Optimizing DNA Extraction for Metagenomic Sequencing: A Guide for Robust Pathogen Detection and Microbiome Analysis
Metagenomic sequencing has revolutionized pathogen detection and microbiome analysis, but its success is critically dependent on the initial DNA extraction step.
Optimizing DNA Extraction for Metagenomic Sequencing: A Guide for Robust Pathogen Detection and Microbiome Analysis
Abstract
Metagenomic sequencing has revolutionized pathogen detection and microbiome analysis, but its success is critically dependent on the initial DNA extraction step. This article provides a comprehensive guide for researchers and drug development professionals on selecting and optimizing DNA extraction methods for metagenomic applications. It covers foundational principles, tailored methodological protocols for diverse sample types, advanced troubleshooting strategies, and rigorous validation techniques. By synthesizing current research, we demonstrate how optimized DNA extraction minimizes biases, enhances sequencing accuracy, and ensures reliable results for downstream biomedical and clinical research, ultimately supporting advancements in diagnostics, therapeutics, and One Health surveillance.
The Critical Foundation: Why DNA Extraction Dictates Metagenomic Sequencing Success
The Pivotal Role of High-Quality DNA in Metagenomic Next-Generation Sequencing (mNGS)
Metagenomic next-generation sequencing (mNGS) has emerged as a transformative, hypothesis-free approach for infectious disease diagnosis and microbiome research, capable of simultaneously detecting bacteria, viruses, fungi, and parasites without prior knowledge of the infectious agent [1] [2]. This unbiased high-throughput sequencing technology directly characterizes microbial genomes from clinical samples, providing unparalleled insights into microbial communities compared to traditional culture-based methods [1]. However, the diagnostic accuracy and analytical sensitivity of mNGS are fundamentally dependent on the quality and integrity of the input nucleic acids. High-quality DNA extraction serves as the critical foundation for successful mNGS applications, influencing everything from library preparation efficiency to taxonomic classification accuracy and the reliable detection of low-abundance pathogens [3] [4] [5].
The necessity for high-quality DNA in mNGS stems from multiple technical considerations. First, the detection of rare, novel, or unculturable pathogens requires DNA of sufficient molecular weight to enable comprehensive genomic coverage [1] [5]. Second, the elimination of contaminants and inhibitors during extraction is essential for maximizing sequencing efficiency and reducing false-positive results [2]. Third, the unbiased representation of complex microbial communities demands extraction methods that equally lyse both Gram-positive and Gram-negative bacteria without introducing taxonomic biases [4] [5]. As mNGS moves from research to clinical laboratories, standardized protocols for obtaining high-quality DNA have become increasingly important for ensuring reproducible and clinically actionable results [6].
Quantitative Impact of DNA Extraction Methods on mNGS Performance
Comparative Performance of DNA Extraction Methods
The selection of appropriate DNA extraction methods significantly influences the yield, integrity, and purity of recovered nucleic acids, which subsequently affects mNGS performance metrics including read depth, genome coverage, and taxonomic classification accuracy [4] [5]. Different DNA extraction protocols employ varying mechanical, chemical, and enzymatic approaches to cell lysis and nucleic acid purification, each with distinct advantages and limitations for specific sample types and research applications.
Table 1: Comparison of DNA Extraction Method Performance Across Sample Types
| Extraction Method | DNA Yield | Fragment Size | Purity (A260/280) | Gram-positive Lysis Efficiency | Best-suited Applications |
|---|---|---|---|---|---|
| Enzymatic Lysis (MetaPolyzyme) | Moderate | High (2.1-fold increase) [3] | Good | Excellent | Urine samples, long-read sequencing [3] |
| Mechanical Bead-Beating | High | Low to Moderate | Variable | Good | Fecal samples, diverse communities [4] [5] |
| Quick-DNA HMW MagBead Kit | High | High | Good | Excellent | Nanopore sequencing, mock communities [5] |
| DNeasy PowerLyzer PowerSoil + SPD | High | High | Excellent (1.8) [4] | Excellent | Gut microbiome studies [4] |
| ZymoBIOMICS DNA Mini Kit | Moderate | Moderate | Good | Good | Standard microbiome analyses [4] |
Impact of DNA Quality on Sequencing Metrics
The quality of input DNA directly correlates with critical mNGS performance metrics, influencing the diagnostic utility and analytical sensitivity of the assay. Methodological comparisons demonstrate that DNA extraction approaches significantly affect the proportion of usable sequencing reads, taxonomic classification accuracy, and limit of detection for low-abundance pathogens.
Table 2: Impact of DNA Quality on mNGS Performance Metrics
| Performance Metric | High-Quality DNA Impact | Compromised DNA Impact | Clinical Significance |
|---|---|---|---|
| Host DNA Background | 10-fold enrichment of microbial reads with effective host depletion [7] | >99% of sequences may be host-derived in blood samples [7] | Enables pathogen detection in sepsis without excessive sequencing costs |
| Taxonomic Resolution | Long reads enable accurate species-level classification [3] | Short, fragmented reads limit classification resolution [3] | Critical for distinguishing pathogenic from commensal organisms |
| Limit of Detection | 100% detection of expected pathogens in clinical samples [7] | Reduced sensitivity for low-abundance pathogens [5] | Essential for early infection diagnosis when pathogen burden is low |
| Community Representation | Preservation of correct microbial abundance profiles [4] | Skewed community structure due to differential lysis [4] | Accurate representation of polymicrobial infections |
Experimental Protocols for High-Quality DNA Extraction in mNGS
Enzymatic Lysis Protocol for Long-Read Sequencing
For applications requiring long-read sequencing technologies such as Nanopore or PacBio, enzymatic lysis methods provide superior DNA fragment length preservation compared to mechanical disruption approaches [3]. The following protocol has been optimized for urine samples but can be adapted to other sample types with appropriate modifications:
Reagents Required:
- MetaPolyzyme (Sigma-Aldrich, reconstituted in PBS)
- Lytic enzyme solution (Qiagen)
- IndiSpin Pathogen Kit (Indical Bioscience) or equivalent purification system
- Phosphate-Buffered Saline (PBS)
Procedure:
- Sample Preparation: Centrifuge 1 mL of urine sample at 20,000 × g for 5 minutes. Discard 800 μL of supernatant and resuspend the pellet in the remaining 200 μL by gentle vortexing [3].
- Enzymatic Lysis: Add 5 μL of lytic enzyme solution and 10 μL of reconstituted MetaPolyzyme to the 200 μL sample. Mix by gentle pipetting to avoid DNA shearing [3].
- Incubation: Incubate the mixture at 37°C in a shaker for 1 hour to enable complete microbial cell lysis [3].
- DNA Purification: Extract DNA from the post-lysed sample using the IndiSpin Pathogen Kit according to the manufacturer's instructions, with careful attention to gentle pipetting throughout [3].
- Quality Assessment: Quantify DNA yield using fluorometric methods (e.g., Qubit dsDNA HS Assay) and assess fragment size distribution via pulsed-field gel electrophoresis or TapeStation analysis [3] [5].
Technical Notes: This enzymatic approach has been shown to increase the average length of microbial reads by a median of 2.1-fold (IQR: 1.7-2.5) and improve the mapped reads proportion of specific species by a median of 11.8-fold (IQR: 6.9-32.2) compared to direct extraction methods without pre-lysis [3].
Host DNA Depletion Protocol for Blood Samples
Blood samples present a unique challenge for mNGS due to the overwhelming abundance of human DNA, which can comprise >99% of sequencing reads without effective depletion strategies [7]. The following protocol utilizes a novel Zwitterionic Interface Ultra-Self-assemble Coating (ZISC)-based filtration device to selectively remove host cells while preserving microbial integrity:
Reagents Required:
- ZISC-based fractionation filter (Micronbrane Medical)
- ZymoBIOMICS Spike-in Control I (High Microbial Load)
- ZISC-based Microbial DNA Enrichment Kit (Micronbrane Medical)
Procedure:
- Sample Preparation: Collect 3-13 mL of whole blood in EDTA tubes. Add ZymoBIOMICS Spike-in Control I (104 genome copies/mL) as an internal process control [7].
- Host Cell Depletion: Transfer 4 mL of whole blood to a syringe connected to the ZISC-based filter. Gently depress the plunger to push the blood sample through the filter into a clean collection tube [7].
- Plasma Separation: Centrifuge the filtered blood at 400 × g for 15 minutes at room temperature to isolate plasma [7].
- Microbial Pellet Recovery: Subject the plasma to high-speed centrifugation at 16,000 × g to pellet microbial cells [7].
- DNA Extraction: Extract DNA from the pellet using the ZISC-based Microbial DNA Enrichment Kit according to the manufacturer's instructions [7].
- Quality Control: Verify host DNA depletion efficiency by qPCR targeting human β-globin gene and assess microbial DNA recovery using the spike-in control [7].
Technical Notes: This filtration method achieves >99% white blood cell removal across various blood volumes while allowing unimpeded passage of bacteria and viruses [7]. When implemented in a gDNA-based mNGS workflow, this approach detects all expected pathogens in 100% of clinical samples with an average microbial read count of 9,351 reads per million (RPM), representing a tenfold improvement over unfiltered samples (925 RPM) [7].
Standardized Fecal DNA Extraction with Stool Preprocessing Device
The complex composition and variable consistency of fecal samples present challenges for reproducible DNA extraction. The integration of a stool preprocessing device (SPD) upstream of DNA extraction improves both standardization and quality of microbial DNA recovery from gut microbiome samples [4]:
Reagents Required:
- Stool preprocessing device (SPD, bioMérieux)
- DNeasy PowerLyzer PowerSoil Kit (QIAGEN)
- Liquid handling reagents for the SPD
Procedure:
- Sample Homogenization: Process the fecal sample using the SPD according to the manufacturer's instructions to generate a homogeneous suspension [4].
- Aliquot Transfer: Transfer 200 μL of the homogenized suspension to a PowerBead Tube provided in the kit [4].
- Mechanical Lysis: Secure the PowerBead Tubes in a vortex adapter and vortex at maximum speed for 10 minutes to ensure complete lysis of both Gram-positive and Gram-negative bacteria [4].
- DNA Purification: Continue with the standard DNeasy PowerLyzer PowerSoil protocol as recommended by the manufacturer [4].
- Elution: Elute DNA in 100 μL of elution buffer and store at -20°C until library preparation [4].
Technical Notes: The SPD combined with the DNeasy PowerLyzer PowerSoil protocol (S-DQ protocol) demonstrates optimal performance for gut microbiome studies, providing high DNA yield, excellent purity (A260/280 ratio of 1.8), and improved recovery of Gram-positive bacteria compared to the standard protocol without preprocessing [4].
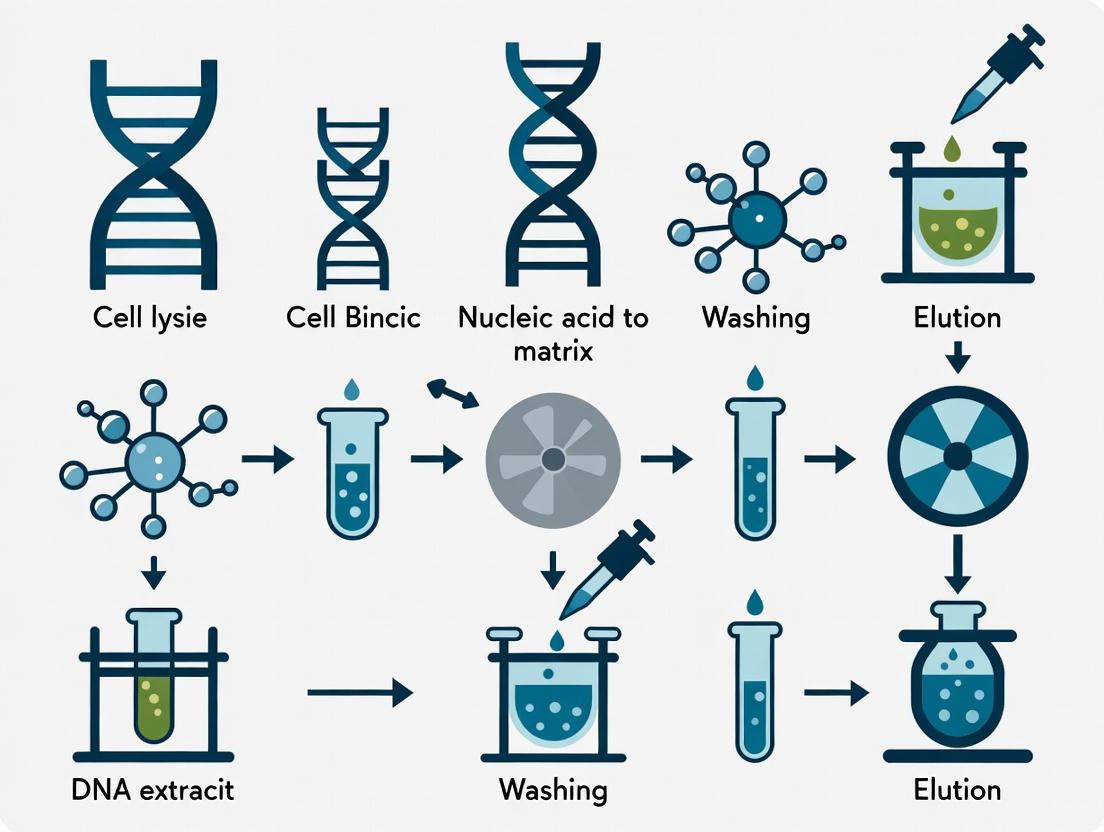
Workflow Visualization: High-Quality DNA in mNGS Analysis
The following diagram illustrates the complete mNGS workflow, highlighting the critical role of high-quality DNA extraction and its impact on downstream analytical steps:
Diagram 1: Comprehensive mNGS Workflow Highlighting DNA Quality Dependencies. This workflow illustrates the sequential steps in metagenomic next-generation sequencing, with the initial sample processing and DNA extraction steps (yellow) serving as critical determinants of final data quality. High-quality DNA extraction influences every downstream analytical component, from library preparation efficiency to taxonomic classification accuracy.
The Scientist's Toolkit: Essential Reagents for High-Quality mNGS DNA Extraction
Table 3: Essential Research Reagent Solutions for mNGS-Quality DNA Extraction
| Reagent/Kit | Primary Function | Key Applications | Performance Considerations |
|---|---|---|---|
| MetaPolyzyme Enzyme Mix | Enzymatic lysis of microbial cell walls | Urine samples, long-read sequencing | Increases read length 2.1-fold; improves species mapping 11.8-fold [3] |
| ZISC-Based Filtration Device | Selective host cell depletion | Blood samples, sepsis diagnostics | >99% WBC removal; 10x microbial read enrichment [7] |
| Quick-DNA HMW MagBead Kit | Gentle isolation of high molecular weight DNA | Nanopore sequencing, mock communities | Optimal yield of pure HMW DNA; accurate detection in complex communities [5] |
| DNeasy PowerLyzer PowerSoil Kit | Mechanical lysis of diverse microbes | Fecal samples, gut microbiome | High DNA yield and purity (A260/280=1.8); effective Gram-positive lysis [4] |
| Stool Preprocessing Device (SPD) | Standardized fecal sample homogenization | Gut microbiome studies | Improves DNA yield and alpha-diversity; enhances Gram-positive recovery [4] |
The critical importance of high-quality DNA in mNGS applications cannot be overstated, as it fundamentally influences the sensitivity, specificity, and diagnostic utility of this powerful technology. As evidenced by the comparative data and optimized protocols presented, DNA extraction methodology must be carefully matched to both sample type and research objectives to maximize mNGS performance. Enzymatic lysis approaches offer distinct advantages for long-read sequencing applications, while mechanical methods combined with standardized preprocessing provide superior results for complex matrices like fecal samples [3] [4]. For challenging sample types such as blood, innovative host depletion strategies are essential for reducing background and enhancing pathogen detection sensitivity [7].
Looking forward, several emerging trends are likely to shape the future of DNA extraction for mNGS applications. First, the development of integrated systems that combine sample preparation with microfluidic technologies may enable more standardized and automated DNA extraction workflows [7]. Second, as long-read sequencing technologies continue to mature with decreasing error rates, the demand for high-molecular-weight DNA extraction methods will increase accordingly [3] [5]. Third, the establishment of validated reference standards and quality control metrics for DNA extraction will be essential for clinical translation of mNGS assays [6]. Finally, the creation of comprehensive databases of high-quality metagenome-assembled genomes (MAGs) will provide improved reference materials for benchmarking DNA extraction performance and its impact on downstream analyses [8].
As mNGS continues to evolve from a research tool to a clinical diagnostic platform, the pivotal role of high-quality DNA extraction will remain at the foundation of its success. By implementing the optimized protocols and quality considerations outlined in this document, researchers and clinical laboratory professionals can ensure that their mNGS applications achieve the sensitivity, reproducibility, and diagnostic accuracy required for both scientific discovery and patient care.
Metagenomic sequencing has revolutionized the study of microbial communities, offering unparalleled insights into diverse ecosystems from the mammalian gut to agricultural waste. However, the accuracy of these analyses is entirely dependent on the initial quality of the extracted nucleic acids. Sample preparation from complex matrices presents three fundamental challenges: effective removal of PCR inhibitors, preservation of nucleic acid integrity, and minimization of biological bias. These challenges are particularly acute in environmental and clinical samples rich in organic and inorganic compounds that interfere with downstream molecular applications. This application note details the core challenges and provides optimized, practical protocols validated for complex sample types to support reliable metagenomic research and diagnostic development.
Core Challenges and Comparative Data
Challenge 1: Effective Inhibitor Removal
Complex matrices such as soil, manure, and wastewater contain substances like humic acids, fulvic acids, and complex polysaccharides that co-purify with nucleic acids and inhibit enzymatic reactions in PCR and sequencing [9] [10] [11]. The efficiency of their removal varies significantly between DNA extraction methods.
Table 1: Inhibitor Removal and DNA Purity Across Kits and Sample Types
| Sample Matrix | DNA Extraction Kit | Key Inhibitor Removed | 260/280 Ratio (Mean ± SD) | 260/230 Ratio (Mean ± SD) | PCR Inhibition Observed? |
|---|---|---|---|---|---|
| Piggery Wastewater [12] | QIAGEN PowerFecal Pro | Humic acids, organic matter | 1.88 ± 0.05 | 2.15 ± 0.08 | No |
| Piggery Effluent [9] | NucleoSpin Soil (Modified Elution) | Humic substances, proteins | 1.85 ± 0.04 | 2.20 ± 0.10 | No |
| Marine Sediment [13] | DNeasy PowerSoil Pro | Humic acids, salts | 1.82 ± 0.07 | 2.10 ± 0.12 | No |
| Mammalian Feces [14] | QIAamp Fast DNA Stool Mini | Bilirubin, complex polysaccharides | 1.90 ± 0.03 | 2.05 ± 0.09 | No |
| Inhibitor-Rich Soil [10] | Phenol-Chloroform (Custom) | Humic/fulvic compounds | 1.80 ± 0.10 | 1.95 ± 0.15 | With dilution |
Challenge 2: Preservation of Nucleic Acid Integrity
Obtaining DNA that is sufficiently intact and high-molecular-weight is crucial, especially for long-read sequencing technologies like Oxford Nanopore Technologies (ONT). The method of cell lysis and subsequent handling are primary determinants of DNA fragmentation.
Table 2: DNA Yield and Quality for Downstream Sequencing
| Sample Matrix | Extraction Method | Average Yield (ng/μL) | DNA Integrity (Gel Electrophoresis) | Suitability for ONT | Suitability for Illumina |
|---|---|---|---|---|---|
| Piggery Wastewater [12] | QIAGEN PowerFecal Pro | 45.2 ± 5.8 | High (≥20 kb) | Excellent | Excellent |
| Marine Sediment [13] | PowerSoil Kit | 38.9 ± 6.5 | High (≥20 kb) | Excellent | Excellent |
| Ovine Blood [15] | Silica-Membrane Kit | 125.0 ± 15.0 | High (≥48.5 kb) | Excellent | Excellent |
| Marine Water [13] | DNeasy Blood & Tissue | 15.3 ± 3.2 | Moderate (5-10 kb) | Good | Excellent |
| Broiler Feces [16] | Hotshot Method | 25.0 ± 4.5 | Low (1-3 kb) | Poor | Good (for PCR) |
Challenge 3: Minimization of Biological Bias
A critical goal of metagenomics is to obtain a nucleic acid pool that accurately represents the true biological community. Different extraction methods can introduce significant bias by preferentially lysing certain cell types over others.
Table 3: Taxonomic Bias Introduced by DNA Extraction Methods
| Extraction Method | Lysis Principle | Gram-Positive Recovery (vs. Expected) | Gram-Negative Recovery (vs. Expected) | Reported Bias | Source | | :--- | :--- | :--- | :--- | :--- | ::--- | | Bead-beating + Enzymatic | Mechanical & Chemical | 92% | 105% | Lowest overall bias; most representative | [17] | | Bead-beating only | Mechanical | 65% | 115% | Under-represents tough Gram-positives | [12] [17] | | Silica Kit (QBT) | Chemical/Enzymatic | ~40-60% | ~110-130% | Significantly under-represents Gram-positives | [14] [17] | | Phenol-Chloroform | Chemical | ~70% | ~95% | Moderate under-representation of Gram-positives | [10] |
A benchmark study on piggery wastewater revealed that the choice of extraction protocol could create a 10-fold difference in the measured proportion of a given taxon from the same original sample [12] [11]. This technical variation can account for 20–30% of the total observed variation in a study, at times exceeding the biological signal of interest [17].
Optimized Experimental Protocols
Protocol 1: Optimized DNA Extraction from Complex Environmental Matrices
This protocol is adapted from the optimized QIAGEN PowerFecal Pro method, identified as superior for piggery wastewater and other complex environmental samples [12].
Application: For extracting high-quality, inhibitor-free genomic DNA from complex matrices (wastewater, manure, soil) for metagenomic sequencing. Sample Types: Piggery wastewater, lagoon effluent, raw manure, soil. Reagent Solutions:
- QIAGEN QIAamp PowerFecal Pro DNA Kit (Cat. No. 51804): Provides lysis buffers and inhibitor removal technology.
- CD1 Lysis Buffer: Contains guanidine thiocyanate for denaturation.
- Proteinase K (provided): Digests proteins and nucleases.
- Ethanol (96-100%): For precipitating DNA onto the membrane.
- Merck Milli-Q Water: For final elution.
Workflow:
- Sample Preparation: Centrifuge 10-40 mL of wastewater sample at 46 g for 1 min to settle heavy solids. Transfer supernatant to a new tube and centrifuge at 4,550 g for 30 min. Discard supernatant and weigh pellet. Resuspend 0.3 g of pellet in 500 μL Milli-Q water [12].
- Cell Lysis: Transfer the entire 500 μL of homogenate to a PowerBead Pro tube. Add 500 μL of CD1 lysis buffer (note: modified from the manufacturer's 800 μL recommendation) and 100 μL of Proteinase K. Vortex thoroughly.
- Mechanical Lysis: Lysate cells using a Vortex-Genie 2 at maximum speed for 10 min. This mechanical beating is critical for disrupting tough Gram-positive bacterial cell walls [12] [17].
- Incubation: Incubate the lysate at 56°C for 30 min with agitation at 300 rpm in a thermomixer.
- Binding: Centrifuge the bead tube at 13,000 g for 1 min. Transfer up to 600 μL of supernatant to a clean microcentrifuge tube.
- Inhibitor Removal: Add 600 μL of solution CD2, vortex for 5 s, and incubate on ice for 5 min. Centrifuge at 13,000 g for 5 min. Transfer up to 600 μL of supernatant to a new tube.
- DNA Binding: Load the supernatant onto an MB Spin Column and centrifuge at 13,000 g for 1 min. Discard the flow-through.
- Wash 1: Add 500 μL of solution EA. Centrifuge at 13,000 g for 1 min. Discard the flow-through.
- Wash 2: Add 500 μL of solution C5. Centrifuge at 13,000 g for 1 min. Discard the flow-through. Perform a second wash with 250 μL of solution C5, incubate on ice for 5 min, then centrifuge at 13,000 g for 1 min.
- Dry Membrane: Leave the spin column lid open for 10 min to evaporate residual ethanol.
- Elution: Add 50 μL of solution C6 to the center of the membrane. Incubate at room temperature for 5 min. Centrifuge at 13,000 g for 1 min to elute pure, high-quality DNA.
Troubleshooting:
- Low Yield: Ensure the bead-beating step is performed at maximum speed for the full duration. Increase the starting sample volume.
- PCR Inhibition: If inhibition is detected (e.g., via qPCR), perform an additional wash with solution C5 and ensure the 10-minute drying step is not skipped.
- DNA Fragmentation: Avoid over-vortexing after the initial lysis step. Do not use a water bath for incubation.
Protocol 2: Unbiased Metagenomic Sequencing from Clinical Samples
This protocol provides a method for generating DNA for sequencing directly from clinical samples, such as swabs, with minimal bias, incorporating key steps to remove host DNA and amplify microbial nucleic acids [18].
Application: For non-targeted detection of DNA and RNA microorganisms in clinical samples (e.g., nasal swabs, serum) for shotgun metagenomics. Sample Types: Nasal swabs, serum, viral culture isolates. Reagent Solutions:
- TURBO DNA-free Kit (Thermo Fisher, AM1907): For complete removal of contaminating DNA from RNA samples.
- SuperScript IV First-Strand Synthesis System (Thermo Fisher, 18091050): For high-efficiency cDNA synthesis.
- NEBNext Ultra II Non-Directional RNA Second Strand Synthesis Module (NEB, E6111S): For dsDNA synthesis.
- GenomiPhi V2 DNA Amplification Kit (Cytiva, 25660031): For whole-genome amplification of DNA samples.
- Agencourt AMPure XP beads (Beckman Coulter, A63881): For purification and size selection.
Workflow:
Figure 1: Unbiased Metagenomic Protocol for Clinical Samples.
Detailed Steps:
- Nucleic Acid Quantification: Quantify extracted RNA/DNA using a Qubit fluorometer with the RNA HS or dsDNA HS assay [18].
- DNA Removal (For RNA targets): Treat ~10 µg of RNA in a 50 µL reaction with 1 µL of TURBO DNase (2U) and 1x buffer. Incubate at 37°C for 30 min. Add DNase Inactivation Reagent, incubate for 5 min at room temperature with occasional mixing, and pellet the reagent [18].
- RNA Precipitation: Bring the DNase-treated RNA to 500 µL with nuclease-free water. Add 50 µL of 3M sodium acetate (pH 5.2) and 500 µL of 2-propanol. Mix and incubate at room temperature for 20 min. Centrifuge at 12,000 rpm for 15 min. Wash the pellet twice with 500 µL of 70% ethanol, air-dry, and resuspend in 30 µL nuclease-free water [18].
- First-Strand cDNA Synthesis: Using the SuperScript IV system, combine RNA, random hexamers, dNTPs, and nuclease-free water. Heat to 65°C for 5 min, then place on ice. Add DTT, RNase inhibitor, and SuperScript IV RT. Incubate at 55°C for 10 min, then inactivate at 80°C for 10 min [18].
- Second-Strand Synthesis: Convert the cDNA to double-stranded DNA using the NEBNext Ultra II Second Strand Synthesis Module according to the manufacturer's instructions [18].
- Purification (For both DNA and RNA paths): Purify the resulting dsDNA using AMPure XP beads at a 1:1 ratio. Elute in nuclease-free water.
- Whole-Genome Amplification (For DNA targets): If the target is microbial DNA, take the purified DNA from Step 1 and amplify it using the GenomiPhi V2 kit according to the manufacturer's protocol to increase mass for sequencing [18].
- Library Preparation and Sequencing: Prepare sequencing libraries from the final dsDNA (from step 5/6 or step 7) using the Illumina Nextera XT DNA Library Preparation Kit. Sequence on the appropriate Illumina platform [18].
Troubleshooting:
- Low cDNA Yield: Ensure the RNA is not degraded and that the SuperScript IV reverse transcriptase is active. Check the integrity of the RNA on a fragment analyzer.
- High Host Background: Increase the DNase treatment incubation time or perform a double DNase treatment.
- Low Library Diversity: Ensure the whole-genome amplification step is not over-cycled, which can lead to amplification bias.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Metagenomic Nucleic Acid Extraction
| Reagent / Kit Name | Manufacturer | Primary Function | Ideal Sample Matrix |
|---|---|---|---|
| QIAamp PowerFecal Pro DNA Kit | QIAGEN | Lysis & inhibitor removal | Feces, wastewater, soil [12] [13] |
| NucleoSpin Soil Kit | Macherey-Nagel | Lysis & inhibitor removal | Soil, sediment, manure [9] [14] |
| TURBO DNA-free Kit | Thermo Fisher | Genomic DNA removal | RNA extracts from any matrix [18] |
| MetaPolyzyme | Sigma-Aldrich | Enzymatic lysis of Gram+ cells | All matrices (supplement) [17] |
| AMPure XP Beads | Beckman Coulter | Nucleic acid purification & size selection | All matrices [18] |
| SuperScript IV RT | Thermo Fisher | High-efficiency cDNA synthesis | RNA viruses, metatranscriptomics [18] |
The fidelity of any metagenomic study is determined at the earliest stage: nucleic acid extraction. The challenges of inhibitor removal, integrity preservation, and bias minimization are interconnected and must be addressed concurrently. As demonstrated, the optimal extraction method is highly dependent on the sample matrix. For environmental samples like wastewater and soil, a robust mechanical lysis protocol combined with validated inhibitor removal technology (e.g., QIAGEN PowerFecal Pro or NucleoSpin Soil) is critical. For clinical applications aiming to detect a broad range of pathogens, a flexible protocol that handles both DNA and RNA and efficiently removes host background is essential. By adopting these optimized protocols and understanding the sources of bias, researchers can significantly improve the accuracy and reproducibility of their metagenomic analyses, thereby generating more reliable data for both scientific research and diagnostic development.
Impact of extraction bias on microbial community representation and pathogen detection
Metagenomic sequencing has revolutionized microbial ecology and clinical diagnostics by enabling culture-free analysis of complex microbial communities. However, the accuracy of these analyses is fundamentally compromised by inherent biases introduced during DNA extraction, leading to distorted microbial community profiles and potentially misleading biological conclusions. The differential lysis efficiency of diverse microbial cell walls results in the over-representation of easily-lysed organisms and the under-detection of tough-to-lyse pathogens, directly impacting diagnostic sensitivity and therapeutic decisions [19]. This application note systematically evaluates the impact of DNA extraction bias on microbial community representation and pathogen detection, providing validated protocols to minimize these effects in both research and clinical settings.
Quantitative Comparison of DNA Extraction Method Performance
The following table summarizes the performance of various DNA extraction methods across different sample types, as reported in recent studies:
Table 1: Performance comparison of DNA extraction methods across sample types
| Sample Type | Extraction Method | Key Performance Findings | Reference |
|---|---|---|---|
| Whole Blood (Sepsis Diagnostics) | Magnetic bead-based (K-SL) | 77.5% accuracy for E. coli detection | [20] |
| Whole Blood (Sepsis Diagnostics) | Magnetic bead-based (GraBon) | 77.5% accuracy for S. aureus detection | [20] |
| Whole Blood (Sepsis Diagnostics) | Column-based (QIAamp) | 65.0% accuracy for E. coli detection | [20] |
| Human Gut Microbiome | SPD + DNeasy PowerLyzer PowerSoil (S-DQ) | Best overall performance for microbial diversity | [4] |
| Human Gut Microbiome | Standard commercial kits | Significantly lower Gram-positive bacteria recovery | [4] |
| Diverse Fermented Foods | Enzymatic lysis methods | Higher eubacterial and yeast DNA yield | [21] |
| Urine (Nanopore Sequencing) | Enzymatic lysis | 2.1-fold increase in read length; 100% clinical concordance | [3] |
| Low Biomass Samples (Sputum, Dust) | Various methods | Extraction accounted for 9-16% of variability | [22] |
Fundamental Mechanisms of Bias
DNA extraction bias primarily stems from differential cell lysis efficiency across microbial taxa with varying cell wall structures. Gram-positive bacteria with thick peptidoglycan layers require more vigorous lysis conditions compared to Gram-negative bacteria with thinner cell walls [19]. This fundamental difference leads to systematic under-representation of Gram-positive organisms in protocols optimized for rapid DNA extraction or those relying solely on chemical lysis.
The physical and chemical composition of sample matrices further exacerbates extraction bias. Complex materials like stool, food, and blood contain inhibitors that differentially affect DNA recovery from various microbial species [21]. Additionally, DNA shearing during extraction, particularly with vigorous mechanical disruption methods, reduces read lengths and impacts assembly quality in downstream sequencing applications [3].
Impact on Microbial Community Profiles
The choice of extraction method significantly alters observed microbial community composition. Studies demonstrate that different DNA extraction kits can produce dramatically different results from identical samples, with error rates from bias exceeding 85% in some cases [23]. The effect is particularly pronounced in low-biomass samples where extraction method accounted for 9-16% of the observed variability in microbial community structure [22].
In gut microbiome studies, protocols incorporating a stool preprocessing device (SPD) significantly improved DNA extraction yield, sample alpha-diversity, and recovery of Gram-positive bacteria compared to standard commercial protocols [4]. Similarly, in fermented food analysis, different extraction principles (enzymatic, mechanical, chemical) recovered distinct fractions of the true eubacterial community, with methods sharing only 29.9-52.0% of the total operational taxonomic units (OTUs) detected [21].
Diagram 1: Sources and impacts of DNA extraction bias
Optimized Protocols for Bias-Reduced DNA Extraction
Magnetic Bead-Based Protocol for Blood Samples (Sepsis Diagnostics)
This protocol optimized for whole blood samples demonstrates superior performance for sepsis pathogen detection compared to traditional column-based methods [20]:
Reagents and Equipment:
- K-SL DNA Extraction Kit or GraBon system
- Proteinase K
- Lysis buffer with guanidine hydrochloride
- Wash buffers (typically ethanol-based)
- Elution buffer (TE or nuclease-free water)
- Magnetic stand
- Thermonmixer or water bath
- Microcentrifuge
Procedure:
- Sample Preparation: Mix 1-3 mL of whole blood with equal volume of lysis buffer containing Proteinase K.
- Incubation: Incubate at 56°C for 30 minutes with occasional mixing.
- Binding: Add magnetic beads and incubate for 10 minutes at room temperature.
- Washing: Place on magnetic stand, discard supernatant. Wash twice with wash buffer 1, once with wash buffer 2.
- Elution: Air-dry beads for 5-10 minutes, elute DNA in 50-100 μL elution buffer.
- Quality Control: Quantify DNA using fluorometric methods, assess fragment size if required for downstream applications.
Performance Notes: This protocol achieved 77.5% accuracy for pathogen detection in clinical blood samples, significantly outperforming column-based methods (65.0% accuracy) [20]. The automated nature of magnetic bead systems reduces hands-on time and improves reproducibility.
Enhanced Gut Microbiome DNA Extraction with Stool Preprocessing
This protocol combines mechanical preprocessing with optimized lysis for superior representation of gut microbial diversity [4]:
Reagents and Equipment:
- Stool preprocessing device (SPD, bioMérieux)
- DNeasy PowerLyzer PowerSoil Kit (QIAGEN)
- PBS buffer
- Bead-beating tubes
- Centrifuge
- Vortex adapter for bead beating
Procedure:
- Sample Preprocessing: Homogenize 100-200 mg stool sample using SPD according to manufacturer's instructions.
- Initial Lysis: Transfer 100 μL homogenate to PowerBead Tubes, add solution CD1.
- Mechanical Lysis: Vortex vigorously using bead beater for 10 minutes.
- Incubation: Incubate at 65°C for 10 minutes.
- Binding: Transfer supernatant to MB Spin Column, centrifuge.
- Washing: Wash with solutions CB and EB.
- Elution: Elute DNA in 100 μL solution C6.
Performance Notes: The SPD preprocessing step significantly improved DNA extraction yield, alpha-diversity measurements, and recovery of Gram-positive bacteria compared to standard protocols [4]. This protocol showed the best overall performance for gut microbiome studies among four tested commercial methods.
Enzymatic Lysis Protocol for Long-Read Metagenomic Sequencing
This gentle enzymatic lysis protocol preserves DNA integrity for long-read sequencing technologies [3]:
Reagents and Equipment:
- MetaPolyzyme (Sigma Aldrich)
- Lytic enzyme solution
- IndiSpin Pathogen Kit (Indical Bioscience)
- Phosphate Buffered Saline (PBS)
- Thermonmixer or water bath
- Microcentrifuge
Procedure:
- Sample Preparation: Concentrate 1 mL urine by centrifugation at 20,000 × g for 5 minutes, resuspend in 200 μL residual volume.
- Enzymatic Lysis: Add 5 μL lytic enzyme solution and 10 μL MetaPolyzyme to 200 μL sample.
- Incubation: Incubate at 37°C for 1 hour with gentle shaking.
- DNA Purification: Extract DNA using IndiSpin Pathogen Kit according to manufacturer's instructions.
- Elution: Elute DNA in 50-100 μL elution buffer.
Performance Notes: This protocol increased the average length of microbial reads by 2.1-fold compared to mechanical lysis methods and achieved 100% concordance with clinical culture results [3]. The gentle lysis preserves DNA integrity crucial for long-read sequencing technologies.
Research Reagent Solutions
Table 2: Essential research reagents for bias-minimized DNA extraction
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Mechanical Lysis Kits | DNeasy PowerLyzer PowerSoil (QIAGEN) | Bead-beating optimized for soil/fecal samples; effective for Gram-positive bacteria |
| Magnetic Bead Kits | K-SL DNA Extraction Kit, GraBon system | Automated processing; superior for blood samples [20] |
| Enzymatic Lysis Reagents | MetaPolyzyme, Lysozyme | Gentle cell wall degradation; preserves DNA integrity [3] |
| Stool Preprocessing | SPD (bioMérieux) | Standardizes homogenization; improves yield and diversity [4] |
| Mock Communities | ZymoBIOMICS Microbial Community Standard | Validation and bias quantification [5] [23] |
| Inhibition Removal | Polyvinylpyrrolidone (PVP-40), Sodium metabisulfite | Reduces polyphenol and polysaccharide interference [24] |
Validation and Quality Control Strategies
Mock Community-Based Bias Assessment
The use of defined mock communities provides essential quality control for quantifying extraction bias:
Protocol for Bias Quantification Using Mock Communities [23]:
Materials:
- ZymoBIOMICS Microbial Community Standard or similar
- Selected DNA extraction methods to evaluate
- PCR reagents for 16S rRNA amplification
- Sequencing platform
Procedure:
- Experimental Design: Create a D-optimal mixture design with 65 unique treatment combinations and 15 replicates for statistical robustness.
- Sample Processing: Extract DNA from mock communities using each method in triplicate.
- Sequencing and Analysis: Sequence 16S rRNA amplicons and classify reads taxonomically.
- Bias Calculation: Compare observed proportions to expected composition using mixture effect models.
- Model Application: Develop correction models for environmental samples based on mock community results.
Interpretation: This approach allows researchers to quantify bias specific to their chosen protocols and develop appropriate correction factors. Studies demonstrate that DNA extraction introduces the largest technical variation in microbiome studies, exceeding PCR amplification and sequencing biases [23].
Diagram 2: Mock community workflow for extraction bias quantification
DNA extraction methodology significantly impacts microbial community representation and pathogen detection accuracy in metagenomic studies. Magnetic bead-based systems demonstrate superior performance for clinical blood samples, while protocols incorporating mechanical preprocessing and bead-beating provide more comprehensive representation of gut microbiome diversity. For long-read sequencing applications, enzymatic lysis methods preserve DNA integrity while maintaining representative community profiles. Implementation of mock community-based quality control enables researchers to quantify and correct for extraction-specific biases. Selection of appropriate extraction protocols based on sample type and research objectives is essential for obtaining accurate, reproducible results in microbial metagenomic studies.
The fidelity of metagenomic sequencing is fundamentally contingent on the initial DNA extraction process. Obtaining nucleic acids that are both quantitatively sufficient and qualitatively representative of the original microbial community is paramount for unbiased downstream analysis. This application note delineates the three core principles—maximizing yield, ensuring purity, and guaranteeing representativeness—that underpin effective DNA extraction protocols for metagenomic sequencing research. Within the context of a broader thesis on methodological optimization, we provide a detailed examination of these principles, supported by comparative data and standardized protocols, to guide researchers and drug development professionals in selecting and optimizing extraction methods for diverse sample types, from complex environmental matrices to clinical specimens.
Core Principles and Comparative Analysis of DNA Extraction Methods
The pursuit of high-quality metagenomic DNA involves balancing often-competing demands. The following principles provide a framework for evaluation:
- Maximizing Yield: The goal is to extract a sufficient quantity of DNA for subsequent library preparation and sequencing, particularly for low-biomass samples. This requires efficient cell lysis of all microorganisms present. Inefficient lysis directly leads to the underrepresentation of certain taxa in the final data.
- Ensuring Purity: Co-extracted substances from samples (e.g., humic acids from soil, bile salts from stool, or organic matter from wastewater) can act as potent inhibitors of downstream enzymatic processes, including PCR and sequencing. Effective removal of these contaminants is crucial for success.
- Guaranteeing Representativeness: The extraction method must lyse all microbial cells equally, without introducing bias against specific groups (e.g., Gram-positive versus Gram-negative bacteria). Furthermore, the method should minimize DNA shearing to preserve high-molecular-weight (HMW) DNA, which is essential for long-read sequencing and accurate genome assembly.
A comparison of several DNA extraction methods, as evaluated in recent studies, is summarized in the table below.
Table 1: Comparison of DNA Extraction Method Performance for Metagenomics
| Method / Kit Name | Core Lysis Mechanism | Performance for HMW DNA | Key Advantages | Reported Limitations | Suitability for Long-Read Sequencing |
|---|---|---|---|---|---|
| Bead Beating + SDS-Chloroform [25] | Mechanical & Chemical | Good (16-20 kb) | High yield; effective for diverse cells [25] | Co-extracts inhibitors (e.g., humic acids) [25] | Good (after purification) |
| Quick-DNA HMW MagBead [5] | Mechanical (Beads) & Magnetic Purification | Excellent | Best yield of pure HMW DNA; accurate species detection [5] | - | Recommended |
| Enzymatic Lysis (MetaPolyzyme) [3] | Enzymatic | Excellent (2.1x longer reads) | Gentle lysis; superior DNA integrity; reduced shearing [3] | May require optimization for diverse cell walls [3] | Highly Suitable |
| QIAGEN PowerFecal Pro [12] | Mechanical & Chemical | Good | Reliable for complex matrices (e.g., wastewater); effective inhibitor removal [12] | - | Suitable and Reliable |
| Phenol-Chloroform (In-house) [12] [5] | Chemical & Physical | Good (Gentle) | Gentle; customizable | Time-consuming; uses hazardous chemicals [5] | Moderate |
Detailed Experimental Protocols
Optimized Bead Mill Homogenization Protocol for Soils and Sediments
This protocol, adapted from a foundational evaluation, is designed for maximum DNA recovery from challenging environmental samples like soils and sediments with high organic matter content [25].
3.1.1 Research Reagent Solutions
- Lysis Buffer: Phosphate-Tris buffer (pH 8), containing SDS, NaCl, and chloroform.
- Sephadex G-200: For spin column purification to remove PCR-inhibitory substances.
- CD1 Lysis Buffer: A component of the QIAGEN PowerFecal Pro kit, used for chemical lysis.
3.1.2 Step-by-Step Procedure
- Sample Preparation: Lyophilize and grind approximately 0.5 g of soil/sediment to a fine powder.
- Cell Lysis: Transfer the sample to a tube containing the lysis buffer and glass beads. Homogenize using a bead mill homogenizer at a low speed for a short duration (30-120 seconds) to maximize DNA size and yield [25].
- Incubation: Incubate the lysate at a elevated temperature (e.g., 60°C) for a period to facilitate complete lysis.
- Centrifugation: Centrifuge to pellet debris and transfer the supernatant to a new tube.
- Purification: Purify the crude DNA extract using a Sephadex G-200 spin column to effectively remove humic acids and other inhibitors while minimizing DNA loss [25].
- DNA Elution: Elute the purified DNA in a suitable buffer (e.g., TE buffer or nuclease-free water). Assess DNA concentration, purity (A260/A280 ratio), and fragment size using agarose gel electrophoresis.
Optimized Enzymatic Lysis Protocol for Clinical Urine Samples
This protocol, derived from a 2022 clinical study, prioritizes the recovery of long, intact DNA fragments from pathogens in urine samples, making it ideal for long-read sequencing applications [3].
3.2.1 Research Reagent Solutions
- MetaPolyzyme: A lytic enzyme solution reconstituted in PBS for gentle microbial cell wall degradation.
- IndiSpin Pathogen Kit: Used for DNA binding, washing, and elution after the enzymatic lysis step.
- Proteinase K: An enzyme used to degrade proteins and nucleases.
3.2.2 Step-by-Step Procedure
- Sample Enrichment: Centrifuge 1 ml of urine at 20,000 × g for 5 min. Discard 800 µl of supernatant and resuspend the pellet in the remaining 200 µl.
- Enzymatic Lysis: Add 10 µl of MetaPolyzyme to the enriched sample. Mix by gentle pipetting and incubate at 37°C for 1 hour in a shaker [3].
- DNA Extraction: Apply the post-lysed sample to the IndiSpin Pathogen Kit (or similar silica-membrane kit). Add Proteinase K and the kit's lysis buffer.
- Binding and Washing: Transfer the lysate to the spin column, centrifuge, and wash the membrane with the provided wash buffers.
- Elution: Elute the high-integrity DNA in 50-100 µl of elution buffer.
Workflow Visualization and the Scientist's Toolkit
The following diagram and table summarize the key decision points and tools for successful DNA extraction.
Diagram 1: DNA Extraction Decision Workflow
Table 2: The Scientist's Toolkit: Essential Reagents for DNA Extraction
| Reagent / Kit | Function | Key Application Note |
|---|---|---|
| Sodium Dodecyl Sulfate (SDS) | Ionic detergent that disrupts lipid membranes and lyses cells [25]. | Core component of direct lysis buffers for environmental samples [25]. |
| MetaPolyzyme | Enzyme cocktail that digests microbial cell walls gently [3]. | Ideal for clinical samples where preserving long DNA fragments is critical [3]. |
| Sephadex G-200 Resin | Gel filtration matrix that separates DNA from smaller inhibitor molecules [25]. | Superior to other methods for removing PCR inhibitors from soil extracts with minimal DNA loss [25]. |
| Magnetic Beads (e.g., MagBead) | SPRI beads that bind DNA for purification and size selection [5]. | Enables efficient washing and elution of HMW DNA; suitable for automation. |
| Phenol-Chloroform | Organic solvent mixture that denatures and removes proteins. | A traditional, gentle method for HMW DNA extraction, though hazardous [5]. |
| PowerFecal Pro Kit | Commercial kit optimized for inhibitor-laden fecal and environmental samples [12]. | A reliable, standardized method for complex matrices like piggery wastewater [12]. |
Methodology in Practice: Selecting and Applying DNA Extraction Kits and Protocols
Comparative Analysis of Commercial DNA Extraction Kits for Metagenomics
Metagenomic sequencing has revolutionized our understanding of microbial communities across diverse environments, from the human gut to complex soil ecosystems. The critical first step in any metagenomic study—DNA extraction—profoundly influences sequencing outcomes, microbial community representation, and ultimately, the biological conclusions that can be drawn. The selection of an appropriate DNA extraction method must balance multiple factors: efficient cell lysis across diverse microbial taxa, effective removal of PCR inhibitors, preservation of DNA integrity, and compatibility with downstream sequencing platforms.
This application note provides a comprehensive comparative analysis of leading commercial DNA extraction kits specifically designed for challenging metagenomic samples. We evaluate kits from prominent manufacturers including QIAGEN's PowerFecal Pro series and Macherey-Nagel's NucleoSpin Soil series, focusing on their performance characteristics, methodological considerations, and suitability for various sample types and sequencing applications. By synthesizing data from recent independent evaluations alongside manufacturer specifications, this document serves as a practical resource for researchers selecting optimal DNA extraction strategies for their metagenomic studies.
Commercial DNA extraction kits employ varied biochemical and mechanical approaches to address the fundamental challenges of metagenomic DNA isolation. The QIAGEN PowerFecal Pro DNA Kit utilizes a novel bead tube system combined with optimized chemistry for efficient lysis of bacteria and fungi, followed by streamlined inhibitor removal technology (IRT) to purify DNA from complex samples like stool and gut material [26]. The Macherey-Nagel NucleoSpin Soil Kit employs a dual-buffer system with specialized enhancers and mechanical disruption using included ceramic beads, coupled with a dedicated inhibitor removal column to eliminate humic acids and other contaminants common in environmental samples [27] [28].
Table 1: Key Specifications of Commercial DNA Extraction Kits for Metagenomics
| Kit Name | Target Sample Types | Lysis Method | Inhibitor Removal | Typelyield (Varies by Sample) | Downstream Applications | Automation Compatibility |
|---|---|---|---|---|---|---|
| QIAGEN QIAamp PowerFecal Pro DNA Kit | Stool, gut samples | Chemical + mechanical (bead beating) | Inhibitor Removal Technology (IRT) | Up to 20-fold more DNA compared to alternative methods [26] | NGS, PCR, sequencing | QIAcube Connect [26] |
| Macherey-Nagel NucleoSpin Soil Kit | Soil, sediment, sludge, peat | Chemical + mechanical (bead tubes) | NucleoSpin Inhibitor Removal Column | 2-10 µg (from 500 mg soil) [27] [28] | PCR, qPCR, microarrays, Southern blotting | Most open robotic platforms (96-well format) [29] |
| QIAGEN DNeasy PowerSoil Pro Kit | Soil, complex environmental samples | Chemical + mechanical | IRT technology | Varies by soil type | Long-read WGS metagenomics | Not specified |
| ZymoBIOMICS DNA Miniprep Kit | Various microbial communities | Bead beating | Proprietary purification | Varies by sample | Short- and long-read sequencing | Not specified |
Performance Comparison in Metagenomic Studies
DNA Yield, Purity, and Microbial Diversity Representation
Independent comparative studies provide critical insights into the performance characteristics of various DNA extraction kits. In evaluations for long-read shotgun metagenomics using Oxford Nanopore sequencing, the QIAGEN PowerFecal Pro DNA kit demonstrated excellent performance, correctly identifying all bacterial species present in both Zymo Mock Community (8/8) and ESKAPE Mock (6/6) communities at read and assembly levels [30]. The combination of chemical and mechanical lysis in this kit proved particularly effective for Gram-positive species, which often resist lysis by purely enzymatic methods.
A comprehensive 2023 preprint comparing four commercially available DNA extraction kits for whole metagenome shotgun sequencing found that kits differentially biased the percentage of reads attributed to microbial taxa across samples and body sites [31]. The PowerSoil Pro kit performed best in approximating expected proportions of mock communities, while the HostZERO kit, though biased against gram-negative bacteria, outperformed other kits in extracting fungal DNA [31].
In soil metagenomics, a 2024 study comparing five commercial soil DNA extraction kits for long-read sequencing found significant differences in extracted DNA length, read length, and detected microbial communities between kits [32]. The QIAGEN DNeasy PowerSoil Pro Kit displayed the best suitability for reproducible long-read whole genome shotgun metagenomic sequencing across diverse soil types [32].
Impact on Sequencing Performance and Community Representation
The choice of DNA extraction method significantly influences downstream sequencing metrics and microbial community representation. Research indicates that extraction kits not only affect DNA yield and purity but also introduce specific biases in microbial community composition that can impact biological interpretations [31].
In a study of clinical samples from oral, vaginal, and rectal sites, extraction kits showed significant differences in the fraction of reads assigned to host versus microbial DNA, with HostZERO yielding a smaller fraction of reads assigned to Homo sapiens across sites [31]. However, this kit also demonstrated the most dispersion in microbial community representation, particularly for vaginal and rectal samples, highlighting the trade-offs between different performance characteristics [31].
For long-read sequencing technologies, DNA extraction methods significantly impact read length and assembly quality. A 2024 evaluation of DNA extraction kits for Nanopore sequencing found that the Nanobind CBB Big DNA kit yielded the longest raw reads, while the Fire Monkey HMW-DNA Extraction Kit and automated Roche MagNaPure 96 platform outperformed in genome assembly, particularly for gram-negative bacteria [33].
Table 2: Performance Characteristics of DNA Extraction Kits in Independent Studies
| Performance Metric | PowerFecal Pro / PowerSoil Pro | NucleoSpin Soil | ZymoBIOMICS Miniprep | HostZERO Microbial DNA |
|---|---|---|---|---|
| Gram-positive lysis efficiency | High (mechanical lysis) [30] | Moderate to High (bead tubes) [27] | High (bead beating) [33] | Variable [31] |
| Gram-negative lysis efficiency | High [30] | High [27] | High [33] | Biased against [31] |
| Inhibitor removal | Efficient (IRT) [26] | Efficient (specialized column) [27] | Proprietary method [33] | Not specified |
| Fungal DNA recovery | Efficient [26] | Not specifically reported | Not specifically reported | Excellent [31] |
| Suitable for long-read sequencing | Yes [30] [32] | Limited data | Yes [33] | Limited data |
| Community representation accuracy | High for mock communities [31] [30] | Varies by soil type [32] | Variable [33] | Biased representation [31] |
Detailed Experimental Protocols
QIAamp PowerFecal Pro DNA Extraction Protocol
Principle: This protocol combines mechanical and chemical lysis through bead beating and optimized buffer systems, followed by efficient inhibitor removal and DNA purification on silica membranes [26].
Procedure:
- Sample Preparation: Weigh approximately 200 mg of stool sample and transfer to the PowerFecal Pro bead tube included in the kit.
- Lysis: Add 750 µL of PowerFecal Pro Solution CF1 to the bead tube. Secure the tube in a vortex adapter and vortex vigorously for 10-15 minutes, or process using a tissue lyser at 25 Hz for 5 minutes [30].
- Inhibitor Removal: Centrifuge the lysate and transfer supernatant to a clean microcentrifuge tube. Add 300 µL of Solution IR1 and mix by pulse-vortexing. Incubate at 4°C for 5 minutes.
- DNA Binding: Centrifuge the mixture and transfer supernatant to a new tube containing 900 µL of Solution PB. Mix and load onto the QIAamp spin column. Centrifuge at 17,000 x g for 1 minute.
- Washing: Add 650 µL of Solution PE to the column and centrifuge at 17,000 x g for 1 minute. Repeat with 650 µL of Solution AW1 and centrifuge. Transfer column to a new collection tube.
- Elution: Add 50-100 µL of Solution EB to the center of the membrane and incubate for 5 minutes at room temperature. Centrifuge at 17,000 x g for 1 minute to elute DNA.
Quality Control: Assess DNA concentration by fluorometric quantification (e.g., Qubit) and purity by A260/A280 ratio (typically ~1.8) [26].
NucleoSpin Soil DNA Extraction Protocol
Principle: This method uses mechanical disruption with ceramic beads combined with specialized lysis buffers tailored to different soil types, followed by purification through an inhibitor removal column and silica membrane [27] [28].
Procedure:
- Sample Preparation: Transfer up to 500 mg of soil sample to a MN Bead Tube Type A.
- Lysis Selection: Based on soil type, add 700 µL of either Lysis Buffer SL1 or SL2. For difficult soils, add Enhancer SX (100 µL).
- Homogenization: Secure tubes in a vortex adapter and vortex vigorously for 10 minutes, or process using a homogenizer.
- Centrifugation: Centrifuge the lysate at 11,000 x g for 1 minute.
- Inhibitor Removal: Transfer supernatant to a NucleoSpin Inhibitor Removal Column placed in a collection tube. Centrifuge at 11,000 x g for 1 minute.
- DNA Binding: Add 650 µL of Binding Buffer SB to the flow-through and mix by vortexing. Load onto a NucleoSpin Soil Column and centrifuge at 11,000 x g for 1 minute.
- Washing: Add 500 µL of Wash Buffer SW1 and centrifuge at 11,000 x g for 1 minute. Replace collection tube, add 500 µL of Wash Buffer SW2, and centrifuge at 11,000 x g for 1 minute.
- Elution: Transfer column to a clean microcentrifuge tube. Add 30-100 µL of pre-warmed (50°C) Elution Buffer SE to the center of the membrane. Incubate for 5 minutes at room temperature then centrifuge at 11,000 x g for 1 minute.
Quality Control: Typical yields range from 2-10 µg DNA from 500 mg soil with A260/A280 ratios of 1.6-1.8 [27] [28].
Workflow Visualization
Diagram 1: Comparative Workflow of DNA Extraction Kits. This diagram illustrates the parallel processes for the PowerFecal Pro and NucleoSpin Soil kits, highlighting their shared workflow structure with different implementations at each step. Both methods begin with sample collection, proceed through specialized lysis and inhibitor removal steps, then through purification and quality assessment before downstream applications.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagent Solutions for Metagenomic DNA Extraction
| Reagent/Kit Component | Function | Example Kits |
|---|---|---|
| Lysis Buffers (SL1/SL2) | Chemical disruption of cell membranes; SL1 for standard soils, SL2 for humic acid-rich soils | NucleoSpin Soil [27] |
| Bead Tubes (Ceramic/Silica) | Mechanical disruption of tough cell walls through bead beating | NucleoSpin Soil (Type A), PowerFecal Pro [26] [27] |
| Inhibitor Removal Technology (IRT) | Selective binding and removal of PCR inhibitors (humic acids, bilirubin, etc.) | PowerFecal Pro [26] |
| Enhancer SX | Additional chemical treatment for difficult-to-lyse microorganisms in complex soils | NucleoSpin Soil [27] |
| Silica Membranes/Columns | Selective binding of DNA based on size and salt conditions | Both kits [26] [27] |
| Binding Buffer SB | Creates optimal salt conditions for DNA binding to silica membrane | NucleoSpin Soil [28] |
| Wash Buffers (SW1/SW2) | Remove contaminants while retaining bound DNA | NucleoSpin Soil [28] |
| Elution Buffer (SE/EB) | Low-salt solution that releases purified DNA from membrane | Both kits [26] [28] |
The optimal selection of DNA extraction methods for metagenomic studies depends on sample type, target microorganisms, and downstream sequencing applications. Based on current comparative evaluations:
For stool and gut microbiome studies, the QIAGEN PowerFecal Pro DNA kit demonstrates superior performance in DNA yield, purity, and microbial diversity representation, particularly for long-read sequencing applications [26] [30]. Its integrated mechanical and chemical lysis efficiently handles both Gram-positive and Gram-negative bacteria, while the proprietary inhibitor removal technology effectively eliminates common PCR inhibitors present in stool samples.
For soil and environmental samples, both the QIAGEN PowerSoil Pro and Macherey-Nagel NucleoSpin Soil kits offer robust solutions, with the former showing advantages in long-read sequencing applications [32] and the latter providing flexibility through its dual-buffer system for different soil types [27]. The NucleoSpin Soil kit's availability in 96-well format makes it particularly suitable for high-throughput studies [29].
Researchers should consider that no extraction method is completely unbiased, and kit selection introduces specific alterations in microbial community representation that must be considered in data interpretation [31]. For comparative studies, consistency in extraction methodology is essential, and the inclusion of mock communities is strongly recommended to quantify technical variability and bias [31] [5].
As sequencing technologies continue to evolve toward longer reads and single-molecule applications, further optimization of DNA extraction protocols will be necessary to preserve DNA integrity while maintaining representative lysis across diverse microbial communities.
Within metagenomic sequencing research, the efficacy of DNA extraction is a pivotal determinant of downstream success. The initial step of cell lysis—the disruption of the cellular envelope to release genetic material—introduces a significant potential for bias, particularly in complex samples containing a mixture of organisms with diverse cell wall structures. The fundamental challenge lies in the starkly different resistance levels exhibited by Gram-positive bacteria, Gram-negative bacteria, and fungi, largely dictated by the biochemical composition of their walls. Inadequate lysis leads to under-representation of robust organisms, while excessively harsh methods can shear DNA and co-extract inhibitors, thereby skewing the apparent taxonomic composition and functional potential of the microbial community [30] [34].
This application note provides a structured comparison between two core lysis strategies: mechanical (with a focus on bead-beating) and enzymatic lysis. We detail the principles, advantages, and limitations of each method, providing definitive protocols and data to guide researchers in selecting and optimizing the lysis step for unbiased DNA extraction in metagenomic studies.
Understanding Cell Wall Architecture and Its Impact on Lysis
The efficiency of any lysis method is inherently linked to the architecture of the cell wall it aims to disrupt. The three primary cellular morphologies encountered in metagenomics present distinct challenges.
- Gram-Positive Bacteria: These cells possess a thick, multi-layered mesh of peptidoglycan fortified with teichoic acids, forming a formidable physical barrier that is highly resistant to simple chemical or osmotic lysis [34] [35].
- Gram-Negative Bacteria: These have a more complex envelope with a thin peptidoglycan layer sandwiched between an inner cytoplasmic membrane and an outer membrane composed of lipopolysaccharides (LPS). While the peptidoglycan layer is thinner, the outer membrane acts as a robust permeability barrier, often requiring chelating agents like EDTA to create pores before lytic enzymes can access their substrate [36] [35].
- Fungi (e.g., Yeasts): Fungal cell walls are robust structures primarily composed of chitin and β-glucans (polymers of glucose). This rigid, carbohydrate-rich matrix necessitates particularly aggressive disruption methods for efficient nucleic acid release [37] [35].
The following workflow diagram outlines a decision-making process for selecting an appropriate lysis strategy based on sample composition and research goals.
Mechanical Lysis: Bead-Beating
Principle and Applications
Bead-beating is a mechanical homogenization method that utilizes rapid, high-energy shaking of a sample with dense, microscopic beads. This action subjects cells to solid shear forces, grinding, and impaction, which physically tears apart tough cell walls [36] [38]. It is exceptionally effective for organisms that are recalcitrant to other methods, making it the gold standard for lysing Gram-positive bacteria and fungi [39] [37]. Its non-selectivity also ensures a more balanced lysis across diverse community members in a metagenomic context, although parameters must be optimized to prevent excessive DNA shearing.
Key Experimental Protocol: Bead-Beating for DNA Extraction
Title: Optimization of Bead-Beating for Maximal DNA Yield from Gram-Positive Bacteria and Fungi.
Objective: To efficiently disrupt tough cell walls in a mixed sample for subsequent metagenomic DNA extraction.
Materials & Reagents:
- Sample: Bacterial pellet or fungal biomass.
- Lysis Buffer: Commercially available buffer (e.g., from QIAamp PowerFecal Pro DNA kit) or Tris-EDTA-SDS buffer.
- Beads: A mixture of 0.1 mm glass beads and 0.5 mm zirconium/silica beads is recommended for comprehensive lysis of different cell sizes and types [30] [38].
- Equipment: High-throughput bead beater (e.g., FastPrep-96) or vortex adapter with a standard vortex mixer set to maximum speed.
Method:
- Preparation: Transfer up to 200 mg of sample (or pellet from 1-2 mL culture) to a 2 mL lysing matrix tube containing the beads.
- Buffer Addition: Add 800 µL - 1 mL of lysis buffer and any required proteinase K to the tube.
- Homogenization: Secure tubes in the bead beater.
- Clarification: Centrifuge the tubes at >12,000 × g for 5 minutes to pellet cell debris and beads.
- Recovery: Carefully transfer the supernatant (containing the released DNA) to a clean tube for subsequent purification steps.
Critical Parameters:
- Bead Composition: The size, shape, and material of beads drastically impact efficiency. Smaller beads provide more surface area for grinding, while angular beads provide higher shear forces. Zirconium oxide beads are particularly effective for tough samples [38].
- Cycle Optimization: Excessive beating can fragment genomic DNA into sizes too short for long-read sequencing. The number and duration of cycles should be empirically determined for each sample type [30].
Enzymatic Lysis
Principle and Applications
Enzymatic lysis employs specific enzymes to catalytically degrade key structural components of the cell wall. This method is gentle, operates under mild conditions (e.g., 37°C), and preserves the integrity of high-molecular-weight DNA and intracellular organelles [36] [40]. Its selectivity, however, can be a source of bias if the sample contains organisms resistant to the enzyme used.
Common enzymes include:
- Lysozyme: Hydrolyzes β-1,4-glycosidic bonds between N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) in bacterial peptidoglycan. It is most effective against Gram-positive bacteria but requires pre-treatment with EDTA to permeabilize the outer membrane of Gram-negative species [36] [35].
- Zymolyase: A commercial enzyme preparation with β-1,3-glucanase activity, which targets the primary structural glucan in the cell walls of yeasts and fungi [37] [35].
- Mutanolysin: Effective for degrading peptidoglycan in Gram-positive bacteria, often used in combination with lysozyme.
Key Experimental Protocol: Enzymatic Lysis for Gram-Negative Bacteria
Title: Enzymatic Lysis of Gram-Negative Bacteria using Lysozyme and EDTA.
Objective: To gently lyse Gram-negative bacterial cells while maximizing DNA length.
Materials & Reagents:
- Lysozyme Solution: 20-50 mg/mL in Tris-EDTA (TE) buffer.
- EDTA Solution: 0.5 M, pH 8.0.
- Other Reagents: Proteinase K, SDS solution.
Method:
- Resuspension: Suspend the bacterial pellet in TE buffer containing 20 mM EDTA.
- Permeabilization: Add lysozyme to a final concentration of 1-2 mg/mL. Mix thoroughly and incubate at 37°C for 30-60 minutes.
- Lysis: Add SDS to a final concentration of 1% and Proteinase K to 100 µg/mL. Invert tubes gently to mix.
- Digestion: Incubate at 56°C for 60 minutes or until the solution becomes clear and viscous.
- Inactivation: Proceed to a standard phenol-chloroform extraction or use a commercial DNA purification kit.
Critical Parameters:
- EDTA is Crucial: For Gram-negative bacteria, EDTA chelates divalent cations (Mg²⁺) that stabilize the LPS layer, creating pores that allow lysozyme to access the underlying peptidoglycan [36] [35].
- Enzyme Specificity: Enzymatic methods are highly specific. A metagenomic sample with unknown diversity will likely require a cocktail of enzymes (e.g., lysozyme, mutanolysin, zymolyase) for complete community representation, which can be costly and complex.
Comparative Data and Strategic Selection
Quantitative Comparison of Lysis Methods
The table below summarizes the performance of mechanical and enzymatic lysis across key criteria relevant to metagenomic sequencing.
Table 1: Comparative Analysis of Mechanical Bead-Beating vs. Enzymatic Lysis
| Criterion | Mechanical Bead-Beating | Enzymatic Lysis |
|---|---|---|
| Lysis Principle | Physical shearing and grinding [36] [38] | Catalytic degradation of cell wall polymers [36] [35] |
| Efficiency on Gram-Positive Bacteria | High (e.g., >15-fold RNA yield increase in L. lactis) [39] | Moderate to Low (thick peptidoglycan is a barrier) [36] |
| Efficiency on Gram-Negative Bacteria | High | High (when combined with EDTA) [36] [35] |
| Efficiency on Fungi/Yeast | High (100% lysis for C. albicans with optimized protocol) [37] | Moderate (requires specific enzymes like Zymolyase) [37] [35] |
| DNA Shearing Risk | Higher (must be optimized to prevent fragmentation) [30] | Lower (gentle process preserves high molecular weight DNA) |
| Potential for Community Bias | Lower (non-specific, broad-range disruption) [30] | Higher (selective for susceptible organisms) [34] |
| Throughput & Automation | High (compatible with 96-well formats) [38] | Moderate (incubation steps lengthen workflow) |
| Cost & Complexity | Moderate (requires specialized equipment) | Low to High (simple setup, but enzyme cocktails can be costly) |
Reagent and Solution Toolkit for Lysis
Table 2: Essential Research Reagents for Cell Lysis
| Reagent / Kit | Function / Principle | Example Application |
|---|---|---|
| QIAamp PowerFecal Pro DNA Kit (Qiagen) | Utilizes chemical and mechanical lysis (bead-beating) with an inhibitor removal technology [30]. | Optimal for soil, stool, and complex samples for balanced Gram-positive/negative lysis in metagenomics [30]. |
| Lysing Matrix Tubes (MP Bio) | Pre-filled tubes with a blend of bead sizes/materials (e.g., zirconium silicate, ceramic) for optimized mechanical disruption [38]. | Standardized bead-beating for diverse sample types, from bacteria to seeds and bone [38]. |
| Lysozyme (from hen egg white) | Glycoside hydrolase that breaks down peptidoglycan in bacterial cell walls [36] [35]. | Core enzyme for lysing Gram-positive bacteria; used with EDTA for Gram-negative bacteria [36] [35]. |
| Zymolyase | Enzyme mixture with β-1,3-glucanase activity that degrades the glucan layer in yeast cell walls [37] [35]. | Essential for efficient lysis of yeast and fungal cells (e.g., C. albicans, S. cerevisiae) [37]. |
| EDTA (Ethylenediaminetetraacetic acid) | Chelating agent that binds Mg²⁺ and Ca²⁺, destabilizing the outer membrane of Gram-negative bacteria [36]. | Used as a pre-treatment to permeabilize Gram-negative cells prior to enzymatic lysis [36] [35]. |
The choice between mechanical and enzymatic lysis is not a matter of superiority but of strategic application. For a typical metagenomic study where the sample composition is unknown or known to contain tough-walled organisms, bead-beating is the recommended default method due to its broad efficacy and lower potential for community bias [30]. However, for projects targeting primarily Gram-negative bacteria or requiring extremely high-molecular-weight DNA, a gentle enzymatic approach may be preferable.
For the most challenging and diverse samples, a hybrid strategy that combines a brief mechanical lysis step with a subsequent enzymatic treatment can offer the most comprehensive disruption, ensuring all cell types are efficiently lysed for a truly representative metagenomic analysis [30]. The protocols and data provided herein serve as a foundation for researchers to tailor their lysis strategy, thereby laying the groundwork for robust and unbiased metagenomic insights.
Effective DNA extraction is the cornerstone of reliable metagenomic sequencing, yet the optimal methodology is highly dependent on sample type. Complex matrices such as wastewater, blood, and sputum present unique challenges, including the presence of PCR inhibitors, difficult-to-lyse cell walls, and low microbial biomass. Inefficient nucleic acid recovery or failure to remove inhibitors can significantly bias sequencing results and impact downstream analyses. This application note provides a consolidated guide of optimized, sample-specific DNA extraction protocols to support researchers and drug development professionals in obtaining high-quality genetic material for metagenomic research.
Sample-Specific DNA Extraction Methodologies
The following section details optimized protocols for various sample types, with key performance metrics summarized for comparison.
Table 1: Comparison of Optimized DNA Extraction Methods Across Sample Types
| Sample Type | Optimized Method / Kit | Key Modifications / Notes | Performance Metrics | Primary Challenge Addressed |
|---|---|---|---|---|
| Wastewater (Piggery) | QIAGEN QIAamp PowerFecal Pro DNA Kit [12] | Reduced CD1 buffer volume (500 µL); mechanical lysis (10 min vortex); extended ice incubation (5 min) during wash [12]. | Most suitable/reliable for pathogen detection via ONT sequencing [12]. | Inhibitor removal; representative pathogen recovery [12]. |
| Blood (Dried Blood Spots) | Chelex-100 Boiling Method [41] | Single 6 mm punch; elution volume of 50 µL [41]. | Significantly higher DNA yield vs. column-based methods (p<0.0001) [41]. | Low DNA yield from limited sample input [41]. |
| Blood (Liquid Whole Blood in EDTA) | QIAamp DNA Blood Kit (for DNA); NucleoSpin RNA Kit (for RNA) [42] | Thawing samples on aluminum blocks at room temperature instead of 37°C water bath [42]. | ~20% increase in DNA yield; higher RNA integrity numbers (RINs) [42]. | Nucleic acid degradation during thawing [42]. |
| Urine (Microbiome) | Quick-DNA Urine Kit with Water Dilution Protocol (WDP) [43] | Pre-dilution of 6 mL urine with 4 mL UltraPure water prior to conditioning buffer [43]. | Superior DNA purity (260/280 ratio: 1.53); reduced contamination; higher microbial abundance (p<0.0001) [43]. | Low bacterial concentration; presence of PCR inhibitors [43]. |
| Sputum (Bacteria) | High Pure PCR Template Preparation Kit (Roche) [44] | Pre-treatment with Dithiothreitol (DTT) and enzymatic digestion (Lysozyme & Lysostaphin) [44]. | Highest DNA yield; lower coefficient of variation between replicates [44]. | Sample heterogeneity; robust bacterial cell walls (e.g., S. aureus) [44]. |
| Sputum (Mycobacterium tuberculosis) | Chelex-100 Resin Boiling Method [45] | Optimized for paucibacillary specimens; targets multi-copy IS6110 element [45]. | High sensitivity (95.1%) and specificity (100%); superior to Xpert MTB/RIF for low bacterial load (75% vs 55%, p=0.03) [45]. | Tough mycobacterial cell wall; low bacillary load in samples [45]. |
Detailed Experimental Protocols
Optimized Protocol for Piggery Wastewater Pathogen Surveillance
This protocol, optimized for Oxford Nanopore Technology (ONT) sequencing, is designed for effective pathogen detection from a complex environmental matrix [12].
- Sample Preparation: Centrifuge 10-40 mL of wastewater (volume dependent on particulate content) at 46 g for 1 min. Transfer supernatant and centrifuge at 4,550 g for 30 min. Discard supernatant and weigh pellet [12].
- Homogenate Reconstitution: Thaw pellet and reconstitute in 500 µL Milli-Q water. Use 0.3 g of homogenate for extraction [12].
- Cell Lysis: Add 500 µL of CD1 lysis buffer (instead of recommended 800 µL) to the homogenate. Mechanically lyse using a vortex adapter at maximum speed for 10 min [12].
- DNA Binding & Washing: Follow kit instructions with a modified wash step: perform two washes with 250 µL of solution C5, each followed by incubation on ice for 5 min and centrifugation at 13,000 g [12].
- DNA Elution: After final wash, leave column lid open for 10 min to evaporate residual ethanol. Add solution C6 and elute DNA in 50 µL elution buffer [12].
Optimized Protocol for Microbial DNA from Urine
The Water Dilution Protocol (WDP) significantly improves DNA purity from urine samples for microbiome studies [43].
- Sample Dilution: Mix 6 mL of urine with 4 mL of UltraPure distilled water in a sterile tube [43].
- Conditioning: Add the recommended volume of Urine Conditioning Buffer to the diluted sample and mix thoroughly [43].
- DNA Extraction: Continue with the standard protocol for the Quick-DNA Urine Kit as per the manufacturer's instructions [43].
- Storage: Store the extracted DNA at -80°C. Assess concentration and purity using spectrophotometry (e.g., NanoDrop) [43].
Optimized Protocol for Sputum Microbiota Analysis
This protocol combines chemical, enzymatic, and mechanical lysis to maximize bacterial DNA recovery from sputum [44].
- Homogenization: Treat sputum sample with Dithiothreitol (DTT) to break down mucoprotein disulfide bonds. This step reduces sample heterogeneity and improves reproducibility [44].
- Enzymatic Lysis: Incubate the homogenized sample with a cocktail of lytic enzymes. Use lysostaphin (0.18-0.36 mg/mL) and lysozyme (3.6 mg/mL) to effectively lyse robust Gram-positive cell walls (e.g., Staphylococcus aureus) [44].
- DNA Extraction: Extract DNA using the High Pure PCR Template Preparation Kit (Roche) according to the manufacturer's protocol [44].
- Downstream Application: The extracted DNA is suitable for 16S rRNA gene sequencing and other metagenomic applications [44].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for DNA Extraction Optimization
| Reagent / Kit | Primary Function | Application Notes |
|---|---|---|
| Chelex-100 Resin | Chelating resin that binds metal ions, inhibiting nucleases; used in simple boiling protocols [41] [45]. | Ideal for cost-effective, high-throughput screening from DBS [41] and efficient lysis of tough cells like M. tuberculosis [45]. |
| Dithiothreitol (DTT) | Reducing agent that breaks disulfide bonds in mucoproteins [44]. | Critical for homogenizing viscous sputum samples prior to DNA extraction, improving yield and reproducibility [44]. |
| Lytic Enzymes (Lysozyme, Lysostaphin) | Enzymatically degrade specific bacterial cell wall components [44]. | Essential for lysing challenging Gram-positive bacteria (e.g., S. aureus) in sputum; significantly improves detection [44]. |
| QIAGEN QIAamp PowerFecal Pro DNA Kit | Silica-membrane based technology for DNA purification from complex, inhibitor-rich samples [12]. | Demonstrated superior performance for pathogen detection from piggery wastewater via ONT sequencing [12]. |
| EDTA (Ethylenediaminetetraacetic acid) | Chelating agent that binds calcium and other metal ions [43] [46]. | Helps dissolve urinary crystals, reducing PCR inhibition and improving DNA yield from urine [43]. |
Workflow Visualization
Optimizing DNA extraction for the specific sample matrix is a critical first step in any robust metagenomic sequencing pipeline. As demonstrated, this often requires more than simply selecting a commercial kit; it involves strategic pre-treatment steps, such as DTT for sputum or water dilution for urine, and protocol modifications like mechanical lysis duration or elution volume adjustment. The protocols detailed herein provide a validated foundation for researchers to obtain high-quality, unbiased nucleic acid extracts from challenging sample types, thereby ensuring the reliability and reproducibility of downstream sequencing data and analyses.
The reliability of metagenomic sequencing research is fundamentally dependent on the initial quality and purity of extracted DNA. The choice of extraction methodology can introduce significant biases, affecting the apparent composition of microbial communities and the downstream ability to assemble genomes [13]. For modern, high-throughput laboratories, the decision often centers on two dominant technologies: silica spin columns and magnetic beads. Both methods exploit the affinity of DNA for silica in the presence of chaotropic salts, but their mechanisms and practical applications differ substantially [47]. Spin columns offer a simple, low-equipment pathway suitable for moderate throughput, while magnetic beads provide a scalable, automation-friendly platform ideal for processing hundreds to thousands of samples with minimal hands-on time and greater consistency [48] [49]. This application note provides a detailed comparison of these methods, supported by quantitative data and standardized protocols, to guide researchers in selecting and implementing the optimal DNA extraction strategy for their metagenomic sequencing projects.
Technology Comparison and Performance Benchmarking
Core Principles and Comparative Advantages
The operational divergence between these two methods leads to distinct performance characteristics, cost structures, and suitability for different laboratory workflows.
Silica Spin Columns utilize a silica membrane embedded in a plastic column. Under high-salt conditions, DNA binds to the silica, and contaminants are removed through a series of wash steps via centrifugation. Purified DNA is then eluted in a low-ionic-strength buffer [47]. This method is renowned for its simplicity and does not require specialized equipment beyond a standard microcentrifuge, making it accessible for low-to-moderate throughput labs. However, its scalability is limited by the need for sequential tube handling and centrifugation, and it often exhibits higher DNA loss, particularly with low-concentration samples or small fragments [49].
Magnetic Bead methods rely on paramagnetic particles coated with a silica surface. When added to a lysed sample, the beads bind DNA. A magnetic field is then applied to immobilize the bead-DNA complexes, allowing the supernatant containing impurities to be removed. After washing, the purified DNA is eluted [47]. This solid-phase reversible immobilization (SPRI) mechanism is inherently scalable and automation-compatible, enabling parallel processing of 96- or 384-well plates [50]. A key advantage is the tunable binding chemistry, where adjusting the polyethylene glycol (PEG) and salt concentration allows for precise size selection of DNA fragments, a critical feature for optimizing various sequencing platforms [49].
Quantitative Performance and Cost Analysis
The following tables summarize key performance metrics and cost considerations, synthesized from comparative evaluations.
Table 1: Performance and Operational Comparison
| Feature | Magnetic Bead Method | Silica Spin Column Method |
|---|---|---|
| Recovery Yield | 94–96% [49] | 70–85% [49] |
| DNA Size Range | 100 bp – 50 kb [49] | 100 bp – 10 kb [49] |
| Size Selection | Yes (via bead-to-sample ratio) [49] | No |
| Throughput & Automation | High (96-well & full automation) [48] [49] | Low (manual, single-tube) |
| Protocol Time (for 96 samples) | ~15 minutes [49] | 4–6 hours (sequential) |
| Typical Cost per Sample | ~$0.90 [49] | ~$1.75 [49] |
Table 2: Application-Specific Performance in Metagenomic Studies
| Sample Type / Application | Magnetic Bead Performance | Silica Spin Column Performance | Key Citation |
|---|---|---|---|
| Vertebrate Faecal Samples (Hologenomics) | Comparable host genome coverage and microbial community profiles to commercial spin column kits [48] | Effective but may have cost and reproducibility limitations for high-throughput workflows [48] | [48] |
| Marine Metagenomics (Water, Sediment) | High purity and effective inhibitor removal; performance varies by specific kit [13] | Varies significantly by kit; some show good purity but lower efficiency for tough cells [13] | [13] |
| Piggery Wastewater (Pathogen Surveillance) | Optimized PowerFecal Pro (magnetic bead-based) identified as most suitable and reliable method [12] | Not top-performing in this complex matrix for pathogen detection [12] | [12] |
| Inhibitor-Rich Samples (e.g., Plant) | High tolerance to viscous lysates; effective removal of polysaccharides and polyphenols [50] | Can be overwhelmed by inhibitors without extensive protocol modifications [50] | [50] |
| Automated Library Prep | Minimal impact on community structure; slightly higher alpha diversity and classification rate vs. manual [51] | Not suitable for automated liquid handling platforms [49] | [51] |
Workflow Visualization
The following diagram illustrates the key procedural differences between the two DNA extraction methods, highlighting the parallel processing advantage of magnetic beads.
Detailed Experimental Protocols
Protocol 1: High-Throughput DNA Extraction Using Magnetic Beads
This protocol is adapted from the open-source DREX procedure benchmarked by the Earth Hologenome Initiative for hologenomic data generation from vertebrate faecal samples [48]. It is optimized for a 96-well plate format.
Research Reagent Solutions & Essential Materials
| Item | Function/Benefit |
|---|---|
| Lysing Matrix E Tubes | Mechanically disrupts tough cell walls via bead beating [48]. |
| DNA/RNA Shield | Preservation buffer that stabilizes nucleic acids and inhibits RNases until extraction [48]. |
| Guanidinium Thiocyanate-based Lysis/Binding Buffer | Chaotropic salt that denatures proteins, releases nucleic acids, and enables binding to silica [48]. |
| Silica-Coated Magnetic Beads | Solid phase for reversible nucleic acid binding (e.g., MagMAX, HighPrep) [52] [49]. |
| Wash Buffer (with Ethanol) | Removes salts, proteins, and other contaminants while keeping DNA bound. |
| Elution Buffer (TE or water) | Low-ionic-strength solution to release pure DNA from the beads. |
| Automated Liquid Handler | Platform for high-throughput, reproducible pipetting (e.g., Agilent Bravo, Thermo Fisher KingFisher) [51] [49]. |
Step-by-Step Methodology
Sample Homogenization and Lysis:
- Transfer up to 100 mg of preserved faecal material to a tube containing Lysing Matrix E and lysis/binding buffer [48].
- Homogenize using a bead beater (e.g., TissueLyser II) at 30 Hz for two 6-minute intervals. Invert tubes between runs [48].
- Centrifuge briefly to pellet debris and transfer the clarified lysate to a deep-well 96-well plate.
Nucleic Acid Binding:
- Add a calculated volume of silica-coated magnetic beads to each well of the lysate plate. For total nucleic acid recovery, a 1.8x bead-to-sample ratio is a robust starting point [49].
- Mix thoroughly by pipetting or plate vortexing. Incubate at room temperature for 5 minutes to allow DNA binding to the beads [49].
Magnetic Separation and Washing:
- Transfer the plate to a magnetic stand and wait until the supernatant is clear and the beads form a pellet (approximately 2-5 minutes).
- Carefully aspirate and discard the supernatant without disturbing the bead pellet.
- With the plate positioned on the magnetic stand, add 200 µL of freshly prepared 80% ethanol to each well. Incubate for 30 seconds, then aspirate and discard the ethanol. Repeat this wash step a second time [49].
- Air-dry the bead pellet for 3-5 minutes at room temperature. Ensure all residual ethanol has evaporated, but avoid over-drying, which can reduce DNA elution efficiency [49].
DNA Elution:
- Remove the plate from the magnetic stand.
- Add 50-100 µL of elution buffer (e.g., TE buffer or nuclease-free water) to each well and resuspend the beads thoroughly by pipetting.
- Incubate at room temperature for 2-5 minutes to allow DNA to dissociate from the beads.
- Return the plate to the magnetic stand. Once the beads have pelleted, transfer the supernatant containing the purified DNA to a new plate.
Protocol 2: Manual DNA Extraction Using Silica Spin Columns
This protocol is based on methodologies used in comparative studies for DNA purification from complex samples like faeces and soil [13] [52].
Research Reagent Solutions & Essential Materials
| Item | Function/Benefit |
|---|---|
| Proteinase K | Digest proteins and inactivate nucleases during lysis. |
| Chaotropic Salt Binding Buffer | Creates high-salt conditions necessary for DNA to bind the silica membrane [47]. |
| Silica Spin Column | Contains a membrane that selectively binds DNA. |
| Wash Buffer (often ethanol-based) | Removes contaminants without eluting DNA from the membrane. |
| Elution Buffer | Low-salt buffer (TE or water) used to release purified DNA from the membrane. |
Step-by-Step Methodology
Sample Lysis:
- Add 200 mg of sample to a microcentrifuge tube with lysis buffer and Proteinase K.
- Vortex thoroughly and incubate at 56°C for 30 minutes to several hours, depending on sample toughness [13].
- Centrifuge at full speed for 1-2 minutes to pellet insoluble debris.
DNA Binding:
- Transfer the supernatant to a new tube and add the appropriate volume of binding buffer.
- Load the mixture onto the silica spin column and centrifuge at ≥ 10,000 g for 1 minute. Discard the flow-through [47].
Washing:
- Add the first wash buffer to the column. Centrifuge for 1 minute and discard the flow-through.
- Add a second, often ethanol-based, wash buffer. Centrifuge for 1 minute and discard the flow-through.
- Centrifuge the empty column for an additional 1-2 minutes to ensure all residual ethanol is removed.
DNA Elution:
- Place the column in a clean 1.5 mL microcentrifuge tube.
- Apply 50-100 µL of pre-warmed (55-65°C) elution buffer directly to the center of the silica membrane.
- Allow it to incubate for 1-5 minutes, then centrifuge for 1 minute to collect the purified DNA.
The choice between magnetic beads and silica spin columns is strategic and should be driven by the project's scale, budget, and required data quality. Magnetic bead technology is unequivocally superior for high-throughput metagenomic studies where throughput, reproducibility, and cost-efficiency are paramount. Its automation compatibility and high recovery yields make it the preferred choice for large-scale projects like the Earth Hologenome Initiative [48] and automated environmental metagenomics [51]. Silica spin columns remain a viable and straightforward option for low-throughput pilot studies, individual sample analysis, or laboratories with limited capital equipment budgets.
For researchers transitioning to high-throughput workflows, initiating the process with a validated magnetic bead kit, such as the QIAamp PowerFecal Pro for complex matrices [12] or the DREX protocol for faecal hologenomics [48], on a manual magnetic rack is a recommended first step. Subsequent integration with an automated liquid handling platform will ultimately unlock the full potential of magnetic bead extraction, enabling robust, reproducible, and scalable metagenomic sequencing.
Protocol Modifications for Long-Read Sequencing Technologies (Oxford Nanopore, PacBio)
Long-read sequencing technologies from Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) have revolutionized genomic analysis by enabling the sequencing of DNA and RNA fragments spanning thousands to hundreds of thousands of bases. These technologies provide unparalleled access to repetitive regions, structural variants, and complex genomic architectures that were previously inaccessible with short-read technologies [53]. The successful application of these platforms, however, is profoundly dependent on initial sample preparation, particularly the quality and integrity of input DNA. This protocol outlines optimized methodologies for sample preparation across diverse research contexts, with specific emphasis on metagenomic sequencing of complex environmental samples.
The fundamental difference between long-read and short-read technologies lies in their analysis of native DNA molecules. While short-read methods require DNA fragmentation and amplification, long-read technologies typically sequence molecules directly extracted from biological samples, preserving epigenetic modifications and eliminating amplification biases [54] [55]. This direct approach places exceptional importance on extraction methods that maximize DNA length, purity, and molecular weight. For metagenomic research involving complex matrices such as soil or wastewater, the challenge is further compounded by the presence of inhibitors that can interfere with downstream sequencing [12].
Oxford Nanopore Technology (ONT)
ONT sequencing is based on the translocation of nucleic acids through protein nanopores embedded in an electro-resistant membrane. Each nanopore is associated with its own electrode and sensor chip that measures changes in ionic current as DNA or RNA passes through the pore. These current changes produce characteristic "squiggles" that are decoded into sequence data in real-time using basecalling algorithms [56] [57]. Key features of ONT sequencing include:
- Real-time analysis: Data can be analyzed as sequencing occurs
- Direct RNA and DNA sequencing: Capable of sequencing native nucleic acids without conversion
- Modification detection: Can identify base modifications simultaneously with nucleotide sequence
- Scalability: Platforms range from pocket-sized MinION to high-throughput PromethION systems [56]
Pacific Biosciences (PacBio) HiFi Sequencing
PacBio's HiFi (High Fidelity) sequencing utilizes Single Molecule, Real-Time (SMRT) technology based on a nanofluidic chip called a SMRT Cell containing millions of zero-mode waveguides (ZMWs). Within each ZMW, a single DNA polymerase enzyme incorporates fluorescently labeled nucleotides into a complementary strand. The light pulses emitted during nucleotide incorporation are detected and used to determine the sequence [54]. The technology employs circular consensus sequencing (CCS), where the polymerase repeatedly traverses the same circularized DNA molecule, generating multiple subreads that are consolidated into one highly accurate HiFi read [54] [55].
Table 1: Comparative Analysis of Long-Read Sequencing Technologies
| Parameter | Oxford Nanopore Technologies | PacBio HiFi Sequencing |
|---|---|---|
| Read Length | 20 bp to >4 Mb [55] | 500 bp to 20 kb [55] |
| Accuracy | ~Q20 (99%) [55] | Q33 (99.9%) [55] |
| Input Material | DNA, RNA [56] [55] | DNA, cDNA [55] |
| Epigenetic Detection | Direct detection of 5mC, 5hmC, 6mA [58] [59] | Detection of 5mC, 6mA [54] |
| Typical Run Time | Up to 72 hours [55] | 24 hours [55] |
| Key Advantage | Portability, ultra-long reads, direct RNA sequencing | Very high accuracy, uniform coverage |
| Key Limitation | Lower raw read accuracy, high file storage requirements | Higher system cost, requires more input DNA [53] |
Optimized DNA Extraction Protocol for Complex Metagenomic Samples
The following protocol has been optimized specifically for long-read sequencing of complex environmental samples, based on rigorous comparative studies [12].
Sample Collection and Pre-processing
- Sample Collection: Collect wastewater or soil samples in sterile containers. Transport on ice and store at -20°C until processing.
- Pre-processing: Centrifuge 10-40 mL of wastewater at 46 × g for 1 minute to sediment heavy solids. Transfer supernatant to a new tube and centrifuge at 4,550 × g for 30 minutes. Discard supernatant and weigh the pellet [12].
- Homogenization: Reconstitute pellet in 500 μL Milli-Q water. Use 0.3 g of homogenized material for DNA extraction [12].
DNA Extraction Using Optimized QIAGEN PowerFecal Pro Protocol
Based on comparative evaluation of six extraction methods for piggery wastewater (a complex matrix rich in inhibitors), the optimized QIAGEN PowerFecal Pro protocol demonstrated superior performance for long-read sequencing [12].
Table 2: Research Reagent Solutions for DNA Extraction
| Reagent/Kit | Function | Optimization Notes |
|---|---|---|
| QIAGEN QIAamp PowerFecal Pro DNA Kit | Primary DNA extraction | Modified protocol showed best performance for complex samples [12] |
| CD1 Lysis Buffer | Cell lysis | Use 500 μL instead of recommended 800 μL [12] |
| Vortex-Genie 2 | Mechanical disruption | 10 min at maximum speed [12] |
| Solution C5 | Wash buffer | Split into two 250 μL steps with 5 min ice incubation [12] |
| Solution C6 | Elution buffer | Add after 10 min column drying [12] |
Procedure:
- Add 500 μL of CD1 lysis buffer to 0.3 g homogenate (modified from manufacturer's 800 μL recommendation).
- Mechanically lyse samples using Vortex-Genie 2 at maximum speed for 10 minutes.
- Follow manufacturer's instructions with the following modifications:
- Perform wash step with C5 solution in two steps of 250 μL each
- Incubate on ice for 5 minutes between washes
- Centrifuge at 13,000 × g after each wash
- After final wash, open spin column lids for 10 minutes to ensure ethanol evaporation
- Add Solution C6 to column after 5 minutes of drying
- Elute DNA in 50 μL volume [12].
Quality Assessment for Long-Read Sequencing
- Quantity: Use fluorometric methods (Qubit) rather than spectrophotometry for accurate DNA quantification.
- Quality: Assess DNA integrity via pulsed-field gel electrophoresis or Fragment Analyzer systems.
- Purity: Ensure A260/A280 ratio between 1.8-2.0 and A260/A230 >2.0.
Technology-Specific Library Preparation Workflows
Oxford Nanopore Sequencing Workflow
Figure 1: Oxford Nanopore sequencing workflow emphasizing native DNA input and real-time analysis.
ONT library preparation maintains DNA in its native state, preserving epigenetic modifications. Key steps include:
- DNA Repair: Using NEBNext FFPE DNA Repair mix or similar for damaged DNA
- End-prep: End-repair and dA-tailing in a single step
- Adapter Ligation: Ligation of sequencing adapters containing motor proteins
- PrimeFlow Cell: Loading of library onto nanopore-containing flow cells [56] [57]
The prepared library is loaded onto a flow cell containing nanopores embedded in an electro-resistant membrane. Application of a voltage bias creates an ionic current through each pore. As DNA strands pass through the pores, characteristic disruptions in current are decoded into sequence data in real time [56].
PacBio HiFi Sequencing Workflow
Figure 2: PacBio HiFi sequencing workflow highlighting the importance of high molecular weight DNA.
PacBio HiFi sequencing requires high molecular weight DNA for optimal performance:
- Size Selection: Use BluePippin or similar systems to select fragments >15 kb
- SMRTbell Library Preparation: Create blunt-ended, double-stranded DNA fragments with hairpin adapters to form circular templates
- Damage Repair: Critical step for removing nicks and breaks that terminate sequencing
- Polymerase Binding: Binding of DNA polymerase to SMRTbell templates
- Sequencing: Loading onto SMRT Cells for sequencing by synthesis [54] [55]
The circular consensus sequencing approach generates multiple passes of each molecule, resulting in HiFi reads with >99.9% accuracy [54].
Applications in Metagenomic Research
Microbial Diversity Discovery
Long-read sequencing has dramatically expanded our ability to discover novel microbial species from complex environments. Recent research applying deep long-read Nanopore sequencing to 154 soil and sediment samples recovered 15,314 previously undescribed microbial species, expanding the phylogenetic diversity of the prokaryotic tree of life by 8% [60]. The long reads enabled by optimized extraction protocols allowed for:
- Recovery of complete ribosomal RNA operons
- Identification of biosynthetic gene clusters
- Characterization of CRISPR-Cas systems
- Resolution of previously inaccessible repetitive regions [60]
Pathogen Surveillance in Complex Matrices
The optimized DNA extraction protocol described in section 3.2 was validated through spike-in experiments with known pig pathogens. Researchers demonstrated that extraction method significantly influences pathogen detection sensitivity in complex matrices like wastewater [12]. This approach has important implications for:
- Early disease detection in livestock
- One Health initiatives connecting animal, human, and environmental health
- Monitoring antimicrobial resistance genes in environmental samples
- Outbreak investigation and containment [12]
Optimized protocol modifications for long-read sequencing technologies are essential for maximizing data quality, particularly when working with complex metagenomic samples. The key considerations include:
- Sample-specific optimization: Extraction methods must be validated for each sample type
- DNA quality over quantity: While sufficient DNA is necessary, integrity and purity are more critical
- Technology matching: Extraction protocols may need adjustment based on chosen sequencing platform
- Inhibitor removal: Critical for complex matrices like soil and wastewater
As long-read technologies continue to evolve, with improvements in accuracy, throughput, and read length, the importance of optimized sample preparation will only increase. Future developments will likely include integrated extraction-to-sequencing workflows and standardized quality metrics specific to long-read applications. The protocols outlined here provide a foundation for researchers to build upon as these technologies mature and find new applications in metagenomic research and beyond.
Troubleshooting and Optimization: Solving Common DNA Extraction Problems
In metagenomic sequencing research, the success of downstream analyses is fundamentally dependent on the initial quality and quantity of the extracted DNA. Incomplete cell lysis and inefficient DNA binding during purification represent two predominant obstacles that compromise DNA yield, particularly from complex microbial communities containing tough-to-lyse microorganisms [5]. These challenges are especially pronounced in samples with low microbial biomass or high host DNA contamination, such as sputum, dust, and clinical specimens, where host DNA can constitute up to 73.3% of the sequenced material, effectively drowning out the microbial signal [61]. The selection of an appropriate DNA extraction method is therefore critical, as it introduces significant variability in observed microbial community composition and functional profiles, accounting for 3-22.3% of the observed variation depending on sample type [61]. This application note systematically addresses the diagnostic and remedial strategies for overcoming low DNA yield within the broader context of optimizing DNA extraction methods for metagenomic sequencing research.
Diagnosing the root causes of low DNA yield
Assessing lysis efficiency
Incomplete cell lysis, particularly of resilient Gram-positive bacteria, represents a major source of low DNA yield. Different lysis methods exhibit distinct efficacies and biases. Mechanical methods like bead-beating are highly effective for disrupting tough cell walls but can cause significant DNA shearing, compromising downstream applications requiring high molecular weight (HMW) DNA [5]. Enzymatic lysis methods using lysozyme are gentler and better preserve DNA integrity but may be insufficient for some resistant microorganisms [5]. The performance of various DNA extraction methods differs significantly between sample types, as shown in Table 1, with no single method performing optimally across all sample matrices [61].
Table 1: Performance comparison of DNA extraction methods across different sample types
| Extraction Method | Fecal Samples | Sputum Samples | Dust Samples | DNA Yield | Host DNA Removal | HMW DNA Recovery |
|---|---|---|---|---|---|---|
| Phenol-Chloroform | Moderate | Moderate | Moderate | Variable | Moderate | Good |
| Promega Maxwell | Good | Good | Good | High | Good | Moderate |
| Qiagen PowerSoil | Good | Moderate | Moderate | Moderate | Good | Moderate |
| Zymo Magbead | Moderate | Poor | Poor | Low | Poor | Poor |
| Zymo HMW MagBead | Excellent | Excellent | Excellent | High | Excellent | Excellent |
Evaluating binding efficiency
Inefficient DNA binding during purification represents another critical failure point. Traditional silica spin columns can selectively lose shorter fragments during washing steps, while magnetic bead-based systems offer more consistent recovery across fragment sizes [5]. The Solid-Phase Reversible Immobilization (SPRI) system allows for selective purification of long fragments but requires optimization of bead-to-sample ratios [5]. Purification methods also vary in their ability to remove inhibitors commonly found in environmental and clinical samples, such as humic acids, bile salts, and mucin, which can interfere with downstream enzymatic reactions [5].
Experimental protocols for troubleshooting
Protocol for systematic evaluation of lysis efficiency
Materials:
- Lysis buffers (commercial or formulated in-house)
- Lytic enzymes (e.g., lysozyme, mutanolysin)
- Mechanical disruptor (bead beater or homogenizer)
- Proteinase K
- Synthetic stool matrix or other relevant sample matrix
- ZymoBIOMICS Microbial Community Standard (as positive control)
Procedure:
- Sample Preparation: Aliquot identical samples of a defined microbial community (e.g., ZymoBIOMICS Microbial Community Standard) or test samples into separate tubes for each lysis condition to be tested [5].
- Enzymatic Pre-treatment: For conditions involving enzymatic lysis, resuspend samples in appropriate buffer containing lysozyme (10-20 mg/mL) and/or other lytic enzymes. Incubate at 37°C for 30-60 minutes with occasional mixing [5].
- Mechanical Lysis: Subject samples to bead-beating using different bead sizes (e.g., 0.1mm, 0.5mm) and durations (30s-180s). Include samples without bead-beating as controls [5].
- Chemical Lysis: Add lysis buffer containing SDS or other detergents to all samples. Incubate at appropriate temperature (typically 56-65°C) for 30-60 minutes.
- Inhibitor Removal: Proceed with DNA purification using the selected method(s). Include additional wash steps if necessary.
- Quantification and Quality Assessment: Measure DNA concentration using fluorometric methods (e.g., Qubit) and assess fragment size using pulse-field or standard gel electrophoresis.
Protocol for optimizing DNA binding and purification
Materials:
- Selected binding buffers (e.g., guanidine hydrochloride, isopropanol, PEG)
- Silica columns or magnetic beads
- Ethanol (70-80%)
- Elution buffer (TE or nuclease-free water)
- DNA size selection beads (if performing HMW selections)
Procedure:
- Binding Condition Optimization: After complete lysis, split samples into equal aliquots for testing different binding conditions:
- Vary the ratio of binding buffer to sample (1:1, 1.5:1, 2:1)
- Test different incubation times (5-30 minutes)
- Evaluate binding at different temperatures (room temperature vs. 37-50°C)
- Washing Optimization: Perform wash steps with freshly prepared 70-80% ethanol:
- Test number of wash steps (1-3 washes)
- Evaluate volume of wash buffer
- Ensure complete removal of ethanol before elution
- Elution Optimization: Elute DNA in appropriate buffer:
- Test different elution volumes
- Evaluate double elution vs. single elution
- Compare elution with pre-warmed buffer (65°C) vs. room temperature
- Allow 5-10 minute incubation before centrifugation
Workflow for comprehensive problem diagnosis
The following workflow provides a systematic approach for diagnosing and resolving low DNA yield issues:
The scientist's toolkit: Research reagent solutions
Table 2: Essential reagents and kits for optimizing DNA extraction
| Product Name | Type | Primary Application | Key Features |
|---|---|---|---|
| Quick-DNA HMW MagBead Kit [5] | Magnetic Bead | HMW DNA Isolation | Gentle lysis, HMW DNA preservation, high yield |
| ZymoBIOMICS Microbial Community Standard [5] | Quality Control | Method Validation | Defined composition, Gram-positive and negative species |
| QIAamp DNA Microbiome Kit [7] | Spin Column | Host DNA Depletion | Differential lysis, effective host removal |
| NEBNext Microbiome DNA Enrichment Kit [7] | Enzymatic | Host DNA Depletion | CpG-methylated host DNA removal |
| Phenol-Chloroform [5] | Organic | HMW DNA Extraction | High yield, but hazardous and time-consuming |
| Lysozyme [5] | Enzyme | Gram-positive Lysis | Gentle cell wall degradation, combinable with other methods |
| Proteinase K [5] | Enzyme | Protein Digestion | Comprehensive protein removal, enhanced yield |
Successful metagenomic sequencing depends critically on overcoming the twin challenges of incomplete lysis and inefficient DNA binding. The protocols and workflows presented here provide a systematic approach for diagnosing and resolving low DNA yield issues, with the Quick-DNA HMW MagBead Kit demonstrating particularly strong performance for HMW DNA isolation [5]. The optimal solution varies significantly by sample type, emphasizing the importance of empirical optimization using relevant controls and metrics [61]. By implementing these evidence-based strategies, researchers can significantly improve DNA yield and quality, thereby enhancing the reliability and depth of their metagenomic analyses.
Preventing and Identifying DNA Degradation from Nucleases and Improper Storage
In metagenomic sequencing research, the integrity of extracted DNA is a foundational determinant of data quality and reliability. DNA degradation, primarily driven by enzymatic activity and improper storage conditions, introduces significant biases in microbial community representation and compromises downstream analyses [62]. This application note provides a detailed framework for preventing nuclease-mediated DNA degradation and identifying its occurrence within the context of DNA extraction workflows for metagenomic sequencing. The protocols and data presented are essential for researchers, scientists, and drug development professionals aiming to generate robust and reproducible metagenomic data.
Understanding DNA Degradation Mechanisms
DNA degradation is a natural process that can severely impact the quality of genetic material, making it difficult to analyze or amplify. Effective management of this degradation requires a thorough understanding of its primary mechanisms [62].
- Oxidation: This occurs when DNA is exposed to environmental stressors like heat, UV radiation, or reactive oxygen species (ROS), leading to modified nucleotide bases and strand breaks.
- Hydrolysis: Water molecules can break the chemical bonds in the DNA backbone, leading to depurination (loss of adenine and guanine bases) and fragmentation, especially if the sample's pH is not properly controlled.
- Enzymatic Breakdown: Endogenous nucleases, present in biological samples like tissue and blood, are highly effective at degrading nucleic acids and can rapidly dismantle DNA if not inactivated during collection or extraction.
- Mechanical Shearing: Overly aggressive mechanical disruption during homogenization can physically fragment DNA into short, unusable pieces.
The following diagram illustrates the pathways and interventions related to DNA degradation.
Figure 1: DNA Degradation Pathways and Prevention Strategies. The diagram outlines primary degradation mechanisms (orange) and corresponding preventive interventions (green) that inhibit, slow, or minimize damage.
Preventing DNA Degradation
Strategic Sample Preservation
Preservation begins immediately after sample collection. The chosen method must rapidly halt metabolic and nuclease activity.
- Flash Freezing: The gold standard for preservation is rapid freezing using liquid nitrogen, followed by storage at -80°C. This method effectively halts all biochemical activity, preserving DNA in a near-native state [62].
- Chemical Preservation: When immediate freezing is not feasible, chemical preservatives provide a reliable alternative. Ethylenediaminetetraacetic acid (EDTA) is highly effective due to its ability to chelate divalent cations (like Mg²⁺ and Ca²⁺) that are essential cofactors for nucleases [63]. Recent evidence indicates that using EDTA at an alkaline pH (e.g., pH 10) significantly enhances its efficacy in preserving high-molecular-weight DNA compared to neutral buffers [63]. While ethanol (95%) is commonly used, it has been shown to be less effective than alkaline EDTA in preventing DNA degradation in frozen-thawed tissues [63].
Optimized DNA Extraction and Handling
The extraction protocol itself must be designed to inactivate nucleases and minimize mechanical and thermal stress.
- Lysis and Homogenization: Mechanical homogenization must balance efficient cell lysis with the preservation of DNA integrity. Using instruments like the Bead Ruptor Elite allows for precise control over parameters such as speed, cycle duration, and bead type. This ensures effective disruption of tough samples (e.g., bone, bacterial spores, stool) while minimizing DNA shearing [62]. Processing samples on a pre-chilled block or using a cryo-cooling unit helps mitigate heat-induced degradation during homogenization.
- Buffer Composition: Lysis and extraction buffers should be optimized with nuclease inhibitors. A key ingredient is EDTA (typically 5-50 mM), which chelates metal ions. Additional components may include proteinase K to digest nucleases and other proteins, and detergents to disrupt cellular membranes without damaging DNA [62] [64]. Maintaining a stable, optimal pH throughout the extraction is critical to reduce hydrolytic damage.
- Thawing Protocol for Frozen Samples: A critical, often overlooked, step is the thawing of frozen samples. Research demonstrates that thawing tissue samples directly in 250 mM EDTA, pH 10, and maintaining them in this solution overnight at 4°C before extraction, significantly improves the recovery of high-molecular-weight DNA compared to thawing in ethanol or without any preservative [63]. This simple step safeguards DNA from nucleases that become active during the thawing process.
Table 1: Comparative Analysis of DNA Preservation and Extraction Methods
| Method / Reagent | Key Function | Optimal Conditions / Concentration | Impact on DNA Integrity |
|---|---|---|---|
| Flash Freezing | Halts biochemical activity | Liquid nitrogen, then -80°C storage | Preserves high-molecular-weight (HMW) DNA; gold standard [62]. |
| EDTA (pH 10) | Chelates nuclease cofactors | 250 mM, pH 10 | Significantly improves HMW DNA recovery, especially during thawing [63]. |
| Ethanol (95%) | Dehydrates and denatures proteins | 95% concentration | Less effective than alkaline EDTA for preserving HMW DNA in thawed tissues [63]. |
| Silica-Binding Buffers | Binds DNA for purification | High concentration of guanidinium salts | Protects DNA and removes inhibitors; different formulations (QG, PB) can affect short fragment recovery [64]. |
| Controlled Homogenization | Physically lyses cells | Optimized speed, duration, and bead type | Maximizes DNA yield while minimizing mechanical shearing and thermal degradation [62]. |
Identifying and Quantifying DNA Degradation
Real-Time Fluorescence-Based Nuclease Assay
Beyond prevention, it is crucial to detect and quantify nuclease activity. A real-time fluorescence-based assay provides a sensitive and quantitative method for this purpose [65].
Experimental Protocol: Real-Time Nuclease Assay
- Principle: The assay relies on the loss of fluorescence signal as a double-stranded DNA (dsDNA) substrate is cleaved by nucleases. The fluorescence dye PicoGreen intercalates into dsDNA; upon cleavage, the dye is released, leading to a decrease in fluorescence that can be measured in real-time [65].
- Materials:
- Black-bottom 96-well plates (to minimize background fluorescence and crosstalk).
- Fluorescent dsDNA dye (e.g., Quant-iT PicoGreen dsDNA Assay Kit).
- Annealed oligonucleotide substrates (e.g., 80-mers, with or without biotin blocks).
- Purified nuclease enzyme or protein extract.
- Reaction buffer (e.g., 1x Tango Buffer with appropriate divalent cations).
- Plate reader capable of fluorescence measurement (e.g., excitation 483 nm, emission 530 nm).
- Procedure:
- Prepare Reaction Mix: In each well of the black-bottom plate, combine:
- 10 µl of dsDNA oligonucleotide substrate (500 nM final concentration).
- 10 µl of 10x reaction buffer.
- 25 µl of nuclease-free water.
- 2 µl of streptavidin (if using biotin-blocked oligonucleotides).
- 50 µl of PicoGreen reagent (diluted as per manufacturer's instructions).
- Baseline Reading: Place the plate in a pre-heated plate reader (e.g., 37°C) and take an initial fluorescence reading to establish a baseline.
- Initiate Reaction: Add 5 µl of the test nuclease enzyme to each well. Use a multi-channel pipette for consistency if processing multiple samples.
- Data Acquisition: Immediately begin kinetic measurements, reading the fluorescence every 45-60 seconds for 15 minutes to 2 hours. The plate reader should shake the plate briefly before each read to mix.
- Controls: Always include control reactions without enzyme (to measure background signal loss/photobleaching) and with a well-characterized nuclease (e.g., Exonuclease III) for calibration [65].
- Prepare Reaction Mix: In each well of the black-bottom plate, combine:
- Data Analysis: Plot fluorescence versus time. The initial rate of fluorescence decrease is proportional to the nuclease activity. This assay can be used to characterize substrate preference, the effect of cofactors, and the catalytic rate of the enzyme.
Quality Control in Metagenomics
For metagenomic samples, rigorous QC is non-negotiable. The choice of DNA extraction and library preparation methods can profoundly impact the apparent microbial composition [64].
- Fragment Analysis: Techniques such as the Agilent Bioanalyzer or Fragment Analyzer provide a DNA integrity number (DIN) or a detailed electrophoretogram of DNA fragment size distribution. A high-quality DNA sample will show a predominant peak of high-molecular-weight fragments, while a degraded sample will show a smear of low-molecular-weight fragments [62].
- Metagenomic QC Metrics: In shotgun metagenomic sequencing, several metrics can indicate degradation:
- Endogenous DNA Content: The proportion of sequencing reads that map to the expected microbial or host genomes. Degraded samples often have lower endogenous content.
- Fragment Length Recovery: The distribution of sequenced fragment lengths. Ancient DNA and highly degraded samples are dominated by very short fragments (<100 bp) [64].
- Clonality: An over-representation of identical sequencing reads, which can be a sign of PCR amplification bias from low-input, degraded DNA templates [64].
Table 2: Impact of Wet-Lab Protocols on DNA Recovery from Challenging Samples
| Protocol Step | Protocol Option | Key Consideration | Effect on Degraded DNA / Microbial Recovery |
|---|---|---|---|
| DNA Extraction | QG Method (Rohland & Hofreiter) | Silica-binding with guanidinium thiocyanate [64]. | Efficient for general use; may be less effective for very short fragments. |
| DNA Extraction | PB Method (Dabney et al.) | Binding buffer with sodium acetate, isopropanol, and guanidinium HCl [64]. | Enhances recovery of ultra-short DNA fragments (<50 bp); ideal for highly degraded samples. |
| Library Prep | Double-Stranded (DSL) | Ligation of adapters to double-stranded molecules [64]. | Standard method; can have higher clonality with degraded DNA. |
| Library Prep | Single-Stranded (SSL) | Ligation of adapters to single-stranded molecules [64]. | Higher conversion efficiency of short, single-stranded fragments; better for highly degraded DNA. |
The Scientist's Toolkit
The following table lists essential reagents and kits for managing DNA degradation in research.
Table 3: Research Reagent Solutions for DNA Integrity Management
| Reagent / Kit | Function | Specific Example |
|---|---|---|
| EDTA (Ethylenediaminetetraacetic acid) | A chelating agent that binds divalent cations (Mg²⁺, Ca²⁺), inactivating metal-dependent nucleases [63]. | Prepare a 250 mM stock solution at pH 10.0 for optimal preservation during tissue thawing [63]. |
| PicoGreen dsDNA Quantitation Reagent | A fluorescent dye used to quantify dsDNA and monitor its degradation in real-time nuclease activity assays [65]. | Part of the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, P7589) [65]. |
| Proteinase K | A broad-spectrum serine protease used in lysis buffers to digest nucleases and other contaminating proteins. | Commonly included in DNA extraction kits (e.g., Qiagen DNeasy Blood and Tissue Kit) [63]. |
| Silica-Membrane Mini-Columns | Purify DNA from lysates by selectively binding DNA in the presence of chaotropic salts, removing contaminants and inhibitors. | Qiagen DNeasy Blood and Tissue Kit [63]. |
| Specialized Bead Tubes | Used with homogenizers for mechanical lysis; different bead materials (ceramic, steel) are optimized for different sample types. | Used with the Omni Bead Ruptor Elite for efficient lysis with minimal DNA shearing [62]. |
Preventing and identifying DNA degradation is not a single step but an integrated practice spanning from sample collection to data analysis. For metagenomic sequencing research, where the integrity of the DNA template directly dictates the fidelity of the resulting microbial community profile, this practice is paramount. Adopting a rigorous workflow that combines strategic preservation (e.g., alkaline EDTA), optimized extraction methods (e.g., PB method for degraded samples), and stringent quality control (e.g., fragment analysis and real-time nuclease assays) is essential. By systematically implementing these protocols, researchers can safeguard their most valuable asset—high-quality DNA—and ensure the generation of reliable, reproducible, and meaningful metagenomic data.
The accuracy of microbial community surveys based on metagenomic sequencing is critically dependent on the purity of the isolated DNA. Contaminants such as proteins, salts, hemoglobin, and polysaccharides can severely compromise downstream applications by inhibiting enzymatic reactions, interfering with accurate DNA quantification, and reducing sequencing reliability [66] [67]. The presence of these impurities is particularly problematic in low-biomass environments where contaminant DNA can comprise a significant fraction of sequenced material, potentially leading to false positive associations and obscuring true biological signals [66] [68]. Effective decontamination is therefore essential for generating accurate profiles of microbial communities, especially in sensitive research applications such as drug development and clinical diagnostics.
This application note outlines standardized protocols for identifying and removing major contaminants encountered during DNA extraction for metagenomic sequencing. We provide detailed methodologies, quantitative comparisons of efficiency, and practical tools to integrate robust decontamination procedures into existing workflows, enabling researchers to produce highly pure DNA suitable for demanding downstream applications.
Contaminant Challenges and Removal Strategies
Different contaminants interfere with DNA extraction and downstream applications through distinct mechanisms. The table below summarizes the primary challenges posed by each contaminant type and the recommended strategies for their removal.
Table 1: Common Contaminants in DNA Extraction: Challenges and Removal Strategies
| Contaminant | Impact on Downstream Applications | Primary Removal Methods |
|---|---|---|
| Proteins | Inhibit enzyme activity in PCR and restriction digestion; can bind to DNA, reducing yield [69] [67]. | Proteinase K digestion; Phenol-chloroform extraction; Salting-out method [70] [67]. |
| Salts | Interfere with enzymatic reactions and spectrophotometric DNA quantification [67]. | Ethanol or isopropanol precipitation with washing; dialysis; spin column purification [70] [67]. |
| Hemoglobin | A potent PCR inhibitor commonly found in DNA extracted from blood samples [70]. | Red Blood Cell (RBC) Lysis Buffer; multiple washing steps; column-based purification [70]. |
| Polysaccharides | Co-precipitate with DNA, inhibiting enzymes and resulting in viscous, hard-to-pipette samples [67]. | CTAB (Cetyltrimethylammonium bromide) extraction; high-salt precipitation buffers [69] [67]. |
Detailed Decontamination Protocols
Comprehensive Protocol for Protein and Polysaccharide Removal Using CTAB
The CTAB method is particularly effective for plant and environmental samples rich in polysaccharides and polyphenols, which are challenging contaminants that often co-precipitate with DNA [67].
Materials and Reagents:
- CTAB Extraction Buffer (2% CTAB, 100 mM Tris-HCl, 20 mM EDTA, 1.4 M NaCl, pH 8.0) [69]
- Proteinase K (optional for enhanced protein removal) [67]
- β-Mercaptoethanol (add 100 μL per 5 mL of CTAB buffer just before use) [69]
- Chloroform:Isoamyl Alcohol (24:1) [69]
- Isopropanol (chilled)
- 70% Ethanol (chilled)
- TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) or nuclease-free water
Procedure:
- Cell Lysis: Grind 100-500 mg of frozen tissue to a fine powder in liquid nitrogen using a mortar and pestle. Transfer the powder to a microfuge tube and add 5 mL of pre-warmed (65°C) CTAB extraction buffer. Mix by inversion and incubate at 65°C for 30-60 minutes, inverting tubes periodically [69].
- Organic Extraction: Add an equal volume of Chloroform:Isoamyl Alcohol (24:1). Mix thoroughly by inversion for 5 minutes to form an emulsion. Centrifuge at 12,000 × g for 15 minutes at room temperature [69].
- Aqueous Phase Recovery: Carefully transfer the upper aqueous phase (containing DNA) to a new tube, avoiding the intermediate proteinaceous layer and lower organic phase.
- DNA Precipitation: Add 0.6-0.7 volumes of room-temperature isopropanol to the aqueous phase. Mix gently by inversion until DNA precipitates as a stringy white mass. Centrifuge at 12,000 × g for 10 minutes to pellet the DNA [69].
- Wash: Discard the supernatant. Wash the DNA pellet with 1 mL of 70% ethanol to remove residual salts and CTAB. Centrifuge at 12,000 × g for 5 minutes and carefully discard the supernatant [69].
- Resuspension: Air-dry the pellet for 10-15 minutes (do not over-dry). Resuspend the DNA in 50-100 μL of TE buffer or nuclease-free water. Incubate at 65°C for 10 minutes to aid dissolution [69].
Salting-Out Protocol for Protein and Hemoglobin Removal from Blood
This non-toxic, cost-effective method is ideal for extracting genomic DNA from fresh or frozen whole blood, effectively removing hemoglobin and soluble proteins [70].
Materials and Reagents:
- RBC Lysis Buffer (0.155 M NH₄Cl, 10 mM KHCO₃, 0.1 M EDTA, pH 7.6) [70]
- Extraction Buffer (1.5 M Tris pH 7.6, 0.4 M Na₂EDTA, 2.5 M NaCl, 2% CTAB, pH 8.0) [70]
- 10% SDS (Sodium Dodecyl Sulfate)
- β-Mercaptoethanol
- Saturated NaCl solution (~6 M)
- Chloroform:Isoamyl Alcohol (24:1)
- Absolute Ethanol and 70% Ethanol (chilled)
Procedure:
- Red Blood Cell Lysis: Transfer 500 μL of whole blood to a microfuge tube. Centrifuge at 2,664 × g for 7 minutes at 4°C. Aspirate the plasma. Add 1 mL of RBC Lysis Buffer to the pellet, mix gently, and incubate at room temperature for 1-2 minutes. Centrifuge at 2,664 × g for 6 minutes. Discard the supernatant. Repeat until a white pellet of white blood cells is obtained [70].
- Protein Lysis and Denaturation: Add 500 μL of pre-warmed (56°C) Extraction Buffer to the pellet. Add 30 μL of 10% SDS and 2 μL of β-Mercaptoethanol. Mix gently and incubate at 56-60°C for 1 hour [70].
- Protein Precipitation: Add 500 μL of Chloroform:Isoamyl Alcohol (24:1), shake vigorously, and centrifuge at 10,656 × g for 12 minutes at 4°C. Transfer the upper aqueous phase to a new tube. Alternatively, add 150-200 μL of saturated NaCl solution to the lysate, shake vigorously for 15 seconds, and centrifuge at 12,000 × g for 10-15 minutes. This precipitates proteins [70].
- DNA Precipitation: Transfer the supernatant to a new tube containing 1 mL of chilled absolute ethanol. Shake gently until white DNA threads appear. Centrifuge at 10,656 × g for 12 minutes at 4°C to pellet the DNA [70].
- Wash and Resuspension: Discard the supernatant. Wash the pellet with 500 μL of 70% ethanol, centrifuge again, and discard the supernatant. Air-dry the pellet and resuspend in 100 μL of TE Buffer overnight at 4°C or for 2-3 hours at 37°C. Store at -20°C [70].
Quality Control and Contaminant Detection
Quantitative Assessment of DNA Purity and Yield
Rigorous quality control is essential to confirm the success of decontamination. The table below outlines standard methods for evaluating DNA purity and concentration.
Table 2: Methods for Assessing DNA Purity and Yield After Decontamination
| Assessment Method | Target Metric | Ideal Value for Pure DNA | Interpretation of Results |
|---|---|---|---|
| Spectrophotometry (A₂₆₀/A₂₈₀) | Protein Contamination | ~1.8 [71] [70] | A lower ratio indicates protein or phenol contamination. |
| Spectrophotometry (A₂₆₀/A₂₃₀) | Salt/Solvent Contamination | >2.0 [67] | A lower ratio indicates salt, EDTA, or carbohydrate contamination. |
| Agarose Gel Electrophoresis | DNA Integrity/RNA Contamination | Sharp, high molecular weight band [71] [69] | Smearing indicates degradation; a discrete low molecular weight band suggests RNA contamination. |
| Fluorometry (e.g., Qubit) | Accurate DNA Quantification | N/A | Provides a highly accurate concentration, unaffected by common contaminants [69]. |
In Silico Contaminant Identification for Metagenomics
For metagenomic sequencing data, computational tools like the decontam R package can identify contaminant sequences post-sequencing. Decontam uses two statistical strategies: frequency-based identification, which exploits the inverse correlation between contaminant frequency and sample DNA concentration, and prevalence-based identification, which identifies sequences more common in negative controls than in true samples [66]. Integrating these in silico methods with rigorous laboratory decontamination protocols provides the most robust approach for generating accurate metagenomic profiles [66] [68].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Decontamination Protocols and Their Functions
| Reagent | Primary Function | Contaminants Targeted |
|---|---|---|
| CTAB (Cetyltrimethylammonium bromide) | Cationic detergent that complexes with polysaccharides and acidic polysaccharides in high-salt buffers to precipitate them [67]. | Polysaccharides, Polyphenols |
| Proteinase K | Broad-spectrum serine protease that digests and inactivates nucleases and other proteins [67]. | Proteins |
| Phenol-Chloroform-Isoamyl Alcohol | Organic mixture that denatures and partitions proteins into the organic phase or interphase, leaving nucleic acids in the aqueous phase [69] [67]. | Proteins, Lipids |
| Chloroform:Isoamyl Alcohol (24:1) | Isoamyl alcohol reduces foaming; chloroform aids in protein denaturation and lipid removal [69]. | Proteins, Lipids |
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent that disrupts cell membranes and denatures proteins by breaking hydrogen bonds [70] [69]. | Proteins, Lipids |
| β-Mercaptoethanol | Reducing agent that breaks disulfide bonds in proteins, aiding denaturation, and inhibits tannins and polyphenols [69]. | Proteins, Polyphenols |
| Sodium Chloride (NaCl) | High salt concentrations precipitate proteins (salting-out) and are used in CTAB to complex with polysaccharides [70] [67]. | Proteins, Polysaccharides |
| Isopropanol/Ethanol | Alcohols reduce the solvation of DNA molecules, causing them to precipitate out of solution, thereby separating from soluble contaminants [70] [69]. | Salts, Soluble Contaminants |
Workflow Visualization
DNA Extraction and Decontamination Workflow
This workflow outlines the core steps for extracting DNA while integrating specific branches for targeted contaminant removal, ensuring high-quality output for metagenomic sequencing.
Effective removal of proteins, salts, hemoglobin, and polysaccharides is a critical determinant of success in metagenomic sequencing research. The protocols detailed in this application note provide researchers with robust, reproducible methods for purifying high-quality DNA from complex samples. By combining rigorous laboratory techniques, such as the CTAB and salting-out methods, with modern computational tools like the decontam package, scientists can significantly reduce technical noise and enhance the biological accuracy of their findings. Adherence to these standardized decontamination protocols ensures that DNA samples are of the highest purity, thereby maximizing the reliability of data generated in downstream applications, from biomarker discovery to drug development.
The success of metagenomic sequencing research hinges on the initial quality and purity of extracted nucleic acids. Difficult samples, characterized by either high concentrations of endogenous nucleases, potent PCR inhibitors, or both, present a formidable barrier to reliable downstream analysis. DNase-rich tissues rapidly degrade target genetic material, while inhibitor-laden matrices—ranging from humic substances in soil to polyphenols in plants and heme in blood—can compromise enzymatic reactions during library preparation and sequencing. Within the context of a broader thesis on DNA extraction methods, this application note details targeted optimization strategies and robust protocols designed to overcome these specific challenges, ensuring the integrity of metagenomic data derived from the most recalcitrant sample types.
The molecular challenges are multifaceted. In DNase-rich environments, such as pancreatic tissue or certain microbial communities, endogenous nucleases catalyze the hydrolysis of DNA phosphodiester bonds, leading to significant fragmentation and loss of informational content [62]. Concurrently, samples like plant tissues (rich in polyphenols and polysaccharides), forensic bone material (containing calcium hydroxyapatite and collagen), and fecal matter (with complex bile salts and bacterial metabolites) introduce substances that inhibit downstream enzymatic processes like PCR and sequencing [72] [73]. The overarching goal, therefore, is to implement extraction strategies that simultaneously inactivate degradative enzymes and sequester or remove inhibitory compounds, all while maximizing the yield of pure, high-molecular-weight DNA suitable for metagenomic applications.
Optimization Strategies and Core Principles
Effective handling of difficult samples requires a strategic combination of pre-processing, tailored lysis, and meticulous purification. The following core principles underpin successful protocol optimization.
Pre-Lysis Sample Preparation
Steps taken before cell lysis are critical for preserving nucleic acid integrity and removing contaminants.
Sorbitol Pre-Wash for Inhibitor-Rich Plant and Fungal Matrices: A pre-wash buffer containing 0.35 M sorbitol, 100 mM Tris-HCl, 5 mM EDTA, and 1% PVP-40 can be employed prior to standard CTAB lysis. This solution helps to leach out water-soluble interfering metabolites like polysaccharides and polyphenols from tissue macerates. The PVP (polyvinylpyrrolidone) binds specifically to polyphenols, preventing their oxidation and subsequent co-precipitation with DNA. This step, which adds only 10-20 minutes to a protocol, results in DNA of significantly higher purity and compatibility with sensitive downstream applications like SNP genotyping and long-read sequencing [73].
Host DNA Depletion for Microbiome Studies: When analyzing the microbiome of host-associated tissues (e.g., colon biopsies), enriching for bacterial DNA is essential. A protocol using a low concentration of saponin (0.0125%) can selectively lyse mammalian cells without disrupting bacterial cell walls. Following lysis, a DNase treatment degrades the released host DNA. This method has been shown to achieve a 4.5-fold enrichment of bacterial DNA without distorting the relative bacterial abundance at the phylum level, thereby dramatically improving the efficiency of shotgun metagenomic sequencing [74].
Particle Removal and Nuclease Treatment for Virome Analysis: For viral metagenomics from clinical samples such as plasma or respiratory secretions, enriching viral particles is key. An optimized approach involves filtration (0.22 µm), centrifugation, and treatment with a cocktail of DNase and RNase enzymes. This process removes cellular debris and degrades free-floating nucleic acids not contained within intact viral capsids. The result is a significant enrichment of viral sequences, allowing for the detection of low-abundance viruses and the assembly of more complete viral genomes [75] [76].
Enhanced Lysis and Extraction
The lysis method must be powerful enough to disrupt tough structures while minimizing DNA shearing and further exposure to inhibitors.
Mechanical Homogenization with Parameter Control: Bead-beating is highly effective for tough samples like bone, plant roots, and bacterial spores. However, over-aggressive homogenization can cause DNA shearing. Using an instrument like the Bead Ruptor Elite allows for precise control over speed, cycle duration, and temperature. Employing cryo-cooling during homogenization minimizes heat-induced degradation. The choice of bead material (e.g., ceramic for tough tissues, glass for standard cells) is also critical for maximizing yield while preserving DNA integrity [62].
Chemical and Enzymatic Demineralization and Digestion: For highly mineralized tissues like bone and teeth, a demineralization step is indispensable. This typically involves incubation in a solution of 0.5 M EDTA for 24-72 hours, which chelates calcium ions and softens the inorganic matrix. This is followed by an extended proteinase K digestion (often overnight) to break down the collagenous organic matrix and fully release DNA sequestered within osteocytes. This combination approach is fundamental to recovering DNA from forensic and ancient skeletal remains [72].
Table 1: Summary of Optimization Strategies for Different Sample Types
| Sample Type | Primary Challenges | Recommended Strategy | Key Additives/Techniques |
|---|---|---|---|
| Plant Tissues | Polyphenols, Polysaccharides | Sorbitol Pre-Wash [73] | PVP-40, 2-mercaptoethanol, High-salt CTAB |
| Bone & Teeth | Mineralized Matrix, Inhibitors | Demineralization & Digestion [72] | EDTA, Proteinase K, Silica columns |
| Host-Associated Microbiome | Overwhelming Host DNA | Selective Host Cell Lysis [74] | Saponin, DNase treatment |
| Clinical Virome | Low Viral Biomass, Host Contamination | Viral Particle Enrichment [75] | Filtration, Nuclease treatment, Centrifugation |
| Forensic/Ancient | Extreme Degradation, Inhibitors | Silica-in-Suspension [77] | EDTA, Proteinase K, Organic extraction (Phenol/Chloroform) |
Workflow Diagram: Strategic Path for Difficult Sample Processing
The following diagram synthesizes the key decision points and strategies for processing difficult samples, from collection to analysis.
Detailed Experimental Protocols
Protocol 1: DNA Extraction from Inhibitor-Rich Plant and Fungal Matrices
This protocol, adapted from Inglis et al. (2018), is optimized for samples high in polyphenols and polysaccharides, such as oak leaves or fungal mycelium [73].
Materials:
- Sorbitol Wash Buffer: 100 mM Tris-HCl (pH 8.0), 0.35 M Sorbitol, 5 mM EDTA (pH 8.0), 1% (w/v) PVP-40. Add 1% (v/v) 2-mercaptoethanol fresh before use.
- High-Salt CTAB Lysis Buffer: 100 mM Tris-HCl (pH 8.0), 3 M NaCl, 3% (w/v) CTAB, 20 mM EDTA, 1% (w/v) PVP-40. Add 1% (v/v) 2-mercaptoethanol fresh before use.
- Chloroform:Isoamyl Alcohol (24:1)
- Isopropanol
- 70% Ethanol
- TE Buffer
Method:
- Grinding: Lyophilize 100-150 mg of fresh tissue and mechanically disrupt it in a bead beater with stainless steel ball bearings to create a fine powder.
- Sorbitol Pre-Wash: Suspend the powdered tissue in 1 mL of Sorbitol Wash Buffer. Vortex thoroughly and centrifuge at 5,000 x g for 5 minutes. Decant the supernatant. For heavily contaminated samples, repeat this step.
- Lysis: Resuspend the pellet in 700 µL of pre-warmed (65°C) High-Salt CTAB Lysis Buffer. Incubate at 65°C for 30-60 minutes, inverting the tube periodically.
- Purification: Add an equal volume of Chloroform:Isoamyl Alcohol, mix thoroughly, and centrifuge at 5,000 x g for 10 minutes. Transfer the upper aqueous phase to a new tube.
- Precipitation: Precipitate the DNA by adding 0.7 volumes of isopropanol, mix by inversion, and centrifuge. Wash the pellet with 70% ethanol, air-dry, and resuspend in TE Buffer.
Protocol 2: Bacterial DNA Enrichment from Host Tissue for Microbiome Analysis
This protocol, based on Bjerre et al. (2021), is designed for human tissue biopsies (e.g., colon) to deplete host DNA and enrich for bacterial DNA, making it ideal for shotgun metagenomics [74].
Materials:
- Saponin Solution: 0.0125% (w/v) saponin in PBS.
- DNase I
- Proteinase K
- Bead-beating system with mechanical lysing matrix.
- Standard molecular biology reagents for DNA purification (e.g., phenol-chloroform or silica columns).
Method:
- Selective Host Cell Lysis: Incubate the tissue biopsy (~2-5 mm) in 1 mL of Saponin Solution on a rotator for 30 minutes at room temperature. This selectively permeabilizes and lyses mammalian cells.
- Host DNA Digestion: Add DNase I to the lysate and incubate to degrade the released host DNA. Bacterial cells, with their intact cell walls, are protected from lysis and DNase activity at this stage.
- Bacterial Lysis: Pellet the intact bacterial cells by centrifugation. Wash the pellet and then subject it to vigorous mechanical lysis using a bead-beater to break open the robust bacterial cell walls.
- DNA Extraction and Purification: Digest lysates with Proteinase K, followed by standard DNA extraction and purification, such as phenol-chloroform extraction or a column-based method.
Table 2: Key Research Reagent Solutions for Challenging Sample Types
| Reagent / Tool | Function | Application Examples |
|---|---|---|
| PVP-40 (Polyvinylpyrrolidone) | Binds to and co-precipitates polyphenols, preventing them from inhibiting polymerases. | Leaf tissues, herbarium specimens, oak species [73]. |
| CTAB (Cetyltrimethylammonium bromide) | A cationic detergent effective in lysis and in precipitating polysaccharides in high-salt buffers. | Plants, fungi, bacteria [73] [78]. |
| Saponin | A surfactant that selectively lyses mammalian cells by disrupting cholesterol in the cell membrane. | Host tissue biopsies for microbiome analysis [74]. |
| EDTA (Ethylenediaminetetraacetic acid) | Chelates divalent cations (Mg2+, Ca2+), inhibiting nucleases and demineralizing hard tissues. | Bone, teeth, forensic samples [72] [62]. |
| Bead Beater (Mechanical Homogenizer) | Provides physical disruption of tough cell walls and matrices through high-speed shaking with beads. | Bacterial spores, mycelium, plant roots, bone powder [62] [73]. |
| Silica Magnetic Beads/Columns | Binds DNA in high-salt conditions, allowing for efficient washing and elution of inhibitor-free DNA. | Universal, but particularly critical for degraded and inhibitor-rich samples [74] [77]. |
Quality Control and Validation
Rigorous QC is non-negotiable when working with difficult samples. Spectrophotometry (A260/A280 and A260/A230 ratios) provides a preliminary assessment of protein and chemical contamination, respectively. However, fluorometric methods (e.g., Qubit) are more accurate for quantifying double-stranded DNA concentration. For degraded samples, fragment analyzers or TapeStation provide a DNA Integrity Number (DIN) that quantifies the level of fragmentation, which is crucial for determining suitability for long-read or short-read sequencing [62].
In the context of metagenomics, the effectiveness of inhibitor removal can be validated by spiking a known quantity of exogenous DNA into the extraction and performing a qPCR assay. Significant inhibition is indicated by a delay in the quantification cycle (Cq) compared to a control. For clinical metagenomic assays, the use of internal controls, such as the External RNA Controls Consortium (ERCC) RNA Spike-In Mix, allows for the monitoring of extraction efficiency and even enables absolute quantification of pathogen load [79].
Troubleshooting Guide
- Low DNA Yield: Increase the amount of starting material. Extend the lysis incubation time. For tissues, ensure complete grinding to a fine powder. For bone, extend the demineralization step.
- High Degradation (Fragmented DNA): Reduce homogenization speed or time. Perform all pre-lysis steps on ice. Ensure nuclease inhibitors (like EDTA) are present in all solutions before lysis.
- PCR/Sequencing Inhibition: Repeat the pre-wash or purification steps. Increase the number of wash steps during silica-based purification. Use a post-extraction cleanup kit. Dilute the DNA template to reduce the concentration of co-eluted inhibitors.
- Poor Metagenomic Sequencing Results (Low on-target reads): For host-associated samples, increase the rigor of the host DNA depletion step (e.g., optimize saponin concentration). For low-biomass virome samples, ensure effective nuclease treatment and concentration steps are performed.
Best practices for sample collection, storage, and pre-processing to preserve DNA integrity
For metagenomic sequencing research, the integrity of DNA from complex microbial communities is paramount. The steps taken from the moment of sample collection directly determine the quality, reliability, and reproducibility of downstream sequencing data. Proper sample handling preserves the true structure of the microbial community and minimizes biases that can arise from DNA degradation or the selective loss of certain microbial groups. This document outlines standardized protocols for collecting, storing, and pre-processing environmental and human-associated samples to ensure the highest DNA integrity for metagenomic applications.
Sample Collection and Primary Preservation
The initial stabilization of samples is critical, especially when collection occurs in the field or in clinical settings without immediate access to laboratory processing.
Core Principles
- Minimize Post-Collection Metabolic Activity: Rapid preservation is necessary to prevent shifts in microbial community structure due to changes in temperature, oxygen, or nutrient availability.
- Avoid Contamination: Use sterile, single-use collection equipment and aseptic techniques.
- Document Comprehensively: Record metadata such as time of collection, environmental parameters (e.g., pH, temperature), and clinical data (if applicable) [80].
Preservation Methods in the Field
The choice of preservation method depends on the sample type, available infrastructure, and downstream analytical goals. A comparative overview is provided in Table 1.
Table 1: Comparison of Sample Preservation Methods for Metagenomic Studies
| Preservation Method | Protocol Details | Optimal Sample Types | Advantages | Limitations |
|---|---|---|---|---|
| Flash Freezing | Immediate immersion in liquid nitrogen or placement on dry ice [81]. | Stool, soil, water, tissue. | Halts biological activity instantly; considered the gold standard [82]. | Requires access to cryogenic materials; transport logistics are complex. |
| Chemical Preservation (Ethanol) | Storage in 75% ethanol at room temperature [81]. | Tissue, environmental solids. | Cost-effective; no continuous freezing required [81]. | Risk of DNA degradation over time; not ideal for community composition [81] [82]. Ethanol is a flammable liquid, and transport may be restricted [81]. |
| Freeze-Drying (Lyophilization) | Samples are frozen and vacuum-dried (e.g., -50°C, 30 mTorr for two days) [81]. | Tissue, stable microbial communities. | Samples can be stored at room temperature; ideal for long-distance shipping [81]. | Requires specialized freeze-drying equipment; potential for DNA fragmentation in some samples [81]. |
| Commercial Preservation Buffers | Sample is mixed with a proprietary buffer (e.g., DNA/RNA Shield) in the field. | Stool, saliva, water. | Stabilizes DNA at room temperature for days or weeks; inhibits nuclease activity [82]. | Cost per sample can be higher; buffer salts may need to be removed during extraction. |
Sample Storage and Transport
After initial preservation, maintaining sample integrity during storage and transport is crucial.
Storage Temperature Guidelines
Long-term storage temperature has a direct impact on the stability of DNA. Recommendations for extracted DNA are summarized in Table 2.
Table 2: Recommended Storage Conditions for Extracted DNA
| Storage Temperature | Use Case | Stability | Best Practices |
|---|---|---|---|
| +4°C | Short-term (less than 24 hours). | Days | Only for temporary holding during active processing. |
| -20°C | Short- to medium-term storage (frequent access) [83] [84]. | Months to a year | Acceptable for purified DNA in TE buffer; risk of degradation from freeze-thaw cycles [83]. |
| -80°C | Long-term archival storage [84] [85]. | Years to decades | Ideal for most biological samples and DNA aliquots; suppresses most degradation reactions [84] [85]. |
| -196°C (Liquid Nitrogen) | Indefinite archival storage [85]. | Indefinite | The gold standard for preserving cell viability and nucleic acid integrity. |
Handling and Transport
- Aliquoting: Divide DNA samples or homogenized tissue into single-use aliquots to avoid repeated freeze-thaw cycles, which cause DNA fragmentation and degradation [83] [84].
- Shipping: For frozen samples, ensure an adequate amount of dry ice is used for the expected transit time. Be aware that international shipping of samples preserved in ethanol may be restricted due to its classification as a dangerous good [81]. Freeze-dried samples pose the fewest logistical problems for shipping [81].
- Packaging: Use validated containers and monitor temperature during transit with data loggers.
Pre-processing and DNA Extraction
The method of DNA extraction is a significant source of bias in metagenomic studies and should be chosen to align with the preservation method.
Sample Homogenization
- Protocol: Homogenize tissue samples or environmental solids with glass beads in an appropriate buffer using a tissue homogenizer (e.g., Precellys) for 1 minute at 5,000 rpm [81]. This ensures a uniform lysate representative of the entire community.
- Considerations: For tough samples (e.g., soil, spores), a combination of mechanical (bead-beating) and chemical lysis may be necessary to access DNA from hard-to-lyse Gram-positive bacteria [82].
DNA Extraction Method Selection
The choice of extraction method significantly influences DNA yield, fragment length, and community representation.
- Silica-Based Kits (e.g., peqGOLD, FastDNA Kit): These are widely used for their convenience and reproducibility. They are particularly effective for freeze-dried samples [81]. However, they can exhibit a bias against Gram-positive bacteria if lysis conditions are not stringent enough [82].
- Phenol-Chloroform Extraction: This traditional method is often considered the most thorough for difficult samples and provides high yields and high-molecular-weight DNA [82] [84]. Its drawbacks include the use of hazardous chemicals, more complex handling, and greater potential for user error [84].
- Chelex 100 Resin: This chelating resin method is fast and inexpensive but may result in lower DNA quality and reduced amplification success, especially for longer DNA fragments, compared to silica-based methods [81]. It is suitable for PCR-based screening but less ideal for whole-metagenome sequencing.
Experimental Validation: Preservation and Extraction
A seminal study highlights the interaction between preservation and extraction methods. Earthworm tissue samples were subjected to different preservation methods (Freezing, Ethanol, Freeze-drying) and subsequently extracted with two different methods (peqGOLD and Chelex 100). The success of PCR amplification for DNA fragments of different lengths was assessed [81].
Key Findings:
- Freeze-drying was the best preservation method when paired with the silica-based peqGOLD extraction kit.
- For samples extracted with Chelex 100, storage in ethanol yielded better results, though the overall amplification success was significantly lower than with peqGOLD.
- The amplification success decreased significantly as the length of the targeted DNA fragment increased, underscoring the impact of preservation and extraction on DNA integrity [81].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Sample Preservation and DNA Extraction
| Item | Function | Application Notes |
|---|---|---|
| Liquid Nitrogen / Dry Ice | For flash freezing and transport of samples. | Essential for preserving the most labile community members and transcripts. |
| Nuclease-Free Water | Preparation of buffers and resuspension of DNA. | Critical for preventing enzymatic degradation of nucleic acids during processing. |
| TE Buffer (Tris-EDTA, pH ~8.0) | Long-term storage buffer for extracted DNA. | The slightly basic pH protects DNA from acid hydrolysis; EDTA chelates metal ions to inhibit nucleases [83]. |
| Proteinase K | Broad-spectrum serine protease. | Digests nucleases and other proteins during cell lysis, protecting the released DNA. |
| Phenol-Chloroform-Isoamyl Alcohol | Organic solvent for liquid-phase DNA extraction. | Separates DNA into the aqueous phase, denaturing and removing proteins. Handle with care. |
| Silica-Based DNA Binding Columns | Selective binding and purification of DNA from lysates. | The core of most modern DNA extraction kits; allows for efficient washing and elution. |
| Commercial Preservation Kits (e.g., Norgen's Stool Kit, DNA/RNA Shield) | Stabilize nucleic acids at room temperature. | Ideal for biobanking and field collections in remote locations [82]. |
Workflow Visualization
Robust metagenomic sequencing data begins long before the sequencing run. A meticulously planned and executed protocol for sample collection, preservation, storage, and DNA extraction is the foundation for accurate and meaningful biological insights. By standardizing these upstream processes and carefully selecting methods that are fit-for-purpose, researchers can significantly reduce technical noise, better reveal true biological variation, and accelerate discoveries in microbiome research and drug development.
Validation and Comparative Analysis: Ensuring Accuracy and Reproducibility
Using Mock Microbial Communities to Evaluate Extraction Efficiency and Bias
In metagenomic sequencing research, the accuracy of microbial community analysis is fundamentally constrained by protocol-dependent biases, with DNA extraction efficiency representing one of the most significant confounding factors [86]. Variations in DNA extraction methodologies can dramatically distort microbial abundance profiles, potentially leading to erroneous biological interpretations [14]. Mock microbial communities—defined mixtures of microorganisms with known composition—serve as essential control reagents that provide a "ground truth" for benchmarking these technical variables [87]. By offering a standardized reference with predetermined abundances, mock communities enable researchers to quantify bias, optimize protocols, and validate methodological performance across different laboratories and platforms [87]. Their systematic application is particularly crucial for translational research and drug development, where reproducible and accurate microbiome characterization is paramount for identifying clinically relevant microbial signatures [86].
The integration of mock controls addresses a persistent challenge in microbiome science: distinguishing true biological signal from technical artifact. As noted in recent methodological studies, extraction bias remains a major unresolved problem that critically limits the comparability of microbiome studies [86]. Without appropriate controls, researchers cannot determine whether observed microbial abundance differences reflect actual ecosystem variation or differential lysis efficiencies of bacterial cells with varying morphological characteristics [86]. This application note details the implementation of mock microbial communities for evaluating DNA extraction efficiency and bias within the broader context of metagenomic sequencing research.
Mock Community Composition and Design Principles
Well-characterized mock communities form the foundation of robust extraction efficiency evaluation. These communities should be formulated to represent relevant microbial lineages with contrasting cellular features that influence lysis efficiency.
Table 1: Exemplary Mock Community Composition for Extraction Efficiency Evaluation
| Species | Phylum | Genome Size (bp) | GC Content (%) | Cell Wall (Gram-type) | Relative Abundance (%) |
|---|---|---|---|---|---|
| Bacteroides uniformis | Bacteroidetes | 4,989,532 | 46.2 | Gram-negative | 4.7 |
| Blautia sp. | Firmicutes | 6,247,046 | 46.7 | Gram-positive | 4.5 |
| Enterocloster clostridioformis | Firmicutes | 5,687,315 | 48.9 | Gram-positive | 5.3 |
| Pseudomonas putida | Proteobacteria | 6,156,701 | 62.3 | Gram-negative | 3.9 |
| Streptococcus mutans | Firmicutes | 2,018,796 | 36.9 | Gram-positive | 6.9 |
| Bifidobacterium longum | Actinobacteriota | 2,594,022 | 60.1 | Gram-positive | 5.7 |
| Staphylococcus epidermidis | Firmicutes | 2,520,735 | 32.2 | Gram-positive | 4.8 |
| Cutibacterium acnes | Actinobacteriota | 2,560,907 | 60.0 | Gram-positive | 5.0 |
The mock community should encompass a diverse range of guanine-cytosine (GC) content (e.g., 32-62%) and include bacteria with both Gram-positive and Gram-negative cell walls [87]. This diversity is crucial because GC content significantly influences sequencing coverage uniformity [88], while cell wall structure directly impacts lysis efficiency during DNA extraction [86]. Commercially available mock communities like the ZymoBIOMICS series provide standardized reference materials with even or staggered abundance distributions to assess both qualitative and quantitative accuracy [86]. These communities typically include 8-20 bacterial strains prevalent in the target ecosystem (e.g., human gastrointestinal tract), with some formulations additionally incorporating fungal species for cross-domain evaluations [86].
Experimental Protocol: DNA Extraction Efficiency Assessment
Sample Preparation and Experimental Design
To evaluate DNA extraction efficiency, employ a factorial design that tests multiple extraction protocols against the same mock community. This approach systematically isolates the impact of individual protocol components:
Mock Community Standardization: Prepare dilution series of cell mock communities with even or staggered compositions. For low-biomass applications, include dilution points ranging from 10^8 to 10^4 cells to simulate different biomass inputs [86]. Spike with a defined quantity of alien species (e.g., ZymoBIOMICS spike-in community D6321) not typically found in the target microbiome to control for cross-contamination [86].
Extraction Protocol Variables: Test a minimum of two commercially available DNA extraction kits specifically designed for microbial community analysis (e.g., QIAamp UCP Pathogen Mini Kit vs. ZymoBIOMICS DNA Microprep Kit) [86]. For each kit, evaluate different lysis conditions:
Replication and Controls: Process eight replicates per mock community dilution and extraction protocol combination to account for stochastic variability [86]. Include extraction-negative controls (empty tubes with swabs if applicable) and PCR-negative controls to identify contamination sources.
DNA Extraction and Sequencing
The DNA extraction and sequencing workflow follows a standardized pathway to ensure consistent evaluation across experimental conditions:
Figure 1: Experimental workflow for evaluating DNA extraction efficiency using mock microbial communities.
Cell Lysis: Apply the designated mechanical lysis conditions using a homogenizer (e.g., Precellys Evolution Touch) with zirconia beads (0.1 mm and 0.5 mm) [86]. For protocols including enzymatic treatment, incubate samples with lysozyme (20 mg/mL) at 37°C for 30 minutes prior to mechanical lysis [14].
DNA Extraction and Purification: Follow manufacturer protocols for the respective extraction kits with the following modifications:
- Use consistent sample input masses (e.g., 25 mg for soil/solid samples) [89]
- Elute DNA in a standardized volume (e.g., 50-100 μL) of elution buffer
- Quantify DNA yield using fluorometric methods (e.g., Qubit dsDNA HS Assay)
Library Preparation and Sequencing:
- For 16S rRNA gene sequencing: Amplify the V3-V4 hypervariable region using primers 341F and 805R [14]
- For shotgun metagenomic sequencing: Use Illumina-compatible library preparation kits with fragmentation to 100-1000 bp fragments [89] [90]
- Sequence on an appropriate platform (e.g., Illumina MiSeq/HiSeq) with sufficient depth (minimum 50,000 reads per sample for 16S; 10 million reads for shotgun)
Data Analysis: Quantifying Extraction Bias
Bioinformatic Processing
Process raw sequencing data through standardized bioinformatic pipelines:
16S rRNA Data:
Shotgun Metagenomic Data:
Extraction Bias Quantification
Calculate bias metrics by comparing observed abundances to expected values:
Relative Abundance Deviation:
- For each species i, compute: [ \text{Deviation}i = \frac{\text{Observed Abundance}i - \text{Expected Abundance}i}{\text{Expected Abundance}i} \times 100\% ]
Extraction Efficiency Ratio:
- Calculate the ratio between observed ASV abundance of Gram-positive to Gram-negative control species (e.g., A. halotolerans to I. halotolerans) [14]
- Compare against the expected ratio based on mock community formulation
Population Fraction Change:
- Quantify PCR amplification bias using the metric: [ Qi = \frac{xi^{(k)}}{xi^{(0)}} ] where (xi^{(k)}) is the population fraction of sequence i after k PCR cycles and (x_i^{(0)}) is the initial fraction [91]
Table 2: Quantitative Evaluation of DNA Extraction Methods Using Mock Communities
| Extraction Method | Mean DNA Yield (ng/μL) | 260/280 Ratio | Gram+/Gram- Ratio | Bias Deviation from Expected | Alpha Diversity Bias |
|---|---|---|---|---|---|
| Mechanical Lysis | 45.2 ± 3.1 | 1.82 ± 0.04 | 0.71 ± 0.08 | 15.3% | High |
| Trypsin Treatment | 38.7 ± 2.8 | 1.85 ± 0.03 | 1.40 ± 0.15 | 8.7% | Moderate |
| Saponin Treatment | 36.9 ± 4.2 | 1.79 ± 0.05 | 1.35 ± 0.19 | 9.2% | Moderate |
| NucleoSpin Soil Kit | 52.1 ± 5.3 | 1.88 ± 0.02 | 1.31 ± 0.25 | 6.5% | Low |
| DNeasy PowerSoil Pro | 48.6 ± 4.7 | 1.84 ± 0.03 | 1.39 ± 0.19 | 7.1% | Low |
Interpretation and Bias Correction
Analysis of Extraction Bias Patterns
The quantitative data derived from mock community analysis reveals systematic patterns of extraction bias:
Gram Status Bias: Methods without enzymatic lysis (e.g., mechanical lysis alone) consistently underrepresent Gram-positive bacteria, with Gram+/Gram- ratios as low as 0.71 compared to expected values near 1.40 [14]. This reflects differential lysis efficiency between cell wall types.
GC Content Bias: All technologies exhibit coverage biases in extreme GC regions, with GC-rich regions (≥75%) and AT-rich regions (≤10% GC) showing significantly lower coverage [88]. This impacts the detection of taxa with atypical genomic GC content.
Morphological Predictability: Extraction bias per species is strongly predicted by bacterial cell morphology, with cell size, shape, and wall structure accounting for significant variance in observed abundance deviations [86].
Computational Bias Correction
The relationship between bacterial morphology and extraction efficiency enables computational correction:
Figure 2: Computational correction of extraction bias using morphological properties.
Implement morphology-based correction using the following approach:
Bias Parameter Estimation: Using mock community data, compute extraction efficiency coefficients for each species based on morphological properties: [ \text{Efficiency}i = f(\text{Cell Wall Thickness}i, \text{GC Content}i, \text{Cell Volume}i) ]
Abundance Correction: Apply efficiency coefficients to environmental samples: [ \text{Corrected Abundance}i = \frac{\text{Observed Abundance}i}{\text{Efficiency}_i} ]
Validation: Verify correction efficacy using staggered mock communities with different taxonomic composition than the training set [86].
Implementation Guide for Research Applications
Research Reagent Solutions
Table 3: Essential Research Reagents for Extraction Efficiency Evaluation
| Reagent/Kit | Manufacturer | Primary Function | Application Notes |
|---|---|---|---|
| ZymoBIOMICS Microbial Community Standards | ZymoResearch | DNA and cell mock communities with even/staggered compositions | Provides ground truth for >8 bacterial species; includes Gram+/Gram- species |
| NucleoSpin Soil Kit | MACHEREY–NAGEL | DNA extraction from challenging samples | Highest alpha diversity recovery in comparative studies [14] |
| QIAamp UCP Pathogen Mini Kit | Qiagen | DNA extraction with bead-based lysis | Compatible with different lysis conditions and buffers [86] |
| ZymoBIOMICS DNA Microprep Kit | ZymoResearch | Low-biomass DNA extraction | Includes dedicated inhibitors removal steps |
| ZymoBIOMICS Spike-in Control | ZymoResearch | Alien species for contamination tracking | Contains species not found in human microbiome |
Protocol Selection Guidelines
Based on comprehensive evaluations:
For fecal samples where host DNA contamination is minimal, standard mechanical lysis without pre-treatment provides satisfactory results [89].
For low-biomass samples (e.g., skin, tissue), the trypsin extraction method significantly reduces host DNA contamination (80.53% vs 89.11% eukaryotic DNA with mechanical lysis) while maintaining microbial diversity [89].
For complex environmental samples containing PCR inhibitors (e.g., soil), the NucleoSpin Soil Kit demonstrates superior performance in DNA purity and diversity representation [14].
For studies requiring absolute quantification, implement morphology-based computational correction using mock community-derived efficiency parameters [86].
Mock microbial communities provide an indispensable tool for quantifying and correcting DNA extraction bias in metagenomic research. Through systematic implementation of the protocols outlined herein, researchers can significantly improve the accuracy and reproducibility of microbiome analyses. The integration of mock controls across experimental workflows—coupled with morphology-based computational correction—represents a critical advancement toward standardized microbiome measurement, particularly for translational applications in drug development and clinical diagnostics. Consistent application of these practices will enhance cross-study comparability and strengthen the biological validity of microbiome research findings.
Deoxyribonucleic acid (DNA) extraction represents a critical first step in metagenomic sequencing research, with the chosen methodology directly influencing downstream results including genomic yield, DNA integrity, and the accurate representation of microbial community structures. Variations in extraction techniques can introduce significant biases, particularly in complex samples where the efficient lysis of diverse microbial taxa is required. This application note establishes a standardized comparative framework for the assessment of DNA extraction methods, providing detailed protocols and quantitative data to guide researchers in selecting optimal protocols for specific sample matrices within metagenomic investigations. The reliability of subsequent next-generation sequencing (NGS) data and metagenome-assembled genomes (MAGs) is fundamentally dependent on the initial DNA extraction quality [92] [93].
Theoretical Background and Key Considerations
The fundamental goal of DNA extraction in metagenomics is to obtain a nucleic acid sample that is both quantitatively sufficient and qualitatively representative of the entire microbial community present in the original specimen. Different methodological approaches can favor the recovery of certain microbial groups over others. For instance, protocols incorporating mechanical lysis, such as bead-beating, are often more effective at disrupting the tough cell walls of Gram-positive bacteria, whereas enzymatic lysis may be sufficient for Gram-negative species [14] [94]. This differential lysis efficiency can lead to a skewed representation of the actual microbial composition if not properly accounted for.
The presence of co-extracted compounds that act as PCR inhibitors—such as humic substances in soil or bile salts in fecal samples—poses another significant challenge, potentially affecting library preparation and sequencing efficiency [14]. Furthermore, in samples with high host DNA background, such as blood or tissue, effective host depletion strategies are essential to enhance the detection sensitivity for microbial pathogens [95]. A robust comparative framework must therefore evaluate methods based on a multi-faceted approach, considering not only the sheer quantity of DNA recovered but also its purity, the integrity of the nucleic acid molecules, and the fidelity of the resulting microbial community profile.
Comparative Data Analysis of DNA Extraction Methods
Performance Across Sample Types
The optimal DNA extraction method is highly dependent on the sample matrix. A comprehensive 2024 study compared five commercial kits across various terrestrial ecosystem samples, revealing that no single kit universally outperformed all others for every sample type [14]. For instance, the QIAamp Fast DNA Stool Mini Kit was best for hare feces, while the QIAamp DNA Micro Kit provided high yields for invertebrates and soil. The NucleoSpin Soil Kit consistently produced the best DNA purity based on the 260/230 ratio across most sample types [14].
Table 1: DNA Extraction Kit Performance Across Different Sample Types
| Sample Type | Recommended Kit | Key Performance Metric | Alternative Kit |
|---|---|---|---|
| Hare Feces | QIAamp Fast DNA Stool Mini | Highest DNA concentration | QIAamp DNA Micro |
| Soil & Invertebrates | QIAamp DNA Micro | High DNA concentration | NucleoSpin Soil |
| General Purity | NucleoSpin Soil | Best 260/230 ratio | - |
| Subgingival Biofilm | DNeasy Blood & Tissue | Highest total & bacterial DNA yield | - |
Efficiency in Gram-Positive vs. Gram-Negative Bacterial Recovery
The ability to lyse different bacterial cell types varies substantially among extraction methods. The same terrestrial ecosystem study incorporated a mock community (MC) containing Imtechella halotolerans (Gram-negative) and Allobacillus halotolerans (Gram-positive) to quantitatively assess this bias [14]. The DNeasy Blood & Tissue Kit, which utilizes an extended enzymatic lysis step, produced a mean MC ratio (A. halotolerans/I. halotolerans) of 0.71 ± 0.08, indicating the highest efficiency for lysing the Gram-positive bacterium compared to other kits, which yielded ratios closer to 1.4 [14]. This finding highlights that kits employing enzymatic or combined lysis strategies are more effective for breaking down robust Gram-positive cell walls.
Impact on Microbial Community Profiles
The choice of DNA extraction method directly influences downstream microbial diversity metrics (alpha and beta diversity). Research has demonstrated that different kits can significantly alter the observed abundance of hundreds of Amplicon Sequence Variants (ASVs) within the same sample [14]. These kit-induced variations can be of a magnitude that leads to statistically significant differences in diversity estimates, potentially confounding biological interpretations. Therefore, maintaining methodological consistency within a study is paramount, and cross-study comparisons should account for the DNA extraction protocol used.
Host DNA Depletion for Clinical Metagenomics
In clinical samples like blood, where microbial DNA can be dwarfed by host genetic material, pre-analytical host depletion is crucial. A 2025 study evaluated a novel Zwitterionic Interface Ultra-Self-assemble Coating (ZISC)-based filtration device, which achieved >99% white blood cell removal while allowing unimpeded passage of bacteria and viruses [95]. When integrated into a genomic DNA (gDNA)-based mNGS workflow for sepsis diagnosis, this method resulted in a tenfold enrichment of microbial reads compared to unfiltered samples (9,351 vs. 925 reads per million), enabling 100% detection of culture-positive pathogens [95]. This performance surpassed that of other host depletion techniques, such as differential lysis or methylated DNA removal.
Table 2: Quantitative Comparison of DNA Extraction and Host Depletion Methods
| Method / Kit | Key Feature | Best For | Performance Data |
|---|---|---|---|
| NucleoSpin Soil | High purity & diversity | Overall ecosystem studies | Highest alpha diversity estimates [14] |
| DNeasy Blood & Tissue | Enzymatic lysis | Gram-positive bacteria; small biopsies | MC ratio: 0.71 ± 0.08; highest yield from paper points [14] [94] |
| Chelex Boiling | Rapid & cost-effective | Large cohort screening (DBS) | Significantly higher DNA yield vs. column kits (p < 0.0001) [41] |
| ZISC-based Filtration | Host cell depletion | Blood samples for mNGS | >99% WBC removal; 10x microbial read increase [95] |
Detailed Experimental Protocols
Protocol 1: DNA Extraction from Complex Environmental Samples using the NucleoSpin Soil Kit
This protocol is adapted for processing diverse samples like soil, rhizosphere soil, and invertebrate taxa, based on its performance in recovering high-purity DNA and supporting high alpha diversity estimates [14].
Reagents and Materials:
- NucleoSpin Soil Kit (MACHEREY–NAGEL)
- Lysis Buffer SL1 and SL2
- Proteinase K
- Bead Tubes (provided)
- Ethanol (96-100%)
- Elution Buffer BE
- Microcentrifuge
- Vortexer with adapter for 2 ml tubes
- Water bath or incubator set to 70°C
Procedure:
- Homogenization and Lysis: Transfer up to 500 mg of soil or a single invertebrate specimen to a Bead Tube. Add 700 µl of Buffer SL1 and 100 µl of Buffer SL2. Secure the cap tightly and vortex vigorously for 5 minutes to homogenize. For effective lysis of Gram-positive bacteria, this mechanical disruption is critical.
- Incubation: Centrifuge the tube for 1 minute at 11,000 x g. Transfer the supernatant to a new 2 ml microcentrifuge tube.
- Protein Digestion: Add 100 µl of Proteinase K to the supernatant. Mix by vortexing briefly and incubate at 70°C for 10 minutes.
- Binding: Centrifuge the tube for 1 minute at 11,000 x g to pellet any residual debris. Transfer the supernatant to a new 2 ml tube. Add 450 µl of Buffer SB and mix by vortexing.
- Column Purification: Apply the mixture to a NucleoSpin Soil Column placed in a collection tube. Centrifuge for 1 minute at 11,000 x g. Discard the flow-through.
- Wash Steps: Add 700 µl of Buffer SW1 to the column. Centrifuge for 1 minute at 11,000 x g and discard the flow-through. Then, add 500 µl of Buffer SW2 and centrifuge for 1 minute at 11,000 x g. Discard the flow-through. Repeat this step with another 500 µl of Buffer SW2.
- Elution: Place the column in a clean 1.5 ml microcentrifuge tube. Apply 50-100 µl of pre-warmed Elution Buffer BE directly onto the silica membrane. Incubate at room temperature for 1 minute, then centrifuge for 1 minute at 11,000 x g to elute the purified DNA.
Protocol 2: Efficient DNA Extraction from Low-Biomass Subgingival Biofilm using a Single Paper Point
This protocol, optimized for minimal sample input, uses the DNeasy Blood & Tissue Kit, which demonstrated superior efficiency for small sample volumes [94].
Reagents and Materials:
- DNeasy Blood & Tissue Kit (QIAGEN)
- Lysozyme (optional, for enhanced Gram-positive lysis)
- PBS (Phosphate Buffered Saline)
- Nuclease-free water
- Glass beads (1.7–2.1 mm)
- 5 mL centrifuge tubes
- Microcentrifuge
Procedure:
- Sample Wash-Off: Place a single paper point containing subgingival biofilm into a 1.5 ml tube. Add 1 mL of nuclease-free water and 12 glass beads. Shake at 14,000 rpm for 5 minutes.
- Lysate Collection: Pierce the bottom of the 1.5 ml tube and place it inside a 5 ml centrifuge tube. Centrifuge the assembly at 4,000 x g for 1 minute. The flow-through containing the washed-off biofilm will collect in the 5 ml tube.
- Pellet Formation: Transfer the flow-through to a new 1.5 ml tube. Centrifuge at 10,000 x g for 15 minutes to pellet the microbial material. Carefully discard the supernatant.
- Enzymatic Lysis: Resuspend the pellet in 180 µl of Buffer ATL. Add 20 µl of Proteinase K. Mix by vortexing and incubate at 56°C until the sample is completely lysed (1-3 hours). For enhanced Gram-positive lysis, a pre-incubation with 20 mg/ml Lysozyme at 37°C for 30 minutes is recommended before adding Proteinase K.
- Follow the standard DNeasy Blood & Tissue protocol from the point of adding 200 µl of Buffer AL.
Protocol 3: Host Depletion from Blood Samples using ZISC-Based Filtration for mNGS
This protocol describes a pre-extraction method to deplete human host cells, significantly improving pathogen detection in sepsis [95].
Reagents and Materials:
- ZISC-based Filtration Device (e.g., "Devin" from Micronbrane)
- Sterile syringes (5-10 mL)
- Whole blood sample (3-5 mL)
- Low-speed and high-speed centrifuges
- ZISC-based Microbial DNA Enrichment Kit or standard DNA extraction kit
Procedure:
- Filtration Setup: Aseptically connect the ZISC-based filter unit to a sterile syringe.
- Host Depletion: Draw 4-5 mL of whole blood into the syringe. Gently depress the plunger to pass the entire blood volume through the filter into a sterile 15 mL collection tube. The filter will retain >99% of white blood cells.
- Plasma Separation: Transfer the filtered blood to a centrifuge tube. Centrifuge at 400 x g for 15 minutes at room temperature to separate plasma.
- Microbial Pellet Isolation: Transfer the plasma to a new tube. Centrifuge at 16,000 x g for 10 minutes to pellet microbial cells and debris.
- DNA Extraction: Proceed with DNA extraction from the pellet using your kit of choice (e.g., ZISC-based Microbial DNA Enrichment Kit or a standard microbial DNA kit). The resulting DNA will be significantly enriched for microbial content.
Workflow Visualization
DNA Extraction Method Selection Workflow: This diagram outlines the decision-making process for selecting an appropriate DNA extraction method based on sample type, leading to the assessment of key performance metrics prior to metagenomic analysis.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for DNA Extraction in Metagenomics
| Reagent / Kit Name | Primary Function | Key Application Note |
|---|---|---|
| NucleoSpin Soil Kit | DNA purification from soil and complex environmental samples. | Effective for high-humic acid content samples; recovers diverse taxa. |
| DNeasy Blood & Tissue Kit | DNA isolation from tissues, blood, and low-biomass samples. | Superior for small samples (e.g., single paper points); good for Gram-positives with extended lysis. |
| ZymoBIOMICS DNA Miniprep Kit | Comprehensive DNA extraction with mechanical lysis. | Includes bead-beating for robust cell disruption across diverse cell types. |
| QIAamp DNA Micro Kit | DNA purification from very small samples. | Ideal for limited material like invertebrate specimens. |
| Chelex-100 Resin | Rapid, low-cost DNA purification by chelation of metal ions. | Suitable for high-throughput DBS screening; yields are adequate for qPCR. |
| ZISC-based Filtration Device | Physical depletion of host white blood cells from blood. | Critical for enhancing microbial signal in clinical mNGS from blood. |
| Proteinase K | Broad-spectrum serine protease for enzymatic cell lysis. | Essential for digesting proteins and degrading nucleases. |
| Lysozyme | Enzyme that breaks down bacterial cell walls. | Used as a pre-treatment to improve lysis of Gram-positive bacteria. |
This application note provides a systematic framework for evaluating DNA extraction methods, underscoring that the choice of protocol is not trivial but a fundamental determinant of data quality in metagenomic studies. Key findings indicate that NucleoSpin Soil Kit is recommended for broad ecosystem studies, the DNeasy Blood & Tissue Kit excels for low-biomass and Gram-positive-rich samples, and novel ZISC-based filtration is transformative for clinical blood samples. Researchers are strongly advised to validate their chosen method using mock communities and sample-specific metrics for yield, integrity, and unbiased community representation to ensure the generation of robust and reliable metagenomic data.
Correlating Extraction Methods with Downstream Sequencing Metrics and Diagnostic Accuracy
Deoxyribonucleic acid (DNA) extraction is a critical pre-analytical step in metagenomic next-generation sequencing (mNGS) that significantly influences downstream sequencing metrics and ultimate diagnostic accuracy [96] [97]. The transformative potential of mNGS in clinical diagnostics lies in its culture-independent, hypothesis-free detection of a broad spectrum of pathogens directly from clinical specimens [96]. However, technical variations in DNA extraction methodologies introduce substantial biases in microbial community representation, impacting the reliability of taxonomic profiling and antimicrobial resistance (AMR) gene detection [97] [14]. This protocol examines the correlation between extraction method selection and subsequent analytical performance, providing a framework for optimizing metagenomic workflows within the broader thesis of standardizing DNA extraction for robust microbial metagenomics.
Comparative Performance of DNA Extraction Kits
The selection of a DNA extraction method involves balancing DNA yield, fragment size, purity, and the efficient lysis of diverse microbial cell walls without introducing significant taxonomic bias. The following section quantitatively compares the performance of various commercially available kits.
Table 1: Comparison of DNA Extraction Kit Performance Across Studies
| Kit Name | Lysis Method | Purification Method | Key Findings / Optimal Use Case | Source |
|---|---|---|---|---|
| Quick-DNA HMW MagBead Kit (Zymo Research) | Mechanical & Chemical | Magnetic Beads | Produced the highest yield of pure HMW DNA; most suitable for accurate bacterial detection in complex mock communities via Nanopore sequencing. [5] | [5] |
| QIAamp PowerFecal Pro DNA Kit (Qiagen) | Chemical & Mechanical (Bead Beating) | Spin Column | Identified all bacterial species (8/8 and 6/6) in Zymo and ESKAPE mock communities; best for rapid taxonomy and AMR identification with ONT. [30] | [30] |
| PureLink Microbiome DNA Purification Kit (Thermo Fisher) | Vigorous Bead-Beating | Spin Column | Manual protocol with vigorous bead-beating necessary for stool to avoid erroneous taxa proportions (e.g., under/over-representation of Blautia, Faecalibacterium). [97] | [97] |
| NucleoSpin Soil Kit (MACHEREY–NAGEL) | Not Specified | Spin Column | Associated with the highest alpha diversity estimates and highest contribution to overall sample diversity in terrestrial ecosystem samples. [14] | [14] |
| QIAamp DNA Mini Kit (Qiagen) | Enzymatic (Lysozyme, Proteinase K) | Spin Column | Fewer aligned bases for Gram-positive species compared to mechanical lysis methods. [30] | [30] |
Sample-Type-Specific Considerations
The optimal DNA extraction protocol is highly dependent on the sample matrix, which influences the microbial community's structure and the concentration of PCR inhibitors [97] [14].
Table 2: Impact of Sample Type on DNA Extraction Method Performance
| Sample Type | Considerations & Recommended Methods | Key Findings |
|---|---|---|
| Stool | Complex matrix with high biomass and PCR inhibitors; requires vigorous bead-beating. [97] | Manual kits with bead-beating (ZymoBIOMICS, PureLink) are necessary. Automated kits not designed for stool (e.g., Maxwell Tissue) underrepresent Gram-positive taxa like Clostridia. [97] |
| Swab Samples (Cervical, Skin) | Less complex matrix; easier to process. [97] | Similar taxonomic results were obtained with both targeted and non-targeted automated protocols, allowing for greater workflow flexibility. [97] |
| Clinical Swabs (Rectal, Nasopharyngeal) | Mixed Gram-positive and Gram-negative bacteria, host DNA. [30] | The QIAamp PowerFecal Pro DNA kit (mechanical lysis) enabled reliable species and AMR gene identification from pooled clinical eSwabs. [30] |
| Soil & Terrestrial Ecosystems | High inhibitor content (e.g., humic substances). [14] | The NucleoSpin Soil Kit yielded the highest alpha diversity estimates. The DNeasy Blood & Tissue kit showed the highest extraction efficiency for the Gram-positive bacterium in a mock community. [14] |
Detailed Experimental Protocol for Kit Comparison
This protocol provides a methodology for comparing DNA extraction kits using a defined mock community, suitable for both Nanopore and Illumina sequencing platforms.
Materials and Reagents
- Mock Community: ZymoBIOMICS Microbial Community Standard (Zymo Research) or an in-house ESKAPE pathogen mix. [5] [30]
- DNA Extraction Kits: Selected based on lysis and purification mechanisms (e.g., Quick-DNA HMW MagBead Kit, QIAamp PowerFecal Pro DNA Kit, QIAamp DNA Mini Kit, NucleoSpin Soil Kit). [5] [30] [14]
- Equipment: TissueLyser or similar bead-beating instrument, centrifuge, thermomixer, Qubit Fluorometer, NanoDrop spectrophotometer. [5] [30]
- Sequencing & Analysis: Oxford Nanopore Technologies (ONT) MinION or PromethION, GridION sequencer, Guppy basecaller, Kraken2, Minimap2, CARD database. [30]
Procedure
Sample Preparation:
- Resuspend the commercial mock community according to the manufacturer's instructions. [5]
- For an in-house mock community (e.g., ESKAPE strains), culture each strain individually, mix in equal volumes (e.g., 200 µL each), pellet by centrifugation (5000 × g for 15 min), and use the pellet for extraction. [30]
DNA Extraction:
DNA Quality and Quantity Assessment:
- Quantity: Measure DNA concentration using a fluorescence-based method (e.g., Qubit dsDNA HS Assay). [30]
- Quality/Purity: Assess using spectrophotometric ratios (A260/280 and A260/230) via NanoDrop. [30]
- Fragment Size: Analyze a subset of extracts on an agarose gel or bioanalyzer to determine the distribution of DNA fragment sizes. [5]
Library Preparation and Sequencing:
Bioinformatic Analysis:
- Basecalling and QC: Perform basecalling (e.g., with Guppy in HAC mode) and assess raw read quality (e.g., with NanoPlot). [30]
- Host Depletion: If applicable, remove host-derived reads by alignment to a host genome (e.g., Minimap2 vs. Hg38). [30]
- Taxonomic Profiling: Assign taxonomy from raw reads using a classifier (e.g., Kraken2) and/or from assembled contigs (e.g., Minimap2). [30]
- AMR Gene Detection: Identify AMR genes by aligning reads and contigs to a reference database (e.g., CARD using Minimap2). [30]
Data Comparison Metrics:
- Completeness: Percentage of expected species identified.
- Accuracy: Deviation from the expected abundance in the mock community.
- Bias: Ratio of recovered Gram-positive to Gram-negative bacteria.
- Sequencing Metrics: Mean read length, N50, number of reads.
Impact on Diagnostic Accuracy
The choice of DNA extraction method directly influences diagnostic conclusions. Inadequate lysis, particularly from Gram-positive bacteria with robust cell walls, leads to false negatives and distorted microbial abundance profiles [97] [14]. For instance, without vigorous bead-beating, stool samples can show significant underrepresentation of genera like Blautia and Faecalibacterium [97]. Furthermore, the accurate detection of plasmid-mediated AMR genes (e.g., mcr-1, blaNDM-5), which are crucial for guiding antimicrobial therapy, is dependent on extraction methods that effectively lyse the host bacteria and preserve the integrity of mobile genetic elements [96] [30]. Standardizing the extraction protocol is therefore essential for achieving reproducible and clinically actionable results.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for DNA Extraction in Metagenomic Studies
| Item | Function / Application | Example Product Names |
|---|---|---|
| Mock Communities | Standardized controls for evaluating extraction bias, lysis efficiency, and bioinformatic pipeline accuracy. | ZymoBIOMICS Microbial Community Standard [5] [30] |
| HMW DNA Extraction Kits | Isolation of long DNA fragments optimal for long-read sequencing, improving genome assembly. | Quick-DNA HMW MagBead Kit [5] |
| Inhibitor Removal Kits | Purification of DNA from complex matrices (stool, soil) by removing humic acids, bile salts, and other PCR inhibitors. | QIAamp PowerFecal Pro DNA Kit [30] |
| Automated Nucleic Acid Extractors | Standardization and high-throughput processing of clinical samples, reducing hands-on time and inter-operator variability. | Maxwell RSC System [97] [30] |
| Enzymatic Lysis Reagents | Gentle, specific digestion of cell walls, often used for difficult-to-lyse Gram-positive bacteria. | Lysozyme, Proteinase K [30] |
Workflow Diagram for Method Selection
The following diagram illustrates the decision-making workflow for selecting an appropriate DNA extraction method based on sample type and research objectives.
Diagram 1: A workflow for selecting DNA extraction methods based on sample type and research goals.
Within the broader scope of metagenomic sequencing research, effective pathogen surveillance is a cornerstone of public health and veterinary medicine. As demonstrated during the SARS-CoV-2 pandemic, wastewater-based epidemiology (WBE) provides a powerful, non-invasive tool for monitoring community-level disease outbreaks [12] [98]. The application of WBE to livestock settings, such as pig farms, is particularly valuable. Due to intensive housing conditions, infectious diseases can spread rapidly within pig herds, resulting in significant economic losses and threatening food security [12].
The successful implementation of a metagenomic surveillance strategy hinges on the initial recovery of high-quality microbial DNA from complex environmental samples. Piggery wastewater presents a formidable challenge for nucleic acid extraction due to its diverse composition of microorganisms, organic matter, metals, and other substances that can inhibit downstream molecular analyses [12] [99]. This case study evaluates multiple DNA extraction methods for their efficacy in recovering high-quality genetic material from piggery wastewater suitable for pathogen detection using Oxford Nanopore Technology (ONT) sequencing.
Materials and methods
Sample collection and preparation
Wastewater samples were collected from multiple piggeries (designated as farms A, C, D, and E) in Queensland, Australia [12]. Samples from farms C, D, and E were collected from wastewater collection ponds, while samples from farm A were collected from drains under pig sheds [12]. Samples were transported on ice and stored at -20°C until processing.
For DNA extraction, samples underwent a preparatory concentration step [12]. Briefly, 10-40 mL of wastewater (volume adjusted based on particulate content) was centrifuged at 46 g for 1 minute to settle heavier solids. The supernatant was then centrifuged at 4,550 g for 30 minutes. The resulting pellet was weighed (typically 0.37-0.67 g) and stored at -20°C. For extraction, the pellet was thawed and reconstituted in 500 μL of Milli-Q water, and 0.3 g of this homogenized material was used for DNA extraction [12].
DNA extraction methods evaluated
Six DNA extraction protocols were initially screened based on DNA yield and quality. The three best-performing commercial kits were selected for further evaluation using wastewater spiked with a mock community of known pig pathogens [12].
Table 1: DNA Extraction Kits and Protocols Evaluated
| Kit Name | Manufacturer | Key Features | Modifications from Manufacturer Protocol |
|---|---|---|---|
| QIAamp PowerFecal Pro DNA Kit (PF) | QIAGEN | Designed for difficult environmental samples | 500 μL CD1 lysis buffer used; 10 min mechanical lysis; modified wash steps; 10 min air-dry before elution [12] |
| DNeasy PowerLyzer PowerSoil Kit | QIAGEN | Effective lysis for diverse soil microbes | Specific protocol details were not provided in the search results [12] |
| NucleoSpin Soil Kit | Macherey-Nagel | Efficient inhibitor removal | Specific protocol details were not provided in the search results [12] |
| PureGene Tissue Core Kit (PG) | QIAGEN | Traditional phenol/chloroform method | Addition of Proteinase K and 2h incubation at 56°C; DNA precipitation overnight [12] |
| In-house Method (IH) | N/A | Custom developed for piggery effluent | Lysis with EDTA-Tris-NaCl buffer and SDS; multiple 98°C incubation steps [12] |
Downstream analysis
To rigorously test the selected methods, a mock microbial community composed of known pig pathogens was spiked into piggery wastewater samples from farm E [12]. DNA extracted using the three best-performing kits was sequenced on an Oxford Nanopore Technologies (ONT) MinION platform. The resulting sequencing data was analyzed using the kraken2 taxonomic classifier and an in-house database to evaluate the recovery of the spiked organisms and the overall microbial community profile [12].
Results and discussion
Performance comparison of extraction methods
The evaluation of the six extraction methods revealed significant discrepancies in their ability to recover high-quality bacterial DNA from the complex piggery wastewater matrix [12]. Based on yield and quality metrics, three commercial kits consistently outperformed the others: the QIAGEN QIAamp PowerFecal Pro DNA Kit, the QIAGEN DNeasy PowerLyzer PowerSoil Kit, and the Macherey-Nagel NucleoSpin Soil Kit [12].
The optimized QIAamp PowerFecal Pro (PF) protocol was identified as the most suitable and reliable method. When tested with the spiked mock community, this method demonstrated superior performance in downstream sequencing analysis, providing a more accurate representation of the known microbial composition and enabling more effective pathogen detection [12].
Table 2: Key Findings from the Extraction Kit Evaluation
| Evaluation Criteria | QIAamp PowerFecal Pro (PF) | DNeasy PowerLyzer PowerSoil | NucleoSpin Soil |
|---|---|---|---|
| DNA Yield | High | High | High |
| DNA Quality/Purity | High | High | High |
| Inhibitor Removal | Effective | Effective | Effective |
| Sequencing Performance | Most suitable and reliable | Good | Good |
| Pathogen Detection | Most accurate representation | Biases observed | Biases observed |
| Key Advantage | Optimized protocol for this matrix | Effective lysis | Efficient inhibitor removal |
Impact of extraction method on metagenomic analysis
The study demonstrated that the choice of DNA extraction method introduces specific biases that significantly influence the outcome of metagenomic analyses [12]. Different extraction protocols exhibited varying efficiencies in lysing diverse bacterial cell types and recovering DNA from different microbial taxa. This variability can lead to distorted representations of the microbial community in subsequent sequencing data, potentially affecting the sensitivity and accuracy of pathogen detection.
These findings underscore a critical principle for metagenomic research: the optimal DNA extraction method must be determined empirically for each specific sample matrix [12]. A method validated for human wastewater or soil may not perform optimally for piggery effluent. This optimization is a prerequisite for establishing robust, reproducible metagenomic surveillance systems to be used for routine early disease detection and intervention in agricultural settings [12].
The scientist's toolkit
Table 3: Essential Research Reagent Solutions for Wastewater Metagenomics
| Item | Function/Application | Example Products/Models |
|---|---|---|
| DNA Extraction Kits | Isolation of inhibitor-free microbial DNA from complex matrices | QIAGEN QIAamp PowerFecal Pro, Macherey-Nagel NucleoSpin Soil [12] |
| Concentration Tools | Concentrating diluted microbial particles from large water volumes | Dynabeads Magnetic Beads, Filtration devices [100] [99] |
| Automated Purification | High-throughput, reproducible nucleic acid extraction | KingFisher instruments with MagMAX kits [100] |
| Sequencing Platform | Long-read sequencing for metagenome-assembled genomes (MAGs) | Oxford Nanopore Technologies (ONT) MinION [12] [60] |
| Bioinformatics Tools | Taxonomic classification and genome analysis | kraken2, custom databases, mmlong2 workflow [12] [60] |
Experimental workflow
The following workflow diagrams the optimized protocol for pathogen detection in piggery wastewater, from sample collection to data analysis.
This case study demonstrates that successful pathogen surveillance in complex environments like piggery wastewater is highly dependent on the initial DNA extraction step. The optimized QIAGEN PowerFecal Pro protocol proved to be the most effective method for recovering high-quality DNA suitable for nanopore sequencing and accurate metagenomic analysis. The findings emphasize that rigorous, matrix-specific optimization of DNA extraction methods is not merely a preliminary step but a critical factor in generating reliable, actionable data for disease surveillance. This work provides a validated framework for implementing metagenomic monitoring as a practical tool for safeguarding animal health and, by extension, public health within a One Health context.
Urinary Tract Infections (UTIs) are among the most prevalent bacterial infections globally, necessitating rapid and accurate diagnostic methods for effective treatment and antimicrobial stewardship [101]. The emergence of long-read sequencing technologies, particularly Oxford Nanopore Technology (ONT), has revolutionized pathogen identification by enabling better genome assembly and resolution of complex genomic regions [5]. However, the pre-analytical step of DNA extraction, specifically the cell lysis method, presents a critical bottleneck that significantly influences downstream sequencing success and diagnostic accuracy [3] [5].
This case study systematically evaluates different lysis methodologies within the broader context of metagenomic sequencing research. The recovery of high-quality, high-molecular-weight (HMW) DNA is paramount for leveraging the full advantages of long-read sequencing platforms [5]. We examine mechanical, enzymatic, and chemical lysis approaches, assessing their performance in terms of DNA yield, integrity, microbial diversity representation, and compatibility with downstream long-read sequencing applications for UTI diagnostics.
Literature Review and Performance Comparison
Quantitative Comparison of Lysis Methods
Recent comparative studies have illuminated the significant performance differences between various DNA extraction and lysis methods. The table below summarizes key quantitative findings from the literature.
Table 1: Performance Metrics of Different Lysis and DNA Extraction Methods
| Method / Kit | Primary Lysis Mechanism | DNA Yield/Quality | Detection Sensitivity | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| Mechanical (Bead Beating) [3] | Physical shearing | High yield but fragmented DNA | Broad pathogen detection | Efficient for tough cell walls; unbiased for Gram-positive bacteria [5] | Excessive DNA fragmentation; reduced read lengths [3] |
| Enzymatic Lysis [3] | Enzyme-based cell wall degradation | High integrity DNA (2.1-fold increase in read length) [3] | 100% concordance with culture [3] | Gentle lysis; preserves long fragments; representative microbial profiles [3] | Longer incubation time; cost of enzymes |
| Ionic Liquid-Based (IL-DEx) [102] | Chemical/Detergent | Yields comparable to commercial kits [102] | ~10²–10⁴ CFU/ml [102] | Fast (under 30 min); minimal equipment; no hazardous chemicals [102] | Lower recovery for Gram-positive bacteria (0.7–8%) [102] |
| Quick-DNA HMW MagBead Kit [5] | Not specified | High yield of pure HMW DNA [5] | Accurate mock community detection [5] | Optimized for HMW DNA; suitable for complex metagenomics [5] | Performance may vary with sample matrix |
| QIAGEN PowerFecal Pro (Optimized) [12] | Mechanical and chemical | High-quality, inhibitor-free DNA [12] | Effective in complex wastewater [12] | Reliable for complex samples; effective inhibitor removal [12] | Protocol requires optimization |
Impact on Diagnostic Outcomes
The choice of lysis method directly influences diagnostic accuracy in clinical settings. A 2025 multicenter comparative study demonstrated that targeted Next-Generation Sequencing (tNGS), which relies on efficient DNA extraction, showed a 96.5% concordance with culture-positive UTI samples and significantly outperformed traditional culture and metagenomic NGS (mNGS) in detecting polymicrobial infections (55.4% of samples vs. 27.7% for mNGS) [101]. The study highlighted tNGS's superior ability to identify fastidious organisms and antibiotic resistance genes, underscoring the importance of the upstream DNA extraction process [101].
Furthermore, the enzymatic lysis method, which provides gentler cell wall degradation, was shown to increase the average length of microbial reads by a median of 2.1-fold and the mapped reads proportion of specific species by a median of 11.8-fold compared to control methods, making it particularly suitable for long-read sequencing platforms [3].
Detailed Experimental Protocols
Principle: Utilizes lytic enzymes to digest bacterial cell walls, preserving DNA integrity for long-fragment sequencing.
Reagents and Equipment:
- Lytic enzyme solution (e.g., from Qiagen)
- MetaPolyzyme (Sigma-Aldrich), reconstituted in PBS
- IndiSpin Pathogen Kit (Indical Bioscience)
- Phosphate Buffer Saline (PBS)
- Thermostatic shaker or water bath
- Microcentrifuge
Procedure:
- Sample Preparation: Centrifuge 1 ml of urine sample at 20,000 × g for 5 min. Discard 800 μl of supernatant and resuspend the pellet in the remaining 200 μl by gentle vortexing.
- Enzymatic Lysis: Add 5 μl of lytic enzyme solution and 10 μl of reconstituted MetaPolyzyme to the 200 μl sample. Mix by gentle pipetting.
- Incubation: Incubate at 37°C in a shaker for 1 hour to lyse microbial cells.
- DNA Extraction: Proceed with DNA extraction using the IndiSpin Pathogen Kit or similar, following the manufacturer's instructions.
- DNA Elution: Elute DNA in 100 μl of elution buffer. Measure concentration using a fluorometer (e.g., Qubit 4.0).
Principle: Uses an ionic liquid and magnetic beads for rapid DNA recovery, eliminating hazardous reagents.
Reagents and Equipment:
- Ionic Liquid Lysis Buffer
- Magnetic Beads
- Wash Buffers
- Elution Buffer
- Magnetic rack
- Thermonixer or water bath
Procedure:
- Lysis: Mix 1 ml of urine sample with ionic liquid lysis buffer. Incubate at room temperature for 10 min.
- Binding: Add functionalized magnetic beads and incubate for 5 min with mixing to allow DNA binding.
- Washing: Place the tube on a magnetic rack. Discard the supernatant once clear. Wash the beads with wash buffer while on the magnet.
- Elution: Resuspend the beads in elution buffer. Incubate at 65°C for 5 min to release DNA.
- Recovery: Place the tube on the magnetic rack and transfer the DNA-containing supernatant to a new tube. The entire process is completed in under 30 minutes.
Principle: Uses bead-beating for vigorous physical disruption of cells, effective for tough cell walls.
Reagents and Equipment:
- Pathogen Lysis Tubes with glass beads (Qiagen)
- Buffer ATL (containing Reagent DX, Qiagen)
- Vortex mixer with horizontal platform
- IndiSpin Pathogen Kit (Indical Bioscience)
Procedure:
- Sample Preparation: Prepare enriched urine sample as in Section 3.1, Step 1.
- Bead-Beating: Transfer 200 μl of enriched sample to a Pathogen Lysis Tube. Add 50 μl of Buffer ATL.
- Mechanical Lysis: Attach tubes to a horizontal platform vortex mixer and vortex at maximum speed for 10 min.
- DNA Extraction: Briefly centrifuge tubes to collect liquid. Extract DNA from the supernatant using the IndiSpin Pathogen Kit.
- DNA Elution: Elute DNA in 100 μl of elution buffer.
Workflow and Decision Pathway
The following diagram illustrates the logical workflow for evaluating and selecting an appropriate lysis method for long-read sequencing of UTIs.
Diagram 1: Lysis method selection for long-read sequencing. This workflow guides the selection of an optimal lysis method based on primary objective and sample characteristics.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents and Their Applications in Lysis Protocols
| Reagent / Kit | Primary Function | Application Context |
|---|---|---|
| MetaPolyzyme [3] | Enzymatic cell wall degradation for Gram-positive and Gram-negative bacteria | Enzymatic lysis protocols for generating long DNA fragments |
| Pathogen Lysis Tubes (Glass Beads) [3] | Physical disruption of microbial cell walls via bead-beating | Mechanical lysis for efficient recovery from tough Gram-positive bacteria |
| Ionic Liquid Lysis Buffer [102] | Chemical lysis and DNA binding in a single rapid step | IL-DEx protocol for fast, equipment-minimal DNA extraction |
| Quick-DNA HMW MagBead Kit [5] | Combined lysis and HMW DNA purification using magnetic beads | Optimized protocol for obtaining high-yield, long-fragment DNA |
| DNase I & Proteinase K [103] | Elimination of extracellular DNA from dead/injured cells | Sample pretreatment for live/dead discrimination in molecular analyses |
| QIAamp PowerFecal Pro DNA Kit [12] | Lysis and purification designed for complex samples | Effective DNA extraction from inhibitor-rich matrices |
Discussion and Implementation Guidelines
Method Selection Criteria
Choosing an appropriate lysis method requires balancing multiple factors, including target pathogens, required turnaround time, available equipment, and downstream applications. For comprehensive pathogen detection in polymicrobial UTIs, where preserving the relative abundance of community members is crucial, enzymatic lysis offers superior performance by providing more representative microbial profiles and longer DNA fragments [3]. In contrast, for rapid diagnostics where speed is paramount, the ionic liquid-based IL-DEx method provides results in under 30 minutes with minimal equipment [102].
For laboratories handling diverse sample types or focusing on antibiotic resistance gene detection, mechanical lysis with optimized kits like QIAamp PowerFecal Pro may be advantageous due to more consistent lysis across different bacterial cell wall structures [12]. This is particularly relevant for UTI diagnostics, where tNGS has demonstrated 100% sensitivity for detecting vancomycin and methicillin resistance genes in Gram-positive pathogens [101].
Troubleshooting and Optimization Tips
- Low DNA Yield: For enzymatic protocols, ensure proper enzyme activity by verifying storage conditions and expiration dates. Increase incubation time if processing samples with high Gram-positive content [3].
- Short Read Lengths: If using mechanical lysis, optimize bead-beating duration and intensity to balance between cell disruption and DNA shearing. Consider transitioning to enzymatic or hybrid approaches for HMW DNA [5].
- Inhibition in Downstream Applications: For complex urine samples, incorporate additional wash steps or use kits specifically designed for inhibitor removal, such as those optimized for environmental samples [12].
- Bias in Microbial Community Representation: Validate lysis efficiency across different bacterial species using mock communities and adjust lysis parameters accordingly. Enzymatic methods generally show less bias compared to harsh mechanical disruption [3] [5].
The selection of an appropriate lysis method is a critical determinant of success in long-read sequencing applications for UTI diagnostics. While mechanical lysis offers robustness for difficult-to-lyse pathogens, enzymatic approaches provide superior DNA integrity for long-read sequencing platforms. Emerging technologies like ionic liquid-based extraction enable rapid processing suitable for point-of-care applications. Researchers must carefully consider their specific diagnostic needs, sample characteristics, and available resources when selecting and optimizing lysis methodologies. As molecular diagnostics continue to evolve, further refinement of these techniques will undoubtedly enhance our ability to rapidly and accurately diagnose UTIs, ultimately improving patient outcomes and supporting antimicrobial stewardship efforts.
Within metagenomic sequencing research, the quality of input genomic DNA (gDNA) is the foundational determinant of all downstream analytical success. This application note details the critical protocols and validation metrics that tether pre-analytical DNA extraction procedures to definitive bioinformatic outcomes, including taxonomic classification accuracy and genome assembly integrity. Evidence confirms that suboptimal DNA quality directly propagates into substantial biases, obscuring true biological signals and compromising the reliability of genomic catalogs [104] [60]. By establishing a rigorous framework for DNA quality assessment and validation, researchers can ensure that high-quality, actionable data is generated for downstream drug development and scientific discovery.
The Impact of DNA Quality on Metagenomic Data
The journey from sample to sequence is fraught with potential biases, many of which originate during DNA extraction. Inadequate lysis of diverse cell types or the co-extraction of inhibitors can severely skew the apparent structure of a microbial community.
- Biased Microbial Representation: Complex samples contain organisms with vastly different cell wall structures, such as Gram-positive bacteria and fungi, which are more difficult to lyse than Gram-negative bacteria. Protocols that fail to aggressively disrupt these resistant cells will under-represent them in subsequent sequencing data, generating a distorted profile of the community [104].
- Inhibition of Downstream Applications: The presence of contaminants like humic acids, proteins, or lipids from the sample matrix can inhibit enzymatic reactions in library preparation and PCR amplification. This can lead to low sequencing yields or even complete amplification failure, particularly in samples with low microbial biomass like human milk [71] [104].
- Compromised Assembly Metrics: High-quality, high-molecular-weight (HMW) DNA is a prerequisite for long-read sequencing and the generation of contiguous assemblies. Fragmented DNA produces short contigs, hampering the ability to reconstruct complete genes or genomes, a challenge acutely observed in highly complex environments like soil [60] [71].
Table 1: DNA Quality Issues and Their Downstream Effects on Metagenomic Data
| DNA Quality Issue | Impact on Classification Accuracy | Impact on Assembly Metrics |
|---|---|---|
| Incomplete Cell Lysis | Skewed taxonomic abundance; under-representation of hardy taxa (e.g., Gram-positives, spores) [104] | Reduced recovery of genomes from difficult-to-lyse organisms [60] |
| Fragmented DNA | N/A | Lower contig N50; failure to assemble complete genes or operons [60] |
| Co-purified Inhibitors | Reduced sequencing depth; increased rate of sample dropout | Poor assembly due to low sequence coverage; increased assembly fragmentation |
| Low DNA Yield | Inability to sequence; or, high technical noise in low-biomass samples [105] | Inadequate coverage for confident genome binning [60] |
Experimental Protocols for DNA Extraction and Validation
An Improved Method for High-Quality Metagenomic DNA Extraction
The following protocol, adapted from the THSTI method, is designed for efficient lysis of a broad spectrum of microorganisms and is applicable to diverse human and environmental samples [71].
1. Sample Pre-processing
- Environmental Samples (e.g., soil, sediment): Homogenize 0.5 g of material in 1 mL of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0).
- Human Milk/Biological Samples: Centrifuge at 2,500 × g for 20 minutes at 4°C. Discard the fat layer and supernatant. Wash the cell pellet with TE buffer and re-concentrate at 20,000 × g for 20 minutes. Resuspend the final pellet in 300 μL of TE buffer [104].
2. Spheroplast Formation
- Add the following to the sample suspension:
- 50 μL of lysozyme (50 mg/mL)
- 10 μL of lysostaphin (1 mg/mL)
- 10 μL of mutanolysin (1 mg/mL)
- Incubate at 37°C for 60 minutes with intermittent mixing.
3. Comprehensive Cell Lysis
- Add 4 mL of Lysis Buffer (4 M Guanidinium Thiocyanate, 0.1 M Tris-HCl, pH 7.5) and 600 μL of 10% N-Lauroylsarcosine to the spheroplast mixture. Vortex thoroughly.
- Transfer the solution to a tube containing sterile silica/zirconia beads (0.1 mm and 0.5 mm).
- Perform bead-beating for 3 minutes at high speed.
- Incubate the lysate at 95°C for 10 minutes.
4. DNA Precipitation and Purification
- Centrifuge the lysate at 10,000 × g for 5 minutes to remove debris.
- Transfer the supernatant to a new tube. Add 0.7 volumes of isopropanol and 0.1 volumes of 3 M sodium acetate (pH 5.2).
- Incubate at -20°C for 30 minutes to precipitate the DNA.
- Pellet the DNA by centrifugation at 15,000 × g for 20 minutes at 4°C.
- Wash the pellet twice with 1 mL of 70% ethanol.
- Air-dry the pellet and resuspend in 50-100 μL of nuclease-free water or TE buffer.
DNA Quality Assessment Workflow
Post-extraction, DNA must be rigorously quantified and qualified before proceeding to sequencing.
1. Spectrophotometric Analysis
- Use a Nanodrop or similar spectrophotometer to determine DNA concentration and purity via absorbance at 260 nm and the A260/A280 ratio, respectively. High-quality DNA should have an A260/A280 ratio between ~1.6 and 1.9 [71].
2. Gel Electrophoresis
- Visualize 1 μL of the extracted DNA on a 0.8% agarose gel. High-quality metagenomic DNA should appear as a tight, high-molecular-weight band with minimal smearing below 10 kb, indicating minimal fragmentation [71].
3. PCR Amplification
- Validate the quality and amplifiability of the DNA by performing PCR targeting a ubiquitous marker gene, such as the V3-V4 region of the bacterial 16S rRNA gene or the fungal ITS region. Successful amplification and a clean amplicon band on a gel confirm the DNA is free of potent PCR inhibitors [104].
Bioinformatic Validation Metrics
Connecting DNA Quality to Classification and Assembly
Once sequencing data is generated, specific bioinformatic metrics serve as a final validation of the input DNA's quality.
Table 2: Key Bioinformatic Metrics for Validation
| Bioinformatic Metric | Definition and Measurement | What It Validates |
|---|---|---|
| Reads Assembled | Proportion of raw sequencing reads that are successfully incorporated into contigs during assembly. | Purity and integrity of DNA; high levels of contamination or fragmentation result in a low proportion of assembled reads [60]. |
| Contig N50 | The length of the shortest contig at which 50% of the total assembled sequence is contained in contigs of that length or longer. Measured in kilobases (kb). | Molecular weight of the input DNA; HMW DNA produces long, contiguous assemblies with a high N50 [60]. |
| MAG Quality (Completeness/Contamination) | Assessed using tools like CheckM. Completeness estimates the percentage of single-copy core genes present; contamination measures the percentage present in multiple copies. | Effectiveness of lysis and uniformity of sequence coverage across the community. Incomplete lysis can lead to uneven coverage, hampering MAG recovery [60]. |
| Taxonomic Classification Rate | The percentage of sequencing reads or assembled contigs that can be confidently assigned to a taxonomic group. | Comprehensiveness of cell lysis and absence of severe biases. High rates of "unclassified" sequences may indicate technical artifacts or novel diversity. |
Case Study: Validation in Soil Metagenomics
The critical link between input DNA quality and output is exemplified in large-scale terrestrial metagenomic studies. Research involving deep long-read sequencing of 154 soil and sediment samples demonstrated that samples yielding higher quantities of HMW DNA produced significantly more high-quality Metagenome-Assembled Genomes (MAGs). The study's custom "mmlong2" binning workflow recovered over 15,000 previously undescribed species-level MAGs, an achievement contingent upon the initial quality of the extracted DNA. Notably, samples from agricultural fields with lower DNA assembly efficiency (median 45.0% of reads assembled into contigs) produced far fewer MAGs (median 56) compared to coastal samples, underscoring how sample-specific challenges and DNA quality directly dictate genomic discovery [60].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Metagenomic DNA Validation
| Reagent / Kit / Tool | Function in Validation Workflow |
|---|---|
| Lytic Enzymes (Lysozyme, Lysostaphin, Mutanolysin) | Enzymatic lysis of diverse bacterial cell walls to ensure unbiased representation in the microbial community profile [71] [104]. |
| Guanidinium Thiocyanate (GTC) | A potent chaotropic agent used for chemical lysis of cells and inactivation of nucleases to preserve DNA integrity [71] [104]. |
| Silica/Zirconia Beads (0.1 & 0.5 mm) | Mechanical lysis via bead-beating to disrupt hardy cell types, such as Gram-positive bacteria and spores, that are resistant to chemical lysis alone [104]. |
| Quick-DNA Fecal/Soil Microbe Kit (Zymo Research) | A commercial silica-membrane-based kit optimized for isolating DNA from complex and inhibitor-rich samples [104]. |
| CheckM / similar tools | Bioinformatic software for assessing the completeness and contamination of Metagenome-Assembled Genomes (MAGs), a key downstream validation metric [60]. |
Workflow and Data Relationships
The following diagram illustrates the logical progression from sample collection to bioinformatic validation, highlighting how DNA quality metrics directly influence downstream data outcomes.
DNA Quality to Data Validation Workflow
This workflow demonstrates the linear process from sample to data and, crucially, the direct correlative relationships (dashed lines) between initial DNA quality checks and final bioinformatic validation metrics. High molecular weight DNA is a prerequisite for a high contig N50, while DNA purity is essential for achieving high taxonomic classification rates.
Conclusion
The choice and optimization of DNA extraction methods are not merely preliminary steps but are foundational to the success of any metagenomic sequencing study. As this review underscores, a one-size-fits-all approach is ineffective; the optimal protocol must be tailored to the specific sample matrix and research question. Methodological rigor, coupled with rigorous validation using mock communities and comparative analysis, is essential to minimize biases and generate biologically accurate data. Future directions point towards greater standardization, the development of more gentle extraction methods to preserve long DNA fragments for advanced sequencing technologies, and the integration of automated, high-throughput workflows. For biomedical and clinical research, these advancements will directly translate into more reliable pathogen detection, accelerated drug discovery, and robust microbiome-based diagnostics, ultimately fulfilling the promise of metagenomics in precision medicine and public health.
References
- 1. Application of metagenomic next-generation sequencing in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11604630/]
- 2. Metagenomic Next Generation Sequencing: How Does It ... [https://asm.org/articles/2019/november/metagenomic-next-generation-sequencing-how-does-it]
- 3. Comparison Analysis of Different DNA Extraction Methods on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9273838/]
- 4. Comparison of DNA extraction methods for 16S rRNA gene ... [https://www.nature.com/articles/s41598-023-33959-6]
- 5. Comparison of 6 DNA extraction methods for isolation of high ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09537-5]
- 6. Clinical metagenomics | Nature Reviews Genetics [https://www.nature.com/articles/s41576-019-0113-7]
- 7. Optimization of Metagenomic Next-Generation Sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12436562/]
- 8. MAGdb: a comprehensive high quality MAGs repository for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12421750/]
- 9. Comparison of DNA extraction kits and modification of DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4287173/]
- 10. Transparent DNA/RNA Co-extraction Workflow Protocol ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5067521/]
- 11. Metagenomic data integrity [https://www.qiagen.com/us/applications/next-generation-sequencing/dna-sequencing/metagenomic-sequencing/bias-and-data-integrity]
- 12. Optimization of DNA extraction methods from pig farm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12551764/]
- 13. Benchmarking DNA isolation methods for marine ... [https://www.nature.com/articles/s41598-023-48804-z]
- 14. Comparison of DNA extraction methods on different ... [https://www.nature.com/articles/s41598-024-59086-4]
- 15. Comparison of Eleven Methods for Genomic DNA Extraction ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0115960]
- 16. Evaluation of different DNA extraction methods for the ... [https://www.sciencedirect.com/science/article/pii/S1056617125000844]
- 17. Addressing Bias in Metagenomic Studies [https://blog.omni-inc.com/blog/addressing-bias-in-metagenomic-studies]
- 18. Unbiased Metagenomic Protocol: From RNA to Sequencing [https://www.protocols.io/view/unbiased-metagenomic-protocol-from-rna-to-sequenci-d6xi9fke.html]
- 19. Resolving bias in metagenomic - The Blog - Tecan analysis [https://www.tecan.com/blog/resolving-bias-in-metagenomic-analysis]
- 20. Comparison evaluation of bacterial DNA extraction ... [https://pubmed.ncbi.nlm.nih.gov/39905084/]
- 21. Quantifying the biases in metagenome mining for realistic assessment... [https://www.nature.com/articles/srep34155?error=cookies_not_supported]
- 22. of Impact Method on Variation in Human and Built... DNA Extraction [https://pubmed.ncbi.nlm.nih.gov/32528434/]
- 23. The truth about metagenomics : quantifying and counteracting bias in... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-015-0351-6]
- 24. An optimized DNA extraction protocol for reliable PCR-based ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-025-01460-y]
- 25. Evaluation and Optimization of DNA Extraction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC91634/]
- 26. QIAamp PowerFecal Pro DNA Kit | Stool DNA Isolation [https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/dna-purification/genomic-dna/qiaamp-powerfecal-pro-dna-kit]
- 27. NucleoSpin Soil, Mini kit for DNA from soil [https://www.mn-net.com/nucleospin-soil-mini-kit-for-dna-from-soil-740780.50]
- 28. Macherey-Nagel™ NucleoSpin™ Soil, Mini kit for DNA ... [https://www.labmartgh.com/shop/macherey-nagel-tm-nucleospin-tm-soil-mini-kit-for-dna-from-soil-29605]
- 29. NucleoSpin 96 Soil, 96-well kit for DNA from soil [https://www.mn-net.com/nucleospin-96-soil-96-well-kit-for-dna-from-soil-740787.2]
- 30. Evaluation of DNA extraction kits for long-read shotgun ... [https://www.nature.com/articles/s41598-024-80660-3]
- 31. Comparison of commercial DNA extraction kits for whole ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9915679/]
- 32. Comparative evaluation of soil DNA extraction kits for long ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11432601/]
- 33. Comparative evaluation of commercial DNA isolation ... [https://www.nature.com/articles/s41598-024-78066-2]
- 34. Impact of DNA Extraction and 16S rRNA Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12154973/]
- 35. Why Lysozyme and Zymolyase are Important in Cell Lysis ... [https://www.goldbio.com/blogs/articles/why-lysozyme-and-zymolyase-are-important-in-cell-lysis-buffers?srsltid=AfmBOopSiA4HsWeVinoZDZw4kYZvlw0XoRdkma5vf6XrjFOMQgDKmfr4]
- 36. Cell Disruption- Definition, Methods, Types, Significance [https://microbenotes.com/cell-disruption-methods/]
- 37. Comparison of the efficiency of different cell lysis methods and different commercial methods for RNA extraction from Candida albicans stored in RNAlater | BMC Microbiology | Full Text [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-019-1473-z]
- 38. Bead Beating Technology Explained [https://www.mpbio.com/bs/bead-beating-technology-explained?srsltid=AfmBOora3pc4pbtpEPwkkYtSQWgU0pzokL7DjPv9fJX-UtBe-sXkBnue]
- 39. An optimised bead beating RNA extraction method for ... [https://www.sciencedirect.com/science/article/pii/S2950194625000731]
- 40. 8 Cell Lysis Methods Explained [https://bitesizebio.com/13536/cell-lysis-methods/]
- 41. Comparison and Optimization of DNA Extraction Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932244/]
- 42. Optimized protocol for the extraction of RNA and DNA from ... [https://www.nature.com/articles/s41598-021-96567-2]
- 43. Optimizing extraction of microbial DNA from urine [https://pmc.ncbi.nlm.nih.gov/articles/PMC12058534/]
- 44. How to Process Sputum Samples and Extract Bacterial DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6214103/]
- 45. Optimized Lysis-Extraction Method Combined With IS6110- ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2018.02224/full]
- 46. Optimization of DNA extraction from human urinary samples ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0210306]
- 47. DNA Extraction Kits Compared: Spin Columns vs. Magnetic ... [https://synapse.patsnap.com/article/dna-extraction-kits-compared-spin-columns-vs-magnetic-beads]
- 48. Robust, Open‐Source and Automation‐Friendly DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550468/]
- 49. SPRI Beads vs Spin Columns [https://www.magbiogenomics.com/latest/post/magnetic-bead-dna-cleanup-protocols-optimizing-pcr-cleanups-with-highprep-vs-spin-columns?srsltid=AfmBOopF8b3lfNnYU1kO_v1btCG9FR70qOH15-Lwu4mARl8nApBnlXcg]
- 50. High-Throughput Plant Genotyping and Metagenomics ... [https://gentelbio.com/high-throughput-plant-genotyping-and-metagenomics-with-magnetic-beads-dna-kits/]
- 51. Automated environmental metagenomics using Oxford ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11989-w]
- 52. Efficiency of DNA Isolation Methods Based on Silica ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7697941/]
- 53. A Hitchhiker's Guide to long-read genomic analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12047252/]
- 54. Sequencing 101: long-read sequencing [https://www.pacb.com/blog/long-read-sequencing/]
- 55. Comparing long-read sequencing technologies [https://www.pacb.com/blog/sequencing-101-comparing-long-read-sequencing-technologies/]
- 56. How nanopore sequencing works [https://nanoporetech.com/platform/technology]
- 57. Modification mapping by nanopore sequencing [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2022.1037134/full]
- 58. Direct sequencing of cytosine modification states from ... [https://nanoporetech.com/resource-centre/direct-sequencing-of-cytosine-modification-states-from-nanopore-long-read-data]
- 59. Epigenetics and methylation analysis [https://nanoporetech.com/applications/investigations/epigenetics-and-methylation-analysis]
- 60. Genome-resolved long-read sequencing expands known ... [https://www.nature.com/articles/s41564-025-02062-z]
- 61. 宏基因组测序中不同DNA提取方法对人类和环境微生物群落 ... [https://www.sohu.com/a/449633507_120055884]
- 62. How to Work with Challenging or Limited Genomic ... [https://blog.omni-inc.com/blog/how-to-work-with-challenging-or-limited-genomic-samples-updated-for-2025]
- 63. Perish the thawed? EDTA reduces DNA degradation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12132941/]
- 64. Wet Lab Protocols Matter: Choice of DNA Extraction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550470/]
- 65. Measuring Real-time DNA/RNA Nuclease Activity through ... [https://en.bio-protocol.org/en/bpdetail?id=4206&type=0]
- 66. Simple statistical identification and removal of contaminant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6298009/]
- 67. DNA extraction and purification - an overview [https://www.abcam.com/en-us/knowledge-center/dna-and-rna/dna-extraction]
- 68. Identification and Removal of Contaminant Sequences From ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5945997/]
- 69. DNA Extraction - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/dna-extraction]
- 70. A rapid and efficient DNA extraction protocol from fresh and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6817243/]
- 71. An Improved Method for High Quality Metagenomics DNA ... [https://www.nature.com/articles/srep26775]
- 72. Challenging biological samples and strategies for DNA ... [https://pubmed.ncbi.nlm.nih.gov/40033560/]
- 73. Fast and inexpensive protocols for consistent extraction of ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0206085]
- 74. Optimized bacterial DNA isolation method for microbiome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8208965/]
- 75. Optimization and validation of sample preparation for ... [https://pubmed.ncbi.nlm.nih.gov/28789678/]
- 76. An Optimized Metagenomic Approach for Virome Detection ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.01552/full]
- 77. A comparison of five DNA extraction methods from ... [https://www.sciencedirect.com/science/article/pii/S0379073823001809]
- 78. Rapid and economical protocols for genomic and ... [https://annforsci.biomedcentral.com/articles/10.1007/s13595-018-0705-y]
- 79. Laboratory validation of a clinical metagenomic next ... [https://www.nature.com/articles/s41467-024-51470-y]
- 80. Recommendations on Collecting and Storing Samples for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6400449/]
- 81. Storage and shipping of tissue samples for DNA analyses [https://pmc.ncbi.nlm.nih.gov/articles/PMC4461180/]
- 82. Best practices for sample processing and storage prior to ... [https://microbe.net/2015/01/31/best-practices-for-sample-storage-prior-to-microbiome-dna-analysis-freeze-buffer-process/]
- 83. DNA sample storage: best practices after extraction [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/dna/handling-dna/storage-of-dna]
- 84. How Are Genomic DNA Samples Collected & Stored? [https://www.azenta.com/learning-center/blog/how-are-genomic-dna-samples-collected-stored]
- 85. Different Ways Professionals Store DNA Samples [https://www.celestis.com/blog/different-ways-professionals-store-dna-samples/]
- 86. De-biasing microbiome sequencing data: bacterial ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-024-01998-4]
- 87. Characterization and Demonstration of Mock Communities as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8941912/]
- 88. Characterizing and measuring bias in sequence data - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4053816/]
- 89. Comparison of Three DNA Isolation Methods and Two ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12028492/]
- 90. Protocol for DNA extraction and shotgun metagenomic ... [https://www.protocols.io/view/protocol-for-dna-extraction-and-shotgun-metagenomi-c8wqzxdw.html]
- 91. Quantifying molecular bias in DNA data storage [https://www.nature.com/articles/s41467-020-16958-3]
- 92. Metagenomic sequencing and reconstruction of 82 ... [https://www.nature.com/articles/s41597-024-03332-x]
- 93. Unlocking the Potential of Metagenomics with the PacBio ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11728442/]
- 94. Comparative pilot study of three commercial kits ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12372513/]
- 95. Optimization of Metagenomic Next-Generation Sequencing ... [https://link.springer.com/article/10.1007/s40291-025-00797-3]
- 96. Metagenomic Next-Generation Sequencing in Infectious ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384723/]
- 97. Pragmatic Considerations When Extracting DNA for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10379426/]
- 98. Wastewater surveillance for viral pathogens: A tool ... [https://www.sciencedirect.com/science/article/pii/S2405844024099043]
- 99. Effective isolation of bacterial and viral DNA/RNA from ... [https://www.takarabio.com/learning-centers/nucleic-acid-purification/viral-dna-and-rna-purification/nucleic-acid-detection-from-wastewater?srsltid=AfmBOoqxljdmTEZGRX1hMBzvskfh3HpDHzh4-ZCsJaQMwAB2VVKpn8wX]
- 100. Wastewater Surveillance [https://www.thermofisher.com/us/en/home/life-science/dna-rna-purification-analysis/automated-purification-extraction/automated-magmax-kits-nucleic-acid-extraction/magmax-wastewater-kits.html]
- 101. Rapid and accurate diagnosis of urinary using... tract infections [https://pubmed.ncbi.nlm.nih.gov/40058503/]
- 102. A rapid ionic liquid-based DNA extraction method for... | CoLab [https://colab.ws/articles/10.1101%2F2025.09.30.679528]
- 103. DNase I and Proteinase K eliminate DNA from injured or ... [https://pubmed.ncbi.nlm.nih.gov/23811209/]
- 104. Improvement and Validation of a Genomic DNA Extraction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10144544/]
- 105. Precision and Accuracy in Quantitative Measurement of Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11042208/]