Phage vs. Conjugative Plasmid Delivery for CRISPR Antimicrobials: A Comparative Analysis of Efficiency and Application
This article provides a comprehensive comparison of two primary bacterial delivery systems for CRISPR-based antimicrobials: engineered bacteriophages and conjugative plasmids.
Phage vs. Conjugative Plasmid Delivery for CRISPR Antimicrobials: A Comparative Analysis of Efficiency and Application
Abstract
This article provides a comprehensive comparison of two primary bacterial delivery systems for CRISPR-based antimicrobials: engineered bacteriophages and conjugative plasmids. Aimed at researchers and drug development professionals, it explores the foundational mechanisms, methodological approaches for engineering and application, key challenges with optimization strategies, and a direct comparative analysis of in vitro and in vivo efficacy. The scope includes delivery efficiency, host range, killing specificity, and clinical translation potential, synthesizing recent advances to guide the selection and development of these innovative antibacterial platforms.
Foundational Principles: Mechanisms of Phage and Conjugation Delivery
The Antibiotic Resistance Crisis and the Need for Precision Antimicrobials
The relentless rise of antimicrobial resistance (AMR) represents one of the most severe global public health threats of our time. In 2019 alone, antibiotic-resistant bacteria were responsible for over 1 million deaths worldwide, with projections suggesting this number could reach 10 million annually by 2050 without effective interventions [1]. The ESKAPE pathogens—Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species—epitomize this crisis as the leading causes of multidrug-resistant (MDR) hospital-acquired infections [2]. Conventional broad-spectrum antibiotics increasingly fail against these pathogens, creating an urgent need for precision antimicrobials that can target resistant bacteria specifically while preserving beneficial microbiota.
The CRISPR-Cas system, an adaptive immune mechanism in prokaryotes, has emerged as a revolutionary platform for developing sequence-specific antimicrobials [3] [4]. These systems can be programmed to disable antibiotic resistance genes or eliminate bacterial virulence factors with remarkable precision [5]. However, the therapeutic potential of CRISPR-based antimicrobials depends entirely on efficient delivery to target bacteria. This guide provides a comprehensive comparison of the two primary delivery strategies—phage-mediated delivery and conjugative plasmid delivery—evaluating their efficiency, applications, and practical implementation for researchers and drug development professionals.
Delivery Mechanism Comparison: Phage vs. Conjugative Plasmid Systems
Fundamental Operating Principles
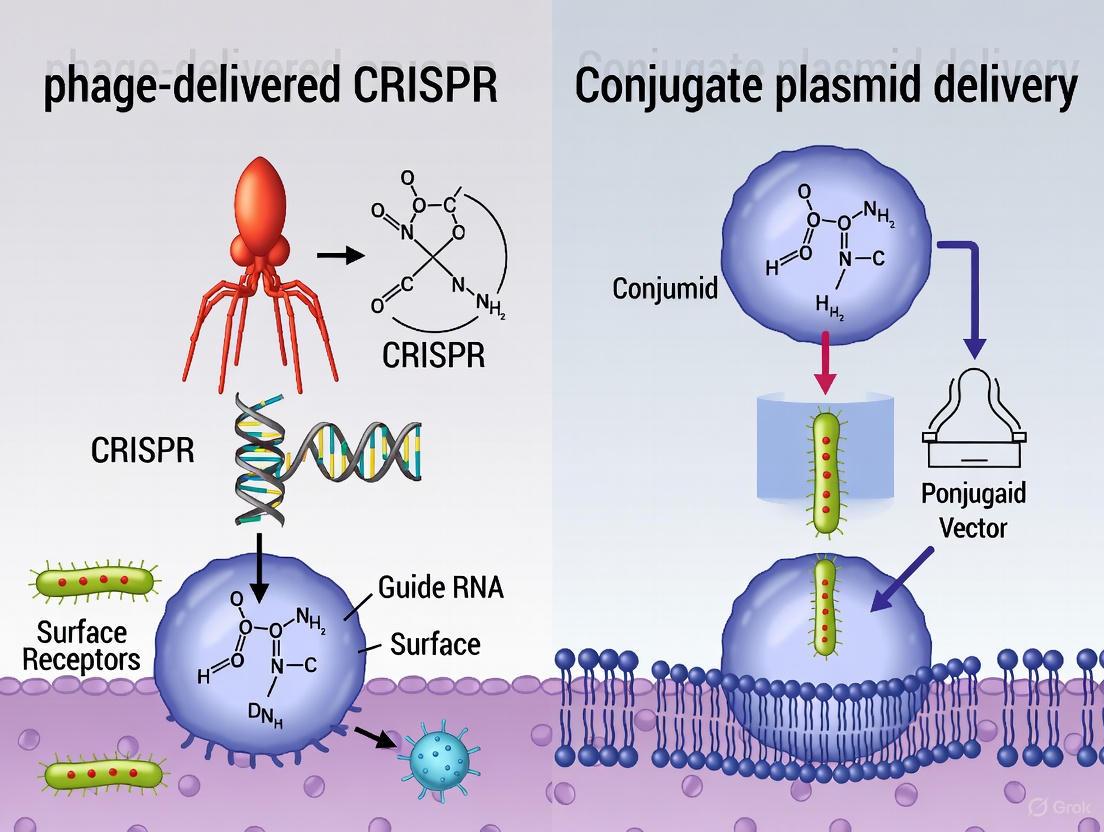
Phage-mediated delivery utilizes bacteriophages—natural viruses that infect bacteria—as vectors to inject CRISPR-Cas machinery directly into target bacterial cells. Engineered phage genomes are modified to carry genes encoding Cas nucleases and specifically designed guide RNAs (gRNAs). Upon infection, these components are expressed inside the host bacterium, where they form functional complexes that cleave targeted genetic elements [6] [7]. Different phage classes offer distinct advantages: temperate phages (like λ) can integrate into the host genome for sustained activity, while lytic phages cause immediate bacterial lysis after executing their CRISPR payload [2].
Conjugative plasmid delivery employs self-transmissible plasmids that can transfer between bacterial cells through direct cell-to-cell contact. These systems harness the natural bacterial mating apparatus to disseminate CRISPR-Cas components throughout a bacterial population [4] [8]. The CRISPR machinery is encoded on a plasmid containing the necessary origin of transfer (oriT) and conjugation genes, enabling it to mobilize from donor to recipient strains. This approach leverages the same horizontal gene transfer mechanisms that bacteria use to spread antibiotic resistance genes, effectively turning the enemy's weapon against them [4].
Quantitative Efficiency Comparison
The table below summarizes key performance metrics for both delivery systems, compiled from recent experimental studies:
Table 1: Efficiency Metrics for CRISPR-Cas Delivery Systems
| Delivery Metric | Phage-Mediated Delivery | Conjugative Plasmid Delivery |
|---|---|---|
| Editing Efficiency | 50-100% population editing in monoculture [6] | 4.7-100% resensitization efficiency [8] |
| Payload Capacity | ~15.4 kb with type I-F CAST; up to 30 kb with type V-K CAST [9] | Limited primarily by plasmid size and stability [4] |
| Host Specificity | High (species to strain-level) [2] | Moderate to high (depends on conjugation compatibility) [4] |
| Delivery Speed | Minutes to hours (single infection cycle) [7] | Hours (dependent on cell encounters and engagement time) [10] |
| Transmission Type | Density-independent within host range [1] | Density-dependent with engagement time limitations [10] |
Targeting Precision and Specificity
Phage-delivered CRISPR systems demonstrate exceptional precision, capable of distinguishing single-nucleotide variations in bacterial genomes. A recent study with Cas12a-programmed λ phages successfully eliminated target cells with perfect crRNA complementarity while sparing those with single-nucleotide mismatches in mixed cultures [7]. This nucleotide-level discrimination enables unprecedented targeting specificity for microbiome modulation applications where preserving beneficial strains is crucial.
Conjugative plasmid systems typically target broader bacterial groups based on conjugation compatibility rather than genetic sequences. Their specificity derives from the limited host range of particular conjugation systems rather than the CRISPR targeting itself [4]. For instance, pheromone-responsive plasmids in Enterococcus faecalis demonstrate high transfer efficiency within this species but limited inter-species transfer [4].
Experimental Protocols for Efficiency Assessment
Phage-Delivered CRISPR-Cas Workflow
Table 2: Key Research Reagents for Phage-Delivered CRISPR Systems
| Reagent/Component | Function | Example Applications |
|---|---|---|
| λ Phage Vector | Delivery chassis with well-characterized genetics | CRISPR-Cas12a programming for precise lysogeny control [7] |
| Cas Nucleases | CRISPR effectors that cleave target DNA | Cas9, Cas12a, Cas3 for different cleavage patterns [3] [7] |
| Guide RNA Plasmids | Encoding sequence-specific crRNAs | pHL027 for FnCas12a; pCK055 for LbCas12a [7] |
| Amber-Suppressor Hosts | Conditional control of phage infection | E. coli LE392MP for Sam7 mutant phage propagation [6] |
| Homology-Directed Repair Templates | For precise phage genome engineering | λ-DART system with CRISPR-associated transposases [6] |
Diagram 1: Experimental workflow for phage-delivered CRISPR-Cas systems
Step 1: Phage Engineering – The selected phage genome (commonly λ phage) is modified using homologous recombination coupled with Cas13a-based counterselection. For the λ-DART system, this involves replacing the b2 region with the LbCas12a gene and appropriate promoters [6] [7].
Step 2: CRISPR Payload Integration – The complete CRISPR system (Cas nuclease and guide RNAs targeting specific resistance genes) is incorporated into the phage genome. For temperate phages, genes essential for lysogeny may be deleted to ensure lytic behavior and prevent lysogenic stability [6].
Step 3: Phage Propagation – Engineered phages are amplified in permissive amber-suppressor hosts (e.g., E. coli LE392MP for Sam7 mutant phages) under controlled conditions (30-37°C with shaking) [6] [7].
Step 4: Infection and Delivery – Target bacteria are infected at specific multiplicities of infection (MOIs), typically ranging from 0.01 to 10, depending on the application. Higher MOIs generally produce faster and more pronounced effects [6].
Step 5: Efficiency Assessment – Editing efficiency is quantified by measuring the percentage of the bacterial population that undergoes successful genome modification or the reduction in viable counts for bactericidal approaches. In mixed cultures, selective plating or fluorescence-activated cell sorting (FACS) distinguishes targeted from non-targeted strains [7].
Conjugative Plasmid Delivery Protocol
Diagram 2: Conjugative plasmid delivery experimental workflow
Step 1: Plasmid Construction – Design and assemble the conjugative plasmid containing: (1) an origin of transfer (oriT) recognized by the conjugation machinery, (2) Cas nuclease genes (Cas9, Cas12a, etc.), (3) guide RNA sequences targeting specific antibiotic resistance genes, and (4) selectable markers for donor and transconjugant selection [4].
Step 2: Donor Strain Preparation – Introduce the constructed plasmid into an appropriate donor strain (e.g., E. coli WM3064 or other diaminopimelic acid [DAP] auxotrophs for controlled conjugation) via transformation or electroporation [4].
Step 3: Conjugation Assay – Mix donor and recipient strains at standardized ratios (typically 1:1 to 1:10 donor:recipient) in liquid media or on solid filters. Incubate for a predetermined mating period (usually 2-24 hours) to allow conjugation [4] [10].
Step 4: Transconjugant Selection – Plate the conjugation mixture on selective media containing antibiotics that counter-select against the donor strain while selecting for transconjugants that have received the plasmid. For DAP-dependent donors, use media lacking DAP [4].
Step 5: Efficiency Calculation – Quantify transfer efficiency by counting transconjugant colonies and calculating the transconjugant-to-recipient ratio. Account for density-dependent effects, as conjugation follows a Holling's Type II functional response with engagement time limitations at higher cell densities [10].
Applications and Pathogen Targeting
Therapeutic Applications Against Priority Pathogens
Both delivery platforms have demonstrated efficacy against critical MDR pathogens:
Phage-delivered CRISPR systems have successfully targeted:
- Klebsiella pneumoniae: Endogenous CRISPR-Cas3 system achieved ~100% elimination of resistance plasmids in vivo [2]
- Escherichia coli: Engineered phages with antibacterial CRISPR-Cas selectively reduced E. coli burden in mice [3]
- Clostridioides difficile: ϕCD24-2 phage delivering CRISPR-Cas3 effectively eradicated infections in a mouse model [3]
- Staphylococcus aureus: Genetic engineering of temperate phages created CRISPR-carrying antibacterial agents that cured infections in mice [3]
Conjugative plasmid delivery has shown promise against:
- Enterococci: Conjugative delivery of CRISPR-Cas9 for selective depletion of antibiotic-resistant enterococci [3]
- Carbapenem-resistant Enterobacteriaceae: pCasCure system removed blaNDM and blaKPC carbapenemase genes, resensitizing bacteria to carbapenems [4]
- Colistin-resistant E. coli: Conjugative CRISPR/Cas9 system targeting mcr-1 eliminated resistant plasmids and restored antibiotic sensitivity [4]
Advantages and Limitations in Clinical Translation
Phage-delivered CRISPR advantages include:
- Exceptional specificity with single-nucleotide discrimination capability [7]
- Self-replication at infection sites, potentially lowering required doses [1]
- Ability to penetrate biofilms and reach spatial niches inaccessible to antibiotics or bacteria [6]
- Natural effectiveness against metabolically dormant persister cells [1]
Phage-delivered CRISPR limitations include:
- Narrow host range requiring custom engineering for different bacterial strains [1]
- Potential for rapid evolution of bacterial resistance through receptor modification [1]
- Immune system recognition and neutralization in human applications [2]
- Technical challenges in large-scale production and purification [2]
Conjugative plasmid advantages include:
- Broader dissemination within compatible bacterial communities [4]
- Ability to transfer large genetic payloads without size constraints of phage capsids [8]
- Potential for "auto-dissemination" where transconjugants become donors [4]
- Utilization of natural bacterial mating processes without special equipment [4]
Conjugative plasmid limitations include:
- Density-dependent transfer efficiency with engagement time constraints [10]
- Requirement for direct cell-to-cell contact, limiting spatial range [10]
- Host range restrictions based on plasmid compatibility and receptor availability [4]
- Competition with endogenous plasmids and potential for unstable maintenance [4]
The escalating antibiotic resistance crisis demands a paradigm shift from broad-spectrum antimicrobials to precision approaches that selectively target resistance mechanisms while preserving beneficial microbiota. Both phage-delivered CRISPR and conjugative plasmid systems offer promising pathways toward this next generation of antimicrobials, with complementary strengths and applications.
Phage-based delivery excels in scenarios requiring maximal precision and single-strain targeting, particularly for established infections with known resistance profiles. Its ability to distinguish single-nucleotide variations and penetrate complex bacterial communities makes it ideal for precision microbiome engineering [7]. Conjugative plasmid systems offer advantages for community-wide interventions where broader dissemination among bacterial populations is desirable, such as decolonizing resistant strains from hospital environments or preventing resistance spread in agricultural settings [4].
Future clinical translation will likely leverage both platforms synergistically, with selection based on specific therapeutic contexts, target pathogens, and resistance mechanisms. As delivery efficiency continues to improve through engineering advances in phage host range expansion and plasmid conjugation systems, CRISPR-based precision antimicrobials hold exceptional promise for restoring the efficacy of existing antibiotics and changing the fundamental dynamics of the resistance crisis.
The escalating crisis of antimicrobial resistance (AMR) demands transformative solutions that move beyond conventional antibiotic discovery. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas system, derived from a prokaryotic adaptive immune system, has emerged as a highly precise and programmable antibacterial tool [2] [11]. Its primary mechanism of action involves the guided introduction of lethal DNA cleavage in target bacterial pathogens, leading to irreversible elimination of resistant or virulent strains [12]. The efficacy of this approach, however, is profoundly influenced by the method used to deliver the CRISPR-Cas machinery into bacterial cells.
This guide objectively compares the two predominant delivery strategies—engineered bacteriophage delivery and conjugative plasmid delivery—within the context of a broader thesis comparing their efficiency. We synthesize current research data, provide detailed experimental protocols, and present quantitative comparisons to inform researchers, scientists, and drug development professionals in selecting the optimal platform for their specific applications.
Delivery Platform Mechanisms and Workflows
The fundamental difference between phage and conjugative plasmid delivery lies in their mechanism of host entry and payload delivery. The workflows for both systems are designed to exploit these natural processes for precise genetic targeting.
Phage Delivery Mechanism
Engineered bacteriophages are natural viruses that infect bacteria. The process begins with the reprogramming of phage genomes to carry CRISPR-Cas systems, as demonstrated with phage λ [6]. The engineered phage specifically attaches to receptors on the target bacterium's surface, injecting its genetic material, which includes the CRISPR machinery. Inside the cell, the CRISPR system becomes active, leading to guided cleavage and destruction of essential bacterial genes or antimicrobial resistance (AMR) genes [2] [13].
Conjugative Plasmid Delivery Mechanism
Conjugative plasmids are mobile genetic elements that can transfer between bacteria via direct cell-to-cell contact. In this approach, the CRISPR-Cas system is cloned into a conjugative plasmid within a donor bacterium [12]. Through the natural process of conjugation, the plasmid is transferred to recipient target bacteria. Once inside, the CRISPR system is expressed and directs lethal cleavage toward specific genetic targets, such as plasmid-borne resistance genes, effectively resensitizing the bacteria to conventional antibiotics [2].
Quantitative Performance Comparison
The choice between delivery platforms involves critical trade-offs in editing efficiency, payload capacity, and host range. The data below provides a direct, objective comparison based on recent experimental findings.
Table 1: Direct Comparison of Phage vs. Conjugative Plasmid Delivery Systems
| Performance Metric | Phage-Delivered CRISPR | Conjugative Plasmid-Delivered CRISPR |
|---|---|---|
| Reported Editing Efficiency | >50% in targeted E. coli within mixed communities [6] | Up to 100% of counter-selected clones in K. pneumoniae [14] |
| Payload Capacity | ~10 kb for the entire DART system in phage λ [6] | Primarily limited by plasmid size and stability; can accommodate standard CRISPR systems |
| Primary Applications | • Flexible in-situ genome editing• Manipulation of mixed communities• Targeted gene knockouts and large insertions [6] | • High-efficiency, markerless genome editing• Re-sensitization to antibiotics• Multiplexed targeting [2] [14] |
| Key Strengths | • High target specificity• Can access spatial niches inaccessible to bacteria• Does not require donor-recipient compatibility [6] | • High editing efficiency in clones• Broad host range based on plasmid transferability• Suitable for complex genetic operations (e.g., point mutations) [14] |
| Notable Limitations | • Host range constrained by phage tropism• Potential for bacterial resistance to phage infection [13] | • Requires donor-recipient compatibility for conjugation• Risk of unintended plasmid spread [6] |
Detailed Experimental Protocols
To ensure reproducibility and provide a clear basis for comparison, we outline the core methodologies for implementing each delivery system, as described in the cited literature.
Protocol for Phage-Delivered CRISPR (λ-DART System)
The following protocol is adapted from studies engineering phage λ to deliver CRISPR-associated transposases (DART) for genome editing in Escherichia coli [6].
- Phage Engineering: Engineer a modified phage λ (e.g., λ cI857 Sam7) to carry the DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) system. This is achieved using homologous recombination paired with Cas13a-based counterselection to replace over 10 kb of the phage genome with the DART payload while eliminating lysogeny components.
- Phage Propagation and Purification: Amplify the engineered λ-DART phages in a permissive amber-suppressor host strain (e.g., E. coli LE392MP) under controlled conditions (e.g., 37°C to induce the lytic cycle in cI857 mutants). Purify phage particles using standard methods like polyethylene glycol (PEG) precipitation and cesium chloride gradient centrifugation.
- Infection and Editing: Infect the target bacterial population (monoculture or mixed community) with the purified λ-DART phages at a defined Multiplicity of Infection (MOI). Critical parameters to optimize include MOI, promoter strength driving the DART system, and incubation time.
- Assessment of Editing Efficiency: After a suitable incubation period, assess editing efficiency by measuring the frequency of successful gene knockouts or insertions, for example, via plating assays, fluorescence-activated cell sorting (FACS) if a reporter is inserted, or PCR-based genotyping. Editing efficiencies of >50% in the targeted population within a mixed community have been reported [6].
Protocol for Conjugative Plasmid Delivery (RECKLEEN System)
The following protocol is based on the RECKLEEN system, a single-plasmid platform for enhanced genome editing in Klebsiella pneumoniae [14].
- Donor Strain Preparation: Clone the CRISPR-Cas9 system (e.g., using the near PAM-less SpG Cas9 variant) and lambda Red recombineering genes (gam, exo, beta) into a single, conjugative plasmid backbone. The expression of these components should be under tightly regulated, inducible promoters (e.g., Ptac for lambda Red, Ptet for Cas9/sgRNA).
- Conjugation: Mix the donor E. coli strain harboring the conjugative plasmid with the recipient target bacterium (e.g., a multidrug-resistant K. pneumoniae strain) on a solid filter membrane. Incubate to allow for conjugative transfer of the plasmid.
- Recombineering and Counterselection: Isolate transconjugants and induce the lambda Red system with IPTG. Transform these cells with a linear editing DNA (e.g., single-stranded or double-stranded DNA with ~50 nt homology arms) to introduce the desired mutation via homologous recombination. Subsequently, induce the Cas9/sgRNA system with anhydrotetracycline (ATc). The sgRNA is designed to target the wild-type sequence, resulting in lethal double-strand breaks in unedited cells, thereby counterselecting for successfully edited clones.
- Validation: Screen surviving clones for the desired genetic modification. The RECKLEEN system reports efficiencies reaching 100% for single edits (deletions, point mutations, integrations) and up to 72% for simultaneous multi-target deletions in the counter-selected population [14].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these technologies relies on a core set of reagents. The table below details key solutions and their functions.
Table 2: Key Research Reagent Solutions for CRISPR Antibacterial Platforms
| Research Reagent / Tool | Function in Experiment | Example Use Case |
|---|---|---|
| Engineered Phage λ (e.g., λ cI857 Sam7) | Delivery chassis for CRISPR payload; Sam7 mutation provides control over lysis in non-suppressor hosts [6]. | Controlled infection and editing in specific E. coli strains without widespread lysis in non-target bacteria. |
| CRISPR-Associated Transposase (DART) | All-in-one system for RNA-guided DNA insertion without double-strand breaks, enabling gene knockouts and integrations [6]. | Inserting large genetic payloads (>1 kb) into the chromosome of targeted bacteria within a mixed community. |
| Single Plasmid System (e.g., RECKLEEN) | Combines lambda Red recombineering and CRISPR-Cas9 counterselection on a single, modular vector for streamlined workflows [14]. | Efficient, markerless, and scarless genome editing in MDR pathogens like K. pneumoniae, avoiding multi-plasmid coordination. |
| Near PAM-less SpG Cas9 | Cas9 variant with broadened PAM recognition (5'-NGN-3'), significantly expanding the range of targetable sequences [14]. | Targeting genomic sites that are inaccessible to wild-type SpCas9 (5'-NGG-3' PAM), crucial for precise point mutations. |
| Anti-CRISPR Proteins | Inhibits Cas9 activity, mitigating its toxicity in bacteria prior to counterselection and improving plasmid stability [14]. | Used in the RECKLEEN system to prevent premature Cas9 activity, allowing for efficient plasmid maintenance and recombineering. |
Both phage-delivered and conjugative plasmid-delivered CRISPR systems represent powerful, programmable tools for antibacterial applications with distinct operational profiles. The selection of an optimal platform is context-dependent.
Phage delivery excels in scenarios requiring high target specificity within complex communities and where the objective is flexible in-situ genome editing without the need for donor-recipient compatibility. Its current limitations in host range can be mitigated through ongoing phage engineering efforts [6] [13].
Conjugative plasmid delivery demonstrates superior absolute editing efficiency in isolated strains and offers greater flexibility for complex genetic manipulations, including point mutations and multiplexed editing, as evidenced by platforms like RECKLEEN [14]. It is particularly potent for recalibrating antibiotic susceptibility by eliminating resistance plasmids [2].
Future advancements will likely focus on hybrid approaches, expanding the host range of phage vectors, and refining the control of conjugative transfer. The continued development and direct comparison of these platforms are crucial for translating programmable antibacterial tools from laboratory research into viable therapeutic and biotechnological applications.
The precise genetic manipulation of microbial communities, particularly within complex native environments, represents a significant hurdle in microbiology and therapeutic development. Two primary strategies have emerged for delivering genetic cargo like CRISPR systems to target bacteria: conjugative plasmid delivery and bacteriophage-mediated delivery. Conjugation involves the transfer of genetic material between bacterial cells via direct contact, a process leveraged by tools like the DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) system for community editing [6]. In contrast, bacteriophage (phage) delivery harnesses the natural infectious mechanisms of viruses that specifically target bacteria. Phages offer several inherent advantages as delivery vehicles, including high host specificity that enables targeted editing of specific bacterial strains within mixed communities, natural proficiency in infecting hard-to-transfect bacteria, and potentially higher delivery efficiency to spatial niches not efficiently accessible by donor bacteria [6]. This guide provides a systematic comparison of these competing delivery modalities, focusing on their application in delivering CRISPR-based editing tools, with experimental data and protocols to inform research development.
Mechanism of Action: Fundamental Delivery Pathways
Conjugative Plasmid Delivery
Conjugative plasmid delivery utilizes the natural bacterial mating mechanism for horizontal gene transfer. The process involves donor bacteria carrying a specialized plasmid vector that encodes both the CRISPR editing machinery and the conjugation apparatus. This plasmid is transferred to recipient bacteria through a specialized pore structure after cell-to-cell contact is established. Tools like the DART system employ non-replicative plasmids to limit persistence within the population after editing occurs [6]. The reliance on donor-recipient compatibility can restrict the range of targetable bacteria, and the transfer efficiency is influenced by multiple factors including plasmid size, the metabolic state of recipient cells, and the presence of physical barriers in complex communities.
Bacteriophage-Mediated Delivery
Bacteriophages deliver genetic cargo by hijacking the bacterial host's cellular machinery. The process begins with phage attachment to specific bacterial surface receptors, which determines host specificity [15]. Following attachment, the phage injects its genetic material (containing the editing system) into the bacterial cytoplasm. Engineered phage λ systems, such as λ-DART, replace native pathogenic genes with CRISPR-transposase constructs while retaining infection capabilities but eliminating components essential for lysogeny to prevent persistent phage maintenance [6]. This receptor-mediated targeting allows for extremely specific delivery to particular bacterial strains, even within complex multispecies environments.
The diagram below illustrates the core operational logic distinguishing these two delivery mechanisms:
Performance Comparison: Quantitative Delivery Efficiency Analysis
Direct comparative studies between phage and conjugation delivery systems reveal significant differences in editing efficiency, specificity, and practical implementation. The data below summarizes key performance metrics from recent experimental studies:
Table 1: Comparative Performance of Phage vs. Conjugative Plasmid Delivery
| Performance Metric | Phage λ-DART Delivery | Conjugative Plasmid Delivery | Experimental Context |
|---|---|---|---|
| Editing Efficiency | >50% of targeted population [6] | Not explicitly quantified (varies by system) | E. coli monoculture editing |
| Editing Specificity | Precise editing in targeted E. coli within mixed community [6] | Requires donor-recipient compatibility [6] | Three-genera mixed community |
| Cargo Capacity | Can accommodate large CRISPR-transposase systems (>10 kb) [6] | Limited by plasmid size and conjugation efficiency | Full DART system delivery |
| Host Range | Narrow (strain-specific targeting) [6] [15] | Broader (determined by plasmid host range) | Defined by receptor vs. compatibility |
| Delivery Speed | Rapid (single infection cycle: ~20-30 min latent period) [15] | Slower (requires conjugation apparatus assembly) | Based on phage replication dynamics |
| Practical Implementation | Requires phage engineering | Requires conjugation donor strain | Laboratory workflow complexity |
Experimental Protocols: Key Methodologies for Delivery Assessment
Phage Engineering and Delivery Validation Protocol
The engineering of bacteriophages for delivery applications involves precise genetic modifications to incorporate cargo while maintaining infectivity. The following protocol outlines the key steps for creating and validating phage delivery systems:
Phage Genome Modification:
- Utilize homologous recombination paired with Cas13a-based counterselection for precise phage modifications [6].
- For λ phage, incorporate CRISPR-guided transposases (DART system) via recombination, replacing non-essential pathogenic genes.
Phage Propagation and Purification:
- Culture host bacteria (e.g., E. coli BW25113 or amber-suppressor strain LE392MP) to mid-log phase [6].
- Infect with engineered phage at appropriate multiplicity of infection (MOI: 0.1-10).
- Recover phage particles through polyethylene glycol (PEG) precipitation or CsCl gradient ultracentrifugation [16].
- Remove endotoxins using Triton X-100 or column purification [16].
Infection and Delivery Assay:
- Grow target bacteria to OD600 ≈ 0.3-0.4.
- Add engineered phage at varying MOI (0.1, 1, 10) and incubate at 37°C.
- For controlled infection, use phage with Sam7 mutation in amber-suppressor hosts to constrain lysis [6].
- Measure editing efficiency via antibiotic selection or fluorescence activation after 4-24 hours.
Conjugative Plasmid Delivery Protocol
Conjugative plasmid delivery requires optimization of bacterial mating conditions for efficient transfer:
Donor and Recipient Preparation:
- Grow donor strain (carrying conjugative plasmid) and recipient strain separately to mid-log phase.
- Use antibiotic selection to maintain plasmid in donor strain.
Conjugation Assay:
- Mix donor and recipient cells at ratios from 1:1 to 1:10.
- Concentrate cells on filters or plate on solid surface for close contact.
- Incubate for 1-24 hours to allow mating.
- Resuspend and plate on selective media to enumerate transconjugants.
Efficiency Calculation:
- Calculate conjugation efficiency as transconjugants per recipient cell.
- Compare to phage delivery efficiency using isogenic systems when possible.
The experimental workflow for comparing these systems can be visualized as follows:
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of bacteriophage delivery systems requires specific reagents and genetic tools. The following table details essential components for developing and testing phage-based delivery platforms:
Table 2: Essential Research Reagents for Phage Delivery Systems
| Reagent / Tool | Function | Example Applications |
|---|---|---|
| Phage λ with cI857 & Sam7 mutations | Temperature-sensitive repressor and lysis control for controlled infection [6] | Controlled infection in amber-suppressor hosts |
| Cas13a Counterselection System | Precise selection of recombinant phages during engineering [6] | Phage genome modification |
| DART (DNA-editing all-in-one RNA-guided CRISPR-Cas transposase) | CRISPR-guided transposase system for large DNA insertions [6] | Targeted gene knockouts and insertions |
| Amber-Suppressor E. coli Strains | Permissive hosts for phages with amber mutations [6] | Propagation of engineered λ phages |
| PEG Precipitation Reagents | Concentration and partial purification of phage particles [16] | Phage preparation and storage |
| Triton X-100 | Efficient endotoxin removal from phage preparations [16] | Preparation of phages for sensitive applications |
| Homologous Recombination System | Genetic engineering of phage genomes [6] | Cargo insertion into phage delivery vectors |
The comparative analysis reveals that both phage-mediated and conjugative plasmid delivery offer distinct advantages for different research scenarios. Phage λ-DART systems excel in applications requiring high specificity within complex microbial communities, demonstrated by their ability to achieve >50% editing efficiency in targeted E. coli within mixed cultures without affecting non-target species [6]. The implementation requires significant engineering but offers unparalleled targeting precision. Conjugative plasmid delivery provides broader host range capabilities and may be more suitable for applications where moderate efficiency across multiple bacterial types is preferred over strain-specific precision.
Selection between these platforms should be guided by specific research requirements: phage systems for precision editing in complex environments, and conjugation for broader-range delivery where donor-recipient compatibility is established. Future directions will likely focus on expanding the host range of engineered phages through receptor engineering and optimizing conjugation systems for enhanced efficiency in challenging environments, further advancing our ability to manipulate microbial communities for therapeutic and industrial applications.
Horizontal gene transfer (HGT) is a fundamental driver of bacterial evolution, allowing microbes to rapidly acquire adaptive traits, including antibiotic resistance. Among the various HGT mechanisms, bacterial conjugation—the contact-dependent transfer of DNA via a type IV secretion system (T4SS)—serves as a powerful vehicle for disseminating genetic material across microbial populations [17] [18]. In the context of microbiome engineering, conjugative plasmids have emerged as promising delivery vectors for introducing desired genetic modifications into complex bacterial communities.
This guide provides a comparative analysis of conjugative plasmid delivery, with particular emphasis on how its efficiency measures against alternative methods such as phage-mediated delivery. By synthesizing current research and experimental data, we aim to offer researchers, scientists, and drug development professionals a clear framework for selecting and optimizing gene delivery systems for specific applications.
Conjugative Plasmid Delivery: Mechanisms and Key Players
Conjugative plasmid transfer involves a sophisticated multi-step process that begins with the formation of a mating pair between donor and recipient cells. The plasmid-encoded T4SS assembles a conjugative pilus that initiates contact with a recipient bacterium. Following retraction of the pilus, a stable mating junction is formed through a process termed mating pair stabilization (MPS), which is critical for efficient DNA transfer [18].
The specificity of conjugation is largely determined by plasmid-encoded donor outer membrane protein TraN and its interaction with distinct outer membrane receptors in recipient cells. Based on structural similarities and receptor specificity, TraN variants are classified into four categories: TraNα (binds OmpW), TraNβ (binds OmpK36), TraNγ (binds OmpA), and TraNδ (binds OmpF) [18]. These specific pairings effectively mediate the species specificity of conjugation and influence the distribution of resistance plasmids within clinical Enterobacteriaceae isolates.
Table 1: Key Receptor Pairings in Conjugative Plasmid Transfer
| TraN Variant | Receptor Protein | Example Plasmids | Bacterial Host Specificity |
|---|---|---|---|
| TraNα | OmpW | R100-1 (Shigella flexneri), pSLT (Salmonella Typhimurium) | Shigella, Salmonella |
| TraNβ | OmpK36 | pKpQIL (Klebsiella pneumoniae) | Klebsiella pneumoniae |
| TraNγ | OmpA | F plasmid (Escherichia coli) | Escherichia coli |
| TraNδ | OmpF | Unspecified variants | Enterobacteriaceae |
Beyond the conjugation machinery, successful plasmid establishment in recipient cells depends on several additional factors:
- Replication compatibility: The plasmid must replicate using the host's cellular machinery [17]
- Host defense evasion: Plasmids employ strategies to circumvent restriction-modification systems and CRISPR-Cas immunity [17]
- Stability maintenance: Partitioning systems and toxin-antitoxin modules ensure plasmid persistence during cell division [17]
Diagram 1: Mechanism of Conjugative Plasmid Transfer. The process initiates with pilus formation and culminates in stable plasmid maintenance, with mating pair stabilization serving as the critical specificity determinant.
Comparative Delivery Efficiency: Conjugative Plasmids vs. Phage-Based Systems
Quantitative Efficiency Metrics
The efficiency of gene delivery systems varies considerably based on the method, target environment, and specific vectors employed. The table below summarizes key efficiency metrics from recent studies.
Table 2: Efficiency Comparison of Bacterial Gene Delivery Systems
| Delivery System | Specific Example | Efficiency Range | Experimental Context | Key Factors Affecting Efficiency |
|---|---|---|---|---|
| Conjugative Plasmids | TP114 (Incl2) | ~100% of recipient population | Mouse gut microbiota [19] | Type IV pilus for mating pair stabilization |
| Conjugative Plasmids | pKpQIL (IncF) | Varies with OmpK36 variants | In vitro conjugation [18] | Recipient porin structure (GD insertion reduces efficiency) |
| Conjugative Plasmids | RTCS with pKpGFP-D | 2-log increase over wild type | Real-time conjugation system [18] | Derepression of transfer genes (finO deletion) |
| Phage-Delivered CRISPR | λ-DART phages | >50% of population | E. coli monoculture [6] | MOI, promoter strength, incubation time |
| Phage-Delivered CRISPR | CAPs (SNIPR001) | 1-6 log10 reduction | E. coli panel [20] | Tail fiber engineering, CRISPR-Cas arming |
| Phage-Delivered CRISPR | Cas12a-programmed λ | Selective killing with SNV resolution | Mixed bacterial cultures [7] | Truncated crRNA for enhanced specificity |
Key Determinants of Delivery Efficiency
For conjugative plasmids, transfer efficiency is governed by both density-dependent and frequency-dependent factors. While traditional models assumed conjugation depended solely on donor-recipient encounter rates, recent research reveals a more complex dynamic. At higher cell concentrations, conjugation becomes limited by the engagement time—the interval required between two successful matings—following a Holling's Type II functional response [10].
For phage-based delivery, efficiency depends on factors including phage-host specificity, receptor availability, multiplicity of infection (MOI), and the stability of the engineered genetic payload [6] [20]. Phage λ with CRISPR-associated transposases (λ-DART) has demonstrated editing efficiencies surpassing 50% in mixed bacterial communities, highlighting its potential for complex environments [6].
Experimental Protocols for Assessing Delivery Efficiency
Standardized In Vitro Conjugation Assay
Purpose: To quantify plasmid transfer rates under controlled laboratory conditions [19].
Method:
- Strain preparation: Grow donor and recipient strains to mid-exponential phase (OD₆₀₀ ≈ 0.4-0.6)
- Mixing: Combine equal volumes of donor and recipient cultures (typically 1:1 ratio)
- Incubation:
- Liquid mating: Incubate mixture for 2 hours at 37°C with gentle agitation
- Solid support mating: Filter mixture through membrane filter, place on pre-warmed non-selective agar plate, incubate 2 hours at 37°C
- Selection: Resuspend cells and plate on selective media containing antibiotics that inhibit donor and recipient while allowing transconjugants to grow
- Calculation: Determine transfer frequency as (number of transconjugants)/(number of recipient cells)
Key considerations: Control for spontaneous mutation rates by plating each strain individually on selective media.
In Situ Conjugation Mouse Model
Purpose: To quantify plasmid transfer rates in the complex gut environment [19].
Method:
- Mouse pretreatment: Administer streptomycin (1 g/L) in drinking water for 3 days to deplete endogenous enterobacteria
- Recipient administration: Orally gavage mice with recipient strain (e.g., EcN KN02)
- Donor administration: After 2-12 hours, orally gavage with donor strain carrying conjugative plasmid
- Monitoring: Collect fecal samples daily for 3 days post-infection
- Quantification: Homogenize samples, plate serial dilutions on selective media to determine donor, recipient, and transconjugant counts
Key considerations: Streptomycin treatment facilitates reproducible colonization by engineered strains while maintaining relevant gut conditions for conjugation assessment.
Real-Time Conjugation System (RTCS)
Purpose: To monitor conjugation dynamics without selective pressure [18].
Method:
- Reporter construction: Engineer conjugative plasmid with fluorescent reporter gene (e.g., sfGFP) under control of inducible promoter
- Donor engineering: Introduce repression system (e.g., LacI) in donor to suppress reporter expression
- Conjugation monitoring: Mix donor and recipient cells, measure fluorescence emission over time
- Data analysis: Correlate fluorescence increase with conjugation events, normalized to cell density
Key considerations: RTCS enables detection of conjugation as early as 150 minutes after mixing, providing temporal resolution of transfer dynamics.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Conjugative Plasmid Research
| Reagent/Cell Line | Key Features | Research Application | Example Source/Reference |
|---|---|---|---|
| E. coli Nissle 1917 (EcN) derivatives | Engineered with distinct antibiotic resistance markers | Standardized host for conjugation assays [19] | KN01 (streptomycin, spectinomycin), KN02 (streptomycin, chloramphenicol), KN03 (streptomycin, tetracycline) |
| TP114 (Incl2 plasmid) | Exceptional transfer efficiency in gut microbiota | Broad-host-range delivery vector [19] | Natural isolate from enteric bacteria |
| pKpQIL (K. pneumoniae) | TraNβ variant binding OmpK36 | Studying species-specific conjugation [18] | Clinical K. pneumoniae isolate |
| pKpGFP/pKpGFP-D | GFP reporter, finO deletion for derepression | Real-time conjugation monitoring [18] | Engineered from pKpQIL |
| F plasmid (E. coli) | TraNγ variant binding OmpA | Prototypical conjugation studies [18] | Laboratory E. coli strains |
| R100-1 (S. flexneri) | TraNα variant binding OmpW | Comparative conjugation specificity [18] | Shigella flexneri isolate |
| C57 BL/6 mouse model | Streptomycin treatment enables EcN colonization | In situ conjugation assessment [19] | Commercial suppliers |
Conjugative plasmid delivery represents a highly efficient mechanism for horizontal gene transfer, with systems like the Incl2 plasmid TP114 achieving nearly complete recipient population coverage in the gut environment [19]. The specificity of these systems is governed by precise molecular interactions between plasmid-encoded TraN proteins and recipient outer membrane receptors [18].
When compared to phage-based delivery systems, conjugative plasmids generally offer broader host range within taxonomic groups and can achieve remarkable efficiency in complex environments like the gut microbiota [19]. In contrast, phage-based systems provide exceptional targeting specificity and can be engineered with CRISPR payloads for precision genome editing [6] [20] [7].
The selection between these delivery platforms ultimately depends on the research or therapeutic objectives: conjugative plasmids excel at population-wide dissemination, while phage systems offer precision targeting of specific genetic variants. As both technologies continue to advance, their complementary strengths will likely expand the toolbox available for microbiome engineering and antibacterial therapies.
The development of advanced biotechnological tools for precisely targeting and eliminating bacterial populations or modifying cellular functions represents a frontier in biomedical science. Two distinct modalities have emerged as particularly powerful platforms: lytic phage action and plasmid establishment. While both can be harnessed to deliver genetic cargo such as CRISPR components for bacterial killing or gene editing, their fundamental mechanisms, efficiency profiles, and practical applications differ substantially. Lytic phages, natural bacterial viruses, exploit the host's cellular machinery to replicate and ultimately lyse bacterial cells through direct physical destruction. In contrast, plasmid-based systems introduce extracellular DNA that establishes itself as an episomal entity within cells, enabling persistent expression of therapeutic genes or antibacterial agents. Understanding the core distinctions between these modalities is essential for researchers and drug development professionals seeking to select the optimal platform for specific therapeutic applications, particularly within the growing field of phage-delivered CRISPR versus conjugate plasmid delivery efficiency research.
Fundamental Mechanisms of Action
Lytic Phage Action Mechanism
Lytic bacteriophages, or virulent phages, operate through a strictly lytic cycle that results in the destruction of the bacterial host cell. The process begins when phage particles recognize and adsorb to specific receptors on the bacterial surface, which dictates their host range. Following attachment, the phage injects its genetic material (DNA or RNA) into the bacterial cytoplasm, effectively hijacking the host's transcriptional and translational machinery. The phage genome redirects cellular resources to express early proteins that shut down host metabolism, followed by the synthesis of viral components including structural proteins and genomic copies. These components self-assemble into progeny virions, a process facilitated by phage-encoded enzymes. The cycle culminates in cell lysis and phage release, mediated by holins and lysins [21]. Holins are small proteins that accumulate in the cytoplasmic membrane and form pores at a genetically determined time, allowing endolysins to access and degrade the peptidoglycan layer of the cell wall. This enzymatic breakdown compromises the structural integrity of the cell wall, leading to osmotic lysis and the release of new infectious phage particles that can infect neighboring bacteria [21]. The entire lytic cycle is remarkably efficient, typically completing within 20-60 minutes and yielding dozens to hundreds of new virions from a single infected cell.
Plasmid Establishment Mechanism
Plasmid establishment represents a fundamentally different approach to genetic modification and bacterial killing. Plasmids are extrachromosomal DNA elements that replicate independently within a host cell following introduction via conjugation, transformation, or transfection. The establishment process begins with the delivery of plasmid DNA into the bacterial cytoplasm, after which it must escape degradation by host restriction enzymes and navigate to the replication machinery. Successful establishment requires replication initiation proteins that recognize the plasmid's origin of replication (ori), directing the host's DNA synthesis apparatus to propagate the plasmid DNA. These systems maintain stable inheritance through partitioning mechanisms that ensure equitable distribution to daughter cells during cell division [22]. Unlike lytic phages, plasmids can persist indefinitely within bacterial populations without immediately killing the host cell. Their killing potential is realized through the expression of encoded toxic proteins, antimicrobial agents, or CRISPR-Cas systems programmed to target essential bacterial genes. This enables controlled, specific bactericidal activity without the immediate physical disruption characteristic of phage-induced lysis. The plasmid-based killing is conditional and programmable, depending on the induction of expression systems or the constitutive production of lethal genetic elements.
Quantitative Comparison of Performance Parameters
Table 1: Comparative Performance Metrics of Lytic Phage Action vs. Plasmid Establishment
| Performance Parameter | Lytic Phage Action | Plasmid Establishment |
|---|---|---|
| Killing Kinetics | Rapid (20-60 minutes post-infection) | Variable (hours to days, dependent on expression system) |
| Bacterial Killing Efficiency | High (up to 5.9 log10 reduction in studies with phage-antibiotic synergy) [23] | Moderate to high (dependent on delivery efficiency and copy number) |
| Host Specificity | Narrow (strain or species-specific) [21] | Broad (can be designed with various promoters for different hosts) |
| Cargo Capacity | Limited (~40 kb for some phages) | Moderate (varies with plasmid system, typically 5-20 kb) |
| Transfer Efficiency | Natural high-efficiency infection process | Variable (requires delivery optimization: electroporation, conjugation) |
| Persistence in Population | Self-replicating and self-limiting (dependent on host availability) | Can be stable or require selection pressure |
| Resistance Development | Common through receptor modification | Common through plasmid loss or target mutation |
Table 2: CRISPR Delivery Efficiency Comparison: Phage vs. Plasmid Vectors
| Delivery Attribute | Phage-Delivered CRISPR | Plasmid-Delivered CRISPR |
|---|---|---|
| Onset of Activity | Immediate upon infection | Delayed (requires transcription and translation) [24] |
| Editing Duration | Transient to sustained (dependent on phage type) | Prolonged (due to plasmid stability) [24] |
| Off-Target Effects | Lower (transient expression) | Higher (persistent Cas9 expression) [24] |
| Insertional Mutagenesis Risk | Low (for non-integrating phages) | Moderate (risk of random integration) [24] |
| Delivery Efficiency to Difficult Cells | High (natural infection mechanisms) | Variable (requires optimization) |
| Clinical Safety Profile | Favorable (no genomic integration in lytic phages) | Concerns (potential immunogenicity, integration) [24] |
Experimental Protocols for Efficacy Assessment
Protocol for Evaluating Lytic Phage Killing Efficacy
Objective: To quantify the bactericidal activity of lytic phages against target bacterial strains.
Materials:
- Bacterial host strains (e.g., Pseudomonas aeruginosa, Escherichia coli)
- Purified lytic phage stocks with known titer (PFU/mL)
- LB broth and LB agar plates
- Double-layer agar plates for phage quantification [21]
- Automated growth analyzer (e.g., Bioscreen C Pro) [21]
- Shaking incubator
- Sterile filtration units (0.45 µm)
Methodology:
- Bacterial Culture Preparation: Grow overnight cultures of target bacteria in LB broth at 37°C with shaking (200 rpm).
- Phage Infection: Dilute bacterial cultures to approximately 10^5 CFU/mL in fresh medium. Add phage at varying multiplicities of infection (MOI: 0.1, 1, 10) to experimental tubes while maintaining an uninfected control.
- Kinetic Growth Analysis: Transfer 180 µL of phage-bacteria mixture to honeycomb plates for automated growth analysis. Monitor optical density (OD600) every 15 minutes for 24 hours at 37°C [21].
- Viability Assessment: At predetermined intervals (0, 2, 4, 6, 8, 24 hours), remove aliquots, perform serial dilutions in sterile saline, and plate on LB agar using the spread plate technique. Enumerate colony-forming units (CFU/mL) after 24 hours incubation at 37°C.
- Phage Quantification: Use the double-layer agar method to determine phage titer throughout the experiment [21]. Mix 100 µL of bacterial culture with 100 µL of phage dilution, add to 3-4 mL of soft agar (0.7% agar), and pour over pre-poured LB agar plates. Count plaques after overnight incubation.
- Data Analysis: Calculate bacterial reduction (log10 CFU/mL) compared to untreated controls. Determine phage amplification (increase in PFU/mL) over time.
This protocol allows researchers to simultaneously monitor bacterial lysis and phage replication kinetics, providing comprehensive data on the efficacy of lytic phage action.
Protocol for Assessing Plasmid Establishment and CRISPR-Mediated Killing
Objective: To evaluate the efficiency of plasmid delivery and subsequent target bacterial killing via CRISPR-Cas systems.
Materials:
- Bacterial target strains with appropriate antibiotic sensitivity profile
- CRISPR plasmid constructs (with Cas9 and target-specific gRNA)
- Conjugation donor strain (if using conjugative plasmids) or electroporation equipment
- Selective media with appropriate antibiotics
- DNA extraction and purification kits
- PCR reagents for verification of plasmid establishment
- Agarose gel electrophoresis equipment
Methodology:
- Plasmid Delivery:
- Conjugation Method: Mix donor (carrying plasmid) and recipient strains at 1:2 ratio on sterile filters placed on non-selective agar. Incubate 6-18 hours. Resuspend cells and plate on selective media containing antibiotics that inhibit donor growth while selecting for transconjugants [22].
- Electroporation Method: Make electrocompetent cells by washing log-phase cultures in cold 10% glycerol. Mix with plasmid DNA and electroporate at optimized conditions. Recover cells in SOC medium for 1-2 hours before plating on selective media.
Quantification of Establishment Efficiency:
- Count transconjugant or transformant colonies after 24-48 hours incubation.
- Calculate establishment efficiency as CFU of transconjugants per recipient or per input plasmid DNA.
CRISPR-Mediated Killing Assessment:
- Spot single colonies of established strains on inductive and non-inductive media.
- Monitor growth reduction on inductive media compared to controls.
- Perform viability counts before and after induction of CRISPR-Cas system.
Molecular Verification:
- Extract plasmid DNA from transconjugants/transformants and verify by restriction digestion or PCR.
- Sequence target loci to confirm CRISPR-induced mutations.
This protocol enables precise quantification of plasmid establishment efficiency and subsequent CRISPR-mediated bacterial killing, facilitating direct comparison between different plasmid systems.
Visualization of Mechanisms and Workflows
Diagram 1: Comparative Mechanisms of Lytic Phage Action versus Plasmid Establishment. The lytic phage pathway (upper) shows the sequential steps from initial attachment to cell lysis, highlighting the critical role of holins and lysins in the final lytic stage. The plasmid establishment pathway (lower) illustrates the process from delivery to functional expression, with optional CRISPR-Cas mediated killing as a programmable bactericidal approach.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Phage and Plasmid Studies
| Reagent / Tool | Function | Application Context |
|---|---|---|
| Double-Layer Agar | Plaque formation and phage quantification | Lytic phage titer determination and isolation [21] |
| LB Broth & Agar | Standard bacterial culture medium | Routine cultivation of bacterial hosts |
| Phage Storage Buffer (SM Buffer) | Maintain phage viability during storage | Long-term preservation of phage stocks |
| Plasmid Purification Kits | Isolation of high-quality plasmid DNA | Preparation of plasmid constructs for delivery experiments [25] |
| Electroporator | Physical method for plasmid introduction | Efficient plasmid transformation into bacterial cells |
| Antibiotic Selection Markers | Selective pressure for plasmid maintenance | Isolation and maintenance of plasmid-containing strains |
| CRISPR Plasmid Constructs | Delivery of Cas9 and guide RNA | Programmable targeting of bacterial genes |
| Automated Growth Analyzer (e.g., Bioscreen C) | High-throughput kinetic growth monitoring | Real-time assessment of bacterial killing kinetics [21] |
| PCR Reagents | Verification of genetic constructs and edits | Confirmation of plasmid establishment and target modifications |
The strategic selection between lytic phage action and plasmid establishment as killing modalities depends fundamentally on the specific research or therapeutic objectives. Lytic phages offer unparalleled efficiency for direct bacterial killing with rapid kinetics and self-amplifying capabilities, making them ideal for acute infection scenarios and applications where immediate bactericidal activity is prioritized. The recent demonstration of phage-antibiotic synergy achieving up to 5.9 log10 reduction in lung bacterial load highlights their therapeutic potential [23]. Conversely, plasmid establishment systems provide programmable, controlled approaches to bacterial killing, particularly when engineered with CRISPR-Cas systems. While suffering from slower onset and requiring efficient delivery optimization, plasmid-based approaches enable precise genetic targeting and can be designed for conditional activation. The emerging understanding of phage-plasmids as natural hybrids that promote gene flow between these elements [22] suggests future engineered systems may harness advantages of both modalities. For researchers and drug development professionals, the decision framework should integrate considerations of kinetic requirements, specificity, delivery efficiency, and regulatory pathway, with lytic phages favoring rapid direct killing and plasmid systems enabling sophisticated programmable approaches.
Engineering and Deployment: Building and Applying Delivery Systems
The escalating crisis of antimicrobial resistance (AMR) necessitates the development of novel therapeutic strategies that can precisely target resistant pathogens. Among the most promising alternatives are CRISPR-Cas systems, which can be programmed to selectively disable antibiotic resistance genes or directly target bacterial viability. A critical challenge, however, lies in efficiently delivering these CRISPR payloads to the target bacterial population. This guide objectively compares the two primary delivery modalities under investigation: engineered phage delivery and conjugate plasmid delivery. We will evaluate their performance based on key metrics such as editing efficiency, host range specificity, payload capacity, and practical implementation, providing a clear comparison of their respective advantages and current limitations for researchers and drug development professionals.
Performance Comparison: Phage-Delivered CRISPR vs. Conjugate Plasmid Delivery
The choice of delivery vehicle significantly impacts the efficacy, specificity, and practical application of CRISPR-based antimicrobials. The following table provides a direct, data-driven comparison of the two main strategies.
Table 1: Performance Comparison of Phage-Delivered CRISPR and Conjugate Plasmid Systems
| Performance Metric | Engineered Phage Delivery | Conjugate Plasmid Delivery |
|---|---|---|
| Reported Editing Efficiency | >50% gene knockout/insertion in targeted E. coli [6]; 3.5 log10 CFU/mL reduction [20] | 4.7% to 100% resensitization efficiency, varies by target [8] |
| Host Range & Specificity | High, inherent phage species/strain-level specificity; can be broadened via tail fiber engineering [20] | Moderate, limited by conjugation compatibility (donor-recipient) [6] |
| Payload Capacity | Large; can deliver entire transposase systems (DART, >10 kb) [6] or Cas nucleases with arrays [20] | Limited by plasmid size; larger plasmids reduce conjugation efficiency |
| Primary Delivery Mechanism | Viral transduction via receptor binding; natural predation [6] | Bacterial conjugation; direct cell-to-cell transfer [26] |
| Key Advantage | Narrow targeting avoids microbiome disruption; access to spatial niches [6] | Broad applicability across genetically diverse bacterial populations [8] |
| Primary Limitation | Complex phage engineering and potential for resistance via receptor mutation [27] | Requires physical proximity and compatible conjugation machinery [6] |
Experimental Protocols for Phage Engineering and Delivery
Protocol 1: Engineering Phage λ with CRISPR-Associated Transposases
This protocol, adapted from a 2025 study, details the process of creating a λ-DART phage for large DNA insertions [6].
Step 1: Phage Engineering via Homologous Recombination and Cas13a Counterselection
- Objective: To embed the entire DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) system into the λ phage genome.
- Procedure:
- Design an insert containing the DART system flanked by homology arms targeting the desired integration site in the λ genome.
- Clone this insert into a replicative plasmid and transform it into a permissive E. coli host strain (e.g., an amber-suppressor strain like LE392MP).
- Infect the host with a modified λ phage (e.g., λ cI857 Sam7) that is constrained for lysis and viral spread to the permissive host.
- Within the host cell, homologous recombination occurs between the plasmid and the infecting phage genome, integrating the DART system.
- To isolate successfully recombined phage, use a Cas13a-based counterselection. Co-express Cas13a programmed to target the wild-type phage sequence. Cas13a's trans-cleavage activity induces cellular dormancy upon recognizing and cleaving target RNA, halting the production of non-edited phage. This enriches the progeny for the desired recombinant λ-DART phages [6].
Step 2: Phage Propagation and Purification
- Procedure:
- Plate the progeny phage from the previous step on a lawn of the permissive host to form plaques.
- Pick individual plaques and amplify them through liquid infection cultures.
- Sequence the phage genome to confirm the precise integration of the DART payload and the absence of wild-type contaminants.
- Procedure:
Step 3: In Vitro Editing Assay in Mixed Communities
- Procedure:
- Grow a monoculture of the target E. coli strain or a mixed bacterial community (e.g., containing three different genera).
- Infect the culture with the purified λ-DART phage at a defined Multiplicity of Infection (MOI). Studies have shown that adjusting MOI and incubation periods is critical for enhancing final editing efficiency [6].
- Plate the infected culture on selective media or use fluorescence-activated cell sorting (FACS) after several hours to isolate edited cells.
- Quantify editing efficiency by calculating the percentage of the population that has acquired the desired knockout or insertion, which has been shown to surpass 50% in targeted E. coli populations [6].
- Procedure:
Protocol 2: Tail Fiber Engineering to Broaden Host Range
This protocol outlines the method for engineering phage tail fibers to alter receptor specificity, thereby overcoming a key limitation of narrow host range [20].
Step 1: Identification and Selection of Heterologous Adhesin
- Procedure:
- From a library of wild-type phages, identify those with broad and complementary host ranges. Characterize their receptor dependencies using efficiency of plating (EoP) assays on bacterial strains with knocked-out surface proteins (e.g., Tsx, LamB, OmpC) or deep-core LPS mutants [20].
- Select a phage with a desirable receptor affinity. For example, phage α17 utilizes the conserved nucleoside transporter Tsx.
- Identify the gene encoding the tail fiber or monomeric adhesin protein responsible for receptor binding in the donor phage.
- Procedure:
Step 2: Genetic Engineering of Phage Tail Fibers
- Procedure:
- Using homologous recombination or CRISPR-Cas-assisted counter-selection in a bacterial host, replace the native tail fiber/adhesin gene of your base phage (e.g., the LPS-dependent α15) with the heterologous one from the donor phage (e.g., the Tsx-binding adhesin from α17) [20].
- This creates a chimeric phage (e.g., α15.2) whose virions carry a stochastic combination of tail fibers with both native and new receptor affinities.
- Procedure:
Step 3: Validation of Host Range Expansion
- Procedure:
- Perform lawn kill assays on a panel of clinical bacterial strains using the wild-type phage and the engineered phage.
- Compare the number of surviving bacterial colonies (resisters). The engineered phage α15.2 demonstrated a substantially reduced number of survivors compared to its wild-type ancestor [20].
- Test the sensitivity of survivors from the wild-type phage challenge to the engineered phage. Resisters to the wild-type LPS-dependent phage often remain sensitive to the engineered phage with dual receptor affinity, confirming a clear benefit of the engineering approach [20].
- Procedure:
Visualization of Phage Engineering Workflows
The following diagrams illustrate the core concepts and experimental workflows for engineering CRISPR-loaded phages.
Figure 1: A high-level workflow for creating therapeutic CRISPR phages, involving sequential engineering steps to improve host range, killing specificity, and payload delivery efficiency.
Figure 2: The phagemid-based capsid system for producing CRISPR-loaded antimicrobial capsids (AB-capsids). A phagemid vector carrying CRISPR-Cas and a phage packaging site is transformed into a host containing a helper phage or prophage. Induction triggers capsid assembly, which packages the phagemid DNA. The resulting AB-capsids transduce the CRISPR system into target bacteria [28].
The Scientist's Toolkit: Essential Research Reagents
Successful execution of the protocols above requires a suite of specialized reagents and genetic tools.
Table 2: Key Research Reagent Solutions for Phage-Delivered CRISPR Experiments
| Reagent / Material | Function and Application | Specific Examples / Notes |
|---|---|---|
| Amber-Suppressor Host Strains | Permissive host for propagating engineered phages with amber mutations (e.g., in lysis gene S), enabling controlled infection and biocontainment [6]. | E. coli LE392MP [6]. |
| Temperate Phage Vectors | Well-characterized chassis for engineering; allows for integration of large payloads and controlled induction. | Phage λ with cI857 (thermolabile repressor) and Sam7 (amber mutation in lysis gene S) mutations [6]. |
| CRISPR-Cas Systems for Arming | The antimicrobial payload; provides sequence-specific targeting of bacterial genomes or resistance genes. | Type I-E CRISPR-Cas3 system from E. coli for potent chromosomal cleavage [20]; DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) for insertions [6]. |
| Phagemid Vectors | Plasmid-based system containing a phage packaging site (e.g., terL-terS-rinA-rinB); simplifies CRISPR cargo construction and is packaged into phage capsids [28]. | E. coli-S. aureus shuttle phagemid vectors; copy number can be optimized using different replication origins (e.g., repC, repM) to boost AB-capsid yield [28]. |
| Packaging Signal Constructs | Genetic elements that commandeer the phage capsid assembly machinery for packaging of non-native DNA. | Phage-inducible chromosomal islands (PICIs) like SaPI [28] or synthetic phage packaging signals (e.g., terL-terS-rinA-rinB) [28]. |
| Inducing Agents | Chemical triggers to initiate the lytic cycle in lysogenic strains or prophage-containing hosts for phage/production. | Mitomycin C [28]. |
The experimental data and protocols presented in this guide underscore that both engineered phages and conjugate plasmids are viable, yet distinct, strategies for delivering CRISPR-Cas systems. The optimal choice is context-dependent. Engineered phages excel in scenarios demanding high specificity to avoid microbiome disruption, when targeting bacteria in challenging spatial niches, or when delivering large, complex payloads like transposases. Their ability to be engineered for broader host ranges and their high transduction efficiency in permissive hosts make them powerful, targeted missiles. Conjugate plasmids, while less specific, offer a broad and versatile delivery mechanism capable of functioning across diverse bacterial populations where conjugation is possible, making them excellent for broader population-level interventions. For researchers, the decision pathway is clear: prioritize phage delivery for precision and large payloads in defined targets, and consider conjugate plasmids for broader, population-wide applications. As both fields advance, hybrid approaches that leverage the strengths of each system may offer the most robust solutions for combating antimicrobial resistance.
The efficacy of CRISPR-Cas gene editing is profoundly influenced by the delivery mechanism employed to introduce editing components into target cells. Within microbial systems, two primary delivery strategies have emerged as powerful platforms for introducing CRISPR machinery: conjugative plasmids and phage-based vectors. Conjugative plasmids facilitate horizontal gene transfer between bacterial cells through direct cell-to-cell contact, relying on specialized conjugative machinery [26]. In contrast, phage-based delivery utilizes bacteriophages—viruses that infect bacteria—as natural vectors to transduce CRISPR payloads into specific bacterial hosts [6]. The fundamental distinction between these systems lies in their configuration and helper function requirements, particularly in how CRISPR components are arranged on mobile genetic elements (cis configuration) versus separated across multiple elements (trans configuration) with helper functions providing essential components in trans.
The strategic decision between cis and trans configurations carries significant implications for editing efficiency, specificity, payload capacity, and biocontainment. This guide provides a systematic comparison of these delivery modalities, focusing on their performance in bacterial genome editing within the broader context of advancing CRISPR-based antimicrobial interventions. As antimicrobial resistance (AMR) continues to pose a critical global health threat, with an estimated 4.95 million deaths attributed to AMR in 2019 alone, developing precise genetic interventions has become increasingly urgent [29]. Both conjugative and phage-delivered CRISPR systems offer promising avenues to address this challenge by selectively targeting resistance genes in World Health Organization priority pathogens.
Performance Comparison: Phage-Delivered vs. Conjugative Plasmid CRISPR Systems
Quantitative data from recent studies reveal distinct performance characteristics between phage-delivered and conjugative plasmid CRISPR systems. The table below summarizes key performance metrics across multiple parameters.
Table 1: Performance Comparison of Phage vs. Conjugative Plasmid CRISPR Delivery Systems
| Performance Parameter | Phage-Delivered CRISPR Systems | Conjugative Plasmid CRISPR Systems |
|---|---|---|
| Editing Efficiency | >50% of population in E. coli monocultures and mixed communities [6] | 2-3 logs of protection against AMR gene acquisition [26] |
| Payload Capacity | ~10 kb for CAST systems (e.g., DART) [6] | Limited mainly by plasmid size and conjugation efficiency |
| Host Specificity | High (species- or strain-level specificity based on phage receptor recognition) [6] | Moderate (dependent on conjugative machinery compatibility between donor and recipient) [26] |
| Delivery Speed | Rapid (single infection cycle, minutes to hours) | Slower (requires cell growth and contact, hours) |
| Application in Mixed Communities | Efficient and specific editing in mixed bacterial communities comprising three genera [6] | Protection against horizontal gene transfer in probiotic E. coli Nissle 1917 [26] |
| Escape Rate | Low with optimized MOI and promoter strength [6] | Variable (depends on target and delivery efficiency) |
| Key System Components | λ phage with cI857 and Sam7 mutations, DART system [6] | CRISPR-Cas9, tracrRNA, conjugative machinery [26] |
The performance data reveal that phage-delivered systems achieve remarkable editing efficiencies exceeding 50% in targeted populations, enabling precise genome manipulations including gene knockouts and large insertions [6]. The DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) system, when delivered via engineered phage λ, facilitates CRISPR-guided transposition events in host genomes, accomplishing insertions of kilobase-scale genetic payloads [6]. This system has demonstrated particular utility in mixed microbial community contexts, where it can manipulate specific bacterial strains without disrupting the broader ecological balance.
Conjugative plasmid systems excel in providing broad protection against antimicrobial resistance gene acquisition, offering 2-3 logs of protection against horizontal gene transfer via transformation, transduction, and conjugation [26]. These systems have been successfully implemented in probiotic strains such as E. coli Nissle 1917, protecting these beneficial bacteria from acquiring virulence factors and AMR genes within the human gut environment [26]. The versatility of conjugative systems allows them to target various resistance mechanisms, including those associated with β-lactam antibiotics (bla genes) and colistin resistance (mcr-1 gene) [8].
Experimental Protocols for Key Applications
Phage-Delivered CRISPR Genome Editing
Objective: To achieve precise gene knockouts and insertions in targeted bacterial cells within mixed microbial communities using engineered λ-DART phages.
Table 2: Key Reagents for Phage-Delivered CRISPR Editing
| Reagent | Function/Description | Source/Reference |
|---|---|---|
| λ phage with cI857 and Sam7 mutations | Temperature-sensitive repressor and lysis mutation for controlled infection | [6] |
| DART (DNA-editing all-in-one RNA-guided CRISPR-Cas transposase) system | Type I-F CRISPR-associated transposon for guided DNA integration | [6] |
| E. coli amber-suppressor strain LE392MP | Permissive host for Sam7-containing phage propagation | [6] |
| Cas13a counterselection system | RNA-targeting CRISPR system for precise phage engineering | [6] |
| Homologous recombination substrates | DNA templates for phage genome engineering | [6] |
Protocol:
- Phage Engineering: Modify phage λ using homologous recombination coupled with Cas13a-based counterselection to embed the entire DART system into the phage genome [6]. The DART system incorporates CRISPR, gene, and transposon components of a type I-F CRISPR-associated transposon within a single vector [6].
- Phage Propagation: Amplify the engineered λ-DART phages in the permissive E. coli amber-suppressor host LE392MP under controlled conditions (30°C for lysogenic maintenance, 37°C for lytic induction) [6].
- Infection Conditions: Apply engineered λ-DART phages to target bacteria at optimized multiplicity of infection (MOI) values, typically ranging from 0.1 to 10, based on empirical determination [6].
- Incubation and Selection: Incubate infected cultures for 6-8 hours post-infection to allow for CRISPR-guided transposition events. Monitor population dynamics through optical density measurements [6].
- Editing Verification: Assess editing efficiency through selective plating, PCR screening, and sequencing to confirm precise gene integrations or disruptions [6].
The following diagram illustrates the core workflow for engineering and applying phage-delivered CRISPR systems:
Conjugative Plasmid Delivery for AMR Protection
Objective: To protect probiotic bacteria from acquiring antimicrobial resistance genes using engineered CRISPR-Cas9 systems delivered via conjugation.
Table 3: Key Reagents for Conjugative Plasmid Systems
| Reagent | Function/Description | Source/Reference |
|---|---|---|
| pWEB-TNC::pJ23104-tracr-pJ23101-cas9-CRISPRsynt | Medium copy plasmid with synthetic CRISPR array targeting AMR genes | [26] |
| Donor strains (E. coli MG1655 derivatives) | Conjugative donor cells with integrated antibiotic markers | [26] |
| Probiotic E. coli Nissle 1917 | Recipient strain for conjugation and AMR protection assessment | [26] |
| pBAD18-kan with target genes | Donor plasmids for transformation assays | [26] |
| Diaminopimelic acid (DAP) | Essential nutrient for donor strain growth in conjugation assays | [26] |
Protocol:
- Strain Construction: Clone the CRISPR-Cas9 system targeting specific AMR genes (e.g., blaNDM, vanA, mcr-1) into a medium-copy plasmid with appropriate conjugative machinery [26]. The system should include Cas9, tracrRNA, and a synthetic CRISPR array with spacers targeting the AMR genes of interest.
- Donor Preparation: Introduce the constructed plasmid into donor E. coli strains (typically E. coli MG1655 derivatives) and culture in LB medium supplemented with appropriate antibiotics and diaminopimelic acid (DAP) at 37°C with shaking [26].
- Conjugation Assay: Mix donor and recipient strains (e.g., probiotic E. coli Nissle 1917) at optimal ratios (typically 1:1 to 1:10 donor:recipient) on solid media or in liquid culture, incubating for 2-24 hours to allow for conjugation [26].
- Selection and Screening: Plate conjugation mixtures on selective media containing appropriate antibiotics to select for transconjugants while counterselecting against donor strains [26].
- Efficiency Assessment: Calculate conjugation efficiency by comparing transconjugant counts to recipient counts. Evaluate AMR protection by challenging transconjugants with relevant antibiotics and measuring survival rates [26].
The following diagram illustrates the molecular configuration of a typical conjugative plasmid system for AMR protection:
Technical Configurations: Cis vs. Trans Plasmid Arrangements
The organization of CRISPR components on genetic elements significantly impacts system performance and functionality. The table below compares the key characteristics of cis versus trans configurations.
Table 4: Comparison of Cis vs. Trans Plasmid Configurations
| Characteristic | Cis Configuration | Trans Configuration |
|---|---|---|
| Definition | All CRISPR components (Cas proteins, guide RNAs, accessory elements) encoded on a single plasmid | CRISPR components distributed across multiple plasmids or genetic elements |
| Editing Efficiency | Generally higher due to coordinated expression and delivery | Variable; depends on co-delivery/co-expression efficiency |
| Experimental Flexibility | Lower; requires rebuilding plasmid for changes | Higher; components can be mixed and matched |
| Payload Size | Limited by plasmid size constraints | Larger potential payload through distribution |
| Helper Functions | Self-sufficient; no helper plasmids required | Requires helper functions in trans |
| Assembly Complexity | More complex cloning for large constructs | Simplified individual component construction |
| Applications | Therapeutic delivery, in vivo editing | Library screening, complex pathway engineering |
| Example Systems | DART system in phage λ [6] | Conjugative systems with separate Cas9 and gRNA plasmids [26] |
In cis configurations, all necessary components for CRISPR editing reside on a single genetic element. This arrangement ensures coordinated delivery and expression, typically resulting in higher editing efficiencies. The phage λ-DART system represents an advanced cis-configured system, where the entire CRISPR-associated transposase system is embedded within the phage genome, enabling efficient, single-vector delivery [6]. This configuration is particularly advantageous for therapeutic applications where simplicity and reliability are paramount.
Trans configurations distribute CRISPR components across multiple genetic elements, offering greater experimental flexibility. For example, a typical conjugative system might separate Cas9 expression, gRNA transcription, and conjugative machinery onto different plasmids [26]. This modularity facilitates component swapping and optimization without reconstructing entire systems. However, trans configurations require efficient co-delivery of all elements to target cells, which can reduce overall efficiency and increase experimental variability. Helper functions provided in trans must be carefully balanced to ensure proper function while minimizing unnecessary genetic baggage.
The comparative analysis of conjugative plasmid and phage-delivered CRISPR systems reveals a complementary relationship rather than a strict superiority of either approach. Phage-delivered systems excel in environments requiring high specificity and efficiency in complex microbial communities, while conjugative plasmids offer broader protection against horizontal gene transfer and modular design flexibility. The decision between cis and trans configurations further refines this landscape, with cis arrangements providing streamlined efficiency for defined applications, and trans configurations enabling complex, multifunctional approaches.
Future research directions will likely focus on hybrid systems that combine the advantageous features of both platforms. The expanding CRISPR toolbox—now including Cas9, Cas12a, Cas13a, and other variants—provides additional dimensions for optimization [30] [31]. As delivery challenges remain a significant bottleneck in therapeutic applications, innovations in vector engineering, payload optimization, and host-pathogen interaction management will be crucial for advancing these technologies toward clinical implementation. Standardized assessment methodologies for comparing delivery efficiency across platforms will enable more systematic evaluation of emerging systems and accelerate the development of next-generation CRISPR delivery platforms for combating antimicrobial resistance and other pressing microbial challenges.
The rise of antimicrobial resistance (AMR) represents a critical global health threat, driving the need for precision weapons against bacterial pathogens. CRISPR-Cas systems have emerged as highly specific, programmable tools to address this challenge. A pivotal decision in designing these antimicrobials is selecting the target sequence, which dictates the mechanism of bacterial elimination: chromosomal killing or plasmid curing. This guide objectively compares the efficiency of the two primary delivery platforms for these CRISPR cargos—engineered phages versus conjugative plasmids—by synthesizing current research data and experimental outcomes.
Target Selection: Mechanism and Strategic Choice
The choice of target within the bacterial cell dictates the primary mechanism of action. This strategic decision influences both the effectiveness and the potential for resistance.
Targeting the Bacterial Chromosome for Cell Killing: This approach programs the CRISPR-Cas system to target essential genes or sequences within the bacterial chromosome. When the Cas nuclease creates double-strand breaks, the bacterium's attempt to repair this lethal damage often fails, leading to cell death [32] [8]. This is highly effective for eliminating a specific bacterial strain.
Targeting Plasmid DNA for Curing and Resensitization: An alternative strategy involves targeting genes located on plasmids, particularly those conferring antibiotic resistance (e.g.,
blagenes for β-lactam resistance ormcr-1for colistin resistance) [8]. The cleavage and loss of the plasmid—a process known as curing—resensitizes the bacterium to conventional antibiotics without necessarily killing it, potentially reducing selective pressure [32].
The table below summarizes the core strategic differences.
Table 1: Comparison of CRISPR Target Strategies for Combating Bacterial Resistance
| Feature | Chromosomal Killing | Plasmid Curing |
|---|---|---|
| Primary Mechanism | Lethal DNA cleavage of essential genes | Selective elimination of resistance plasmids |
| Primary Outcome | Bacterial cell death | Resensitization to antibiotics |
| Efficiency Range | Up to 99.9% load reduction (phage-delivered) [20] | 4.7% to 100% resensitization (plasmid-delivered) [8] |
| Advantage | Potent, direct pathogen removal | Restores efficacy of traditional antibiotics |
| Disadvantage | Strong selective pressure for escape mutants | Does not directly kill the pathogen |
Delivery Platform Efficiency: Phage vs. Conjugative Plasmid
The therapeutic success of a CRISPR antimicrobial is fundamentally linked to its delivery vehicle. Phages and conjugative plasmids represent the two most advanced delivery modalities, each with distinct performance profiles.
Engineered Phage Delivery
Bacteriophages, viruses that infect bacteria, are engineered to deliver CRISPR-Cas systems directly into the bacterial cytoplasm. A prominent example is SNIPR001, a cocktail of four CRISPR-Cas-armed phages (CAPs) developed to target E. coli in the gut [20].
- Efficiency Data: Preclinical studies demonstrate that engineered phages like SNIPR001 can reduce the bacterial load by over 3.5 log~10~ CFU/mL (99.9%) in vitro and significantly outcompete wild-type phages [20]. In a mixed microbial community context, phage λ-DART systems have achieved editing efficiencies surpassing 50% of the targeted E. coli population [6].
- Key Advantage: Specificity. Phages offer narrow host-range specificity, minimizing collateral damage to the commensal microbiome. Their natural life cycle is exploited for highly efficient, targeted delivery [6] [20].
Conjugative Plasmid Delivery
Conjugative plasmids are self-transmissible genetic elements that can transfer DNA from a donor to a recipient bacterium via direct cell-to-cell contact.
- Efficiency Data: The efficacy of conjugative plasmids in delivering CRISPR-Cas9 for resensitizing bacteria to antimicrobials is highly variable, with reported efficiencies ranging from 4.7% to 100% depending on the target and bacterial strain [8]. Conjugation efficiency can be influenced by donor-recipient compatibility and environmental factors [6].
- Key Advantage: Broad Host Range. Certain conjugative plasmids can transfer genes across a wider range of bacterial strains and species compared to the narrow specificity of most phages.
Table 2: Quantitative Comparison of Phage vs. Conjugative Plasmid Delivery Platforms
| Performance Metric | Engineered Phage Delivery | Conjugative Plasmid Delivery |
|---|---|---|
| Reported Editing/Killing Efficiency | >50% in mixed communities [6]; >99.9% load reduction [20] | 4.7% - 100% resensitization [8] |
| Delivery Specificity | High (strain- or species-level) [20] | Moderate to Broad (depends on plasmid host range) [6] |
| Typical Cargo Format | DNA-encoding Cas and guide RNA [6] [20] | DNA-encoding Cas and guide RNA [8] |
| Suitability for In Situ Editing | Excellent (natural delivery vehicle) [6] | Limited (requires donor-recipient compatibility) [6] |
Experimental Protocols and Workflows
To objectively compare the two platforms, the following are generalized protocols for key experiments measuring their efficacy.
Protocol 1: Assessing Phage-Delivered CRISPR Killing Efficiency
This protocol outlines the steps to engineer a phage and test its efficacy [6] [20].
- Phage Engineering: Select a lytic phage with a broad host range. Use homologous recombination and Cas13a-based counterselection to replace a non-essential genomic region with a CRISPR-Cas module (e.g., Type I-E with guides targeting essential chromosomal genes of the pathogen) [6] [20].
- Phage Propagation: Amplify the engineered CRISPR-Cas-armed phage (CAP) in a permissive, laboratory-safe host strain under controlled conditions.
- In Vitro Killing Assay:
- Culture Target Bacteria: Grow the target pathogen (e.g., E. coli) to mid-log phase.
- Infect: Infect the culture with the engineered CAP at a specific Multiplicity of Infection (MOI).
- Incubate & Quantify: Incubate for a set period (e.g., 4-24 hours). Serially dilute the culture and plate on agar to determine the bacterial titer (CFU/mL). Compare to a control treated with a non-targeting phage.
- Data Analysis: Calculate the reduction in bacterial load (log~10~ CFU/mL).
Protocol 2: Assessing Plasmid-Delivered CRISPR Curing Efficiency
This protocol measures the efficiency of a conjugative plasmid in delivering a CRISPR system to cure a resistance plasmid [8].
- Donor Strain Preparation: Engineer a donor bacterial strain (e.g., E. coli) to harbor a conjugative plasmid carrying the CRISPR-Cas9 system programmed with a guide RNA targeting an antibiotic resistance gene (e.g.,
bla_{NDM-1}) on a mobile genetic element. - Conjugation Assay:
- Mix Cultures: Combine mid-log phase cultures of the donor and the recipient (target MDR pathogen) at a defined ratio on a filter placed on solid media.
- Conjugate: Allow conjugation to proceed for a set time (e.g., 4-18 hours).
- Select Transconjugants: Resuspend the cells and plate on selective media containing antibiotics that counterselect against the donor and select for the recipient. Also include the antibiotic for which resistance is being targeted.
- Efficiency Analysis:
- Count the number of transconjugants that grow on the general selection media.
- Count the number of transconjugants that fail to grow on media containing the antibiotic whose resistance gene was targeted.
- Calculate Curing Efficiency:
Curing Efficiency (%) = [1 - (CFU on target antibiotic / CFU on general selection)] * 100.
The Scientist's Toolkit: Key Research Reagent Solutions
The table below lists essential materials and their functions for conducting research in phage and plasmid-delivered CRISPR antimicrobials.
Table 3: Essential Research Reagents for CRISPR Antimicrobial Development
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| CRISPR-Cas System | The core effector for programmed DNA cleavage. | Type I-E (e.g., from E. coli for chromosomal killing [20]); Type II Cas9 (for plasmid curing [8]). |
| Delivery Vector | Vehicle to deliver CRISPR cargo into target bacteria. | Engineered phage λ [6] [20]; Conjugative plasmids (e.g., F-plasmid derivatives) [8]. |
| Target Bacterial Strains | Pathogens to be targeted; require phylogenetic diversity for host range testing. | E. coli clinical isolates from bloodstream infections [20]; MDR strains harboring bla or mcr-1 plasmids [8]. |
| Selection & Counterselection Systems | For engineering phages and selecting for successful conjugation. | Cas13a for phage counterselection [6]; Antibiotic markers for plasmid selection. |
| In Vitro Culture Models | To assess efficacy under controlled conditions. | Planktonic growth kinetic assays; Biofilm models (e.g., peg lids) [20]. |
| In Vivo Model Systems | To evaluate efficacy and safety in a complex biological environment. | Mouse models for gut colonization or infection [20]. |
The choice between phage and conjugative plasmid delivery for CRISPR antimicrobials is not a matter of one being universally superior, but rather which is optimal for a specific application. The data indicate that engineered phages excel in highly specific and potent bacterial killing, achieving high efficiency even in complex environments like the gut microbiome. In contrast, conjugative plasmids offer a versatile tool for plasmid curing and resensitization across a potentially broader range of strains, though with more variable efficiency.
The most effective strategy may ultimately be guided by the clinical objective: use phage-delivered CRISPR for precise pathogen eradication in a complex community, and employ plasmid-delivered curing to restore the efficacy of an existing antibiotic arsenal against resistant infections. Future clinical success will depend on continued optimization of cargo design, delivery efficiency, and safety profiles for both platforms.
The rise of antibiotic-resistant pathogens necessitates the development of precision antimicrobials. Among the most promising strategies are those utilizing CRISPR-based systems to selectively target and eliminate bacterial pathogens. Two primary delivery vectors have emerged as front-runners for in vitro applications: engineered bacteriophages and conjugative plasmids. This guide objectively compares the performance of phage-delivered CRISPR systems against conjugate plasmid delivery for targeting pathogens in both planktonic cultures and biofilms, providing researchers with experimental data and methodologies to inform their therapeutic development.
Performance Comparison: Phage vs. Conjugative Plasmid Delivery
The table below summarizes key performance metrics for phage-delivered and conjugative plasmid-delivered CRISPR systems against bacterial pathogens, based on recent experimental findings.
Table 1: Performance Comparison of CRISPR Delivery Systems for Pathogen Targeting
| Feature | Phage-Delivered CRISPR | Conjugative Plasmid-Delivered CRISPR |
|---|---|---|
| Delivery Mechanism | Viral infection and transduction of CRISPR machinery [6] [20] | Bacterial mating and plasmid transfer via type 4 secretion system [33] |
| Targeting Specificity | High; inherent phage tropism for specific bacterial receptors can be engineered [20] | Broad; relies on plasmid host range, which can be wide (e.g., IncP RK2) [33] |
| Editing Efficiency in Planktonic Culture | High; >50% population editing efficiency in E. coli monocultures reported [6] | Very high; approaches 100% conjugation frequency under optimized contact conditions [33] |
| Efficacy in Biofilms | Effective; demonstrated ability to reduce E. coli biofilm burden, with engineered phages targeting bacteria within biofilms [34] [20] | Variable; mature biofilm architecture can limit donor cell access to inner layers, hindering plasmid transfer [35] |
| Key Advantage | Natural affinity for bacterial hosts; ability to penetrate biofilms; can be engineered to reduce phage-tolerant mutants [20] | Extremely high transfer rates under cell contact-promoting conditions; donor cells can become new donors, amplifying transfer [33] |
| Primary Challenge | Potential for bacterial resistance to phage infection; limited cargo capacity [20] | Spatial constraints in structured communities like mature biofilms can severely limit dissemination [35] |
| Notable Experimental Payload | CRISPR-associated transposases (DART) for gene knockouts/insertions; Type I-E CRISPR-Cas system for targeted killing [6] [20] | TevSpCas9 dual nuclease for targeted chromosomal cleavage and bacterial killing [33] |
Experimental Protocols for Key Studies
Protocol 1: Phage λ-DART for Bacterial Genome Editing
This protocol details the methodology for using engineered phage λ to deliver the DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) system, as described in the 2025 PNAS study [6].
- Phage Engineering: Phage λ was engineered using homologous recombination paired with a Cas13a-based counterselection strategy. The entire DART system, which incorporates the components of a type I-F CRISPR-associated transposon (CAST), was embedded into the λ phage genome to create λ-DART phages. These phages were rendered non-lysogenic to prevent persistent phage maintenance [6].
- Bacterial Culture and Infection: Target E. coli strains are grown in appropriate liquid media. The λ-DART phages are added to the bacterial culture at a specific multiplicity of infection (MOI). The culture is incubated to allow phage infection and delivery of the DART system [6].
- Editing Verification: After infection, the bacterial population is assessed for genetic edits. The λ-DART system has been shown to generate precise CRISPR RNA-guided transposition events, leading to gene knockouts and insertions with efficiencies surpassing 50% of the targeted population, even within a mixed bacterial community [6].
Protocol 2: Cis-Conjugative Plasmid Delivery of CRISPR Nuclease
This protocol is derived from the 2019 Nature Communications study that demonstrated highly efficient inter-species conjugative transfer of a CRISPR nuclease [33].
- Plasmid Construction: A cis-conjugative plasmid (e.g., pNuc-cis) is constructed based on a broad-host-range plasmid like IncP RK2. This plasmid encodes both the conjugative machinery (relaxase, type IV coupling protein, type IV secretion system) and the CRISPR nuclease (e.g., TevSpCas9) under an inducible promoter (e.g., pBAD), along with guide RNA(s) targeting essential or species-specific genes in the pathogen [33].
- Donor and Recipient Preparation: Donor E. coli strains are transformed with the cis-conjugative plasmid. Recipient pathogenic bacteria (e.g., Salmonella enterica) are cultured separately.
- Conjugation Assay:
- Filter Mating: Donor and recipient cells are mixed at a desired ratio (e.g., 1:10), concentrated on a filter membrane, and incubated on a non-selective solid surface for several hours (e.g., 24h) to allow cell-to-cell contact and plasmid transfer [33].
- Liquid Mating with Beads: To maximize conjugation efficiency, the mating is performed in a low-salt LB medium with the addition of 0.5 mm glass beads, which provide a surface for biofilm-like cell-to-cell contact. This method has been shown to increase conjugation frequencies to nearly 100% [33].
- Selection and Killing Assessment: Transconjugants (recipient cells that have received the plasmid) are selected on agar plates containing antibiotics that counter-select against the donor strain and to which the plasmid confers resistance. The killing efficiency is determined by comparing the number of transconjugants with a control plasmid lacking the CRISPR nuclease or by monitoring the reduction in viable pathogen counts after induction of the nuclease [33].
Mechanism and Workflow Visualization
The following diagram illustrates the core mechanisms and experimental workflows for both delivery systems, highlighting the logical relationship between their components and processes.
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and their functions as used in the cited studies, providing a resource for experimental design.
Table 2: Key Research Reagents for Phage and Conjugative Plasmid CRISPR Delivery
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Phage λ with cI857 & Sam7 mutations | Temperature-sensitive control of lysogeny; host-range restriction to amber-suppressor strains for safer, controlled experiments [6]. | Creating a controllable phage delivery chassis (λ-DART) for precise genome editing [6]. |
| CRISPR-associated Transposase (DART) | Enables RNA-guided insertion of large DNA fragments or gene knockouts without relying on host repair mechanisms [6]. | Phage-mediated stable gene integration and targeted gene disruption in E. coli [6]. |
| Type I-E CRISPR-Cas System (EcCas) | A multi-protein complex (CasA-E, Cas3) native to E. coli that provides potent, targeted cleavage of bacterial chromosomes [20]. | Arming phages (e.g., in SNIPR001 development) for enhanced and specific killing of diverse E. coli strains [20]. |
| IncP RK2 Plasmid Backbone | A broad-host-range conjugative plasmid system that facilitates efficient DNA transfer between a wide array of bacterial species [33]. | Building cis-conjugative plasmids (e.g., pNuc-cis) for high-frequency delivery of CRISPR nucleases [33]. |
| TevSpCas9 Nuclease | A dual nuclease fusion protein (I-TevI domain + SpCas9) that introduces targeted double-strand breaks in bacterial DNA, leading to cell death [33]. | Sequence-specific killing of bacterial pathogens like Salmonella enterica upon conjugative delivery [33]. |
| PbolA Promoter | A bacterial promoter that remains active under restricted growth conditions, such as those found in biofilms or the gut [20]. | Driving expression of CRISPR-Cas systems in engineered phages to ensure functionality within biofilm environments [20]. |
Both phage-delivered and conjugative plasmid-delivered CRISPR systems present powerful, yet distinct, approaches for targeting pathogens in vitro. Phage-based systems offer natural precision and biofilm-penetrating potential, making them suitable for applications targeting specific pathogens within complex communities. Conjugative plasmids excel in environments that promote high cell-to-cell contact, achieving near-total transfer efficiency in planktonic and early-stage biofilm cultures. The choice between these two potent delivery mechanisms ultimately depends on the specific pathogen, the nature of the microbial community (planktonic vs. mature biofilm), and the desired genetic outcome.
Antimicrobial resistance (AMR) poses a critical global health threat, with projections estimating 10 million annual deaths by 2050 without effective interventions [36] [37]. Traditional antibiotic treatments detrimentally affect the microbiome and contribute to resistance, creating an urgent need for precision approaches that selectively reduce pathogenic bacterial burdens while preserving commensal communities [20]. This comparison guide evaluates two advanced therapeutic modalities for reducing bacterial burden in the gut microbiome: phage-delivered CRISPR systems and conjugate plasmid delivery. Both approaches represent paradigm shifts from broad-spectrum antimicrobials to targeted genetic interventions, yet they differ significantly in delivery mechanisms, editing efficiencies, and practical implementation. Framed within a broader thesis comparing delivery efficiency, this analysis synthesizes current experimental data to objectively assess the performance, applications, and limitations of each strategy for researchers, scientists, and drug development professionals.
Phage-Delivered CRISPR Systems
Bacteriophages (phages), the natural viruses of bacteria, can be engineered as precision vectors to deliver CRISPR-Cas systems to specific bacterial targets. These engineered phages offer species- or strain-level specificity, leveraging natural phage infection pathways to introduce CRISPR machinery that selectively eliminates antibiotic-resistant pathogens [6] [20]. The technology involves modifying phage genomes to incorporate genes encoding Cas nucleases and CRISPR arrays, creating CRISPR-Cas-armed phages (CAPs). Upon infection, these components are expressed in the target bacterium, where the CRISPR system introduces lethal double-strand breaks in chromosomal DNA or antibiotic resistance genes [20].
Recent advances have demonstrated the feasibility of engineering well-characterized temperate phages like λ to deliver not only standard CRISPR-Cas systems but also more complex CRISPR-associated transposases (CASTs). These λ-DART phages can perform flexible bacterial genome manipulation, including large insertions and targeted gene disruptions, achieving editing efficiencies surpassing 50% in both monocultures and mixed bacterial communities [6]. The technology has been successfully scaled for in vivo applications, with studies showing that engineered phages can reduce target bacterial loads in mouse models while minimizing collateral damage to commensal microbiota [20].
Conjugate Plasmid Delivery
Conjugative plasmids are self-transmissible genetic elements that naturally mediate horizontal gene transfer between bacteria. This innate capability has been harnessed to deliver CRISPR-Cas systems to target bacteria through a process requiring direct cell-to-cell contact [8] [35]. The delivery system typically involves donor bacteria carrying conjugative plasmids equipped with CRISPR-Cas components, which are transferred to recipient pathogens through specialized conjugation machinery.
The process initiates when donor bacteria produce sex pili that attach to recipient cells, forming stable mating pairs. A plasmid-encoded type IV secretion system then facilitates the transfer of single-stranded DNA containing the CRISPR-Cas construct into recipient bacteria [35]. Inside the transconjugant, the single-stranded DNA is converted to double-stranded form and expressed, enabling CRISPR-mediated targeting of chromosomal genes or antibiotic resistance determinants. Research has demonstrated that the RP4 broad host range conjugative plasmid can transfer genetic cargo at remarkable speeds, with DNA translocation rates estimated at 556±267 nucleotides per second, and recipient cells typically converted to transconjugants within 20 minutes of donor contact [35].
Table 1: Core Mechanism Comparison Between Delivery Systems
| Feature | Phage-Delivered CRISPR | Conjugate Plasmid Delivery |
|---|---|---|
| Primary Delivery Vector | Engineered bacteriophage | Conjugative plasmid |
| Infection/Transfer Mechanism | Viral infection via receptor binding | Conjugation pilus-mediated DNA transfer |
| Target Specificity | Phage tropism (species/strain-level) | Donor-recipient compatibility |
| Genetic Cargo Capacity | Moderate (~10 kb for λ-DART) [6] | Larger (plasmids + inserts) |
| Transfer Speed | Single infection cycle (hours) | Rapid DNA transfer (minutes) [35] |
| Self-Replication in Target | Yes (lytic cycle amplification) | No (requires separate replication origin) |
| Primary Applications | Pathogen-specific killing, microbiome editing [6] [38] | Antibiotic resensitization, community editing [8] |
Visualizing Core Mechanisms
The following diagrams illustrate the fundamental mechanisms and experimental workflows for both therapeutic approaches, highlighting key differences in their operational paradigms.
Diagram 1: Core therapeutic delivery mechanisms
Experimental Protocols and Methodologies
Phage-Delivered CRISPR Workflow
The development and implementation of phage-delivered CRISPR therapeutics follows a systematic workflow from phage selection to in vivo validation:
Phase 1: Phage Library Screening and Selection
- Source diverse lytic phages from wastewater, environmental samples, or phage banks [20]
- Screen against target pathogen panels (e.g., 429 phylogenetically diverse E. coli strains) using growth kinetics assays
- Select phages based on broad host range, complementary receptor binding, and engineerability
- Assess efficiency of plating (EoP) on target strains and receptor knockout mutants
Phase 2: Phage Engineering
- Employ homologous recombination with Cas13a-based counterselection for precise phage genome modifications [6]
- Incorporate CRISPR-Cas systems (type I-E or type I-F CAST) under optimized promoters (PbolA for biofilm activity) [20]
- For λ phage engineering: utilize amber mutations (Sam7) and thermolabile repressors (cI857) for controlled infection [6]
- Implement tail fiber engineering to expand receptor tropism and reduce escape mutants [20]
Phase 3: In Vitro Validation
- Conduct kinetic assays in monocultures and mixed bacterial communities
- Quantify editing efficiency via selective plating and fluorescence reporters
- Assess biofilm penetration using confocal microscopy and metabolic activity assays
- Evaluate emergence of phage-resistant mutants through lawn kill assays
Phase 4: In Vivo Testing
- Administer phage cocktails (e.g., SNIPR001) to mouse models via oral gavage or other relevant routes
- Monitor bacterial load reduction in gut homogenates over time
- Assess microbiome impact through 16S rRNA sequencing and metagenomic analysis
- Evaluate safety and pharmacokinetics in higher organisms (e.g., minipigs) [20]
Conjugate Plasmid Delivery Workflow
The implementation of conjugative plasmid delivery for bacterial burden reduction involves distinct methodological phases:
Phase 1: Plasmid Design and Construction
- Select appropriate conjugative plasmid backbone (IncP, IncF, etc.) based on host range requirements
- Clone CRISPR-Cas system with specific guide RNAs targeting essential genes or antibiotic resistance determinants
- Incorporate fluorescent reporters (sfGFP, mCherry) or selective markers for transconjugant tracking
- Verify plasmid stability and transfer efficiency in model strains
Phase 2: Donor Strain Preparation
- Introduce conjugative plasmid into appropriate donor strain (often E. coli)
- Optimize donor growth conditions to enhance conjugation efficiency
- Validate CRISPR functionality in donor background before conjugation assays
Phase 3: Conjugation Assays
- Mix donor and recipient cells at optimized ratios (typically 1:10 donor:recipient) [35]
- Allow mating on filters or in liquid medium for specified durations (1-24 hours)
- Plate on selective media to quantify transconjugants, donors, and recipients
- Calculate conjugation frequency as transconjugants per recipient
Phase 4: In Vivo Application
- Introduce donor strains to animal models (often mice) via oral gavage or injection
- Monitor plasmid dissemination through fecal sampling and selective plating
- Quantify target pathogen reduction and potential off-target effects
- Assess horizontal transfer to non-target bacteria in gut microbiome
Diagram 2: Therapeutic development workflow comparison
Efficiency and Performance Data Comparison
Quantitative Efficacy Metrics
Both phage-delivered CRISPR and conjugate plasmid delivery systems have demonstrated significant efficacy in reducing bacterial burden, though through different mechanistic approaches and with distinct efficiency profiles.
Table 2: Efficiency and Performance Comparison
| Parameter | Phage-Delivered CRISPR | Conjugate Plasmid Delivery | References |
|---|---|---|---|
| Editing Efficiency | >50% population editing in targeted E. coli [6] | 4.7% to 100% resensitization efficiency [8] | [6] [8] |
| Bacterial Reduction | >3.5 log10 CFU mL-1 reduction of E. coli in mouse gut [20] | 1-6 log10 reduction with conjugative delivery [35] | [35] [20] |
| Delivery Speed | Single infection cycle (varies by phage) | ~20 min for recipient conversion post-contact [35] | [35] |
| DNA Transfer Rate | N/A (varies by phage) | 556±267 nt/s for RP4 plasmid [35] | [35] |
| Target Specificity | High (phage receptor-dependent) | Moderate (conjugation compatibility) | [6] [35] |
| Payload Capacity | ~10 kb for λ-DART system [6] | Larger capacity (plasmid-based) | [6] |
| Biofilm Activity | Significant killing in E. coli biofilms with PbolA promoter [20] | Limited by spatial constraints in mature biofilms [35] | [35] [20] |
| Escape Mutant Frequency | Reduced with tail fiber engineering [20] | Varies with target and selection pressure | [20] |
In Vivo Performance Data
Recent advances in both technologies have yielded promising in vivo results for reducing bacterial burden in the gut microbiome:
Phage-Delivered CRISPR Performance:
- The SNIPR001 cocktail, comprising four CRISPR-Cas-armed phages (CAPs), significantly reduced E. coli loads in mouse models, demonstrating better efficacy than individual component phages [20].
- Engineered λ-DART phages achieved precise gene knockouts and insertions in targeted E. coli cells within mixed bacterial communities, with editing efficiencies surpassing 50% of the target population [6].
- Phages engineered with the PbolA promoter showed enhanced activity under restricted bacterial growth conditions similar to the gut environment and within biofilms [20].
Conjugate Plasmid Delivery Performance:
- Conjugative delivery of CRISPR-Cas systems successfully resensitized bacterial strains to various antimicrobials, with efficiency rates ranging from 4.7% to 100% depending on the target and delivery conditions [8].
- Studies tracking RP4 plasmid dissemination revealed that in high-density, two-dimensional cell monolayers, 92±2% of recipients are converted into transconjugants within 20 minutes after contacting a donor cell [35].
- However, spatial constraints in three-dimensional mature biofilms limited donor cell ability to establish direct contacts with recipients, impeding plasmid transfer efficiency in structured environments [35].
Research Reagent Solutions
Successful implementation of these therapeutic approaches requires specialized reagents and materials. The following table details key research tools referenced in the literature.
Table 3: Essential Research Reagents and Materials
| Reagent/Material | Function/Application | Examples/Specifications | References |
|---|---|---|---|
| Bacteriophage Vectors | Delivery chassis for CRISPR systems | Phage λ (temperate), T-even phages (lytic) | [6] [20] |
| Conjugative Plasmids | DNA transfer vehicles for CRISPR systems | IncP-1 (RP4), IncF plasmids | [8] [35] |
| CRISPR-Cas Systems | Targeted DNA cleavage or manipulation | Type I-E (E. coli), Type I-F CAST (DART) | [6] [20] |
| Fluorescent Reporters | Tracking DNA transfer and gene expression | Ssb-Ypet (ssDNA), mCherry-ParB (dsDNA) | [35] |
| Promoter Systems | Controlling CRISPR expression in bacteria | PbolA (biofilm/gut conditions), PrelB | [20] |
| Bacterial Strains | Target pathogens and donor constructs | E. coli BW25113, LE392MP (amber-suppressor) | [6] [35] |
| Animal Models | In vivo efficacy and safety testing | Mouse models, minipigs | [20] |
| Induction Agents | Prophage induction studies | Mitomycin C, hydrogen peroxide, Stevia | [39] |
Discussion and Comparative Analysis
Advantages and Limitations
Phage-Delivered CRISPR Advantages:
- High Specificity: Phage receptor binding provides natural targeting precision, potentially reaching species- or strain-level specificity [6] [20].
- Self-Amplification: Lytic phage replication at infection sites enables dose amplification and enhanced bacterial killing [20].
- Biofilm Penetration: Engineered phages can access and kill bacteria within biofilms, a significant challenge for conventional antibiotics [20].
- Programmability: CRISPR guidance allows targeting of specific genetic sequences, including antibiotic resistance genes [20].
Phage-Delivered CRISPR Limitations:
- Host Range Constraints: Limited to bacteria susceptible to the engineered phage, though tail fiber engineering can expand tropism [20].
- Bacterial Resistance: Phage resistance can develop through receptor modification, though combination approaches mitigate this risk [20].
- Manufacturing Complexity: Phage production and quality control present scaling challenges for clinical applications [20].
- Immune Recognition: Potential for neutralization by host immune systems in therapeutic applications.
Conjugate Plasmid Delivery Advantages:
- Broad Host Range: Many conjugative plasmids transfer across species boundaries, enabling wider application [35].
- Large Payload Capacity: Plasmid systems can deliver substantial genetic cargo, including multiple CRISPR components [8].
- Established Methodology: Conjugation protocols are well-established in microbiology research [35].
- Persistence Potential: Plasmid maintenance can provide sustained CRISPR activity in target populations.
Conjugate Plasmid Delivery Limitations:
- Spatial Constraints: Transfer efficiency decreases significantly in structured environments like mature biofilms [35].
- Off-Target Effects: Potential for unintended transfer to non-target bacteria in complex communities [35].
- Donor Competition: Donor strains may not persist or compete effectively in complex gut microbiota [35].
- Regulatory Concerns: Uncontrolled spread of engineered genetic elements raises safety considerations.
Applications and Future Directions
The complementary strengths of these technologies suggest potential application spaces where each excels. Phage-delivered CRISPR systems appear particularly suited for:
- Targeted pathogen elimination in defined infections
- Biofilm-associated infections where penetration is critical
- Applications requiring minimal disruption to commensal microbiota
- Situations where phage tropism aligns with therapeutic targets
Conjugate plasmid delivery may be preferable for:
- Broad-spectrum approaches targeting resistance genes across multiple species
- Engineering complex functions into microbial communities
- Applications where donor strain colonization is desirable
- Research settings where established conjugation protocols offer practical advantages
Future developments will likely focus on overcoming current limitations through engineering approaches. For phage systems, this includes expanding host range through synthetic biology and evading immune responses. For conjugate systems, research may focus on enhancing spatial dissemination and controlling transfer specificity. The emerging paradigm of combining both approaches—using phages to deliver conjugation systems or vice versa—represents a promising frontier for precision microbiome engineering.
Both phage-delivered CRISPR and conjugate plasmid delivery systems represent transformative approaches for reducing bacterial burden in the gut microbiome with distinct efficiency profiles and application landscapes. Phage systems offer exceptional targeting precision and biofilm penetration, achieving >50% editing efficiency in target populations and >3.5 log10 CFU reductions in vivo. Conjugate plasmids provide broader host range and efficient DNA transfer, with resensitization efficiencies ranging from 4.7% to 100% but facing limitations in structured environments.
The choice between these modalities depends critically on the specific therapeutic objectives, target pathogens, and environmental context. Phage-delivered CRISPR excels in precision applications against defined pathogens, while conjugate plasmid systems offer advantages for broader community interventions. As both technologies advance through clinical development, they hold significant promise for addressing the urgent threat of antimicrobial resistance while establishing new paradigms for precision microbiome engineering.
Challenges and Solutions: Overcoming Delivery Limitations
The efficacy of any antibacterial agent or delivery vector is fundamentally constrained by its host range—the spectrum of bacterial strains it can recognize, infect, or manipulate. This specificity, while advantageous for precision, poses a significant limitation for applications requiring broad-spectrum activity, such as combating complex infections or manipulating diverse microbial communities. The field of precision antibacterial strategies is currently dominated by two powerful delivery platforms: engineered bacteriophages and conjugative plasmids. Both leverage natural biological processes—viral infection and bacterial conjugation, respectively—to introduce genetic cargo, such as CRISPR-Cas systems, into target bacteria.
This guide provides a direct comparison of these platforms, focusing on the inherent host range limitations of each and the engineered strategies developed to overcome them. The core thesis is that while both platforms face distinct host range barriers, the solutions employed—ranging from molecular engineering of viral attachment machinery to the exploitation of plasmid anti-defence systems—are defining the next generation of precision antimicrobials and microbiome editing tools. We will dissect the experimental data, methodologies, and material toolkits that are enabling researchers to broaden bacterial targeting, moving from narrow, strain-specific interventions to broader, community-wide applications.
Platform Comparison: Phage-Delivered CRISPR vs. Conjugative Plasmids
The choice between phage and conjugative plasmid delivery involves trade-offs between specificity, cargo capacity, and ease of host range modification. The table below summarizes a direct comparison based on recent research.
Table 1: Comparative Analysis of Delivery Platform Efficiency and Host Range
| Feature | Phage-Delivered CRISPR | Conjugative Plasmid Delivery |
|---|---|---|
| Natural Host Range | Typically narrow, strain-specific; determined by receptor-binding proteins (RBPs) [13] [20] | Often broader; dependent on the plasmid's transfer machinery and compatibility with recipient cells [40] |
| Primary Host Range Limitation | Bacterial surface receptor availability and compatibility [20] | Plasmid incompatibility groups, restriction barriers, and host defence systems (e.g., CRISPR-Cas) [40] |
| Key Engineering Strategy | Tail fiber engineering & RBP swapping [20] | Exploiting natural anti-defence "islands" in plasmid leading regions [40] |
| Editing Efficiency (Representative Data) | >50% of targeted population in mixed communities [6]; Significant reduction of E. coli burden in mouse models [20] | Conjugative delivery of CRISPR resensitized bacteria with 4.7% to 100% efficiency [8] |
| Cargo Capacity | Limited by phage capsid size [6] | Larger, can deliver extensive systems like entire CRISPR-associated transposases (DART) [6] |
| Advantage | High specificity; can be engineered to reduce phage-tolerant survivors [20] | Broad mobilization potential; can be delivered to hard-to-transform bacteria [8] |
Experimental Strategies for Broadening Host Range
Phage Engineering: Tail Fiber Modification
Objective: To overcome the narrow specificity of phage receptor-binding proteins (RBPs) and redirect phage tropism towards new bacterial hosts.
Detailed Protocol:
- Identify Donor and Recipient Phages: Select a donor phage possessing the desired RBP (e.g., a Tsx-binding adhesin) and a recipient phage with a broad host range but a potentially expandable receptor profile (e.g., an LPS-dependent phage) [20].
- Genetic Modification via Homologous Recombination: Engineer the recipient phage genome using homologous recombination. This involves co-transforming the phage DNA with a repair plasmid containing the donor RBP gene, flanked by homology arms matching the sequences upstream and downstream of the recipient's native RBP gene.
- Counterselection with Cas13a: To isolate successfully recombined phages, employ a Cas13a-based counterselection strategy. A CRISPR RNA (crRNA) is designed to target the wild-type RBP sequence of the recipient phage. When Cas13a and this crRNA are expressed in a bacterial host, they cleave the RNA of infected cells harboring the wild-type phage, inducing dormancy and preventing plaque formation. Engineered phages with the swapped RBP evade this targeting and form clear plaques, allowing for their selection [6].
- Validation: Verify the engineered phage's new host range through efficiency of plating (EoP) assays on bacterial strains expressing the new target receptor. Confirm the genetic modification by sequencing the tail fiber region [20].
Plasmid Engineering: Leveraging Leading Region Anti-Defence Systems
Objective: To enhance plasmid survival and establishment in a new host by countering its innate immune defences immediately upon entry.
Detailed Protocol:
- Bioinformatic Identification: Analyze the leading region (the first segment transferred during conjugation) of a conjugative plasmid for known anti-defence genes. This involves using profile hidden Markov models (pHMMs) to identify homologs of anti-CRISPR (Acr), anti-restriction (e.g., ArdA), and anti-SOS (e.g., PsiB) genes [40].
- Characterization of Promoter Activity: Identify and validate single-strand promoters (e.g., Frpo) within the leading region. These promoters are active on the single-stranded DNA state during conjugation, enabling immediate expression of downstream anti-defence genes before the plasmid is fully established as double-stranded DNA [40].
- Functional Assays: To test the efficacy of an identified anti-defence system:
- Anti-Restriction Assay: Conjugate the plasmid into a recipient strain with a known restriction-modification system. The conjugation efficiency is compared against a control plasmid lacking the anti-restriction gene.
- Anti-CRISPR Assay: Attempt conjugation into a recipient strain possessing a functional CRISPR-Cas system that targets a sequence on the incoming plasmid. A successful anti-CRISPR system will yield significantly higher conjugation efficiency by preventing CRISPR-mediated cleavage [40].
- Engineering Application: The identified anti-defence genes and their single-strand promoters can be synthesized as a modular "anti-defence cassette" and cloned into the leading region of non-conjugative delivery plasmids to improve their stability and success in diverse microbial communities [40].
Visualizing the Core Engineering Workflows
The following diagrams illustrate the logical flow and key components of the two primary host-range broadening strategies discussed.
Phage Host Range Expansion
Plasmid Anti-Defense Strategy
The Scientist's Toolkit: Essential Research Reagents
Successful experimentation in broadening bacterial targeting requires a suite of specialized reagents. The following table details key materials and their functions as derived from the cited protocols.
Table 2: Key Research Reagent Solutions for Host Range Expansion Studies
| Reagent / Solution | Function / Application | Example from Research |
|---|---|---|
| Cas13a Counterselection System | Precisely selects for engineered phages by targeting and eliminating wild-type genomes during phage engineering [6]. | Used in engineering phage λ with CRISPR-associated transposases [6]. |
| Amber-Suppressor Bacterial Strains | Provides a genetic biocontainment system for phages with amber mutations (e.g., Sam7), controlling lysis and enabling safe experimentation [6]. | E. coli strain LE392MP used to control λ cI857 Sam7 phage infection [6]. |
| Profile Hidden Markov Models (pHMMs) | Bioinformatics tool for identifying anti-defence gene homologs in plasmid leading regions through sequence homology searches [40]. | Used to discover anti-CRISPR and anti-restriction genes in plasmid databases [40]. |
| Synthetic Anti-Defence Cassette | A modular genetic component containing an anti-defence gene (e.g., Acr, ArdA) under a single-strand promoter, used to arm delivery vectors [40]. | Proposed strategy to enhance plasmid establishment in diverse hosts [40]. |
| CRISPR-Guided Vector (CGV) | A plasmid vector encoding a CRISPR-Cas system (proteins and guide array) for targeted bacterial killing or gene editing upon delivery [20]. | CGV-EcCas used to confer specificity and potency against E. coli [20]. |
| Denial-of-Spread (DoS) Plasmid | An engineered, mobilizable plasmid that acts as a "predatory gene drive" to eliminate specific conjugative plasmids from a microbial community [41]. | DoS 1.0 plasmid designed to target and eliminate the broad-host-range RP4 plasmid [41]. |
The escalating crisis of antimicrobial resistance (AMR) necessitates the development of next-generation, precision weapons. Among the most promising are advanced biological platforms that can precisely target and eliminate resistance mechanisms within bacterial pathogens. This guide provides a comparative analysis of two leading-edge strategies: phage-delivered CRISPR systems and conjugate plasmid delivery. Both systems harness the natural capabilities of mobile genetic elements—bacteriophages and conjugative plasmids—to transport payloads, such as CRISPR-Cas machinery, into bacterial cells with the goal of dismantling AMR genes. However, their efficacy is critically challenged by two major evolutionary counter-strategies employed by bacteria: phage insensitivity (the ability of bacteria to evolve resistance to phage infection) and plasmid escape (the ability of bacteria to avoid accepting or to eliminate a therapeutic plasmid).
Understanding the performance, strengths, and limitations of these platforms is essential for researchers and drug development professionals aiming to select the right tool for specific therapeutic or biocontrol applications. This guide objectively compares their efficiency, supported by recent experimental data, and provides detailed protocols and resource toolkits to facilitate their application in research and development.
Performance Comparison: Phage-Delivered CRISPR vs. Conjugate Plasmid Delivery
The table below summarizes key performance metrics for phage-delivered CRISPR and conjugate plasmid systems, based on recent experimental studies. These metrics are critical for evaluating their potential to overcome bacterial resistance mechanisms.
Table 1: Performance Comparison of Phage-Delivered CRISPR and Conjugate Plasmid Systems
| Performance Metric | Phage-Delivered CRISPR | Conjugate Plasmid Delivery | Key Supporting Experimental Data |
|---|---|---|---|
| Editing Efficiency | High (>50% of targeted population in mixed communities) [6] | Variable; can be highly efficient in specific contexts (e.g., ~100% plasmid elimination reported) [2] | λ-DART phages achieved >50% knockout/insertion in E. coli within monocultures and mixed communities [6]. A native CRISPR-Cas3 system delivered via conjugation achieved ~100% elimination of resistance plasmids in K. pneumoniae [2]. |
| Host Specificity | Very High (Largely determined by phage receptor recognition) [6] [1] | Moderate to High (Depends on conjugative machinery compatibility and recipient cell physiology) [2] [35] | Phage λ-DART specifically edited E. coli within a mixed-genera community without affecting non-targets [6]. Conjugation efficiency in biofilms is highly constrained by spatial architecture and physical access to recipients [35]. |
| Payload Capacity | Limited by phage capsid size (Typically <10-20 kbp for λ-derived systems) [6] | Large (Plasmids can be >100 kbp, carrying multiple resistance genes) [42] | The entire DART system (~10 kb CRISPR-guided transposase system) was incorporated into phage λ [6]. Multidrug-resistant "mega-plasmids" over 100 kb are commonly found in wastewater effluent [42]. |
| Overcoming Physical Barriers (e.g., Biofilms) | Promising (Phages can evolve to produce biofilm-degrading enzymes like depolymerases) [1] | Limited (Mature biofilm architecture physically impedes donor cell entry and plasmid transfer) [35] | In mature 3D biofilms, community architecture limits donor cell access to high-density regions, severely hindering conjugation [35]. Phages can be adaptively evolved to overcome barriers like receptor masking via capsules [1]. |
| Primary Resistance Challenge | Phage Insensitivity: Bacteria can rapidly evolve surface receptor modifications or CRISPR-Cas defenses to block infection [1]. | Plasmid Escape: Transfer is physically blocked in structured populations; plasmids can be lost or targeted by host nucleases [2] [35]. | Bacterial resistance to phages has been reported in up to 82% of in vivo studies [1]. Plasmid transfer is optimal in 2D monolayers but drops sharply in structured 3D biofilms [35]. |
Experimental Protocols for Assessing Delivery Efficiency and Overcoming Resistance
Protocol 1: Engineering and Validating Phage-Delivered CRISPR (λ-DART System)
This protocol outlines the methodology for creating a phage capable of delivering a CRISPR-associated transposase (DART) for precise genome editing, as detailed in recent pioneering research [6].
- Step 1: Phage Engineering via Homologous Recombination and Cas13a Counterselection
- Objective: To embed the large DART payload into the genome of phage λ.
- Procedure:
- A donor plasmid containing the DART system (CRISPR arrays, cas genes, and transposon) flanked by homology arms to the λ genome is introduced into an E. coli host cell.
- The host is infected with wild-type phage λ, allowing homologous recombination to occur between the donor plasmid and the phage genome in a small subset of the viral progeny.
- To isolate these successfully recombined phages, a powerful counterselection is applied. The phage lysate is used to infect a new bacterial host that expresses the RNA-targeting nuclease Cas13a, programmed with a crRNA that targets the original, wild-type λ genome sequence.
- Cas13a cleaves the wild-type phage RNA, triggering dormancy and death in cells infected with unmodified phage. Cells infected with the successfully recombined λ-DART phage, whose RNA sequence no longer matches the crRNA, survive and produce plaques.
- Step 2: Phage Propagation in a Controlled Host System
- Objective: To produce high-titer stocks of the engineered phage without allowing for lysogeny or uncontrolled spread.
- Procedure: The λ-DART phage carries two key mutations:
cI857(a thermolabile repressor) andSam7(an amber mutation in the lysis gene S). Phages are propagated in a permissive amber-suppressor host (e.g., E. coli LE392) at 30°C to maintain the lysogenic state. To induce the lytic cycle and produce phage particles, the temperature is shifted to 37°C, destabilizing the repressor [6].
- Step 3: Editing Efficiency Assay in Complex Communities
- Objective: To quantify the ability of λ-DART to edit a target bacterium within a mixed-species community.
- Procedure:
- A defined microbial community is constructed, including the target bacterium (e.g., E. coli) and non-target genera.
- The community is infected with the λ-DART phage at a specific Multiplicity of Infection (MOI).
- After a defined incubation period, the target bacteria are isolated and screened for the desired genetic edit (e.g., knockout via transposon insertion) using PCR, sequencing, or phenotypic assays (e.g., restored antibiotic sensitivity).
- Editing efficiency is calculated as the percentage of the target population that has been successfully modified [6].
Protocol 2: Evaluating Conjugate Plasmid Delivery and Spread in Biofilms
This protocol, derived from high-resolution microscopy studies, measures how bacterial community structure, particularly biofilms, impacts the dissemination of conjugative plasmids [35].
- Step 1: Plasmid and Strain Engineering for Single-Cell Visualization
- Objective: To genetically tag donors, recipients, and the plasmid for real-time tracking.
- Procedure:
- The broad-host-range RP4 plasmid (or another conjugative plasmid of interest) is engineered to carry a constitutively expressed fluorescent reporter gene (e.g.,
sfGFP). - The donor strain carries this RP4-sfGFP plasmid.
- The recipient strain is engineered with a chromosomal marker for a fluorescent protein of a different color (e.g.,
mRuby2).
- The broad-host-range RP4 plasmid (or another conjugative plasmid of interest) is engineered to carry a constitutively expressed fluorescent reporter gene (e.g.,
- Step 2: Establishing Structured Bacterial Communities
- Objective: To create different community architectures for testing.
- Procedure:
- 2D Monolayers: Donor and recipient cells are mixed in a defined ratio (e.g., 1:10) and seeded into microfluidic chambers or on thin agar pads, forming a densely packed, quasi-2D layer where all cells are in direct contact [35].
- 3D Mature Biofilms: Recipient cells are first allowed to form a mature biofilm over 24-48 hours in a flow cell or microfluidic device. Donor cells are then introduced to the pre-formed biofilm [35].
- Step 3: Live-Cell Imaging and Quantification of Conjugation Dynamics
- Objective: To capture the spatiotemporal dynamics of plasmid transfer at the single-cell level.
- Procedure:
- Time-lapse fluorescence microscopy is performed on the samples at short intervals (e.g., 1 minute).
- Transconjugant identification: A recipient cell (mRuby2+) is classified as a transconjugant once a significant increase in intracellular sfGFP signal (e.g., >15%) is detected, indicating it has received the plasmid [35].
- Image analysis software (e.g., BiofilmQ, StarDistOPP) is used to segment individual cells and quantify key parameters: the percentage of donor cells that produce transconjugants, the time between donor-recipient contact and plasmid transfer, and the spatial distribution of transconjugants within the biofilm architecture [35].
Visualizing Mechanisms and Workflows
Phage-Delivered CRISPR-Cas Mechanism
The following diagram illustrates the stepwise mechanism by which an engineered bacteriophage delivers a CRISPR-based payload to a target bacterial cell, leading to the disruption of an antimicrobial resistance (AMR) gene.
Diagram Title: Mechanism of Phage-Delivered CRISPR to Combat AMR
Conjugate Plasmid Transfer in Biofilms
The following diagram contrasts the efficiency of conjugative plasmid transfer between different bacterial community structures, highlighting the physical barrier posed by mature biofilms.
Diagram Title: Conjugation Efficiency in 2D vs 3D Bacterial Communities
The Scientist's Toolkit: Key Research Reagent Solutions
The table below catalogues essential materials and reagents used in the featured experiments, providing a resource for researchers seeking to implement these protocols.
Table 2: Essential Research Reagents for Phage and Plasmid Delivery Studies
| Reagent/Tool | Function/Application | Specific Examples |
|---|---|---|
| Engineered Phage λ | Delivery chassis for CRISPR payloads to specific bacterial hosts. | λ phage with cI857 and Sam7 mutations for controlled infection; λ-DART phage with integrated CRISPR-transposase system [6]. |
| CRISPR-Cas Systems | Precision targeting and disruption of bacterial genes, including AMR genes. | DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) [6]. Cas9 for gene knockouts; Cas13a for counterselection in phage engineering [6] [2]. |
| Specialized Bacterial Strains | Provide specific genetic backgrounds for propagation, selection, or assay readouts. | Amber-suppressor host (e.g., E. coli LE392) for propagating phage with amber mutations; recipient strains with chromosomal fluorescent markers (e.g., mRuby2) [6] [35]. |
| Conjugative Plasmids | Self-transmissible vectors to deliver genetic cargo between bacterial cells. | Broad-host-range IncP-1α RP4 plasmid engineered with fluorescent reporters (e.g., RP4-sfGFP) for tracking conjugation dynamics [35]. |
| Live-Cell Imaging & Analysis Software | Real-time, single-cell quantification of dynamic processes like infection and conjugation. | Microfluidic chambers for growth; Confocal/fluorescence microscopy; Analysis software BiofilmQ and StarDistOPP [35]. |
| Machine Learning Prediction Tools | In silico prediction of phage-host interactions to guide experimental design. | Models using protein-protein interactions (PPI) to predict strain-specific phage infectivity with high accuracy [43]. |
The efficacy of CRISPR-Cas systems in combating antimicrobial resistance (AMR) and enabling precise bacterial genome engineering is fundamentally constrained by the efficiency and specificity of their delivery vectors. Within this landscape, phage-based delivery and conjugative plasmid delivery represent two of the most prominent strategies. This guide provides an objective, data-driven comparison of their performance, with a focused analysis on how promoter selection and vector engineering enhance in vivo stability and overall editing efficiency. Understanding these parameters is critical for researchers and drug development professionals aiming to select the optimal system for specific applications, whether for foundational genetic research or the development of novel antimicrobial therapies.
Direct Comparison of Delivery System Performance
The table below summarizes key performance metrics for phage-based and conjugative plasmid delivery systems, based on recent experimental findings.
Table 1: Performance Comparison of Phage-Delivered CRISPR vs. Conjugate Plasmid Delivery
| Performance Metric | Phage-Delivered CRISPR | Conjugative Plasmid Delivery | Experimental Context |
|---|---|---|---|
| Editing Efficiency | >50% of targeted population [6] | 4.7% to 100% [8] | In vitro bacterial monoculture editing. |
| Delivery Specificity | High (Leverages natural phage tropism and engineered tail fibers) [20] | Moderate (Dependent on bacterial conjugation compatibility) [8] | Targeting E. coli within a mixed microbial community [6] [20]. |
| Payload Capacity | Large (Capable of delivering CRISPR-guided transposases like DART, ~10+ kb) [6] | Large (Can deliver complex CRISPR-Cas systems) [8] | Delivery of the DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) system [6]. |
| In Vivo Stability & Efficacy | Reduces bacterial burden in mouse gut; well-tolerated in mouse and minipig models [20] | Efficacy can be limited by plasmid stability and host compatibility in complex environments [29] | Prophylactic use against E. coli in immunocompromised mouse models [20]. |
| Key Advantages | • High specificity and efficiency• Self-replicating within host• Can penetrate biofilms [20] | • Broadly applicable across susceptible strains• High efficiency in permissive conditions [8] |
Detailed Experimental Protocols and Data
Protocol for Phage-Delivered CRISPR (λ-DART System)
The engineering and application of phage λ to deliver CRISPR-associated transposases (DART) involves a multi-stage process, as detailed below [6].
1. Phage Engineering:
- Method: Homologous recombination in E. coli coupled with Cas13a-based counterselection.
- Procedure: The ~10 kb DART system, which incorporates a type I-F CRISPR-associated transposon (CAST), is inserted into the genome of phage λ via homologous recombination. To isolate successfully recombined phage, a CRISPR-Cas13a system targeting the wild-type λ sequence is employed. Cas13a's RNA-guided, trans-cleavage activity induces host dormancy, effectively counterselecting against unmodified phage and enriching for the engineered λ-DART.
- Key Modification: The engineered λ phages are made nonlysogenic by removing components essential for lysogeny, ensuring a lytic cycle and preventing persistent phage maintenance in the host [6].
2. Bacterial Infection and Editing Assay:
- Host Strain: E. coli (e.g., BW25113 or amber-suppressor strain LE392MP).
- Infection Conditions: Phage infection is conducted at a controlled temperature (37°C to induce the lytic cycle in phages with the cI857 mutation) and at varying multiplicities of infection (MOI).
- Efficiency Assessment: Editing efficiency is quantified by measuring the percentage of the population that acquires the desired gene knockouts or insertions, often via antibiotic resistance markers or phenotypic assays. In mixed community contexts, specificity is confirmed by assessing editing only in the targeted E. coli strain [6].
Protocol for Conjugative Plasmid Delivery
The delivery of CRISPR-Cas systems via conjugative plasmids is a well-established method for bacterial genome editing [8] [29].
1. Donor-Recipient Setup:
- Donor Strain: A laboratory strain (e.g., E. coli S17-1) containing the conjugative plasmid that carries the CRISPR-Cas machinery and a CRISPR RNA (crRNA) array targeting specific bacterial genes (e.g., β-lactamase
blagenes or colistin resistancemcr-1). - Recipient Strain: The target bacterial strain possessing the AMR genes to be disrupted.
2. Conjugation Process:
- Procedure: Donor and recipient bacteria are mixed on a filter placed on solid media or combined in liquid broth, typically at a donor-to-recipient ratio between 1:1 and 1:10.
- Incubation: The mixture is incubated for several hours (often 4-24 hours) to allow for cell-to-cell contact and plasmid transfer.
- Selection: The mixture is then plated on selective media containing antibiotics that inhibit the donor strain and select for recipient cells that have successfully received the plasmid.
3. Efficiency Assessment:
- Calculation: Conjugation efficiency is reported as the number of transconjugants (successful recipients) per donor cell. The efficacy of the CRISPR-Cas system is measured by the reduction in viable counts of the recipient strain or by the restoration of antibiotic susceptibility, with reported efficiencies ranging from 4.7% to 100% [8].
Critical Factor Analysis: Promoter Selection and In Vivo Stability
Promoter Selection for Enhanced Activity
The choice of promoter driving the expression of CRISPR-Cas components is a critical determinant of editing efficiency, especially in challenging in vivo conditions like the gut or within biofilms.
Table 2: Comparison of Promoter Performance in Phage-Delivered CRISPR
| Promoter | Proposed Function/Context | Reported Performance |
|---|---|---|
| PbolA | Stress-induced promoter; active in slow-growing or non-dividing cells and biofilms [20]. | Superior performance in biofilms and under restricted bacterial growth conditions; significant reduction in metabolic activity of E. coli biofilms [20]. |
| PrelB | Constitutive-like promoter, often used for stable expression in standard lab conditions [20]. | Effective under standard planktonic growth conditions (LB, 37°C), but outperformed by PbolA in biofilms and other complex environments [20]. |
Experimental data from engineered phages demonstrates that driving CRISPR-Cas system expression with the PbolA promoter resulted in significantly greater killing efficacy in E. coli biofilms compared to the PrelB promoter. This establishes PbolA as the preferred choice for applications targeting bacteria within complex, slow-growth environments similar to in vivo states [20].
Engineering for In Vivo Stability
Both delivery systems require engineering to ensure stability and function within a live host.
Phage Stability Enhancements:
- Tail Fiber Engineering: To combat the evolution of phage-resistant bacteria, the receptor-binding adhesins of phage tail fibers can be engineered. For example, a Tsx-binding adhesin from one phage was engineered into another, creating a phage capable of using two different bacterial surface receptors. This dual-affinity approach substantially reduced the number of bacterial survivors in challenge assays [20].
- Control of Lifecycle: Incorporating mutations like
Sam7(an amber mutation in the lysis gene S) confines productive infection and lytic spread to specific permissive hosts, enhancing control and safety in complex environments [6].
Conjugative Plasmid Stability:
- The stability of conjugative plasmids in vivo can be variable and is highly dependent on the compatibility between the donor and recipient strains. A significant limitation is that conjugative delivery requires direct cell-to-cell contact and functional conjugation machinery, which may not be efficiently maintained in the complex and dynamic environment of a host organism [29].
Research Reagent Solutions
The table below lists key reagents and their functions for implementing the discussed delivery systems.
Table 3: Essential Research Reagents for Phage and Plasmid Delivery Systems
| Reagent/Solution | Function in Research |
|---|---|
| Phage λ with cI857 & Sam7 mutations | Temperature-controlled lytic cycle; host-range restricted delivery chassis for CRISPR components [6]. |
| CRISPR-associated Transposase (DART) System | All-in-one system for CRISPR-guided DNA transposition; enables large insertions and gene knockouts [6]. |
| Cas13a Protein & crRNA | Provides counterselection capability during phage engineering via RNA-targeting and trans-cleavage activity [6]. |
| Conjugative Plasmid with CRISPR-Cas9/Cas3 | Vehicle for delivering CRISPR machinery via bacterial mating; used for gene disruption and plasmid curing [8] [29]. |
| Amber-Suppressor E. coli Strain (e.g., LE392MP) | Permissive host for propagating and titrating phages with amber mutations (e.g., Sam7) [6]. |
| Lipid Nanoparticles (LNPs) | An alternative delivery vehicle for in vivo CRISPR therapy, particularly for liver accumulation, allowing for systemic administration and potential redosing [44]. |
Experimental Workflow and System Mechanism
The following diagrams illustrate the core workflows and mechanisms of the two delivery systems.
The therapeutic application of CRISPR-Cas technology is fundamentally constrained by the challenge of delivering genome-editing machinery to target cells efficiently and safely. Two primary delivery strategies have emerged as promising vehicles for bacterial genome engineering: phage-based delivery and conjugate plasmid delivery. These systems represent divergent approaches with distinct advantages and limitations for research and therapeutic development. Phage delivery leverages the natural infectious machinery of bacteriophages to introduce CRISPR components into specific bacterial hosts, while conjugate plasmid delivery utilizes engineered plasmids that transfer between bacterial cells via direct contact. This guide provides an objective comparison of these technologies, focusing on their scalability, immune responses, and off-target effects, with supporting experimental data from current research.
The fundamental difference between these delivery systems lies in their mechanism of cellular entry and genetic cargo delivery. Phage-delivered CRISPR systems package editing machinery into bacteriophage capsids, which leverage natural phage infection pathways to introduce CRISPR components into specific bacterial hosts. In contrast, conjugate plasmid delivery relies on engineered plasmids that facilitate direct cell-to-cell transfer through specialized conjugation machinery, allowing the CRISPR system to spread horizontally through a bacterial population.
Table 1: Fundamental Characteristics of Phage and Conjugate Plasmid Delivery Systems
| Characteristic | Phage-Delivered CRISPR | Conjugate Plasmid Delivery |
|---|---|---|
| Delivery Mechanism | Viral capsid fusion with cell membrane and DNA injection [6] [27] | Direct cell-to-cell transfer via conjugation machinery [8] |
| Primary Applications | Targeted bacterial genome editing, microbiome engineering, antimicrobial therapy [6] [27] | Bacterial strain resensitization, antimicrobial resistance reversal [8] |
| Key Advantage | Natural target specificity, high transduction efficiency, biofilm penetration [6] [27] | Broad host range compatibility, self-disseminating capability [8] |
| Editing Persistence | Transient (lytic phage) or stable (lysogenic phage) expression [6] | Stable plasmid maintenance with continuous expression [8] |
| Payload Capacity | ~10-15 kb for λ phage variants [6] | Virtually unlimited through plasmid design [8] |
Figure 1: Comparative delivery pathways for phage and conjugate plasmid CRISPR systems. Phage delivery typically results in cell lysis, while conjugate plasmids enable continuous spread and maintenance.
Quantitative Efficiency Comparison
Direct comparison of editing efficiency reveals significant differences between these delivery modalities. Phage-delivered CRISPR systems demonstrate remarkably high efficiency in mixed microbial communities, while conjugate plasmid systems show more variable but still substantial efficacy across bacterial strains.
Table 2: Experimental Efficiency Data for Phage vs. Conjugate Plasmid Delivery
| Delivery System | Specific Technology | Target Organism | Editing Efficiency | Key Experimental Findings |
|---|---|---|---|---|
| Phage-Delivered CRISPR | λ-DART phage [6] | E. coli in monoculture | >50% | Efficient gene knockouts and insertions achieved |
| Phage-Delivered CRISPR | λ-DART phage [6] | E. coli in mixed community | >50% | Specific editing maintained despite competition |
| Conjugate Plasmid Delivery | Plasmid vectors [8] | E. coli and other Enterobacterales | 4.7%-100% | High variability depending on target gene |
| Conjugate Plasmid Delivery | Plasmid vectors [8] | Multiple resistant pathogens | ~90% (average) | Effective resensitization to antimicrobials |
Recent research with engineered λ-DART phages demonstrates how phage delivery achieves high efficiency. When applied to monocultures and mixed bacterial communities comprising three genera, these modified phages led to efficient, precise, and specific gene knockouts and insertions in the targeted E. coli cells, achieving editing efficiencies surpassing 50% of the population [6]. The efficiency remained consistently high even in complex microbial environments, highlighting the targeting specificity of phage delivery systems.
In contrast, conjugate plasmid delivery shows remarkable breadth but inconsistent efficiency across studies. A comprehensive review of CRISPR-Cas9 delivery via conjugative plasmids reported efficacy ranging from 4.7% to 100% in resensitizing bacterial strains to various antimicrobials [8]. The extreme variability highlights how conjugate plasmid efficiency depends heavily on factors including bacterial strain, target gene, and specific plasmid design.
Manufacturing and Scalability Assessment
Scalability and manufacturing considerations present distinct challenges for both delivery systems, impacting their translational potential and practical application.
Phage Manufacturing Complexities
Phage-based CRISPR delivery systems face significant scaling challenges due to their biological nature and specificity requirements:
- Production Complexity: Engineered phage variants like λ-DART require sophisticated host systems (e.g., amber-suppressor E. coli strains) for propagation, adding layers of complexity to manufacturing [6].
- Purification Challenges: Removal of host cell contaminants and endotoxins requires multiple purification steps, increasing production costs and limiting scale-up potential.
- Titration Standardization: Phage titer quantification lacks standardized methods across laboratories, creating reproducibility challenges for clinical translation [27].
Conjugate Plasmid Production
Plasmid-based systems benefit from more established manufacturing frameworks but face their own limitations:
- Fermentation-Based Production: Large-scale plasmid DNA production utilizes bacterial fermentation, a well-established but costly process requiring specialized equipment [45].
- Purification Requirements: Plasmid purification must eliminate genomic DNA, RNA, and protein contaminants, requiring multiple chromatography steps [45].
- Storage and Stability: Plasmid DNA demonstrates superior shelf-life stability compared to phage preparations, simplifying storage and distribution logistics.
Immune Response and Safety Profiles
The safety profiles of these delivery systems diverge significantly, particularly regarding host immune recognition and potential toxicities.
Immune Recognition of Phage Delivery
Phage delivery systems trigger complex immune interactions that impact both safety and efficacy:
- Bacterial Defense Activation: CRISPR-Cas systems naturally function as bacterial immune defenses against phage infection, creating potential resistance mechanisms that may limit editing efficiency [46].
- Anti-CRISPR Responses: Phages are vulnerable to anti-CRISPR proteins produced by bacteria, which can inhibit the CRISPR-Cas system's functionality [46].
- Nutrient-Dependent Efficacy: Research demonstrates that phage-delivered CRISPR efficacy can be modulated by environmental conditions, with complete protection against some phages only occurring in low-nutrient conditions [46].
Plasmid-Induced Immune Responses
Conjugate plasmid systems present different immune challenges:
- Horizontal Gene Transfer Concerns: Conjugative plasmids may potentially transfer to non-target bacterial species, raising safety concerns about unintended genetic dissemination [8].
- Mobile Element Interference: Natural plasmid incompatibility groups can interfere with engineered conjugate plasmids, limiting their stability and spread in certain bacterial hosts.
Figure 2: Immune recognition pathways affecting phage and conjugate plasmid delivery systems. Both face distinct cellular defense mechanisms that can impact editing efficiency.
Off-Target Editing Analysis
Precision and specificity represent critical safety parameters for CRISPR delivery systems. Both phage and conjugate plasmid delivery demonstrate generally high specificity, though through different mechanisms.
Phage-delivered CRISPR systems achieve high specificity through dual targeting mechanisms. First, phage host range naturally limits editing to specific bacterial species or strains. Second, CRISPR-Cas systems themselves provide sequence-specific targeting. Research with λ-DART phages demonstrated precise gene integrations in targeted E. coli cells within mixed communities with minimal off-target effects on non-target species [6].
Conjugate plasmid systems achieve specificity through conjugative transfer machinery that typically functions between related bacterial species. The CRISPR-Cas system then provides additional sequence-specific targeting within recipient cells. Studies show that conjugative plasmids resensitize specific bacterial strains to antimicrobials by precisely targeting resistance genes, indicating high on-target activity [8].
Detailed Experimental Protocols
Phage-Delivered CRISPR Workflow
The development and implementation of engineered phage CRISPR systems involves a multi-stage process:
Phase 1: Phage Engineering
- Vector Design: CRISPR-Cas transposase (DART) system is engineered into λ phage genome using homologous recombination [6].
- Counterselection: Cas13a-based counterselection isolates successfully modified phage variants [6].
- Validation: Engineered λ-DART phages are sequenced and functionally validated for cargo delivery.
Phase 2: Delivery and Editing
- Host Preparation: Target bacterial cultures are grown to mid-log phase (OD~600 = 0.4-0.6) in appropriate media.
- Infection Parameters: Phages are applied at optimized multiplicity of infection (MOI) ranging from 0.1 to 10 [6].
- Incubation: Cultures are incubated for 6-24 hours to allow for complete editing cycles.
- Analysis: Editing efficiency is quantified via selective plating, sequencing, or functional assays.
Phase 3: Specificity Validation
- On-target Verification: PCR amplification and sequencing of target loci confirm precise editing.
- Off-target Screening: Whole-genome sequencing of edited strains identifies potential off-target effects.
- Community Impact: In mixed cultures, species-specific probes quantify editing specificity.
Conjugate Plasmid Delivery Protocol
Conjugate plasmid delivery follows a different experimental pathway:
Phase 1: Donor-Recipient Setup
- Donor Strain Preparation: E. coli donor strains harboring conjugative CRISPR plasmids are cultured with selective antibiotics [8].
- Recipient Strain Preparation: Target bacterial recipients are cultured without selection pressure.
- Mixing Ratio Optimization: Donor and recipient strains are mixed at ratios typically between 1:1 and 1:10.
Phase 2: Conjugation Process
- Filter Mating: Donor and recipient cells are mixed, concentrated on filters, and incubated on non-selective media for 2-24 hours [8].
- Liquid Mating: Alternative approach where strains are mixed in liquid culture with mild agitation.
- Selection: Transconjugants are selected using appropriate antibiotic resistance markers.
Phase 3: Editing Analysis
- Efficiency Quantification: Transconjugant counts are compared to recipient counts to determine conjugation efficiency.
- Functional Assessment: Antimicrobial susceptibility testing verifies resensitization phenotype [8].
- Stability Testing: Edited strains are passaged without selection to assess genetic stability.
Essential Research Reagents
Successful implementation of these delivery systems requires specific reagent systems and specialized materials.
Table 3: Essential Research Reagents for Phage and Conjugate Plasmid Studies
| Reagent Category | Specific Examples | Function | Applications |
|---|---|---|---|
| Engineering Tools | Cas13a counterselection system [6] | Precise phage genome editing | λ-DART phage construction |
| Engineering Tools | Homologous recombination systems [27] | Insert foreign DNA into phage genomes | Initial phage vector development |
| Bacterial Strains | Amber-suppressor E. coli (e.g., LE392MP) [6] | Propagate engineered phage with Sam7 mutation | Phage production and titration |
| Bacterial Strains | Conjugation-proficient donors (e.g., S17-1) | Enable plasmid transfer to recipients | Conjugate plasmid delivery assays |
| Selection Agents | Antibiotic resistance markers (kanamycin, ampicillin) | Select for successful transconjugants | Both delivery systems |
| Selection Agents | Phage resistance mutations (e.g., Sam7) [6] | Control phage infection timing | Phage delivery optimization |
| Delivery Vectors | Conjugative plasmids with mobilizable origins | Enable interbacterial DNA transfer | Conjugate plasmid systems [8] |
| Delivery Vectors | Engineered phage genomes (e.g., λ-DART) [6] | Package and deliver CRISPR machinery | Phage editing systems |
Phage-delivered CRISPR and conjugate plasmid systems offer complementary strengths for bacterial genome editing. Phage systems provide superior targeting specificity and efficiency in complex microbial communities, achieving >50% editing efficiency even in mixed-species environments [6]. Conversely, conjugate plasmid delivery demonstrates remarkable versatility across bacterial species, with efficacy ranging from 4.7% to 100% depending on the target organism and genetic context [8].
The manufacturing and safety profiles of these systems present distinct considerations for research and therapeutic development. Phage systems face scaling challenges due to their biological complexity but offer natural targeting specificity that minimizes off-target effects. Conjugate plasmid systems benefit from more established DNA production methodologies but raise concerns about horizontal gene transfer and broader ecological impact.
Selection between these technologies should be guided by specific application requirements: phage delivery excels in precision editing of specific bacterial targets within complex communities, while conjugate plasmids offer broader coverage across bacterial populations. Future developments in phage engineering and plasmid design will likely address current limitations in scalability and immune recognition, further enhancing the translational potential of both delivery platforms.
The rise of multidrug-resistant (MDR) bacteria poses a critical challenge to global public health, driving the urgent need for precision antimicrobial strategies that can target specific pathogens without disrupting commensal microbiota [47] [48]. Two innovative platforms have emerged as particularly promising for this purpose: bacteriophage (phage)-based delivery systems and conjugative plasmid vectors. These systems enable the precise introduction of CRISPR-Cas machinery into target bacteria, facilitating the selective elimination of antimicrobial resistance (AMR) genes or the disruption of essential bacterial functions [49] [8]. Phage-delivered CRISPR systems leverage the natural specificity of bacteriophages—viruses that infect bacteria—to deliver genetic cargo to particular bacterial strains or species [20] [50]. In contrast, conjugative plasmids utilize bacterial mating mechanisms to transfer DNA between cells, offering a broader host range within a bacterial population [51]. Understanding the relative efficiencies, specificities, and practical applications of these platforms is essential for advancing the field of precision antimicrobials and developing effective therapies against MDR pathogens.
Phage-Delivered CRISPR Systems: Mechanisms and Workflows
Phage-delivered CRISPR systems represent a sophisticated synergy of viral delivery and programmable gene editing. This approach typically involves engineering temperate or lytic phages to carry CRISPR-Cas components, which can include Cas nucleases and guide RNA arrays targeting specific bacterial genes [6] [20]. The process begins with the natural infection cycle of the bacteriophage, which initiates with the specific binding of phage tail fibers to bacterial surface receptors such as lipopolysaccharides (LPS) or outer membrane proteins (e.g., LamB, Tsx) [20] [48]. Following adsorption, the phage injects its genetic material into the bacterial cell, delivering the engineered CRISPR system. Once inside, the CRISPR-Cas machinery becomes active and directs sequence-specific cleavage of target DNA, which may include antibiotic resistance genes or essential bacterial genomic regions [49] [5]. This DNA damage typically leads to bacterial cell death by overwhelming the host's repair mechanisms [20].
Recent advances have enhanced the specificity and efficacy of phage-delivered CRISPR systems. For instance, researchers have developed CRISPR-associated transposase (DART) systems embedded in phage genomes, enabling both gene disruption and large DNA insertions [6]. Additionally, tail fiber engineering has been employed to expand or alter phage host range, combining adhesins from different phages to create variants capable of targeting multiple bacterial receptors [20]. The selection of promoters active in specific bacterial growth conditions (e.g., PbolA for biofilm populations) has further improved performance in complex environments [20].
Figure 1. Workflow of phage-delivered CRISPR systems, from engineering to bacterial cell death. The process begins with phage modification to incorporate CRISPR-Cas components, followed by specific stages of bacterial infection and lethal DNA targeting.
Conjugative Plasmid Delivery: Mechanisms and Workflows
Conjugative plasmid delivery offers an alternative mechanism for introducing CRISPR-Cas systems into bacterial populations. This approach leverages natural bacterial mating processes to transfer genetic material from donor to recipient cells [51]. The conjugation process initiates when donor bacteria detect pheromones or signaling molecules secreted by recipient cells, triggering the synthesis of aggregation substances that facilitate direct cell-to-cell contact [51]. A key advantage of conjugative plasmids is their capacity to transfer large genetic payloads, including entire CRISPR-Cas systems with multiple guide RNAs, making them suitable for multiplexed targeting of various genes or pathways [51].
The conjugative transfer process is followed by the establishment of the CRISPR-Cas system in the recipient cell, where it can employ different mechanisms for eliminating target bacteria. When targeting antibiotic resistance genes, the system introduces double-strand breaks in plasmids or chromosomes carrying resistance determinants, effectively resensitizing bacteria to antibiotics [51]. For essential gene targeting, the CRISPR-Cas system can disrupt vital bacterial functions, leading to direct cell death. Additionally, the conjugative plasmid itself can encode bacteriocins or other lethal factors that enhance the elimination of non-target bacteria, providing a dual selection mechanism [51].
Figure 2. Workflow of conjugative plasmid delivery for CRISPR-Cas systems. The process involves plasmid design, bacterial mating, and subsequent CRISPR-mediated targeting of antibiotic resistance genes or essential bacterial functions.
Comparative Efficiency Analysis: Quantitative Data
Direct comparison of delivery efficiency between phage and conjugation systems reveals distinct performance profiles across multiple parameters. The following tables summarize key quantitative findings from recent studies, highlighting the strengths and limitations of each approach.
Table 1: Editing Efficiency and Specificity in Different Environments
| Delivery Method | Editing Efficiency | Target Bacteria | Specificity | Mixed Community Performance | Reference |
|---|---|---|---|---|---|
| Phage λ-DART | >50% editing efficiency in targeted E. coli | Escherichia coli | High (strain-level) | Efficient and precise editing in mixed communities of three genera | [6] |
| Conjugative Plasmid pKH88 | Reduced erythromycin-resistant E. faecalis by several orders of magnitude | Enterococcus faecalis | Moderate (species-level) | Effective in murine intestinal microbiota | [51] |
| SNIPR001 Phage Cocktail | Significant reduction in E. coli burden in mouse gut | Escherichia coli | High (strain-level) | Selective killing without major microbiota disruption | [20] |
| Conjugative Delivery | 4.7%-100% resensitization efficiency range | Various Enterobacterales | Varies with conjugation efficiency | Dependent on donor-recipient compatibility | [8] |
Table 2: Practical Implementation Characteristics
| Parameter | Phage Delivery | Conjugative Plasmid Delivery |
|---|---|---|
| Host Range | Narrow (strain/species-specific) [20] | Broader (often genus-level) [51] |
| Payload Capacity | Limited by phage head size [6] | Larger capacity for multiple cassettes [51] |
| Delivery Speed | Rapid (minutes to hours post-infection) [48] | Slower (requires cell growth and conjugation) [51] |
| Resistance Development | Receptor mutations common [20] | Restriction systems can limit transfer [51] |
| Regulatory Considerations | Complex (biologics framework) [48] | More established (antibiotic framework) [51] |
Experimental Protocols for Efficiency Assessment
Phage Delivery Efficiency Protocol
Objective: Quantify the editing efficiency of phage-delivered CRISPR systems in target bacteria within mixed microbial communities.
Phage Engineering:
- Modify phage λ using homologous recombination coupled with Cas13a-based counterselection to embed the DART (DNA-editing all-in-one RNA-guided CRISPR-Cas transposase) system, creating λ-DART phages [6].
- For enhanced targeting, engineer phage tail fibers by combining adhesins from different phages (e.g., Tsx-binding adhesin from phage α17 into phage α15) to expand receptor tropism [20].
Infection and Editing:
- Culture target bacteria (e.g., E. coli) in monoculture or in mixed communities with other genera (e.g., 3-species model communities).
- Infect cultures with engineered λ-DART phages at varying multiplicities of infection (MOIs: 0.1, 1, 10) and incubate at 37°C for 6-24 hours [6].
- For in vivo assessment, administer SNIPR001 (a 4-phage cocktail) to mouse models and quantify E. coli burden in the gut over 2-5 days [20].
Efficiency Quantification:
- Plate bacterial dilutions on selective media to determine viable counts.
- Use PCR and sequencing to verify precise gene knockouts or insertions in surviving colonies.
- Calculate editing efficiency as: (number of edited colonies / total surviving colonies) × 100% [6].
Conjugative Plasmid Delivery Efficiency Protocol
Objective: Assess the ability of conjugative plasmids to deliver CRISPR-Cas systems and eliminate antibiotic resistance in target bacterial populations.
Plasmid Construction:
- Engineer pheromone-responsive plasmids (PRPs) like pPD1 to encode Cas9 and guide RNAs under constitutive promoters (e.g., bacA promoter) targeting specific antibiotic resistance genes (e.g., ermB, tetM) [51].
- Include selective markers (e.g., chloramphenicol resistance) for donor selection and bacteriocin genes (e.g., bac-21) to enhance plasmid maintenance.
Conjugation Assay:
- Mix donor strain (e.g., E. faecalis CK135 carrying engineered pPD1) and recipient strain (e.g., E. faecalis OG1SSp with target resistance plasmid) at approximately 1:1 ratio.
- Co-culture on solid media (BHI agar) or in broth for 16-24 hours to allow conjugation [51].
- For in vivo assessment, colonize mice with donor and recipient strains and track plasmid transfer and resistance elimination in the intestinal tract over 3-7 days.
Efficiency Assessment:
- Plate serial dilutions on media containing appropriate antibiotics to enumerate total recipients, transconjugants, and resistant populations.
- Calculate conjugation frequency as: (number of transconjugants / total recipients).
- Determine resistance elimination efficiency as: 1 - (resistant transconjugants / total transconjugants) × 100% [51].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Phage and Conjugative Delivery Systems
| Reagent Category | Specific Examples | Function/Application | Source/Reference |
|---|---|---|---|
| Engineered Phages | λ-DART phages, SNIPR001 components (α15, α17, α20, α31) | CRISPR-Cas delivery with large insertion capability or specific bacterial killing | [6] [20] |
| Conjugative Plasmids | pPD1 derivatives (pKH88[sp-ermB], pKH88[sp-tetM]) | High-efficiency delivery of CRISPR-Cas to Enterococcus species | [51] |
| CRISPR Systems | Type I-E CRISPR-Cas (EcCas3), Type I-F CAST, Type II Cas9 | Programmable DNA targeting and cleavage | [6] [20] [51] |
| Bacterial Strains | E. coli BW25113, E. faecalis OG1SSp, E. faecalis V583 | Model strains for efficiency testing and in vivo models | [6] [51] |
| Specialized Promoters | PbolA, PrelB | Conditional CRISPR expression in biofilms or slow-growth conditions | [20] |
| Animal Models | Mouse intestinal colonization models, Minipig models | In vivo efficacy and safety assessment | [20] [51] |
The comparative analysis of phage-delivered CRISPR systems and conjugative plasmid delivery reveals complementary strengths that may be strategically leveraged for different therapeutic scenarios. Phage systems offer superior specificity for targeted elimination of particular bacterial strains within complex communities, making them ideal for precision editing applications [6] [20]. Conversely, conjugative plasmids provide broader population-level coverage within a bacterial species, making them potentially more effective for decolonization scenarios where eliminating an entire resistant population is desired [51].
Future developments in this field will likely focus on hybrid approaches that combine the advantages of both systems, such as engineering phages with expanded host ranges or designing conjugative systems with enhanced specificity. Additionally, addressing delivery challenges through nanoparticle formulations or chemical modifications may further improve the stability and efficacy of both platforms [8]. As these technologies mature, they hold tremendous promise for revolutionizing our approach to combating antimicrobial resistance, potentially enabling a new class of precision antimicrobials that can selectively eliminate problematic bacteria while preserving beneficial microbiota. The ongoing clinical development of phage-based therapies like SNIPR001 represents an important milestone in this journey from laboratory concept to clinical reality [20].
Efficacy and Comparison: Validating Performance in Models
The development of effective CRISPR-based antimicrobials and microbiome editing tools hinges on one critical factor: the efficient delivery of CRISPR machinery to target bacterial cells. Two primary delivery methods have emerged at the forefront of this research: bacteriophage-mediated delivery and conjugative plasmid transfer. Understanding the quantitative efficiency of these methods is essential for selecting appropriate strategies for specific research or therapeutic applications. This guide provides an objective comparison of these technologies, focusing on direct experimental metrics and methodologies to inform researchers and drug development professionals.
Table 1: Core Characteristics of Delivery Modalities
| Feature | Phage-Delivered CRISPR | Conjugative Plasmid Delivery |
|---|---|---|
| Primary Mechanism | Viral infection and DNA injection | Bacterial mating pilus formation and DNA transfer |
| Natural Target Specificity | High (species/strain-level, often using chromosomally-encoded receptors) [52] [20] | Lower (dependent on plasmid host range and recipient compatibility) [53] |
| Delivery Speed | Minutes to hours (single infection cycle) | Hours (requires establishment in recipient) |
| Cargo Capacity | Limited by phage capsid size (can be a constraint for large Cas proteins) [6] | Large (can deliver entire CRISPR systems and donor DNA) [53] |
| Quantitative Efficiency Metric | Infectivity Rate, Plaque-Forming Units (PFU), Editing Efficiency in Population | Conjugation Frequency (transconjugants per recipient), Editing Efficiency |
Quantitative Efficiency Data
Direct, head-to-head comparisons of phage and conjugation delivery within a single study are rare. However, data from independent studies on each method provide strong indicators of their performance. The table below summarizes key quantitative findings from recent high-impact research.
Table 2: Experimental Efficiency Metrics from Recent Studies
| Study & Delivery Method | Target Bacteria | Experimental Editing Efficiency | Key Quantitative Metric |
|---|---|---|---|
| Phage λ with DART (CAST System) [6] | E. coli in monoculture and a mixed community | >50% of targeted population | Editing efficiency surpassing 50% |
| Engineered CAPs (SNIPR001) [20] | E. coli in mouse gut | Significant reduction in bacterial load | Better reduction than wild-type phages |
| Conjugative Plasmid with CRISPRi [53] | Recombinant and clinical E. coli isolates | Successful re-sensitization; conjugation efficiency is a prerequisite | Conjugative delivery achieved in 75% of a 82-strain panel |
Detailed Experimental Protocols
To ensure the reproducibility of the quantified efficiencies, the following sections detail the core methodologies employed in the cited studies.
Protocol for Phage-Delivered CRISPR Efficiency
This protocol is adapted from studies engineering phage λ to deliver CRISPR-associated transposases (DART system) [6].
- 1. Phage Engineering: The phage genome is modified using homologous recombination in an E. coli host. A Cas13a-based counterselection system is employed to efficiently isolate successfully recombined phages by exploiting Cas13a's ability to induce dormancy upon detecting unmodified phage RNA.
- 2. Phage Propagation & Quantification: Engineered phages are propagated in a permissive, amber-suppressor host strain (e.g., E. coli LE392) to control the lytic cycle via the Sam7 mutation. Phage lysates are titrated using a standard double-layer agar method to determine the plaque-forming unit (PFU) concentration, which defines the infectious dose.
- 3. Infection & Editing Assay: Target bacteria (e.g., a monoculture of E. coli or a defined mixed-species community) are infected with the engineered phage at a specific Multiplicity of Infection (MOI). The culture is incubated under appropriate conditions (e.g., 37°C with aeration) for a set period to allow for infection, CRISPR machinery delivery, and editing.
- 4. Efficiency Quantification: After incubation, the bacterial population is harvested. Editing efficiency is quantified by:
- For gene knockouts: Plating dilutions on selective media and calculating the percentage of antibiotic-resistant colonies.
- For insertions: Using flow cytometry or PCR-based assays on the population to determine the percentage of cells carrying the desired genetic modification.
Protocol for Conjugative Plasmid Delivery Efficiency
This protocol is based on methods used to deliver CRISPRi systems for antibiotic re-sensitization [53].
- 1. Donor Strain Preparation: A donor E. coli strain (e.g., a laboratory strain with a conjugation-efficient background) is transformed with the conjugative plasmid carrying the CRISPR machinery (e.g., dCas9 and gRNA expression cassettes).
- 2. Conjugation Assay: Donor and recipient bacterial strains are grown separately to mid-log phase. They are then mixed at a standardized ratio (e.g., 1:1 donor-to-recipient) on a solid filter membrane placed on non-selective agar. This facilitates cell-to-cell contact.
- 3. Mating & Selection: The mating mixture is incubated for a set period (typically 1-2 hours) to allow for conjugation. The cells are then removed from the filter, diluted, and plated on selective media that only allows the growth of transconjugants (successful recipient cells). The media contains antibiotics that counter-select against the donor strain and select for the plasmid marker in the recipient.
- 4. Efficiency Calculation: Conjugation frequency is calculated as the number of transconjugant colonies divided by the number of recipient cells at the start of the mating period. The functional editing efficiency (e.g., rate of antibiotic re-sensitization) among the transconjugants is then assessed through subsequent experiments, such as determining the minimum inhibitory concentration (MIC).
Diagram Title: Experimental Workflows for Delivery Methods
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and their functions, as utilized in the studies cited in this guide, to facilitate experimental design.
Table 3: Key Research Reagent Solutions
| Reagent / Tool | Function in Research | Example Use Case |
|---|---|---|
| Temperate Phage λ (e.g., λ cI857 Sam7) | Engineerable viral delivery chassis; Sam7 mutation allows controlled lysis in amber-suppressor hosts [6]. | Controlled delivery of CRISPR-guided transposases (DART) to E. coli [6]. |
| CRISPR-associated Transposase (DART) | All-in-one system for RNA-guided DNA insertion, delivered by phage to enable large genetic edits [6]. | Phage-mediated gene knockouts and insertions in mixed microbial communities [6]. |
| Type I-E CRISPR-Cas System (from E. coli) | A multi-protein Cas effector complex used for targeted bacterial killing when delivered by phage [20]. | Arming CAPs (CRISPR-Cas Armed Phages) in the SNIPR001 cocktail to selectively kill E. coli [20]. |
| dead Cas9 (dCas9) | Catalytically inactive Cas9 used for gene repression (CRISPRi) without cleaving DNA, reducing escape mutant risk [53]. | Delivery via conjugation to repress antibiotic resistance genes (ARGs) and re-sensitize bacteria [53]. |
| Conjugative Plasmid (e.g., IncP, IncF groups) | Self-transmissible DNA vector that enables transfer of CRISPR cargo from donor to recipient bacteria [52] [53]. | Delivering dCas9 and gRNA constructs to clinical isolates for antibiotic re-sensitization [53]. |
| Amber-Suppressor E. coli Strain (e.g., LE392) | Permissive host for propagating phage with amber mutations (e.g., Sam7), controlling lytic activity [6]. | Production of high-titer lysates of engineered λ phages for infection assays [6]. |
Both phage-mediated and conjugation-based delivery methods for CRISPR systems demonstrate significant efficacy in laboratory settings, albeit with distinct performance profiles and applications. Phage delivery excels in its high, specific infectivity rates and is particularly powerful for targeted editing within complex microbial communities. Conjugative plasmid delivery offers a robust and broad-host-range mechanism for delivering large CRISPR payloads to a wide array of recipient cells. The choice between these methods should be guided by the specific experimental or therapeutic goals, including the target bacterium, required cargo size, desired community context, and the need for temporal control over delivery.
The selective manipulation of bacterial populations within complex microbial communities is a cornerstone of modern microbiome research and therapeutic development. Two powerful methods for achieving targeted bacterial killing have emerged: phage-delivered CRISPR-Cas systems and conjugative plasmid delivery. Both leverage the precision of CRISPR nucleases to induce lethal double-strand breaks in bacterial chromosomes, but their delivery mechanisms and performance in co-culture environments differ significantly. This guide provides an objective comparison of these technologies, focusing on their killing efficiency as measured by log-reduction in bacterial load, with supporting experimental data from key studies. Understanding the comparative strengths of each approach is critical for researchers selecting the optimal strategy for specific applications, from microbiome editing to combating antimicrobial-resistant pathogens.
The table below summarizes the core characteristics and reported efficacies of phage-delivered CRISPR and conjugative plasmid systems.
Table 1: Key Characteristics and Performance Metrics of Bacterial Killing Technologies
| Feature | Phage-Delivered CRISPR | Conjugative Plasmid Delivery |
|---|---|---|
| Delivery Vector | Engineered bacteriophages (e.g., λ phage, T-even phages) [6] [20] | Conjugative plasmids (e.g., IncP RK2-based) [54] |
| Primary Mechanism | Viral infection and injection of CRISPR-Cas payload [6] | Bacterial mating pilus formation and DNA transfer [54] |
| Targeting Specificity | High (species/strain-level via phage receptor binding) [6] [20] | Moderate to High (broad-host-range plasmids can be tailored with specific sgRNAs) [54] |
| Reported Killing Efficiency (Log-Reduction) | 1–6 log10 CFU mL-1 [20] | Up to ~3.5 log10 CFU mL-1 [54] |
| Key Advantage | Narrow targeting, high payload delivery efficiency, effective in biofilms [20] | Broad-host-range, self-replicating donors, does not require viral receptor [54] |
Experimental Data and Methodologies
Phage-Delivered CRISPR-Cas Systems
Experimental Workflow: The general workflow for assessing phage-delivered CRISPR killing efficiency involves engineering a lytic phage to carry a CRISPR nuclease and targeting guide RNA, then applying this construct to bacterial cultures or co-cultures.
Diagram 1: Phage CRISPR Assay Workflow
- Engineering and Assay Setup: Researchers selected bacteriophages with broad host ranges, such as T-even phages, and engineered them to lack genes for lysogeny, ensuring a purely lytic cycle. The phages were armed with a CRISPR-Cas system (e.g., Type I-E) and guide RNAs (gRNAs) targeting essential genes in the pathogen of interest, such as E. coli [20].
- Co-culture and Infection: Target bacteria were cultured, either in monoculture or in mixed-species co-cultures, to simulate a complex environment. The engineered phages were added at a specific Multiplicity of Infection (MOI), which is the ratio of phage particles to bacterial cells [6] [20].
- Quantification and Efficacy: After incubation, surviving bacteria were quantified by plating and counting Colony Forming Units (CFU). The killing efficiency was calculated as the log10-reduction in CFU mL-1 compared to a control. One study using a cocktail of four CRISPR-Cas-armed phages (CAPs) demonstrated a reduction of 1–6 log10 CFU mL-1 across a panel of E. coli strains. The same system also showed significant killing of bacteria within biofilms, a challenging environment for many antimicrobials [20].
Conjugative Plasmid Delivery Systems
Experimental Workflow: This method relies on the bacterial conjugation process to deliver a plasmid encoding a CRISPR nuclease into target cells.
Diagram 2: Conjugative Plasmid Assay Workflow
- Plasmid Design: A critical variable is the plasmid configuration. A "cis" configuration, where the plasmid encodes both the CRISPR-Cas system and the conjugation machinery (e.g., the pNuc-cis plasmid based on the IncP RK2 system), is significantly more effective than a "trans" configuration that relies on a helper plasmid. The cis setup allows bacteria that receive the plasmid to become new donors, leading to an exponential increase in conjugation events [54].
- Mating Assays: Donor E. coli carrying the conjugative plasmid were mixed with recipient Salmonella enterica in defined ratios. Mating was conducted either on solid filters or in liquid culture. To enhance cell-to-cell contact and dramatically boost conjugation frequency (up to nearly 100%), some experiments included surfaces for biofilm formation, such as the addition of glass beads to liquid cultures [54].
- Killing Assessment: After conjugation, the CRISPR-Cas system was induced in the recipient cells. The resulting chromosome cleavage led to cell death. Conjugation of a cis-configured plasmid expressing the TevSpCas9 nuclease into S. enterica resulted in a reduction of up to 3.5 log10 CFU mL-1 [54].
Table 2: Summary of Key Experimental Outcomes from Literature
| Study System | Target Bacterium | Experimental Context | Key Efficiency Metric |
|---|---|---|---|
| λ-DART Phage [6] | Escherichia coli | Monoculture & mixed community | >50% population editing (knockouts/insertions) |
| CAPs (SNIPR001) [20] | Escherichia coli | Monoculture & Biofilm | 1–6 log10 CFU mL-1 reduction |
| pNuc-cis Plasmid [54] | Salmonella enterica | Co-culture with E. coli | ~3.5 log10 CFU mL-1 reduction; Conjugation frequency up to 100% with biofilm |
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and their functions for implementing these bacterial-killing technologies.
Table 3: Key Reagent Solutions for Phage and Conjugation-Based Killing Assays
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Engineered λ Phage (e.g., λ-DART) | Delivery vector for CRISPR-guided transposases or nucleases [6] | Precise gene knockouts and insertions in E. coli within mixed communities [6] |
| CRISPR-Cas-Armed Phages (CAPs) | Phages engineered to deliver a CRISPR-Cas system for targeted bacterial killing [20] | Creating a phage cocktail (e.g., SNIPR001) for selective reduction of E. coli [20] |
| Cis-Conjugative Plasmid (e.g., pNuc-cis) | All-in-one plasmid encoding conjugation machinery and CRISPR nuclease [54] | High-frequency delivery of TevSpCas9 for efficient killing of Salmonella [54] |
| Amber-Suppressor E. coli Strain | Permissive host for controlling phage infection using amber mutations (e.g., Sam7) [6] | Enables controlled propagation of engineered λ phages for research [6] |
| Type I-E CRISPR-Cas System (EcCas) | Native E. coli CRISPR system used for engineering phages or plasmids [20] | Provides potent, species-specific killing when delivered to target E. coli [20] |
Both phage-delivered CRISPR and conjugative plasmid systems are potent technologies for targeted bacterial killing in co-culture assays. The current data indicates that phage-delivered systems can achieve a broader range of log-reduction (1–6 log10) and are particularly effective in challenging environments like biofilms, leveraging the natural efficiency of viral infection. Conjugative plasmids, especially in a cis-configuration, offer the advantage of exponentially spreading through a population and can achieve very high, near-complete conjugation frequencies under optimized conditions, resulting in substantial (~3.5 log10) killing. The choice between these methods depends on the specific research requirements, including the need for narrow versus broad targeting, the complexity of the microbial community, and the desired balance between killing efficiency and the potential for horizontal gene transfer.
The emergence of antimicrobial resistance (AMR) represents a critical global health threat, driving the development of next-generation antibacterial strategies that move beyond broad-spectrum antibiotics. Among these, CRISPR-Cas systems have emerged as powerful, programmable tools capable of selectively targeting and eliminating bacterial pathogens or resensitizing them to antibiotics. A pivotal consideration in deploying these systems is the choice of delivery vector, which directly determines the host range and strain specificity of the antimicrobial activity. This guide objectively compares the two primary in vivo delivery modalities—phage-based delivery and conjugative plasmid delivery—evaluating their performance based on experimental data for researchers and drug development professionals.
The core trade-off often lies between specificity and breadth. Phage-derived systems can be engineered for exceptional strain specificity, making them ideal for precision manipulation of complex microbial communities. In contrast, conjugative plasmids, particularly those using broad-host-range conjugation machinery, can extend their activity across diverse and phylogenetically distant bacterial species, offering a broader therapeutic reach.
Performance Comparison: Phage vs. Conjugate Plasmid Delivery
The following table summarizes key performance characteristics of both delivery systems, based on published experimental evidence.
Table 1: Comparative Performance of Phage-Delivered and Conjugative Plasmid-Delivered CRISPR-Cas Systems
| Feature | Phage-Delivered CRISPR-Cas | Conjugative Plasmid-Delivered CRISPR-Cas (TAPs) |
|---|---|---|
| Mechanism of Delivery | Viral infection and transduction of CRISPR machinery into targeted bacteria [6] [20]. | Bacterial conjugation to transfer mobilizable plasmids carrying CRISPR-Cas systems [55] [56]. |
| Inherent Host Range Determinant | Phage receptor specificity (e.g., LPS, LamB, Tsx); can be broadened via tail fiber engineering [20]. | Donor-recipient conjugation compatibility; can be expanded using different conjugation systems (e.g., F, RP4) [55] [56]. |
| Demonstrated Specificity | High strain-specificity; capable of targeting a single strain within a mixed microbial community without affecting non-targeted species [57] [20]. | Strain-selective killing within multi-species populations, directed by bioinformatically designed gRNAs [55]. |
| Demonstrated Breadth | Effective against a diverse spectrum of a single species (e.g., E. coli) or engineered for related genera [27] [20]. | Broad activity across Gram-negative Enterobacteriaceae and, with RP4 machinery, distant species like Pseudomonas aeruginosa and Vibrio cholerae [56]. |
| Reported Editing/Efficiency | >50% population editing in monocultures and mixed communities for genomic insertions/deletions [6]. Efficient reduction of E. coli burden in mouse models [20]. | Highly efficient killing, resensitizing strains to antimicrobials with efficacy ranging from 4.7% to 100% [8]. |
| Key Experimental Workflows | Engineering of phage λ with DART system using Cas13a counterselection; infection at controlled MOI [6]. Screening of wild-type phages, tail fiber engineering, and CRISPR-Cas arming (CAPs) [20]. | Construction of Targeted-Antibacterial-Plasmids (TAPs) with constitutive Cas9/dCas9; mating assays with donor and recipient strains [55]. |
Experimental Protocols and Methodologies
Protocol for Phage-Delivered CRISPR-Cas Editing
The following workflow, termed λ-DART, enables large genomic insertions and deletions using engineered bacteriophage λ [6].
Figure 1: Workflow for Phage-Delivered CRISPR-Cas Editing
Key Steps:
- Phage Engineering: The temperate phage λ (harboring the
cI857andSam7mutations for thermo-inducible control and host-dependent lysis, respectively) is engineered via homologous recombination to carry the entire DNA-editing all-in-one RNA-guided CRISPR-Cas transposase (DART) system. Precise modifications are achieved using Cas13a-based counterselection to isolate successfully recombined phage particles [6]. - Phage Amplification and Preparation: The engineered λ-DART phages are propagated in a permissive amber-suppressor host strain (e.g., E. coli LE392) under controlled conditions (30°C for lysogeny, 37°C to induce the lytic cycle) [6].
- Infection and Editing: Target bacteria (e.g., E. coli in monoculture or a mixed community) are infected with the λ-DART phage at a defined Multiplicity of Infection (MOI). The phage delivers the DART system, which performs CRISPR RNA-guided transposition, leading to precise gene knockouts or kilobase-scale insertions in the host genome with reported efficiencies exceeding 50% [6].
Protocol for Conjugative Plasmid Delivery (TAPs)
Targeted-Antibacterial-Plasmids (TAPs) leverage bacterial conjugation to deliver CRISPR-Cas systems with strain-specific activity [55].
Figure 2: Workflow for Conjugative Plasmid Delivery (TAPs)
Key Steps:
- TAPs Design: A mobilizable plasmid is constructed containing a constitutively expressed CRISPR-Cas system (e.g., Cas9 for lethal double-strand breaks or dCas9 for CRISPR interference). The spacer sequence in the gRNA is designed using a bioinformatic tool like the CRISPR Search Tool for Bacteria (CSTB) to ensure it is unique to the target strain's genome or resistance plasmid [55].
- Bacterial Mating: Donor bacteria (e.g., an laboratory E. coli strain) carrying the TAP are mixed with recipient cells (the target pathogen) at a standardized ratio (e.g., 1:3 donor-to-recipient) and co-cultured to allow conjugation [55].
- Selection and Killing: The mixture is plated on selective media to isolate transconjugants (recipients that have received the TAP). Inside the target recipient, the constitutive CRISPR-Cas system is activated. If the target genome contains the sequence complementary to the gRNA, the Cas nuclease is directed to induce lethal double-strand breaks, specifically eliminating that strain from the population [55] [56].
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and their functions for implementing these delivery strategies.
Table 2: Essential Reagents for Phage and Conjugative Plasmid Research
| Reagent / Tool | Function / Description | Relevance |
|---|---|---|
| Phage λ (cI857 Sam7) | Engineered temperate phage; cI857 allows thermo-inducible lytic cycle, Sam7 restricts lysis to amber-suppressor hosts for safety [6]. |
Phage Delivery Chassis |
| CRISPR-associated Transposase (DART) | All-in-one system incorporating Type I-F CAST for RNA-guided DNA insertion without double-strand breaks [6]. | Payload for Large Edits |
| Cas13a Protein | RNA-targeting Cas protein used for counterselection during phage engineering to isolate recombinant phages [6]. | Phage Engineering Tool |
| Targeted-Antibacterial-Plasmid (TAP) | Mobilizable plasmid carrying CRISPR-Cas9/dCas9 and a specific gRNA expression cassette [55]. | Conjugative Delivery Payload |
| CRISPR Search Tool for Bacteria (CSTB) | Bioinformatics algorithm to identify strain-specific gRNA sequences for precise targeting [55]. | Specificity Design Tool |
| F and RP4 Conjugation Systems | Conjugation machinery; F-plasmid derived for Enterobacteriaceae, RP4 for broader host range including P. aeruginosa and V. cholerae [55] [56]. | Conjugation Machinery |
| Amber-Suppressor E. coli (e.g., LE392) | Permissive host strain allowing propagation of phage λ with the Sam7 mutation [6]. |
Phage Propagation Host |
The choice between phage and conjugative plasmid delivery for CRISPR-Cas antimicrobials is not a matter of superiority, but of strategic alignment with research or therapeutic goals.
- Prioritize Phage-Delivered Systems when the objective requires extreme strain-specific precision within a complex microbiome (e.g., gut microbiome modeling, targeting a specific pathogen without collateral damage) or when the goal involves large-scale genomic integrations using systems like DART [6] [57].
- Opt for Conjugative Plasmid Delivery (TAPs) when the goal is to target a broader range of bacterial species simultaneously, including phylogenetically distant pathogens, leveraging the natural promiscuity of conjugation systems like RP4 [55] [56].
Future advancements will likely focus on combining the strengths of both systems—for instance, engineering phages with expanded host ranges or developing more efficiently mobilizing plasmids—to create a versatile and powerful arsenal against antimicrobial resistance.
The rising global threat of antimicrobial resistance (AMR) has spurred the development of novel antibacterial strategies that move beyond conventional antibiotics. Two such advanced approaches are phage-delivered CRISPR-Cas systems and conjugate plasmid delivery. These technologies enable precise targeting of bacterial pathogens through distinct mechanisms and delivery vectors. Phage-delivered CRISPR utilizes engineered bacteriophages—viruses that naturally infect bacteria—as vehicles to transport CRISPR-Cas genetic machinery directly to target bacterial cells within a host. This system can be programmed to induce lethal DNA breaks in specific bacterial sequences, selectively eliminating target pathogens while preserving commensal microbiota. In contrast, conjugate plasmid delivery often leverages bacterial mating mechanisms, such as the F pilus system in Escherichia coli, to transfer genetic material between bacterial cells, though this method may require additional selection pressures, such as antibiotics, to establish the transferred genes effectively. This guide objectively compares the in vivo performance of these innovative approaches, drawing on experimental data primarily from murine and minipig infection models that are critical for preclinical therapeutic development.
Comparative Performance Data from Animal Models
The in vivo efficacy of phage-delivered CRISPR and conjugate plasmid systems has been evaluated in multiple animal models, providing critical quantitative data for comparison. The tables below summarize key performance metrics from these studies.
Table 1: Performance of Phage-Delivered CRISPR Systems in Animal Models
| Delivery Vector / Product | Animal Model | Target Bacterium | Key Performance Metrics | Reference |
|---|---|---|---|---|
| SNIPR001 (4 CAPs cocktail) | Mouse & Minipig | Escherichia coli | Well tolerated; reduced E. coli load in mouse gut better than individual components. | [20] |
| M13 Phagemid | Mouse | Escherichia coli | Selective reduction of GFP-marked E. coli by ~1-3 log; required antibiotic selection for efficient plasmid establishment. | [58] [59] |
| P1 Phagemid | Zebrafish Larva | Shigella flexneri | Significant reduction in bacterial load and improved host survival in an infection model. | [60] |
Table 2: Performance of Conjugate Plasmid Delivery in Animal Models
| Delivery Method | Animal Model | Target Bacterium | Key Performance Metrics | Reference |
|---|---|---|---|---|
| Conjugative Delivery (CGV-EcCas) | In Vitro (Relevant to In Vivo) | Escherichia coli | Average reduction of 3.5 log10 CFU/mL; reduction to below limit of detection (200 CFU/mL) in 75% of isolates where delivery was successful. | [20] |
The data reveals that phage-delivered systems have demonstrated efficacy in live animal models, with SNIPR001 showing promising results in both mice and the more translationally relevant minipigs [20]. The M13 system achieved a modest but specific reduction in targeted bacterial populations within the complex gut environment of mice [58] [59]. In contrast, the summarized data for conjugate plasmid delivery of a CRISPR system, while showing potent killing in vitro, primarily underscores a critical limitation: its efficiency is highly dependent on the successful conjugative transfer of the plasmid into the target bacterial population, which may not be universally achievable for all strains in vivo [20].
Detailed Experimental Protocols
Understanding the methodologies behind the generated data is crucial for interpreting results and designing future experiments. Below are the detailed protocols for key studies in murine and minipig models.
Murine Model: M13 Phage Delivery to Gut E. coli
- Bacterial Strain and Colonization: Mice are first treated with streptomycin in their drinking water to decrease native microbiota complexity. This is followed by colonization with a streptomycin-resistant (SmR) F+ E. coli strain, which allows for engraftment and high-level colonization (reaching a median of 18% of the gut microbiota) [58].
- Phagemid Construction: An M13-compatible phagemid vector is engineered to carry the CRISPR-Cas9 system, including the cas9 gene and a guide RNA (gRNA) targeting a specific gene (e.g., gfp or a bacterial genomic locus) [58] [59].
- Phage Propagation and Dosing: The engineered phagemid is packaged into M13 phage particles by infecting a producer E. coli strain and collecting phage from the culture supernatant. Mice are orally gavaged with a high dose of these phage particles (e.g., 1 × 10^14 plague-forming units) [58].
- Selection and Tracking: For the delivery of antibiotic resistance genes, mice are transferred to water containing carbenicillin to select for successfully transduced E. coli. For CRISPR-based killing, fecal samples are collected over time, and the targeted bacterial population is tracked via fluorescence (if targeting a fluorophore gene) or by plating and sequencing [58] [59].
Murine Model: SNIPR001 against E. coli
- Phage Cocktail Design: A cocktail of four natural phages (α15, α17, α20, α51) are selected from a large library based on broad and complementary coverage of clinically relevant E. coli strains. These phages are then engineered to carry a type I-E CRISPR-Cas system targeting E. coli genomic DNA, creating CRISPR–Cas-armed phages (CAPs) [20].
- CRISPR System Engineering: The CRISPR-Cas system is optimized for activity under gut conditions by using the bacterial starvation-induced PbolA promoter, which shows significant killing in biofilm models [20].
- Dosing and Efficacy Testing: The engineered CAP cocktail, SNIPR001, is administered to mice. The E. coli burden in the gut is quantified and compared to control groups or groups treated with the individual constituent phages to demonstrate superior efficacy of the combination [20].
Minipig Model: SNIPR001 Tolerability
- Animal Model: Göttingen minipigs are used as a large animal model to better predict responses in humans due to physiological similarities [20] [61].
- Experimental Procedure: The SNIPR001 phage cocktail is administered to minipigs. The primary outcome measured is the tolerability and safety profile of the treatment, establishing its preclinical safety for clinical development [20].
Figure 1: Generalized workflow for evaluating antibacterial therapies in murine and minipig infection models, highlighting the common pre-conditioning, colonization, and treatment steps, followed by key analytical endpoints.
Key Signaling Pathways and Mechanisms of Action
The therapeutic action of phage-delivered CRISPR systems involves a cascade of molecular events that culminate in the death of the target bacterium. Understanding these pathways is key to appreciating the specificity and efficacy of this approach.
Figure 2: Mechanism of phage-delivered CRISPR-Cas antimicrobial activity. The engineered phage injects the CRISPR system into the target bacterium, leading to a targeted DNA double-strand break, activation of the bacterial SOS response, and ultimately cell death. Bacteria can escape via several mechanisms [58] [59].
The mechanism begins with the specific attachment of the engineered phage to bacterial surface receptors (e.g., LPS, Tsx, LamB), initiating the injection of its genetic material, which includes the engineered CRISPR-Cas system [20]. Inside the bacterium, the CRISPR-Cas system is expressed. The Cas nuclease (e.g., Cas9, Cas3), guided by a programmable RNA (gRNA), forms a complex that identifies and binds to a complementary DNA sequence adjacent to a protospacer adjacent motif (PAM) in the bacterial chromosome [27] [62]. The bound Cas nuclease then induces a lethal double-strand break (DSB) in the bacterial DNA. In response to this damage, the bacterium activates its SOS response pathway, a stress response system for DNA repair. However, in the absence of efficient error-prone repair mechanisms like non-homologous end joining, this damage typically leads to irreversible cell death [20] [27]. Studies note that bacteria can develop resistance through escape mechanisms such as mutations in the target DNA sequence, loss of the CRISPR spacer, or even loss of the entire CRISPR-Cas system, highlighting an ongoing evolutionary arms race [58] [59].
The Scientist's Toolkit: Essential Research Reagents
Advancing these technologies from concept to in vivo validation relies on a suite of specialized reagents and tools. The following table details key components used in the featured studies.
Table 3: Key Reagent Solutions for Phage-Delivered CRISPR Research
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Tevenvirinae Phages (α15, α17, etc.) | Natural phage vectors; broad host range for E. coli; engineerable to carry cargo. | Selected from a 162-phage library and engineered to create the SNIPR001 cocktail [20]. |
| M13 Bacteriophage | Filamentous phage; exploits F-pilus for conjugation; effective for DNA delivery to E. coli. | Used to deliver CRISPR-Cas9 system to F+ E. coli in the mouse gut [58] [59]. |
| P1 Bacteriophage | Broad-host-range transducing phage; suitable for delivery to Enterobacteriaceae. | Delivered CRISPR-Cas9 phagemid to Shigella flexneri in a zebrafish infection model [60]. |
| Type I-E CRISPR-Cas System | Native E. coli system; uses Cas3 for DNA degradation. | Engineered into SNIPR001 phages for targeted killing of E. coli [20]. |
| Type II CRISPR-Cas9 System | RNA-guided endonuclease (Cas9); creates double-strand breaks. | Delivered via M13 and P1 phages for sequence-specific killing in E. coli and Shigella [58] [60]. |
| PbolA Promoter | Stress-induced promoter; active under slow-growth conditions (e.g., gut, biofilms). | Used to drive CRISPR-Cas expression in SNIPR001 for enhanced in vivo activity [20]. |
| Animal Models | Preclinical testing of safety and efficacy. | Mouse: Gut colonization models. Minipig: Large animal model for tolerability [20] [61]. |
Direct comparison of in vivo data reveals distinct profiles for phage-delivered CRISPR and conjugate plasmid systems. Phage-delivered CRISPR has demonstrated measurable efficacy in reducing specific bacterial pathogens within the complex gut microbiota of live animals, including mice and minipigs, without the mandatory need for antibiotic co-selection [20] [58]. This approach benefits from the natural efficiency of phages as bacterial delivery vectors. In contrast, while conjugative delivery can be highly efficient in vitro, its in vivo performance in animal models for direct antimicrobial killing is less documented in the provided literature; it often relies on antibiotic selection to establish the delivered genetic construct in the target population, which may limit its standalone therapeutic application [20] [58]. The choice between these platforms depends heavily on the specific therapeutic objective: phage-delivered CRISPR offers a promising all-in-one targeted antimicrobial strategy, whereas conjugate delivery may serve other specialized genetic manipulation purposes in complex microbial communities.
The development of precise microbial genome editing tools is crucial for advancing functional studies and therapeutic applications within complex native contexts, such as the human microbiome. Two primary strategies have emerged for delivering CRISPR-based machinery to bacterial populations: phage-mediated delivery and conjugative plasmid delivery. The former leverages the natural specificity of bacteriophages, while the latter exploits bacterial mating mechanisms for horizontal gene transfer. This guide provides a head-to-head comparison of these delivery modalities, focusing on their engineering strategies, performance parameters, and experimental protocols to inform researchers and drug development professionals.
Performance Comparison at a Glance
The table below summarizes the key performance parameters of phage-delivered CRISPR and conjugate plasmid delivery systems, based on recent experimental findings.
Table 1: Key Performance Parameters of Phage and Conjugative Plasmid Delivery Systems
| Performance Parameter | Phage-Delivered CRISPR | Conjugative Plasmid-Delivered CRISPR |
|---|---|---|
| Reported Editing Efficiency | >50% gene knockout/insertion in targeted E. coli population in mixed community [6] | Up to 100% conjugation frequency to S. enterica under optimized liquid culture conditions [33] |
| Payload Capacity | Delivery of large CRISPR-associated transposase (DART) system (~10 kb+) demonstrated [6] | Limited primarily by plasmid size and stability; broad-host-range IncP plasmids used [33] |
| Primary Delivery Mechanism | Viral infection via bacteriophage (e.g., λ, M13) [6] [58] | Bacterial conjugation via type IV secretion system [33] |
| Key Advantage | High natural species/strain-level specificity; can access spatial niches not efficiently accessible by donor bacteria [6] | Extremely high transfer frequency under conditions that enhance cell-to-cell contact (e.g., biofilm formation) [33] |
| Key Engineering Strategy | Engineered non-lysogenic phage (e.g., λ cI857 Sam7) lacking components for lysogeny to prevent persistent phage maintenance [6] | Use of cis-acting conjugative plasmid (e.g., pNuc-cis) where the plasmid encodes both conjugative machinery and CRISPR nuclease [33] |
| Specificity & Targeting | Narrow targeting range dictated by phage host specificity; enables strain-specific editing in mixed communities [6] [58] | Broader host range possible (e.g., IncP RK2 plasmid); targeting dictated by sgRNA sequence and recipient compatibility [33] |
| Critical Experimental Parameter | Multiplicity of Infection (MOI) and incubation period [6] | Donor-to-recipient ratio and culture conditions (e.g., low-salt media, glass beads) [33] |
Detailed Experimental Protocols
To ensure reproducibility and provide a practical framework for researchers, this section outlines the core methodologies employed in the cited studies for both delivery systems.
Protocol for Phage-Delivered CRISPR-Cas
The following workflow is adapted from studies using engineered phage λ to deliver CRISPR-associated transposases (DART) for genome editing in E. coli [6].
1. Phage Engineering:
- Tool: Cas13a-based counterselection following homologous recombination.
- Process: Use homologous recombination in the phage λ genome. Subsequently, employ Cas13a, a CRISPR RNA-guided RNA-targeting nuclease, as a counterselection tool. Cas13a induces cellular dormancy upon target RNA binding, halting the infection cycle of unedited phages, thereby allowing for the selective isolation of successfully recombined phage particles.
- Objective: To embed the entire DART system into the phage genome, creating a λ-DART phage, while removing genes essential for lysogeny to create a non-lysogenic, editing-focused vector.
2. Phage Propagation and Quantification:
- Host Strains: Use an amber-suppressor host (e.g., E. coli LE392MP) for propagating phage with the Sam7 mutation. Use a non-suppressor host (e.g., E. coli BW25113) for subsequent editing assays to control viral spread.
- Infection Assay: Conduct phage infection at 37°C for the cI857 mutant, which renders the repressor protein thermolabile, inducing the lytic cycle.
3. In Vitro/In Vivo Editing Assay:
- Infection: Mix the engineered λ-DART phages with the target bacterial culture (monoculture or mixed community) at a specific Multiplicity of Infection (MOI).
- Incubation: Incubate the culture for a defined period (e.g., 8-24 hours) to allow for phage infection, payload delivery, and the CRISPR-guided transposition event.
- Analysis: Plate the cultures on selective media or use flow cytometry to quantify the percentage of the population exhibiting the desired genetic modification (e.g., gene knockout or insertion).
Protocol for Conjugative Plasmid-Delivered CRISPR-Cas
The following methodology is based on studies using a cis-acting IncP RK2 conjugative plasmid (pNuc-cis) to deliver a TevSpCas9 nuclease for targeted bacterial killing [33].
1. Plasmid Construction:
- Backbone: Use a broad-host-range conjugative plasmid backbone like pTA-Mob 2.0 or IncP RK2.
- Configuration: Clone the origin of transfer (oriT), the CRISPR nuclease (e.g., TevSpCas9 under an inducible pBAD promoter), and the sgRNA expression cassette (e.g., under a constitutive pTet promoter) into the conjugative backbone to create a cis-acting system.
- Optimization: A cluster mutation in the promoter of the conjugative gene traJ has been shown to significantly improve conjugation efficiency to diverse yeast species, a strategy that may also enhance bacterial conjugation [63].
2. Donor and Recipient Preparation:
- Donor Strain: Transform the constructed cis-conjugative plasmid into the donor E. coli strain.
- Recipient Strain: Use the target bacterium (e.g., Salmonella enterica).
3. Conjugation Assay:
- Mixing: Combine donor and recipient cells at various ratios (e.g., 1:1, 10:1 donor-to-recipient) in liquid low-salt LB (LSLB) media.
- Condition Optimization: To maximize cell-to-cell contact, add 0.5 mm glass beads to the liquid culture and incubate with mild agitation (60 RPM) for up to 72 hours.
- Quantification: Plate the conjugation mixture on selective antibiotics to which only the transconjugants (successful recipients) are resistant. Calculate the conjugation frequency as the number of transconjugants divided by the total number of recipient cells.
Visualizing the Core Mechanisms
The diagrams below illustrate the fundamental workflows and logical relationships for each delivery system.
Phage-Delivered CRISPR Workflow
Conjugative Plasmid Delivery Workflow
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these technologies relies on a set of core reagents. The following table details key materials and their functions in the featured experiments.
Table 2: Essential Research Reagents for Phage and Conjugative Delivery Systems
| Reagent/Solution | Function/Application | Example/Specification |
|---|---|---|
| Engineered Phage λ | Delivery chassis for CRISPR payload to specific bacterial hosts. | λ cI857 Sam7 mutant (thermolabile repressor, lysis-deficient in non-suppressor hosts) [6]. |
| Amber-Suppressor Host | Propagation host for phage with amber mutations (e.g., Sam7). | E. coli LE392MP [6]. |
| Non-Suppressor Host | Target host for controlled editing assays where phage lysis is restricted. | E. coli BW25113 [6]. |
| cis-Conjugative Plasmid | A single plasmid encoding both conjugation machinery and CRISPR effectors for high-frequency transfer. | pNuc-cis (based on IncP RK2 backbone) [33]. |
| Optimized Conjugation Media | Liquid culture medium that enhances cell-to-cell contact and conjugation frequency. | Low-Salt LB (LSLB) with 0.25% NaCl, sometimes supplemented with 0.5mm glass beads [33]. |
| CRISPR-Associated Transposase (DART) | All-in-one system for RNA-guided DNA insertion without double-strand breaks. | DNA-editing all-in-one RNA-guided CRISPR-Cas transposase system delivered by phage [6]. |
| TevSpCas9 Nuclease | A dual nuclease fusion protein for targeted bacterial killing. | I-TevI nuclease domain fused to S. pyogenes Cas9, delivered by conjugation [33]. |
Conclusion
Both phage-delivered CRISPR and conjugative plasmid systems represent powerful, yet distinct, platforms for next-generation antimicrobials. Phage delivery offers unparalleled, strain-specific targeting and potent, direct lytic activity, making it ideal for precision elimination of defined pathogens, as demonstrated by clinical candidates like SNIPR001. Conjugative plasmids excel with their exceptionally broad host range and self-amplifying delivery mechanism in bacterial populations, enabling widespread dissemination of CRISPR cargo. The choice between them is context-dependent, hinging on the requirement for precision versus breadth. Future directions will involve combining the strengths of both systems, further optimizing in vivo delivery efficiency using novel nanoparticles, and advancing the most promising candidates through regulatory pathways to address the urgent threat of antimicrobial resistance.
References
- 1. Phage Therapy as a Novel Alternative to Antibiotics Through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562026/]
- 2. CRISPR-Cas mediated targeting of resistance genes for ... [https://www.sciencedirect.com/science/article/pii/S0141813025097375]
- 3. –Cas therapies targeting... | Nature Reviews Bioengineering CRISPR [https://www.nature.com/articles/s44222-025-00311-8?error=cookies_not_supported&code=c025f623-50cb-419d-82a5-148ec13a01af]
- 4. The Application of the CRISPR -Cas System in Antibiotic Resistance... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9356603/]
- 5. -Cas9 mediated CRISPR therapy as an alternative to antibiotics phage [https://animaldiseases.biomedcentral.com/articles/10.1186/s44149-023-00065-z]
- 6. Phage-based delivery of CRISPR-associated transposases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12318184/]
- 7. Precision targeting of genetic variations in mixed bacterial ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1575339/full]
- 8. Unravelling the advances of CRISPR-Cas9 as a precise ... [https://www.sciencedirect.com/science/article/pii/S2213716525000360]
- 9. Advances in large-scale DNA engineering with the ... [https://www.nature.com/articles/s12276-025-01530-0]
- 10. Encounter rates and engagement times limit the ... [https://pubmed.ncbi.nlm.nih.gov/39919124]
- 11. Harnessing bacterial immunity: CRISPR-Cas system as a ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1588446/full]
- 12. CRISPR-Cas Systems: Transformative Precision Tools to ... [https://www.genesispub.org/crispr-cas-systems-transformative-precision-tools-to-combat-antimicrobial-resistance-in-multidrug-resistant-gram-negative-pathogens]
- 13. CRISPR-Cas-assisted phage engineering for personalized ... [https://www.sciencedirect.com/science/article/abs/pii/S0255085724002469]
- 14. RECKLEEN is a lambda Red/CRISPR-Cas9 based single ... [https://www.nature.com/articles/s42003-025-08934-8]
- 15. Characterization of a plasmid dependent DNA phage ... [https://www.nature.com/articles/s41598-025-04494-3]
- 16. Our Phage Picks for March 2025 [https://phage.directory/capsid/phage-picks-march-2025]
- 17. Characterizing conjugative plasmids from an antibiotic- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10301749/]
- 18. Mating pair stabilization mediates bacterial conjugation ... [https://www.nature.com/articles/s41564-022-01146-4]
- 19. Highly efficient gene transfer in the mouse gut microbiota is ... [https://www.nature.com/articles/s42003-020-01253-0]
- 20. Engineered phage with antibacterial CRISPR–Cas ... [https://www.nature.com/articles/s41587-023-01759-y]
- 21. Application of Lytic Bacteriophages and Their Enzymes to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9865725/]
- 22. Phage-plasmids promote recombination and emergence of ... [https://www.nature.com/articles/s41467-024-45757-3]
- 23. Phage therapy: Innovative approaches for refractory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12613065/]
- 24. CRISPR This Way: Choosing Between Delivery System [https://en.vectorbuilder.com/resources/vector-academy/lessons/crispr-delivery-systems.html]
- 25. Plasmid Purification Market Size, YoY Growth Rate, 2025- ... [https://www.coherentmarketinsights.com/industry-reports/plasmid-purification-market]
- 26. A CRISPR-Cas9 system protecting E. coli against ... [https://www.nature.com/articles/s41598-025-85334-2]
- 27. Phage engineering and phage‐assisted CRISPR‐Cas ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10013820/]
- 28. Phagemid-based capsid system for CRISPR-Cas13a ... [https://www.nature.com/articles/s42003-024-06754-w]
- 29. Reinvigorating AMR resilience: leveraging CRISPR–Cas ... [https://tropmedhealth.biomedcentral.com/articles/10.1186/s41182-025-00728-2]
- 30. Broad-spectrum CRISPR-Cas13a enables efficient phage ... [https://www.nature.com/articles/s41564-022-01258-x]
- 31. CRISPR-Cas12a REC2–Nuc interactions drive target-strand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507515/]
- 32. Current Knowledge on CRISPR Strategies Against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11672446/]
- 33. Efficient inter-species conjugative transfer of a CRISPR ... [https://www.nature.com/articles/s41467-019-12448-3]
- 34. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12366227/]
- 35. Biofilm architecture determines the dissemination of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12054802/]
- 36. Beyond antibiotics: exploring multifaceted approaches to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11962305/]
- 37. Advancements in alternative approaches to address ... [https://www.frontiersin.org/articles/10.3389/fmicb.2025.1704931]
- 38. Applications of bacteriophages in precision engineering ... [https://www.sciencedirect.com/science/article/pii/S2667370325000013]
- 39. Isolation, engineering and ecology of temperate phages ... [https://www.nature.com/articles/s41586-025-09614-7]
- 40. Diverse anti-defence systems are encoded in the leading ... [https://www.nature.com/articles/s41586-024-07994-w]
- 41. A predatory gene drive for targeted control of self ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11963995/]
- 42. Communities of plasmids as strategies for antimicrobial ... [https://www.nature.com/articles/s44259-025-00151-x]
- 43. A machine learning approach to predict strain-specific ... [https://www.nature.com/articles/s41598-025-22075-2]
- 44. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 45. Nucleic Acid Spotlights 2025 [https://www.insights.bio/nucleic-acid-insights/spotlights/year/2025]
- 46. Phage susceptibility to a minimal, modular synthetic CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12409346/]
- 47. Genomic insights into bacteriophages: a new frontier in AMR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12302716/]
- 48. A Comprehensive Review on Phage Therapy and ... [https://www.mdpi.com/2079-6382/13/9/870]
- 49. Phage delivered CRISPR-Cas system to combat multidrug ... [https://www.sciencedirect.com/science/article/pii/S075333222200511X]
- 50. Applications of bacteriophages in precision engineering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12173823/]
- 51. Conjugative Delivery of CRISPR-Cas9 for the Selective ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6811441/]
- 52. Diverse and abundant phages exploit conjugative plasmids [https://www.nature.com/articles/s41467-024-47416-z]
- 53. Harnessing CRISPR interference to resensitize laboratory ... [https://www.nature.com/articles/s41598-024-81989-5]
- 54. Efficient inter-species conjugative transfer of a CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6778077/]
- 55. Targeted-antibacterial-plasmids (TAPs) combining ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8034655/]
- 56. Reprogramming Targeted-Antibacterial-Plasmids (TAPs) to ... [https://www.sciencedirect.com/science/article/pii/S0147619X23000112]
- 57. Phage-delivered CRISPR-Cas9 for strain-specific ... [https://www.sciencedirect.com/science/article/pii/S2211124721014030]
- 58. Phage-delivered CRISPR-Cas9 for strain-specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8591988/]
- 59. First CRISPR Manipulation of a Mammalian Microbiome ... [https://crisprmedicinenews.com/news/first-crispr-manipulation-of-a-mammalian-microbiome-via-a-phage-bacteria-pair/]
- 60. P1 Bacteriophage-Enabled Delivery of CRISPR-Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10028697/]
- 61. Establishment of a Pig Influenza Challenge Model for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7372317/]
- 62. Armed Phages: A New Weapon in the Battle Against ... [https://www.mdpi.com/1999-4915/17/7/911]
- 63. Superior Conjugative Plasmids Delivered by Bacteria to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10521675/]