Optimized AFM Protocols for Bacterial Cell Immobilization: A Guide for Reliable Single-Cell Analysis
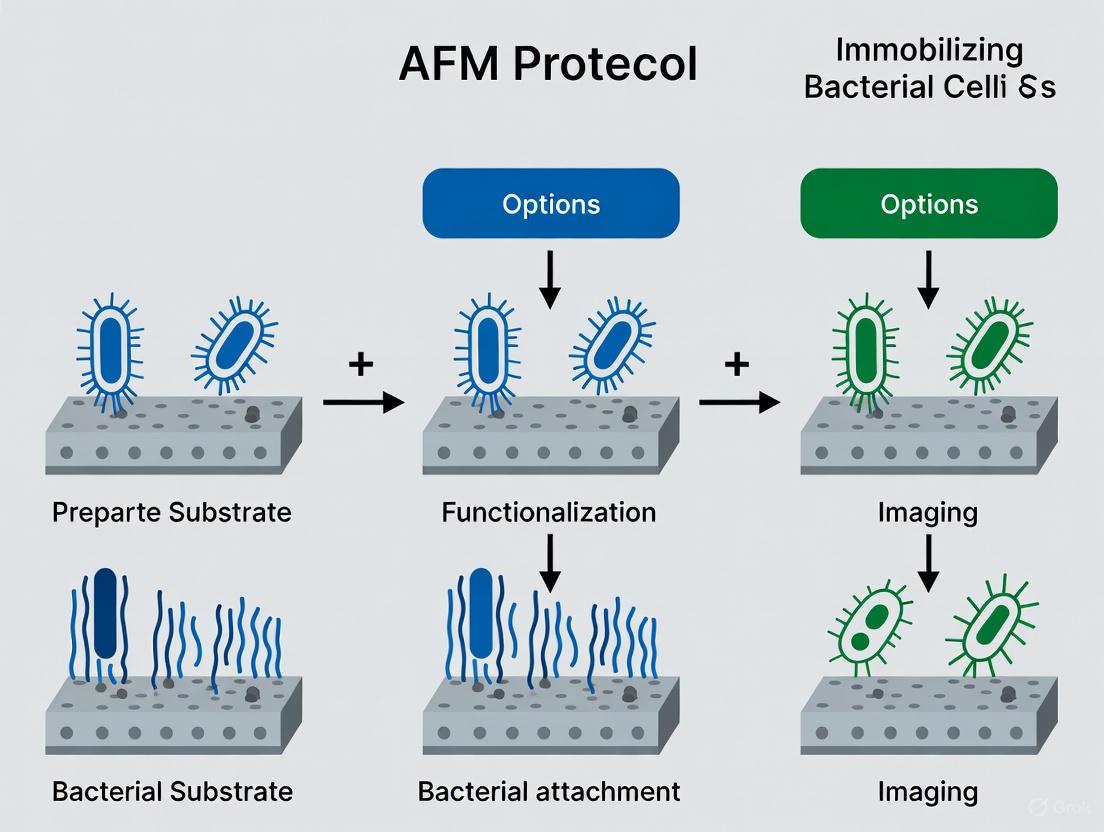
This article provides a comprehensive guide to Atomic Force Microscopy (AFM) protocols for immobilizing bacterial cells, a critical step for obtaining reliable nanoscale data on cell morphology, adhesion, and mechanics.
Optimized AFM Protocols for Bacterial Cell Immobilization: A Guide for Reliable Single-Cell Analysis
Abstract
This article provides a comprehensive guide to Atomic Force Microscopy (AFM) protocols for immobilizing bacterial cells, a critical step for obtaining reliable nanoscale data on cell morphology, adhesion, and mechanics. Tailored for researchers and drug development professionals, it covers the fundamental principles of bacterium-surface interactions, details step-by-step methodologies for various immobilization techniques, and offers troubleshooting advice for common pitfalls. By comparing the performance of different strategies and presenting validation methods, this resource aims to standardize sample preparation, enhance data reproducibility, and support advancements in antimicrobial development and biofilm research.
Why Immobilization Matters: The Foundation of reliable Bacterial AFM
The Critical Role of Firm Immobilization in AFM Imaging and Force Spectroscopy
Atomic Force Microscopy (AFM) has emerged as a premier tool for investigating bacterial cells at the nanoscale, enabling researchers to resolve topographical features and measure nanomechanical properties under physiological conditions. However, the accuracy of both AFM imaging and force spectroscopy is critically dependent on effectively immobilizing bacterial cells to prevent displacement by the scanning probe. Successful immobilization must be firm enough to resist scanning forces yet minimally invasive to preserve native cell structure and function. This application note details the foundational principles, validated protocols, and practical considerations for immobilizing bacterial cells, providing a critical framework for reliable AFM data acquisition in microbiological research.
Immobilization Principles and Method Comparison
The fundamental goal of bacterial immobilization is to secure cells firmly to a substrate through adhesion forces that exceed the lateral forces exerted by the AFM cantilever during scanning. Optimal immobilization strategies achieve a balance between firm attachment and preserved cell viability and function, avoiding chemical fixation unless the specific research question permits altered mechanical properties.
The following table summarizes the primary immobilization methods, their mechanisms, and their key characteristics for bacterial AFM studies:
Table 1: Comparison of Bacterial Immobilization Methods for AFM
| Method | Immobilization Mechanism | Key Advantages | Key Limitations | Best Suited For |
|---|---|---|---|---|
| Gelatin-Coated Mica [1] [2] | Electrostatic interaction between negatively charged bacteria and positively charged gelatin | Generally applicable to many microbial cells; suitable for liquid imaging; preserves cell viability | Effectiveness depends on gelatin source (porcine recommended) and bacterial strain; sensitive to buffer salts | Live-cell imaging and force spectroscopy of Gram-negative and Gram-positive bacteria |
| APTES-Glutaraldehyde [3] | Covalent bonding between glutaraldehyde and primary amines on cell surface | Extremely firm attachment; low fluorescence background; generic for cells with surface amines | Chemical modification of cell surface; may affect physiology for long-term studies | Super-resolution imaging and single-particle tracking requiring absolute immobilization |
| Mechanical Entrapment [4] | Physical confinement in porous membrane filters | Avoids chemical treatment of cells; simple setup | Potential for uneven surface exposure; not suitable for all cell shapes | Stiffness measurements where chemical cross-linking is undesirable |
| Cell-Tak [4] | Bio-adhesive from marine mussels | Does not interact with bacterial cell wall | Commercial product with associated cost | Live-cell studies where non-invasive immobilization is critical |
| Substrate Optimization [5] | Exploits inherent adhesion to engineered surfaces (e.g., ITO-coated glass) | No additional immobilization reagents; maintains pristine cell condition | Adhesion strength is strain and substrate dependent | Imaging native bacteria in liquid with minimal sample preparation |
Recommended Protocols for Firm Immobilization
Gelatin-Coated Mica for Live-Cell Imaging
This widely applicable protocol is highly effective for immobilizing both Gram-negative and Gram-positive bacteria for imaging and force spectroscopy in liquid environments [1] [2].
Materials:
- Freshly cleaved mica sheets
- Porcine gelatin (e.g., Sigma G-6144 or G-2625)
- Distilled water
- AFM liquid cell
- Centrifuge
Procedure:
- Prepare Gelatin Solution: Add 0.5 grams of porcine gelatin to 100 mL of boiling distilled water. Swirl gently until completely dissolved. Cool to 60-70°C before use [2].
- Coat Mica Substrate: Briefly submerge a freshly cleaved mica square into the warmed gelatin solution and withdraw quickly. Support the mica on its edge on a paper towel to dry in ambient air. Coated mica can be stored and used for up to two weeks [2].
- Prepare Bacterial Sample: Pellet 1 mL of bacterial culture (OD₆₀₀ ≈ 0.5-1.0) by centrifugation. Wash the pellet in filtered deionized water or a compatible buffer to remove growth media and salts that can interfere with adhesion. Resuspend the pellet in 500 μL of nanopure water or a dilute buffer to create a turbid suspension [2].
- Immobilize Cells: Apply 10-20 μL of the bacterial suspension to the gelatin-coated mica and spread gently without touching the surface. Allow the sample to rest for 10 minutes for cells to adhere.
- Rinse: Gently rinse the surface with a stream of deionized water or imaging buffer to remove loosely bound cells. A simple dryness test will show a cloudy area on the mica if immobilization is successful; a clear surface indicates failure [2].
- Image: Mount the sample in the AFM. For weakly bound cells, use non-contact imaging modes (Tapping Mode, MAC Mode) or contact mode with low spring constant cantilevers to minimize lateral forces [2].
APTES-Glutaraldehyde for Ultra-Firm Attachment
This method provides robust covalent attachment, ideal for long-duration experiments like single-particle tracking, where any cell movement is detrimental [3].
Materials:
- Glass coverslips
- (3-Aminopropyl)triethoxysilane (APTES)
- Glutaraldehyde (EM-grade for low fluorescence)
- Sorbitol solution (150 mM, or other non-ionic osmolyte)
Procedure:
- Silanize Glass: Treat plasma-cleaned glass coverslips with APTES to create an amine-functionalized surface. This results in a hydrophobic surface, confirmed by contact angle measurements [3].
- Activate with Glutaraldehyde: Treat the APTES-coated slides with glutaraldehyde solution for 30 minutes. This step introduces aldehyde groups that will react with primary amines on the bacterial surface. Rise thoroughly with water after treatment [3].
- Immobilize Cells: Suspend bacterial cells in a non-ionic solution like 150 mM sorbitol. Ionic solutions can shield the charge interactions and impair immobilization. Apply the cell suspension to the activated surface, allowing covalent bonds to form between the glutaraldehyde and surface amines of the cells [3].
- Image: Proceed with AFM imaging. This surface provides exceptionally firm attachment, allowing for repeated scanning and long-term measurements.
Immobilization Strategy Selection Workflow
The following diagram outlines a logical decision pathway for selecting the most appropriate immobilization method based on experimental goals and sample characteristics:
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of AFM immobilization protocols requires specific reagents and materials. The following table lists key solutions and their critical functions.
Table 2: Essential Research Reagent Solutions for Bacterial Immobilization
| Reagent/Material | Function in Protocol | Key Considerations |
|---|---|---|
| Porcine Gelatin (e.g., Sigma G-6144) | Creates a positively charged coating on mica to electrostatically immobilize negatively charged bacterial cells. | Gelatin source is critical; porcine is recommended. Bovine gelatin is often ineffective. Test for compatibility with your bacterial strain [2]. |
| APTES ( (3-Aminopropyl)triethoxysilane) | Functionalizes glass surfaces with amine groups for subsequent cross-linking with glutaraldehyde. | Creates a hydrophobic surface. Use in a fume hood. Quality can vary between suppliers [3]. |
| Glutaraldehyde (EM-Grade) | Acts as a cross-linker, forming covalent bonds between APTES-treated surfaces and primary amines on bacterial cell surfaces. | EM-grade is recommended for fluorescence applications due to lower background autofluorescence [3]. |
| Sorbitol Solution (150 mM) | A non-ionic osmolyte used as an attaching and imaging medium for osmotically sensitive cells. | Prevents osmotic shock without introducing ions that compete with cells for binding to charged surfaces like gelatin [3]. |
| Indium-Tin-Oxide (ITO) Coated Glass | Provides a smooth, hydrophobic substrate that promotes adhesion of some bacterial cells without chemical coatings. | Offers chemical stability and excellent compatibility with AFM probes for high-resolution imaging in liquid [5]. |
| Poly-L-Lysine Solution | Creates a positively charged coating on glass or mica to enhance electrostatic cell adhesion. | A common alternative to gelatin; effectiveness can be strain-dependent. |
Firm and reproducible immobilization of bacterial cells is a non-negotiable prerequisite for high-quality, reliable AFM imaging and force spectroscopy. The choice of method must be carefully aligned with the experimental objectives, whether they prioritize the preservation of native mechanical properties or absolute spatial stability. As AFM technology evolves with advancements like large-area automated scanning and machine learning-enhanced analysis [6], the demand for robust, high-throughput immobilization techniques will only grow. Furthermore, the elucidation of complex bacterial behaviors—such as nanotube-mediated communication [5] and biofilm assembly dynamics [6] [7]—will increasingly depend on immobilization strategies that secure cells without perturbing their delicate functional structures. By adhering to the validated protocols and principles outlined in this application note, researchers can lay a solid foundation for groundbreaking discoveries in microbial biophysics and drug development.
Understanding Physicochemical Forces in Bacterium-Surface Interactions
Atomic force microscopy (AFM) has emerged as a powerful tool for investigating the physicochemical forces that govern bacterial adhesion to surfaces, a critical initial step in biofilm formation and microbial infection [8]. This application note details established and emerging protocols for immobilizing live bacterial cells for AFM studies, enabling researchers to quantitatively measure adhesion forces, nanomechanical properties, and surface dynamics under physiologically relevant conditions. The ability to immobilize cells effectively without compromising their viability or surface properties is fundamental to obtaining reliable data on the initial interactions between bacteria and substrates, which can inform strategies for controlling biofilm formation in medical and industrial contexts.
Quantitative Comparison of Bacterial Immobilization Methods
The following table summarizes key parameters for different bacterial immobilization approaches used in AFM studies:
Table 1: Comparison of Bacterial Immobilization Methods for AFM Studies
| Immobilization Method | Typical Adhesion Forces Measured | Relative Throughput | Cell Viability Preservation | Key Applications | Technical Limitations |
|---|---|---|---|---|---|
| Poly-L-Lysine Coating | Not specified | Medium | Moderate (antimicrobial effects noted) | Imaging surface dynamics in nutrient media [9] | Potential alteration of cell physiology; antimicrobial properties may affect viability |
| Gelatin Coating | Not specified | Medium | High | Stable imaging in aqueous conditions; studies of outer membrane vesicle production [9] | Potential obstruction of cell surface features |
| Physical Entrapment | Not specified | Low | High (inert method) | General bacterial imaging [9] | Unpredictible surface obstructions; may exert non-native forces on cells |
| Polydopamine Coating | 1-50 nN (depending on strain) | High | High (maintains cell functionality) | Single-cell force spectroscopy on diverse bacterial isolates [10] | Requires controlled polymerization conditions |
| ITO-Coated Glass Substrates | Not specified | Medium | High (no chemical immobilization) | Nanomechanical mapping of living bacteria in liquid [5] | Requires specific substrate preparation |
| FluidFM Technology | pN to µN range | High (up to 200 cells/day) | Excellent (reversible immobilization) | High-throughput single-cell force spectroscopy; kinetic studies of adhesion [11] [10] | Requires specialized equipment |
Experimental Protocols
Chemical Immobilization Using Poly-L-Lysine or Gelatin for Live Cell Imaging
This protocol describes a method for immobilizing Gram-negative bacteria such as Escherichia coli for AFM studies of surface dynamics, optimized to preserve cell viability during extended imaging sessions in nutrient media [9].
Materials:
- Glass slides or coverslips
- Poly-L-lysine (PLL) solution (0.1% w/v) or gelatin (high, medium, or low bloom strength)
- Bacterial culture in mid-exponential or stationary growth phase
- Immobilization buffer: 0.01× PBS-S (diluted phosphate-buffered saline)
- Imaging media: LB broth or minimal media (MM)
Procedure:
- Substrate Preparation:
- Clean glass substrates thoroughly to remove organic contaminants.
- Coat substrates with 0.5% gelatin solution or 0.1% PLL solution and allow to dry at room temperature.
- For PLL substrates, note that antimicrobial effects may impact cell viability and require careful optimization.
Cell Preparation:
- Grow bacterial cells to mid-exponential or stationary phase based on experimental requirements.
- Harvest cells by gentle centrifugation (4,100 × g for 10 minutes at 10°C).
- Wash cell pellets three times with immobilization buffer (0.01× PBS-S) to remove growth media components.
Immobilization:
- Apply bacterial suspension to coated substrates and incubate for 10-20 minutes.
- Gently rinse with immobilization buffer to remove non-adherent cells.
- For PLL-immobilized cells, allow a recovery period in imaging media (MM or LB broth) before AFM analysis to restore membrane integrity.
Viability Assessment:
- Monitor membrane integrity using fluorescent viability stains (e.g., SYTO 9 and propidium iodide).
- Confirm preserved cell division capability as an indicator of maintained viability during time-lapse experiments.
Applications: This method enables stable immobilization for high-resolution imaging of bacterial surface dynamics, including outer membrane vesicle production and cell division events in nutrient media [9].
Non-Perturbative Immobilization for Nanomechanical Mapping
This protocol describes a method for immobilizing Rhodococcus wratislaviensis and similar bacteria without chemical fixation, enabling nanomechanical characterization of bacterial surfaces and intercellular structures in liquid environments [5].
Materials:
- Indium-tin-oxide (ITO)-coated glass substrates
- Bacterial culture in exponential growth phase
- Liquid culture medium for AFM imaging
- Nanowizard AFM system (JPK Instruments) or comparable equipment
- PPP-CONTPt AFM probes (Nanosensors, stiffness 0.3 N/m)
Procedure:
- Substrate Preparation:
- Use ITO-coated glass substrates without additional functionalization.
- ITO's hydrophobic properties and smooth surface facilitate bacterial adhesion without chemical immobilization.
Sample Preparation:
- Pipette 500 μL of bacterial culture in exponential growth phase directly onto ITO substrate.
- Place substrate in electrochemical liquid cell (ECCell) without rinsing or immobilization steps.
AFM Imaging:
- Perform AFM imaging in liquid culture medium at controlled temperature (24.0 ± 0.2°C).
- Use Quantitative Imaging (QI) mode with the following parameters:
- Total extension: 600 nm
- Constant speed: 125 μm/s
- Indentation speed: 17-175 mN/s
- Image resolution: 64 × 64 pixels
Data Analysis:
- Calculate Young's modulus using Sneddon model fit to force curves:
- F = (2/π) × [E/(1-ν²)] × tan(α) × δ²
- Where E is Young's modulus, ν is Poisson's ratio (0.5), α is tip semi-angle (35°), and δ is indentation.
- Calculate Young's modulus using Sneddon model fit to force curves:
Applications: This method enables real-time nanomechanical mapping of living bacteria, including characterization of bacterial nanotubes and other delicate surface features without immobilization-induced artifacts [5].
Modular Single-Cell Force Spectroscopy with Functionalized Beads
This protocol describes a modular approach for quantifying adhesion forces of diverse bacterial species using functionalized beads immobilized via FluidFM technology, enabling high-throughput single-cell force spectroscopy without chemical fixation of cells to cantilevers [10].
Materials:
- FluidFM system with hollow cantilevers
- C30-functionalized silica beads (to mimic hydrophobic surfaces)
- Polydopamine-coated glass substrates
- Diverse bacterial strains suspended in appropriate buffer
Procedure:
- Bacterial Immobilization:
- Immobilize bacteria on polydopamine-coated glass substrates to prevent displacement during measurement.
- Confirm isolated, viable cell positioning under optical microscopy.
Bead Preparation:
- Apply negative pressure to reversibly immobilize a C30-functionalized silica bead on the tipless aperture of the FluidFM cantilever.
- Exchange beads regularly between measurements using overpressure pulses to maintain consistent surface properties.
Force Spectroscopy:
- Approach a target bacterial cell with the functionalized bead at controlled speed.
- Establish contact with a defined force of 10 nN.
- Maintain contact for 5 seconds.
- Retract cantilever at constant speed while recording deflection.
Data Collection:
- Measure adhesion forces from retraction curves as the maximum recorded force.
- Characterize force curve patterns indicative of cellular appendages (e.g., force jumps suggesting pilus engagement).
- Collect data from multiple cells (typically 10-20 per strain) to assess population heterogeneity.
Applications: This method enables quantitative comparison of hydrophobic adhesion forces across phylogenetically diverse bacterial strains, with demonstrated correlation to bacterial retention on plant surfaces in ecological contexts [10].
Research Reagent Solutions
Table 2: Essential Materials for Bacterial Immobilization and AFM Force Spectroscopy
| Reagent/Equipment | Function | Application Notes |
|---|---|---|
| ITO-coated glass substrates | Provides adhesion-friendly surface without chemical immobilization | Enables nanomechanical mapping of living bacteria in liquid; hydrophobic properties facilitate cell adhesion [5] |
| Poly-L-lysine coating | Electrostatic immobilization of bacterial cells | Useful for imaging in nutrient media; requires viability assessment due to potential antimicrobial effects [9] |
| Gelatin coatings | Non-cytotoxic adhesive layer for cell immobilization | Various bloom strengths available; suitable for Gram-negative and Gram-positive bacteria in aqueous conditions [9] |
| Polydopamine coating | Firm immobilization of bacteria for force spectroscopy | Prevents cell displacement during adhesion measurements; maintains cell functionality [10] |
| C30-functionalized beads | Mimics hydrophobic surfaces like plant cuticles | Used in modular AFM to quantify hydrophobic interaction forces with bacterial cells [10] |
| FluidFM cantilevers | Enables reversible immobilization of beads or cells | Hollow cantilevers allow aspiration-based handling; dramatically increases throughput of single-cell force spectroscopy [11] [10] |
| PPP-CONTPt AFM probes | Standard probes for nanomechanical mapping | 0.3 N/m stiffness suitable for living bacterial cells in liquid [5] |
Experimental Workflow Visualization
The selection of an appropriate bacterial immobilization strategy is critical for obtaining reliable AFM measurements of bacterium-surface interactions. Traditional chemical methods using PLL or gelatin provide stable immobilization for dynamic studies but require careful optimization to maintain cell viability. Emerging approaches such as I-coated substrates enable nanomechanical characterization without potential artifacts from immobilization reagents, while modular FluidFM-based methods dramatically increase throughput for single-cell force spectroscopy. The correlation between measured adhesion forces and bacterial retention in ecological contexts demonstrates the biological relevance of these AFM-based approaches. By selecting immobilization methods aligned with specific research questions—whether investigating fundamental nanomechanical properties, dynamic surface processes, or population-level heterogeneity—researchers can generate meaningful insights into the physicochemical forces governing bacterial adhesion and biofilm formation.
Atomic force microscopy (AFM) provides unparalleled capability for high-resolution imaging and mechanical probing of live bacterial cells under physiological conditions. However, the reliability of AFM data is critically dependent on effective cell immobilization. Insufficient adhesion results in cell displacement by the scanning probe, while overly invasive methods can introduce surface artifacts or alter native biophysical properties, compromising experimental validity. This application note details the prevalent challenges in bacterial immobilization—cell displacement, surface artifacts, and altered biophysical properties—and provides validated protocols to mitigate them, ensuring the acquisition of robust, physiologically relevant data.
Common Immobilization Challenges and Strategic Solutions
The core challenges in bacterial immobilization often involve conflicting requirements; methods that provide strong adhesion to prevent displacement can damage the cell surface or alter its natural state. The table below summarizes the primary challenges and corresponding strategic solutions.
Table 1: Summary of Common Immobilization Challenges and Strategic Solutions
| Challenge | Primary Cause | Recommended Solution | Key Considerations |
|---|---|---|---|
| Cell Displacement [12] [13] | Lateral forces from AFM tip exceeding adhesion strength. | Gelatin-coated mica for electrostatic immobilization [14] [15]; Physical entrapment in porous membranes [12] [13]. | Gelatin origin is critical (porcine recommended); Entrapment best for coccoid cells [14] [13]. |
| Surface Artifacts | Sample drying; Contamination from immobilization coatings. | Immobilization in liquid without drying; Use of pure, biocompatible coatings [14] [13]. | Avoid bovine gelatin; Ensure gelatin is fully dissolved and coating is even [14]. |
| Altered Biophysical Properties [13] [16] | Osmotic stress from low-ionic-strength buffers; Chemical fixation. | Use of physiological buffers supplemented with divalent cations (Mg²⁺, Ca²⁺) [16]; Use of adhesive proteins like Cell-Tak [13] [17]. | Monitor cell viability throughout protocol; Divalent cations help maintain membrane integrity [16]. |
Cell Displacement by Scanning Probe
Cell displacement occurs when lateral forces exerted by the AFM cantilever overcome the adhesive forces tethering the cell to the substrate. This is a frequent obstacle when imaging rod-shaped bacteria, which have a small contact area with the surface [13]. Electrostatic immobilization on gelatin-coated mica is a widely successful strategy. The negatively charged bacterial surface adheres to the positively charged gelatin, sufficiently immobilizing cells for imaging in liquid [14] [15]. The protocol for this method is detailed in Section 4.1. Alternatively, physical entrapment in porous membranes with a pore size similar to the cell dimension can be highly effective, particularly for coccoid cells, and avoids chemical modification of the cell surface [12] [13].
Surface artifacts are artificial features introduced during sample preparation that obscure the native cell surface topography. A common source is the drying and rehydration of cells, which can collapse surface structures like pili and capsules [13]. To preserve native structures, immobilization must be performed in a liquid environment without intermediate drying steps [14]. Another source is chemical contamination from impure or incompatible immobilization reagents. For example, gelatin derived from bovine sources has been shown to be ineffective for immobilizing many bacterial strains, whereas porcine gelatin (e.g., Sigma G-6144, G-2625) is generally effective [14].
Alteration of Native Biophysical Properties
Preserving the native physiological state of the cell is paramount for meaningful data. A frequent pitfall is the induction of osmotic stress when using low-ionic-strength buffers like deionized water for immobilization and imaging [13] [16]. This can destabilize extracellular structures and alter mechanical properties. Supplementing buffers with divalent cations (Mg²⁺, Ca²⁺) and glucose has been shown to stabilize the bacterial membrane, maintaining viability and native surface properties during immobilization on poly-L-lysine [16]. Furthermore, chemical fixation, while enhancing adhesion, drastically alters surface elasticity and should be avoided in live-cell studies [12].
The Scientist's Toolkit: Essential Research Reagents
The selection of appropriate reagents is fundamental to successful immobilization. The following table catalogues key materials and their functions.
Table 2: Key Research Reagents for Bacterial Immobilization
| Reagent/Material | Function in Immobilization | Specific Examples & Notes |
|---|---|---|
| Porcine Gelatin | Creates a positively charged coating on mica for electrostatic binding of cells [14] [15]. | Sigma G-6144 (low Bloom) and G-2625 (medium Bloom) are most effective [14]. |
| Poly-L-Lysine | A positively charged polymer that strongly adheres negatively charged cells to surfaces [16]. | Can compromise membrane integrity unless used with divalent cations [16]. |
| Cell-Tak | A biocompatible, polyphenolic protein adhesive from mussels for strong physical attachment [13] [17]. | Effective for a wide range of cell types under physiological conditions [13]. |
| Polyethylenimine (PEI) | Positively charged polymer used for coating beads in single-cell force spectroscopy [18]. | Used to create a monolayer of cells on silica beads for probe-based force measurements [18]. |
| Mica | An atomically flat, negatively charged substrate that can be freshly cleaved for a clean surface [14]. | Ideal for high-resolution imaging; often used as a base for gelatin or other coatings [14]. |
| Divalent Cations (Mg²⁺, Ca²⁺) | Added to buffers to stabilize the bacterial outer membrane and improve cell viability during immobilization [16]. | Mitigates the harmful effects of low-ionic-strength buffers and poly-L-lysine [16]. |
Validated Experimental Protocols
Protocol: Immobilization on Gelatin-Coated Mica
This protocol is adapted from the highly cited method for immobilizing a broad spectrum of Gram-negative and Gram-positive bacteria [14] [15].
Workflow Overview:
Step-by-Step Methodology:
- Mica Preparation: Cut a piece of mica to approximately 22 x 30 mm. Using adhesive tape, cleave the top layers from both sides until a smooth, unbroken surface is achieved [14].
- Gelatin Solution Preparation:
- Add 0.5 grams of porcine gelatin (e.g., Sigma G-6144) to 100 mL of boiling distilled water.
- Gently swirl until the gelatin is completely dissolved.
- Cool the solution to 60-70°C before use. The solution can be stored refrigerated for up to a month and reheated for future use [14].
- Mica Coating:
- Submerge the freshly cleaved mica square into the warm gelatin solution and withdraw it quickly.
- Place the coated mica on its edge on a paper towel to dry in ambient air. The coated mica is stable for at least two weeks [14].
- Bacterial Preparation:
- Pellet 1 mL of a bacterial culture (OD₆₀₀ ≈ 0.5-1.0) by centrifugation (800 - 4,500 rcf for ~5 minutes).
- Wash the pellet in filtered deionized water or a compatible buffer (e.g., 0.01M PBS) to remove growth media and salts that can interfere with adhesion.
- Resuspend the final pellet in 500 µL of nanopure water or a dilute buffer to create a visibly turbid suspension [14].
- Cell Mounting:
- Apply 10-20 µL of the bacterial suspension to the center of the gelatin-coated mica.
- Gently spread the droplet using a pipette tip, taking care not to touch or scratch the gelatin surface.
- Allow the sample to incubate for 10 minutes at room temperature.
- Rinse gently with a steady, gentle stream of deionized water or imaging buffer to remove loosely attached cells. Critical: Do not allow the sample to dry at any point [14].
- Quality Control: A simple test for successful immobilization is to allow the rinsed sample to air dry. A cloudy area on the mica indicates retained cells, while a clear spot suggests the cells were washed away [14].
Protocol: Immobilization for Live-Cell Dynamics in Nutrient Media
This protocol is optimized for immobilizing less adherent strains for time-lapse imaging and division studies in nutrient-rich media, where maintaining viability is crucial [16].
Workflow Overview:
Step-by-Step Methodology:
- Substrate Coating: Apply a solution of poly-L-lysine (PLL) to a clean glass or mica substrate. After a brief incubation, rinse the surface with water to remove excess PLL and allow it to dry [16].
- Buffer Preparation: Prepare a low-ionic-strength immobilization buffer (e.g., 1-5 mM HEPES or Tris). Supplement this buffer with 1-10 mM MgCl₂ and CaCl₂. The divalent cations are critical for stabilizing the bacterial outer membrane [16].
- Bacterial Preparation:
- Pellet bacterial cells from the growth medium.
- Wash the cells gently in the prepared immobilization buffer (with Mg²⁺/Ca²⁺) to remove media components.
- Resuspend the cells in the same buffer [16].
- Cell Immobilization:
- Apply the bacterial suspension to the PLL-coated surface and allow to incubate for a defined period (e.g., 10-30 minutes).
- Gently rinse with immobilization buffer or the intended imaging medium to remove non-adhered cells [16].
- Viability Check: It is essential to confirm cell viability after immobilization. This can be done using a membrane integrity stain (e.g., propidium iodide exclusion assay) [18] [16]. Only preparations with high viability should be used for dynamic studies.
- Imaging: For live-cell dynamics, replace the immobilization buffer with a nutrient medium (e.g., diluted LB broth) for AFM imaging [16].
Quantitative Data from Immobilization Studies
The effectiveness of different immobilization strategies can be quantified by measuring adhesion forces, cell viability, and imaging success rates.
Table 3: Quantitative Comparison of Immobilization Methods
| Immobilization Method | Reported Adhesion Force | Cell Viability / Integrity | Imaging Success Rate / Notes |
|---|---|---|---|
| Gelatin-coated Mica [14] | Not quantitatively reported, but sufficient for imaging in liquid and dilute buffers. | High, when performed without drying [14]. | Generally applicable to many microbial cells; successful for force measurements [14]. |
| Poly-L-Lysine (with Mg²⁺/Ca²⁺) [16] | Not quantitatively reported, but sufficient for imaging in nutrient media. | High, membrane integrity maintained with cation supplementation [16]. | Enables time-lapse imaging through multiple cell division cycles [16]. |
| Physical Entrapment [12] [13] | N/A | Can exert mechanical stress on cells [13]. | Best for coccoid cells; less suitable for rods [13]. |
| Cell-Tak [13] | Not quantitatively reported, provides strong physical attachment. | High, compatible with physiological conditions [13]. | Effective for diverse cell shapes and sizes under physiological ionic strength [13]. |
| E. coli to Goethite [19] | -97 ± 34 pN (attractive jump-in); Maximum adhesion: -3.0 ± 0.4 nN [19]. | N/A | Measured using single-cell force spectroscopy; bond strengthening observed over 4s [19]. |
Successful AFM investigation of live bacteria hinges on a immobilization strategy that balances the competing demands of mechanical stability, biological preservation, and minimal intervention. While gelatin-coated mica offers a broadly applicable and gentle approach, specific experimental goals—such as long-term imaging in rich media—may require optimized methods like poly-L-lysine with membrane-stabilizing cations. By understanding the sources of major challenges like cell displacement, surface artifacts, and altered biophysics, researchers can select and refine the most appropriate protocol. The rigorous application of these detailed protocols, coupled with systematic quality control like viability testing, will ensure that AFM data truly reflects the native structure and function of the bacterial cell surface.
Preserving Cell Viability and Surface Integrity During the Immobilization Process
Atomic force microscopy (AFM) has emerged as a powerful tool in microbiological research, enabling the investigation of bacterial cells at unprecedented nanoscale resolution. Its capability to operate under physiological conditions provides unique insights into the structural and mechanical properties of living microorganisms. However, a significant challenge persists: securely immobilizing bacterial cells without compromising their viability or structural integrity. This balance is critical for obtaining biologically relevant data, as invasive immobilization techniques can alter cellular physiology, surface properties, and mechanical responses, ultimately leading to experimental artifacts [5] [20].
This application note addresses this fundamental challenge by presenting standardized protocols for bacterial immobilization tailored specifically for AFM studies. We focus on methods that preserve native cell conditions while providing sufficient stability for high-resolution imaging and force spectroscopy. Within the broader context of AFM protocol development for bacterial cell research, mastering this immobilization step is prerequisite for any investigation into bacterial adhesion, biofilm formation, antimicrobial efficacy, or single-cell biomechanics.
Key Immobilization Principles
Successful AFM analysis of bacterial cells requires adherence to several core principles designed to maintain cells in a viable, unperturbed state during scanning procedures.
- Minimizing External Stress: Immobilization must withstand scanning forces while avoiding chemical or physical stress that alters cellular physiology. Chemical cross-linking agents, while providing strong adhesion, often reduce viability and alter nanomechanical properties [20].
- Substrate Biocompatibility: The chosen substrate must facilitate firm cell adhesion without inducing toxicity. Functionalized glass surfaces, particularly indium-tin-oxide (ITO)-coated glass, provide excellent bacterial adhesion while maintaining compatibility with liquid-phase AFM imaging [5].
- Physiological Conditions: Throughout immobilization and imaging, cells should remain in appropriate buffered solutions to prevent osmotic shock or dehydration, which dramatically alter cellular morphology and mechanical properties [19] [21].
Immobilization Methods and Protocols
Non-Immobilization Approach Using ITO-Coated Glass
Recent advancements have challenged the notion that aggressive immobilization is necessary for AFM imaging in liquid. A protocol developed for Rhodococcus wratislaviensis demonstrates that specific substrate properties can eliminate the need for chemical or mechanical immobilization [5].
Workflow: ITO Substrate Preparation and Cell Deposition
Materials and Reagents:
- Indium-Tin-Oxide (ITO)-coated glass slides (e.g., Neyco)
- Oxygen plasma cleaner or UV-ozone treatment system
- Bacterial culture in exponential growth phase
- Appropriate culture medium for rinsing and imaging
Critical Steps and Optimization:
- Substrate Preparation: ITO-coated glass provides an optimal combination of smoothness and controlled hydrophobicity that promotes spontaneous bacterial adhesion without external agents [5].
- Cell Deposition: Pipette 500μL of bacterial culture during exponential growth phase directly onto the ITO substrate.
- Adhesion Period: Allow 30-60 minutes for cells to settle and adhere under controlled temperature conditions.
- Rinsing: Gently rinse with fresh culture medium to remove non-adherent cells while maintaining physiological conditions.
- Imaging: Maintain cells in appropriate culture medium during AFM analysis using Quantitative Imaging mode to minimize lateral forces.
This method successfully enabled the first characterization of bacterial nanotubes in liquid on living bacteria without immobilization, revealing a lower Young's modulus of nanotubes (0.07-0.08 GPa) compared to the cell body (0.15 GPa), which would likely have been altered by chemical fixation [5].
Gelatin-Coated Surfaces for Single-Cell Analysis
For single-cell force spectroscopy studies requiring precise positioning, gelatin coating provides a biocompatible immobilization method that preserves membrane integrity and cellular viability [21].
Protocol: Gelatin Coating for E. coli Immobilization
- Prepare Gelatin Solution: Dissolve gelatin in Milli-Q water to create a 0.1-0.5% w/v solution.
- Coat Glass Slides: Apply the gelatin solution to clean glass slides and allow to air dry completely.
- Cell Deposition: Centrifuge bacterial culture (2151 × g for 5 min), wash twice with Milli-Q water, and resuspend.
- Adjust Concentration: Dilute bacterial suspension to approximately 10⁶ CFU/mL.
- Immobilize Cells: Deposit bacterial suspension on gelatin-coated slides and incubate for 30 minutes.
- Gentle Rinsing: Carefully rinse with appropriate buffer to remove non-adherent cells.
This method has been successfully applied for AFM studies investigating lipopolysaccharide-mediated heterogeneity in bacterial adhesion and mechanics, confirming preservation of native outer membrane structure [21].
Mechanical Entrapment in Porous Membranes
For challenging imaging scenarios requiring extreme stability, mechanical entrapment provides an alternative that avoids chemical modification of cell surfaces.
Protocol: Mechanical Entrapment Using Porous Membranes
- Membrane Selection: Choose polycarbonate or PDMS membranes with pore diameters slightly smaller than the target cells.
- Cell Concentration: Centrifuge bacterial culture and resuspend in appropriate buffer to create a concentrated suspension.
- Filtration Assembly: Assemble a filtration unit with the selected membrane.
- Gentle Filtration: Apply bacterial suspension to the filtration unit using minimal pressure.
- Transfer to Substrate: Carefully transfer the membrane with trapped cells to AFM substrate.
- Hydration Maintenance: Ensure the membrane remains hydrated with appropriate buffer throughout imaging.
While this method provides excellent stability for imaging, it may not be suitable for all bacterial strains, particularly those susceptible to physical stress during the filtration process [20].
Quantitative Comparison of Immobilization Methods
Table 1: Comparative Analysis of Bacterial Immobilization Methods for AFM
| Method | Cell Viability Preservation | Immobilization Strength | Preservation of Nanomechanical Properties | Ease of Implementation | Recommended Applications |
|---|---|---|---|---|---|
| ITO-coated Glass (Non-immobilization) | High | Moderate | Excellent | Moderate | Live cell imaging, Nanomechanical mapping, Dynamic processes |
| Gelatin Coating | High | Moderate-High | Good | High | Single-cell force spectroscopy, Adhesion studies, Population heterogeneity |
| Mechanical Entrapment | Moderate | Very High | Moderate (potential compression artifacts) | Moderate | Topographical imaging of motile strains, High-resolution surface characterization |
| Poly-L-Lysine Coating | Moderate (varies by protocol) | High | Moderate (may alter surface properties) | High | Fixed cell imaging, Rapid screening |
| APTES Functionalization | Low-Moderate | Very High | Poor (significant alterations) | High | Fixed cells only, Structural studies requiring extreme stability |
Table 2: Effects of Immobilization on Bacterial Nanomechanical Properties
| Immobilization Method | Reported Young's Modulus (kPa) | Adhesion Force (nN) | Impact on Membrane Structure | Structural Features Resolvable |
|---|---|---|---|---|
| ITO-coated Glass | 150 (cell body), 70-80 (nanotubes) [5] | Not reported | Minimal alteration | Nanotubes, Surface appendages, Membrane protrusions |
| Gelatin Coating | Cell-specific, maintained heterogeneity [21] | Cell-specific, maintained heterogeneity | Preservation of LPS structure | Native outer membrane organization |
| Chemical Cross-linking | Artificially increased (200-400% higher than native) | Reduced or inconsistent | Significant disruption | Limited to gross cellular morphology |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Bacterial Immobilization
| Reagent/Material | Function | Application Notes | Supplier Examples |
|---|---|---|---|
| ITO-coated glass slides | Provides adhesion-friendly surface without chemical modification | Optimal for liquid-phase AFM; surface hydrophobicity enhances bacterial adhesion | Neyco, Bruker-JPK |
| Gelatin from porcine skin | Creates biocompatible coating for cell adhesion | Maintains viability; suitable for single-cell force spectroscopy | Sigma-Aldrich |
| Polycarbonate membranes | Mechanical entrapment of bacterial cells | Pore size should be 50-80% of cell diameter for optimal trapping | Millipore, Whatman |
| Polydimethylsiloxane (PDMS) | Customizable microstructured stamps for cell immobilization | Enables controlled cell positioning; requires microfabrication expertise | Dow Sylgard |
| Cationic reagents (Mg²⁺, Ca²⁺) | Enhances adhesion to negatively charged surfaces | Can be added to buffers to improve attachment without chemical fixation | Various |
Troubleshooting and Quality Control
Assessing Immobilization Success
Visual Inspection via AFM: Before data collection, perform quick scans to verify:
- Consistent cell orientation and position across multiple scans
- Absence of cellular debris or detachment artifacts
- Maintenance of typical cellular morphology (rod-shaped bacteria maintaining elongation)
Viability Assessment: When viability is crucial, employ:
- Post-imaging culturability: Retrieve cells from substrate and assess growth on agar plates
- Membrane integrity stains: Use fluorescent viability markers after AFM analysis
- Metabolic activity assays: Measure respiration or enzymatic activity following imaging
Addressing Common Challenges
Problem: Cell Detachment During Scanning
- Solution: Optimize adhesion time; increase divalent cation concentration in imaging buffer; verify substrate functionalization.
Problem: Altered Mechanical Properties
- Solution: Shift toward milder immobilization methods (e.g., ITO substrates); reduce chemical cross-linking; ensure physiological buffer conditions.
Problem: Poor Image Resolution
- Solution: Verify immobilization stability; optimize AFM scanning parameters (reduced force, higher oscillation amplitude in tapping mode); check tip quality.
The optimal immobilization strategy for AFM studies of bacterial cells must be carefully selected based on research objectives, balancing the competing demands of mechanical stability against preservation of native cellular properties. The protocols presented herein provide a foundation for reliable bacterial immobilization while maintaining viability and surface integrity. As AFM continues to evolve toward more sophisticated biological applications, particularly in antimicrobial development and single-cell analysis, these immobilization techniques will remain fundamental to generating physiologically relevant data at the nanoscale.
A Practical Guide to Bacterial Immobilization Techniques for AFM
In the field of single-cell analysis, particularly using techniques like Atomic Force Microscopy (AFM), effective cell immobilization is a critical prerequisite. The principle of AFM involves scanning the sample surface with a nanometric tip on a flexible cantilever, requiring precise positioning via piezoelectric scanners [22]. For microbiological applications, this has opened new avenues for describing topographical features and molecular mechanisms at the cell wall [22]. However, microbial cells are mostly round-shaped, making proper immobilization essential to prevent the tip from pushing the cell during scanning rather than accurately scanning the cell surface [22]. Among the commonly used immobilization methodologies—which include embedding in gelatin and electrostatic immobilization on positively charged substrates—mechanical trapping in porous membranes stands out as a particularly robust technique for high-resolution imaging and molecular mapping [22]. This protocol details the application of mechanical trapping within the broader context of AFM-based research on bacterial cells, providing a standardized approach for researchers and drug development professionals seeking to investigate cell surface heterogeneity, adhesion, and mechanics at the single-cell level.
Principle of the Technique
Mechanical trapping involves the physical entrapment of individual microbial cells within the pores of a membrane filter. This method counteracts the lateral forces exerted by the AFM tip during scanning by physically constraining the cells, thereby enabling stable and high-resolution measurements [22]. This approach is especially well-suited for studying a wide range of microbial cells, including both Gram-positive and Gram-negative bacteria, under physiological conditions.
Comparison of Immobilization Techniques
The selection of an appropriate immobilization strategy is critical and depends on the specific experimental goals. The table below summarizes the key characteristics of common methods.
Table 1: Comparison of AFM Cell Immobilization Methods
| Immobilization Method | Key Principle | Best Suited Applications | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Mechanical Trapping | Physical entrapment in membrane pores [22] | High-resolution imaging, molecular mapping [22] | Robust immobilization, suitable for physiological conditions | Can be time-consuming; may select for specific cell sizes [22] |
| Electrostatic Adsorption | Attachment to positively-charged substrates (e.g., PEI, PLL) [22] | Imaging, nanomechanical mapping [22] | Simple and fast procedure | Charged polymers may affect cell viability or denature molecules [22] |
| Gelatin Embedding | Embedding cell volume in a gelatin layer [22] | Observing bacterial growth [22] | Good for time-lapse studies | Gelatin can cause AFM tip contamination [22] |
| PDMS Stamps | Convective/capillary assembly on polydimethylsiloxane stamps [22] | Statistically relevant measurements on multiple cells [22] | Enables array formation for high-throughput analysis | Requires specialized fabrication |
| Microfluidics | Pressure-driven anchoring in microscopic traps [22] | Sequential immobilization/release, combined AFM & fluorescence [22] | Allows for integrated, dynamic experimental setups | Complex device design and operation |
Experimental Protocols
Workflow for Mechanical Trapping and AFM Analysis
The following diagram outlines the comprehensive experimental workflow, from cell culture to data analysis.
Protocol 1: Cell Preparation and Immobilization
Objective: To prepare a bacterial culture and immobilize cells via mechanical trapping for AFM analysis.
Materials:
- Escherichia coli ATCC 25922 (or other relevant strain) [21]
- Luria-Bertani (LB) broth and agar
- Centrifuge
- Phosphate buffer (0.01 M, pH 7.0) [21]
- Porous membrane filters (e.g., polycarbonate membranes with pore sizes comparable to cell dimensions)
- Gelatin-coated glass slides (alternative for viability checks) [21]
- AFM liquid cell
Procedure:
- Cell Culture: Revive the bacterial strain from -80°C storage on LB agar. Inoculate a single colony into LB broth and culture for 24 hours at 37°C with shaking at 150 rpm [21].
- Harvesting: Centrifuge the bacterial culture at 2151 × g for 5 minutes at 24°C. Carefully decant the supernatant [21].
- Washing: Resuspend the cell pellet in Milli-Q water. Repeat the centrifugation and washing step twice to remove residual growth media [21].
- Final Resuspension: Resuspend the final cell pellet in 0.01 M phosphate buffer (pH 7.0). Adjust the cell suspension to an optical density suitable for obtaining a semi-confluent layer on the membrane (e.g., ~10⁶ CFU/ml) [21].
- Mechanical Trapping:
- Place a porous membrane filter on a suitable support.
- Apply the cell suspension to the membrane and allow it to filter by gravity or gentle vacuum.
- Rinse gently with buffer to remove non-adherent or loosely trapped cells.
- Carefully transfer the membrane with trapped cells to the AFM sample stage.
- Assemble the AFM liquid cell and add an appropriate physiological buffer to submerge the sample.
Troubleshooting:
- Low Immobilization Density: Optimize cell concentration and filtration volume.
- Cell Damage: Avoid excessive vacuum pressure during filtration.
- Tip Contamination: Ensure the membrane surface is clean and free of debris before use.
Protocol 2: AFM Imaging and Force Spectroscopy
Objective: To perform topographical imaging and quantify adhesion forces/mechanical properties of immobilized cells.
Materials:
- Atomic Force Microscope
- AFM cantilevers (e.g., silicon nitride tips for imaging in liquid; colloidal probes for single-cell force spectroscopy)
- Liquid cell setup
Procedure:
- AFM Setup: Mount the prepared sample in the AFM. Select an appropriate cantilever based on the experiment (sharp tip for imaging, colloidal probe for full-cell force spectroscopy) [21].
- Engagement: Engage the tip with the surface in liquid using standard procedures for the instrument.
- Imaging: Acquire topographic images of the trapped cells using a gentle imaging mode such as tapping mode or peak force tapping mode in liquid to minimize lateral forces [22].
- Force Spectroscopy:
- Position the AFM tip over the center of a selected cell.
- Record force-distance curves by extending and retracting the tip from the cell surface. Perform a sufficient number of measurements (e.g., 256-1024 curves per cell) across multiple cells to ensure statistical significance [21].
- Analyze the force curves to extract parameters such as adhesion force (from the retraction curve) and elastic modulus (from the indentation part of the extension curve using an appropriate model, e.g., Hertzian).
Troubleshooting:
- Low Image Resolution / Cell Movement: Verify the effectiveness of trapping. Ensure scanning parameters (setpoint, gains) are optimized for minimal force.
- Inconsistent Force Curves: Check for tip contamination and clean or replace the tip if necessary. Ensure the cell surface is fully submerged in buffer.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for AFM Single-Cell Studies
| Item | Function/Application | Examples & Notes |
|---|---|---|
| Porous Membranes | Physical scaffold for mechanical trapping of cells [22] | Polycarbonate membranes; pore size is critical and must be matched to cell dimensions. |
| Functionalized Substrates | Electrostatic immobilization of negatively-charged cells [22] | Poly-L-Lysine or Polyethylenimine (PEI) coated glass/silicon. |
| PDMS Stamps | Patterned immobilization of cell arrays for high-throughput analysis [22] | Polydimethylsiloxane stamps fabricated via soft lithography. |
| AFM Cantilevers | Probing surface topology and nanomechanical forces. | Sharp tips for imaging; colloidal probes for single-cell force spectroscopy [21]; fluidic probes (FluidFM) for injection/extraction [23]. |
| Chemical Perturbation Agents | Modifying cell surface properties to study function. | EDTA for partial removal of Lipopolysaccharides (LPS) in Gram-negative bacteria [21]. |
Application in Research: Quantifying Phenotypic Heterogeneity
Mechanical trapping provides the stability required to investigate phenotypic heterogeneity within clonal bacterial populations. The following diagram and data table illustrate a typical application studying the effect of LPS removal on E. coli.
Table 3: Example AFM Data: Effect of LPS Removal on E. coli Biophysical Properties
| Experimental Group | Surface Roughness (nm) | Adhesion Force (nN) | Elastic Modulus (MPa) | Heterogeneity Index | Key Interpretation |
|---|---|---|---|---|---|
| Control Cells (Untreated) | Data needed | Data needed | Data needed | Data needed | Representative of a native, heterogeneous population. |
| EDTA-Treated Cells (LPS Removed) | Smoother, featureless [21] | Diminished [21] | Diminished [21] | Markedly Reduced [21] | LPS is a key determinant of surface architecture, mechanics, and phenotypic diversity. |
Interpretation: As shown in the conceptual data above, partial removal of LPS via EDTA treatment homogenizes the outer membrane, leading to a significant reduction in cell-to-cell variability of biophysical properties. This demonstrates the critical role of LPS in generating phenotypic heterogeneity, which has implications for bacterial adhesion and adaptation [21]. Mechanical trapping enables such single-cell analyses that would be masked in population-averaged measurements.
Troubleshooting and Best Practices
- Pore Size Selection: The pore size of the membrane is critical. It must be small enough to securely trap cells but not so small as to cause deformation or prevent entry. Empirical testing is required for different cell types [22].
- Viability Considerations: While mechanical trapping is physical, ensure that the buffer and environmental conditions (temperature, osmolarity) within the AFM liquid cell maintain cell viability throughout the experiment.
- Data Robustness: To account for inherent biological variability and draw meaningful conclusions about population heterogeneity, analyze a statistically relevant number of cells (e.g., n > 20) [21].
Electrostatic Adsorption onto Chemically Modified Surfaces (e.g., Poly-L-Lysine, APTES)
The precise immobilization of bacterial cells on substrates is a critical prerequisite for successful atomic force microscopy (AFM) investigations in liquid environments. AFM, a powerful scanning probe technique, is ideally suited for investigating the surface properties of bacteria at nanoscale resolution while maintaining physiological conditions. A significant obstacle, however, is preventing cell displacement from lateral forces exerted by the AFM probe, necessitating firm adhesion to the substrate. Electrostatic adsorption onto chemically modified surfaces presents a robust solution, maximizing the cell surface area accessible to the AFM probe and enabling high-resolution topographical and mechanical studies. This protocol details methodologies for preparing poly-L-lysine (PLL) and amine-functionalized surfaces (e.g., using APTES) for the effective electrostatic immobilization of bacterial cells, framed within the broader context of developing reliable AFM protocols for microbiological research.
Surface Chemistry and Immobilization Principles
The foundation of this immobilization strategy lies in manipulating the electrostatic interactions between the bacterial cell wall and the functionalized substrate. For Gram-negative bacteria like Escherichia coli, the outer membrane serves as the critical binding interface. The success of electrostatic immobilization hinges on creating a strong, attractive force between this surface and the substrate.
- Poly-L-Lysine (PLL) Coating: PLL, a cationic polymer, adsorbs to negatively charged surfaces such as glass or mica, presenting a uniform layer of primary amine groups. These protonated amines create a persistent positive charge, facilitating strong electrostatic attraction to the generally negatively charged components of the bacterial cell wall [9].
- Amine-Functionalized Surfaces (e.g., APTES): (3-Aminopropyl)triethoxysilane (APTES) reacts with hydroxylated surfaces (e.g., glass, silicon wafer), forming a self-assembled monolayer that terminates in primary amine groups. This provides a covalently attached, stable surface for electrostatic cell capture [24].
- Buffer Considerations: The ionic strength and pH of the immobilization and imaging buffer are crucial. High ionic strength can shield the electrostatic charges, reducing the binding efficacy. For PLL, the use of a diluted phosphate-buffered saline (e.g., 0.01× PBS) has been shown to enhance stable attachment without compromising cell viability, allowing for subsequent exchange to nutrient media for live-cell imaging [9].
Table 1: Comparison of Chemical Immobilization Strategies for Bacterial AFM
| Method | Chemical Basis | Key Advantages | Potential Limitations | Optimal Use Case |
|---|---|---|---|---|
| Poly-L-Lysine (PLL) | Electrostatic adsorption of cationic polymer | Readily available, inexpensive, easy to prepare, maximizes accessible cell surface [9] | May have antimicrobial properties; requires optimization of buffer conditions [9] | General-purpose immobilization for high-resolution surface imaging |
| APTES | Covalent silane monolayer with terminal amine groups | Stable, covalently attached layer; well-defined surface chemistry | Requires rigorous surface cleaning and controlled reaction conditions [24] | Experiments requiring extreme surface stability or specific chemical linkage |
| Gelatin Coating | Physical entrapment and electrostatic interactions | Non-cytotoxic, naturally derived, preserves cell viability [9] | Can create unpredictable obstructions of the cell surface [9] | Long-term live-cell imaging where physiology is paramount |
Detailed Experimental Protocols
Protocol A: Substrate Coating with Poly-L-Lysine
This protocol describes the coating of glass substrates with PLL to create a positively charged surface for bacterial adsorption.
Materials & Reagents
- Clean Glass Substrates: Glass slides, cover slips, or glass-bottom Petri dishes.
- Poly-L-Lysine Solution: 0.1% (w/v) aqueous solution.
- Purified Water: Deionized or distilled water.
- Plasma Cleaner (optional, for enhanced coating uniformity).
Procedure
- Substrate Cleaning: Thoroughly clean glass substrates. Optionally, use an oxygen plasma cleaner for 1–2 minutes to remove organic contaminants and enhance hydrophilicity [25].
- Coating Application: Apply a sufficient volume of the 0.1% PLL solution to completely cover the substrate surface.
- Incubation: Allow the substrate to incubate at room temperature for a minimum of 30 minutes.
- Rinsing: Carefully rinse the substrate three times with purified water to remove any non-adsorbed PLL polymer.
- Drying: Let the coated substrate air-dry completely in a clean environment. The PLL-coated substrates can be stored dry at 4°C for several weeks.
Protocol B: Bacterial Immobilization via Electrostatic Adsorption
This protocol outlines the procedure for immobilizing bacterial cells onto PLL-coated substrates.
Materials & Reagents
- Bacterial Culture: Late exponential or stationary phase culture, typically E. coli.
- PLL-coated Substrate: From Protocol A.
- Immobilization Buffer: 0.01× PBS [9].
- Growth or Imaging Media: e.g., LB broth or Minimal Media (MM).
Procedure
- Cell Harvesting: Harvest bacterial cells by centrifuging a liquid culture (e.g., 5 mL) at a moderate speed (e.g., 3000–5000 × g for 5 minutes).
- Washing: Gently resuspend the cell pellet in 0.01× PBS to remove growth media components. Repeat centrifugation and resuspension in 0.01× PBS.
- Immobilization: Pipette a small volume (e.g., 20–50 µL) of the washed cell suspension onto the center of the PLL-coated substrate. Allow the cells to settle and adsorb for 15–30 minutes at room temperature.
- Rinsing: Gently rinse the substrate with 0.01× PBS to remove any non-adhered cells.
- Media Exchange (for live-cell imaging): Carefully add the appropriate growth or imaging media (e.g., LB broth) to the sample chamber. If using a hermetically sealed chamber for pathogenic organisms, follow specific biosafety procedures for assembly [26].
- Viability Check: Assess cell membrane integrity using viability stains like propidium iodide if required [9].
The following diagram illustrates the core workflow and underlying electrostatic mechanism for immobilizing bacterial cells on a PLL-coated surface.
Key Results and Data Interpretation
Successful immobilization is characterized by cells that are firmly attached to the substrate, withstand lateral forces from the AFM probe, and remain viable for dynamic studies. The table below summarizes critical parameters that require optimization and their typical values or outcomes.
Table 2: Critical Experimental Parameters and Expected Outcomes for Bacterial Immobilization
| Parameter | Recommended Conditions / Expected Outcome | Impact on Experiment |
|---|---|---|
| PLL Concentration | 0.01% - 0.1% (w/v) | Lower may yield insufficient adhesion; higher may be cytotoxic or create a soft polymer layer. |
| Adsorption Time | 15 - 60 minutes | Shorter times may lead to low density; longer times may not increase yield significantly. |
| Immobilization Buffer | Low ionic strength (e.g., 0.01× PBS) [9] | Enhances electrostatic interaction strength compared to physiological buffers. |
| Cell Viability | >90% membrane integrity post-immobilization [9] | Essential for live-cell imaging and studying dynamic physiological processes. |
| Imaging Stability | Cells remain fixed during contact mode scanning in liquid | Unstable immobilization results in cell displacement and failed imaging. |
| AFM Image Quality | Clear, high-resolution topography with recognizable cell morphology | The ultimate validation of a successful sample preparation protocol. |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Electrostatic Immobilization and AFM
| Reagent / Material | Function / Role in Protocol | Specific Example / Note |
|---|---|---|
| Poly-L-Lysine (PLL) | Creates a cationic coating on substrates for electrostatic cell adhesion [9]. | Use 0.1% (w/v) solution; molecular weight 150,000-300,000 is common [25]. |
| (3-Mercaptopropyl)trimethoxysilane | Silane used for functionalizing surfaces (e.g., AFM tips) with thiol groups for further chemistry [24]. | Critical for covalent attachment of biomolecules in force spectroscopy. |
| Sulfo-LC-SPDP | Heterobifunctional crosslinker for covalently linking amines to thiols [24]. | Used to attach streptavidin to functionalized AFM cantilevers. |
| Streptavidin | Protein that binds biotin with high affinity, used as a bridge in functionalization [24]. | Allows attachment of any biotinylated ligand (antibodies, peptides) to surfaces. |
| Indium-Tin-Oxide (ITO) Coated Glass | Conductive, hydrophobic substrate that promotes bacterial adhesion without chemical immobilization [5]. | Enables AFM imaging of living bacteria in liquid without potentially stressful immobilization protocols. |
| Propidium Iodide | Membrane-impermeant fluorescent dye for assessing cell viability post-immobilization [9]. | Cells with compromised membranes stain positive, indicating loss of viability. |
Advanced Applications and Integration
The basic principle of electrostatic adsorption enables a wide range of advanced AFM applications. Beyond simple topography, a stably immobilized sample is the foundation for nanomechanical mapping, where the Young's modulus of the cell surface is calculated from force-indentation curves, providing insights into cell wall stiffness and its alterations [5]. Furthermore, this immobilization strategy is crucial for single-cell force spectroscopy, which quantifies adhesion forces between a bacterial cell and a surface, and for molecular recognition mapping, which locates specific receptors on the cell surface using functionalized AFM tips [24].
For research involving pathogenic microorganisms, the immobilization protocol must be integrated with biosafety-compliant AFM chambers. These hermetically sealed chambers confine the biohazardous material while allowing for high-resolution, time-lapse nano-characterization, ensuring user and environmental safety [26]. The combination of robust electrostatic immobilization and advanced AFM techniques thus provides a powerful platform for uncovering the nanoscale world of bacteria.
Atomic force microscopy (AFM) has emerged as a powerful tool in cellular biology, enabling the investigation of microbial surfaces at nanometer resolution. A critical prerequisite for successful AFM analysis is the effective immobilization of cells without altering their native structural or mechanical properties. Chemical fixation, particularly using glutaraldehyde, is a widely employed method to achieve this stability. However, the very process of cross-linking that provides stabilization can also introduce nanoscale artefacts on the cell surface. This application note details the use of glutaraldehyde fixation for AFM studies on bacterial cells, providing a balanced examination of its benefits for cellular stabilization against its potential pitfalls in surface preservation. We present optimized protocols, quantitative data on fixation effects, and strategic recommendations to guide researchers in obtaining reliable, high-quality AFM data.
Table: Key Effects of Glutaraldehyde Fixation on Cells for AFM Analysis
| Parameter | Effect of Glutaraldehyde Fixation | Implication for AFM Studies |
|---|---|---|
| Cellular Stiffness | Increases Young's modulus significantly (from ~27 kPa in living cells to ~535 kPa) [27] | Enhances mechanical stability, reduces tip-induced deformation |
| Nanoscale Topography | Creates larger protrusions (median area increases from ~102.5 nm² to ~187.8 nm²) [27] | May introduce clustering artefacts on membrane surfaces |
| Fixation Speed | Fast fixation of cytoplasmic proteins (within 4 minutes) [28] | Rapid preservation of intracellular components |
| Protein Mobility | Halts cytoplasmic protein diffusion effectively [28] | Preserves spatial organization of proteins when fixation is rapid |
| Structural Preservation | Excellent preservation of surface ultrastructures (e.g., flagella, pili) [29] | Superior to alcohol-based fixatives for surface feature integrity |
Glutaraldehyde Mechanism and Key Considerations
Glutaraldehyde functions as a bifunctional crosslinking agent, with aldehyde groups at either end of the molecule that react primarily with the amino groups of lysine and other nucleophiles in proteins [30] [31]. This creates covalent bonds between neighboring proteins, resulting in a extensively cross-linked cellular structure that stabilizes the cell against degradation and mechanical deformation [31]. This extensive cross-linking is particularly advantageous for AFM as it increases cellular rigidity, thereby reducing indentation artefacts during scanning and yielding more reliable topographical and mechanical measurements [27].
However, several critical factors must be managed to avoid artefacts:
- Osmolality: Glutaraldehyde solutions can dramatically alter the osmolality of the fixation medium in a concentration-dependent manner, potentially causing cell shrinkage or swelling and resulting in distorted cell shapes [30]. The osmolality of the vehicle (buffer) is therefore crucial and should be adjusted to resemble that of the bacterial culture medium.
- Batch Variation: Commercial glutaraldehyde batches can differ significantly in their properties, particularly in the ratio of monomers to polymers, which affects fixation efficiency [30]. UV-absorption spectroscopy can determine the monomer-polymer ratio, with extinction peaks at 280 nm for monomers and around 235 nm for polymers [30].
- Concentration Effects: While increased glutaraldehyde concentration generally enhances cross-linking and stiffness, the fixation event itself is described as a rather "digital" (all-or-none) process, whereas the increase in rigidity is more analog and concentration-dependent [30].
Quantitative Effects of Fixation on Cellular Properties
Impact on Nanoscale Surface Topography
Recent high-resolution AFM studies utilizing microporous silicon nitride membranes have revealed that chemical fixatives, including glutaraldehyde, can induce nanoscale clustering of membrane proteins. These studies quantified the size distribution of protrusions on cell surfaces before and after fixation, demonstrating a significant increase in median protrusion area from 102.5 nm² in living cells to 187.8 nm² after glutaraldehyde treatment [27]. This aggregation of membrane proteins creates pseudo-clusters that were not present in the living state, highlighting a critical artefact that researchers must consider when interpreting AFM images of fixed cells.
Changes in Cellular Mechanical Properties
The mechanical stiffening induced by glutaraldehyde fixation has been quantitatively measured through AFM force spectroscopy. Studies report that Young's modulus of the cell surface increases dramatically—from approximately 27 kPa in living cells to 535 kPa after glutaraldehyde treatment, representing a nearly 20-fold increase in stiffness [27]. This substantial alteration in mechanical properties means that AFM measurements on fixed cells do not reflect the native mechanical state of living cells, limiting the applicability of such data for biomechanical studies focused on physiological conditions.
Table: Comparison of Common Chemical Fixatives for Bacterial AFM
| Fixative | Concentration & Duration | Preservation of Surface Ultrastructures | Induced Protrusion Size (Median Area) | Young's Modulus After Fixation | Recommended Application |
|---|---|---|---|---|---|
| Glutaraldehyde | 1-2.5%, 1-2 hours [29] [28] | Excellent (preserves flagella, pili) [29] | 187.8 nm² [27] | ~535 kPa [27] | High-resolution ultrastructural studies |
| Paraformaldehyde | 4%, 30 minutes [27] | Moderate | 162.1 nm² [27] | ~449 kPa [27] | General morphology and immunolabeling |
| Methanol/Acetone | 100%, -20°C, 10 min [27] | Poor (detaches surface filaments) [29] | 213.1 nm² [27] | ~165 kPa [27] | When alcohol fixation is specifically required |
| Formalin | 10%, 10 minutes [29] | Moderate | Not quantified | Not quantified | Routine histology when glutaraldehyde unavailable |
Experimental Protocols
Optimized Glutaraldehyde Fixation Protocol for Bacterial AFM
The following protocol has been optimized for immobilizing bacterial cells for AFM analysis, balancing structural preservation with artefact minimization:
Sample Preparation:
Fixation Solution Preparation:
- Prepare fixation solution containing 2.5% glutaraldehyde in PBS [29].
- Use electron microscopy-grade glutaraldehyde from sealed ampoules to ensure quality and minimize polymerization [31].
- Consider adding formaldehyde to reduce glutaraldehyde-induced autofluorescence without significantly compromising fixation speed [28].
Fixation Procedure:
- Resuspend bacterial pellet in fixation solution using a large solution-to-cell volume ratio (approximately 65:1) to ensure sufficient volume for fixation as individual cells and prevent aggregation [30].
- Fix for 1-2 hours at room temperature with gentle agitation on a tube roller [30] [29].
- For thicker samples or biofilms, ensure tissue dimensions do not exceed 1mm in thickness to allow adequate fixative penetration [31].
Post-Fixation Processing:
Diagram Title: Bacterial Fixation Workflow for AFM
APTES-Glutaraldehyde Surface Functionalization for Cell Immobilization
For studies requiring particularly stable immobilization, covalently binding cells to surfaces through APTES-glutaraldehyde functionalization provides exceptional stability:
Surface Preparation:
Glutaraldehyde Activation:
- Incubate APTES-coated surfaces with 2% glutaraldehyde in PBS for 30 minutes [3].
- Wash thoroughly with distilled water to remove unbound glutaraldehyde.
Cell Attachment:
- Apply bacterial suspension in a non-ionic solution such as 150 mM sorbitol (rather than growth media) to prevent reaction of glutaraldehyde with primary amines in the medium [3].
- Allow attachment for 30-60 minutes.
- Gently rinse with appropriate buffer to remove non-adherent cells before AFM analysis.
The Scientist's Toolkit: Essential Research Reagents
Table: Essential Reagents for Glutaraldehyde Fixation in AFM Studies
| Reagent | Function | Application Notes |
|---|---|---|
| Electron Microscopy-Grade\nGlutaraldehyde | Primary cross-linking fixative | Use from sealed ampoules; concentration typically 1-3% in buffer; check monomer/polymer ratio [30] [31] |
| Phosphate Buffered Saline (PBS) | Buffer vehicle for fixative | Maintains physiological pH (7.2-7.4); adjust osmolality to match bacterial culture conditions [30] [29] |
| APTES (3-Aminopropyl-\ntriethoxysilane) | Surface functionalization | Creates amine groups on glass surfaces for covalent cell attachment [3] |
| Sorbitol Solution | Non-ionic attaching medium | Used for cell immobilization without competing primary amines (150 mM) [3] |
| Sodium Borohydride (NaBH₄) | Autofluorescence reduction | Quenches glutaraldehyde-induced fluorescence (5 mg/mL for 2 h) [28] |
| ITO-Coated Glass Slides | AFM substrate | Provides superior cell adhesion without chemical immobilization for living cell AFM [5] |
Troubleshooting and Best Practices
Mitigating Common Artefacts
Successful application of glutaraldehyde fixation for AFM requires careful attention to potential artefacts:
- Minimizing Nanoscale Clustering: While some protein aggregation may be unavoidable, using the lowest effective concentration of glutaraldehyde (1-2% rather than higher concentrations) can reduce the extent of clustering artefacts [27]. Whenever possible, validate key findings on living cells to confirm that observed structures are not fixation artefacts.
- Controlling Osmotic Effects: Always adjust the osmolality of the fixation buffer to match that of the bacterial growth medium. This prevents cell shrinkage or swelling that can distort cellular morphology [30].
- Managing Autofluorescence: For correlative fluorescence-AFM studies, glutaraldehyde-induced autofluorescence can be reduced through treatment with 100 mM NH₄Cl for 40 minutes followed by 5 mg/mL NaBH₄ for 2 hours, or by using a combination of glutaraldehyde and formaldehyde [28].
Validation Strategies
- Live-Cell Correlations: Whenever feasible, perform comparative AFM imaging on living cells to establish baseline topography and mechanical properties [27] [5].
- Multiple Fixation Assessment: Compare results obtained with glutaraldehyde fixation against other fixation methods (e.g., paraformaldehyde) to identify method-dependent artefacts [29] [27].
- Independent Method Verification: Confirm critical findings using alternative imaging techniques such as cryo-electron microscopy when possible.
Diagram Title: Fixation Optimization Balance
Glutaraldehyde fixation remains a valuable method for preparing bacterial cells for AFM analysis, particularly when ultrastructural preservation and cellular stability are prioritized. The cross-linking action of glutaraldehyde provides exceptional stabilization of cellular components and preserves delicate surface structures such as flagella and pili better than alcohol-based fixatives. However, researchers must remain cognizant of its significant limitations, including the induction of nanoscale protein clustering and alteration of native mechanical properties. By implementing the optimized protocols outlined herein—including careful concentration control, osmolality adjustment, and appropriate validation strategies—researchers can effectively balance the competing demands of structural stability and authentic surface preservation. This approach enables the acquisition of reliable, high-resolution AFM data while maintaining awareness of potential fixation-induced artefacts that might influence biological interpretations.
Fluid force microscopy (FluidFM) represents a transformative technological advancement that integrates atomic force microscopy (AFM) with microfluidic channels embedded within the cantilever [32] [33]. This synergy creates a powerful tool for single-cell manipulation and analysis, with reversible immobilization standing as one of its most significant capabilities for biomedical and microbiological research. Unlike traditional AFM methods that require permanent chemical or physical fixation of cells—often affecting their viability and physiological state—FluidFM enables the gentle, reversible trapping of individual cells onto the cantilever using precisely controlled suction [34]. This technique allows researchers to use a single living cell as a probe for multiple force spectroscopy measurements, dramatically increasing throughput and reproducibility while maintaining the cell in a near-native state [34] [33].
The core principle of reversible immobilization addresses a fundamental challenge in single-cell force spectroscopy (SCFS): the need to firmly attach a cell to the AFM cantilever without altering its natural properties. Traditional SCFS assays involve gluing cells to the cantilever, resulting in complex handling, potential chemical damage, and low experimental throughput [34]. In contrast, FluidFM uses a hollow cantilever with a nanoscale aperture connected to a pressure controller. By applying a brief negative pressure pulse, a single cell is aspirated and reversibly immobilized onto the aperture; a subsequent positive pressure pulse releases the cell unharmed after measurements are complete [32] [34]. This "pick-measure-release" cycle enables researchers to probe up to 200 individual cells per day—a more than tenfold increase in throughput compared to conventional methods [34].
Key Principles and Mechanism of Operation
Core Components of the FluidFM System
The FluidFM system's capability for reversible immobilization relies on several integrated components working in concert. The heart of the system is the micro-channeled cantilever, which features an internal fluidic channel terminating in a precisely defined aperture at the tip. These apertures range in size from 300 nm to 8 μm, allowing for the immobilization of various biological specimens from bacterial cells to mammalian cells [35]. The cantilever is connected to a pressure control system capable of generating both negative and positive pressure with millibar precision, enabling the gentle aspiration and release of delicate biological samples [32] [35]. The entire system operates under optical control,
allowing real-time monitoring of the immobilization process, while the AFM component provides exquisite force sensitivity from piconewtons to micronewtons [34].
The Reversible Immobilization Process
The reversible immobilization process follows a precise sequence that maintains cellular viability and function. First, a cell is selected from the substrate or attracted from suspension via liquid influx through the FluidFM probe's aperture [34]. Next, negative pressure is applied to immobilize the cell reversibly against the aperture. The quality of the seal can be monitored in real-time using electrochemical methods that measure changes in the internal electrical resistance of the micro-channel; a successful immobilization event typically doubles the electrical resistance at low ionic strengths [35]. Once immobilized, the cell can be used as a probe for force spectroscopy measurements to quantify adhesion forces, mechanical properties, or interaction kinetics. Finally, the cell is released via a pressure pulse, leaving it viable for further culture or analysis while the same cantilever can be used to immobilize another cell [34].
Table 1: Key Steps in FluidFM Reversible Immobilization Protocol
| Step | Process | Parameters | Purpose |
|---|---|---|---|
| 1. Cell Selection | Approach target cell with FluidFM probe | Aperture size matched to cell diameter (0.5-100 μm) | Select specific cell for analysis |
| 2. Immobilization | Apply negative pressure | -50 to -300 mbar pressure range | Reversibly secure cell to cantilever |
| 3. Measurement | Perform force spectroscopy | Force range: pN to μN; Multiple approach-retract cycles | Quantify adhesion, stiffness, or molecular interactions |
| 4. Release | Apply positive pressure pulse | +100 to +1000 mbar pressure pulse | Gently release cell maintaining viability |
| 5. Probe Reuse | Continue to next cell | No probe change required | High-throughput data collection |
Experimental Protocols
Protocol for Bacterial Adhesion Force Measurements
Principle: This protocol enables the quantification of adhesion forces between bacterial cells and surfaces or other cells using FluidFM's reversible immobilization. The approach is particularly valuable for studying biofilm formation, bacterial pathogenesis, and microbial ecology [34].
Materials and Reagents:
- FluidFM system with micro-channeled cantilevers (2-8 μm aperture recommended)
- Bacterial culture in exponential growth phase
- Appropriate growth medium and measurement buffer
- Indium-tin-oxide (ITO) coated glass substrates or other relevant surfaces
- Pressure controller system
Procedure:
- Cantilever Preparation: Select a cantilever with an aperture size slightly smaller than the bacterial cells to ensure secure immobilization. Flush the microchannel with appropriate buffer to remove air bubbles and ensure proper fluidic function [35].
- Substrate Preparation: Coat ITO glass substrates with a thin layer of porcine gelatin to enhance bacterial immobilization during imaging, leveraging electrostatic interactions between negatively charged bacteria and the positively charged gelatin [1].
- Bacterial Immobilization: Apply a negative pressure of -100 to -300 mbar to aspirate a single bacterial cell onto the cantilever aperture. Verify successful immobilization by monitoring the increase in electrical resistance through the microchannel [35].
- Adhesion Measurement: Approach the bacterial cell, immobilized on the cantilever, toward the target surface at a controlled speed of 0.5-1.0 μm/s. Upon contact, apply a defined force (typically 0.5-1 nN) for a controlled contact time (0.1-10 s). Retract the cantilever at the same speed while recording the force-distance curve.
- Data Collection: Collect a minimum of 50-100 force curves from different locations on the surface to ensure statistical significance.
- Cell Release: Apply a positive pressure pulse (+500 to +1000 mbar) to release the bacterial cell. The same cantilever can then be used to immobilize another cell for subsequent measurements [34].
Troubleshooting Tips:
- If cells cannot be properly immobilized, verify the aperture is not clogged by measuring baseline electrical resistance.
- If adhesion forces appear inconsistent, ensure consistent contact time and force across measurements.
- For weak adhesion signals, increase the sensitivity of the photodetector and reduce thermal noise by allowing the system to thermally equilibrate.
Protocol for Single-Cell Force Spectroscopy with Mammalian Cells
Principle: This protocol measures adhesion forces between individual mammalian cells and substrates or other cells, with applications in cancer research, immunology, and tissue engineering [34] [36].
Materials and Reagents:
- FluidFM OMNIUM platform or equivalent FluidFM system
- Mammalian cells (adherent or suspension cultures)
- Cell culture medium and appropriate measurement buffer
- Functionalized substrates (e.g., extracellular matrix proteins, engineered materials)
- FluidFM probes with 8-12 μm apertures for mammalian cells
Procedure:
- System Calibration: Calibrate the cantilever sensitivity and spring constant using thermal tuning or contact-based methods before measurements.
- Cell Immobilization: Select a target cell and carefully approach with the FluidFM probe. Apply gentle negative pressure (-50 to -150 mbar) to immobilize the cell. Verify immobilization by observing a slight deformation of the cell membrane at the aperture site under optical microscopy [34].
- Single-Cell Probing: Position the cell-probe assembly above the target surface or cell. Program the AFM to perform multiple approach-retract cycles with varying contact times (1-60 s) and forces (0.1-2 nN) to characterize the time and force dependence of adhesion.
- Adhesion Force Quantification: Analyze force-distance curves to determine maximum detachment force, work of adhesion, and rupture events. Use the Johnson-Kendall-Roberts (JKR) or Hertz model depending on the cell type and contact mechanics.
- Cell Viability Verification: After release, collect the cell and assess viability using trypan blue exclusion or calcein-AM staining to confirm the non-destructive nature of the procedure.
Applications: This protocol has been successfully applied to study tumor progression and metastasis through cell-cell adhesion measurements [34], investigate cardiomyocyte mechanics [33], and optimize stent surface design by measuring endothelial cell adhesion forces [34].
Research Reagent Solutions and Materials
Table 2: Essential Research Reagents and Materials for FluidFM Reversible Immobilization
| Item | Specifications | Function | Application Examples |
|---|---|---|---|
| FluidFM Probes | Hollow cantilevers with 300 nm - 12 μm apertures; spring constants: 0.2-2 N/m | Reversible cell immobilization and force sensing | Single-cell force spectroscopy, nanomechanical measurements |
| Pressure Controller | Precision: ±0.1 mbar; Range: -1000 to +1000 mbar | Controlled aspiration and release of cells | Gentle cell handling, maintaining viability |
| ITO-coated Substrates | Coated with indium-tin-oxide; smooth surface (RMS roughness <1 nm) | Optimal substrate for cell adhesion and AFM imaging | High-quality imaging in liquid environments [5] |
| Gelatin Coating | Porcine gelatin (0.1-1% w/v in water) | Electrostatic immobilization of bacterial cells | Microbial cell imaging and force measurements [1] |
| Measurement Buffers | Ionic strength: 0.1 mM - 150 mM; physiological pH | Control electrostatic interactions and maintain cell viability | Adhesion force measurements at various ionic strengths [35] |
Data Interpretation and Analysis
Analysis of Force-Distance Curves
Force-distance curves obtained from FluidFM experiments contain rich information about cell-surface interactions. The adhesion force is determined from the maximum negative force during retraction, while the work of adhesion is calculated from the area under the retraction curve [34]. Specific binding events often appear as distinct rupture peaks in the retraction curve, with the number and magnitude of these peaks providing information about the density and strength of molecular bonds [33].
For bacterial adhesion studies, researchers have employed FluidFM to quantify the hydrophobic adhesion properties of different bacterial strains, revealing cell-cell heterogeneity and correlations with in planta retention [34]. In cancer research, force spectroscopy has enabled the measurement of intercellular adhesion forces between cancer cells and fibroblasts at different contact times, providing insights into tumor progression and metastasis [34].
Statistical Analysis and Data Reproducibility
The high-throughput capability of FluidFM reversible immobilization enables robust statistical analysis of single-cell properties. A typical experiment should include measurements from at least 30-50 individual cells per condition, with multiple force curves collected per cell. Data should be presented as mean ± standard error of the mean, and statistical significance between conditions determined using appropriate tests (e.g., Student's t-test, ANOVA). The reproducibility of FluidFM measurements is enhanced by the consistent positioning of objects on the cantilever, dictated by the aperture location [34].
Table 3: Typical Force Ranges Measurable with FluidFM in Different Applications
| Application | Force Range | Measured Parameters | Biological Significance |
|---|---|---|---|
| Bacterial Adhesion | 10 pN - 5 nN | Adhesion force, work of adhesion | Biofilm formation, antimicrobial resistance [34] |
| Cell-Cell Interactions | 50 pN - 20 nN | Detachment force, rupture length | Tumor metastasis, immune recognition [34] |
| Nanomechanical Properties | 0.1 - 10 nN | Young's modulus, deformation | Cell differentiation, disease states [37] [33] |
| Single-Molecule Interactions | 50 - 500 pN | Unbinding force, bond lifetime | Receptor-ligand kinetics, drug targeting [33] |
Applications in Biomedical Research
Microbial Cell Studies
FluidFM with reversible immobilization has revolutionized the study of microbial cells by enabling the quantification of adhesion forces at the single-cell level. Researchers have applied this technology to investigate bacterial adhesion forces to various surfaces, including stainless steel reactor materials, providing crucial insights for industrial biofilm prevention [34]. The technology has also been used to profile the hydrophobic adhesion properties of diverse bacterial strains from leaf isolates, revealing significant heterogeneity that correlates with environmental retention [34]. Furthermore, FluidFM has enabled the study of amyloid bonds between microbial cells, which play crucial roles in community organization and resilience [34].
Cancer Research and Drug Development
In cancer research, FluidFM's reversible immobilization has provided unprecedented insights into cellular mechanisms underlying tumor progression and metastasis. Researchers have employed the technology to measure cell-cell adhesion forces between cancer cells and stromal cells, revealing how contact time influences adhesion strength—a critical factor in metastatic dissemination [34]. The technology has also facilitated studies of drug resistance mechanisms, such as research that identified a potential approach to overcome resistance to the drug Midostaurin in acute myeloid leukemia [34]. Additionally, the ability to measure adhesion forces between cells and engineered materials has supported the optimization of medical implant surfaces, including stent design optimization through precise quantification of endothelial cell adhesion [34].
Biomaterials and Tissue Engineering
The reversible immobilization capability of FluidFM has proven invaluable in biomaterials research and tissue engineering applications. Scientists have utilized the technology to characterize novel materials, such as melt-electrowritten hydrogels for tissue engineering, by measuring their interaction with cells using the fast colloidal probe technique [34]. The capacity to measure single-cell adhesion forces to nano-engineered implant surfaces enables the rational design of bioactive implants that promote tissue integration while preventing bacterial colonization [36]. Furthermore, the technology allows for the assessment of mature intercellular adhesion forces in physiological settings, providing insights relevant to developmental biology, tissue regeneration, and fibrotic diseases [34].
Visualizing the FluidFM Workflow
Diagram 1: FluidFM Reversible Immobilization Workflow. This diagram illustrates the cyclic process of reversible cell immobilization, enabling high-throughput single-cell force spectroscopy.
Advantages and Future Perspectives
FluidFM's reversible immobilization technology offers several significant advantages over conventional single-cell force spectroscopy methods. The most prominent benefit is the dramatically increased throughput—up to 200 cells per day compared to approximately 10-20 cells with traditional methods [34]. This high-throughput capability enables researchers to obtain statistically robust data on cellular heterogeneity, which is often masked in population-average measurements. Additionally, the gentle, non-destructive nature of the immobilization process preserves cell viability and function, allowing for subsequent analysis of the same cells if needed [34]. The technology also provides exceptional versatility, accommodating a wide range of biological specimens including mammalian cells, microbes, colloids, bubbles, and droplets ranging from 0.5 to 100 μm in size [34].
Future developments in FluidFM reversible immobilization are likely to focus on increasing automation and integration with other analytical techniques. The combination of FluidFM with advanced microscopy methods, such as super-resolution fluorescence imaging, could provide simultaneous topological, mechanical, and molecular information from single cells. Further miniaturization of apertures may enable the manipulation of subcellular organelles and nanoscale particles. As the technology becomes more accessible and user-friendly, its application in clinical settings may expand, potentially contributing to personalized medicine approaches through the mechanical characterization of patient-derived cells for diagnostic and therapeutic monitoring purposes [37].
Within the broader scope of developing robust Atomic Force Microscopy (AFM) methodologies for bacterial surface research, sample immobilization represents a critical foundational step. Successful AFM imaging and force spectroscopy of live bacterial cells in physiological liquids depend on firmly anchoring the cells to a substrate to prevent displacement by lateral forces exerted by the scanning AFM tip [14] [9]. This protocol details the use of gelatin-coated mica surfaces, a method that exploits electrostatic interactions to immobilize both Gram-negative and Gram-positive bacteria effectively and with minimal impact on cell viability, enabling high-resolution imaging and accurate mechanical property measurements [14] [38].
Materials and Reagents
Research Reagent Solutions
The following table lists essential materials and their specific functions in the immobilization protocol.
Table 1: Key Research Reagents and Materials
| Item Name | Function/Application | Critical Notes |
|---|---|---|
| Mica Sheets | Provides an atomically flat, negatively charged substrate for coating. | Must be freshly cleaved before coating [14]. |
| Porcine Gelatin | Forms a positively charged coating to immobilize negatively charged bacterial cells. | Sigma G-6144 (low Bloom) or G-2625 (medium Bloom) are recommended. Bovine gelatin is ineffective [14] [9]. |
| Silicon Nitride Cantilevers | AFM probes for imaging in liquid. | Use cantilevers with low spring constants (e.g., 0.01–0.1 nN/nm) to minimize imaging forces [14] [38]. |
| Poly-L-Lysine (PLL) | Alternative positively charged polymer for electrostatic immobilization. | Note: May have antimicrobial properties; viability must be confirmed [9]. |
| 0.01× PBS-S Buffer | A dilute phosphate-buffered saline used for washing and immobilization. | Reduces ionic strength to minimize competition for binding sites on the gelatin surface [9]. |
| Isopore Membrane Filters | (For mechanical trapping) Immobilizes cells by physical entrapment in pores. | Pore size should be slightly smaller than the bacterial dimensions (e.g., 0.8 μm) [39]. |
Gelatin-Coated Mica Immobilization Protocol
This section outlines the definitive, step-by-step protocol for immobilizing bacteria using gelatin-coated mica, a method valued for its general applicability and minimal invasiveness to the sample [14] [38].
The following diagram illustrates the complete experimental workflow from sample preparation to AFM imaging.
Step-by-Step Procedure
Part 1: Substrate Preparation
- Mica Preparation: Cut a mica sheet to the required size (approx. 22 × 30 mm to fit a standard AFM stage). Using adhesive tape, cleave the top layers from both sides of the mica until a smooth, unbroken surface is achieved [14] [38].
- Gelatin Solution Preparation:
- Add 100 mL of distilled water to a laboratory bottle and heat until boiling (using a microwave or hot plate) [14].
- Weigh out 0.5 g of porcine gelatin (e.g., Sigma G-6144) and add it to the hot water. Gently swirl the bottle until the gelatin is completely dissolved [14] [38].
- Cool the solution to 60–70 °C. Pour approximately 15 mL into a small beaker (20 mL) for dipping [14].
- Mica Coating:
- Using fine tweezers, completely submerge a cleaved mica square into the warm gelatin solution and withdraw it quickly [38].
- Immediately place the mica on its edge on a paper towel to dry in ambient air overnight. The coated mica is stable for at least two weeks when stored in a covered Petri dish at room temperature [14] [38].
Part 2: Bacterial Sample Preparation
- Culture and Harvest:
- Grow a bacterial culture to mid-exponential or stationary phase (OD₆₀₀ of 0.5–1.0 is optimal) [14] [38].
- Transfer 1 mL of culture to a microcentrifuge tube and pellet the cells by centrifugation (800–4,500 rcf for ~5 minutes). Note: Use the minimum force required to pellet your specific strain [14].
- Wash and Resuspend:
- Carefully remove the supernatant and wash the pellet in 1 mL of filtered deionized water or a compatible low-ionic-strength buffer (e.g., 0.01× PBS-S) [14] [9].
- Centrifuge again and promptly resuspend the final pellet in 500 μL of nanopure water or buffer. The suspension should be visibly turbid [14] [38].
Part 3: Immobilization and Imaging
- Mounting Bacteria:
- Apply 10–20 μL of the bacterial suspension to the center of the dry, gelatin-coated mica surface [14].
- Gently spread the droplet over the surface in both X and Y directions using a pipette tip, taking care not to touch or scratch the gelatin surface [38].
- Allow the sample to incubate for 10 minutes at room temperature [14] [38].
- Rinsing:
- After incubation, rinse the mica thoroughly with a gentle stream of water or the imaging buffer (e.g., 0.005 M PBS) to remove loosely attached cells [14].
- Wick away excess liquid by touching the edge of the sample to a paper towel. Crucially, do not allow the sample to dry if it is to be imaged in liquid [14] [38].
- Quality Check: A quick visual check can be performed by drying one slide. A cloudy, opaque spot after rinsing indicates successful immobilization, whereas a clear spot suggests the bacteria were washed away [14].
- AFM Imaging: Mount the hydrated sample into the AFM liquid cell, add appropriate imaging buffer, and begin imaging. Use non-contact modes (Tapping/MAC Mode) or contact mode with low spring constant cantilevers to minimize lateral forces on the cells [14] [8].
Comparative Analysis of Immobilization Methods
While gelatin-coated mica is a widely applicable method, researchers should be aware of alternative techniques. The choice of immobilization strategy can significantly impact the outcome of AFM experiments, including the measured interaction forces [39].
Table 2: Comparison of Bacterial Immobilization Methods for AFM
| Method | Mechanism | Advantages | Disadvantages/Limitations | Best For |
|---|---|---|---|---|
| Gelatin-Coated Mica | Electrostatic adsorption [14] | Minimally invasive, preserves viability, suitable for liquid imaging, disperses cells for individual analysis [14] [9] | Growth media and salts can interfere with binding; requires strain-specific optimization [14] | General live-cell imaging and force spectroscopy in physiological liquids [14] [38] |
| Mechanical Trapping | Physical entrapment in porous membrane [39] | No chemical treatment; preserves native cell surface properties [39] [9] | Can exert non-native forces on cell; may obstruct parts of the cell surface [9] | Studies where chemical fixation is undesirable; robust immobilization for force measurements [39] |
| Poly-L-Lysine (PLL) Coating | Electrostatic adsorption [39] [9] | Strong, irreversible adhesion; simple and fast preparation [39] | Can have antimicrobial effects, potentially compromising cell physiology and viability [9] | Applications where maximum adhesion strength is critical and viability is less concern [39] |
| Glutaraldehyde Fixation | Covalent cross-linking to AFM tip or surface [39] | Extremely firm anchoring, allows for single-cell probe creation [8] [39] | Chemically alters cell surface, kills cells, may change physicochemical properties [39] | Single-cell force spectroscopy (SCFS) where the cell is used as a probe [8] [39] |
Advanced Applications and Downstream Analysis
Successful immobilization is the gateway to a suite of advanced AFM techniques that probe the functional and mechanical properties of bacteria.
- Single-Molecule Force Spectroscopy (SMFS): The AFM tip can be functionalized with specific molecules (e.g., ligands, antibodies) to map and measure interaction forces with single receptors on the immobilized bacterial surface [8] [40]. This involves coating the tip with a linker molecule like PEG, which is then conjugated to the protein of interest, allowing specific unbinding events to be distinguished from non-specific adhesion [40].
- Single-Cell Force Spectroscopy (SCFS): A single bacterial cell can be attached to the cantilever to quantitatively probe its adhesive interactions with various substrates, such as biotic surfaces, abiotic materials, or other cells [8] [41]. This has revealed, for instance, that hydrophobic adhesion forces among leaf microbiota can vary by orders of magnitude, correlating with their retention in plants [41].
- Real-Time Dynamic Studies: Stable immobilization of live cells enables the investigation of dynamic processes such as cell division, the effect of antimicrobial agents, and the production of outer membrane vesicles (OMVs) in real time and under native conditions [9].
Troubleshooting and Best Practices
- Poor Immobilization: If bacteria are washed away during rinsing, confirm you are using a recommended porcine gelatin. Ensure the bacterial suspension medium is free of growth media constituents or high salt concentrations that can compete for binding sites on the gelatin [14]. Using overnight-grown cells can also improve the stability of attachment [9].
- Low Cell Viability: If maintaining cell viability is paramount, avoid poly-L-lysine and confirm that the imaging buffer is physiologically compatible. The addition of osmotic protectants like 0.25 M sucrose to the imaging buffer can help preserve osmotically sensitive cells [14] [9].
- Weak Image Resolution or Cell Displacement: This is often caused by excessive lateral scanning forces. Switch to a non-contact imaging mode (Tapping or MAC Mode) or use cantilevers with a lower spring constant to minimize applied forces [14] [8]. Ensure cells are firmly immobilized by optimizing the incubation time and rinse stringency.
Troubleshooting AFM Immobilization: Overcoming Common Pitfalls
Optimizing Surface Chemistry for Different Bacterial Strains and Cell Envelopes
The application of Atomic Force Microscopy (AFM) in microbiology has revolutionized our ability to study the nanoscale surface structures of living bacterial cells in physiological conditions. A critical prerequisite for successful AFM imaging is the effective immobilization of bacterial cells to a flat surface without altering cell surface properties or viability. The bacterial cell envelope, being the first line of defense and environmental gatekeeper, varies significantly between Gram-positive and Gram-negative strains, necessitating tailored immobilization strategies. This application note provides a comprehensive guide to optimizing surface chemistry for immobilizing diverse bacterial strains, framed within broader AFM protocol development for bacterial cell research.
Bacterial Cell Envelope Architecture: Implications for Immobilization
The bacterial cell envelope is a complex structure essential for viability, maintaining turgor pressure, and mediating environmental interactions. Its composition varies significantly between major bacterial groups, which directly influences how cells should be immobilized for AFM studies [42].
- Gram-Negative Envelopes: Characterized by a thin peptidoglycan layer surrounded by an outer membrane consisting of phospholipids on the inner leaflet and lipopolysaccharides (LPS) on the outer leaflet. This structure presents a significant permeation barrier and serves as the primary binding interface for immobilization [42].
- Gram-Positive Envelopes: Feature a thick, mechanically strong peptidoglycan layer that provides an exoskeleton, sometimes interwoven with teichoic acids. This thick external layer offers different chemical groups for surface attachment [42].
- Mycobacterial and Other Envelopes: Exceptions like Mycobacterium tuberculosis possess a thin peptidoglycan layer surrounded by an arabinogalactan layer to which mycolic acids are covalently bound, creating a waxy, hydrophobic surface that demands specialized immobilization approaches [42].
The following diagram illustrates the logical decision process for selecting an appropriate immobilization strategy based on bacterial envelope type and experimental requirements:
Comparative Evaluation of Immobilization Methods
Quantitative Comparison of Immobilization Techniques
The following table summarizes the performance characteristics of major immobilization methods based on comprehensive studies:
Table 1: Quantitative Comparison of Bacterial Immobilization Methods for AFM
| Immobilization Method | Success Rate (%) | Firmness of Attachment | Impact on Viability | Buffer Compatibility | Best Suited Envelope Types |
|---|---|---|---|---|---|
| Polyphenolic Proteins [43] | >95 | Excellent (Minimal detachment) | Minimal effect | Broad (PBS, MOPS, nutrient media) | Gram-positive, Gram-negative |
| Covalent Binding [43] | 85-90 | Very Good (Some detachment) | Moderate effect | Physiological buffers | Gram-negative, Mycobacterial |
| Gelatin Coating [9] | 80-85 | Good in specific buffers | Minimal effect | Low ionic strength (0.01× PBS) | Gram-negative |
| Poly-L-Lysine [9] | 75-80 | Variable (Detachment in PBS) | Antimicrobial effects | Complex media after recovery | Gram-negative (with caution) |
| Physical Confinement [43] | 70-75 | Moderate | Minimal effect | Most aqueous buffers | Gram-positive, Cocci |
| Electrostatic Adsorption [43] | 65-70 | Poor (Significant detachment) | Minimal effect | Limited (not in PBS/MOPS) | Gram-positive |
Method-Specific Performance Metrics
Table 2: Detailed Performance Metrics by Bacterial Type
| Method | Parameter | E. coli (Gram-Negative) | B. subtilis (Gram-Positive) | M. smegmatis (Mycobacterial) |
|---|---|---|---|---|
| Polyphenolic Proteins | Detachment Rate | <5% | <3% | 8% |
| Viability Retention | >90% | >95% | >85% | |
| Optimal Buffer | PBS, MOPS, LB broth | PBS, MOPS, Nutrient media | PBS, MOPS | |
| Covalent Binding | Detachment Rate | 10-15% | 20% | 12% |
| Viability Retention | 70-80% | 60% | 75% | |
| Optimal Buffer | MOPS, HEPES | MOPS, HEPES | MOPS | |
| Gelatin Coating | Detachment Rate | 8% (in 0.01× PBS) | 25% | 30% |
| Viability Retention | >90% | 80% | 70% | |
| Optimal Buffer | 0.01× PBS-S | Not recommended | Not recommended |
Detailed Experimental Protocols
Optimal Immobilization Protocol Using Polyphenolic Proteins
Principle: Mussel-derived polyphenolic proteins (e.g., Cell-Tak) provide a fast, reproducible, and generally applicable scheme for immobilizing living bacteria through strong, non-specific adhesion that doesn't compromise cell viability [43].
Materials:
- Polyphenolic protein solution (e.g., Cell-Tak, BD Biosciences)
- Acidified water (pH 2.0-3.0)
- Glass slides or mica disks
- Sterile phosphate-buffered saline (PBS)
- Bacterial culture in mid-logarithmic phase
Procedure:
- Surface Preparation:
- Clean glass slides or mica disks with oxygen plasma or strong oxidizers for 30 minutes
- Rinse thoroughly with deionized water and dry under nitrogen stream
Coating Application:
- Prepare polyphenolic protein solution at 30-40 µg/mL in acidified water (pH 2.0-3.0)
- Apply 20-50 µL to the center of cleaned substrate
- Incubate for 20 minutes at room temperature in a humidified chamber
- Remove excess solution by gentle pipetting
- Rinse twice with sterile deionized water to remove unbound adhesive
- Air dry for 5-10 minutes before use
Cell Immobilization:
- Harvest bacterial cells at mid-logarithmic phase by gentle centrifugation (2,000 × g for 5 minutes)
- Wash twice with appropriate physiological buffer (PBS, MOPS, or imaging buffer)
- Resuspend to moderate density (OD₆₀₀ ≈ 0.4-0.6)
- Apply 10-20 µL cell suspension to coated surface
- Allow attachment for 15-30 minutes in a humidified chamber
- Gently rinse with imaging buffer to remove non-adhered cells
- Proceed immediately with AFM imaging
Critical Notes:
- Coated surfaces can be prepared in advance and stored at 4°C for up to 2 weeks
- Avoid excessive drying of coated surfaces before cell application
- Optimal cell density may require empirical determination for different strains
Gram-Negative Specific Protocol: Gelatin Coating with Buffer Optimization
Principle: Gelatin provides a non-cytothermic immobilization matrix that facilitates stable imaging of Gram-negative bacteria in nutrient media when used with optimized low-ionic strength buffers [9].
Materials:
- High bloom strength gelatin (G2500, Sigma-Aldrich)
- Glass slides
- 0.01× PBS-S buffer (1:100 dilution of standard PBS)
- Complex growth media (e.g., LB broth)
- Bacterial culture (overnight growth recommended)
Procedure:
- Gelatin Substrate Preparation:
- Prepare 0.5% high bloom gelatin solution in warm deionized water (40-45°C)
- Apply to clean glass slides and allow to dry vertically at room temperature
- Store coated slides in a desiccator until use
- Cell Preparation and Immobilization:
- Use overnight grown cultures for synchronized cell populations
- Harvest cells by gentle centrifugation and wash with 0.01× PBS-S
- Resuspend in 0.01× PBS-S at moderate density
- Apply cell suspension to gelatin-coated slides
- Allow attachment for 20 minutes
- Gently replace 0.01× PBS-S with complex growth media
- Incubate for 30-60 minutes recovery time before imaging
Critical Notes:
- Low ionic strength buffer (0.01× PBS-S) is essential for initial attachment
- Recovery period in growth media significantly enhances viability during extended imaging
- Protocol has been specifically validated for E. coli strains [9]
Covalent Immobilization Protocol for Challenging Strains
Principle: Chemical functionalization of surfaces with amine or carboxyl groups enables covalent bonding to bacterial surface proteins, providing strong attachment for high-resolution imaging [43].
Materials:
- Aminosilane or carboxyl-terminated surfaces
- Cross-linkers (e.g., EDC, NHS)
- Coupling buffers (MES, pH 5.5-6.0)
- Bacterial culture
Procedure:
- Surface Functionalization:
- Activate cleaned surfaces with oxygen plasma
- Incubate with 2% aminosilane solution in ethanol for 1 hour
- Rinse thoroughly with ethanol and cure at 110°C for 15 minutes
- Cell Immobilization:
- Harvest and wash bacterial cells with coupling buffer
- Activate functionalized surfaces with cross-linker solution
- Apply cell suspension and incubate for 1 hour
- Rinse vigorously with imaging buffer to remove loosely bound cells
Critical Notes:
- May affect cell viability more than other methods
- Essential for imaging under high lateral forces
- Some cell detachment may still occur during AFM imaging [43]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Bacterial Immobilization
| Reagent/Chemical | Supplier Examples | Function | Application Notes |
|---|---|---|---|
| Cell-Tak | BD Biosciences | Polyphenolic adhesive for firm cell attachment | Most versatile; works across bacterial types; minimal viability impact [43] |
| Gelatin (High Bloom) | Sigma-Aldrich (G2500) | Non-cytotoxic matrix for cell entrapment | Gram-negative specific; requires low ionic strength buffers [9] |
| Poly-L-Lysine | Sigma-Aldrich | Positively charged polymer for electrostatic binding | Use with caution due to antimicrobial effects; viability concerns [9] |
| Aminosilanes | Gelest, Sigma-Aldrich | Surface functionalization for covalent binding | Strong attachment but may affect surface properties [43] |
| Silicon Nitride AFM Cantilevers | Bruker Corporation | AFM imaging with minimal sample damage | Spring constant ~14.4 pN/nm; tip diameter <40nm [44] |
| HEPES Buffer | Sigma-Aldrich | Physiological imaging buffer | Maintains pH during extended imaging sessions [44] |
Advanced Applications and Method Integration
High-Speed AFM Integration
Recent advances in HS-AFM modalities enable tracking of single bacterial proteins and cells at high temporal resolution. Successful implementation requires particularly firm immobilization, making polyphenolic proteins or covalent binding methods essential. Kumar et al. developed a specialized HS-AFM protocol using high-resonance frequency cantilevers, optimized scanning parameters, and imaging buffers that allows visualization of single proteins in curved membranes of living cells [42].
Multiparametric AFM Measurements
Modern AFM technologies allow simultaneous recording of ultrastructure, adhesion, and mechanical properties at nanoscale resolution on bacterial surfaces. These applications demand immobilization strategies that neither alter native surface properties nor constrain natural cell dynamics. Physical confinement methods or gentle polyphenolic immobilization are preferred for such studies as they minimize alteration of mechanical properties [42].
Troubleshooting Guide
Table 4: Common Immobilization Problems and Solutions
| Problem | Potential Causes | Solutions |
|---|---|---|
| Cell Detachment During Imaging | Insufficient attachment strength; inappropriate buffer; excessive imaging forces | Switch to polyphenolic proteins; optimize buffer ionic strength; reduce AFM contact forces |
| Poor Viability | Cytotoxic immobilization method; inadequate recovery time; nutrient deprivation | Use gelatin or polyphenolic proteins; incorporate recovery period in growth media; image in nutrient media |
| Inadequate Resolution | Cell movement during scanning; excessive surface roughness; probe contamination | Improve immobilization firmness; use smoother substrates; clean/replace AFM probes |
| Inconsistent Results Across Strains | Failure to account for envelope differences; one-size-fits-all approach | Tailor method to envelope type: gelatin for Gram-negative, polyphenolic proteins for broad application |
Optimizing surface chemistry for immobilizing bacterial cells requires careful consideration of strain-specific envelope architectures and experimental objectives. Polyphenolic proteins like Cell-Tak provide the most generally applicable solution, offering firm immobilization with minimal impact on viability across diverse bacterial types. For Gram-negative specific applications, gelatin coating with low-ionic strength buffers provides an excellent balance of attachment strength and preservation of physiological function. The protocols and comparative data presented herein provide researchers with a rational framework for selecting and implementing the optimal immobilization strategy for their specific AFM applications in bacterial cell research.
Preventing and Identifying Salt Artifacts through Proper Washing Protocols
Within the broader context of developing robust atomic force microscopy (AFM) protocols for immobilizing bacterial cells, managing salt artifacts is a critical, yet often overlooked, prerequisite. Successful high-resolution AFM imaging and accurate force spectroscopy of live bacteria are contingent on effective sample immobilization. This immobilization, however, can be severely compromised by residual salts from culture media or buffers, which form crystalline deposits upon drying [45] [1]. These deposits introduce significant topographical artifacts, adversely affecting image quality, compromising nanomechanical property measurements, and potentially interfering with the intended bacterium-substrate interactions.
This application note details standardized protocols for preventing salt crystallization through effective washing procedures and provides a framework for identifying common salt artifacts in AFM data. The methodologies are framed within the context of immobilizing Gram-negative bacteria like Escherichia coli for AFM studies, with a specific focus on preserving cell viability and surface integrity while eliminating confounding salt contaminants.
The Impact of Salt Artifacts on AFM Analysis
Salt crystals exhibit mechanical properties starkly different from biological samples. Their high stiffness and irregular shapes can lead to tip contamination, damage, and completely non-representative force-distance curves [46] [47]. During imaging, the probe may interact with these crystals instead of the bacterial surface, producing images with sharp, angular features that mask true surface morphology. Furthermore, the presence of a salt layer between the bacterium and the immobilization substrate can weaken adhesion, causing cells to be dislodged during scanning and resulting in incomplete or missing data [45]. For studies investigating biophysical properties such as cell elasticity or adhesion forces—where the structural and chemical diversity of the outer membrane is a key determinant of phenotypic heterogeneity—these artifacts can render data unreliable and irreproducible [21].
Table 1: Common Salt Artifacts and Their Signatures in AFM Data
| Artifact Type | Typical Morphology | Key Identifying Features | Impact on AFM Analysis |
|---|---|---|---|
| Cubic NaCl Crystals | Sharp, geometric, cube-like structures [47] | High stiffness, uniform angularity; appears in "salt and pepper" noise patterns [46] | Obscures bacterial surface, causes tip convolution, risks tip damage [47] |
| Amorphous Salt Layers | Fine, granular, or uniform film coating the surface | Unusually high and uniform adhesion forces; smoothens native topography | Alters measured adhesion forces and nanomechanical properties; masks true surface roughness |
| Salt Particulates | Small, scattered, irregularly shaped particles | Random distribution across the substrate and cell surface; high contrast in phase imaging | Leads to overestimation of feature dimensions (tip convolution); produces outlier force spectroscopy curves |
Experimental Protocols for Effective Washing and Immobilization
The following protocols are designed to integrate seamlessly with common bacterial immobilization procedures, such as the use of gelatin-coated mica or poly-L-lysine-coated glass [21] [45] [1].
Protocol 1: Standardized Washing for Gelatin-Coated Mica Immobilization
This protocol is adapted from established methods for immobilizing E. coli and is highly effective for removing media-derived salts [21] [1].
Principle: Gelatin coating provides a positively charged surface that electrostatically immobilizes negatively charged bacterial cells. Using low-ionic-strength washing buffers is crucial to prevent charge shielding, which would weaken this interaction and reduce immobilization efficiency [1].
Materials:
- Centrifuge
- Gelatin-coated mica substrates [1]
- Low-ionic-strength buffer (e.g., 2 mM Tris-HCl, pH 7.0) or Milli-Q water [21] [45]
- Bacterial culture (e.g., E. coli ATCC 25922)
Procedure:
- Culture Harvesting: Centrifuge a 1-2 mL aliquot of bacterial culture at 2151 × g for 5 minutes at 24°C to pellet the cells [21].
- Primary Wash: Carefully decant the supernatant. Resuspend the cell pellet in 1 mL of a low-ionic-strength buffer or Milli-Q water. Vortex gently to ensure complete resuspension.
- Secondary Wash: Repeat the centrifugation and resuspension steps for a total of two washes. A second wash is critical to dilute and remove salts effectively.
- Immobilization: Adjust the optical density of the final suspension as required (e.g., to ~10⁶ CFU/mL) and deposit a small volume (e.g., 20-50 µL) onto a gelatin-coated mica surface [21].
- Incubation and Rinsing: Allow the cells to adhere for a designated period (e.g., 30 minutes). Gently rinse the substrate with a stream of low-ionic-strength buffer or Milli-Q water to remove any non-adherent cells and residual salts. The prepared sample should be kept hydrated for immediate AFM analysis.
Protocol 2: Washing for Membrane-Sensitive Studies with Poly-L-Lysine Immobilization
For studies where outer membrane integrity is paramount, such as those involving lipopolysaccharide (LPS) characterization, this protocol incorporates divalent cations to stabilize the membrane during washing [45].
Principle: Poly-L-lysine provides strong electrostatic binding in both low and high ionic strength buffers. However, low-ionic-strength conditions can impose hypoosmotic stress on bacteria. The addition of divalent cations (Mg²⁺, Ca²⁺) and glucose to the immobilization buffer helps preserve membrane integrity without promoting salt crystallization [45].
Materials:
- Poly-L-lysine-coated glass or mica substrates
- Low-ionic-strength immobilization buffer (e.g., 2 mM HEPES, pH 7.0) supplemented with 5 mM MgCl₂, 1 mM CaCl₂, and 10 mM glucose [45]
- Bacterial culture
Procedure:
- Culture Harvesting: Pellet bacteria via centrifugation as in Protocol 1.
- Cation-Stabilized Wash: Resuspend the pellet in the supplemented low-ionic-strength immobilization buffer. The divalent cations mitigate the hypoosmotic stress and help maintain outer membrane organization [45].
- Immobilization: Deposit the cell suspension onto a poly-L-lysine-coated surface.
- Final Rinse: After incubation, perform a final, gentle rinse with the same supplemented buffer to remove loosely bound material while keeping the sample in a stabilized, hydrated state.
Protocol 3: Artifact Identification and Verification
A simple control experiment can confirm the presence of salt artifacts.
Procedure:
- Prepare an AFM sample following your standard protocol but omit the bacterial cells, depositing only the final wash suspension buffer onto the substrate.
- Allow the droplet to air-dry completely.
- Image the dried substrate in tapping mode. The presence of crystalline or granular structures confirms that your washing procedure is insufficient to remove salts, and the protocol requires further optimization (e.g., more wash cycles, use of purer water).
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Bacterial Immobilization and Salt Mitigation
| Reagent / Material | Function / Purpose | Key Considerations |
|---|---|---|
| Gelatin-Coated Mica | Positively charged substrate for electrostatic immobilization of bacteria [1]. | Optimal for low-ionic-strength environments; binding capacity reduced in high-salt buffers [45]. |
| Poly-L-Lysine-Coated Surfaces | Provides a strong, positively charged coating for immobilizing a wide range of cell types [45]. | Can compromise membrane integrity under hypoosmotic stress; requires stabilization with cations [45]. |
| Milli-Q Water (Ultrapure) | A low-ionic-strength washing solvent to dissolve and remove soluble salts [21]. | Avoids introduction of new ions; its low osmolarity may stress cells without proper stabilization. |
| Divalent Cations (Mg²⁺, Ca²⁺) | Membrane stabilizers added to low-ionic-strength buffers to preserve cell viability and integrity [45]. | Critical for studies on outer membrane mechanics (e.g., LPS-related research) [21] [45]. |
| HEPES or Tris Buffer | Low-ionic-strength buffering agents to maintain physiological pH during washing without salt precipitation. | Prevents pH fluctuation-induced stress on cells while minimizing salt content. |
Workflow for Salt-Free Bacterial AFM
The following diagram summarizes the decision-making pathway and procedural steps for preparing a bacterial AFM sample free from salt artifacts, integrating both washing and immobilization strategies.
Figure 1: A comprehensive workflow for preparing bacterial AFM samples while preventing salt artifacts. The process guides the user through key decisions, such as selecting a washing protocol based on the need for membrane integrity, and concludes with a verification step.
Integrating rigorous and appropriate washing protocols is a foundational step in any AFM study of bacterial cells. By systematically removing salts that lead to crystalline artifacts, researchers can ensure that the data obtained—whether topographical images or nanomechanical properties—accurately reflect the native state of the bacterial sample. The protocols outlined here, when combined with the artifact identification guide, provide a reliable framework for enhancing the reproducibility and biological relevance of AFM-based microbiological research.
Addressing Excessive Adhesion Forces that Compromise Measurement
Excessive adhesion forces between the atomic force microscope (AFM) probe and bacterial samples represent a significant challenge in nanobiotechnology, often leading to compromised data, sample displacement, or even damage to delicate cellular structures [48]. Successful AFM analysis hinges on effectively immobilizing bacterial cells to a surface, securing them against the lateral forces exerted by the scanning probe tip, yet doing so in a way that minimizes alterations to their native physiological state [1]. This document outlines standardized protocols and application notes for researchers, detailing methods to mitigate excessive adhesion forces during AFM investigations of bacterial cells, thereby ensuring the acquisition of reliable topographical and nanomechanical data.
Surface Immobilization Strategies for Bacterial Cells
A critical step in preparing bacterial samples for AFM is their effective immobilization onto a substrate. The optimal method should be minimally invasive, preserving the bacterium's native structure and function, while providing sufficient adhesion to withstand scanning forces [1]. The following protocols describe two effective approaches.
Protocol: Immobilization on Gelatin-Coated Mica
This method utilizes a gelatin film to create an electrostatic interface for immobilizing negatively charged bacteria [1].
- Step 1: Substrate Preparation. Freshly cleave a thin sheet of mica using a sharp blade to obtain an atomically flat, clean surface.
- Step 2: Gelatin Coating. Prepare a 0.1% (w/v) solution of porcine gelatin in deionized water. Apply a small volume (e.g., 100 µL) of this solution onto the cleaved mica surface and incubate for 10 minutes at room temperature.
- Step 3: Rinsing. After incubation, gently rinse the coated mica with deionized water to remove any excess or unbound gelatin. Carefully dry the surface under a gentle stream of inert gas (e.g., nitrogen or argon).
- Step 4: Cell Deposition. Apply a bacterial suspension (in an appropriate buffer) onto the gelatin-coated mica and allow it to incubate for a defined period, typically 15-30 minutes, to facilitate adhesion.
- Step 5: Final Rinse. Gently rinse the surface with the imaging buffer to remove any non-adherent cells before placing the substrate into the AFM liquid cell.
Protocol: Immobilization on Indium-Tin-Oxide (ITO)-Coated Glass
This method leverages the hydrophobic and smooth properties of ITO for imaging without chemical immobilization [5].
- Step 1: Substrate Selection. Use commercially available ITO-coated glass slides.
- Step 2: Surface Cleaning. Clean the ITO surface with solvents like ethanol and deionized water in a sequence, followed by drying.
- Step 3: Cell Deposition. Pipette a bacterial suspension directly onto the ITO surface and allow the cells to adhere under physiological conditions for a predetermined time.
- Step 4: Assemble AFM Liquid Cell. Transfer the substrate with adhered bacteria into the AFM liquid cell and add the appropriate imaging buffer for analysis.
Table 1: Comparison of Bacterial Immobilization Substrates
| Feature | Gelatin-Coated Mica | ITO-Coated Glass |
|---|---|---|
| Immobilization Mechanism | Electrostatic interaction [1] | Hydrophobic adhesion & surface properties [5] |
| Sample Preparation | Requires coating and rinsing steps [1] | Simple, uses pre-coated slides [5] |
| Invasiveness | Minimal chemical invasiveness [1] | Non-perturbative, no chemical treatment [5] |
| Primary Advantage | Generally applicable for many microbial cells [1] | Excellent for imaging living, native bacteria in liquid [5] |
| Considerations | Adhesion can be influenced by suspension buffer chemistry [1] | Relies on inherent bacterial adhesion properties [5] |
Research Reagent Solutions
Table 2: Essential Materials for Bacterial AFM Immobilization
| Item | Function/Description |
|---|---|
| Freshly Cleaved Mica | Provides an atomically flat and clean substrate for coating [1]. |
| Porcine Gelatin | Forms a positively charged coating on mica to immobilize negatively charged bacteria [1]. |
| ITO-coated Glass Slides | A smooth, hydrophobic substrate that facilitates bacterial adhesion without chemical treatments [5]. |
| AFM Liquid Cell | A sealed chamber that allows for imaging in buffer solutions under physiological conditions [5]. |
Quantifying and Interpreting Adhesion Forces
A fundamental advantage of AFM is its ability to quantitatively measure interaction forces via force-distance curves [48]. These measurements are crucial for diagnosing and understanding excessive adhesion.
Protocol: Acquiring Force-Distance Curves
- Step 1: Cantilever Selection. Choose a cantilever with an appropriate spring constant (e.g., ~0.08-0.3 N/m for soft biological samples) [49] [5].
- Step 2: System Calibration. Calibrate the cantilever's sensitivity and spring constant using standard methods (e.g., thermal tune).
- Step 3: Approach-Retract Cycle. Position the probe over the area of interest. The piezoelectric scanner extends the sample towards the probe until contact is made (approach), then retracts it (retract). The cantilever deflection is recorded throughout this cycle.
- Step 4: Data Conversion. Convert the raw photodetector voltage versus piezo displacement data into a force-distance curve using the calibrated spring constant and sensitivity.
Adhesion Force Analysis
On the retraction curve, a characteristic "pull-off" event signifies the rupture of the bond between the tip and the sample. The minimum force of this retraction curve is the measured adhesion force [48] [49]. Analyzing these curves provides insights into the nanomechanical properties of the sample.
Diagram 1: Force-distance curve cycle.
Instrumental and Operational Modifications to Mitigate Adhesion
When excessive adhesion persists despite optimal sample preparation, instrumental and operational adjustments are necessary.
Cantilever and Probe Modifications
- Use of Spherical Probes: Replacing sharp pyramidal tips with colloidal probes (e.g., a silica bead glued to a cantilever) increases the contact area but can be useful for quantifying average surface adhesion properties, as forces scale with the radius of the spherical probe [49].
- Functionalized Probes: Covalently bonding specific molecules (e.g., biotin) to the cantilever allows for the measurement of specific binding forces (e.g., biotin-streptavidin), distinguishing them from nonspecific adhesion [49].
Advanced Imaging Modes and Detection Upgrades
- Tapping Mode Operation: In tapping mode, the cantilever is oscillated at its resonance frequency, and the tip only intermittently contacts the sample. This significantly reduces lateral forces and minimizes sample damage and displacement compared to contact mode [50].
- Enhanced Detection System: The standard four-quadrant photodiode has a limited detection range for large cantilever deflections, which is problematic when measuring strong adhesive forces. Replacing it with a linear Position Sensitive Detector (PSD) improves the sensitivity, linearity, and dynamic range of force measurements [49].
Diagram 2: Solutions for excessive adhesion.
A Case Study: Investigating Bacterial Nanotubes
The application of these optimized protocols is exemplified in the study of bacterial nanotubes in Rhodococcus wratislaviensis [5]. This research successfully visualized these delicate intercellular structures by:
- Utilizing the non-immobilization protocol on ITO-coated glass slides to maintain native cell physiology.
- Employing Quantitative Imaging (QI) mode, a high-speed force mapping technique, to acquire topographical and nanomechanical data simultaneously with minimal perturbation.
- Calculating the Young's modulus from approach curves, revealing that the nanotubes have a lower modulus (i.e., are more flexible) than the main bacterial cell body [5].
This case demonstrates that a combination of thoughtful sample preparation and advanced AFM operational modes is essential for probing delicate biological features without compromising their structural integrity through excessive adhesive forces.
Adapting Protocols for High-Resolution Imaging of Appendages like Flagella and Pili
Atomic Force Microscopy (AFM) has emerged as a cornerstone technique in biophysical research, enabling the investigation of microbial surfaces at nanometer resolution under physiological conditions. For researchers studying bacterial appendages such as flagella and pili, AFM offers unparalleled capability to visualize these delicate structures and quantify their functional properties without the need for extensive sample preparation that can alter native morphology. This protocol details specialized methodologies for immobilizing bacterial cells to facilitate high-resolution imaging of surface appendages, addressing the critical challenge of maintaining cell viability and structural integrity while achieving sufficient adhesion to withstand scanning forces. The procedures outlined here are particularly optimized for the investigation of flagellar organization and pili interactions that are essential for bacterial motility, adhesion, and biofilm formation, providing a framework for reliable and reproducible AFM analysis in bacterial cell surface research.
Key Considerations for Appendage Imaging
Imaging bacterial appendages presents unique challenges that require careful methodological adaptation. Flagella and pili are nanoscale structures with diameters typically ranging from 10-50 nm, requiring exceptional resolution and minimal sample disturbance for accurate visualization [6]. These structures are not only delicate but also dynamic, often involved in continuous cycles of attachment, detachment, and rearrangement. Successful imaging must therefore preserve their native conformation while preventing displacement during scanning.
The fundamental requirement for reliable AFM imaging is effective cell immobilization that anchors cells sufficiently to resist lateral forces exerted by the AFM probe. This immobilization must be achieved without compromising cell viability or structural integrity, particularly for time-series studies of dynamic processes. Selection of appropriate substrates, immobilization agents, and imaging buffers must be tailored to the specific bacterial strain and the experimental objectives, whether for topographical mapping, nanomechanical property assessment, or real-time observation of surface dynamics.
Research Reagent Solutions
The following table summarizes essential materials and their specific functions in bacterial immobilization for AFM imaging:
Table 1: Key Research Reagents for Bacterial Immobilization and AFM Imaging
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Gelatin | Electrostatic immobilization of negatively charged bacteria on coated surfaces | Porcine gelatin; Varying bloom strengths (high, medium, low) for adhesion tuning [1] [9] |
| Poly-L-Lysine (PLL) | Chemical immobilization via electrostatic interaction with bacterial surfaces | α-poly-l-lysine for live cell imaging [9] |
| Indium-Tin-Oxide (ITO) | Substrate for imaging without aggressive immobilization; hydrophobic properties enhance cell adhesion | ITO-coated glass substrates for stable imaging in liquid [5] |
| EDTA | Outer membrane disorganization for LPS removal studies | 100 mM EDTA solution (pH 8.0) for controlled LPS removal [21] |
| Buffers | Imaging environment and sample preparation | 0.01× PBS-S, modified PBS, nutrient media (e.g., LB broth) [9] |
Substrate Selection and Preparation Protocol
Gelatin-Coated Surfaces
Gelatin coating creates a positively charged surface that electrostatically immobilizes negatively charged bacterial cells. This method is particularly suitable for Gram-negative bacteria and enables imaging in liquid environments [1].
Procedure:
- Prepare a 0.5% gelatin solution (porcine gelatin, Sigma-Aldrich G2500, G2625, or G6144) in purified water.
- Clean glass slides or mica surfaces to ensure uniform coating.
- Apply the gelatin solution to the cleaned surface and allow it to dry at room temperature (approximately 30-60 minutes).
- Verify coating uniformity before use.
- Apply bacterial suspension to the gelatin-coated surface and incubate for 30 minutes to facilitate adhesion.
- Gently rinse with appropriate buffer to remove non-adherent cells before AFM imaging.
Poly-L-Lysine Coated Surfaces
PLL provides strong electrostatic immobilization suitable for extended imaging sessions, though its potential antimicrobial effects must be considered for live cell studies [9].
Procedure:
- Prepare a 0.1% w/v α-poly-l-lysine solution in purified water.
- Apply to cleaned glass slides and incubate for 30 minutes.
- Remove excess solution and rinse gently with purified water.
- Air dry the coated slides before use.
- Apply bacterial suspension and incubate for 15-30 minutes.
- Rinse gently with imaging buffer to remove loosely attached cells.
ITO-Coated Substrates
ITO substrates provide an alternative for imaging without chemical immobilization agents, leveraging their hydrophobic properties to enhance bacterial adhesion [5].
Procedure:
- Clean ITO-coated glass substrates with appropriate solvents.
- Sterilize if required for live cell imaging.
- Apply bacterial suspension directly to the ITO surface.
- Allow cells to adhere for specified duration (typically 30-60 minutes).
- Gently rinse with growth medium or compatible buffer before mounting for AFM.
Bacterial Immobilization Strategies
Strain Selection and Culture Conditions
The choice of bacterial strain and growth conditions significantly influences surface properties and immobilization efficiency. For appendage imaging, Pantoea sp. YR343 serves as an excellent model organism as it possesses peritrichous flagella and forms structured biofilms, enabling studies of flagellar organization and function [6]. Alternatively, Escherichia coli ATCC 25922 provides a well-characterized system for investigating lipopolysaccharide (LPS)-mediated surface properties that affect adhesion [21].
Culture Protocol:
- Grow bacterial strains in appropriate medium (e.g., LB broth) at optimal temperature (typically 37°C for E. coli).
- Harvest cells during mid-exponential growth phase (OD660 ≈ 0.5-0.8) to ensure uniform population and maximal appendage expression.
- Centrifuge culture at 2,151 × g for 5 minutes at 24°C and resuspend in appropriate buffer.
- Adjust cell density to approximately 10^6 CFU/mL for immobilization.
Immobilization Optimization
Different immobilization approaches offer distinct advantages depending on experimental goals. The following workflow outlines the decision process for selecting and optimizing immobilization strategies:
Viability and Integrity Assessment
Maintaining cell viability and membrane integrity is crucial for live cell imaging, particularly when investigating dynamic processes involving appendages.
Assessment Protocol:
- Membrane Integrity Testing: Use propidium iodide exclusion assay to confirm membrane integrity after immobilization.
- Metabolic Activity: Monitor continued growth and division potential after immobilization.
- Morphological Inspection: Verify normal cell shape and absence of deformation using optical microscopy before AFM.
- Control Experiments: Compare with untreated cells to ensure immobilization does not alter native physiology.
For PLL immobilization, specific optimization is required due to potential antimicrobial effects. Research indicates that using overnight grown cells immobilized on PLL in diluted PBS-S (0.01×) with subsequent recovery time in nutrient media produces stably attached cells with preserved membrane integrity and viability [9].
AFM Imaging Parameters for Appendage Visualization
Instrumentation Configuration
Optimizing AFM parameters is essential for resolving delicate nanostructures like flagella and pili while preserving sample integrity.
Table 2: AFM Imaging Parameters for Appendage Visualization
| Parameter | Recommended Setting | Rationale |
|---|---|---|
| Imaging Mode | Quantitative Imaging (QI) mode or Tapping mode in liquid | Minimizes lateral forces on delicate structures [5] |
| Scan Size | Variable, with automated large-area capability | Captures both cellular and community context (up to mm scale) [6] |
| Scan Rate | 0.5-1.5 Hz | Balances resolution and stability for high-magnification imaging |
| Cantilever | PPP-CONTPt (Nanosensors), k = 0.3 N/m | Soft cantilever minimizes sample deformation [5] |
| Resolution | 64 × 64 pixels to 512 × 512 pixels | Optimizes detail capture while managing file size |
| Image Processing | Machine learning-based stitching and analysis | Enables large-area reconstruction from high-res tiles [6] |
Large-Area Automated AFM
Traditional AFM is limited by small scan areas (<100 µm), restricting the ability to contextualize nanoscale features within larger biofilm architectures. Automated large-area AFM addresses this limitation through:
Implementation Protocol:
- Integrate motorized stage with precise positional control.
- Implement automated tile selection with minimal overlap (5-10%) between adjacent images.
- Utilize machine learning algorithms for seamless image stitching.
- Apply cell detection and classification algorithms for quantitative analysis of spatial distributions.
- Combine with gradient-structured surfaces to study surface property effects on attachment in a single experiment [6].
This approach has revealed previously obscured structural details, such as the honeycomb pattern in Pantoea sp. YR343 biofilms and the coordinated role of flagella in biofilm assembly beyond initial attachment [6].
Liquid Imaging Conditions
Imaging in liquid preserves native conformation of appendages and enables real-time observation of dynamic processes.
Buffer Considerations:
- Use physiological buffers (e.g., modified PBS at appropriate dilution) for structural studies.
- Employ nutrient media (e.g., LB broth) for time-lapse studies of dynamic processes.
- Optimize ionic strength to balance immobilization stability and physiological relevance.
- Maintain temperature control (24.0 ± 0.2°C) for consistent imaging conditions [5].
Data Analysis and Interpretation
Structural Analysis of Appendages
AFM enables quantitative assessment of appendage dimensions and distribution patterns that are critical for understanding their functional roles.
Flagellar Characterization:
- Diameter: 20-50 nm, measured from height in AFM cross-section [6]
- Length: Tens of micrometers, extending across multiple cells
- Distribution: Peritrichous arrangement visible around bacterial cells
- Patterns: Network formation between cells, suggesting coordination in biofilm assembly
Analytical Protocol:
- Apply segmentation algorithms to identify individual appendages.
- Measure dimensional parameters (height, length, curvature) from topographic data.
- Quantify spatial distribution and orientation relative to cell body.
- Analyze connectivity patterns in cellular networks.
Nanomechanical Property Mapping
Beyond topography, AFM enables quantification of mechanical properties through force spectroscopy, providing insights into the functional state of bacterial surfaces.
Mechanical Mapping Protocol:
- Acquire force-volume maps using colloidal probes instead of sharp tips for whole-cell assessment.
- Calculate Young's modulus using Hertz/Sneddon model fitting to force-indentation curves.
- Compare mechanical properties of cell body versus appendages.
- Assess population heterogeneity in mechanical properties [21] [5].
Research indicates that bacterial nanotubes exhibit lower Young's modulus compared to the cell body, suggesting flexibility that facilitates intercellular communication and material transfer [5].
Applications and Case Studies
Flagellar Organization in Biofilm Formation
Large-area AFM imaging of Pantoea sp. YR343 has revealed intricate patterns of flagellar organization during early biofilm development. Studies show that flagella not only facilitate initial surface attachment but also form coordinated networks between cells, creating bridging structures that contribute to biofilm architecture [6]. These flagellar interactions appear to guide the development of characteristic honeycomb patterns observed in mature biofilms, suggesting a structural role beyond motility.
LPS-Mediated Surface Heterogeneity
Single-cell AFM analysis of E. coli ATCC 25922 has demonstrated that lipopolysaccharides (LPS) are key determinants of surface heterogeneity, influencing adhesion forces and cellular elasticity [21]. Partial removal of LPS through EDTA treatment reduces cell-to-cell variability in biophysical properties, creating a more homogeneous population with diminished adhesive capability. This approach enables investigation of structure-function relationships in bacterial adhesion and surface interactions.
Nanotube-Mediated Intercellular Connections
AFM imaging in liquid has enabled visualization of bacterial nanotubes connecting individual cells of Rhodococcus wratislaviensis, revealing these intercellular bridges have distinct mechanical properties compared to the cell body [5]. Their lower Young's modulus suggests structural flexibility that may facilitate molecular transfer between cells, representing a previously uncharacterized communication pathway in bacterial communities.
Troubleshooting Guide
Table 3: Troublescommon AFM Imaging Issues and Solutions
| Problem | Potential Causes | Solutions |
|---|---|---|
| Poor appendage resolution | Excessive scanning force, inappropriate cantilever, sample drift | Softer cantilever (0.1-0.5 N/m), reduce setpoint, optimize feedback parameters |
| Cell detachment during scanning | Inadequate immobilization, excessive lateral forces | Optimize substrate coating, increase adhesion time, reduce scan size/speed |
| Loss of viability | Toxic immobilization agents, non-physiological buffers | Switch to gelatin instead of PLL, use diluted buffers with nutrient supplementation |
| Inconsistent results | Population heterogeneity, variable growth conditions | Standardize culture conditions, use synchronized cultures, increase sample size |
| Image artifacts | Tip contamination, improper calibration, vibration | Clean/replace tip, recalibrate instrument, improve vibration isolation |
The protocols presented here provide a comprehensive framework for high-resolution AFM imaging of bacterial appendages, with specific adaptations for visualizing flagella, pili, and intercellular connections. The integration of advanced immobilization strategies with optimized AFM parameters enables researchers to overcome the traditional challenges associated with nanoscale imaging of these delicate structures. By implementing these methodologies, investigators can reliably capture both structural details and functional properties of bacterial appendages, opening new avenues for understanding their roles in adhesion, motility, community formation, and antimicrobial resistance. The continuing development of automated large-area AFM combined with machine learning analytics promises to further enhance our ability to contextualize nanoscale features within broader biological systems, advancing both fundamental knowledge and applied research in microbial biophysics.
Benchmarking Immobilization Strategies: Validation and Comparative Analysis
Within the framework of a broader thesis on Atomic Force Microscopy (AFM) protocols for immobilizing bacterial cells, the critical step of sample preparation directly dictates the quality, reliability, and biological relevance of the acquired data. Successful AFM analysis of microbial cells hinges on effectively immobilizing the specimen to the mounting surface to prevent displacement by the scanning cantilever tip, while simultaneously maintaining the cells in a viable, unperturbed physiological state [51] [1]. This application note provides a comparative analysis of common bacterial immobilization strategies, focusing on their impact on force-distance curve measurements and the introduction of image artefacts. We detail standardized protocols for two distinct methods—one based on mechanical entrapment and the other on electrostatic adhesion—and provide a quantitative framework for evaluating their performance in AFM-based microbiological research.
Comparative Analysis of Immobilization Methods
A fundamental challenge in AFM of bacterial cells is balancing immobilization strength with minimal cellular perturbation. Suboptimal immobilization can lead to cellular detachment or deformation during scanning, resulting in unreliable topographical data and force spectroscopy measurements [1]. The choice of substrate and immobilization chemistry is paramount. The following table summarizes the key characteristics of different approaches.
Table 1: Comparison of Bacterial Immobilization Methods for AFM
| Immobilization Method | Mechanism of Adhesion | Typical Substrate | Relative Immobilization Strength | Risk of Image Artefacts | Preservation of Native State | Key Applications |
|---|---|---|---|---|---|---|
| Gelatin-Coated Mica | Electrostatic interaction | Freshly cleaved mica | Medium | Low | High | Imaging in liquid, live-cell dynamics, single-cell force spectroscopy [1] |
| Mechanical Entrapment | Physical confinement | Porous membrane (e.g., polycarbonate) | High | Medium (due to pore-induced deformation) | Medium | Imaging of poorly adherent cells in liquid [51] |
| Poly-L-Lysine Coating | Cationic polymer adhesion | Glass, Mica, Silicon | Very High | High (cellular stress, surface flattening) | Low | Fixed cells or robust imaging where viability is not a priority |
| Non-Immobilization (Adhesion-promoting substrate) | Native adhesion to engineered surface | Indium-Tin-Oxide (ITO)-coated glass | Low to Medium | Very Low | Very High | High-resolution nanomechanical mapping on living, native bacteria [5] |
| Covalent Linkage | Chemical cross-linking | Functionalized (e.g., APTES) surfaces | Very High | High (chemical alteration of surface) | Low | Single-molecule force spectroscopy on specific receptors |
The selection of an immobilization method is a trade-off. Gelatin coating offers a generally applicable, minimally invasive method that leverages the natural negative charge of bacterial cells [1]. In contrast, methods like poly-L-lysine provide strong adhesion but at the cost of potentially inducing cellular stress and surface deformation, leading to artefacts in nanomechanical property measurements [5] [52]. A recent advanced approach bypasses external immobilization altogether by using substrates like Indium-Tin-Oxide (ITO)-coated glass, which promotes sufficient adhesion for stable imaging due to its smooth and hydrophobic properties, thereby preserving the native state of the bacteria for real-time nanomechanical mapping [5].
Impact on Force Curves and Image Artefacts
The immobilization method directly influences the two primary data outputs of AFM: topographical images and force-distance curves.
Analysis of Force-Distance Curves
Force spectroscopy is highly sensitive to immobilization quality. An improperly immobilized cell may detach or move during the approach-retract cycle, producing force curves that reflect the cell-substrate bond instead of the intended tip-cell interaction.
Table 2: Impact of Immobilization on Force-Curve Parameters
| Force-Curve Feature | Well-Immobilized Cell | Poorly-Immobilized Cell |
|---|---|---|
| Approach Curve | Well-defined, reproducible. Can show a monotonic deflection or a "jump-in" due to attractive forces [19]. | Irregular, non-reproducible. May show multiple, ill-defined jump-in events. |
| Adhesion Force (Retraction) | Clear, single or multiple rupture events reflecting specific (e.g., ligand-receptor) or non-specific interactions [52]. | Very high, broad adhesion peak often indicating the detachment of the entire cell from the substrate. |
| Elastic Modulus (Young's Modulus) | Consistent, reliable values calculated from indentation curves using Sneddon or Hertz models [5]. | Inconsistent, erroneously high or low values due to cell movement or subsurface contributions. |
| Work of Adhesion | Quantifiable area under the retraction curve, representing the energy needed to separate tip and sample. | Unquantifiable or vastly overestimated due to cell displacement. |
For example, when measuring the interaction between E. coli and goethite, a well-immobilized cell will show characteristic jump-in events with attractive forces around 97 ± 34 pN, and adhesion forces of several nN, reflecting the true molecular interaction [19]. If the cell is poorly immobilized, the force curves will be dominated by the detachment of the bacterium from the gelatin or underlying mica.
Common Image Artefacts
Immobilization-induced artefacts can severely compromise image interpretation.
- "Trapping" Artefacts: Cells immobilized in porous membranes can appear flattened or deformed, obscuring true surface topography [51].
- "Spreading" Artefacts: Strong cationic coatings like poly-L-lysine can cause cells to spread and flatten on the surface, altering their native spherical or rod-like shape and leading to an overestimation of cell volume and contact area.
- "Carpet Effect": A thick or uneven immobilization layer (e.g., a lumpy gelatin coating) can create a soft, compliant "carpet" underneath the cell, which the AFM tip indents simultaneously with the cell. This leads to an underestimation of the cell's true Young's modulus [5] [52].
- Background Contamination: Residual chemicals from immobilization protocols (e.g., glutaraldehyde) can coat the substrate or cell surface, creating particulate or fibrous contaminants in the image that are mistaken for surface structures.
The following diagram illustrates the logical decision-making process for selecting and validating an immobilization method to minimize such artefacts.
Detailed Experimental Protocols
Protocol A: Immobilization on Gelatin-Coated Mica
This protocol is adapted from established methodologies for imaging bacterial cells in liquid environments [1].
4.1.1 Research Reagent Solutions
Table 3: Essential Materials for Gelatin-Coated Mica Protocol
| Item | Function/Description |
|---|---|
| Freshly Cleaved Mica Discs | Provides an atomically flat, negatively charged surface for coating. |
| Porcine Skin Gelatin | Forms a positively charged coating to electrostatically immobilize negatively charged bacterial cells. |
| Centrifuge | For pelleting and washing bacterial cells from growth medium. |
| Appropriate Liquid Medium | For resuspending bacteria and as imaging buffer to maintain physiological conditions. |
| AFM Liquid Cell | Allows imaging in a controlled liquid environment. |
4.1.2 Step-by-Step Procedure
- Gelatin Coating: Prepare a 0.1% (w/v) solution of porcine skin gelatin in deionized water. Heat gently to dissolve completely. Pipette 50-100 µL of the gelatin solution onto a freshly cleaved mica surface. Incubate for 30 minutes at room temperature.
- Rinse: After incubation, gently rinse the mica surface with copious amounts of deionized water to remove any unbound gelatin. Blot the edges dry with a clean tissue, ensuring the coated surface remains hydrated. Air dry completely.
- Bacterial Preparation: Grow the bacterial strain of interest to the mid-exponential growth phase. Pellet the cells by centrifugation (e.g., 4100 g for 10 minutes). Decant the growth medium and resuspend the pellet gently in deionized water or an appropriate imaging buffer. Repeat this wash step three times to remove residual media and extracellular polymers that might interfere with adhesion [19].
- Immobilization: Pipette 50 µL of the washed bacterial suspension onto the prepared, dry gelatin-coated mica. Allow the cells to adhere for 30 minutes in a humid chamber to prevent evaporation.
- Rinse and Assemble: Gently rinse the surface with imaging buffer to remove any non-adherent cells. Carefully place the mica into the AFM liquid cell, fill with the appropriate liquid medium, and proceed with imaging.
Protocol B: Direct Imaging on ITO-Coated Glass without Immobilization
This advanced protocol avoids chemical or mechanical immobilization, ideal for nanomechanical mapping [5].
4.2.1 Research Reagent Solutions
| Item | Function/Description |
|---|---|
| ITO-Coated Glass Substrates | Provides a smooth, hydrophobic surface that promotes native bacterial adhesion without chemical treatments. |
| Electrochemical Cell (EC Cell) | A specialized AFM liquid cell that accommodates the ITO substrate and allows for controlled imaging conditions. |
| Quantitative Imaging (QI) Mode AFM | A fast, force-mapping AFM mode that minimizes lateral forces on poorly immobilized samples. |
4.2.2 Step-by-Step Procedure
- Substrate Preparation: Use commercially available ITO-coated glass slides. Clean the ITO surface with solvents (e.g., ethanol) and plasma cleaning if available, to ensure a clean, uniform surface.
- Bacterial Preparation: As in Protocol A, harvest bacteria at the desired growth phase and wash gently to remove growth media.
- Sample Loading: Pipette a volume of the bacterial suspension (e.g., 500 µL) directly onto the ITO substrate seated in the electrochemical cell. Allow a short period (e.g., 10-15 minutes) for the cells to settle and adhere via native interactions.
- AFM Imaging: Fill the cell with the liquid culture medium. Employ a high-speed, force-mapping mode such as Quantitative Imaging (QI) mode. Use a soft cantilever (e.g., 0.3 N/m) and a large extension/retraction distance (e.g., 600 nm) to stably image the non-immobilized cells [5].
The following workflow diagram summarizes the key steps for both protocols.
Data Processing and Analysis
Post-acquisition data processing is essential for accurate interpretation. The free, open-source software Gwyddion is a powerful tool for this task [53] [54]. It supports a vast array of SPM data formats and provides essential processing functions.
- Leveling: Apply line-by-line leveling and plane subtraction to correct for sample tilt and scanner bow.
- Filtering: Use median or Gaussian filters to remove high-frequency noise, but apply cautiously to avoid obscuring real nanoscale features.
- Analysis: For force curves, use the software's tools to fit the retraction curve with appropriate models (e.g., Sneddon model for a conical tip) to calculate the Young's modulus, as demonstrated in studies of bacterial nanotubes [5]. For topography, use grain analysis and roughness parameters to quantify surface properties.
Correlating Immobilization Efficiency with Biophysical Data Quality
Within the broader context of developing robust atomic force microscopy (AFM) protocols for immobilizing bacterial cells, the choice of immobilization strategy is not merely a preparatory step but a critical determinant of data quality and biological relevance. AFM enables the investigation of bacterial surface structures and interaction forces at the nanoscale under physiological conditions [55]. However, its application in microbiology is challenging because bacterial cells must be firmly adhered to a substrate to prevent displacement by the AFM tip, without altering cell surface properties or viability [43]. This document details standardized protocols and quantitative comparisons of common immobilization methods, correlating their efficiency with the quality of the resulting biophysical data, to guide researchers in selecting the most appropriate technique for their specific biological questions.
Comparative Analysis of Immobilization Methods
An extensive comparative study evaluated multiple surface functionalization strategies to immobilize living bacteria for AFM imaging in liquid environments [43]. The success of each method was judged based on the strength of cell attachment (preventing detachment during scanning) and the preservation of cell viability and native surface properties.
Table 1: Quantitative Comparison of Bacterial Immobilization Methods for AFM
| Immobilization Method | Immobilization Strength | Impact on Cell Viability | Preservation of Surface Chemistry | Best Suited For |
|---|---|---|---|---|
| Physical Confinement (Microwells) | Moderate | High | High | Studies where chemical alteration of the cell surface must be avoided. |
| Physisorption (Positively Charged Surfaces) | Weak (cells often detach in PBS/MOPS) | High | High | Preliminary scans or with bacterial strains that have strong natural adhesion. |
| Covalent Binding | Strong | Moderate | Low (surface chemistry is altered) | Experiments requiring the highest immobilization strength, where surface properties are not the focus. |
| Mussel-Adhesive Proteins (e.g., Cell-Tak) | Very Strong | High | High | High-resolution imaging of live cells in physiological buffers; force spectroscopy. |
The most successful method identified was the use of mussel-adhesive proteins (e.g., from Mytilus edulis). This approach provided firm immobilization, did not affect cell viability, and offered a fast, reproducible, and generally applicable scheme [43]. Another study on Escherichia coli confirmed that immobilization on poly-L-lysine (PLL) in a diluted phosphate buffer allowed for stable imaging in nutrient media for extended periods, with preserved membrane integrity and even recorded cell division events [9].
Detailed Experimental Protocols
Immobilization with Mussel-Adhesive Proteins
This protocol is adapted from the method highlighted as highly effective for firm immobilization of living bacteria [43].
Key Reagent Solutions:
- Mussel-Adhesive Protein Solution: Commercially available Cell-Tak or a purified solution of Mytilus edulis polyphenolic proteins.
- Glass Substrates: Freshly cleaned mica or glass coverslips.
- Imaging Buffer: A physiologically compatible buffer such as PBS or MOPS.
Step-by-Step Procedure:
- Surface Coating: Apply a small volume (e.g., 20-50 µL) of the mussel-adhesive protein solution onto the center of a clean glass substrate.
- Air-Drying: Allow the coated substrate to air dry completely at room temperature.
- Rinsing: Gently rinse the coated surface with the imaging buffer to remove any unbound adhesive material.
- Cell Deposition: Apply a concentrated suspension of bacterial cells in the imaging buffer onto the coated, rinsed surface.
- Adsorption: Allow the cells to adsorb for 15-30 minutes at room temperature.
- Washing: Carefully rinse the substrate with imaging buffer to remove any non-immobilized cells.
- AFM Imaging: Immediately mount the substrate in the AFM liquid cell, submerge in the appropriate imaging buffer, and commence imaging.
Immobilization on Poly-L-Lysine for Live-Cell Dynamics
This protocol, derived from a study on E. coli, is optimized for imaging in nutrient media and observing dynamic processes [9].
Key Reagent Solutions:
- Poly-L-Lysine (PLL) Solution: 0.1% (w/v) aqueous solution.
- Immobilization Buffer: 0.01x Phosphate-Buffered Saline with Sucrose (PBS-S). The dilute buffer and sucrose help maintain osmotic balance with minimal interference from ions that can weaken electrostatic binding.
- Recovery/Nutrient Media: The desired growth media for the bacteria, e.g., LB Broth or Minimal Media (MM).
Step-by-Step Procedure:
- Surface Coating: Treat a clean glass substrate with the 0.1% PLL solution for 30 minutes.
- Rinsing and Drying: Rinse the substrate thoroughly with deionized water and allow it to air dry.
- Cell Preparation: Use bacterial cells from an overnight culture for a synchronized population.
- Immobilization Step: Resuspend the bacterial pellet in the 0.01x PBS-S buffer and deposit onto the PLL-coated surface. Allow cells to immobilize for 10-20 minutes.
- Buffer Exchange: Gently replace the PBS-S buffer with the recovery/nutrient media (e.g., LB broth). Let the sample equilibrate for at least 30 minutes to ensure cell viability and adaptation.
- AFM Imaging: Proceed with AFM imaging in the nutrient media.
The following workflow diagram illustrates the critical decision points and steps for preparing a robust bacterial sample for AFM imaging.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Bacterial Immobilization
| Reagent / Material | Function / Purpose | Key Considerations |
|---|---|---|
| Mussel-Adhesive Protein (e.g., Cell-Tak) | Provides a strong, biocompatible, and general-purpose adhesive surface for firm cell immobilization. | Highly effective for both Gram-positive and Gram-negative bacteria; preserves viability [43]. |
| Poly-L-Lysine (PLL) | A polycation that promotes cell adhesion via electrostatic interactions with negatively charged bacterial surfaces. | Can have antimicrobial properties; effectiveness is buffer-dependent; use diluted ionic strength for better results [9]. |
| Gelatin (Varying Bloom Strength) | Creates a porous, non-cytotoxic matrix for physical entrapment and adsorption of cells. | Different bloom strengths offer varying rigidity; may partially obstruct the cell surface [9]. |
| Dilute Phosphate Buffered Saline with Sucrose (0.01x PBS-S) | Facilitates electrostatic immobilization on PLL by minimizing charge shielding while maintaining osmolarity. | Critical for stable immobilization on PLL prior to switching to nutrient media [9]. |
| Physiological Imaging Buffers (e.g., MOPS, LB Broth) | Maintains cell viability and native state during AFM imaging. | The choice between simple buffer and rich media depends on the experimental goal (viability vs. activity). |
Correlating Method with Biophysical Data Quality
The immobilization method directly impacts the quality and type of biophysical data that can be reliably acquired.
- High-Resolution Topography: For imaging nanoscale structures like outer membrane vesicles or protein domains, mussel-adhesive proteins are superior. Their strong attachment prevents tip-induced cell displacement, which blurs images and creates artifacts [43].
- Single-Molecule Force Spectroscopy (SMFS): This technique measures specific interaction forces (e.g., receptor-ligand binding) and requires exceptionally stable immobilization. The strong, non-disruptive attachment from mussel-adhesive proteins is ideal, as it prevents whole-cell detachment during force curve acquisition and ensures that measured rupture forces are from the molecule of interest, not cell movement [55].
- Long-Term Live-Cell Dynamics: To monitor processes like cell division or membrane remodelling over time in nutrient media, the PLL-based protocol is highly effective. It balances sufficient adhesion with preserved cell viability and function, as evidenced by the recording of division events [9].
The following diagram summarizes the decision-making process for selecting an immobilization method based on the primary research objective.
Atomic Force Microscopy (AFM) has become an indispensable tool in microbiology for probing the nanoscale surface topography and mechanical properties of bacterial cells. However, a significant limitation of AFM is its inability to easily confirm the biological identity of structures or provide internal structural context. This limitation is effectively addressed through correlative microscopy approaches that integrate AFM with complementary techniques, primarily Scanning Electron Microscopy (SEM) and Optical Microscopy. The fusion of these techniques creates a powerful analytical framework where the high-resolution, real-time imaging capabilities of AFM under physiological conditions are validated and enriched by the extensive field of view and compositional analysis offered by SEM and optical methods.
The critical importance of these validation techniques is particularly evident when studying complex bacterial systems, such as biofilms and cellular nanostructures. For instance, recent research on bacterial nanotubes—membrane-derived filaments connecting bacterial cells—has demonstrated how AFM and SEM provide complementary structural information that would be incomplete with either technique alone [5] [56]. Similarly, studies of bacterial adhesion and biofilm formation benefit immensely from correlative approaches that contextualize nanoscale AFM measurements within larger spatial organizations visible through optical techniques [6]. This application note establishes detailed protocols for implementing these validation techniques specifically within the context of AFM studies on immobilized bacterial cells, with an emphasis on practical implementation for researchers in microbiology and drug development.
Technical Comparisons and Capabilities
Comparative Analysis of Microscopy Techniques
Table 1: Technical comparison of AFM, SEM, and Optical Microscopy for bacterial studies
| Characteristic | Atomic Force Microscopy (AFM) | Scanning Electron Microscopy (SEM) | Optical Microscopy |
|---|---|---|---|
| Resolution | Sub-nanometer to nanometer [5] | Nanometer range [56] | Diffraction-limited (~200 nm) [6] |
| Imaging Environment | Liquid, air, vacuum (physiological conditions possible) [9] [5] | High vacuum typically required [56] | Liquid, air (physiological conditions possible) |
| Sample Preparation | Minimal for live cells; may require immobilization [9] [5] | Extensive (fixation, dehydration, coating) [56] | Minimal to moderate (may require staining) |
| Information Type | Topography, mechanical properties, adhesion forces [5] [57] | Surface morphology, composition with EDX [56] | Morphology, fluorescence localization, dynamic processes |
| Field of View | Limited (typically <100 μm) [6] | Large area capability | Large area capability |
| Live Cell Imaging | Yes, in physiological buffers [9] [5] | No (except with specialized environmental SEM) | Yes, with phase contrast or fluorescence |
Information Synergy Between Techniques
The power of correlative microscopy lies in the complementary information provided by each technique, creating a comprehensive understanding of bacterial systems that transcends the capabilities of individual methods. AFM provides exceptional nanoscale resolution of surface structures under physiological conditions, enabling quantification of mechanical properties through force spectroscopy measurements [5] [57]. This is particularly valuable for assessing bacterial response to antimicrobial agents, where surface modifications and stiffness changes can be precisely quantified [56]. Additionally, AFM enables the visualization of delicate extracellular structures such as flagella and pili that are crucial for bacterial adhesion and biofilm formation [6].
SEM complements AFM by providing high-resolution imaging of bacterial surface ultrastructure with greater field of view and depth of field [56]. While typically requiring sample fixation and dehydration that precludes live cell imaging, SEM offers superior visualization of surface details and can be combined with energy-dispersive X-ray spectroscopy (EDX) for elemental analysis of bacterial surfaces or associated particles [56]. The correlation between AFM and SEM is particularly effective for validating nanoscale features observed with AFM, as the techniques provide different perspectives on similar structural attributes.
Optical microscopy, especially fluorescence-based techniques, provides critical contextual information about molecular specificity and dynamic processes through live-cell imaging [6]. While limited by diffraction in spatial resolution, optical microscopy enables researchers to identify specific bacterial components through fluorescent labeling and monitor temporal changes in bacterial behavior [6]. The integration of optical microscopy with AFM has been significantly advanced through the development of combined instruments that permit simultaneous data acquisition, bridging the gap between nanoscale topography and molecular localization [58].
Experimental Protocols
Sample Preparation for Correlative Imaging
Bacterial Immobilization Protocols
Successful correlative microscopy requires sample preparation strategies that accommodate the distinct requirements of each technique while maintaining structural integrity across imaging sessions. For AFM imaging, firm immobilization of bacterial cells is essential to prevent displacement by scanning forces. Multiple approaches have been validated for bacterial immobilization:
Gelatin Coating: Prepare 0.5% gelatin solutions (varying bloom strengths) in distilled water. Apply to substrate surface and allow to dry at room temperature. Bacterial suspension is then deposited on coated surface and allowed to adhere for 15-30 minutes before gentle rinsing with appropriate buffer [9].
Poly-L-Lysine (PLL) Coating: Use concentration of 0.1-0.01% PLL in distilled water. Apply to substrate for 5-10 minutes, rinse with distilled water, and air dry. Bacterial adhesion occurs within 10-20 minutes of application [9].
Physical Entrapment: For more challenging specimens, porous membrane filters can be used to physically trap bacteria against the substrate surface. This method is particularly useful for rod-shaped bacteria that may not adhere strongly to chemically coated surfaces [9].
ITO Coated Substrates: Indium-tin-oxide (ITO) coated glass substrates provide superior bacterial adhesion for liquid-phase AFM imaging without chemical immobilization. The hydrophobic properties and smooth surface of ITO facilitate stable cell adhesion, preserving native physiological states [5].
Substrate Selection for Multi-Technique Imaging
The choice of substrate is critical for successful correlative microscopy. The ideal substrate must accommodate the requirements of all imaging techniques in the workflow:
Glass Coverslips: Standard thickness (0.17 mm) for high-resolution optical microscopy. Compatible with AFM when properly coated and with SEM if conductive coating is applied.
Mica Sheets: Excellent for AFM due to atomically flat surface but problematic for optical microscopy due to opacity. Can be used with specially designed holders for correlative workflows.
SEM Stubs with Coverslip Mounting: Specialized holders that allow mounting of standard coverslips on SEM stubs, facilitating transfer between instruments without sample relocation.
FindER Grids: Patterned grid systems with coordinate registration marks that enable precise relocation of specific cells or regions between different microscopy platforms.
Workflow for Sequential AFM-SEM Correlation
Table 2: Protocol for correlated AFM-SEM analysis of bacterial cells
| Step | Procedure | Notes & Critical Parameters |
|---|---|---|
| 1. Sample Preparation | Immobilize bacteria on appropriate substrate using preferred method (chemical or physical). | Maintain bacterial viability if live imaging is required; ensure uniform distribution for statistical relevance |
| 2. Optical Survey | Acquire low-magnification optical images to map sample areas of interest. | Record coordinate positions; identify features for correlation; document using phase contrast or DIC |
| 3. AFM Imaging | Perform AFM analysis in desired mode (contact, tapping, quantitative imaging). | Acquire high-resolution topography and mechanical properties; note scan areas relative to optical reference points |
| 4. Fixation | Fix samples with 2.5% glutaraldehyde in buffer for 1-2 hours at 4°C. | Maintain structural integrity; use appropriate buffer for bacterial type; gradual dehydration recommended |
| 5. Dehydration | Ethanol series (30%, 50%, 70%, 90%, 100%) with 10-15 minutes per step. | Critical for SEM preparation; incomplete dehydration causes structural collapse in vacuum |
| 6. Drying | Critical point drying or hexamethyldisilazane (HMDS) treatment. | Preserves delicate structures better than air drying; minimizes deformation of extracellular features |
| 7. Sputter Coating | Apply thin conductive layer (5-15 nm gold/palladium) using sputter coater. | Required for conventional SEM imaging; minimizes charging effects; should be as thin as possible to preserve topology |
| 8. SEM Imaging | Acquire SEM images at comparable magnifications to AFM data. | Use landmarks for precise correlation; image same regions analyzed by AFM |
| 9. Data Correlation | Overlay and compare AFM and SEM datasets using software alignment tools. | Use distinctive features as registration points; account for dimensional changes from processing |
Simultaneous AFM-Optical Microscopy Integration
The development of integrated AFM-optical systems has enabled simultaneous data acquisition, providing perfect temporal registration between nanoscale topography and fluorescence information. Implementation requires:
System Configuration: Combined AFM-optical instruments with precisely aligned optical paths that allow simultaneous imaging without interference between detection systems.
Sample Compatibility: Use of coverslip-bottomed dishes compatible with high numerical aperture objectives for optimal optical resolution.
Fluorescent Labeling: Application of appropriate fluorescent markers (membrane stains, fluorescent proteins, specific molecular labels) that do not interfere with AFM tip-sample interactions.
Synchronized Data Acquisition: Coordination of AFM scanning parameters with optical image acquisition rates to ensure temporal alignment of datasets.
Data Fusion Software: Utilization of software platforms capable of overlaying and analyzing correlated AFM and optical data with precise pixel registration.
Recent advances in large-area automated AFM have significantly enhanced correlative approaches by enabling the acquisition of high-resolution AFM data over millimeter-scale areas, effectively bridging the scale gap between single-cell AFM analysis and population-level optical observations [6]. This approach, aided by machine learning for image stitching and analysis, has revealed previously inaccessible structural details in bacterial biofilms, including preferred cellular orientation patterns and flagellar coordination during surface attachment [6].
Research Reagent Solutions
Table 3: Essential research reagents for correlative microscopy of bacterial cells
| Reagent/Category | Specific Examples | Function in Correlative Microscopy |
|---|---|---|
| Immobilization Agents | Gelatin (varying bloom strength), Poly-L-Lysine (PLL), APTES silane | Secure bacterial cells to substrates for AFM scanning; maintain viability for live-cell imaging [9] |
| Fixatives | Glutaraldehyde (2.5%), Formaldehyde (4%), Paraformaldehyde | Preserve cellular structure for SEM processing; maintain structural integrity between techniques [56] |
| Conductive Coatings | Gold/Palladium (5-15 nm), Carbon (10-20 nm) | Prevent charging in SEM; should be applied after AFM analysis to preserve native surface properties [56] |
| Fluorescent Labels | SYTO dyes, FM lipophilic stains, GFP transfection, Immunofluorescence tags | Enable optical localization of specific structures; verify biological identity in correlation [6] |
| Buffers & Media | Phosphate Buffered Saline (PBS), LB broth, M9 minimal media | Maintain physiological conditions during live-cell AFM; compatibility with immobilization chemistry [9] |
| Dehydration Reagents | Ethanol series (30-100%), Hexamethyldisilazane (HMDS) | Prepare samples for SEM imaging; preserve delicate structures with minimal distortion [56] |
| Specialized Substrates | ITO-coated glass, FindER grids, Patterned substrates | Facilitate sample relocation and transfer between instruments; provide coordinates for correlation |
Data Interpretation and Validation Metrics
Quantitative Correlation Approaches
Successful validation through correlative microscopy requires rigorous quantitative approaches to establish correspondence between datasets from different techniques. Key validation metrics include:
Dimensional Consistency: Comparison of feature dimensions (cell length, width, appendage diameters) measured across techniques. Agreement within 10-15% is typically achievable, accounting for preparation-induced variations [56]. For example, E. coli cells should measure approximately 2 μm in length and 0.25-1.0 μm in diameter across AFM, SEM, and optical measurements [56].
Morphological Correspondence: Quantitative shape descriptors (aspect ratio, surface roughness, contour parameters) should show consistent patterns across modalities. Distinctive morphological features induced by treatments, such as the elongation observed in ethanol-adapted E. coli, should be evident in both AFM and SEM datasets [56].
Spatial Distribution Analysis: For biofilm studies, spatial statistics including nearest-neighbor distances, clustering parameters, and orientation order should demonstrate correlation between AFM and optical datasets [6]. Recent studies of Pantoea sp. YR343 revealed a distinctive honeycomb pattern during biofilm assembly that was quantifiable across resolution scales [6].
Mechanical-Structural Correlation: Linking nanomechanical properties measured by AFM (elastic modulus, adhesion forces) with structural features visible in SEM and optical microscopy. For instance, bacterial nanotubes show lower Young's modulus compared to cell bodies, suggesting flexibility that facilitates intercellular communication [5].
Addressing Technique-Specific Artifacts
A critical aspect of validation is recognizing and accounting for technique-specific artifacts that may influence interpretation:
AFM Artifacts: Tip convolution effects that exaggerate lateral dimensions, force-induced deformation of soft samples, and scanner nonlinearities that distort geometry. These can be minimized through appropriate probe selection, optimized imaging forces, and regular scanner calibration.
SEM Artifacts: Shrinkage and collapse from dehydration procedures, charging effects in non-conductive regions, and surface detail masking by excessive metal coating. These are addressed through optimized critical point drying, appropriate conductive coating thickness, and charge reduction strategies.
Optical Artifacts: Diffraction-limited resolution boundaries, photobleaching of fluorescent markers, and spherical aberration in thick samples. These are mitigated through super-resolution techniques when needed, optimized illumination, and correction collar adjustments.
Advanced Applications and Future Directions
Emerging Applications in Bacterial Cell Research
Correlative AFM-SEM-optical approaches are enabling groundbreaking applications in microbiology and antimicrobial development:
Antibiotic Mechanism Studies: Investigating nanoscale structural modifications in bacterial membranes and cell walls following antibiotic exposure, correlating morphological changes with mechanical property alterations [56]. AFM reveals subtle surface modifications and stiffness changes, while SEM provides comprehensive ultrastructural context, and fluorescence microscopy localizes specific cellular targets.
Bacterial Nanotube Characterization: Visualization and mechanical analysis of intercellular connections that mediate nutrient exchange and communication in bacterial communities [5]. The flexible nature of these structures (evidenced by lower Young's modulus) combined with their membrane-derived composition creates a comprehensive understanding of their function in horizontal gene transfer, particularly relevant for antibiotic resistance dissemination.
Biofilm Architecture Analysis: Multi-scale investigation of biofilm development from initial attachment to mature community formation [6]. Large-area AFM approaches now enable high-resolution mapping over millimeter scales, revealing organizational patterns like the honeycomb structure observed in Pantoea sp. YR343, while fluorescence microscopy identifies distinct subpopulations within the biofilm matrix.
Antimicrobial Surface Evaluation: Assessing the interaction between bacterial pathogens and engineered surfaces designed to prevent adhesion and biofilm formation [59] [6]. Correlative approaches quantify adhesion forces through AFM while simultaneously visualizing attachment patterns through optical microscopy and surface coverage through SEM.
Technological Innovations Enhancing Correlative Approaches
The field of correlative microscopy continues to evolve through several technological advancements:
Large-Area Automated AFM: Systems capable of automated acquisition over millimeter-scale areas with minimal user intervention, effectively bridging the resolution gap between single-cell AFM and population-level optical microscopy [6]. These systems incorporate machine learning for intelligent region selection, image stitching, and feature analysis.
Environmental Control Systems: Specialized sample chambers that maintain physiological conditions during correlated imaging, including temperature regulation, gas control, and perfusion capabilities for nutrient delivery [26]. Hermetically sealed chambers have been developed specifically for imaging pathogenic microorganisms under biosafety requirements [26].
Artificial Intelligence Integration: Machine learning algorithms that enhance every aspect of correlative microscopy, from automated scan region selection based on optical surveys to distortion correction and feature recognition across modalities [58] [6]. AI-driven approaches are particularly valuable for analyzing the vast datasets generated by large-area AFM and identifying subtle correlations between structural, mechanical, and compositional parameters.
Standardized Correlation Frameworks: Emerging software platforms and sample holder systems that facilitate precise relocation and data fusion between different microscopy platforms. These include coordinate registration systems, multimodal fiducial markers, and standardized data formats that enable seamless correlation between commercial instruments from different manufacturers.
The continued development and application of these correlative validation techniques will undoubtedly expand our understanding of bacterial physiology, pathogenesis, and response to antimicrobial agents, providing critical insights for drug development and infectious disease management.
Within the broader scope of developing robust Atomic Force Microscopy (AFM) protocols for bacterial immobilization, this case study examines a critical methodological variable: how cell immobilization influences the quantitative assessment of lipopolysaccharide (LPS)-mediated adhesion heterogeneity. The outer membrane of Gram-negative bacteria, with LPS as a primary component, is a key determinant of cellular mechanics and adhesion [21]. A foundational thesis in this field posits that the method of surface immobilization must preserve the native state of these structures to obtain accurate, physiologically relevant biophysical measurements. This application note details a controlled investigation into how a specific immobilization protocol impacts the observed heterogeneity in adhesion forces and elasticity within a clonal population of Escherichia coli.
Background
Lipopolysaccharide (LPS) and Phenotypic Heterogeneity
The outer membrane of Gram-negative bacteria is a complex structure wherein Lipopolysaccharides (LPS) play a critical role in determining surface properties. LPS molecules contribute significantly to the structural and chemical diversity of the cell envelope, which in turn governs bacterial adhesion and cell elasticity [21]. Even within a genetically identical, clonal population, individual bacterial cells can exhibit marked phenotypic heterogeneity. This diversity manifests as subpopulations of cells with varying biophysical properties, such as adhesion strength and rigidity [21] [60]. Such heterogeneity is a strategic advantage, enhancing the population's ability to colonize diverse surfaces and survive environmental stressors [21].
Atomic Force Microscopy (AFM) in Single-Cell Biophysics
AFM has emerged as a powerful tool in microbiology for its ability to probe the surface of biological samples at the nanoscale. Its key advantages include:
- High-resolution imaging and force measurement under physiological conditions [5] [61].
- Quantitative mapping of nanomechanical properties like elasticity (Young's modulus) and adhesion forces [5] [21].
- Single-cell analysis, enabling the detection of phenotypic heterogeneity that is often masked in bulk, averaged measurements [21] [60].
A critical, yet often overlooked, aspect of AFM studies on bacterial cells is the method of cell immobilization on a substrate. The immobilization technique must be robust enough to hold the cell stationary during scanning but non-perturbative to avoid altering the very surface properties under investigation. Studies have highlighted the development of protocols that avoid aggressive chemical or mechanical entrapment to prevent inducing stressful conditions and altering bacterial cell physiology [5].
Experimental Protocol
Bacterial Strain and Culture Conditions
- Bacterial Strain: Escherichia coli ATCC 25922 [21].
- Culture Medium: Luria-Bertani (LB) broth.
- Growth Conditions: Incubate for 24 hours at 37°C with shaking at 150 rpm [21].
LPS Removal via EDTA Treatment
- Harvesting: Centrifuge bacterial culture at 2151 × g for 5 minutes at 24°C. Wash cell pellets with Milli-Q water.
- EDTA Treatment: Resuspend cells in a 100 mM EDTA solution (pH 8.0).
- Incubation: Incubate the suspension at 37°C for 30 minutes with gentle shaking (20 rpm).
- Washing: Recentrifuge, wash twice with Milli-Q water, and resuspend in 0.01 M phosphate buffer (pH 7.0) for analysis [21].
- Validation: Confirm via AFM imaging that EDTA treatment did not cause visible membrane rupture or lysis, preserving cell viability and normal cylindrical shape [21].
Critical Step: Bacterial Immobilization for AFM
The following protocol is adapted for studying E. coli and is identified as a key variable in this case study.
- Substrate Preparation: Use gelatin-coated glass surfaces.
- Immobilization Procedure:
- Centrifuge bacterial suspension at 2151 × g for 5 minutes at 24°C.
- Wash cells twice with Milli-Q water.
- Adjust suspension to 10⁶ CFU/ml.
- Deposit the suspension on gelatin-coated slides and allow to adhere for 30 minutes [21].
AFM Imaging and Force Spectroscopy
- Instrumentation: A suitably calibrated Atomic Force Microscope.
- Mode: Force spectroscopy with a colloidal probe (to assess whole-cell properties rather than local, nanoscale diversity) [21].
- Environment: Perform measurements in a liquid cell using phosphate buffer.
- Data Acquisition: Acquire force-distance curves on multiple cells (a sufficiently large sample size is crucial for heterogeneity analysis) for both untreated and EDTA-treated populations.
- Data Analysis:
- Adhesion Force: Extract from the retraction force curves.
- Elasticity (Young's Modulus): Fit approach curves using an appropriate contact mechanics model (e.g., Sneddon model) [5].
- Heterogeneity Quantification: Analyze data at the single-cell level and calculate a heterogeneity index based on the variability of measurements across the population [21] [60].
Results and Data Analysis
The experimental results are summarized in the following tables, highlighting the quantitative impact of LPS removal and the role of immobilization.
Table 1: Single-Cell AFM Force Spectroscopy Data Summary
| Parameter | Untreated Cells (with LPS) | EDTA-Treated Cells (LPS Removed) |
|---|---|---|
| Average Adhesion Force | Higher | Substantially Diminished [21] |
| Average Cell Elasticity (Young's Modulus) | Higher and more variable | Markedly Reduced [21] |
| Surface Topography | Structurally diverse, rough | Smoother and featureless [21] |
| Presence of Strongly Adherent/Stiff Subpopulation | Yes | No longer observed [21] |
Table 2: Quantification of Population Heterogeneity
| Analysis Level | Heterogeneity Index (Untreated) | Heterogeneity Index (EDTA-Treated) | Implication |
|---|---|---|---|
| Cell-to-Cell (within population) | High | Markedly Reduced [21] | LPS is a key driver of biophysical heterogeneity. A proper immobilization protocol preserves this diversity. |
The following workflow diagram synthesizes the experimental procedure and the core findings of this case study.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents
| Item | Function/Description | Relevance in Protocol |
|---|---|---|
| Gelatin-Coated Glass Slides | Substrate for bacterial immobilization. Provides a non-perturbative, adhesive surface that secures cells for AFM scanning without harsh chemicals. | Critical for preserving native cell surface properties and enabling reliable single-cell force measurements [21]. |
| Colloidal AFM Probe | A spherical tip attached to the AFM cantilever. Measures interaction forces over the entire cell surface, providing averaged mechanical properties for single-cell analysis. | Essential for quantifying whole-cell adhesion and elasticity, minimizing variability from ultra-local surface probes [21]. |
| EDTA (Ethylenediaminetetraacetic acid) | A chelating agent that selectively removes divalent cations, destabilizing the outer membrane and releasing lipopolysaccharides (LPS). | Key reagent for experimentally modulating the primary variable (LPS) to study its role in adhesion and heterogeneity [21]. |
| Indium-Tin-Oxide (ITO) Coated Substrate | An alternative substrate with a smooth, hydrophobic surface that facilitates bacterial adhesion. | Recommended in other AFM studies for stable imaging of living bacteria in liquid without any immobilization agents, serving as a benchmark for minimal-perturbation preparation [5]. |
Discussion
Interpretation of Findings
The results demonstrate that the gelatin-based immobilization protocol successfully preserved the phenotypic heterogeneity inherent to the clonal E. coli population. The observed high cell-to-cell variability in adhesion and elasticity among untreated cells [21] is consistent with the expected structural and chemical diversity conferred by LPS in the outer membrane. The significant reduction of this heterogeneity following EDTA treatment directly confirms LPS as a primary determinant of biophysical diversity [21]. Furthermore, the protocol was sensitive enough to detect the elimination of a distinct subpopulation of strongly adherent and stiff cells post-treatment. This underscores the critical importance of a non-disruptive immobilization method; an aggressive protocol could have artificially homogenized the surface properties of the untreated cells, thereby masking the true biological effect of LPS removal.
Broader Implications for AFM Protocol Development
This case study provides a concrete example of how immobilization is not merely a preparatory step but an integral part of the measurement itself. It validates a specific protocol for studies aiming to quantify genuine single-cell heterogeneity in Gram-negative bacteria. The findings align with the broader thesis that AFM protocols must be meticulously designed to minimize external stress on bacterial cells. As highlighted in other research, avoiding chemical or mechanical entrapment prevents alterations to bacterial cell physiology that could bias experimental outcomes [5]. For future work, comparing data from gelatin-immobilized cells with data from cells adhering natively to optimized substrates like ITO-coated glass [5] could further refine best practices for immobilization in bacterial AFM research.
Atomic force microscopy (AFM) has emerged as a powerful tool in microbiology, enabling the high-resolution imaging of microbial surfaces and the quantification of their biophysical properties under physiological conditions [48] [62]. A critical prerequisite for successful AFM analysis is the effective immobilization of bacterial cells on a substrate, as the scanning forces can otherwise displace cells during imaging [1]. The immobilization strategy must be tailored to the specific research objectives, whether they involve high-resolution topographic imaging, single-cell force spectroscopy, or the assessment of cell-surface interactions. This application note provides a structured decision matrix and detailed protocols for selecting the appropriate bacterial immobilization method based on defined experimental goals, framed within the broader context of AFM protocol development for bacterial cell research.
The Scientist's Toolkit: Essential Research Reagents for Bacterial Immobilization
The following table details key reagents and materials commonly used for immobilizing bacterial cells for AFM studies, along with their primary functions.
Table 1: Key Research Reagent Solutions for Bacterial Immobilization
| Reagent/Material | Function in Immobilization | Key Considerations |
|---|---|---|
| Gelatin (Porcine) | Creates a positively charged coating on negatively charged mica to electrostatically immobilize bacterial cells [1]. | Generally applicable for imaging microbial cells in liquid; immobilization is influenced by suspension buffer and bacterial surface characteristics [1]. |
| APTES ((3-Aminopropyl)triethoxysilane) | Functionalizes surfaces with amino groups for covalent attachment or enhanced electrostatic interaction with cells. | Used in creating chemically defined surfaces; charge can be modulated to control adhesion. |
| PFOTS (Perfluorooctyltrichlorosilane) | Creates a hydrophobic surface to study bacterial attachment and biofilm assembly on specific interfaces [6]. | Useful for studying the effect of surface properties on initial bacterial adhesion and biofilm formation. |
| Poly-L-Lysine | Provides a positively charged coating on glass or mica to promote cell adhesion via electrostatic interactions. | A common and easy-to-use adhesion promoter; potential for multiple, non-specific interactions. |
| Indium-Tin-Oxide (ITO) coated glass | Provides a smooth, hydrophobic substrate that facilitates bacterial adhesion for AFM imaging without chemical immobilization [5]. | Enables imaging of living, native bacteria in liquid without aggressive external immobilization protocols [5]. |
| Porous Membrane Filters | Used for mechanical entrapment of cells by filtration. | Simple and fast; may apply stress to cells and is not suitable for all force measurements. |
Decision Matrix for Immobilization Method Selection
The choice of immobilization method should be guided by the primary research objective. The following matrix aligns common AFM experimental goals with recommended immobilization strategies.
Table 2: Decision Matrix for Selecting an Immobilization Method Based on Research Objectives
| Research Objective | Recommended Immobilization Method | Key Advantages | Limitations & Considerations |
|---|---|---|---|
| High-Resolution Topography of Live Cells | Gelatin-coated mica [1] | Preserves native surface structure; allows imaging in liquid under physiological conditions; generally applicable [1]. | Immobilization efficiency depends on bacterial surface charge and suspension buffer [1]. |
| Single-Cell Force Spectroscopy (Mechanics/Adhesion) | Gelatin-coated mica or ITO-coated glass without immobilization [21] [5] | Secure immobilization for force application; ITO method avoids potential softening from chemical coatings [5]. | Gelatin layer may influence mechanical property measurements; validation of substrate effect is recommended. |
| Study of Cell-Surface Interactions & Biofilm Initiation | Functionalized surfaces (e.g., PFOTS, APTES) [6] | Allows investigation of how specific surface properties (hydrophobicity, charge) influence adhesion and early biofilm assembly [6]. | Requires preparation of specialized surfaces; may not be suitable for all bacterial strains. |
| Imaging of Intercellular Structures (e.g., Nanotubes) | ITO-coated glass without immobilization [5] | Avoids any chemical immobilization that could disrupt delicate, native intercellular connections [5]. | Requires a substrate that promotes natural adhesion; may not work for all bacterial species. |
| Rapid Screening of Bacterial Adhesion Phenotypes | Mechanical entrapment (e.g., porous membranes) | Fast and simple for preparing multiple samples. | Can be too harsh for some cells and may not be suitable for quantitative nanomechanical mapping. |
Detailed Experimental Protocols
Protocol 1: Immobilization on Gelatin-Coated Mica for High-Resolution Imaging
This protocol, adapted from established methodologies, is ideal for high-resolution topographic imaging and single-cell force spectroscopy of living bacteria in liquid [1].
Workflow Overview:
Materials:
- Freshly cleaved mica discs (e.g., 10 mm diameter)
- Porcine skin gelatin (Type A)
- Milli-Q water
- Bacterial culture in mid-exponential growth phase
- Appropriate imaging buffer (e.g., phosphate-buffered saline (PBS) or a defined minimal medium)
- Centrifuge
- AFM with a liquid cell
Step-by-Step Procedure:
- Gelatin Coating Solution: Prepare a 0.1% (w/v) solution of porcine skin gelatin in Milli-Q water. Warm the mixture gently to dissolve the gelatin completely.
- Substrate Coating: Pipette 50-100 µL of the 0.1% gelatin solution onto the surface of a freshly cleaved mica disc. Incubate for 5 minutes at room temperature.
- Rinsing and Drying: Carefully aspirate the excess gelatin solution and rinse the coated mica surface gently with Milli-Q water to remove any unbound gelatin. Allow the substrate to air dry completely.
- Bacterial Sample Preparation: Harvest bacterial cells by centrifuging a liquid culture at 2151 × g for 5 minutes. Wash the cell pellet twice with Milli-Q water to remove growth medium components that could interfere with adhesion [21]. Resuspend the final pellet in an appropriate imaging buffer or Milli-Q water and adjust the cell density to approximately 10^6 CFU/mL.
- Cell Immobilization: Apply 50-100 µL of the bacterial suspension onto the center of the dry, gelatin-coated mica surface. Allow the cells to settle and adhere for 30 minutes in a humidified chamber to prevent evaporation.
- Final Rinse and Mounting: Tilt the substrate and gently rinse with 2-3 mL of imaging buffer to remove any non-adherent cells. The goal is to achieve a monolayer of well-spaced, immobilized cells. Carefully mount the sample into the AFM liquid cell, introduce the imaging buffer, and commence AFM imaging.
Protocol 2: Immobilization on ITO-Coated Glass for Imaging Native Structures
This protocol is designed for imaging living bacteria in their native state, preserving delicate structures like intercellular nanotubes, without chemical fixation or coating [5].
Workflow Overview:
Materials:
- ITO-coated glass substrates
- Bacterial culture in exponential growth phase
- Fresh growth medium
- AFM with a liquid cell (preferably an electrochemical cell)
Step-by-Step Procedure:
- Substrate Preparation: Clean ITO-coated glass substrates by sonication in ethanol and then in Milli-Q water, followed by plasma cleaning or UV-ozone treatment to ensure a clean, hydrophobic surface.
- Bacterial Adhesion: Pipette 500 µL of the bacterial culture, directly from the exponential growth phase, onto the ITO substrate placed in the AFM liquid cell [5].
- Incubation: Allow the bacteria to adhere to the substrate for 15-30 minutes under quiescent conditions. The hydrophobic properties of ITO facilitate this natural adhesion [5].
- Rinsing: Gently perfuse the liquid cell with 2-3 mL of fresh, pre-warmed growth medium to remove planktonic, non-adherent cells. Avoid disturbing the adhered cells with rapid fluid flow.
- AFM Imaging: Proceed with AFM imaging in the liquid culture medium using gentle, high-speed quantitative imaging modes (e.g., JPK's QI mode or Bruker's PeakForce Tapping) to minimize lateral forces on the unsecured cells [5].
Data Analysis and Interpretation
Validating Immobilization Success
Successful immobilization is indicated by the absence of cell movement during consecutive AFM scans. Before data acquisition, perform a quick scan over a large area (e.g., 20 × 20 µm) and then zoom in on a region of interest. Stable cells will remain in the same position and maintain their structural integrity throughout the imaging process. Cells that are poorly immobilized will be pushed by the AFM tip, resulting in smeared or blurred images.
Correlating Biophysical Properties with Biological Function
AFM provides quantitative data that can be linked to biological states. For instance:
- Cell Elasticity (Young's Modulus): Softer cells may indicate a response to environmental stress or the action of antimicrobial agents. Treatment with EDTA to remove LPS in E. coli has been shown to significantly reduce cell elasticity, homogenizing the population [21].
- Adhesion Forces: Mapping adhesion forces can reveal heterogeneity within a clonal population. The presence of highly adherent subpopulations, often linked to specific surface structures like LPS, can be identified through single-cell force spectroscopy [21].
Troubleshooting Common Immobilization Issues
Table 3: Troubleshooting Guide for Bacterial Immobilization
| Problem | Potential Cause | Solution |
|---|---|---|
| Cells are displaced during scanning | Insufficient adhesion force; excessive scanning force. | Optimize incubation time and buffer ionic strength on gelatin-coated mica [1]; use a softer cantilever and reduce the applied force setpoint. |
| Low number of immobilized cells | Bacterial surface charge repels substrate; suspension buffer interferes. | Ensure bacterial wash steps are thorough to remove media [21]; try a different immobilization substrate (e.g., switch from gelatin to poly-L-lysine). |
| Cells appear deformed or lysed | Toxic substrate coating; excessive drying during preparation. | Use a different, more biocompatible coating (e.g., gelatin); ensure the sample is kept hydrated from the immobilization rinse onward. |
| High non-specific background adhesion | Contaminated substrates or buffers. | Implement stricter cleaning protocols for substrates and use filtered buffers. |
Conclusion
The choice of bacterial immobilization protocol is not merely a preliminary step but a decisive factor that directly impacts the quality, reliability, and biological relevance of AFM data. This guide synthesizes key takeaways, demonstrating that while mechanical trapping often best preserves native surface properties, the optimal method depends on the specific application, whether it's high-resolution imaging, single-cell force spectroscopy, or large-area biofilm analysis. Standardizing and carefully selecting immobilization strategies is paramount for generating reproducible results in fundamental research and applied fields. Future directions will likely involve the increased use of automated, high-throughput methods like large-area AFM and FluidFM, further integrating machine learning for analysis. These advancements will deepen our understanding of bacterial adhesion mechanics, accelerating the development of novel anti-fouling surfaces and therapeutic agents in the pharmaceutical and biomedical industries.
References
- 1. Bacterial immobilization for imaging by atomic force ... [https://impact.ornl.gov/en/publications/bacterial-immobilization-for-imaging-by-atomic-force-microscopy]
- 2. Bacterial Immobilization for Imaging by Atomic Force ... [https://www.jove.com/t/2880/bacterial-immobilization-for-imaging-by-atomic-force-microscopy]
- 3. Method for immobilization of living and synthetic cells ... [https://www.nature.com/articles/s41598-018-32166-y]
- 4. Do Immobilization Methods Affect Force Spectroscopy ... [https://www.taylorfrancis.com/books/9788743804024/chapters/10.1007/978-3-031-17457-5_4]
- 5. Atomic Force Microsocopy: Key Unconventional Approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11603319/]
- 6. Analysis of biofilm assembly by large area automated AFM [https://www.nature.com/articles/s41522-025-00704-y]
- 7. Insights into the interfacial dynamics and interaction ... [https://link.springer.com/article/10.1007/s42773-025-00444-4]
- 8. How Microbes Use Force To Control Adhesion - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7253613/]
- 9. Immobilizing live Escherichia coli for AFM studies of ... [https://www.sciencedirect.com/science/article/abs/pii/S0304399113002945]
- 10. A modular atomic force microscopy approach reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6591122/]
- 11. An Overview of Cell Adhesion [https://www.cytosurge.com/applications/single-cell-adhesion]
- 12. Atomic force microscopy – looking at mechanosensors on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3516434/]
- 13. Immobilisation of living bacteria for AFM imaging under physiological conditions - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0304399110001919]
- 14. Bacterial Immobilization for Imaging by Atomic Force Microscopy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3211126/]
- 15. Bacterial immobilization for imaging by atomic force microscopy - PubMed [https://pubmed.ncbi.nlm.nih.gov/21860374/]
- 16. Immobilizing live Escherichia coli for AFM studies of surface dynamics - PubMed [https://pubmed.ncbi.nlm.nih.gov/24286980/]
- 17. Correlative atomic force microscopy quantitative imaging ... [https://www.nature.com/articles/s41598-018-26433-1]
- 18. A novel protocol to prepare cell probes for the quantification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6999235/]
- 19. Atomic force microscopy measurements of bacterial ... [https://www.nature.com/articles/srep16857]
- 20. Atomic Force Microscopy of Biofilms—Imaging, Interactions ... [https://www.intechopen.com/chapters/50739]
- 21. Single-cell analysis of lipopolysaccharide-mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535489/]
- 22. Microbial adhesion and ultrastructure from the single-molecule to the single-cell levels by Atomic Force Microscopy - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7389263/]
- 23. Force-controlled manipulation of single cells: from AFM to FluidFM - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0167779914000857]
- 24. Biochemical stimulation of immune cells and measurement of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6565469/]
- 25. Poly-l-lysine/poly-l-glutamic acid-based layer-by-layer self- ... [https://www.sciencedirect.com/science/article/abs/pii/S1226086X18305963]
- 26. A hermetically closed sample chamber enables time-lapse ... [https://pubs.rsc.org/en/content/articlehtml/2025/na/d4na01053a]
- 27. Chemical fixation creates nanoscale clusters on the cell ... [https://www.nature.com/articles/s42003-022-03437-2]
- 28. Quantification of protein mobility and associated reshuffling ... [https://www.nature.com/articles/s41598-018-36112-w]
- 29. Optimization of fixation methods for observation of bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3181414/]
- 30. Glutaraldehyde – A Subtle Tool in the Investigation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6527840/]
- 31. Factors Influencing Chemical Fixation, Formaldehyde & ... [https://www.leicabiosystems.com/us/knowledge-pathway/fixation-and-fixatives-2-factors-influencing-chemical-fixation-formaldehyde-and-glutaraldehyde/]
- 32. Single-cell manipulation with FluidFM [https://www.cytosurge.com/applications/single-cell-manipulation]
- 33. AFM and FluidFM Technologies: Recent Applications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6057332/]
- 34. An Overview of Mechanobiology with FluidFM [https://www.cytosurge.com/applications/mechanobiology]
- 35. Electrokinetics in Micro-channeled Cantilevers [https://www.nature.com/articles/s41598-019-56716-0]
- 36. Profiling to Probing: Atomic force microscopy ... [https://www.sciencedirect.com/science/article/pii/S1742706123004580]
- 37. applications of atomic force microscopy in disease [https://link.springer.com/article/10.1007/s12551-025-01306-w]
- 38. Bacterial Immobilization for Imaging by Atomic Force ... [https://www.jove.com/v/2880/bacterial-immobilization-for-imaging-by-atomic-force-microscopy]
- 39. Comparison of Atomic Force Microscopy Interaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC520872/]
- 40. Tip Functionalization Approaches for Molecular Recognition [https://www.azonano.com/article.aspx?ArticleID=2801]
- 41. A modular atomic force microscopy approach reveals ... [https://www.nature.com/articles/s41396-019-0404-1]
- 42. Scratching the Surface: Bacterial Cell Envelopes at the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7042696/]
- 43. Immobilisation of living bacteria for AFM imaging under ... [https://pubmed.ncbi.nlm.nih.gov/20619542/]
- 44. Atomic Force Microscopy Protocol for Measurement of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4854888/]
- 45. Immobilizing live Escherichia coli for AFM studies of surface ... [https://inis.iaea.org/records/5xj7z-ekm37]
- 46. How To Tackle Artifacts In Atomic Force Microscopy Imaging [https://eureka.patsnap.com/report-how-to-tackle-artifacts-in-atomic-force-microscopy-imaging-solutions]
- 47. Overcoming Challenges and Limitations Regarding the ... [https://www.mdpi.com/2079-6439/11/10/83]
- 48. Application of atomic force microscopy to microbial surfaces [https://www.sciencedirect.com/science/article/abs/pii/S0968432899001067]
- 49. Adhesion Force Measurements Using an Atomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4130233/]
- 50. 9.2: Atomic Force Microscopy (AFM) [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Physical_Methods_in_Chemistry_and_Nano_Science_(Barron)/09%3A_Surface_Morphology_and_Structure/9.02%3A_Atomic_Force_Microscopy_(AFM)]
- 51. Atomic Force Microscopy in Microbiology: New Structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4120197/]
- 52. Atomic Force Microscopy (AFM) As a Surface Mapping ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2020.517165/full]
- 53. Gwyddion – Free SPM (AFM, SNOM/NSOM, STM, MFM ... [https://gwyddion.net/]
- 54. Free AFM Image Processing Software [https://www.afmworkshop.com/newsletter/free-afm-image-processing-software?srsltid=AfmBOopeegQ-u63hGz7bhs8ATo_sCMIhXHWVKmjknGd3GPT0t--JMchJ]
- 55. The biophysics of bacterial infections: Adhesion events in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7898176/]
- 56. Atomic force microscopy study of morphological ... [https://www.sciencedirect.com/science/article/abs/pii/S0304399116301565]
- 57. Atomic Force Microscopy (AFM) nanomechanical ... [https://www.sciencedirect.com/science/article/pii/S2405665025000125]
- 58. What does 2025 have in store for AFM? - NuNano [https://www.nunano.com/blog/2025/1/26/what-does-2025-have-in-store-for-afm]
- 59. Micro- and nano-scale adhesion of oral bacteria to ... [https://www.sciencedirect.com/science/article/pii/S1882761625000055]
- 60. Atomic force microscopy investigations of heterogeneities ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3172993/]
- 61. unveiling foodborne viruses with biophysical tools - Nature [https://www.nature.com/articles/s44298-025-00107-y]
- 62. Microbial adhesion and ultrastructure from the single- ... [https://www.sciencedirect.com/science/article/pii/S246823301930009X]