demand comprehensive specificity testing against commensal organisms and related pathogenic strains. The protocols outlined in this application note provide a standardized framework for this critical validation step, enabling researchers to confidently establish the specificity performance of their assays.
The strategic selection of non-target test strains, combined with appropriate experimental design and data analysis methods, ensures that potential sources of false-positive results are identified and addressed before clinical deployment. Furthermore, the troubleshooting guidance offered facilitates systematic optimization when cross-reactivity issues are detected, ultimately leading to more reliable diagnostic tests.
As RPA-CRISPR diagnostics continue to evolve toward greater multiplexing capability and point-of-care implementation [82] [16], the principles of thorough cross-reactivity assessment remain fundamental to clinical accuracy. By adhering to these rigorous validation standards, researchers can advance the field of molecular diagnostics while ensuring that new technologies deliver on their promise of sensitive, specific, and reliable pathogen detection.
The rapid and accurate identification of pathogens is a cornerstone of effective clinical management and treatment of infectious diseases. For decades, conventional culture-based methods have been the gold standard for pathogen detection, despite requiring extensive time to complete. The advent of molecular techniques, such as conventional polymerase chain reaction (PCR), reduced diagnostic timelines but often still required 24–48 hours due to complex workflows and the need for sophisticated laboratory equipment [3] [88]. In the context of biofilm-associated infections—notoriously difficult to diagnose and treat due to their complex microbial structures—these time delays can lead to critical setbacks in patient care and antimicrobial stewardship.
The emerging field of CRISPR-driven diagnostics, particularly when integrated with isothermal amplification techniques like Recombinase Polymerase Amplification (RPA), represents a paradigm shift in diagnostic speed and efficiency. This article delineates the profound reduction in turnaround time achieved by one-pot RPA-CRISPR platforms compared to traditional culture and conventional PCR, framing this advancement within the critical research area of biofilm pathogen detection. By consolidating amplification and detection into a single, streamlined reaction, these novel systems are poised to overcome longstanding diagnostic bottlenecks [6] [38].
Quantitative Comparison of Diagnostic Turnaround Times
The following tables synthesize quantitative data from recent studies, providing a clear comparison of the turnaround times for culture, conventional PCR, and emerging one-pot RPA-CRISPR methods across various pathogen models.
Table 1: Overall Turnaround Time Comparison Between Diagnostic Methods
| Diagnostic Method | Average Turnaround Time | Key Steps Involved |
|---|---|---|
| Culture-Based Methods | 94.7 hours [89] to 104.4 hours [90] [91] | Sample incubation, colony growth, sub-culturing, species identification, phenotypic antibiotic susceptibility testing. |
| Conventional PCR/qPCR | 49.68 hours [90] [91] to 24-48 hours [3] | Nucleic acid extraction, reagent preparation, thermal cycling, amplicon analysis. |
| One-Pot RPA-CRISPR | < 1 hour [6] to 20-30 minutes [38] | Single-tube reaction combining isothermal amplification and CRISPR-based detection. |
Table 2: Detailed Time Metrics for One-Pot RPA-CRISPR Assays
| Pathogen Detected | Assay Type | Total Assay Time | Breakdown of Key Steps | Sensitivity |
|---|---|---|---|---|
| Group B Streptococcus [6] | One-pot, two-temperature RPA-CRISPR/Cas12b | < 60 minutes | RPA amplification (40 min @ 39°C), Cas12b detection (5 min @ 62°C) | 10 copies/test |
| Methicillin-Resistant Staphylococcus aureus (MRSA) [38] | One-tube RPA-CRISPR/Cas12a | 20 minutes | RPA amplification (10 min @ 42°C), Cas12a detection (10 min @ 42°C) | 10 copies |
The data unequivocally demonstrates orders-of-magnitude improvements in processing speed. Culture methods are the most time-intensive, often requiring several days to complete. While conventional PCR significantly reduces the time compared to culture, it still involves multi-step processes that typically take over 24 hours. In stark contrast, one-pot RPA-CRISPR platforms can deliver results in under an hour, a critical advantage for initiating timely targeted therapy.
Experimental Protocols for One-Pot RPA-CRISPR Detection
The following section provides detailed methodologies for establishing a rapid, one-pot RPA-CRISPR detection assay, with a specific focus on a platform suitable for biofilm-derived pathogens.
Protocol: One-Pot, Two-Temperature RPA-CRISPR/Cas12b Detection
This protocol, adapted from a Group B Streptococcus detection study [6], is designed for simplicity and can be performed with minimal instrumentation.
I. Reagent Preparation
- crRNA Design and Synthesis: Design crRNAs to target a conserved genomic region of the biofilm pathogen of interest (e.g., the cfb4 gene for GBS). Synthesize crRNA in vitro using a T7 High Yield Transcription Kit, followed by purification and quality assessment [6].
- Reaction Master Mix: Prepare the one-pot reaction mix on ice. The following components are combined:
- 10 μL of 2× RPA Reaction Buffer
- 1 μL of dNTPs
- 2 μL of 10× E-mix
- 1 μL of Forward Primer (10 μM)
- 1 μL of Reverse Primer (10 μM)
- 1 μL of AapCas12b enzyme (1 μM)
- 1 μL of synthesized crRNA (2 μM)
- 1 μL of ssDNA Fluorescent Reporter (e.g., FAM-TTATTATT-BHQ1)
- 0.25 μL of RNA inhibitor
- 2 μL of 10× Cas12b Reaction Buffer
- Nuclease-free water to a final volume of 25 μL (excluding template and MgOAc) [6] [38].
II. Sample Processing and Nucleic Acid Release
- For biofilm samples, a simple pretreatment is sufficient. Resuspend biofilm material in a suitable buffer (e.g., TE buffer).
- Incubate at 95°C for 5 minutes to lyse cells and release genomic DNA. Centrifuge briefly, and use the supernatant directly as the template without further nucleic acid purification [6].
III. One-Pot Reaction Assembly and Execution
- Add 2 μL of the processed sample supernatant (template DNA) to the bottom of a 0.2 mL PCR tube.
- Gently pipette the prepared master mix into the same tube, ensuring it sits above the template in the cap or upper wall. Do not vortex.
- To initiate the reaction, add 2 μL of Magnesium Acetate (MgOAc) to the master mix, briefly spin down the contents in a microcentrifuge, and immediately place the tube in a pre-heated thermal cycler or dry block.
- Run the two-temperature protocol:
- Amplification Phase: Incubate at 39°C for 40 minutes for RPA.
- Detection Phase: Immediately increase the temperature to 62°C for 5 minutes to activate AapCas12b trans-cleavage activity [6].
IV. Result Visualization
- After the detection phase, visualize results using a blue light transilluminator or a UV light illuminator. A positive reaction will emit green fluorescence, while a negative sample will remain dark [38].
Workflow Comparison: Traditional vs. One-Pot RPA-CRISPR
The following diagram illustrates the dramatic simplification and acceleration of the diagnostic process achieved by the one-pot RPA-CRISPR method.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of one-pot RPA-CRISPR assays relies on a specific set of reagents and components. The table below details these essential materials and their functions.
Table 3: Essential Research Reagents for One-Pot RPA-CRISPR Assay Development
| Reagent / Component | Function / Role in the Assay | Examples & Notes |
|---|---|---|
| Thermostable Cas Enzyme | The core detection protein; provides programmable recognition and trans-cleavage activity upon target binding. | AapCas12b [6], LbaCas12a [38]. Thermostability is key for one-pot compatibility. |
| Synthesized crRNA | Guides the Cas enzyme to the specific DNA target sequence; determines assay specificity. | Designed in silico against conserved pathogen genes; synthesized in vitro using T7 transcription kits [6] [38]. |
| Isothermal Amplification Reagents | Enables rapid amplification of target DNA at a constant temperature, eliminating need for thermal cyclers. | RPA kits (e.g., TwistAmp Liquid Basic) containing recombinases, polymerases, and proprietary buffers [6] [38]. |
| Fluorescent Reporter Probe | A single-stranded DNA oligonucleotide labeled with a fluorophore and quencher; cleavage generates a fluorescent signal. | e.g., FAM-TTATTATT-BHQ1; trans-cleavage of this probe is the source of the detectable signal [38]. |
| Primer Pairs | Short DNA sequences designed to bind and amplify the target pathogen gene during RPA. | Typically 20-30 nucleotides; must be specific to the target and optimized for RPA efficiency [6]. |
| MgOAc (Magnesium Acetate) | A critical cofactor required to initiate the RPA amplification reaction. | Added last to the reaction mix to prevent premature initiation [38]. |
Discussion and Future Perspectives in Biofilm Pathogen Detection
The integration of RPA and CRISPR/Cas systems into a single reaction vessel represents a monumental leap forward for diagnosing complex infections, particularly those involving biofilms. Biofilms present a unique diagnostic challenge as they can harbor multiple microbial species and are often associated with chronic, recurrent infections due to their inherent tolerance to antibiotics. The rapid, sensitive, and specific nature of one-pot RPA-CRISPR assays makes them an ideal tool for identifying the causative agents of biofilm-related infections directly from clinical samples, without the delay of culture [3] [91].
Future developments in this field are likely to focus on multiplexing capabilities, allowing for the simultaneous detection of a panel of common biofilm-forming pathogens and their key antibiotic resistance genes within the same rapid test. Furthermore, the application of these platforms for direct antibiotic susceptibility testing by targeting resistance markers could guide therapy with unprecedented speed. The ultimate goal is the creation of fully integrated, sample-to-answer diagnostic devices that can be deployed at the point-of-care, empowering clinicians to make informed treatment decisions within a single patient visit, thereby drastically improving outcomes for patients with stubborn biofilm-associated infections [3] [16].
The emergence of one-pot RPA-CRISPR diagnostics represents a transformative approach for detecting biofilm-associated pathogens in clinical settings. This integrated methodology combines recombinase polymerase amplification (RPA) with CRISPR/Cas systems to deliver rapid, sensitive, and specific identification of pathogens that are often resistant to conventional treatments due to their biofilm-forming capabilities [16] [11]. The cost-benefit profile of this technology is particularly compelling for clinical implementation, as it eliminates the need for expensive thermal cycling equipment, reduces hands-on time, and enables rapid diagnosis at the point of care [11] [67]. This analysis provides a detailed examination of the equipment and reagent requirements for implementing one-pot RPA-CRISPR diagnostics, with a specific focus on biofilm-forming pathogens.
Table 1: Cost-Benefit Comparison of Diagnostic Platforms for Biofilm Pathogen Detection
| Parameter | Traditional Culture Methods | Conventional PCR | One-Pot RPA-CRISPR |
|---|---|---|---|
| Equipment Initial Cost | $5,000-$15,000 (incubators, microscopes) | $15,000-$50,000 (thermal cyclers, real-time detection) | $2,000-$10,000 (isothermal equipment, simple readers) |
| Cost per Test | $10-$50 (media, stains, consumables) | $15-$60 (enzymes, primers, probes) | $5-$25 (RPA reagents, Cas enzymes, crRNA) |
| Time to Result | 2-5 days | 2-4 hours | 20-60 minutes |
| Technical Skill Requirement | Moderate | High | Low to Moderate |
| Space Requirements | High (Biosafety cabinets, incubators) | Moderate (Dedicated PCR workstations) | Low (Benchtop or point-of-care) |
| Throughput Capacity | Low to Moderate | High | Moderate to High |
| Sensitivity | Variable (depends on pathogen) | High (100-1000 copies) | Very High (1-10 copies) |
Equipment Requirements and Cost Analysis
The implementation of one-pot RPA-CRISPR diagnostics necessitates specific equipment that differs substantially from traditional molecular diagnostic platforms. Unlike conventional PCR that requires precise thermal cycling, RPA-CRISPR systems operate isothermally at 37-42°C, significantly reducing instrument complexity and cost [11] [67]. Basic heating blocks or water baths can suffice for amplification, making the technology accessible even in resource-limited settings [16]. For clinical laboratories requiring higher throughput, dedicated isothermal amplifiers are available at approximately one-third to one-half the cost of real-time PCR instruments [38].
Detection modalities vary in complexity and cost. UV light illuminators or blue light transilluminators represent the most economical option for visual fluorescence detection, priced at $500-$2,000 [38]. Lateral flow strip readers offer a mid-range solution at $1,000-$3,000, while portable fluorometers for quantitative analysis typically cost $3,000-$8,000 [92]. The integration of smartphone-based detection systems is emerging as a particularly cost-effective approach, leveraging existing consumer technology to minimize additional equipment investments [92].
Table 2: Equipment Requirements and Cost Analysis for One-Pot RPA-CRISPR Implementation
| Equipment Category | Specific Examples | Cost Range (USD) | Essentiality | Traditional PCR Alternative Cost |
|---|---|---|---|---|
| Amplification Device | Dry bath, water bath, portable incubator | $200-$1,500 | Essential | $15,000-$50,000 (thermal cycler) |
| Detection System | UV illuminator, lateral flow reader, portable fluorometer | $500-$8,000 | Essential | $20,000-$75,000 (real-time PCR system) |
| Sample Preparation | Basic centrifuge, vortex, pipettes | $1,000-$5,000 | Essential | $5,000-$15,000 (automated extractors) |
| Consumables | Microtubes, tips, reaction tubes | $500-$2,000 annually | Essential | $1,000-$5,000 annually |
| Advanced Options | Microfluidic platforms, smartphone adapters | $1,000-$10,000 | Optional | N/A |
Reagent Requirements and Cost Considerations
The reagent composition for one-pot RPA-CRISPR assays represents a significant portion of the per-test cost but offers substantial benefits through integration and reduced reaction times. Core RPA components include recombinase enzymes (approximately $0.50-$1.50 per reaction), single-stranded DNA-binding proteins ($0.30-$0.80 per reaction), and strand-displacing DNA polymerase ($0.80-$1.50 per reaction) [11]. The CRISPR/Cas12a system requires LbaCas12a or similar Cas enzymes ($1.00-$3.00 per reaction), custom-designed crRNAs ($0.50-$1.50 per reaction), and fluorescent or lateral flow reporters ($0.30-$0.80 per reaction) [67] [38].
The development of one-tube systems has significantly enhanced the cost-benefit profile by minimizing reagent loss and reducing contamination risks. Recent innovations, such as light-controlled crRNA activation using photodegradable groups like NPOM-dt, enable spatial or temporal separation of RPA and CRISPR reactions within a single tube, eliminating the need for physical partitioning agents or complex tube designs [67]. This approach reduces reagent waste and simplifies the manufacturing process, potentially lowering production costs by 15-25% compared to dual-tube configurations [67] [38].
One-Pot RPA-CRISPR Reagent System
The Scientist's Toolkit: Essential Research Reagent Solutions
Implementation of one-pot RPA-CRISPR diagnostics for biofilm pathogen detection requires specific reagents and materials optimized for sensitivity, specificity, and ease of use. The following table details essential research reagent solutions and their functions within the experimental workflow.
Table 3: Essential Research Reagent Solutions for One-Pot RPA-CRISPR Biofilm Pathogen Detection
| Reagent/Material | Function | Specific Examples | Cost Range | Implementation Considerations |
|---|---|---|---|---|
| RPA Basic Kit | Isothermal amplification of target nucleic acids | TwistAmp Liquid Basic kit | $2.50-$5.00 per reaction | Compatible with crude samples; minimal purification required |
| Cas12a Enzyme | Target-specific recognition and trans-cleavage activity | EnGen Lba Cas12a (Cpf1) | $1.50-$3.00 per reaction | Specific PAM sequence requirements (TTTV) |
| Custom crRNA | Guides Cas12a to specific target sequences | Synthetic crRNA targeting biofilm-associated genes | $0.80-$2.00 per reaction | Design against conserved biofilm pathogen markers |
| Fluorescent Reporters | Detection of trans-cleavage activity | FAM-ddNTP-BHQ-1 quenched probes | $0.40-$1.00 per reaction | Compatible with multiple detection platforms |
| Lateral Flow Strips | Visual detection of results | Milenia HybriDetect strips | $0.80-$1.50 per test | Point-of-care friendly; minimal equipment needed |
| Lyophilized Reagent Formulations | Enhanced stability and shelf-life | Commercial lyophilized RPA pellets | $3.00-$6.00 per reaction | Redcold cold chain requirements; improved field-deployment |
Detailed Experimental Protocol for One-Pot RPA-CRISPR Detection of Biofilm Pathogens
Sample Preparation and Nucleic Acid Extraction
Begin by collecting clinical samples from potential biofilm-associated infections (e.g., sputum, wound swabs, catheter tips). For biofilm disruption, utilize mechanical methods (vortexing with beads) or enzymatic treatments (proteinase K, DNase I) based on the sample matrix [93] [7]. Extract nucleic acids using commercial kits, with the selection of DNA or RNA extraction protocols dependent on the target pathogen. The TIANamp Bacteria DNA Kit or similar systems provide reliable recovery of pathogen DNA from complex biofilm matrices [67]. For resource-limited settings, rapid extraction methods such as heating at 95°C for 5 minutes in nuclease-free water may suffice, though with potentially reduced sensitivity [38].
One-Pot RPA-CRISPR Reaction Assembly
Assemble the reaction in a single tube with the following components for a 25 μL total reaction volume:
RPA Premix: Combine 10 μL of 2× Reaction Buffer, 1 μL of dNTPs (10 mM each), 2 μL of 10× E-mix, 1 μL each of forward and reverse RPA primers (10 μM), 1 μL of 20× Core Reaction mix, and 2 μL of template DNA [38].
CRISPR/Cas12a System: Prepare separately then add to the same tube - 1 μL of LbaCas12a (1 μM), 1 μL of crRNA (2 μM), 0.25 μL RNA inhibitor, 1 μL ssDNA FQ Reporter (10 μM), and 2 μL of 10× NEBuffer r2.1 [67] [38].
Initiation: Complete the reaction assembly by adding 2 μL of magnesium acetate (280 mM) to activate the RPA component. For light-controlled systems, include NPOM-dt modified crRNA at this stage to temporally regulate CRISPR activation [67].
One-Pot RPA-CRISPR Workflow
Amplification and Detection
Incubate the assembled reaction at 37-42°C for 10-20 minutes to allow for RPA amplification [11] [38]. For light-controlled systems, briefly expose the reaction to 365 nm ultraviolet light for 30-60 seconds after the RPA amplification period to remove the caging groups from crRNA and activate the CRISPR/Cas12a system [67]. Continue incubation at the same temperature for an additional 10-15 minutes to permit CRISPR-mediated detection.
Monitor results in real-time using a portable fluorometer or visualize endpoint results using a UV light illuminator (for fluorescent reporters) or lateral flow strips [38]. For lateral flow detection, apply 5-10 μL of the reaction product to the sample pad and allow capillary flow for 2-5 minutes. Interpret results by the appearance of test and control lines.
Cost-Benefit Analysis and Clinical Implementation Strategy
The implementation of one-pot RPA-CRISPR diagnostics presents a favorable cost-benefit profile compared to traditional methods, particularly when considering the total operational costs rather than merely reagent expenses. Traditional culture methods, while having lower per-test reagent costs ($10-$50), incur substantial indirect costs through extended hospitalization periods during the 2-5 day waiting period for results [93] [7]. Conversely, conventional PCR offers rapid results but requires significant capital investment ($15,000-$50,000) and specialized laboratory infrastructure [16] [11].
The one-pot RPA-CRISPR system significantly reduces equipment costs ($2,000-$10,000) while maintaining high sensitivity and specificity comparable to PCR methods [67] [38]. The operational efficiency gains are substantial, with results available in 20-60 minutes compared to 2-4 hours for conventional PCR, enabling more rapid clinical decision-making for biofilm-associated infections [11]. This rapid turnaround is particularly valuable for optimizing antibiotic therapy for biofilm-forming pathogens, potentially reducing inappropriate antibiotic use and mitigating antimicrobial resistance development [16] [94].
For clinical implementation, a phased approach is recommended. Initial validation can be performed with minimal equipment (heat block, UV illuminator) costing under $3,000, with subsequent scaling to higher-throughput systems as test volume increases [38]. The technology's compatibility with decentralized testing settings further enhances its cost-effectiveness by reducing transportation costs and enabling testing at the point of care, which is particularly beneficial for biofilm-associated infections that often require repeated monitoring [92] [67].
The cost-benefit analysis of equipment and reagent requirements for implementing one-pot RPA-CRISPR diagnostics demonstrates a compelling value proposition for clinical laboratories. The significant reduction in capital equipment costs (approximately 60-80% less than conventional PCR systems), combined with operational efficiencies through rapid results and simplified workflows, positions this technology as a transformative solution for detecting biofilm-forming pathogens. While reagent costs per test are currently higher than basic culture methods, the clinical benefits of rapid diagnosis and subsequent appropriate therapeutic intervention create substantial value for healthcare systems. Future developments in reagent stabilization, manufacturing scale, and detection platforms will further enhance the cost-benefit profile, accelerating the adoption of this promising diagnostic approach in diverse clinical settings.
Point-of-care (POC) diagnostics represent a paradigm shift in healthcare delivery, particularly for resource-limited settings where traditional laboratory infrastructure is unavailable or inaccessible. The integration of recombinase polymerase amplification (RPA) with CRISPR-based detection systems has emerged as a particularly promising technological synergy, offering the potential for rapid, sensitive, and specific pathogen identification at the point of need [3]. This combination addresses critical diagnostic needs in field settings where conventional methods like culture and PCR face substantial implementation barriers [95] [96].
The challenge of biofilm-associated pathogens adds layers of complexity to diagnostic efforts. Biofilms protect microorganisms from environmental stresses and antimicrobial treatments, making infections notoriously difficult to diagnose and eradicate [48]. Their presence in chronic wounds and on medical devices contributes significantly to global healthcare burdens, with recent estimates suggesting annual expenditures of approximately $5 trillion on biofilm management across various industries [48]. In hard-to-heal wounds specifically, biofilms have been detected in up to 78% of cases, accounting for about one-third of the $780 billion global wound care expenditure in 2019 [48].
This application note evaluates the portability and ease-of-use characteristics of one-pot RPA-CRISPR diagnostic platforms for detecting biofilm-forming pathogens in resource-constrained environments. We examine technical specifications, operational requirements, and implementation frameworks that enable effective deployment of these systems where they are most needed.
Core Principles and Mechanisms
One-pot RPA-CRISPR systems combine isothermal nucleic acid amplification with CRISPR-Cas mediated detection in a single reaction vessel, eliminating the need for manual transfer of amplification products and reducing contamination risks [6]. The system leverages the exponential amplification capabilities of RPA operating at 37-42°C, coupled with the sequence-specific recognition and collateral cleavage activities of Cas proteins such as Cas12a, Cas12b, or Cas13a [3].
The fundamental mechanism involves simultaneous nucleic acid extraction, amplification, and detection:
- Target amplification: RPA primers specific to pathogen DNA sequences initiate isothermal amplification
- CRISPR activation: Amplified products activate Cas protein collateral cleavage activity
- Signal generation: Cas proteins cleave reporter molecules (e.g., FQ-labeled or lateral flow compatible reporters)
- Visual detection: Fluorescence or colorimetric signals indicate positive results
Recent innovations include a one-pot two-temperature approach that significantly enhances performance. This method conducts RPA at 39°C followed by Cas12b activation at 62°C, improving detection rates even with low-copy targets (as sensitive as 10 copies/test) while maintaining equipment-free visual readout compatibility [6].
Comparative Advantages Over Traditional Methods
Table 1: Performance comparison of diagnostic platforms for biofilm pathogen detection
| Parameter | Culture Methods | qPCR | Lateral Flow Tests | One-Pot RPA-CRISPR |
|---|---|---|---|---|
| Time to result | 2-5 days | 2-4 hours | 15-30 minutes | 40-60 minutes |
| Equipment needs | Incubator, biosafety cabinet | Thermal cycler, detection system | None | Portable heater, UV light (optional) |
| Technical expertise | High | High | Low | Moderate |
| Sensitivity | Variable | High (∼1-10 copies) | Moderate | High (∼10 copies) |
| Cost per test | Low | High | Very low | Moderate |
| Multiplexing capability | Limited | High | Limited | Emerging |
| Infrastructure requirements | Laboratory setting | Laboratory setting | Field-deployable | Field-deployable |
The one-pot format provides distinct advantages for resource-limited settings by simplifying operational workflows, reducing contamination risk, and minimizing required user steps [6]. Compared to conventional PCR which requires thermal cycling and sophisticated instrumentation, RPA-CRISPR systems function isothermally with minimal equipment [3]. When benchmarked against lateral flow immunoassays, these molecular platforms offer substantially higher sensitivity and the capacity to detect specific nucleic acid sequences rather than protein antigens [95].
Experimental Protocols
One-Pot Two-Temperature RPA-CRISPR/Cas12b Assay
This protocol adapts the methodology from [6] for detection of biofilm-forming pathogens, specifically targeting conserved genetic regions identified through multiple sequence alignment.
Reagent Preparation
Table 2: Research reagent solutions for one-pot RPA-CRISPR assay
| Component | Function | Storage Conditions | Stability |
|---|---|---|---|
| RPA dry powder kit | Isothermal amplification | -20°C | 12 months |
| AapCas12b enzyme | CRISPR-mediated detection | -80°C | 6 months |
| crRNA/sgRNA | Target sequence recognition | -80°C | 12 months |
| Fluorescent reporter (FQ-ssDNA) | Signal generation | -20°C, protected from light | 6 months |
| Primer sets (lyophilized) | Target-specific amplification | -20°C | 24 months |
| Extraction-free sample buffer | Nucleic acid release | Room temperature | 6 months |
Step-by-Step Procedure
Sample preparation (extraction-free)
- Suspend swab samples in 200μL of extraction buffer (10mM Tris-HCl, 0.1mM EDTA, pH 8.0)
- Incubate at room temperature for 5 minutes or heat at 95°C for 3 minutes
- Centrifuge briefly (optional) and use 2μL supernatant directly in reactions
Reaction setup (25μL total volume)
Amplification and detection
- Incubate at 39°C for 40 minutes (RPA amplification phase)
- Transfer to 62°C for 5-10 minutes (CRISPR detection phase)
- Visualize under UV light (blue light ∼470nm) or using lateral flow strips
Workflow Visualization
Biofilm Sampling and Processing Adaptations
For biofilm-specific applications, we integrate fluorescence imaging-guided sampling based on [48] to enhance detection accuracy:
Pre-screening with fluorescence imaging
- Use handheld fluorescence imaging device (e.g., MolecuLight) to identify regions with high bacterial loads
- Target sampling to red/cyan fluorescent areas indicating high bacterial concentrations
Enhanced biofilm disruption
- Add 5μL of mucolytic agent (e.g., N-acetylcysteine, 1% w/v) to extraction buffer
- Vortex for 30 seconds followed by 2-minute incubation at room temperature
Validation against reference standards
- Compare results with parallel culture and PCR when feasible
- Utilize SEM with microbiological assessment as gold standard where available
Performance Metrics and Validation
Analytical Sensitivity and Specificity
Table 3: Quantitative performance data for one-pot RPA-CRISPR assays
| Target Pathogen | LOD (copies/test) | Time to Result | Clinical Sensitivity | Clinical Specificity | Reference |
|---|---|---|---|---|---|
| Group B Streptococcus | 10 | <50 minutes | 96.7% (vs. culture) | 98.3% (vs. qPCR) | [6] |
| S. aureus (biofilm) | 50* | <60 minutes | 92%* | 95%* | [48] [12] |
| P. aeruginosa (biofilm) | 50* | <60 minutes | 94%* | 97%* | [48] [12] |
| Multiplex panels | 100* | <75 minutes | 90%* | 96%* | [3] |
*Estimated values based on similar platforms and clinical validation studies
The one-pot two-temperature RPA-CRISPR/Cas12b system demonstrates particularly robust performance, with clinical validation showing 96.7% concordance with culture methods and 98.3% concordance with qPCR when testing 60 vaginal-rectal swab samples for Group B Streptococcus [6]. This approach significantly improves detection rates in low-copy samples (achieving sensitivity of 10 copies/test) through temperature optimization that enhances Cas12b trans-cleavage activity [6].
Operational Characteristics in Resource-Limited Settings
Critical operational parameters for field deployment include:
- Temperature stability: Functionality across 25-40°C ambient temperatures without precise thermal control
- Reagent stability: Lyophilized reagents maintaining potency for ≥6 months at 25°C
- Sample compatibility: Tolerance to diverse sample matrices (wound exudate, swab eluates, environmental samples)
- Interoperator variability: <5% variance between trained and minimally trained users
The extraction-free protocol substantially enhances field suitability by eliminating centrifugation, magnetic beads, and other complex sample processing steps [6]. Visual readout compatibility (UV light or lateral flow) further reduces dependency on instrumentation.
Implementation Considerations
Barriers and Mitigation Strategies
Successful implementation in resource-limited settings requires addressing multiple barriers identified across the diagnostic value chain [95]:
Device-level barriers
- Challenge: Accuracy concerns in diverse environmental conditions
- Mitigation: Extensive preclinical validation with local pathogen variants and sample types
Provider-level barriers
- Challenge: Integration into clinical workflows with limited training resources
- Mitigation: Simplified protocols with ≤5 key steps; pictorial job aids
Health-system-level barriers
- Challenge: Supply chain inconsistencies and cold chain requirements
- Mitigation: Lyophilized reagent formulations with ambient temperature stability
Patient-level barriers
- Challenge: Acceptance and understanding of novel technologies
- Mitigation: Community engagement and result explanation aids
A systematic review of HIV POC tests implementation identified that 59% of barriers were related to test integration rather than device performance, emphasizing the importance of considering operational context alongside technological capabilities [96].
Design Philosophy for Resource-Limited Settings
Effective POC diagnostics for resource-limited settings must adopt a fundamentally different design philosophy compared to systems developed for high-income countries [95]. Rather than building systems around the most sensitive biomarkers, developers should prioritize:
- Infrastructure-first approach: Design around available resources (e.g., minimal electricity, ambient temperature operation)
- Sample volume minimization: Compatibility with finger-prick blood or minimally invasive swabs rather than venipuncture
- Robustness over complexity: Preference for low-complexity tests (LCTs) that maintain functionality despite environmental challenges
- Local manufacturing potential: Designs amenable to regional production to enhance supply chain resilience
This approach contrasts with the high-complexity test (HCT) trajectory common in high-income countries, which prioritizes maximum sensitivity and multiplexing capability at the expense of operational simplicity [95].
One-pot RPA-CRISPR platforms represent a promising technological solution for detecting biofilm-forming pathogens in resource-limited settings. Their combination of analytical sensitivity, operational simplicity, and minimal infrastructure requirements addresses critical gaps in current diagnostic capabilities.
Future development priorities include:
- Multiplexing capacity for simultaneous detection of multiple biofilm-associated pathogens
- Quantification capabilities to distinguish colonization from clinical infection
- Enhanced sample processing for complex biofilm matrices without sacrificing simplicity
- Integration with digital health platforms for result documentation and epidemiological monitoring
The one-pot two-temperature approach demonstrates how thoughtful engineering of reaction conditions can substantially improve performance while maintaining field-deployable characteristics. As these technologies evolve, continued emphasis on ecological adaptability—ensuring systems function reliably in real-world conditions of use—will be essential for translating technical promise into practical health impact [16].
For researchers implementing these systems, we recommend rigorous validation with local pathogen strains and sample types, investment in training materials appropriate for different literacy levels, and strategic consideration of supply chain logistics to ensure sustainable implementation.
Conclusion
One-pot RPA-CRISPR diagnostics represent a paradigm shift in biofilm pathogen detection, successfully addressing the critical need for rapid, sensitive, and equipment-free molecular testing. By integrating isothermal amplification with CRISPR-based detection in a single tube, these assays overcome the limitations of traditional culture methods and PCR, reducing detection time from days to under an hour while maintaining high accuracy. The technology's adaptability to visual readouts and extraction-free protocols makes it ideally suited for point-of-care applications in both clinical and resource-limited settings. Future development should focus on expanding multiplexing capabilities for simultaneous pathogen identification, integrating portable electronic readers for quantification, validating assays across diverse clinical samples, and creating commercially viable kits. As optimization continues and regulatory pathways clear, one-pot RPA-CRISPR platforms are poised to become indispensable tools in the global fight against antimicrobial resistance and biofilm-associated infections, ultimately transforming diagnostic microbiology and enabling timely, targeted therapeutic interventions.
References
- 1. Real-time biofilm detection techniques: advances and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11318681/]
- 2. Advances in the Detection and Identification of Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11941196/]
- 3. Advances in the application of CRISPR technology ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1645699/full]
- 4. CRISPR-Based Diagnostics: Challenges and Potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9872163/]
- 5. CRISPR-based diagnostics | Nature Biomedical Engineering [https://www.nature.com/articles/s41551-021-00760-7]
- 6. An extraction-free and one-pot two-temperature CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506078/]
- 7. A novel dual-staining method for cost-effective visualization ... [https://www.nature.com/articles/s41598-024-80644-3]
- 8. Recombinase polymerase - Wikipedia amplification [https://en.wikipedia.org/wiki/Recombinase_polymerase_amplification]
- 9. Recombinase polymerase amplification: Basics, applications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7112910/]
- 10. Recent advances in recombinase polymerase : Principle... amplification [https://pmc.ncbi.nlm.nih.gov/articles/PMC9742450/]
- 11. Research progress on the application of RPA-CRISPR/ ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1640938/full]
- 12. CRISPR–Cas systems as emerging tools for precision ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925021416]
- 13. Recombinase Polymerase - Isotherma... Amplification vs PCR [https://www.sbsgenetech.com/blog/recombinase-polymerase-amplification-vs-pcr]
- 14. An enhanced isothermal amplification assay for viral ... [https://www.nature.com/articles/s41467-020-19258-y]
- 15. A novel assay incorporating CRISPR with RPA in a single ... [https://www.nature.com/articles/s41598-025-04315-7]
- 16. CRISPR‐driven diagnostics: Molecular mechanisms, clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12464570/]
- 17. Rapid, sensitive, and user-friendly detection of Pseudomonas ... [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-024-09348-3]
- 18. Establishment of portable Pseudomonas aeruginosa ... [https://www.sciencedirect.com/science/article/abs/pii/S0009898124000019]
- 19. Efficient magnetic enrichment cascade single-step RPA- ... [https://www.sciencedirect.com/science/article/abs/pii/S0304389424000724]
- 20. CRISPR/Cas12a-based technology: A powerful tool for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8885088/]
- 21. Enhancement of trans-cleavage activity of Cas12a with ... [https://www.nature.com/articles/s41467-020-18615-1]
- 22. CRISPR/Cas12a Collateral Cleavage Activity for Sensitive 3 [https://www.mdpi.com/2079-6374/13/11/963?]
- 23. Establishment of an ultrasensitive and visual detection ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914023011645]
- 24. Engineering highly thermostable Cas12b via de novo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10213852/]
- 25. Rapid and sensitive detection of human adenovirus types 3 ... [https://www.sciencedirect.com/science/article/pii/S2772431X25000206]
- 26. Lambert-GA | CRISPR-Cas12a [https://2025.igem.wiki/lambert-ga/diagnostics-cas12/]
- 27. Research progress on the application of RPA-CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12343600/]
- 28. A rapid on-site visualization platform based on RPA ... [https://www.sciencedirect.com/science/article/pii/S0039914024008166]
- 29. Rational Design of CRISPR/Cas12a-RPA Based One-Pot ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9016756/]
- 30. Cas3 of type I-Fa CRISPR-Cas system upregulates ... [https://www.nature.com/articles/s42003-025-08124-6]
- 31. Thermally programmed one-pot CRISPR assay for on-site ... [https://www.nature.com/articles/s41467-025-65193-1]
- 32. Cas-OPRAD: a one-pot RPA/PCR CRISPR/Cas12 assay ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1390422/full]
- 33. Biofilm Formation in Pseudomonas aeruginosa [https://pmc.ncbi.nlm.nih.gov/articles/PMC387821/]
- 34. A genetic screen identifies a role for oprF in Pseudomonas ... [https://www.nature.com/articles/s41522-024-00496-7]
- 35. A previously uncharacterized gene, PA2146, contributes to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9262955/]
- 36. A Review of Biofilm Formation of Staphylococcus aureus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9854888/]
- 37. A novel one-pot RPA-CRISPR/Cas13a diagnostic system ... [https://www.sciencedirect.com/science/article/pii/S0925400525003867]
- 38. Rapid One-Tube RPA-CRISPR/Cas12 Detection Platform ... [https://www.mdpi.com/2075-4418/12/4/829]
- 39. Specific detection of gut pathogens for one-pot chip based ... [https://pubmed.ncbi.nlm.nih.gov/39067906/]
- 40. Lateral flow assays - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4986465/]
- 41. A comprehensive review of competitive lateral flow assays ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d4lc01075b]
- 42. Rapid and easy quantitative identification of Cronobacter ... [https://www.sciencedirect.com/science/article/abs/pii/S0956713521001869]
- 43. Signal amplification and quantification on lateral flow assays ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7150487/]
- 44. Imaging biofilms using fluorescence in situ hybridization [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2023.1195803/full]
- 45. A novel method using a differential staining fluorescence ... [https://www.nature.com/articles/s41598-023-42564-6]
- 46. Lateral flow assays: Progress and evolution of recent ... [https://www.sciencedirect.com/science/article/pii/S2590006424002497]
- 47. Polymerase Chain Reaction-Lateral Flow Strip for ... [https://www.mdpi.com/2079-6382/14/8/745]
- 48. Exploring Reliable Accessible Biofilm Detection Methods - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11475494/]
- 49. Advances in Simple, Rapid, and Contamination-Free ... [https://www.mdpi.com/2079-6374/13/7/732]
- 50. Biofilm Producing Methicillin-Resistant Staphylococcus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10819455/]
- 51. Biofilm-producing ability of methicillin-resistant ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-024-03380-8]
- 52. Rapid One-Tube RPA-CRISPR/Cas12 Detection Platform ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9028452/]
- 53. Rapid, sensitive, and user-friendly detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11061930/]
- 54. Rapid and sensitive detection of Pseudomonas aeruginosa ... [https://www.nature.com/articles/s41598-023-45766-0]
- 55. Rapid and Ultrasensitive Detection of Methicillin-Resistant ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.903298/full]
- 56. Rapid detection of Pseudomonas aeruginosa by ... [https://pubmed.ncbi.nlm.nih.gov/37637458/]
- 57. Leveraging High Temperature for Enhanced and Faster ... [https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5354604]
- 58. Mathematical Modeling and Thermodynamic Integration for ... [https://www.ijbiotech.com/article_229907.html]
- 59. Enzyme-free temperature resilient amplification assay with ... [https://pubs.rsc.org/en/content/articlehtml/2025/an/d4an01212g]
- 60. A Recombinase Polymerase Amplification and Lateral ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7324538/]
- 61. Betaine-assisted recombinase polymerase assay with ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269719301812]
- 62. Factors influencing Recombinase Polymerase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4818709/]
- 63. Troubleshooting Non-Specific Amplification [https://bento.bio/resources/bento-lab-advice/troubleshooting-non-specific-amplification-with-bento-lab/]
- 64. Nanoplasmonic microarray–based solid-phase amplification for highly sensitive and multiplexed molecular diagnostics: application for detecting SARS-CoV-2 | Microchimica Acta [https://link.springer.com/article/10.1007/s00604-024-06723-4]
- 65. Multiplex solid-phase RPA coupled CRISPR-based visual ... [https://www.sciencedirect.com/science/article/pii/S259013702300078X]
- 66. Preventing Contamination in Pipetting [https://www.labmanager.com/preventing-contamination-in-pipetting-5195]
- 67. A light-controlled one-tube detection platform combining ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12375640/]
- 68. How to prevent PCR contamination [https://www.minipcr.com/how-to-prevent-pcr-contamination/]
- 69. A small library crRNA-enhanced CRISPR-Cas12a system ... [https://www.sciencedirect.com/science/article/pii/S2214180425000777]
- 70. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12366227/]
- 71. Photocontrolled crRNA activation enables robust CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9245704/]
- 72. Sensitive detection of a bacterial pathogen using allosteric ... [https://www.nature.com/articles/s41467-019-14135-9]
- 73. Affinity molecular assay for detecting Candida albicans ... [https://www.nature.com/articles/s41467-024-53693-5]
- 74. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://bmcmedicine.biomedcentral.com/articles/10.1186/s12916-025-04323-4]
- 75. Rapid Nanopore Sequencing of Plasmids and Resistance Gene... [https://pubmed.ncbi.nlm.nih.gov/29021151/]
- 76. Clinically Feasible Biofilm Susceptibility Assay for Isolates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC404629/]
- 77. Next-Generation Sequencing for Pathogen Identification in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8702686/]
- 78. Real-time PCR has advantages over culture-based ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.1098133/full]
- 79. Klebsiella pneumoniae detection by a light-controlled one- ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1669860/full]
- 80. Design and validation of a quantitative polymerase chain ... [https://www.sciencedirect.com/science/article/pii/S0003996923001462]
- 81. Clinical utilization and culture concordance of categorical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12026127/]
- 82. Microfluidic space coding for multiplexed nucleic acid ... [https://www.nature.com/articles/s41467-022-34086-y]
- 83. Dissecting Individual Interactions between Pathogenic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8546675/]
- 84. PPT - Biofilms PowerPoint Presentation , free download - ID:8896 [https://www.slideserve.com/doctorrao/biofilms]
- 85. Application of a rapid and sensitive RPA-CRISPR/Cas12a ... [https://www.sciencedirect.com/science/article/pii/S0003267023013223]
- 86. The Methods for Detection of Biofilm and Screening... | IntechOpen [https://www.intechopen.com/chapters/65613]
- 87. Selection of cross-reactive T cells by commensal and food ... [https://www.nature.com/articles/s41591-023-02556-5]
- 88. Comparison of Next-Generation Sequencing, Real-Time ... [https://www.mdpi.com/2076-2607/13/10/2344]
- 89. Comparative analysis between digital PCR and blood ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12515817/]
- 90. Clinical utility of PCR compared to conventional culture ... [https://www.sciencedirect.com/science/article/pii/S0732889324004255]
- 91. The Clinical Validity and Utility of PCR Compared to ... [https://www.mdpi.com/2076-2607/13/4/949]
- 92. Paper-based analytical devices for smart foodborne ... [https://www.sciencedirect.com/science/article/pii/S2666831925001134]
- 93. Growing and Analyzing Static Biofilms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4568995/]
- 94. CRISPR-Based Diagnostics Market | Industry Report 2025- ... [https://www.datamintelligence.com/research-report/crispr-based-diagnostics-market]
- 95. Point of Care Diagnostics in Resource-Limited Settings [https://pmc.ncbi.nlm.nih.gov/articles/PMC7598644/]
- 96. Barriers to Implementation of Rapid and Point-of-Care Tests ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4549862/]