Polyphasic Taxonomy: The Comprehensive Framework for Modern Bacterial Identification and Classification
This article provides a comprehensive overview of the polyphasic taxonomic approach, which integrates phenotypic, genotypic, and phylogenetic data for robust bacterial identification and classification.
Polyphasic Taxonomy: The Comprehensive Framework for Modern Bacterial Identification and Classification
Abstract
This article provides a comprehensive overview of the polyphasic taxonomic approach, which integrates phenotypic, genotypic, and phylogenetic data for robust bacterial identification and classification. Tailored for researchers, scientists, and drug development professionals, it covers the foundational principles of why this consensus approach is necessary beyond traditional methods. The scope extends to detailed methodologies, from 16S rRNA gene sequencing and DNA-DNA hybridization to modern genomic techniques like Average Nucleotide Identity (ANI) and whole-genome sequencing. It further addresses troubleshooting common limitations and validates the approach through comparative analysis with traditional techniques, highlighting its critical applications in clinical diagnostics, probiotic development, and discovering novel microbial diversity.
Beyond Bergey's Manual: The Evolution and Rationale for a Polyphasic Taxonomy
The Limitations of Traditional Phenotypic and Biochemical Methods
Within clinical microbiology and taxonomic research, the accurate identification of bacterial pathogens is a fundamental requirement for diagnosing infections and guiding antimicrobial therapy [1]. For nearly 150 years, the primary tools for this task were traditional phenotypic and biochemical methods, which rely on the visual assessment of microbial physical characteristics, growth patterns on various media, and metabolic capabilities [2] [3]. While these methods laid the foundation for bacteriology, they possess inherent limitations that can compromise their accuracy and utility in modern settings. The shift towards a polyphasic approach, which integrates genotypic, chemotaxonomic, and phenotypic data, has revealed the constraints of relying solely on traditional techniques [4] [5]. This application note delineates the specific limitations of traditional phenotypic and biochemical methods, providing structured data and experimental context to inform researchers and drug development professionals.
Core Limitations of Traditional Methods
The constraints of traditional methods can be categorized into several key areas, encompassing speed, analytical power, and practical laboratory challenges.
Limited Analytical Accuracy and Resolution
Traditional methods often lack the resolution to distinguish between closely related species or strains, leading to misidentification.
- Inability to Differentiate Closely Related Taxa: Biochemical profiling can fail to differentiate organisms with highly similar metabolic pathways. One study found that for a set of unusual aerobic gram-negative bacilli, conventional biochemical methods successfully identified only 84.6% of isolates to the species level, compared to 89.2% identified by 16S rRNA gene sequencing [1].
- Dependence on Subjective Interpretation: The reading of biochemical test results, such as color changes in substrate utilization panels, involves substantial subjective judgment, introducing a source of potential error [1].
- Erroneous and Inconclusive Results: Phenotypic methods like biotyping and antibiogram profiling can yield erroneous or inconclusive results, as they are heavily influenced by environmental conditions and gene expression variability [6].
Lengthy Turnaround Time
The multi-step, growth-dependent nature of these methods results in significant delays in obtaining identification.
- Extended Incubation Periods: Traditional identification based on morphology, physiology, and biochemical characterization is estimated to require 2 to 5 days to complete [2] [6]. This slow turnaround time can delay the initiation of targeted antimicrobial therapy.
- Dependence on Pure Cultures: These methods require the isolation of a pure culture, which itself necessitates a prior 18–24 hour incubation period, adding to the total time-to-identification [2].
Inability to Study Unculturable Microbes
A fundamental constraint is the reliance on the microorganism's ability to proliferate under standard laboratory conditions.
- Non-Culturability: A vast proportion of environmental bacteria cannot be cultured using standard media and conditions [4] [3]. Traditional methods are incapable of identifying these unculturable organisms, leading to a significant gap in our understanding of microbial diversity.
- Fastidious Organisms: Even clinically relevant bacteria that are slow-growing or require specific nutrients (fastidious organisms) are difficult or impossible to identify using conventional phenotypic identification, making it time-consuming and unreliable for these pathogens [1].
Practical and Technical Challenges
The implementation of traditional methods in the laboratory presents several operational difficulties.
- Labor-Intensive Processes: These methods are laborious, requiring significant hands-on time for media preparation, inoculation, and manual reading of results [6].
- Limited Scope of Automated Systems: While automated biochemical systems (e.g., VITEK 2, BD Phoenix) improve efficiency, their accuracy is confined to the database provided by the manufacturer and they remain ineffective for unculturable or highly unusual organisms [2].
- Challenges in Standardization: Classifications based primarily on phenotypic characters lack stability compared to those based on genetic relatedness, as phenotypic expression can be variable and context-dependent [6].
Table 1: Quantitative Comparison of Identification Methods for Unusual Aerobic Gram-Negardive Bacilli
| Identification Method | Genus-Level Identification Rate (n=72) | Species-Level Identification Rate (n=65) | Basis of Identification |
|---|---|---|---|
| Cellular Fatty Acid Analysis (Sherlock) | 77.8% (56/72) | 67.7% (44/65) | Phenotypic (Chemotaxonomic) |
| Carbon Source Utilization (Microlog) | 87.5% (63/72) | 84.6% (55/65) | Phenotypic (Biochemical) |
| 16S rRNA Gene Sequencing (MicroSeq) | 97.2% (70/72) | 89.2% (58/65) | Genotypic |
| Conventional Phenotypic Methods | 100% (72/72) | 100% (65/65) | Phenotypic (Reference Standard) |
Data adapted from a comparative evaluation of 72 clinical isolates [1].
Experimental Protocols for Validation
To empirically demonstrate the limitations of traditional methods, the following protocol outlines a comparison study against a genotypic standard.
Protocol: Comparison of Method Identification Rates
Objective: To quantify the identification accuracy and turnaround time of traditional biochemical methods versus 16S rRNA gene sequencing for a panel of clinical bacterial isolates.
Materials:
- Strains: 40-100 bacterial clinical isolates, preferably including fastidious and unusual organisms [1] [7].
- Growth Media: Tryptic Soy Agar (TSA) plates, 5% Sheep Blood Agar plates [1].
- Biochemical Identification System: Automated system (e.g., VITEK 2, BD Phoenix) or manual kit (e.g., API) [2].
- Genotypic Identification: DNA extraction reagents (e.g., Chelex solution), PCR master mix for 16S rRNA gene amplification, sequencing primers, and access to a DNA sequencer [1].
- Data Analysis Software: Software for sequence alignment and comparison to a validated database (e.g., MicroSeq software) [1].
Method:
- Sample Preparation: Inoculate each isolate onto a Blood Agar plate and incubate at 35°C for 18-24 hours to obtain pure colonies [1].
- Biochemical Identification:
- Prepare a bacterial suspension from pure colonies as per the manufacturer's instructions for the biochemical system.
- Inoculate the identification card or strip.
- Load the card into the automated instrument or incubate the strip manually.
- Record the identification result and the time from initial inoculation to final result [2].
- Genotypic Identification (Reference Method):
- DNA Extraction: Harvest bacterial cells from a pure colony. Lyse cells using a thermal shock protocol with a 5% Chelex solution [1].
- PCR Amplification: Amplify the nearly full-length 16S rRNA gene using universal primers (e.g., 0005F and 1540R). Use a thermal cycler with the following parameters: initial denaturation at 95°C for 10 min; 30 cycles of 95°C for 30s, 60°C for 30s, and 72°C for 45s; final extension at 72°C for 10 min [1].
- DNA Sequencing and Analysis: Purify the PCR product and perform cycle sequencing. Electrophorese the sequencing reactions on a DNA sequencer. Assemble the sequence and compare it to a curated database (e.g., MicroSeq library) for identification [1].
- Data Analysis:
- Calculate the percentage of isolates identified to the genus and species level by each method.
- Compare the results of the biochemical method to the genotypic reference standard. Discrepancies are considered errors in the biochemical method.
- Document the average time to identification for each method.
Workflow Diagram: Traditional vs. Modern Identification
The following diagram contrasts the workflows of traditional biochemical and modern genotypic identification, highlighting the steps contributing to the limitations of the traditional approach.
The Scientist's Toolkit: Key Research Reagents
The following reagents are essential for executing the comparative validation protocol described above.
Table 2: Essential Reagents for Method Comparison Studies
| Reagent / Solution | Function in Protocol | Justification for Use |
|---|---|---|
| Chelex 100 Resin | Rapid preparation of genomic DNA from bacterial colonies for PCR. | A fast, inexpensive method for DNA extraction that is sufficient for PCR amplification of the 16S rRNA gene [1]. |
| Universal 16S rRNA Primers (e.g., 0005F, 1540R) | PCR amplification of a phylogenetically informative genetic target. | The 16S rRNA gene contains conserved regions (for primer binding) and variable regions (for discrimination), making it a standard for bacterial identification [1] [3]. |
| PCR Master Mix | Enzymatic amplification of the target DNA segment. | Provides the necessary components (Taq polymerase, dNTPs, buffer) for robust and specific PCR amplification [1]. |
| Selective & Differential Agar (e.g., MacConkey, Blood Agar) | Isolation and preliminary phenotypic characterization of isolates. | Allows for the selection of specific bacterial groups (e.g., Gram-negatives) and provides early phenotypic data [3]. |
| Biochemical Identification Kit / Cards | Generation of a phenotypic metabolic profile for automated identification. | Represents the standard of practice for traditional, phenotypic identification in many clinical laboratories [2]. |
| Sanger Sequencing Reagents | Determining the nucleotide sequence of the amplified 16S PCR product. | Provides the high-accuracy sequence data required for definitive genotypic identification [1]. |
The evidence demonstrates that traditional phenotypic and biochemical methods for bacterial identification are constrained by limited accuracy for unusual taxa, slow turnaround times, and an inherent inability to characterize unculturable organisms. These limitations can directly impact patient care by delaying appropriate therapy and impede taxonomic research by providing an incomplete picture of microbial diversity. The data and protocols presented herein validate the necessity of moving beyond traditional methods. A polyphasic approach, which integrates genotypic techniques like 16S rRNA sequencing with chemotaxonomic and phenotypic data, is the contemporary solution for achieving rapid, sensitive, and accurate microbial identification [4] [5]. For researchers and drug development professionals, adopting this comprehensive framework is critical for advancing both clinical microbiology and microbial systematics.
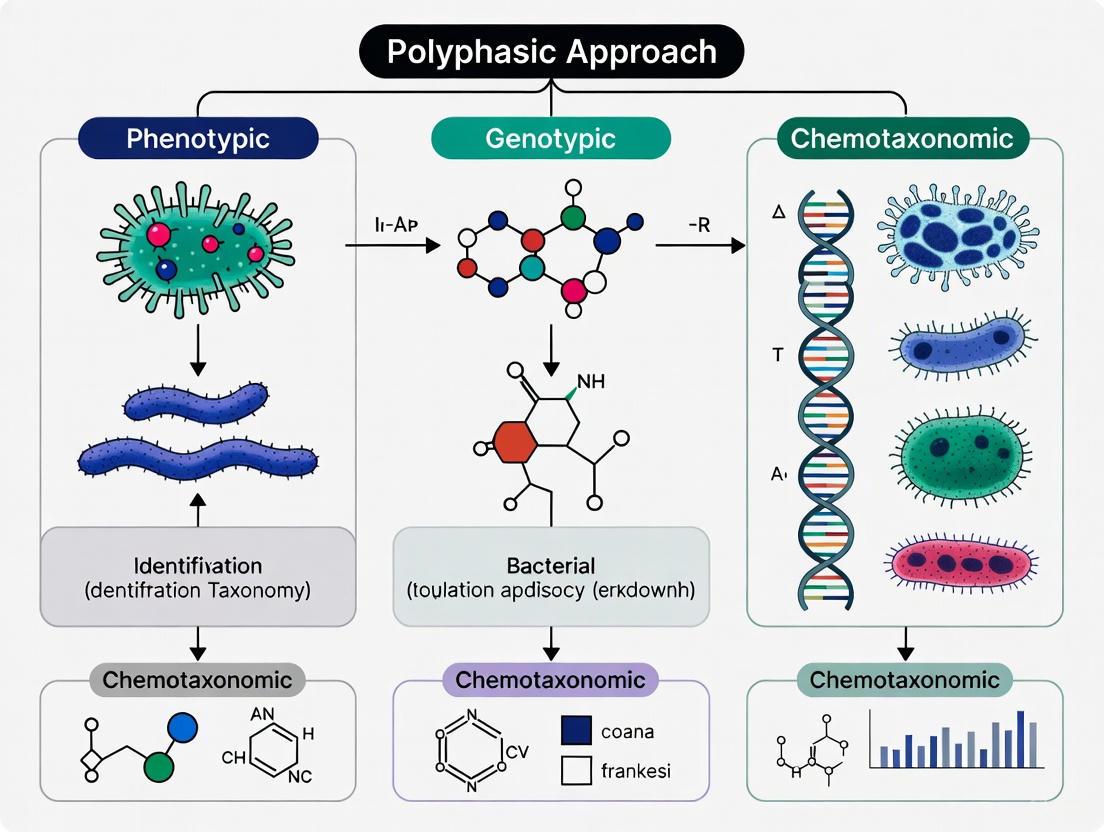
The polyphasic approach represents the foundational framework and consensus standard for the classification and identification of bacteria in modern systematics. This methodology integrates data from genotypic, chemotaxonomic, and phenotypic analyses to provide a comprehensive characterization of microbial taxa, thereby overcoming the limitations inherent in relying on any single method [4]. The approach has evolved from traditional classification systems based primarily on morphological and physiological observations to incorporate advanced molecular techniques and genomic data, revolutionizing our understanding of microbial phylogeny and diversity [8].
The philosophical underpinning of the polyphasic approach acknowledges that a complete understanding of taxonomic relationships requires multiple lines of evidence. As Vandamme et al. articulated, this consensus approach to bacterial systematics creates a robust framework for delineating taxonomic boundaries [4]. This is particularly crucial in an era of rapidly evolving genomic technologies, where the traditional boundaries of bacterial species are constantly being redefined. The polyphasic strategy remains the gold standard for describing novel bacterial species and has become increasingly important in pharmaceutical quality control, clinical diagnostics, and environmental microbiology [9].
Core Components of the Polyphasic Approach
The polyphasic approach systematically integrates data from three primary methodological domains, each contributing essential information for comprehensive taxonomic classification.
Table 1: Core Methodological Components of the Polyphasic Approach
| Component | Key Methods | Primary Taxonomic Application |
|---|---|---|
| Genotypic Analysis | 16S rRNA gene sequencing, DNA-DNA hybridization (DDH), Whole Genome Sequencing (WGS), Multilocus Sequence Analysis (MLSA) | Phylogenetic relationships, species delineation, evolutionary history |
| Chemotaxonomic Analysis | Fatty Acid Methyl Esters (FAME) profiling, Cell wall composition analysis, Lipid analysis | Differentiation at genus and species levels based on chemical markers |
| Phenotypic Analysis | Morphological characterization, Biochemical tests, Physiological profiling | Preliminary grouping and traditional classification |
Genotypic Methods
Genotypic methods form the backbone of modern polyphasic taxonomy by providing direct insights into the genetic relatedness and evolutionary relationships between microorganisms. The 16S rRNA gene sequencing serves as the primary tool for initial phylogenetic placement, allowing researchers to determine the approximate taxonomic position of an unknown isolate [4] [10]. For higher resolution at the species level, DNA-DNA hybridization (DDH) has historically been the gold standard, with a threshold of ≥70% similarity typically indicating that strains belong to the same species [8].
With the advent of accessible whole genome sequencing, genomic data is increasingly being incorporated into taxonomic descriptions. Genome sequences provide the most comprehensive dataset for comparison, including Average Nucleotide Identity (ANI) and in silico DDH values, which offer robust criteria for species delineation [11]. Additional genotypic methods such as rep-PCR fingerprinting and pulsed-field gel electrophoresis (PFGE) provide strain-level differentiation, which is particularly valuable for epidemiological studies and contamination investigation in pharmaceutical settings [4].
Chemotaxonomic Methods
Chemotaxonomic methods analyze the chemical composition of bacterial cells to identify markers that are stable and characteristic for specific taxonomic groups. The analysis of cellular fatty acids through gas chromatographic separation of Fatty Acid Methyl Esters (FAME) is widely used for routine identification in quality control laboratories [9]. This method relies on standardized growth conditions to generate reproducible fatty acid profiles that can be compared against extensive databases.
Other chemotaxonomic markers include cell wall components (e.g., peptidoglycan structure), polar lipids, polyamines, and isoprenoid quinones. These chemical signatures provide complementary data to genotypic methods and are particularly valuable for distinguishing between closely related species that may exhibit high 16S rRNA gene sequence similarity but have distinct metabolic pathways or ecological niches [10].
Phenotypic Methods
Phenotypic characterization encompasses the traditional observations and tests that formed the basis of early bacterial taxonomy. Morphological examination includes colony characteristics, cell shape and size, Gram stain reaction, and motility [4] [9]. Physiological and biochemical profiling assesses metabolic capabilities, including carbon source utilization, enzyme activities, temperature and pH tolerance, and antibiotic susceptibility patterns.
While phenotypic methods are generally insufficient alone for definitive classification, they provide essential contextual information about the functional characteristics of microorganisms and remain crucial for the initial grouping of isolates. Furthermore, phenotypic data can reveal ecologically relevant traits that may not be apparent from genetic sequences alone, thus completing the comprehensive portrait of a microbial strain [10].
Experimental Protocols for Polyphasic Analysis
Protocol 1: 16S rRNA Gene Sequencing and Analysis
Principle: Amplification and sequencing of the highly conserved 16S ribosomal RNA gene allows for phylogenetic placement and preliminary identification of bacterial isolates [10].
Procedure:
- DNA Extraction: Isolate genomic DNA from pure bacterial culture using commercial extraction kits or standard enzymatic lysis methods.
- PCR Amplification: Amplify the nearly full-length 16S rRNA gene using universal primers 27F (5'-AGAGTTTGATCMTGGCTCAG-3') and 1492R (5'-GGTTACCTTGTTACGACTT-3').
- PCR Conditions: Initial denaturation at 95°C for 5 min; 35 cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec, extension at 72°C for 90 sec; final extension at 72°C for 7 min.
- Sequencing and Analysis: Purify PCR products and perform Sanger sequencing. Compare obtained sequences against curated databases (e.g., EzBioCloud, SILVA) using BLAST analysis. Construct phylogenetic trees using neighbor-joining or maximum likelihood methods with related type strains.
Interpretation: ≥98.7% 16S rRNA gene sequence similarity suggests potential membership in the same species, though further confirmation with DDH or ANI is required for novel species description [8].
Protocol 2: Fatty Acid Methyl Esters (FAME) Analysis
Principle: Gas chromatographic separation of cellular fatty acids provides a reproducible chemical fingerprint for bacterial identification [9].
Procedure:
- Standardized Growth: Cultivate isolates on prescribed media (e.g., Tryptic Soy Broth Agar) at standardized temperature (28°C) for 24-48 hours.
- Saponification: Harvest bacterial cells and treat with 1.2 mL of 15% NaOH in 50% aqueous methanol at 100°C for 30 minutes.
- Methylation: Add 2 mL of 6.0 N HCl in 50% aqueous methanol and incubate at 80°C for 10 minutes.
- Extraction: Extract fatty acid methyl esters with 1.25 mL of 1:1 (v/v) hexane:methyl tert-butyl ether.
- Analysis: Inject sample into gas chromatography system equipped with flame ionization detector and microbial identification system software.
Interpretation: Compare resulting FAME profiles with commercial databases (e.g., MIDI System). Similarity indices (SI) >0.6 generally indicate reliable identification to species level when supported by other polyphasic data [9].
Protocol 3: DNA-DNA Hybridization (DDH)
Principle: Measure the reassociation rate between single-stranded DNA from two strains to determine genomic relatedness at the species level [4] [8].
Procedure:
- DNA Preparation: Extract and purify high-molecular-weight DNA from reference and test strains.
- DNA Labeling: Label reference DNA with fluorophores or radioactive isotopes.
- Hybridization: Mix denatured single-stranded DNA from both strains in equimolar concentrations and allow reassociation at optimal renaturation temperature (25-30°C below melting temperature).
- Measurement: Quantify the formation of hybrid double-stranded DNA through spectrophotometric or fluorometric methods.
Interpretation: DDH values ≥70% coupled with ΔTm ≤5°C indicate strains belong to the same species [4].
Figure 1: Workflow of a standard polyphasic taxonomic approach integrating genotypic, chemotaxonomic, and phenotypic data streams.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Research Reagents for Polyphasic Taxonomic Studies
| Reagent/Material | Application | Function in Analysis |
|---|---|---|
| Tryptic Soy Agar/Blood Agar | Microbial cultivation | Standardized growth for phenotypic tests and FAME analysis |
| Gram Staining Reagents | Preliminary differentiation | Basic cell morphology and classification |
| DNA Extraction Kits | Genotypic analysis | High-quality genomic DNA preparation for PCR and sequencing |
| 16S rRNA PCR Primers | Genotypic analysis | Amplification of phylogenetic marker gene |
| PCR Master Mix | Genotypic analysis | Enzymatic amplification of target DNA sequences |
| Sanger Sequencing Reagents | Genotypic analysis | Determination of nucleotide sequences |
| FAME Standards | Chemotaxonomic analysis | Reference compounds for fatty acid identification |
| GC-MS System | Chemotaxonomic analysis | Separation and detection of chemical markers |
| API Test Strips | Phenotypic analysis | Standardized biochemical profiling |
| Salt Tolerance Media | Phenotypic analysis | Determination of physiological parameters |
Recent Developments and Future Perspectives
The field of bacterial taxonomy is experiencing rapid transformation with the integration of genomic data into the polyphasic framework. The so-called "taxono-genomics" approach incorporates whole genome sequences as a fundamental component of taxonomic descriptions, providing unprecedented resolution for discriminating between closely related taxa [11]. This is particularly valuable for resolving complex taxonomic groups where 16S rRNA gene sequence similarity is high but genomic relatedness is low.
The increasing accessibility of next-generation sequencing technologies has accelerated the rate of taxonomic revisions and the description of novel species. As a result, longstanding genera such as Bacillus have been subdivided into multiple new genera (Peribacillus, Cytobacillus, Mesobacillus, etc.) based on robust genomic data [12]. Similarly, clinical important taxa like Propionibacterium acnes have been reclassified as Cutibacterium acnes following comprehensive genomic analyses [12].
Future developments in bacterial taxonomy will likely see increased emphasis on metagenomic data from uncultivated microorganisms and the formal recognition of Candidatus taxa based on sequence information alone. However, the core principles of the polyphasic approach—the integration of multiple data types for comprehensive characterization—will continue to provide the philosophical foundation for microbial systematics, ensuring that taxonomic classifications reflect both evolutionary relationships and functional characteristics [8].
The polyphasic approach remains the gold standard for bacterial taxonomy, providing a robust framework that integrates genotypic, chemotaxonomic, and phenotypic data. This consensus methodology has proven adaptable to technological advances, particularly in genomics, while maintaining the rigor necessary for reliable taxonomic decisions. As bacterial classification continues to evolve, the polyphasic strategy ensures that taxonomic descriptions reflect comprehensive characterization rather than single methodological perspectives, ultimately supporting diverse fields from pharmaceutical quality control to fundamental evolutionary research.
The field of bacterial taxonomy has undergone a profound transformation, shifting from a system based primarily on phenotypic observations to one grounded in evolutionary history. This transition represents a move from numerical taxonomy, which grouped organisms based on overall phenotypic similarity, to a phylogenetic framework that classifies organisms based on their evolutionary relationships and common ancestry [13]. This shift has been accelerated by technological advances, particularly in genomics, enabling a more precise and natural classification of bacterial diversity. Within this modern paradigm, the polyphasic approach has emerged as the standard, integrating phylogenetic data with phenotypic, chemotaxonomic, and genomic information to provide a holistic view of taxonomic relationships [14] [15]. This article details the key protocols and applications of this modern, phylogenetic framework, providing researchers with the methodologies to implement it effectively.
Core Concepts and Definitions
- Numerical Taxonomy: A classification system that groups organisms based on the quantitative analysis of a large number of phenotypic characteristics, operating under the principle that the more characteristics two organisms share, the closer their relationship. This method does not inherently reflect evolutionary history.
- Phylogenetic Taxonomy: A classification system that groups organisms based on their evolutionary relationships, aiming to ensure that all taxonomic groups are monophyletic—meaning they consist of an ancestor and all of its descendants [13].
- Monophyly, Paraphyly, and Polyphyly: A monophyletic group (or clade) is a natural group that includes a common ancestor and all of its descendants. A paraphyletic group includes a common ancestor but not all of its descendants, while a polyphyletic group includes organisms from multiple evolutionary origins without their common ancestor [13]. Modern taxonomy prioritizes the identification of monophyletic groups for their explanatory power.
- Homology vs. Analogy: Homologous traits are similarities due to shared ancestry and are the primary data used for building phylogenetic trees. Analogous traits are similarities due to convergent evolution or other processes and can mislead phylogenetic inference if misinterpreted [13].
Table 1: Key Concepts in Modern Phylogenetic Taxonomy
| Term | Definition | Significance in Taxonomy |
|---|---|---|
| Monophyletic Group (Clade) | A group consisting of a common ancestor and all of its descendants [13] | Forms the basis for natural, evolutionarily valid classification |
| Paraphyletic Group | A group consisting of a common ancestor but not all of its descendants [13] | Considered artificial and avoided in modern taxonomy |
| Homology | Similarity in traits due to shared ancestry [13] | Provides evidence for evolutionary relationships and common descent |
| Phylogeny | The evolutionary history and relationships among individuals, groups, or genes [13] | The framework upon which modern classification is built |
Essential Data Analysis Protocols
Protocol 1: 16S rRNA Gene Sequencing and Phylogenetic Analysis
The 16S rRNA gene remains a cornerstone for initial phylogenetic placement in bacterial taxonomy due to its universal presence and conserved nature.
- Objective: To obtain a preliminary phylogenetic classification of a bacterial isolate.
- Experimental Workflow:
- Detailed Methodology:
- DNA Extraction & Amplification: Extract genomic DNA from a pure bacterial culture using a commercial kit (e.g., DNeasy PowerSoil Pro kit, QIAGEN). Amplify the nearly full-length 16S rRNA gene using universal primers such as 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1429R (5′-TACGGYTACCTTGTTACGACTT-3′) [15].
- Sequencing and Assembly: Sequence the purified PCR amplicons and assemble the resulting sequences into a complete 16S rRNA gene sequence using software like BioEdit [15].
- Sequence Alignment & Tree Construction: Align the query sequence with closely related reference sequences from databases like EzBioCloud or GenBank using CLUSTAL W. Construct phylogenetic trees using neighbor-joining (NJ), maximum-likelihood (ML), and maximum-parsimony (MP) algorithms implemented in software such as MEGA X [15].
- Statistical Validation: Assess the reliability of the tree topology by performing a bootstrap analysis (e.g., 1000 replications) to assign confidence levels to the branches [15].
Protocol 2: Genome Sequencing and Phylogenomic Analysis
For definitive taxonomic resolution, particularly at the species level, whole-genome sequencing and analysis are required. This overcomes the limited resolution of the 16S rRNA gene.
- Objective: To determine the precise genomic relatedness between a novel isolate and its closest phylogenetic neighbors.
- Experimental Workflow:
- Detailed Methodology:
- Genome Sequencing and Assembly: Sequence the bacterial genome using an appropriate platform (e.g., Illumina, PacBio) and assemble the reads into contigs or a complete genome. Assess assembly quality using metrics like N50 and number of contigs.
- Calculation of Genomic Relatedness Indices:
- Average Nucleotide Identity (ANI): Calculate using tools such as OrthoANIU. ANI values ≥95% are the widely accepted threshold for species demarcation [16] [15].
- Digital DNA-DNA Hybridization (dDDH): Calculate using the Genome-to-Genome Distance Calculator (GGDC). A dDDH value ≥70% corresponds to the species boundary [14] [15].
- Average Amino Acid Identity (AAI): Calculate the identity of orthologous proteins, useful for genus-level classification [15].
- Phylogenomic Tree Construction: Identify a set of core genes shared across all genomes under comparison. Create a multiple sequence alignment of the concatenated core gene sequences and infer a phylogeny using maximum-likelihood or Bayesian methods. This produces a highly robust tree reflecting true evolutionary relationships [16].
Table 2: Genomic Standards for Species and Genus Delineation in Bacteria
| Genomic Index | Species Boundary | Genus-Level Boundary | Interpretation & Significance |
|---|---|---|---|
| Average Nucleotide Identity (ANI) | ≥95% [15] | ~70-80% | Replaced wet-lab DDH; primary genomic standard for species definition |
| digital DDH (dDDH) | ≥70% [14] [15] | - | Computational simulation of laboratory DDH experiment |
| Average Amino Acid Identity (AAI) | - | ~60-80% [15] | Useful for inferring genus-level relationships based on protein sequence conservation |
Integrated Application Note: A Polyphasic Taxonomy Workflow
The following case study illustrates the application of a full polyphasic approach to characterize a novel bacterium, Mariniflexile rhizosphaerae sp. nov., isolated from the tomato rhizosphere [16].
- Background: Strain TRM1-10ᴸ was isolated for its role in conferring bacterial wilt resistance. Initial 16S rRNA gene analysis suggested affiliation with the genus Mariniflexile, whose members were previously known only from marine environments [16].
- Integrated Workflow:
- Execution and Results:
- Phylogenetic Analysis: A phylogenomic tree based on 1,347 core genes confirmed the strain's placement within the genus Mariniflexile but as a distinct lineage [16].
- Genomic Analysis: ANI and dDDH values between TRM1-10ᴸ and its closest relatives (M. soesokkakense and M. litorale) were 85.86%/27.8% and 85.42%/27.0%, respectively—both well below the species thresholds [16].
- Phenotypic & Chemotaxonomic Analysis: The strain differed from its marine relatives in its ability to grow without NaCl and in its carbon source utilization profile (e.g., it could utilize D-raffinose, lactose, and melibiose). Genomic analysis revealed adaptations to the soil rhizosphere, such as a unique repertoire of genes for carbohydrate metabolism [16].
- Conclusion: The consistent evidence from all datasets confirmed the strain as a novel species, demonstrating how polyphasic taxonomy can reveal ecological adaptations and clarify taxonomic status.
Table 3: Key Reagents and Software for Polyphasic Taxonomic Studies
| Item Name | Function/Application | Specific Example / Note |
|---|---|---|
| DNeasy PowerSoil Pro Kit | Standardized extraction of high-quality genomic DNA from bacterial cultures [15] | Critical for downstream sequencing applications |
| Universal 16S rRNA Primers | Amplification of the 16S rRNA gene for initial phylogenetic screening [15] | e.g., 27F/1429R; allows for sequencing and comparison with databases |
| Marine Agar (MA) | Cultivation and isolation of marine bacteria [16] [15] | Used for isolation and physiological characterization |
| API ZYM / API 20NE | Standardized strips for assessing physiological and biochemical characteristics [16] | Provides reproducible phenotypic data |
| EzBioCloud Database | High-quality, curated database for 16S rRNA gene and genome sequence comparison [15] | Essential for accurate phylogenetic placement |
| MEGA X Software | Integrated tool for sequence alignment and phylogenetic tree construction using multiple methods (NJ, ML, MP) [15] | User-friendly for molecular phylogenetics |
| GGDC / OrthoANIU | Web servers for calculating dDDH and ANI values from genome sequences [14] | Gold standard for genomic species delineation |
| ggtree R Package | Powerful and highly customizable visualization and annotation of phylogenetic trees [17] [18] | Enables publication-quality tree figures with associated data |
The historical shift from numerical to phylogenetic taxonomy, powered by genomics, has fundamentally refined our understanding of bacterial diversity. The polyphasic approach is the embodiment of this modern framework, robustly integrating data from multiple independent lines of evidence. The protocols and applications detailed herein provide a roadmap for researchers to confidently navigate bacterial identification and classification, ultimately contributing to discoveries in fields ranging from microbial ecology to drug development. As genomic technologies continue to evolve, the phylogenetic framework will only become more resolved, further solidifying its role as the foundation of bacterial taxonomy.
A stable and natural classification system is the cornerstone of microbiology, enabling clear communication and guiding research in evolution, ecology, and drug development. The polyphasic approach, which integrates phenotypic, genotypic, and phylogenetic data, is the established paradigm for constructing a robust taxonomic framework for bacteria. This methodology ensures that classifications reflect true evolutionary relationships, providing a reliable system for identifying novel isolates, understanding microbial ecology, and tracing the origins of pathogenic traits. These Application Notes provide detailed protocols and data analysis frameworks to implement this approach effectively.
Quantitative Data in Polyphasic Taxonomy
Polyphasic taxonomy relies on quantitative thresholds to delineate taxonomic ranks. The following tables summarize the key genomic and phenotypic standards.
Table 1: Genomic Standards for Species and Genus Delineation
| Taxonomic Rank | Genomic Standard | Threshold Value | Interpretation |
|---|---|---|---|
| Species | Average Nucleotide Identity (ANI) | ≥95-96% [16] [19] | Values at or above this threshold indicate organisms belong to the same species. |
| Species | digital DNA-DNA Hybridization (dDDH) | ≥70% [19] | Values at or above this threshold indicate organisms belong to the same species. |
| Species * | 16S rRNA Gene Sequence Identity | ≥98.7-99% [19] | A preliminary screen; higher divergence suggests a novel species, but ANI/dDDH is required for confirmation. |
| Genus | 16S rRNA Gene Sequence Identity | <94-96% [16] | Divergence beyond this level typically indicates a novel genus. |
Table 2: Phenotypic and Chemotaxonomic Characteristics for Differentiation
| Characteristic Category | Examples of Differentiating Tests | Application in Taxonomy |
|---|---|---|
| Physiological & Biochemical | Catalase activity, oxidase test, carbon source utilization (e.g., D-raffinose, lactose), enzyme activity (e.g., α-galactosidase, phosphatase) [16] | Distinguishes between closely related species based on metabolic capabilities. |
| Chemotaxonomic | Cellular fatty acid profiles, polar lipid composition, flexirubin-type pigments [16] | Provides a chemical fingerprint that is often consistent within a genus or species. |
| Morphological & Growth | Cell shape and size, Gram stain, optimum growth temperature and pH, NaCl tolerance [16] | Provides fundamental descriptive data for a novel species or genus. |
Experimental Protocols for a Polyphasic Analysis
Protocol 2.1: 16S rRNA Gene Sequencing and Phylogenetic Analysis
I. Purpose To obtain a preliminary phylogenetic placement of a bacterial isolate using 16S rRNA gene sequencing, a cornerstone of modern microbial classification [19].
II. Materials
- Bacterial genomic DNA
- Universal primers (e.g., 27F and 1492R)
- PCR reagents (polymerase, dNTPs, buffer)
- Agarose gel electrophoresis equipment
- DNA sequencing facility or kit
III. Procedure
- PCR Amplification: Amplify the nearly full-length 16S rRNA gene using universal primers.
- Purification: Purify the PCR amplicon to remove primers and enzymes.
- Sequencing: Submit the purified product for Sanger sequencing or perform sequencing in-house.
- Sequence Assembly: Assemble the forward and reverse sequences into a single consensus sequence.
- Database Comparison: Compare the consensus sequence against a public database (e.g., EzBioCloud, NCBI) using the BLAST algorithm to identify closest relatives.
- Phylogenetic Tree Construction:
- Perform a multiple sequence alignment with closely related type strain sequences.
- Construct a phylogenetic tree using a robust method (e.g., Neighbor-Joining, Maximum Likelihood).
- Evaluate tree robustness with bootstrap analysis (e.g., 1000 replicates).
IV. Data Interpretation The isolate represents a potential novel species if its 16S rRNA gene sequence similarity to all known type strains is below 98.7-99% [19]. A similarity below 94-96% suggests a novel genus [16].
Protocol 2.2: Whole-Genome Sequencing and Phylogenomic Analysis
I. Purpose To establish a high-resolution, evolutionary framework for classification based on whole-genome data, moving beyond the single-gene view of 16S analysis [19].
II. Materials
- High-quality genomic DNA (>20 kb fragment size)
- Whole-genome sequencing platform (e.g., Illumina, PacBio, Oxford Nanopore)
- High-performance computing cluster
- Bioinformatics software (e.g., GTDB-Tk, OrthoFinder, FastANI)
III. Procedure
- DNA Extraction & Sequencing: Extract high-molecular-weight DNA and sequence the genome to an appropriate depth of coverage.
- Genome Assembly: Assemble sequencing reads into contigs or scaffolds.
- Average Nucleotide Identity (ANI) Calculation: Calculate the ANI between the query genome and its closest relatives using a tool like FastANI. ANI values ≥95-96% indicate the same species [16] [19].
- Phylogenomic Tree Construction:
- Identify a set of single-copy core genes present in the query and a panel of closely related reference genomes.
- Create a multiple sequence alignment for each core gene.
- Concatenate the alignments into a supermatrix.
- Infer a phylogenomic tree from the supermatrix using Maximum Likelihood or Bayesian methods.
IV. Data Interpretation A phylogenomic tree provides the most robust framework for genus and family-level classification. The monophyly of a clade (i.e., all members sharing a common ancestor) in a high-confidence tree strongly supports its status as a distinct genus [16] [19].
Visualizing the Polyphasic Taxonomy Workflow
The following diagram illustrates the integrated workflow of the polyphasic approach, from isolation to final classification.
Polyphasic Taxonomy Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Polyphasic Taxonomy
| Item Name | Function / Application | Example / Specification |
|---|---|---|
| Universal 16S rRNA Primers | PCR amplification of the 16S rRNA gene for preliminary phylogenetic identification [19]. | 27F (5'-AGAGTTTGATCMTGGCTCAG-3'), 1492R (5'-GGTTACCTTGTTACGACTT-3') |
| DNA Sequencing Kit | Determining the nucleotide sequence of PCR amplicons or whole genomes. | Sanger sequencing reagents; library prep kits for Illumina/PacBio. |
| Bioinformatics Suite | Software for genome assembly, annotation, phylogenetic tree construction, and ANI calculation. | GTDB-Tk, OrthoFinder, FastANI, MEGA, RAxML. |
| API/BIOLOG Test Strips | Standardized phenotypic assays for carbon source utilization and enzyme activity profiling [16]. | API 20NE, API ZYM, BIOLOG Gen III microplates. |
| Chemotaxonomy Standards | Reagents and protocols for analyzing cellular components that serve as taxonomic markers. | Reagents for analyzing cellular fatty acids (FAME), polar lipids, and respiratory quinones. |
The Polyphasic Toolkit: From 16S rRNA to Genomics in Laboratory and Clinical Practice
Within the framework of modern polyphasic taxonomy, the integration of genotypic data with phenotypic and chemotaxonomic information is paramount for a robust classification and identification of bacteria [4] [20]. This approach acknowledges the limitations of any single method and seeks a consensus by combining multiple datasets to achieve a stable and natural classification system [20]. The 16S ribosomal RNA (rRNA) gene sequencing serves as a foundational genotypic cornerstone in this framework, providing a universal and reliable method for determining the phylogenetic relationships of prokaryotes [21] [22].
The 16S rRNA gene is a approximately 1500 base-pair long genetic element found in all bacteria and archaea, featuring a mosaic of nine hypervariable regions (V1-V9) interspersed between conserved sequences [21] [23]. The conserved regions allow for the design of universal primers, enabling the amplification of the gene from a wide array of organisms, while the variable regions provide the phylogenetic signal necessary for taxonomic discrimination at various levels, from phylum to species [21] [24]. Its essential function in the ribosome, coupled with its evolutionary characteristics, has established it as the most widely used molecular chronometer for bacterial systematics [22].
This application note details standardized protocols for 16S rRNA gene sequencing and subsequent phylogenetic analysis, positioning these methodologies as critical components within a comprehensive polyphasic taxonomic workflow for researchers and drug development professionals.
Principles and Applications of 16S rRNA Gene Sequencing
The 16S rRNA Gene as a Molecular Marker
The 16S rRNA gene is a subunit of the 30S component of the prokaryotic ribosome, and its "S" designation refers to the Svedberg unit, which characterizes sedimentation rates [21] [24]. Its efficacy as a molecular marker stems from several key properties:
- Ubiquitous Distribution: It is present in virtually all bacteria and archaea, often in multiple copies (5-10) per genome, which enhances detection sensitivity [22] [24].
- Functional Stability: Its fundamental role in protein synthesis means that its function has remained constant over evolutionary time, implying that sequence changes are primarily a result of random, neutral evolution [22].
- Sequence Characteristics: The gene is of sufficient length (~1500 bp) to contain a substantial amount of informational data, and its structure of highly conserved regions flanking variable regions provides both anchors for universal PCR primers and sites for phylogenetic differentiation [21] [24].
Applications in Bacterial Identification and Taxonomy
16S rRNA gene sequencing is a culture-free method that has revolutionized microbial ecology and systematics. Its primary applications include:
- Identification of Uncultivable Bacteria: A significant proportion of environmental bacteria cannot be cultured using standard laboratory techniques. 16S sequencing allows for the identification and phylogenetic placement of these organisms directly from complex samples [21] [25].
- Polyphasic Taxonomy: In the description of novel taxa, 16S rRNA gene sequence analysis is the first and essential genotypic step. While a sequence similarity of less than 97% compared to known species often suggests a novel genus, similarities above 97% require confirmation via DNA-DNA hybridization (DDH) or other genomic methods to define species boundaries [4] [22].
- Microbiome Profiling: High-throughput sequencing of 16S rRNA amplicons enables the characterization of microbial community structure from diverse environments, including the human gut, soil, and water [23] [25]. This is critical for understanding host-microbe interactions in health and disease, and for discovering novel microbes with potential industrial or therapeutic value.
Table 1: Key Characteristics of the 16S rRNA Gene
| Characteristic | Description | Implication for Taxonomy |
|---|---|---|
| Universal Presence | Found in all bacteria and archaea. | Allows for a unified phylogenetic framework. |
| Conserved Regions | Sequences shared across broad taxonomic groups. | Enables design of universal PCR primers. |
| Hypervariable Regions | Nine regions (V1-V9) with genus- or species-specific signatures. | Provides the phylogenetic signal for discrimination. |
| Gene Length | ~1500 nucleotides. | Contains sufficient information for robust analysis. |
| Multiple Copies | Often 5-10 copies per genome. | May contain intragenomic sequence variation. |
Experimental Protocol for 16S rRNA Gene Sequencing
The following protocol outlines a standard workflow for 16S rRNA gene amplicon sequencing, from sample preparation to data generation.
Sample Collection and DNA Extraction
Critical Step: The integrity of the sample is paramount for obtaining accurate and reproducible results.
- Sample Collection: Collect samples (e.g., fecal material, soil, water, clinical swabs) using sterile containers to prevent exogenous contamination. For human-associated microbiota, consistent collection methods are vital [25].
- Preservation: Immediately freeze samples at -20°C or -80°C to preserve microbial composition. Avoid repeated freeze-thaw cycles. For field applications, use preservation buffers or liquid nitrogen for snap-freezing [25].
- DNA Extraction: Use commercially available DNA extraction kits suitable for the sample type. The protocol generally involves:
- Quality Control: Assess DNA concentration and purity using spectrophotometry (e.g., Nanodrop) or fluorometry (e.g., Qubit). Include negative (no-template) controls and positive controls (e.g., mock microbial communities) to monitor contamination and PCR efficacy [23] [25].
Library Preparation and Sequencing
This stage involves the targeted amplification of the 16S rRNA gene and preparation of the resulting amplicons for sequencing.
- Hypervariable Region Selection: Choose the target region(s) based on the required taxonomic resolution. While sequencing the full-length gene (V1-V9) provides the highest phylogenetic resolution [26], common partial-gene targets include the V3-V4 regions (~460 bp) for Illumina platforms [23]. The choice of region can influence the observed taxonomic profile due to differential amplification efficiencies across taxa [26].
- PCR Amplification: Perform amplification using primers that target the conserved regions flanking the chosen hypervariable region(s). Universal primer sets, such as 341F and 805R for V3-V4, are commonly used [23].
- Indexing and Library Construction: A second, limited-cycle PCR is used to attach unique dual-index barcodes and sequencing adapters to each sample's amplicons. This allows for the multiplexing of hundreds of samples in a single sequencing run [25].
- Library Clean-up: Purify the final PCR products using magnetic beads to remove primer dimers and other enzymatic reaction components [25].
- Sequencing: Pool the barcoded libraries in equimolar ratios and load onto a high-throughput sequencing platform. The Illumina MiSeq system is widely used for 16S studies due to its read length and output, which are well-suited for paired-end sequencing of regions like V3-V4 [23] [24]. For full-length 16S sequencing, Pacific Biosciences (PacBio) circular consensus sequencing (CCS) or Oxford Nanopore technologies are employed [26] [24].
Diagram 1: 16S rRNA Gene Sequencing and Analysis Workflow.
Bioinformatics and Phylogenetic Analysis Protocol
Raw sequencing data must be processed through a bioinformatics pipeline to derive biological insights. The following protocol is based on tools like QIIME 2 and Phyloseq.
Data Processing and Denoising
- Demultiplexing: Assign raw sequencing reads (in FASTQ format) to their respective samples based on the unique barcode sequences.
- Quality Filtering and Denoising: Use algorithms such as DADA2 to correct sequencing errors, remove chimeric sequences, and infer the exact biological sequences present in the sample, known as Amplicon Sequence Variants (ASVs) [23]. Unlike Operational Taxonomic Units (OTUs), which cluster sequences at an arbitrary identity threshold (e.g., 97%), ASVs discriminate sequences that differ by as little as a single nucleotide, providing higher resolution [23].
- Feature Table Construction: Generate a frequency table (BIOM format) containing the counts of each ASV in every sample.
Taxonomic Assignment and Phylogenetic Tree Building
- Taxonomic Classification: Assign taxonomy to each ASV using a naive Bayesian classifier trained on reference databases such as Greengenes, SILVA, or the Human Oral Microbiome Database (HOMD) [23]. This step links each ASV to a taxonomic lineage (e.g., Phylum, Class, Order, Family, Genus, Species).
- Multiple Sequence Alignment: Align all ASV sequences using tools like MAFFT [27]. This step is crucial for identifying homologous positions for phylogenetic inference.
- Phylogenetic Tree Estimation: Construct a phylogenetic tree from the aligned sequences using Bayesian inference or maximum likelihood methods. This tree represents the evolutionary relationships among the ASVs.
Protocol: Bayesian Phylogenetic Analysis with MrBayes
This protocol details a comprehensive workflow for Bayesian phylogenetic tree estimation [27].
- Software Requirements: Python, JAVA, PAUP*, MEGA X, MrModeltest, MrBayes.
- Procedure:
- A. Robust Sequence Alignment: Use GUIDANCE2 with MAFFT as the alignment tool to account for alignment uncertainty. Upload your multi-sequence FASTA file and select appropriate parameters (e.g.,
genafpairfor global alignment of longer sequences) [27]. - B. Sequence Format Conversion: Convert the resulting alignment (in FASTA format) to NEXUS format using MEGA X or PAUP, as required for downstream analysis [27].
- C. Evolutionary Model Selection: For nucleotide data, use MrModeltest (executed within PAUP) to determine the best-fit model of nucleotide substitution (e.g., GTR+I+Γ) using the Akaike Information Criterion (AIC) [27].
- D. Bayesian Inference in MrBayes: Execute MrBayes with the NEXUS file and the selected model. A typical command block within a MrBayes file includes: This runs a Markov Chain Monte Carlo (MCMC) analysis for 1 million generations, sampling every 1000 generations. Diagnostics are used to ensure chains have converged [27].
- E. Tree Visualization and Validation: The consensus tree generated by MrBayes can be visualized and annotated in tools like FigTree or the R package
ggtree.
- A. Robust Sequence Alignment: Use GUIDANCE2 with MAFFT as the alignment tool to account for alignment uncertainty. Upload your multi-sequence FASTA file and select appropriate parameters (e.g.,
Diagram 2: Bayesian Phylogenetic Tree Construction Workflow.
Statistical and Ecological Analysis
- Data Integration: Use the R package Phyloseq to integrate the feature table, taxonomic assignments, phylogenetic tree, and sample metadata into a single object for streamlined analysis [23].
- Alpha Diversity: Calculate within-sample diversity using metrics like the Shannon index, which combines richness (number of ASVs) and evenness (distribution of their abundances) [23].
- Beta Diversity: Quantify between-sample dissimilarities using metrics such as Bray-Curtis dissimilarity (composition-based) or UniFrac (phylogeny-based). Visualize patterns using ordination methods like Principal Coordinates Analysis (PCoA) [23].
- Differential Abundance Testing: Identify taxa that are statistically associated with specific sample groups (e.g., disease vs. control) using methods like the Linear Decomposition Model (LDM), which controls for multiple testing using the False Discovery Rate (FDR) [23].
Table 2: Comparison of 16S rRNA Sequencing Performance Across Hypervariable Regions
| Target Region | Approximate Length | Relative Taxonomic Resolution | Notes and Common Platforms |
|---|---|---|---|
| V1-V3 | ~510 bp | High | Good for Gram-positive bacteria; used on Roche 454 [24]. |
| V3-V4 | ~428 bp | Moderate-High | Common, well-balanced choice for Illumina MiSeq [23] [24]. |
| V4 | ~252 bp | Moderate | Most common region for Illumina HiSeq; lower resolution [26] [24]. |
| V6-V9 | ~548 bp | Variable | Best for Clostridium and Staphylococcus; used on Roche 454 [24]. |
| Full-Length (V1-V9) | ~1500 bp | Highest | Enables species- and strain-level resolution; requires PacBio or Nanopore [26] [24]. |
Table 3: Key Research Reagent Solutions for 16S rRNA Sequencing and Analysis
| Item | Function | Example Products/Kits |
|---|---|---|
| DNA Extraction Kit | Isolate genomic DNA from complex samples. | DNeasy PowerSoil Kit (Qiagen), MagMAX Microbiome Kit (Thermo Fisher) |
| 16S PCR Primers | Amplify specific hypervariable regions of the 16S gene. | 341F/805R (V3-V4), 27F/534R (V1-V3) |
| Library Prep Kit | Prepare amplicon libraries for sequencing by adding indices and adapters. | Illumina DNA Prep, KAPA HiFi HotStart ReadyMix |
| Positive Control | Mock microbial community with known composition to validate the entire workflow. | ZymoBIOMICS Microbial Community Standard |
| Bioinformatics Pipelines | Process raw sequencing data, perform denoising, taxonomy assignment, and diversity analysis. | QIIME 2, mothur, DADA2 |
| Reference Databases | Curated collections of 16S sequences for taxonomic classification. | Greengenes, SILVA, RDP, HOMD |
Discussion: Integration into a Polyphasic Taxonomy Framework
While 16S rRNA gene sequencing is a powerful tool, its limitations must be recognized within a polyphasic taxonomy paradigm.
- Taxonomic Resolution: 16S sequencing often provides genus-level identification but can fail to distinguish between closely related species, as some species share identical or near-identical 16S sequences (e.g., Bacillus globisporus and B. psychrophilus share >99.5% similarity) [22]. For definitive species assignment, DNA-DNA hybridization (DDH), the historical gold standard, or newer genome-based methods like Average Nucleotide Identity (ANI) are required [28] [22].
- Intragenomic Heterogeneity: The presence of multiple, slightly different copies of the 16S rRNA gene within a single genome can complicate analysis. Modern full-length sequencing platforms are now accurate enough to resolve these intragenomic copy variants, which can potentially be used for strain-level discrimination [26].
- Functional Insights: A significant limitation of 16S sequencing is its inability to directly infer the functional potential of a microbial community. This gap is typically addressed by shotgun metagenomic sequencing, which sequences all the genomic content in a sample and allows for functional profiling [21] [25].
Therefore, in a comprehensive polyphasic approach, 16S rRNA gene sequencing serves as the initial, high-throughput screening tool to determine phylogenetic placement and community structure. Its findings are then validated and supplemented with other genotypic (DDH, ANI, whole-genome sequencing), phenotypic (morphological, physiological, biochemical), and chemotaxonomic (fatty acid analysis, isoprenoid quinones) data to achieve a consensus classification that truly reflects the natural relationships among bacteria [4] [28] [20].
For nearly 50 years, DNA-DNA hybridization (DDH) has served as the gold standard for prokaryotic species circumscriptions at the genomic level, providing a numerical and relatively stable species boundary that has profoundly influenced the construction of modern microbial taxonomy [29]. This methodological cornerstone has enabled taxonomists to establish a pragmatic species concept for Bacteria and Archaea, despite the challenges posed by the limited morphological features available for microbial differentiation [30] [31]. The technique measures the overall genetic similarity between whole genomes, offering a comprehensive genomic comparison that single-gene analyses cannot provide [4]. Within the framework of polyphasic taxonomy, which integrates phenotypic, genotypic, and phylogenetic data, DDH has provided the definitive genomic evidence for species delineation, creating a classification system that remains both operative and predictive for various microbiology disciplines [29] [4].
Principles of DNA-DNA Hybridization
The fundamental principle of DDH relies on the thermodynamic properties of DNA reassociation. When double-stranded DNA from two organisms is denatured by heating and subsequently allowed to reanneal, hybrid duplexes form between complementary strands from different organisms. The stability of these hybrid duplexes, reflected in their melting temperature, directly correlates with the degree of sequence complementarity between the two genomes [30] [32].
The DDH value is expressed as the relative binding ratio compared to the homologous recombination, where 100% represents perfect sequence complementarity (self-hybridization), and decreasing percentages reflect increasing genetic divergence. The generally accepted threshold for species delimitation is 70% DDH similarity, with strains exhibiting values above this threshold considered members of the same species [30] [33]. This 70% boundary was established through extensive empirical studies showing that it generally corresponds to clear-cut clusters of organisms with high phenotypic coherence [29].
Figure 1: DDH Experimental Workflow. The process begins with DNA extraction from both reference and test strains, followed by mechanical shearing, denaturation, hybridization, and melting profile analysis to determine genetic similarity.
Established DDH Methodologies and Protocols
Conventional DDH Methods
Several laboratory methods have been developed to determine DDH values, each with specific technical considerations and applications:
3.1.1 Hydroxyapatite Method This classical approach exploits the differential binding of single-stranded and double-stranded DNA to hydroxyapatite columns. Following hybridization, the column is subjected to stepwise temperature increases, and the amount of DNA eluted at each temperature is quantified to determine the thermal stability of hybrid duplexes [30] [32].
3.1.2 S1 Nuclease Method The S1 nuclease technique utilizes the enzyme's specific activity against single-stranded DNA. After hybridization, S1 nuclease digests any unhybridized single-stranded regions, and the remaining double-stranded DNA is quantified. The proportion of nuclease-resistant hybrid DNA indicates the sequence similarity between the two genomes [30].
3.1.3 Renaturation Kinetics Method This method measures the initial rate of DNA reassociation by monitoring the decrease in absorbance at 260 nm (hypochromic effect) as single-stranded DNA forms double-stranded complexes. The similarity between two genomes is calculated by comparing the renaturation rate of the hybrid mixture to the renaturation rates of the homologous controls [30].
Table 1: Comparison of Major DDH Methodological Approaches
| Method | Principle | Key Steps | Advantages | Limitations |
|---|---|---|---|---|
| Hydroxyapatite | Differential binding to hydroxyapatite based on strandedness | Stepwise temperature elution from columns | Direct measurement of duplex stability | Labor-intensive, requires precise temperature control |
| S1 Nuclease | Enzymatic digestion of single-stranded DNA | Hybridization → S1 nuclease treatment → quantification | Specific for duplex DNA | Enzyme activity variability, optimization required |
| Renaturation Kinetics | Spectrophotometric monitoring of reassociation rate | Absorbance measurement at 260nm over time | No labeling required, continuous monitoring | Lower sensitivity, requires high DNA purity |
| Microplate | Colorimetric detection using biotin-streptavidin | Hybridization in microplates, enzymatic detection | High-throughput, suitable for multiple samples | Requires DNA labeling, additional steps |
Detailed Microplate DDH Protocol
The microplate method, developed in 2004, represents a more recent advancement that increases throughput and reduces the sample processing time [32]. The following protocol provides a detailed methodology for implementing this approach:
Reagents and Materials:
- Purified genomic DNA from reference and test strains
- Photobiotin labeling kit
- Streptavidin-coated microplates
- Peroxidase-conjugated anti-DNA antibody
- Enzyme substrate (e.g., TMB) and stop solution
- Hybridization buffer (e.g., 50% formamide, 10x SSC, 0.1% SDS)
- Washing buffers of varying stringency
Procedure:
- DNA Preparation and Labeling
- Extract high-molecular-weight DNA using standard phenol-chloroform methods.
- Mechanically shear DNA to fragments of 600-800 bp using a sonicator or French press.
- Label reference DNA with photobiotin according to manufacturer's instructions.
Hybridization
- Mix 2 μg of biotinylated reference DNA with 10 μg of unlabeled test DNA in hybridization buffer.
- Denature at 95°C for 10 minutes in a thermal cycler.
- Incubate at optimal hybridization temperature (typically 25-30°C below Tm) for 16 hours.
Capture and Detection
- Transfer hybridization mixture to streptavidin-coated microplates.
- Incubate for 1 hour at room temperature to capture biotinylated hybrids.
- Wash plates with decreasing stringency buffers to remove non-specifically bound DNA.
- Add peroxidase-conjugated anti-DNA antibody and incubate for 1 hour.
- Develop with enzyme substrate and measure absorbance at appropriate wavelength.
Calculation and Interpretation
- Calculate DDH value as (absorbance of heterologous hybrid/absorbance of homologous control) × 100%.
- Include appropriate controls: homologous hybridization (100% reference), unrelated strain hybridization (background), and no-DNA control.
Troubleshooting Notes:
- Ensure DNA fragment size uniformity for reproducible results.
- Optimize hybridization temperature based on the G+C content of the organisms.
- Include replicate hybridizations to assess technical variability.
- Validate method with known related and unrelated strains before testing unknowns.
The Research Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for DDH Experiments
| Reagent/Material | Function | Application Notes |
|---|---|---|
| High-Purity Genomic DNA | Source of genetic material for hybridization | Must be free of contaminants; recommended A260/A280 ratio of 1.8-2.0 |
| Hydroxyapatite | Chromatographic medium for separating single and double-stranded DNA | Requires calibration with control DNA before experimental use |
| S1 Nuclease | Digests single-stranded DNA regions | Activity must be standardized; concentration optimization required |
| Photobiotin | Non-radioactive label for DNA detection | Alternative to radioactive labeling; enables colorimetric detection |
| Streptavidin-Coated Microplates | Solid support for capturing biotinylated DNA complexes | Enables high-throughput processing of multiple samples |
| Formamide | Denaturing agent in hybridization buffer | Reduces melting temperature, allowing lower hybridization temperatures |
| SSC Buffer (Saline-Sodium Citrate) | Regulates stringency in hybridization and washing | Higher concentration increases stringency; critical for specificity |
DDH in the Genomic Era: Transition to Digital Approaches
The advent of rapid and affordable genome sequencing has prompted the development of in silico replacements for wet-lab DDH [29] [33]. These digital approaches overcome several limitations of traditional DDH, including the inability to build cumulative databases and the technical challenges associated with experimental reproducibility [29].
Average Nucleotide Identity (ANI)
The Average Nucleotide Identity approach calculates the average nucleotide-level similarity between homologous regions of two genomes [29]. Initially implemented in the JSpecies software package, ANI can be calculated using BLAST-based (ANIb) or MUMmer-based (ANIm) algorithms [29]. Extensive comparisons have demonstrated that an ANI value of 95-96% generally corresponds to the traditional 70% DDH threshold for species delineation [29] [34].
Digital DDH (dDDH) and Genome-Blast Distance Phylogeny (GBDP)
The Genome-to-Genome Distance Calculator (GGDC) implements digital DDH calculations using the Genome Blast Distance Phylogeny (GBDP) approach [35] [33]. This method infers genome-to-genome distances between entirely or partially sequenced genomes, providing a highly reliable digital estimator for genomic relatedness that closely mimics wet-lab DDH values [33] [31]. The GBDP method has been shown to yield higher correlations with wet-lab DDH than other computational approaches and includes confidence interval estimation for statistical evaluation of results [33].
Figure 2: Modern Genome-Based Taxonomy Workflow. The transition to digital approaches enables cumulative databases, statistical confidence estimates, and reproducible species delineation decisions within the polyphasic taxonomy framework.
Correlation Between Traditional and Digital Methods
Comparative studies have established robust correlations between traditional DDH and genome-based parameters, though these relationships can vary between taxonomic groups. Recent research on Amycolatopsis species revealed that a 70% dDDH value corresponded to approximately 96.6% ANIm, slightly higher than the generally accepted 95-96% threshold [34]. This highlights the importance of considering taxon-specific variations when applying these digital thresholds.
Table 3: Comparison of Species Delineation Methods in Prokaryotic Taxonomy
| Method | Principle | Species Threshold | Advantages | Limitations |
|---|---|---|---|---|
| Traditional DDH | DNA reassociation kinetics | 70% similarity | Established gold standard, whole-genome comparison | No cumulative database, technically demanding, variable results |
| 16S rRNA Sequencing | Single gene sequence similarity | <97% suggests different species | Rapid, extensive databases available | Limited resolution, conservative nature |
| Average Nucleotide Identity (ANI) | Genome-wide average nucleotide identity | 95-96% | Robust, cumulative database | Requires genome sequences |
| Digital DDH (dDDH) | Genome-to-genome distance calculation | 70% similarity | High correlation with DDH, confidence intervals | Requires genome sequences, computational resources |
| Multilocus Sequence Analysis | Concatenated housekeeping gene sequences | Sequence type clusters | Higher resolution than 16S rRNA | Gene selection bias, primer availability |
DDH in Polyphasic Taxonomy: Current Status and Applications
Within the modern polyphasic taxonomy framework, DDH and its genomic equivalents remain crucial elements for species delineation, particularly when 16S rRNA gene sequence similarity exceeds 97% [4] [30]. The polyphasic approach integrates genotypic, phenotypic, and phylogenetic data to obtain a comprehensive characterization of microbial taxa [4]. While wet-lab DDH is still required for the description of novel taxa in some cases, there is increasing acceptance of digital genomic methods such as dDDH and ANI as valid alternatives [32] [11].
The transition to genome-based taxonomy continues to accelerate, with initiatives such as the Type Strain Genome Server (TYGS) providing user-friendly platforms for prokaryote taxonomy using whole-genome sequence data [35]. This evolution from traditional DDH to genomic taxonomy represents a natural progression similar to the earlier transition from DNA:rRNA hybridization to 16S rRNA gene sequencing, enabling the construction of cumulative databases that support incremental advances in microbial systematics [29] [33].
The classification of microorganisms has evolved significantly from reliance on traditional morphological, physiological, and biochemical methods. These classical approaches often create a blurred image about the taxonomic status of microbes and thus require further clarification using more robust techniques [4]. This need has led to the adoption of a polyphasic approach, a consensus method for bacterial systematics that integrates all available genotypic, phenotypic, and chemotaxonomic data to determine the precise taxonomic position of microbes [4] [20]. Within this framework, genetic analysis forms the cornerstone, and Multilocus Sequence Analysis (MLSA) has emerged as a powerful phylogenetic tool for elucidating the relationships between closely related bacterial species and genera [36].
MLSA involves the analysis of partial sequences of multiple housekeeping genes—essential genes with conserved functions that are present in all microbes. Unlike the 16S rRNA gene, which is highly conserved and often lacks resolution at the species level, protein-coding housekeeping genes such as gyrB (DNA gyrase subunit B) and rpoB (RNA polymerase beta subunit) evolve more rapidly, providing a finer taxonomic resolution [37] [38]. By comparing sequences of multiple genes, MLSA minimizes the impact of horizontal gene transfer and recombination events, offering a more stable and reliable phylogenetic reconstruction than single-gene analyses [36] [38]. This protocol outlines the detailed application of MLSA, focusing on gyrB and rpoB, within the context of modern polyphasic taxonomy.
Principles and Applications of MLSA
Theoretical Basis for Gene Selection
The selection of appropriate housekeeping genes is critical for a successful MLSA scheme. Ideal genes are ubiquitously present, functionally conserved, and distributed as single copies in the genome. They should also possess a degree of sequence variability sufficient to discriminate between closely related lineages [36] [38]. The genes gyrB and rpoB meet these criteria effectively.
- gyrB: This gene encodes the B subunit of DNA gyrase, an essential enzyme involved in DNA replication. It has been established as a reliable molecular chronometer due to its sequence conservation and has been widely used for phylogenetic analyses in numerous bacterial genera, including Pseudomonas and Aeromonas [39] [40].
- rpoB: This gene encodes the beta subunit of RNA polymerase. Studies have demonstrated that rpoB offers a higher level of taxonomic discrimination than the 16S rRNA gene, enabling more accurate species-level identification [39] [37]. Its use is particularly valuable in metabarcoding studies, where it shows higher sensitivity and specificity compared to 16S rRNA markers [37].
The following table summarizes the advantages of these genes over the 16S rRNA gene.
Table 1: Comparison of Phylogenetic Markers
| Feature | 16S rRNA Gene | gyrB | rpoB |
|---|---|---|---|
| Evolutionary Rate | Slow, highly conserved | Faster | Faster |
| Taxonomic Resolution | Poor at species level [37] [38] | High at species and sub-species level [39] | High at species and sub-species level [39] [37] |
| Copy Number | Multiple, often heterogeneous [4] [37] | Typically single [37] | Typically single [37] |
| Primary Application | Genus-level phylogeny, initial identification | Species-level phylogeny, MLSA [40] [38] | Species-level phylogeny, MLSA, metabarcoding [37] |
| Example Discriminatory Power | Unable to separate some Thioclava species [38] | Maximum interspecies divergence in Aeromonas: 10% [39] | Maximum interspecies divergence in Aeromonas: 9% [39] |
Applications in Prokaryotic Taxonomy and Clinical Diagnostics
MLSA has become a widely accepted method for clarifying phylogenetic relationships within a genus or family [36]. Its applications are diverse:
- Species Delineation: MLSA has been proposed as a replacement for DNA-DNA hybridization (DDH) in species delineation. It provides a reproducible and standardized framework for defining species boundaries [36] [38].
- Resolution of Complex Taxa: For genera where 16S rRNA gene sequencing is insufficient, such as Thioclava, MLSA schemes based on concatenated housekeeping genes (e.g., gyrB, rpoD, dnaK, trpB, recA) have successfully resolved distinct clades corresponding to established and novel species [38].
- Clinical Identification: Biochemical identification of clinically relevant bacteria like Aeromonas can be unreliable. Multiplex PCR assays targeting gyrB and rpoB enable rapid and accurate identification of species such as A. hydrophila, A. caviae, A. media, and A. veronii from clinical samples [39].
- Microbiome Studies: In metabarcoding, the rpoB gene demonstrates higher specificity and sensitivity compared to the 16S rRNA V3-V4 region, reducing spurious OTU detection and providing more accurate taxonomic affiliation in complex communities [37].
Experimental Protocols
The following diagram illustrates the comprehensive workflow for an MLSA study, from strain selection to phylogenetic inference.
Detailed Laboratory Methods
Genomic DNA Extraction
High-quality, pure genomic DNA is a prerequisite for successful PCR amplification.
- Protocol: Use a commercial bacterial genomic DNA extraction kit. For Gram-negative strains, a simplified preparation for PCR can involve boiling a bacterial colony in 10% Chelex 100 resin, followed by centrifugation and dilution of the supernatant [39].
- Quality Control: Assess DNA concentration and purity using a spectrophotometer (e.g., Nanodrop). Acceptable A260/A280 ratios are typically between 1.8 and 2.0.
PCR Amplification of gyrB and rpoB
This section provides specific primer sequences and cycling conditions for amplifying these genes.
Table 2: Primer Sequences for gyrB and rpoB Amplification
| Gene | Primer Name | Sequence (5' → 3') | Amplicon Size | Target Group | Source |
|---|---|---|---|---|---|
| gyrB | gyrB-F | AGCATYAARGTGCTGAARGG | ~1461-1467 bp | Pseudomonas genus [40] | Designed |
| gyrB-R | GGTCATGATGATGATGTTGTG | ||||
| gyrB | UP-1 | GAAGTCATCATGACCGTTCTGCAYGCNGGNGGNAARTTYGA | ~1273 bp | General / Aeromonas [39] | Literature |
| UP-2r | AGCAGGGTACGGATGTGCGAGCCRTCNACRTCNGCRTCNGTCAT | ||||
| rpoB | Pas-rpoB-L | TGGCCGAGAACCAGTTCCGCGT | ~560 bp | General / Aeromonas [39] | Literature |
| Rpob-R | CGTTGCATGTTGGTACCCAT |
PCR Reaction Mixture (25 µL volume):
- 1x PCR Buffer (containing 2.0 mM MgCl₂)
- 0.2 mM of each dNTP
- 0.2 - 0.5 µM of each primer (see Table 2 for specifics)
- 1 U of DNA Polymerase (e.g., Platinum Taq)
- 5% (v/v) DMSO (for difficult templates, like Pseudomonas [40])
- 1-5 µL of template DNA (10-30 ng/µL)
PCR Cycling Conditions:
A typical thermocycling program is as follows [39] [40]:
- Initial Denaturation: 94-95°C for 2-5 minutes.
- Amplification (30-35 cycles):
- Denaturation: 94°C for 30-40 seconds.
- Annealing: 55-67°C for 40-50 seconds (temperature must be optimized for primer pair).
- Extension: 72°C for 40-90 seconds (1 min/kb).
- Final Extension: 72°C for 5-10 minutes.
Sequencing and Sequence Analysis
- Purification and Sequencing: Purify PCR products using a standard PCR purification kit. Perform Sanger sequencing using the same PCR primers or internal sequencing primers for longer amplicons [39].
- Sequence Assembly and Curation: Use software like CLC DNA Workbench or MEGA to assemble contigs from forward and reverse sequences, visually inspect chromatograms, and trim low-quality ends.
- Multiple Sequence Alignment: Align curated sequences from all strains under study using algorithms like MUSCLE [40] [38].
- Phylogenetic Analysis: Construct phylogenetic trees using methods such as Maximum-Likelihood or Neighbor-Joining. For MLSA, concatenate the aligned sequences of all genes (e.g., gyrB + rpoB) into a single supermatrix before tree construction [39] [38]. Support for tree nodes is typically assessed using bootstrap analysis (e.g., 1000 replicates).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for MLSA
| Reagent / Material | Function / Application | Example / Note |
|---|---|---|
| Chelex 100 Resin | Rapid preparation of crude DNA template for PCR. | Ideal for high-throughput screening [39]. |
| Marine Broth/Agar 2216 | Cultivation of marine bacteria. | Used for growing genera like Thioclava [38]. |
| Nutrient Agar/Broth | Standard medium for cultivation of common bacteria. | Used for Pseudomonas and Aeromonas [39] [40]. |
| High-Fidelity DNA Polymerase | PCR amplification with low error rate. | Critical for obtaining accurate sequences for phylogenetic analysis. |
| DMSO (Dimethyl Sulfoxide) | PCR additive. | Helps amplify GC-rich templates or difficult amplicons [40]. |
| PCR Purification Kit | Removal of primers, dNTPs, and enzymes post-amplification. | Essential step before sequencing. |
| Sanger Sequencing Services | Determination of nucleotide sequence of PCR amplicons. | Outsourcing to specialized companies is common. |
Data Analysis and Interpretation
Quantitative Analysis of Sequence Data
The power of gyrB and rpoB for discrimination can be quantified by calculating sequence divergence. The following table summarizes data from a study on Aeromonas species.
Table 4: Inter- and Intraspecies Sequence Divergence of gyrB and rpoB in Aeromonas
| Species | Maximum Intraspecies Divergence (gyrB) | Maximum Intraspecies Divergence (rpoB) |
|---|---|---|
| A. veronii | 5.0% (53/1113 nt) [39] | 2.3% (9/390 nt) [39] |
| A. hydrophila | 2.3% (26/1113 nt) [39] | 1.5% (6/390 nt) [39] |
| A. caviae | 2.5% (28/1113 nt) [39] | 1.3% (5/390 nt) [39] |
| A. media | 3.1% (35/1113 nt) [39] | Not specified |
| All species | Max Interspecies: 10% [39] | Max Interspecies: 9% [39] |
Integration with Other Taxonomic Methods
The final step in a polyphasic taxonomy is to integrate MLSA results with other data types. The following diagram illustrates this integrative logic.
MLSA phylogenies should be validated against other genomic standards where possible. For instance, in the study of Thioclava, the clades defined by MLSA were reconfirmed by digital DNA-DNA hybridization (dDDH) and Average Nucleotide Identity (ANI) analyses based on whole-genome sequences [38]. A similarity of 97.3% in the MLSA was proposed as a soft threshold for species demarcation in that genus. Furthermore, biochemical profiles and physiological tests remain essential for providing a complete phenotypic characterization that matches the genotypic clustering.
The classification of microbial diversity into species is a keystone for understanding the ecological role of microorganisms and the evolutionary processes that shape them. For decades, the taxonomic framework for prokaryotes relied heavily on phenotypic characteristics and limited genetic methods. DNA–DNA hybridization (DDH) and 16S rRNA gene sequencing provided initial pathways for understanding microbial diversity but came with significant limitations: the former required closely related isolates, while the latter lacked species-level resolution [41]. The genomic revolution has fundamentally transformed this landscape by introducing whole-genome sequencing (WGS) as the highest resolution method for characterizing pathogen evolution, epidemiology, and diagnostics [42].
WGS provides an unbiased and complete view of the microbial genome, enabling the discovery of genetic variation without the technical limitations of other genotyping technologies [43]. This advancement has made genetic methods, particularly Average Nucleotide Identity (ANI) and digital DNA-DNA Hybridization (dDDH), the cornerstone of modern prokaryotic species delineation. These genome-based metrics have established a practical, robust framework for bacterial identification that forms the backbone of modern ecological genomics [41]. The integration of these genomic tools into a polyphasic taxonomy approach—which combines genomic, phenotypic, and phylogenetic data—delivers a comprehensive understanding of microbial relationships and functions, as demonstrated in the recent discovery and classification of Desertivibrio insolitus, a novel psychrotolerant actinobacterium [44].
Core Genomic Concepts and Calculations
Average Nucleotide Identity (ANI)
Average Nucleotide Identity is a simple yet powerful bioinformatic metric for consistently determining the relatedness between two microbial genomes. Introduced in 2005 by Konstantinidis and Tiedje, ANI represents the mean nucleotide identity derived from the sequence-based comparison of orthologous genes or genomic fragments between two genomes [41]. The original implementation involved comparing predicted gene sequences from a query to a closely related reference genome, then determining the mean identity between the selected matches. A subsequent variation introduced in 2007 used 1,020 contiguous nucleotide fragments derived from the query genome to mirror the DNA fragmentation in traditional DDH approaches [41].
The calculation of ANI typically involves one of two primary methods:
- BLAST-based ANI (ANIb): Uses the BLAST algorithm to identify homologous regions and calculates average identity.
- MUMmer-based ANI (ANIm): Uses MUMmer to identify maximal unique matches and calculates identity.
Modern implementations often leverage k-mer-based alignment-free approaches, which considerably accelerate calculations while maintaining accuracy [41]. The established species boundary for prokaryotes using ANI is approximately 95-96%, meaning two isolates sharing ANI values above this threshold are likely members of the same species, a correlation derived from the original genomic studies that laid the foundations for the eco-evolutionary interpretation of microbial genomics [41].
Recent research has explored ANI applications beyond bacterial classification. Studies of bacterial dsDNA viruses revealed a multimodal ANI distribution with a distinct gap around 80%, akin to the bacterial ANI gap (~90%) but shifted, likely due to viral-specific evolutionary processes such as recombination dynamics and mosaicism [45]. This highlights the metric's versatility while underscoring the need for careful interpretation in different biological contexts.
Digital DNA-DNA Hybridization (dDDH)
Digital DDH represents the computational counterpart to the wet-lab DDH method that served as the historical gold standard for prokaryotic species delineation. This method calculates the in silico equivalent of the hybridization similarity between two genomes, providing values that closely correlate with traditional DDH measurements [44]. The dDDH method is typically implemented through web-based platforms like the Genome-to-Genome Distance Calculator (GGDC), which uses models to predict DDH values from genome sequences.
The dDDH approach offers several advantages over its wet-lab predecessor:
- Reproducibility: Eliminates experimental variability between laboratories
- Accessibility: Allows comparison without physical strain availability
- Scalability: Enables rapid comparison of multiple genomes simultaneously
The standard species threshold for dDDH is approximately 70% similarity, corresponding to the traditional DDH cutoff for species definition [44]. This value shows strong correlation with the ANI species boundary of 95-96%, providing complementary evidence for taxonomic decisions.
Comparative Analysis of Genomic Identity Metrics
Table 1: Comparison of Genomic Identity Metrics for Microbial Taxonomy
| Metric | Methodology | Species Threshold | Key Advantages | Limitations |
|---|---|---|---|---|
| ANI | Calculates mean nucleotide identity of orthologous genes or genomic fragments | 95-96% [41] | High resolution; automated calculation; robust for closely related genomes | Limited discrimination at higher taxonomic ranks; requires genome sequences |
| dDDH | Computes in silico equivalent of laboratory DDH using GGDC | ~70% [44] | Direct correlation with traditional method; established historical context | Model-dependent; less suitable for highly divergent genomes |
| 16S rRNA | Compares sequence identity of the 16S ribosomal RNA gene | ~98.7% [41] | Universal; extensive database; rapid preliminary analysis | Limited resolution at species level; single gene does not reflect whole genome |
| AAI | Computes average amino acid identity of orthologous proteins | ~95% (varies by group) | Functional insights; more stable than nucleotide identity | Requires high-quality annotation; computationally intensive |
The relationship between these metrics provides a robust framework for taxonomic decisions. ANI and dDDH show strong correlation, with ANI values of 95-96% approximately equivalent to dDDH values of 70% [41] [44]. In polyphasic taxonomy, these genomic indices are interpreted alongside other genomic relatedness indices such as Percentage of Conserved Proteins (PCP) and Amino Acid Identity (AAI) to build a comprehensive understanding of microbial relationships [44].
Experimental Protocols for Genomic Taxonomy
Whole-Genome Sequencing Workflow
The genomic taxonomy pipeline begins with high-quality whole-genome sequencing. Recent advances have made both short-read and long-read technologies viable for microbial genomics, with each offering distinct advantages [42].
Table 2: Comparison of Sequencing Platforms for Microbial Genomics
| Platform/Technology | Read Length | Key Strengths | Considerations for Taxonomy | Example Systems |
|---|---|---|---|---|
| Short-read Sequencing | 50-300 bp | High accuracy per base; cost-effective for large-scale projects [42] | May struggle with repetitive regions; requires assembly | Illumina NovaSeq X [46] |
| Long-read Sequencing | 10,000+ bp | Spans repetitive regions; produces more complete genomes [42] [47] | Historically higher error rates, though improved in latest platforms [42] | Oxford Nanopore Technologies; PacBio SMRT [47] |
| Hybrid Approaches | Combination of both | Leverages accuracy of short reads with continuity of long reads | Higher cost and computational requirements | Illumina + Nanopore combination [42] |
Protocol: Genome Sequencing and Assembly for Taxonomic Studies
Sample Preparation and Sequencing
- DNA Extraction: Use standardized kits to obtain high-molecular-weight DNA. Assess quality using spectrophotometry (A260/A280 ratio ~1.8-2.0) and gel electrophoresis to ensure high molecular weight without degradation.
- Library Preparation:
- For short-read platforms: Fragment DNA to appropriate size (typically 300-800 bp) using enzymatic or mechanical shearing. Ligate platform-specific adapters, optionally including barcodes for multiplexing [47].
- For long-read platforms: Procedures differ by technology. For Nanopore: Use ligation sequencing kits without fragmentation to maintain read length. For PacBio: Prepare SMRTbell libraries according to manufacturer specifications.
- Sequencing: Execute sequencing run according to platform specifications. For Illumina, aim for ≥50x coverage; for long-read technologies, ≥30x coverage often suffices due to more complete assemblies [42].
Quality Control and Assembly
- Quality Assessment: Process raw data with FastQC to evaluate read quality, GC content, and potential contaminants. Perform quality trimming with tools like Trimmomatic or PRINSEQ to remove low-quality bases and adapter sequences [47].
- Genome Assembly:
- For short reads: Use de Bruijn graph-based assemblers such as Velvet or SPAdes with optimized k-mer sizes [47].
- For long reads: Utilize overlap-layout-consensus assemblers such as SMARTdenovo or Canu [47].
- Hybrid approach: Combine short and long reads using assemblers like Unicycler for optimal continuity and accuracy [42].
- Assembly Assessment: Evaluate assembly quality using QUAST to generate metrics including number of contigs, N50, and total assembly size. Check for completeness with BUSCO or CheckM.
Protocol: Calculating ANI and dDDH for Species Delineation
ANI Calculation
- Data Input: Prepare assembled genomes in FASTA format. Ensure comparable assembly quality between compared genomes.
- Method Selection: Choose ANI calculation method based on needs:
- For highest accuracy with closely related genomes: Use BLAST-based ANI (ANIb) as implemented in tools like pyani or Kostas lab protocols [41].
- For rapid comparison of multiple genomes: Use k-mer-based methods such as FastANI [41].
- For viral genomes or highly divergent sequences: Consider specialized tools like MANIAC, optimized for ANI estimation around 70% [45].
- Execution: Run ANI calculation with default parameters initially. For borderline cases (94-96% ANI), verify with multiple methods and inspect alignment fractions.
- Interpretation: Apply the species boundary threshold of 95-96% ANI. Values above this range indicate organisms likely belong to the same species [41].
dDDH Calculation
- Platform Access: Access the Genome-to-Genome Distance Calculator (GGDC) web service or stand-alone version.
- Genome Submission: Upload genome sequences in FASTA format. Ensure chromosomal molecules are appropriately designated.
- Model Selection: Choose appropriate calculation model based on genome completeness:
- Model 1: Suitable for draft genomes with potential fragmentation
- Model 2: Recommended for complete genomes
- Model 3: Conservative estimate accounting for various uncertainties
- Result Analysis: Review calculated dDDH values with confidence intervals. Values ≥70% support species-level relatedness [44]. Correlate with ANI results for consensus.
Workflow Integration in Polyphasic Taxonomy
The genomic analyses are integrated into a comprehensive taxonomic workflow as demonstrated in the discovery of Desertivibrio insolitus [44]:
Diagram 1: Polyphasic taxonomy workflow integrating genomic and phenotypic approaches
Essential Research Tools and Reagents
Table 3: Research Reagent Solutions for Genomic Taxonomy
| Category | Specific Tools/Reagents | Function in Workflow | Implementation Examples |
|---|---|---|---|
| Sequencing Platforms | Illumina NovaSeq X; Oxford Nanopore MinION | Generate raw sequence data | Illumina for high-accuracy short reads; Nanopore for long reads and rapid turnaround [42] [46] |
| DNA Extraction Kits | High-molecular-weight DNA extraction kits | Obtain pure, undegraded DNA | Critical for long-read sequencing; ensures maximum read length |
| Library Preparation | Illumina DNA Prep; Nanopore Ligation Sequencing Kit | Prepare sequencing libraries | Barcoding enables multiplexing of multiple samples [47] |
| Assembly Software | Velvet Optimiser; SPAdes; SMARTdenovo | reconstruct genomes from reads | Choice depends on read type and project goals [47] |
| Quality Assessment | FastQC; QUAST; CheckM | Evaluate data quality and assembly completeness | Identifies potential issues before analysis [47] |
| ANI Calculation | FastANI; pyani; MANIAC (for viruses) | Compute average nucleotide identity | FastANI for speed; pyani for detailed analysis [41] [45] |
| dDDH Platform | Genome-to-Genome Distance Calculator (GGDC) | Calculate digital DDH values | Web-based or standalone version [44] |
| Annotation Tools | Prokka; RAST | Identify genomic features | Provides functional context for taxonomic decisions [47] [44] |
Applications and Case Studies
Microbial Pathogen Epidemiology
WGS has become indispensable for tracking microbial pathogen evolution and transmission. A 2025 comparative study demonstrated that Oxford Nanopore long-read sequencing now produces sufficiently accurate data for bacterial whole-genome assembly and epidemiology [42]. The research found that assemblies from long reads were more complete than those from short-read data and contained few sequence errors. Importantly, the study established that computationally fragmenting long reads can improve the accuracy of variant calling in population-level studies, allowing researchers to incorporate the advantages of Nanopore sequencing for genome assembly while maintaining high accuracy in epidemiology and population analyses [42].
Novel Species Discovery
The integration of WGS with ANI and dDDH has accelerated the discovery and classification of novel microorganisms. The identification of Desertivibrio insolitus exemplifies the modern polyphasic approach [44]. Researchers sequenced the genome using Illumina technology, assembled it into a draft genome, then calculated ANI and dDDH values against closely related taxa. These genomic indices demonstrated that the strain represented a novel genus and species, which was further supported by phenotypic characterization and metabolic analysis through genome mining [44].
Large-Scale Genomic Initiatives
Massive WGS projects like the UK Biobank, which sequenced 490,640 participants, demonstrate the power of genomic approaches at scale [43]. While focused on human genetics, the methodologies and bioinformatic pipelines developed for such projects directly inform microbial genomics. The UK Biobank effort identified approximately 1.5 billion variants—a greater than 40-fold increase in observed human variation compared to whole-exome sequencing—highlighting how comprehensive WGS captures genetic diversity that targeted approaches miss [43].
The genomic revolution has fundamentally redefined bacterial taxonomy by providing unambiguous, data-driven criteria for species delineation. The integration of WGS with computational metrics like ANI and dDDH has created a robust framework for classifying microbial diversity that surpasses traditional methods in resolution, reproducibility, and scalability. As sequencing technologies continue to evolve—with platforms like the Illumina NovaSeq X delivering higher accuracy and coverage across challenging genomic regions [46]—and bioinformatic tools become more sophisticated, genomic taxonomy will continue to refine our understanding of microbial relationships.
The emerging trends in genomic analysis will further enhance taxonomic practices:
- AI-Enhanced Analysis: Tools like Google's DeepVariant utilize deep learning to identify genetic variants with greater accuracy than traditional methods [48].
- Single-Cell Genomics: Reveals heterogeneity within populations and enables taxonomy of unculturable organisms.
- Integration with Multi-Omics: Combines genomic data with transcriptomic, proteomic, and metabolomic information for functional taxonomy [48].
- Cloud Computing: Provides scalable infrastructure for the massive computational demands of large-scale taxonomic studies [48].
In conclusion, the polyphasic taxonomic approach, with genomic indices at its core, represents the new standard for bacterial identification and classification. The powerful combination of WGS, ANI, and dDDH provides researchers with unambiguous criteria for species delineation, enabling discoveries that advance our understanding of microbial evolution, ecology, and functionality. As these technologies become more accessible and integrated with other data modalities, they will continue to drive the genomic revolution in microbiology, with profound implications for human health, biotechnology, and environmental science.
Within the framework of modern bacterial systematics, the polyphasic approach is the consensus methodology for the accurate identification and classification of microorganisms. This approach integrates genotypic, phenotypic, and chemotaxonomic data to delineate taxonomic relationships reliably [4] [10]. Chemotaxonomy, which involves the chemical analysis of cellular components, and phenotypic profiling provide critical insights into the functional and metabolic characteristics of bacteria, complementing genetic information [20]. Key chemotaxonomic markers include fatty acid profiles, protein patterns, and the outcomes of various biochemical assays. These elements are indispensable for distinguishing between closely related species and for the formal description of novel taxa [10] [49]. The following application notes and protocols detail the standard methodologies for these essential techniques, providing a practical guide for researchers in bacterial taxonomy and drug development.
Application Notes & Experimental Protocols
Cellular Fatty Acid Analysis (CFA)
Application Note: Cellular fatty acid methyl ester (FAME) analysis is a cornerstone of chemotaxonomy. The composition of fatty acids in bacterial cell membranes is a stable, genetically conserved trait that varies between genera and species [10] [50]. Profiling these components using gas chromatography (GC) provides a reproducible fingerprint for bacterial identification and classification. For instance, studies of novel Duganella species have shown that major fatty acids like C~16:0~, C~17:0~ cyclo, and summed feature 3 (C~16:1~ ω7c and/or C~16:1~ ω6c) are critical for their taxonomic delineation [49]. Advanced techniques like comprehensive two-dimensional liquid chromatography (LC×LC) hyphenated to mass spectrometry (MS) offer superior resolution for complex mixtures, including conjugated fatty acid isomers and their oxidation products [51].
Protocol: Fatty Acid Extraction and Analysis via GC-MS
- Objective: To extract, identify, and quantify cellular fatty acids from bacterial biomass for chemotaxonomic profiling.
- Principle: Fatty acids are saponified from cellular lipids, methylated to form volatile Fatty Acid Methyl Esters (FAMEs), and analyzed by Gas Chromatography-Mass Spectrometry (GC-MS) for separation and identification [50].
- Materials & Reagents:
- Saponification Reagent: Aqueous sodium hydroxide (NaOH) in methanol.
- Methylation Reagent: Hydrochloric acid (HCl) in methanol.
- Extraction Solvent: A mixture of hexane and methyl tert-butyl ether (MTBE).
- Wash Solution: Dilute aqueous NaOH.
- GC-MS System: Equipped with a high-resolution capillary column (e.g., 50-60% cyanopropyl-phenyl polysiloxane).
- Reference Standards: Bacterial Acid Methyl Esters (BAME) mix and other quantitative FAME standards.
- Procedure:
- Cultivation and Harvesting: Grow the bacterial strain on a standardized medium (e.g., R2A agar for 24-48 hours). Harvest late-logarithmic phase cells from a single quadrant to ensure consistent physiological age [49].
- Saponification: Resuspend ~40 mg of wet cell mass in 1 mL of saponification reagent. Heat at 100°C for 30 minutes to hydrolyze lipids and release fatty acids.
- Methylation: Cool the tubes. Add 2 mL of methylation reagent and incubate at 80°C for 10 minutes to convert free fatty acids to FAMEs.
- Extraction: Cool the tubes rapidly. Add 1.25 mL of extraction solvent and mix for 10 minutes. Centrifuge to separate phases.
- Washing: Transfer the organic (upper) layer to a vial containing 3 mL of wash solution. Mix and discard the aqueous (lower) layer.
- Chromatography: Inject the purified FAME extract into the GC-MS system. Use a temperature program (e.g., 130°C to 250°C at 4°C/min) for optimal separation [50].
- Data Analysis: Identify fatty acids by comparing retention times and mass spectra to commercial standards. Express results as a percentage of the total fatty acids detected.
Table 1: Common Bacterial Fatty Acids and Their Taxonomic Significance
| Fatty Acid | Structure | Typical Occurrence | Taxonomic Utility |
|---|---|---|---|
| C~16:0~ | Saturated | Ubiquitous | General biomarker; relative abundance varies |
| C~18:1~ ω7c | Monounsaturated | Pseudomonas, Rhizobia | Distinguishes specific Gammaproteobacteria |
| C~17:0~ cyclo | Cyclopropane | Rhizobium, Bradyrhizobium | Characteristic of some Alpha- and Betaproteobacteria |
| C~15:0~ iso | Branched-chain | Bacillus (Gram-positive) | Marker for many Firmicutes |
| C~16:1~ ω11c | Monounsaturated | Campylobacter | Specific to certain Epsilonproteobacteria |
| Summed Feature 3 | C~16:1~ ω7c / C~16:1~ ω6c | Diverse (e.g., Duganella) | Common in many Proteobacteria [49] |
Protein Pattern Analysis (SDS-PAGE)
Application Note: The analysis of whole-cell protein patterns using Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) provides a rapid method for comparing and grouping bacterial strains at the infraspecific level. The banding pattern, or proteotype, reflects the overall protein expression profile and is highly reproducible under standardized conditions [10]. This technique is particularly useful for typing strains below the species level and for preliminary screening to determine genetic relatedness before undertaking more complex genomic analyses.
Protocol: Whole-Cell Protein Profiling via SDS-PAGE
- Objective: To generate and compare whole-cell protein profiles of bacterial isolates for taxonomic grouping.
- Principle: Proteins are denatured and coated with SDS, giving them a uniform negative charge. Separation by electrophoresis through a polyacrylamide gel is based primarily on molecular weight, creating a characteristic banding pattern [10].
- Materials & Reagents:
- Lysis Buffer: Tris-HCl buffer (pH 8.0) containing SDS and β-mercaptoethanol.
- Acrylamide/Bis-acrylamide Solution: For casting resolving and stacking gels.
- Electrophoresis Buffer: Tris-Glycine buffer with SDS.
- Staining Solution: Coomassie Brilliant Blue R-250 or silver stain.
- Destaining Solution: Methanol, acetic acid, and water.
- Molecular Weight Standards: Prestained protein ladder.
- Vertical Gel Electrophoresis Unit.
- Procedure:
- Protein Extraction: Harvest bacterial cells from a pure culture. Lyse cells by boiling in lysis buffer for 10 minutes. Centrifuge to remove cell debris.
- Gel Preparation: Prepare a discontinuous polyacrylamide gel (e.g., 12% resolving gel, 4% stacking gel).
- Sample Loading and Electrophoresis: Load equal amounts of protein supernatant (e.g., 20 µg) and molecular weight standards into the gel wells. Run electrophoresis at constant voltage until the dye front reaches the bottom of the gel.
- Staining and Destaining: Carefully remove the gel and stain with Coomassie Blue for 1-2 hours. Destain until the background is clear and protein bands are visible.
- Pattern Analysis: Digitize the gel image. Use specialized software to normalize band positions and perform numerical analysis (e.g., Dice coefficient, UPGMA clustering) to generate a dendrogram of strain similarity.
Biochemical Assays and Carbon Source Utilization
Application Note: Physiological and biochemical characteristics remain a fundamental component of phenotypic profiling. These tests assess the metabolic capabilities of a bacterium, including enzyme activities, fermentation pathways, and the ability to utilize specific carbon sources [52] [20]. Commercial automated or miniaturized systems allow for the simultaneous testing of dozens of parameters, generating a metabolic fingerprint that can be compared against extensive databases for identification.
Protocol: Biochemical Characterization Using Commercial Kits
- Objective: To determine the enzymatic and carbon source utilization profile of a bacterial isolate.
- Principle: Microplates or strips containing dehydrated substrates detect specific enzymatic activities or acid production from carbon sources. Bacterial growth or metabolism causes a color change, indicating a positive reaction [52].
- Materials & Reagents:
- API / BIOLOG / VITEK Systems: Commercial test strips or panels.
- Inoculation Fluid: Saline or specific media as per kit instructions.
- Incubator.
- Procedure:
- Inoculum Preparation: Prepare a bacterial suspension of standardized turbidity (e.g., 0.5 McFarland standard) in the provided inoculation fluid.
- Inoculation: Dispense the suspension into the wells of the test panel, ensuring each cupule is filled.
- Incubation: Place the panel in a humidified chamber and incubate at the appropriate temperature (e.g., 28°C or 37°C) for the specified time (typically 4-48 hours).
- Reading Results: After incubation, record color changes. Some tests may require the addition of reagents. Compare the profile to the database for identification.
Table 2: Key Biochemical and Physiological Tests for Bacterial Taxonomy
| Test Category | Example Assays | Methodology | Taxonomic Application |
|---|---|---|---|
| Enzyme Activity | Catalase, Oxidase, Urease, β-Galactosidase | Detection of gas production or color change from specific substrates | Differentiates families and genera (e.g., Catalase: Staph vs. Strep) |
| Carbon Utilization | API 50CH, BIOLOG GN2 | Growth assessment in wells with sole carbon sources | Creates metabolic fingerprint for species-level ID |
| Chemical Resistance | Growth in 6.5% NaCl, Optochin susceptibility | Growth inhibition assays | Strain characterization and species delimitation |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Phenotypic and Chemotaxonomic Profiling
| Item / Reagent Solution | Function / Application | Brief Explanation |
|---|---|---|
| R2A Agar | Bacterial Cultivation | A low-nutrient medium ideal for isolating and growing environmental bacteria, including plant-associated strains like Duganella [49]. |
| Methanol with KOH | Saponification | The alkaline methanol solution hydrolyzes lipid esters, releasing free fatty acids from cellular membranes for FAME analysis [50]. |
| Methanol with HCl | Methylation | Acidic methanol catalyzes the esterification of free fatty acids into volatile Fatty Acid Methyl Esters (FAMEs) suitable for GC analysis [50]. |
| BIOLOG Phenotype MicroArrays | Carbon Source Utilization | Pre-configured microplates test an organism's ability to use hundreds of sole carbon sources, generating a metabolic phenotype [52]. |
| API 20E / API 50CH Strips | Biochemical Profiling | Miniaturized, standardized test strips for the determination of enzymatic activities and fermentation profiles of bacteria [52]. |
| MIDI Sherlock Microbial ID System | FAME Analysis | A complete integrated system (software, standards, protocols) for the automated identification of bacteria and yeast based on their FAME profiles. |
| Coomassie Brilliant Blue R-250 | Protein Staining | A dye that binds non-specifically to proteins, allowing visualization of banding patterns after SDS-PAGE separation [10]. |
Workflow and Data Integration
The following diagram illustrates the integrated workflow of a polyphasic taxonomic study, highlighting the role of phenotypic and chemotaxonomic methods.
Diagram 1: Integrated workflow for polyphasic bacterial taxonomy, showing how phenotypic, chemotaxonomic, and genotypic data are combined to reach a consensus classification.
Data Interpretation and Integration
In the polyphasic taxonomy framework, data from fatty acid profiles, protein patterns, and biochemical tests are not used in isolation. Fatty acid data must be compared with profiles from closely related reference strains under identical analytical conditions [49]. Protein banding patterns are highly effective for clustering strains but are generally used for typing below the species level rather than for primary genus or species assignment [10]. Ultimately, the results from these phenotypic and chemotaxonomic analyses are integrated with 16S rRNA gene sequencing and genomic data (such as Average Nucleotide Identity - ANI) to form a robust and consensus-based taxonomic conclusion [4] [49] [20]. This multi-layered approach ensures a comprehensive understanding of microbial relatedness, which is fundamental for research in systematics, ecology, and drug discovery.
The classification of microorganisms has evolved significantly from reliance on traditional microbiological methods to a more comprehensive, pragmatic strategy known as the polyphasic taxonomic approach [4]. This consensus approach integrates genotypic, phenotypic, and phylogenetic data to obtain a complete characterization of microbes, thereby clarifying their taxonomic status and natural phylogenetic relationships [4] [10] [20]. The genotypic analysis includes complete 16S rRNA gene sequencing, DNA-DNA hybridization, and analyses of various molecular markers [4]. The phenotypic and chemotaxonomic analyses encompass morphological observations, physiological and biochemical tests, and chemical marker analysis [4]. This multi-layered methodology is now the gold standard in bacterial systematics, enabling the resolution of previously misclassified organisms into new genera and species and providing a stable, consensus-based classification [20]. This article frames its examination of clinical and probiotic case studies within this foundational thesis of polyphasic taxonomy.
The following workflow diagram illustrates the integrated stages of a standard polyphasic analysis for bacterial identification and characterization:
Case Study 1: Polyphasic Identification of Clinical and Environmental Isolates
Background and Experimental Aims
Conventional techniques for microbial identification—based on morphology, physiology, and biochemistry—often provide an incomplete picture, creating a "blurred image" of taxonomic status [4] [10]. This case study applies the polyphasic approach to identify unknown bacterial isolates from clinical and environmental sources, demonstrating how this methodology resolves the limitations of single-method techniques and ensures accurate classification [4].
Detailed Protocol: A Multi-Layered Methodology
The identification process follows a sequential, multi-phase protocol.
2.2.1 Phase 1: Phenotypic and Biochemical Characterization
- Morphological Examination: Sub-culture the isolate on non-selective solid media (e.g., MRS Agar for Lactic Acid Bacteria). Incubate at appropriate temperatures (e.g., 37°C) for 24-48 hours under suitable atmospheric conditions [53] [54]. Record colony morphology (shape, color, margin, opacity). Perform Gram staining and microscopic examination to determine cell shape, size, and arrangement [53].
- Biochemical Profiling: Using pure cultures, perform a suite of standard tests, including:
- Catalase, oxidase, and urease tests [53].
- MR-VP reaction, Indole production, Citrate utilization [53].
- H₂S and NH₃ production, synthesis from arginine [53].
- Carbohydrate fermentation patterns using substrates like lactose, xylose, glucose, sucrose, and fructose [53]. Automated systems like VITEK 2C can be employed for this step [54].
2.2.2 Phase 2: Genotypic Analysis
- DNA Extraction and 16S rRNA Gene Sequencing: Extract genomic DNA from a fresh, pure culture using a commercial kit. Amplify the nearly full-length 16S rRNA gene via PCR using universal primers (e.g., 27F and 1492R) [4] [10]. Purify the PCR product and perform Sanger sequencing.
- Sequence Analysis and Phylogenetics: Compare the obtained sequence against public databases (e.g., NCBI BLAST). Construct a phylogenetic tree using reference sequences from related type strains to visualize evolutionary relationships [4].
- Strain Typing (if required): For strain-level discrimination, employ DNA-based typing methods such as Repetitive Sequence-based PCR (rep-PCR) or Pulsed-Field Gel Electrophoresis (PFGE) to generate genomic fingerprints [4].
2.2.3 Phase 3: Chemotaxonomic Analysis
- Cellular Fatty Acid Analysis: Analyze cellular fatty acid methyl esters (FAMEs) using Gas Chromatography and compare the profile with a reference database [10].
Key Research Reagents and Solutions
Table 1: Essential Reagents for Polyphasic Bacterial Identification
| Research Reagent | Function/Application |
|---|---|
| MRS Agar/Broth [53] [54] | Non-selective culture medium for the isolation and growth of Lactic Acid Bacteria. |
| Gram Staining Kit [53] | Differentiates bacteria based on cell wall structure (Gram-positive vs. Gram-negative). |
| PCR Reagents (Primers, Taq Polymerase, dNTPs) [4] | Amplification of target genes, such as the 16S rRNA gene, for sequencing. |
| VITEK 2C ANC Cards [54] | Automated, standardized biochemical test panels for bacterial identification. |
| DNA Extraction Kit | Purification of high-quality genomic DNA from bacterial cells for molecular analysis. |
Case Study 2: Probiotic Characterization of Lactic Acid Bacteria
Background and Experimental Aims
Lactic acid bacteria (LAB) are widely used as probiotics in food and healthcare [53]. To be classified as a probiotic, a strain must meet stringent criteria for safety, functionality, and technological applicability [54]. This case study details the polyphasic characterization of LAB isolates from dietary sources for their potential use as probiotics, focusing on stress tolerance, safety, and functionality [53] [54].
Detailed Protocol: Assessing Probiotic Properties
The probiotic characterization follows a structured workflow to evaluate key traits.
3.2.1 Strain Isolation and Basic Characterization
- Sample Collection and Isolation: Collect samples (e.g., from curd, pickle, milk) and homogenize in buffered peptone water [53] [54]. Inoculate into de Man-Rogosa-Sharpe (MRS) broth as an enrichment medium and incubate anaerobically at 37°C for 24-48 hours. Streak on MRS agar plates to obtain single colonies and purify by successive sub-culturing [53] [54].
- Phenotypic and Biochemical Identification: Follow the same protocols outlined in Section 2.2.1 for morphological and biochemical characterization [53].
3.2.2 Functional Probiotic Property Assays
- Acid Tolerance Test: Grow an overnight culture in MRS broth (pH 7). Pellet cells by centrifugation (5000 rpm for 10 min) and re-suspend in PBS buffer adjusted to different pH levels (e.g., 2, 3, 4, 5, and 6). Incubate at 37°C for 2h and 5h. Subsequently, inoculate 1% (v/v) of this suspension into fresh MRS broth (pH 7) and incubate for 24h. Measure the optical density (OD) at 600 nm. Strains with >50% resistance (compared to a pH 7 control) at pH 3 after 2h are considered acid-tolerant [53] [54].
- Bile Salt Tolerance Test: Inoculate 200 µl of a fresh bacterial suspension (10⁷-10⁸ CFU/ml) into 1000 µl of MRS broth supplemented with different concentrations of bile salts (e.g., 0.1% to 0.5% w/v). Incubate at 37°C for 2h and 5h. Measure the OD600. Isolates exhibiting more than 50% resistance at a 0.3% bile concentration are designated bile-tolerant [53] [54].
- NaCl Tolerance Test: Inoculate 1% (v/v) of a fresh culture into modified MRS broth with varying NaCl concentrations (0% to 7% w/v). Incubate anaerobically at 37°C and measure absorbance at 600 nm at intervals (e.g., 2h and 5h) [54].
- Antimicrobial Activity: Evaluate the isolate's cell-free supernatant against common Gram-positive and Gram-negative pathogens (e.g., E. coli, S. aureus) using well-diffusion or disc-diffusion assays on Mueller-Hinton Agar [53] [54].
3.2.3 Safety Assessment
- Antibiotic Susceptibility Test: Using the disc diffusion technique on Mueller-Hinton Agar, test the isolate's susceptibility to a panel of antibiotics (e.g., Ampicillin, Gentamicin, Tetracycline, Erythromycin, etc.) [54]. Interpret the zone of inhibition (ZOI) according to CLSI guidelines [54].
- Hemolytic Activity: Streak the isolate on blood agar plates and incubate for 24-48 hours. Observe for β-hemolysis (clear zones), which is a safety disqualifier [53].
3.2.4 Genomic Analysis for Safety and Function
- Whole Genome Sequencing (WGS): Perform high-throughput sequencing of promising isolates [54].
- Bioinformatic Analysis: Use WGS data to confirm species identity via in silico tools, screen for the absence of virulence factors and acquired antibiotic resistance genes, and identify gene clusters related to beneficial traits (e.g., bile salt hydrolases, stress response genes, bacteriocin production) [54].
Key Research Reagents and Solutions
Table 2: Essential Reagents for Probiotic Characterization
| Research Reagent | Function/Application |
|---|---|
| MRS Broth/Agar [53] [54] | Standard medium for cultivation and maintenance of Lactobacilli and other LAB. |
| PBS Buffer (pH 2-7) [53] [54] | Used in acid tolerance assays to simulate the harsh environment of the human stomach. |
| Bile Salts (e.g., Oxgall) [53] [54] | Used in bile tolerance assays to simulate the intestinal environment. |
| Antibiotic Susceptibility Discs [54] | For determining the antibiotic resistance profile of potential probiotic strains. |
| Mueller-Hinton Agar (with blood) [54] | Standard medium for antibiotic susceptibility testing and antimicrobial assays. |
The following table summarizes quantitative data from probiotic characterization studies, illustrating the performance of potential probiotic strains under stress conditions.
Table 3: Quantitative Summary of Probiotic Strain Stress Tolerance [53] [54]
| Characteristic | Test Condition | Performance Metric / Result |
|---|---|---|
| Acid Tolerance | pH 2 - 3 for 2 hours | Survival rates significantly above 50% for robust strains (e.g., L. acidophilus CM1) [53]. |
| Bile Tolerance | 0.3% Bile Salts for 2 hours | >50% resistance observed in promising isolates (e.g., L. delbrueckii OS1) [53]. |
| NaCl Tolerance | 4% - 6% NaCl | Significant growth observed in tolerant strains (e.g., L. acidophilus CM1 & L. delbrueckii OS1) [53]. |
| Antibiotic Sensitivity | CLSI Disc Diffusion | Variable by strain; sensitive to Ampicillin, Chloramphenicol, Erythromycin; potentially resistant to Nalidixic Acid, Trimethoprim/Sulfamethizole [54]. |
| Genomic Safety | Whole Genome Sequencing | Absence of virulence factors and pathogenic islands confirmed in safe strains (e.g., L. delbrueckii subsp. bulgaricus) [54]. |
The presented case studies demonstrate the critical application of the polyphasic taxonomy framework in both clinical microbiology and industrial probiotic development. This approach, which integrates phenotypic, genotypic, and chemotaxonomic data, moves beyond the limitations of single-method techniques to provide a robust, consensus-based classification of microorganisms [4] [20]. The detailed protocols for identifying clinical isolates and characterizing probiotic strains underscore the practicality and necessity of this comprehensive methodology. As molecular techniques continue to advance, the polyphasic approach will remain the cornerstone of microbial systematics, ensuring accurate taxonomic identification and supporting the development of safe, well-characterized microbial agents for research, food, and health applications [4] [10].
Resolving Taxonomic Ambiguity: Overcoming Pitfalls and Limitations in Microbial Identification
The 16S rRNA gene has served as the cornerstone of bacterial identification and phylogenetic studies for decades, providing a universal framework for classifying microbial life. This ~1,500 base-pair molecular chronometer is present in almost all bacteria and contains a unique combination of highly conserved and variable regions that enables phylogenetic analysis at various taxonomic levels [22]. The explosion in recognized bacterial taxa—from 1,791 species in 1980 to over 8,168 today—is directly attributable to the ease of 16S rRNA gene sequencing compared to more cumbersome DNA-DNA hybridization methods [22]. Despite its widespread adoption and utility, 16S rRNA sequencing possesses inherent limitations that preclude definitive identification in clinically and taxonomically significant scenarios.
The fundamental resolution limit of 16S rRNA gene sequencing stems from its genetic characteristics and evolutionary conservation. While sequences with less than 97% similarity generally represent distinct species, the biological meaning of similarity scores exceeding 97% remains ambiguous [22]. This ambiguity creates a "resolution limit" where 16S rRNA sequencing cannot reliably distinguish between recently diverged species or resolve complex taxonomic relationships. In clinical settings, this translates to genus-level identification rates exceeding 90%, but species-level identification rates ranging from 65% to 91%, with 1-14% of isolates remaining completely unidentified after testing [22]. This application note examines the specific scenarios where 16S rRNA sequencing reaches its resolution limit and outlines a polyphasic framework incorporating advanced genomic techniques to achieve definitive bacterial identification.
Quantitative Assessment of 16S rRNA Sequencing Limitations
Performance Metrics in Clinical and Taxonomic Studies
Comprehensive studies have quantified the performance of 16S rRNA sequencing across diverse bacterial groups, revealing substantial variation in identification success rates. Table 1 summarizes the concordance rates between 16S rRNA sequencing and conventional identification methods for various bacterial groups, highlighting specific taxonomic groups where resolution is particularly problematic.
Table 1: Performance of 16S rRNA Gene Sequencing for Bacterial Identification
| Bacterial Group | Number of Strains | Species Identification Rate (%) | Problematic Taxa/Notes | Citation |
|---|---|---|---|---|
| Broad Clinical Pathogens | 617 | 87.5 | Genus-level concordance higher (96%) | [55] |
| Gram-Negative Bacteria | 72 | 89.2 | Enterobacter, Pantoea | [22] |
| Mycobacteria | 328 | 62.5 | Rapid-growing mycobacteria | [22] |
| Coagulase-Negative Staphylococci | 47 | 87.2 | Staphylococcus species complexes | [22] |
| Gram-Positive Anaerobes | 20 | 65 | Clostridium, Actinomyces | [22] |
| Gram-Negative Nonfermentative Bacteria | 107 | 91.6 | Acinetobacter, Stenotrophomonas | [22] |
Bacterial Taxa with Inherent 16S rRNA Resolution Challenges
Certain bacterial taxa present particular challenges for 16S rRNA-based identification due to high sequence similarity between genetically distinct species or the existence of complex species groups. Table 2 enumerates key genera and species where 16S rRNA sequencing demonstrates limited discriminatory power and requires supplemental methodologies for definitive identification.
Table 2: Bacterial Taxa with Documented 16S rRNA Sequencing Limitations
| Genus | Species with Poor Resolution | Primary Identification Challenge | Citation |
|---|---|---|---|
| Bacillus | B. globisporus, B. psychrophilus | >99.5% 16S similarity but only 23-50% DNA relatedness | [22] |
| Streptococcus | S. mitis, S. oralis, S. pneumoniae | Shared identical or nearly identical 16S sequences | [22] |
| Edwardsiella | E. tarda, E. hoshinae, E. ictaluri | 99.35-99.81% similarity despite clear genetic distinction | [22] |
| Burkholderia | B. pseudomallei, B. thailandensis | High sequence similarity between distinct pathogens | [22] |
| Acinetobacter | A. baumannii, A. calcoaceticus | Forms complexes with minimal 16S variation | [22] |
| Zhongshania/Marortus | Multiple marine species | Taxonomic ambiguity resolvable only with genomic data | [15] |
The Polyphasic Taxonomy Framework
Principles and Components
Polyphasic taxonomy integrates multiple lines of evidence to achieve robust bacterial classification and identification, overcoming the limitations of single-method approaches. This framework combines phenotypic, genotypic, and phylogenetic information to create a comprehensive identification system [15]. The core components include:
- Phenotypic Characterization: Traditional methods including morphology, biochemical profiling, growth requirements, and chemotaxonomic markers (fatty acid analysis, polar lipids, respiratory quinones) [15].
- Genotypic Analysis: DNA-based methods ranging from single-gene sequencing to whole-genome analysis.
- Phylogenetic Placement: Evolutionary relationship determination through sequence comparison and tree-building algorithms.
This integrated approach is particularly valuable for resolving complex taxonomic relationships, such as those observed in the genera Zhongshania and Marortus, where high 16S rRNA similarity (>99%) masked significant genomic and phenotypic differences that were only revealed through polyphasic analysis [15].
Decision Framework for Method Selection
The following diagram outlines a systematic approach for selecting appropriate identification methods when 16S rRNA sequencing reaches its resolution limit:
Advanced Genomic Methods for Bacterial Identification
Whole Genome Sequencing and Analysis
Whole genome sequencing (WGS) represents the most comprehensive approach for overcoming 16S rRNA resolution limits, providing complete genetic information for taxonomic classification [56]. WGS enables several advanced analysis methods:
- Average Nucleotide Identity (ANI): Calculates the percentage of identical nucleotides between homologous regions of two genomes, with values ≥95% indicating species-level relatedness [15].
- Digital DNA-DNA Hybridization (dDDH): Computational simulation of wet-lab DDH with values ≥70% supporting species-level classification [15].
- Phylogenomic Analysis: Construction of robust phylogenetic trees based on conserved, single-copy core genes rather than a single marker gene.
In the reclassification of Zhongshania and Marortus species, genome-based analyses proved essential for resolving taxonomic ambiguities. dDDH values between reference strains were notably lower than 70%, with ANI values ranging from 73.31 to 78.57%, confirming they represented distinct species despite high 16S rRNA similarity [15].
Protocol: Genome-Based Taxonomic Delineation
Objective: To perform definitive species identification and delineation using whole genome sequencing data when 16S rRNA sequencing provides ambiguous results.
Materials:
- Pure bacterial genomic DNA (>10 ng/µL, minimum 20 µL)
- Library preparation kit (Illumina, Oxford Nanopore, or PacBio)
- High-performance computing cluster with bioinformatics tools
- Reference genome databases (NCBI RefSeq, GTDB)
Methodology:
Genome Sequencing and Assembly
- Sequence genomic DNA using an appropriate platform (Illumina for accuracy, long-read technologies for contiguity)
- Assemble reads into contigs using assemblers such as SPAdes or Flye
- Assess assembly quality (completeness, contamination) using CheckM
Average Nucleotide Identity Calculation
- Identify orthologous regions between query and reference genomes using BLAST or MUMmer
- Calculate ANI using established tools such as FastANI or OrthoANI
- Apply species threshold of ≥95% ANI
Digital DNA-DNA Hybridization
- Utilize the Genome-to-Genome Distance Calculator (GGDC) or formula-based dDDH methods
- Interpret results against species threshold of ≥70% dDDH
Phylogenomic Tree Construction
- Extract conserved, single-copy marker genes (e.g., UBCG set, 120 bacterial markers)
- Perform multiple sequence alignment for each marker
- Concatenate alignments and construct maximum-likelihood or Bayesian phylogenetic trees
- Assess tree robustness with bootstrap analysis (≥1000 replicates)
Interpretation: Integrate ANI, dDDH, and phylogenomic results with phenotypic data to make definitive taxonomic assignments. ANI values ≥95% or dDDH values ≥70% indicate members of the same species, while lower values support novel species designation.
Complementary Molecular Methods
Targeted Loci Sequencing
For situations where whole genome sequencing is impractical, sequencing of alternative molecular markers can provide additional resolution:
- Ribosomal Multilocus Sequence Typing (rMLST): Analysis of multiple ribosomal protein genes to improve discrimination.
- Housekeeping Gene Sequencing: Targets such as rpoB, gyrB, dnaK, or recA which may evolve faster than 16S rRNA.
- Core Genome Multilocus Sequence Typing (cgMLST): Analysis of hundreds to thousands of conserved genes across the core genome.
Protocol: Ribosomal Protein Gene Amplification and Sequencing
Objective: To obtain supplemental phylogenetic data through amplification and sequencing of ribosomal protein genes.
Materials:
- Bacterial genomic DNA
- Ribosomal protein gene-specific primers (e.g., rpsL, rpsG, rplB)
- PCR reagents (polymerase, dNTPs, buffer)
- Sanger sequencing reagents
Methodology:
- Design primers targeting conserved ribosomal protein genes
- Perform PCR amplification with optimized cycling conditions
- Verify amplicon size and purity via gel electrophoresis
- Purify PCR products and perform Sanger sequencing
- Analyze sequences against specialized databases
Interpretation: Compare ribosomal protein gene sequences to reference databases. Congruence with 16S rRNA phylogeny supports taxonomic placement, while discordance may indicate horizontal gene transfer or misclassification.
Bioinformatics Tools for Enhanced Classification
Advanced Classification Algorithms
Recent bioinformatics developments have created more robust taxonomic classification tools specifically designed to handle sequences from novel or poorly represented taxa:
- CAT and BAT (Contig/Bin Annotation Tool): Tools that integrate multiple open reading frames to classify contigs and metagenome-assembled genomes, automatically making classifications at low taxonomic ranks when closely related organisms are present and higher ranks otherwise [57].
- PhyloPhlAn: Uses a set of 400 universal marker genes for precise taxonomic assignment.
- GTDB-Tk: Reference tree-based tool for consistent classification of bacterial genomes.
These tools outperform best-hit approaches, especially for sequences from highly unknown organisms, by integrating distributed taxonomic signals across multiple genes rather than relying on single-gene similarity [57].
Protocol: Robust Taxonomic Classification with CAT and BAT
Objective: To employ advanced classification algorithms that integrate multiple genomic signals for accurate taxonomic placement of contigs and genomes.
Materials:
- Assembled contigs or metagenome-assembled genomes (MAGs)
- High-performance computing cluster
- CAT/BAT software installation
- Reference database (e.g., NCBI NR, custom protein database)
Methodology:
- Software Installation and Database Setup
- Install CAT and BAT via conda or from source
- Download and prepare protein reference database
ORF Prediction and Homology Search
- Predict open reading frames using Prodigal
- Perform diamond BLAST searches against reference database
- Include hits within top 10% of best hit score (default r parameter)
Taxonomic Classification
- Classify contigs based on consolidated ORF classifications
- Apply minimum support threshold (default f=0.5)
- Generate taxonomic classification reports
Interpretation: CAT/BAT provides classifications with higher precision than best-hit approaches, particularly for sequences from unknown organisms. The tools automatically determine appropriate taxonomic levels based on available evidence, preventing over-classification of novel taxa.
Essential Research Reagent Solutions
Successful implementation of a polyphasic identification approach requires specific research reagents and tools. Table 3 catalogs essential materials and their applications in advanced bacterial identification workflows.
Table 3: Essential Research Reagents for Polyphasic Bacterial Identification
| Reagent/Tool | Application | Function in Identification Workflow | Specifications |
|---|---|---|---|
| Marine Agar (MA) | Cultivation | Optimal growth medium for marine bacteria including Zhongshania | [15] |
| DNeasy PowerSoil Kit | DNA Extraction | High-quality genomic DNA extraction from environmental samples | [15] |
| Universal 16S Primers (27F/1492R) | 16S Amplification | Initial amplification of 16S rRNA gene for preliminary identification | [15] |
| Ribosomal Protein Gene Primers | Supplemental Gene Amplification | Targets for improved resolution (rpsL, rpsG, rplB) | [22] |
| CheckM | Genome Quality Assessment | Assesses completeness/contamination of assembled genomes | [57] |
| FastANI | Genome Comparison | Calculates Average Nucleotide Identity between genomes | [15] |
| GGDC | Digital DDH | Computes genome-to-genome distances for species delineation | [15] |
| CAT/BAT | Taxonomic Classification | Classifies contigs/MAGs using multiple ORF evidence | [57] |
Integrated Workflow for Comprehensive Identification
The following diagram illustrates the complete integrated workflow for bacterial identification that systematically addresses the limitations of 16S rRNA sequencing:
This integrated workflow emphasizes that 16S rRNA sequencing should serve as an initial screening tool rather than a definitive identification method when working with taxonomically complex bacteria. The polyphasic approach systematically combines data from multiple sources to achieve confident species-level identification, particularly for clinically relevant pathogens and organisms representing novel taxonomic lineages.
The vast majority of prokaryotic life—estimated at over 99% of microorganisms in most environments—resists cultivation under standard laboratory conditions, creating a significant gap in our understanding of microbial diversity and function [58]. This "microbial dark matter" represents an immense reservoir of unexplored biological diversity with profound implications for ecosystem functioning, biotechnology, and human health [59]. For decades, this limitation constrained microbiologists to studying only a tiny fraction of the microbial world, creating a biased understanding of microbial biology skewed toward "easy growers" [60].
The emergence of culture-independent methods, particularly metagenome-assembled genomes (MAGs), has revolutionized microbial ecology by enabling genome-resolved study of uncultured microorganisms directly from environmental samples [59]. MAGs represent complete or near-complete microbial genomes reconstructed entirely from complex microbial communities through bioinformatic approaches, bypassing the need for cultivation [59] [58]. This breakthrough has expanded the known microbial diversity, revealing novel taxa and metabolic pathways involved in key biogeochemical cycles and opening new frontiers in microbial taxonomy, ecology, and biotechnology [59].
This application note explores the integration of MAGs within the framework of polyphasic taxonomic approaches, providing detailed methodologies for researchers seeking to leverage these powerful tools to navigate the uncultured microbial world.
MAGs in the Context of Polyphasic Taxonomy
Polyphasic taxonomy represents a consensus approach that integrates phenotypic, genotypic, and phylogenetic data for comprehensive bacterial classification [4] [20]. This multidimensional methodology combines information from genetic markers, ecological traits, metabolic capabilities, and morphological characteristics to offer a holistic understanding of microbial diversity and relationships [15]. The adoption of polyphasic taxonomy has resolved numerous previously misclassified taxa and continues to refine our understanding of microbial phylogeny [4].
Traditional polyphasic approaches relied heavily on characteristics obtained from cultured isolates, creating an inherent bias toward cultivable microorganisms. The integration of MAGs into polyphasic frameworks addresses this limitation by providing genomic access to the uncultured majority. MAGs serve as genomic anchors for uncultured lineages, enabling their placement within taxonomic structures and facilitating comparisons with cultured relatives through genome-based metrics such as Average Nucleotide Identity (ANI) and digital DNA-DNA Hybridization (dDDH) [15]. When combined with functional predictions from genomic data, this approach enables a more comprehensive taxonomic placement of uncultured lineages, enriching our understanding of microbial diversity and evolution.
Table 1: Core Components of Polyphasic Taxonomy and MAG Integration
| Component Type | Traditional Approach | MAG-Enhanced Approach |
|---|---|---|
| Genotypic | 16S rRNA gene sequencing, DNA-DNA hybridization | Whole-genome sequencing, ANI, dDDH from MAGs |
| Phylogenetic | Single-gene trees (e.g., 16S rRNA) | Genome-scale phylogenies, phylogenomics |
| Phenotypic | Culture-based morphological, physiological tests | Inferred from genomic potential, single-cell imaging |
| Chemotaxonomic | Lipid analysis, quinone profiles from cultures | Predicted from biosynthetic gene clusters |
| Ecological | Limited to cultivable niches | Direct linkage to habitat of origin |
Methodological Framework: From Sample to MAG Analysis
Sample Collection and DNA Extraction Considerations
The foundation of successful MAG generation lies in proper sample handling and nucleic acid preservation. Sampling strategies should be tailored to research objectives, whether aimed at discovering novel taxa, identifying biosynthetic gene clusters, or characterizing specific microbiome functions [59]. Appropriate sampling and storage protocols are crucial for preserving microbial community structure and DNA integrity.
For host-associated microbiomes, particularly gut contents, samples must be collected using sterile tools and placed in sterile, DNA-free containers. Immediate storage at -80°C is ideal, though nucleic acid preservation buffers (e.g., RNAlater or OMNIgene.GUT) provide alternatives when freezing is not feasible [59]. Repeated freeze-thaw cycles must be avoided as they cause DNA shearing and impact downstream assembly quality. For environmental samples with high diversity (e.g., soils, sediments), deeper sequencing is required to capture rare taxa, while less diverse systems may benefit from selective enrichment strategies [59].
DNA extraction should prioritize high-molecular-weight DNA with minimal fragmentation, using protocols optimized for different sample types. For host-associated samples, reduction of host DNA contamination is particularly critical, as host reads can dominate sequencing data and reduce microbial sequence recovery [59].
Sequencing Technology Selection
The choice of sequencing technology significantly influences MAG quality and completeness. The table below compares the primary sequencing approaches for MAG generation:
Table 2: Sequencing Technology Comparison for MAG Generation
| Parameter | Short-Read Sequencing | Long-Read Sequencing | HiFi Long-Read Sequencing |
|---|---|---|---|
| Read Length | 75-300 bp | 10 kb+ | 15-25 kb |
| Accuracy | ~99.9% | ~85-98% | >99.9% |
| MAG Quality | Fragmented, draft genomes | Improved continuity | Single-contig, complete genomes |
| Binning Dependency | High | Moderate | Low |
| Strain Resolution | Limited | Improved | High |
| Cost Efficiency | High | Moderate | Lower throughput |
Recent studies demonstrate that HiFi long-read sequencing produces more total MAGs and higher-quality MAGs compared to both short-read and other long-read technologies [58]. The combination of long read length and high accuracy enables recovery of single-contig complete genomes, overcoming challenges associated with repetitive regions and strain variation that fragment short-read assemblies [58].
Bioinformatics Workflow for MAG Generation
The computational generation of MAGs follows a multi-stage process involving assembly, binning, and quality assessment. The following workflow illustrates the key steps:
Assembly involves stitching sequencing reads into longer contiguous sequences (contigs). This process is computationally challenging due to the presence of multiple species, uneven abundances, conserved regions, and strain-level variation [58]. Advanced assemblers such as metaSPAdes, HiCanu, and hifiasm-meta are specifically designed for metagenomic data and perform better with long-read inputs [58].
Binning groups contigs into discrete bins representing individual genomes based on sequence composition (k-mer frequencies), abundance patterns across samples, and/or phylogenetic markers [59]. Tools like MetaBAT2, MaxBin2, and CONCOCT implement different algorithmic approaches to this clustering problem. The recently developed HiFi-MAG-Pipeline leverages HiFi sequencing data to generate high-quality bins with minimal contamination [58].
Quality assessment is critical for evaluating MAG completeness and contamination. Standards have been established using checkM and other tools that assess the presence of single-copy marker genes [59]. MAGs are typically categorized as high-quality (>90% complete, <5% contaminated) or medium-quality (>50% complete, <10% contaminated) for downstream analyses.
Research Reagent Solutions for MAG Workflows
Table 3: Essential Research Reagents and Platforms for MAG Generation
| Category | Product/Platform | Application Notes |
|---|---|---|
| DNA Preservation | RNAlater, OMNIgene.GUT | Stabilize nucleic acids during sample transport/storage |
| DNA Extraction | DNeasy PowerSoil Pro Kit | Optimized for diverse environmental samples; minimizes inhibitors |
| Library Prep | SMRTbell Express Template Prep | For PacBio HiFi sequencing; requires high-molecular-weight DNA |
| Sequencing | PacBio Revio, Sequel IIe | HiFi sequencing platforms for long-read metagenomics |
| Assembly | metaSPAdes, HiCanu, hifiasm-meta | Metagenome-specific assemblers for short/long reads |
| Binning | MetaBAT2, MaxBin2, CONCOCT | Contig-binning algorithms using composition/abundance |
| Quality Control | CheckM, BUSCO | Assess MAG completeness/contamination via marker genes |
| Taxonomic Classification | GTDB-Tk, CAT/BAT | Genome-based taxonomy using reference databases |
| Functional Annotation | Prokka, DRAM, antiSMASH | Gene prediction, metabolic pathway, and BGC annotation |
Applications and Case Studies
Expanding Pathogen Diversity: Klebsiella pneumoniae Case Study
A recent study integrating 656 human gut-derived K. pneumoniae genomes (317 MAGs, 339 isolates) demonstrated the power of MAGs to reveal hidden pathogen diversity [61]. The analysis revealed that over 60% of MAGs belonged to new sequence types, highlighting a large uncharacterized diversity of K. pneumoniae missing from clinical isolate collections [61]. Integration of MAGs nearly doubled the phylogenetic diversity of gut-associated K. pneumoniae and uncovered 86 MAGs with >0.5% genomic distance compared to 20,792 Klebsiella isolate genomes from various sources [61].
Pan-genome analysis identified 214 genes exclusively detected among MAGs, with 107 predicted to encode putative virulence factors [61]. This expanded genomic landscape enabled more accurate classification of disease and carriage states compared to isolates alone, demonstrating the value of MAGs for public health surveillance and understanding pathogen evolution [61].
Resolving Taxonomic Complexities: Zhongshania and Marortus
A polyphasic study reevaluating the bacterial genera Zhongshania and Marortus combined high-resolution phylogenomics with detailed phenotypic characterization, including cryo-transmission electron microscopy for flagellar visualization [15]. The research demonstrated that Marortus luteolus should be reclassified as a later heterotypic synonym of Zhongshania marina based on dDDH values >70% and ANI/AAI values exceeding 95% [15].
This case study highlights how genome-based metrics (dDDH, ANI, AAI) within a polyphasic framework can resolve taxonomic ambiguities, even when initial classifications suggested separate genera [15]. The researchers further described a novel species, Zhongshania aquatica sp. nov., expanding the known diversity within this genus [15].
Global MAG Repositories and Cross-Ecosystem Discovery
The scale of MAG generation is evidenced by repositories such as gcMeta, which currently compiles over 2.7 million MAGs from 104,266 samples spanning diverse biomes [62]. This resource has established 50 biome-specific MAG catalogues comprising 109,586 species-level clusters, of which 63% (69,248) represent previously uncharacterized taxa [62]. Such databases provide standardized, AI-ready datasets encompassing microbial enzymes, anti-phage defense systems, and other functional modules, enabling advanced machine learning applications and cross-ecosystem comparisons [62].
Metagenome-assembled genomes have fundamentally transformed our approach to microbial taxonomy and ecology, providing genomic access to the vast uncultured majority of microorganisms. When integrated within polyphasic taxonomic frameworks, MAGs enable a more comprehensive understanding of microbial diversity and function, bridging the gap between traditional cultivation-based methods and modern genomic approaches.
Despite these advances, challenges remain in MAG generation, including assembly biases, incomplete metabolic reconstructions, and taxonomic uncertainties [59]. Continued improvements in sequencing technologies, hybrid assembly approaches, and multi-omics integration will further refine MAG-based analyses [59]. Emerging methods such as single-cell sequencing and long-read metagenomics promise to enhance genome completeness and resolve strain-level variation [60] [58].
As methodologies advance, MAGs will remain a cornerstone for understanding microbial contributions to global biogeochemical processes and developing sustainable interventions for environmental resilience [59]. The integration of MAGs with experimental validation through innovative cultivation techniques will further strengthen polyphasic taxonomy, ultimately leading to a more complete understanding of the microbial world.
In the field of bacterial identification taxonomy, the polyphasic approach—which integrates genotypic, phenotypic, and phylogenetic data—represents the consensus methodology for the complete characterization of microbes [4] [10] [20]. This approach is fundamental to establishing a reliable taxonomic framework, yet its application across different research laboratories faces significant reproducibility challenges. The inability to reproduce scientific findings poses a substantial problem, with a survey in the field of biology revealing that over 70% of researchers were unable to reproduce other scientists' findings, and approximately 60% could not reproduce their own results [63]. This reproducibility crisis has tangible financial impacts, estimated at $28 billion annually spent on non-reproducible preclinical research [63]. For researchers and drug development professionals, addressing these challenges is paramount to ensuring the credibility of scientific data, accelerating discovery, and maintaining public trust in research outcomes.
The fundamental principles of reproducibility are defined through specific measurement conditions. Repeatability refers to measurements taken under identical conditions (same method, instruments, personnel, and short time interval), while reproducibility assesses measurements under changed conditions (different locations, operators, measuring systems) [64]. Counterintuitively, excessive standardization within a single laboratory can create a "standardization fallacy," where results become idiosyncratic to specific laboratory conditions and less reproducible elsewhere [65]. This article outlines the major challenges and provides actionable protocols to enhance reproducibility in bacterial taxonomy studies across laboratories.
Core Challenges to Reproducibility
Biological and Methodological Variability
The polyphasic taxonomic approach, while comprehensive, introduces multiple potential failure points in reproducibility. These include the use of misidentified or cross-contaminated cell lines, improper maintenance of biological materials through long-term serial passaging that alters genotype and phenotype, and inability to manage complex datasets [63]. Additionally, variations in cognitive biases (e.g., confirmation bias, selection bias) and a competitive culture that rewards novel findings over negative results further undermine reproducibility [63].
The Standardization Fallacy in Laboratory Practice
Rigorous standardization within a single laboratory often fails to yield reproducible results in other laboratories. This "standardization fallacy" occurs because excessively homogenous study samples produce results that are only valid under the specific standardized conditions [65]. As laboratories inevitably differ in aspects such as animal microbiomes, personnel, environmental factors, and reagent batches, ultra-standardization narrows the range of conditions under which results remain valid, ultimately compromising external validity and reproducibility [65]. Empirical evidence demonstrates that multi-laboratory studies, which incorporate inherent heterogeneity, produce more reproducible results without requiring larger sample sizes [65].
Table 1: Major Factors Affecting Reproducibility in Life Science Research
| Category | Specific Challenge | Impact on Reproducibility |
|---|---|---|
| Materials & Data | Lack of access to methodological details, raw data, research materials | Prevents direct replication and validation of results [63] |
| Biological Materials | Use of misidentified, cross-contaminated, or over-passaged cell lines and microorganisms | Invalidates experimental results and conclusions [63] |
| Data Management | Inability to manage complex datasets; lack of standardized analytical protocols | Introduces variations and biases in data interpretation [63] |
| Experimental Design | Poor research practices and inadequate experimental design | Reduces likelihood of successful replication [63] |
| Cultural Factors | Competitive culture rewarding novel findings; undervaluing negative results | Leads to publication bias and selective reporting [63] |
Quantitative Assessment of Variation
Understanding and quantifying measurement variation is essential for interpreting laboratory data accurately. The total variation in measurement results arises from both analytical variation (from the measurement procedure) and biological variation (inherent to the dynamic nature of metabolism) [64].
Table 2: Types of Measurement Imprecision Under Different Conditions
| Condition Type | Key Variable Factors | Impact on Imprecision | Typical Use Case |
|---|---|---|---|
| Repeatability | None (short time interval, same equipment/reagents) | Minimal imprecision; bias contribution most evident [64] | Instrument performance verification |
| Intermediate Precision | Time (days/months), instruments, reagents, personnel | Moderate imprecision; bias behaves more randomly [64] | Internal quality control procedures |
| Reproducibility | Location, operators, measuring systems | Maximum imprecision; bias contributes as random variable [64] | Multi-laboratory study validation |
The mathematical foundation for quantifying imprecision relies on calculating the standard deviation (SD) across repeated measurements. The conventional equation for SD is:
[ SD = \sqrt{\frac{1}{N} \sum{i=1}^{N} (xi - \mu)^2} ]
where ( \mu ) represents the mean of the measurements, ( x_i ) represents each individual measurement, and ( N ) represents the total number of measurements [64]. This statistical approach allows researchers to objectively compare variability across different experimental setups and laboratories.
Experimental Protocols for Enhanced Reproducibility
Protocol 1: Polyphasic Taxonomic Characterization of Bacterial Isolates
The polyphasic approach integrates multiple lines of evidence to achieve robust classification and identification of bacterial strains [4] [10] [20].
Key Reagent Solutions:
- DNA Extraction Kits: For high-quality, inhibitor-free genomic DNA (e.g., kits with lysozyme for Gram-positive bacteria).
- PCR Reagents: Including primers for 16S rRNA gene (e.g., 27F/1492R), housekeeping genes (e.g., gyrB, rpoB), and high-fidelity DNA polymerase.
- Culture Media: A range of standardized media (e.g., R2A, LB, specific selective media) for physiological characterization.
- Fatty Acid Methyl Ester (FAME) Analysis Standards: For chemotaxonomic profiling.
- Authentication Services: For validation of reference strains (e.g., ATCC).
Methodology:
- Genotypic Analysis:
- Extract genomic DNA using a standardized protocol.
- Amplify and sequence the nearly full-length 16S rRNA gene (~1500 bp). Analyze sequence against curated databases (e.g., EzBioCloud, SILVA) for preliminary genus identification [10] [19].
- For higher resolution, perform Multilocus Sequence Analysis (MLSA) using a set of housekeeping genes (e.g., atpD, gyrB, recA). Concatenate sequences and construct phylogenetic trees [8] [19].
- Calculate Average Nucleotide Identity (ANI) using whole genome sequences for definitive species demarcation (threshold ≥95-96% for species boundary) [19].
Phenotypic Analysis:
- Culture isolates on standardized media and incubate under optimal conditions.
- Perform morphological characterization (cell shape, size, motility, Gram stain).
- Conduct biochemical tests (carbon source utilization, enzyme activities) using standardized microarrays or API strips.
- Analyze cellular fatty acid profiles using FAME analysis.
Phylogenetic Analysis:
- Integrate genotypic and phenotypic data to construct a consensus taxonomy.
- Use phylogenetic trees (based on 16S rRNA, MLSA, or genome sequences) to determine evolutionary relationships.
- Compare results with type strains from culture collections.
Quality Control: Include type/reference strains in each experimental batch. Perform all tests in triplicate. Use negative controls in PCR and biochemical assays.
Protocol 2: Implementing Heterogenization in Study Design
This protocol systematically introduces biological and environmental variation to enhance the generalizability and reproducibility of findings [65].
Key Reagent Solutions:
- Multiple Strains/Isolates: Utilize genetically distinct strains of the same species from different repositories (e.g., ATCC, DSMZ).
- Multiple Reagent Lots: Intentionally use different lots of culture media and critical reagents.
- Environmental Variation Tools: Controlled environmental chambers to vary temperature, humidity, or light cycles if relevant.
Methodology:
- Strain Selection: Instead of a single strain, select multiple bacterial strains representing the genetic diversity of the species under investigation.
- Reagent Variation: For long-term studies, plan to incorporate at least two different lots of key reagents (e.g., growth media, extraction kits).
- Temporal Replication: Conduct experiments across multiple days or weeks rather than within a single session.
- Data Analysis:
- Use statistical models that account for blocking factors (e.g., day, reagent lot, strain).
- Include interaction terms (e.g., treatment-by-laboratory) in the statistical model when analyzing multi-laboratory data.
- Report variances associated with each introduced variable to quantify their impact.
Quality Control: Document all variations meticulously. Use positive controls across all variations to ensure system functionality.
Visualization of Workflows
Polyphasic Taxonomy Workflow
The following diagram illustrates the integrated approach of the polyphasic taxonomy methodology, which combines multiple data types for robust bacterial classification:
Multi-Laboratory Validation Strategy
This diagram outlines a systematic approach for designing reproducible multi-laboratory studies:
Ensuring reproducibility across laboratories in bacterial taxonomy research requires a fundamental shift from extreme standardization to systematic heterogenization. By implementing polyphasic taxonomic approaches with robust protocols, incorporating planned variation in study designs, and embracing multi-laboratory validation, researchers can significantly enhance the reliability and generalizability of their findings. The integration of genomic data with traditional methods provides an increasingly stable taxonomic framework, while quantitative approaches to measuring and reporting variation offer transparency in assessing data quality [19]. For the research community, adopting these practices will strengthen the scientific foundation of microbial taxonomy and accelerate drug development by providing more reliable and reproducible characterization of bacterial isolates.
In the field of bacterial taxonomy and infectious disease management, strain-level identification is paramount. It enables researchers and clinicians to differentiate between harmless commensals and pathogenic variants within the same species, track the spread of antibiotic-resistant clones, and investigate outbreaks with high precision [66]. The polyphasic taxonomic approach, which integrates genotypic, phenotypic, and phylogenetic data, provides the foundational principle for robust bacterial classification [4] [10] [20]. This application note outlines optimized workflows that strategically combine traditional methods, advanced sequencing technologies, and novel culture-independent techniques to achieve efficient and accurate bacterial strain typing within a modern polyphasic framework.
The Strain Typing Toolkit: Methodologies and Mechanisms
A range of techniques is available for strain typing, each with distinct principles, advantages, and appropriate applications. The following section details key methods cited in this note.
Core Genome Multilocus Sequence Typing (cgMLST)
- Principle: cgMLST extends traditional Multi-Locus Sequence Typing (MLST) by analyzing hundreds to thousands of core genes found across the vast majority of isolates within a species. This provides a highly discriminatory and standardized approach for assessing strain relatedness.
- Protocol Outline (as used for manual WGS):
- DNA Extraction: Use a commercial kit (e.g., Quick-DNA Fungal/Bacterial MiniPrep Kit) to extract high-quality genomic DNA from a pure bacterial culture.
- Library Preparation: Employ a library prep kit (e.g., Nextera XT DNA Library Preparation Kit) to fragment DNA and attach sequencing adapters.
- Whole-Genome Sequencing: Sequence the library on an appropriate platform (e.g., Illumina MiSeq with a 2x250 bp cycle kit).
- Bioinformatic Analysis:
- Assemble sequencing reads into contigs using a de novo assembler (e.g., SKESA within SeqSphere+ software).
- Upload the assembled genome to a cgMLST scheme within SeqSphere+.
- The software automatically identifies the target core genes and calls their alleles.
- Compare the allelic profiles of different isolates to construct minimum spanning trees for visualizing phylogenetic relationships [67].
Optical DNA Mapping (ODM) for Direct Strain Typing
- Principle: This single-molecule method visualizes sequence-specific fluorescent patterns on long DNA fragments (~50 kb) stretched in nanofluidic channels. It uses the competitive binding of a fluorescent dye (YOYO-1) and an AT-specific non-fluorescent molecule (netropsin), creating an intensity profile that serves as a "molecular barcode."
- Protocol Outline (for direct patient samples):
- Sample Preparation: Pellet bacteria from liquid culture or uncultured patient samples (e.g., 1-3 mL of urine).
- DNA Extraction (Agarose Plug Method): Encase the pelleted bacteria in an agarose plug to protect long DNA molecules from shear forces. Perform lysis, RNase, and proteinase K treatments within the plug. Melt the plug and digest the agarose with agarase to release high-molecular-weight DNA.
- Fluorescent Labeling: Mix the extracted DNA with YOYO-1, netropsin, and an internal size standard (e.g., λ-DNA). Incubate at 50°C for 30 minutes.
- Imaging and Analysis: Load the sample into a nanofluidic chip and image the stretched DNA molecules using fluorescence microscopy. The resulting intensity profiles are matched against a curated reference database for strain-level identification [68].
Automated Whole Genome Sequencing (WGS)
- Principle: This approach fully automates the steps from nucleic acid extraction to sequencing library preparation, drastically reducing hands-on time and improving reproducibility.
- Protocol Outline (Using the Clear Dx Platform):
- Sample Loading: Resuspend a loopful of bacteria in a resuspension buffer and transfer the supernatant to a 96-well plate.
- Automated Run: Load the plate, reagents, and consumables onto the platform (e.g., Clear Dx instrument).
- Hands-off Processing: The instrument automatically performs cell lysis, nucleic acid extraction, library preparation, and loads the sequencing cartridges onto onboard sequencers (e.g., Illumina iSeq100).
- Data Analysis: The platform's software provides species identification, quality metrics, and downloadable FASTQ files for downstream cgMLST analysis [67].
Quantitative Comparison of Strain Typing Workflows
The choice of strain typing method significantly impacts turnaround time, cost, labor, and applicability to different sample types. The data below, synthesized from recent studies, allows for direct comparison.
Table 1: Comparative Analysis of Bacterial Strain Typing Methods
| Method | Typical Turnaround Time | Key Advantages | Key Limitations | Ideal Use Case |
|---|---|---|---|---|
| Automated WGS (e.g., Clear Dx) | ~28 hours (hands-on time minimal) [67] | High resolution; full genome data; 34-57% cost reduction vs. manual WGS; streamlined workflow [67] | Requires pure culture; high initial equipment cost | High-throughput outbreak investigation in public health labs |
| Manual WGS + cgMLST | ~44-47 hours (includes ~3h hands-on) [67] | Considered gold standard; high resolution; predicts AMR & virulence [67] | Labor-intensive; requires bioinformatics expertise; pure culture needed | Research and reference labs establishing strain relatedness |
| Optical DNA Mapping | <24 hours (from sample) [68] | Works directly on patient samples (e.g., urine); identifies multiple strains in mixtures; no cultivation needed [68] | Emerging technology; requires specialized instrumentation | Rapid diagnostics for polymicrobial infections and urgent cases |
| Polyphasic Taxonomy | Days to weeks [4] [10] | Highly robust; consensus classification; does not rely on a single method [4] [20] | Very time-consuming; requires multiple techniques and expertise | Defining novel bacterial species and comprehensive taxonomic studies |
Table 2: Performance Metrics of Automated vs. Manual WGS
| Parameter | Automated WGS Workflow | Manual WGS Workflow |
|---|---|---|
| Concordance in Isolate Grouping | 99% (222/224 isolates) [67] | (Used as reference method) [67] |
| Average Depth of Coverage | ~88x (range 48x-171x) [67] | Target: 100x-200x [67] |
| Total Turnaround Time | 26-32 hours [67] | 16-19 hours longer than automated [67] |
| Hands-on Technologist Time | Minimal (automated) [67] | ~3 hours [67] |
Integrated Workflow for Strategic Strain Typing
The following diagram synthesizes the discussed methodologies into a strategic, decision-based workflow for efficient strain typing, emphasizing the polyphasic integration of data.
Essential Research Reagent Solutions
The following table catalogs key reagents and kits essential for implementing the strain-typing protocols described in this note.
Table 3: Research Reagent Solutions for Strain Typing Workflows
| Reagent / Kit | Function / Application | Specific Example (if cited) |
|---|---|---|
| DNA Extraction Kit (Bacteria) | Isolation of high-molecular-weight genomic DNA for WGS and other molecular applications. | Quick-DNA Fungal/Bacterial MiniPrep Kit (Zymo Research) [67] |
| WGS Library Prep Kit | Preparation of sequencing libraries from genomic DNA for next-generation sequencing platforms. | Nextera XT DNA Library Preparation Kit (Illumina) [67] |
| cgMLST Analysis Software | Bioinformatic tool for genome assembly, scheme-based allele calling, and phylogenetic analysis. | SeqSphere+ Software (Ridom) [67] |
| Optical DNA Mapping Dyes | Fluorescent and competitive binding molecules for generating sequence-specific intensity profiles on DNA. | YOYO-1 and Netropsin [68] |
| CRISPR-Cas9 Components | Targeted restriction of plasmids for locating antibiotic resistance genes in ODM assays. | crRNA/tracrRNA targeting specific genes (e.g., blaCTX-M) [68] |
| Automated WGS Platform | Integrated system for fully automated nucleic acid extraction, library prep, and sequencing. | Clear Dx Microbial Surveillance WGS v2.0 (Clear Labs) [67] |
Application Notes: Managing Polyphasic Data in Bacterial Taxonomy
The polyphasic approach to bacterial systematics, which integrates genotypic, phenotypic, and chemotaxonomic data, is the established standard for the definitive classification and identification of microorganisms [4]. This methodology resolves taxonomic uncertainties that arise from using single-method characterization, but it generates complex, multi-faceted datasets that present significant data management challenges. Effective handling of this data volume and complexity is crucial for research in bacterial identification, taxonomy, and subsequent drug development.
The core challenge lies in the heterogeneous nature of the data, which typically includes:
- Genotypic Data: Genome sequences, 16S rRNA gene sequences, digital DNA-DNA hybridization (dDDH) values, and Average Nucleotide Identity (ANI) calculations [69] [16].
- Phenotypic Data: Results from morphological, physiological, and biochemical assays (e.g., API ZYM, API 20NE tests) [69] [16].
- Chemotaxonomic Data: Analyses of cellular components like pigments, fatty acids, and other macromolecules [16].
The following table summarizes the key data types and management solutions in polyphasic taxonomy.
Table 1: Data Types and Management Solutions in Polyphasic Taxonomy
| Data Category | Specific Data Types | Data Management Challenges | Proposed Solution |
|---|---|---|---|
| Genotypic Data | 16S rRNA gene sequences, Whole Genome Sequences (WGS), ANI values, dDDH values [69] [16] | Large file sizes, requirement for specialized bioinformatics tools, need for accurate phylogenetic analysis | Use of standardized bioinformatics pipelines (e.g., INNuca for assembly [69]), online services for dDDH/ANI (GGDC, JSpeciesWS) [69], and structured databases for sequence metadata. |
| Phenotypic Data | Gram stain, colony morphology, temperature/pH/salt tolerance, carbon source utilization, enzyme assays [69] [16] | Diverse, non-standardized, often categorical or ordinal data (nominal, ordinal levels of measurement) [70] | Centralization in a structured database with controlled vocabularies; use of numerical coding for categorical data to facilitate comparison. |
| Chemotaxonomic Data | Presence of specific pigments (e.g., flexirubin), cellular fatty acid profiles, other macromolecular compositions [16] | Specialized analytical techniques, complex quantitative results (ratio level of measurement) [70] | Standardized reporting formats and integration with genomic data to link traits with genetic determinants. |
| Strain & Project Metadata | Strain designations, source of isolation, growth conditions, literature references [69] | Incomplete or inconsistent recording, hindering reproducibility | Implementation of mandatory, well-defined fields in project databases, linked to a unique project or strain identifier. |
Experimental Protocols for Key Polyphasic Analyses
Protocol for Phylogenomic Analysis and Species Delineation
This protocol outlines the steps for genome-based phylogenetic analysis and calculating genomic similarity indices for species demarcation, as applied in recent studies [69] [16].
I. Materials
- Bioinformatics Workstation: A computer with high processing power and ample storage for genomic data.
- Software & Pipelines: INNuca pipeline v3.1 (or other robust assembler for Illumina reads), Kraken software 2.0.9 for taxonomic classification [69].
- Online Tools: Access to the Type (Strain) Genome Server (TYGS) for whole-genome-based phylogeny and the Genome-to-Genome Distance Calculator (GGDC) for dDDH. Access to JSpeciesWS for ANI calculation [69].
II. Methodology
- Genome Sequencing and Assembly:
- Extract genomic DNA using a certified kit (e.g., Invitrogen PureLink Genomic DNA mini kit).
- Sequence the genome using an Illumina system (e.g., 2 x 150 bp PE on an Illumina NextSeq 500).
- Assemble the raw reads using a standardized pipeline like INNuca to obtain a draft genome [69].
- Taxonomic Classification:
- Use Kraken software to perform an initial taxonomic classification of the assembled genome [69].
- Phylogenomic Tree Construction:
- Submit the assembled genome to the TYGS online service. This service utilizes the Genome BLAST Distance Phylogeny (GBDP) method to generate a robust, whole-genome-based phylogenetic tree, which will place your strain within the context of related type strains [69].
- Genomic Similarity Calculations:
- Average Nucleotide Identity (ANI): Calculate the ANI using the BLAST algorithm (ANIb) via the JSpeciesWS web service. An ANI value > 95% indicates that the genomes belong to the same species [69].
- Digital DNA-DNA Hybridization (dDDH): Calculate dDDH values using the GGDC online service. Use formula d6. Strains of the same species typically exhibit dDDH values > 70% [69].
III. Data Interpretation
- A novel species is supported when the isolate forms a distinct phylogenetic clade and the ANI and dDDH values with the closest related type strains are below the species threshold (95% and 70%, respectively) [69] [16].
Protocol for Standardized Phenotypic Characterization
This protocol details the phenotypic tests used to differentiate closely related bacterial species, such as Shewanella seohaensis and Shewanella xiamenensis [69].
I. Materials
- Growth Media: Luria-Bertani (LB) agar and broth, or other suitable media (e.g., Marine Agar for marine bacteria) [69] [16].
- Incubators: Capable of maintaining a range of temperatures (e.g., 4°C, 30°C, 37°C, 40°C, 42°C).
- Biochemical Test Kits: Commercial kits such as API ZYM and API 20NE [16].
- Salinity Solutions: NaCl for preparing media with varying salt concentrations (e.g., 0-4% w/v) [16].
II. Methodology
- Morphological Analysis:
- Describe colony morphology (size, color, shape, opacity) on solid media after standard incubation.
- Perform Gram staining and observe cell morphology and arrangement under a microscope [16].
- Physiological Tests:
- Temperature Tolerance: Inoculate bacteria on LB agar and incubate at 4°C, 30°C, 37°C, 40°C, and 42°C. Assess growth after 24-48 hours [69].
- Salinity Tolerance: Inoculate bacteria in broth or on agar containing different NaCl concentrations (e.g., 0%, 1%, 2%, 3%, 4% w/v). Assess growth after standard incubation [16].
- pH Tolerance: Test growth in media adjusted to a pH range (e.g., 6.0-8.0) to determine the optimal pH and range [16].
- Biochemical Profiling:
III. Data Interpretation
- Compile results into a differential table comparing the test isolate to its closest relatives. Positive and negative reactions, as well as differences in growth intensity or optimal ranges, are used for discrimination [69] [16].
Visualization of Workflows and Relationships
Polyphasic Taxonomy Data Management Workflow
This diagram outlines the integrated workflow for managing and analyzing diverse data types in a polyphasic taxonomy study.
Phylogenomic Analysis Decision Pathway
This diagram illustrates the logical decision process for species delineation based on genomic similarity metrics.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents and Materials for Polyphasic Taxonomy
| Item | Function/Application in Protocol |
|---|---|
| Invitrogen PureLink Genomic DNA Mini Kit | For high-quality genomic DNA extraction, which is a prerequisite for whole-genome sequencing and subsequent genotypic analysis [69]. |
| API ZYM and API 20NE Test Kits | Standardized, miniaturized kits for rapid, reproducible biochemical profiling of bacterial isolates, providing crucial phenotypic data [16]. |
| Luria-Bertani (LB) Agar/Broth | A general-purpose growth medium for the cultivation and maintenance of a wide range of bacterial isolates under laboratory conditions [69]. |
| Marine Agar | A specialized growth medium for the cultivation and physiological testing of marine bacteria, such as members of the genus Mariniflexile [16]. |
| Sodium Chloride (NaCl) | For preparing media with defined salinity levels to test an isolate's salt tolerance, a key physiological characteristic [16]. |
| Illumina DNA Sequencing Kits (e.g., Nextera) | Kits for preparing sequencing libraries for platforms like the Illumina NextSeq 500, enabling high-throughput whole-genome sequencing [69]. |
Validating Microbial Identity: Comparative Analysis and the Genomic Gold Standard
The accurate identification and classification of microorganisms are foundational to advancements in microbiology, drug development, and therapeutic discovery. While monophasic methods, relying on a single data type, have been historically used, they often provide an incomplete taxonomic picture. This application note delineates the definitive advantages of the polyphasic taxonomy approach, which integrates genotypic, phenotypic, and phylogenetic data. Through comparative data, detailed protocols, and visual workflows, we demonstrate that polyphasic taxonomy is indispensable for achieving a stable, consensus classification, ensuring the accurate characterization of microbial strains crucial for research and biotechnological applications.
Bacterial systematics has evolved from reliance on morphological and biochemical observations to incorporate powerful molecular techniques. Monophasic methods, which depend on a single line of evidence—such as 16S rRNA gene sequencing—offer speed but can lack resolution and are sometimes misleading for species-level demarcation [10] [4]. This creates a blurred image of taxonomic status and necessitates a more robust framework [4].
The polyphasic taxonomy approach addresses these limitations by integrating all available data—genotypic, phenotypic, and phylogenetic—into a consensus classification [20] [4]. This methodology is not tied to a single theory but is a pragmatic strategy for delineating bacterial taxa with high confidence, a capability critical for discovering novel species, validating probiotics, and ensuring reproducibility in research [20] [71]. This document provides a comparative analysis of these approaches and detailed protocols for implementing polyphasic taxonomy.
Comparative Analysis: Polyphasic vs. Monophasic Methods
The table below summarizes the core differences between monophasic and polyphasic approaches, highlighting the superior diagnostic power of the latter.
Table 1: A Comparative Overview of Monophasic and Polyphasic Taxonomic Approaches
| Feature | Monophasic Approach | Polyphasic Approach |
|---|---|---|
| Definition | Relies on a single type of data (e.g., only 16S rRNA or only morphology) for identification [4]. | Integrates genotypic, phenotypic, and phylogenetic data into a consensus classification [20] [4]. |
| Key Methods | 16S rRNA gene sequencing; conventional biochemical tests [10] [4]. | 16S rRNA, genome sequencing (ANI, dDDH), chemotaxonomy (FAME, MLSA), phenotyping (Biolog) [10] [20] [72]. |
| Resolution | Limited, often to genus level; cannot reliably resolve closely related species [10]. | High, enables differentiation at the species and often strain level [20] [16]. |
| Stability of Classification | Low, can be unstable and change with new single-method data [20]. | High, provides a stable classification resilient to new data [20]. |
| Ability to Identify Novel Taxa | Poor, may misassign or fail to identify novel species [4]. | Excellent, the gold standard for proposing new species and genera [20] [16]. |
| Time & Resource Investment | Lower | Higher |
| Data Integration | None, single-dimensional view. | Holistic, creates a comprehensive profile of the organism. |
| Ideal Application | Preliminary identification, high-throughput screening where approximate identity suffices. | Definitive characterization, taxonomic discovery, quality control for live biotherapeutics [71]. |
The Polyphasic Taxonomy Workflow: A Protocol for Robust Identification
The following section outlines a standard operational protocol for the polyphasic characterization of a bacterial isolate, from cultivation to final classification.
Protocol: Comprehensive Polyphasic Characterization of a Bacterial Isolate
Objective: To accurately identify and characterize a bacterial isolate to the species level using a polyphasic taxonomy framework.
Principle: This protocol combines morphological, biochemical, chemotaxonomic, and genotypic analyses to build a consensus on the taxonomic position of an unknown microorganism [20] [4] [72].
Materials and Reagents
Table 2: Research Reagent Solutions for Polyphasic Taxonomy
| Reagent / Kit | Function / Application |
|---|---|
| Lysogeny Broth (LB) Agar/Media | Routine cultivation and maintenance of bacterial isolates [73]. |
| API ZYM / API 20NE Strips (bioMérieux) | Standardized tests for enzymatic activities and carbohydrate assimilation patterns [16]. |
| BIOLOG ANI MicroPlates | High-throughput phenotypic profiling based on carbon source utilization [72]. |
| Wizard Genomic DNA Purification Kit (Promega) | Extraction of high-quality, high-molecular-weight genomic DNA for sequencing [73]. |
| ThruPLEX DNA-Seq Kit (Takara) | Preparation of paired-end sequencing libraries for next-generation sequencing (NGS) [73]. |
| Primers (27F / 1492R) | Amplification of the nearly full-length 16S rRNA gene for preliminary phylogenetic analysis [72]. |
| MALDI-TOF MS Matrix Solution | Matrix compound for protein profiling using MALDI-TOF Mass Spectrometry [73]. |
Experimental Procedure
Step 1: Phenotypic and Morphological Characterization
- Cultivation: Grow the isolate on appropriate solid and liquid media (e.g., Marine Agar for marine bacteria, LB for common isolates) under optimal temperature and atmospheric conditions [72] [16].
- Colony Morphology: Record colony size, shape, color, elevation, margin, and opacity after 24-72 hours of growth [16].
- Cell Morphology: Perform Gram staining. For detailed ultrastructure, use light microscopy, scanning electron microscopy (SEM), and transmission electron microscopy (TEM) to determine cell shape, size, flagellation, and other structural features [72] [16].
- Physiological Tests: Conduct tests for catalase, oxidase, and H2S production. Determine growth range for temperature, pH, and salinity (NaCl concentration) [16].
Step 2: Chemotaxonomic and Biochemical Profiling
- Fatty Acid Analysis: Extract and analyze cellular fatty acid methyl esters (FAMEs) using Gas Chromatography. Compare the profile with databases (e.g., MIDI) for identification [20].
- Enzyme and Substrate Utilization: Use commercial kits like API ZYM and API 20NE or BIOLOG MicroPlates, following the manufacturer's instructions, to create a metabolic fingerprint of the isolate [72] [16].
Step 3: Genotypic Analysis
- Genomic DNA Extraction: Purify genomic DNA using a commercial kit (e.g., Promega Wizard Kit) [73]. Verify DNA quality and quantity via spectrophotometry and gel electrophoresis.
- 16S rRNA Gene Sequencing: Amplify the 16S rRNA gene using universal primers 27F and 1492R. Sanger-sequence the PCR product. Compare the resulting sequence against public databases (e.g., NCBI, EZBioCloud) for a preliminary phylogenetic affiliation [72].
- Whole-Genome Sequencing (WGS): Prepare a paired-end library from the extracted DNA and sequence the entire genome using an NGS platform (e.g., Illumina) [16] [73]. Assemble the raw reads into contigs using an appropriate assembler (e.g., SPAdes).
Step 4: Phylogenomic and Comparative Genomic Analysis
- Phylogenetic Trees: For isolates with potential novelty, construct a 16S rRNA gene-based phylogenetic tree with related type strains using software like MEGA7 [72]. For higher resolution, perform a phylogenomic analysis based on core genes extracted from the genome sequences [16].
- Genome Similarity Calculations: Calculate the Average Nucleotide Identity (ANI) and digital DNA-DNA Hybridization (dDDH) values between the isolate's genome and its closest phylogenetic neighbors. Use established thresholds (≥95% ANI, >70% dDDH) to confirm novel species status [16] [73].
- Functional Annotation: Annotate the genome to identify key functional genes, biosynthetic gene clusters, and other genomic features that support the phenotypic observations [73].
Step 5: Data Integration and Consensus Classification Synthesize all data from Steps 1-4. A novel species is proposed when the isolate forms a distinct phylogenetic lineage, has genome similarity values below the species thresholds, and possesses unique phenotypic or chemotaxonomic characteristics that distinguish it from its closest relatives [20] [16].
The following workflow diagram visualizes the integration of these multi-layered data streams.
Polyphasic Taxonomy Workflow
Case Study: Application of Polyphasic Taxonomy in Novel Species Identification
A recent study exemplifies the power of this approach. Researchers aimed to characterize strain TRM1-10, isolated from the tomato rhizosphere, which conferred resistance to bacterial wilt [16].
- Phenotypic Data: TRM1-10 was Gram-negative, rod-shaped, and formed yellow colonies. Its growth conditions (0–4% NaCl) differed from known Mariniflexile species, which are predominantly marine [16].
- Genotypic Data: 16S rRNA gene sequencing showed the highest similarity to M. soesokkakense RSSK-9T at only 96.9%, below the common species threshold [16].
- Genomic Evidence: Crucially, whole-genome sequencing revealed ANI values of 85.86% with M. soesokkakense and dDDH values of 27.8%, both significantly below the species delineation thresholds (95% and 70%, respectively). Phylogenomic trees confirmed its placement within the Mariniflexile genus but as a separate lineage [16].
- Consensus Outcome: The integrated data from all analyses provided compelling evidence that TRM1-10 represented a novel species, proposed as Mariniflexile rhizosphaerae sp. nov. A monophasic 16S approach might have misidentified it, while biochemical tests alone could not have confirmed its novelty.
The transition from monophasic to polyphasic taxonomy represents a paradigm shift in microbial systematics. While monophasic methods are useful for rapid, preliminary identification, their limitations in resolution and accuracy make them unsuitable for definitive classification, especially in research and development where strain integrity is paramount [10] [4].
The polyphasic approach, by leveraging multiple, independent data lines, provides a robust and stable taxonomic framework. It is the only method capable of reliably identifying novel species and resolving complex relationships within genera, as demonstrated in the continuous reclassification of groups like the Bacillus subtilis group [73]. For drug development professionals and scientists, adopting this comprehensive approach is critical for ensuring the accurate identification of probiotic candidates [71], the discovery of novel bioactive compound-producing strains [16] [73], and the maintenance of reproducible and reliable research data. The comparative advantage of polyphasic taxonomy is not merely incremental; it is foundational to modern microbiological science.
For nearly 50 years, DNA-DNA hybridization (DDH) served as the gold standard for prokaryotic species circumscriptions at the genomic level, providing a numerical and relatively stable species boundary that has profoundly influenced modern microbial classification systems [29]. This method established that strains showing ≥70% DDH similarity typically belong to the same species [74]. However, in the current genomic era, DDH has revealed significant limitations: it is labor-intensive, difficult to standardize, and impossible to use for building cumulative databases that can be reused for future comparisons [29].
The advent of whole-genome sequencing has facilitated the development of in silico alternatives that overcome these limitations while maintaining correlation with traditional DDH values. Two methods have emerged as the primary genomic metrics for species delineation: Average Nucleotide Identity (ANI) and digital DNA-DNA Hybridization (dDDH) [75]. These genomic metrics now serve as the foundation for a modern, sequence-based taxonomic framework that complements traditional polyphasic approaches.
Establishing the Correlation: From Wet-Lab to In Silico
Quantitative Correlation Between Traditional and Genomic Metrics
Early foundational studies established a clear correlation between wet-lab DDH values and computational genome-based metrics, enabling the transition to in silico methods. The following table summarizes the established correlations and current thresholds for species delineation:
Table 1: Correlation thresholds between traditional and genomic species delineation methods
| Method | Threshold for Species Delineation | Correlated Metric | Correlation Value |
|---|---|---|---|
| DNA-DNA Hybridization (DDH) | ≥70% [74] | N/A | N/A |
| Average Nucleotide Identity (ANI) | 95-96% [29] [75] | DDH 70% | ~95% ANI [29] |
| Digital DDH (dDDH) | ≥70% [75] | DDH 70% | ~70% dDDH [75] |
| 16S rRNA Gene Similarity | ≥98.7% [76] | N/A | N/A |
Research has demonstrated that these thresholds generally hold across diverse taxonomic groups, though some variations exist. In the genus Streptomyces, for instance, a 70% dDDH value corresponds more closely to approximately 96.7% ANI rather than 95-96% [76]. Similarly, for the Enterobacter cloacae complex, the 95-96% ANI threshold effectively delineates species and subspecies when combined with supporting genomic evidence [75].
Advantages of Genomic Metrics
The transition from traditional DDH to genome-based metrics offers several substantial advantages for modern taxonomy:
- Cumulative Databases: Unlike DDH results, ANI and dDDH values generate data that can be stored in reusable, searchable databases for future comparative studies [29]
- Standardization: Computational methods reduce laboratory-specific variations inherent in traditional DDH protocols [74]
- High Resolution: ANI provides superior discriminatory power compared to single-gene approaches like 16S rRNA sequencing, which often lacks resolution at the species level despite its conservation across prokaryotes [29] [76]
- Reproducibility: In silico methods yield identical results when repeated with the same genomic data, facilitating verification and comparison across research groups [77]
Experimental Protocols for Genomic Metrics
Protocol 1: Average Nucleotide Identity (ANI) Analysis
Research Reagent Solutions
Table 2: Essential tools and resources for ANI analysis
| Tool/Resource | Function | Access |
|---|---|---|
| JSpecies | Biologist-oriented interface to calculate ANI and tetranucleotide signatures [29] | Web application or standalone tool |
| fastANI | Rapid alignment-free tool for large-scale ANI calculations [75] | Command-line tool |
| MUMmer | Ultra-rapid genome alignment system used for ANIm calculation [29] | Command-line tool |
| NCBI Genome Database | Source of reference genomes for comparison [29] | Public database |
| Type Strain Genome Server (TYGS) | Comprehensive platform for bacterial species analysis [73] | Web service |
Step-by-Step Workflow
Genome Sequence Acquisition
- Obtain high-quality genome sequences of the query strain and reference type strains. For novel species description, compare against type strains of most closely related species [16]
- Ensure data quality by removing poor-quality reads and assembling with appropriate tools (e.g., SPAdes). Filter contigs shorter than 200 bp [75]
Method Selection
- Choose appropriate ANI calculation method based on research needs:
- ANIm (MUMmer-based): Faster computation, ideal for large datasets or highly similar genomes (>90% identity) [76]
- ANIb (BLAST-based): More robust for divergent genomes, fragments genomes into 1020 nt fragments mimicking DDH [29] [74]
- fastANI (alignment-free): Ultrafast computation for large-scale comparisons [75]
- Choose appropriate ANI calculation method based on research needs:
Calculation Execution
Interpretation of Results
Protocol 2: Digital DNA-DNA Hybridization (dDDH) Analysis
Research Reagent Solutions
Table 3: Essential tools and resources for dDDH analysis
| Tool/Resource | Function | Access |
|---|---|---|
| GGDC (Genome-to-Genome Distance Calculator) | Primary tool for dDDH calculation using multiple formulas [76] | Web service |
| Type Strain Genome Server (TYGS) | Integrated platform for dDDH analysis and phylogenetic placement [73] | Web service |
| Reference Genome Database | Curated collection of type strain genomes for comparison | DSMZ / NCBI |
Step-by-Step Workflow
Data Preparation
- Obtain complete or draft genome sequences of query and reference strains
- Ensure sequences are in appropriate format (FASTA)
- Verify strain identities, particularly when using publicly available genomes, as misidentified strains are common in databases [29]
GGDC Analysis
- Access the GGDC web server
- Upload query and reference genome sequences
- Select appropriate calculation formula:
- Formula d0: Considers all matching k-mers
- Formula d4: Primarily considers identity of k-mers
- Formula d6: Corresponds most closely to conventional DDH values [74]
- Execute analysis
TYGS Analysis
- Alternative comprehensive platform for dDDH calculation
- Provides additional phylogenetic context through whole-genome-based trees [73]
- Offers access to a specialized database of type strain genomes
Interpretation of Results
Integration with Polyphasic Taxonomy
The Role of Genomic Metrics in a Polyphasic Framework
While ANI and dDDH provide robust genomic boundaries for species delineation, they function most effectively within a comprehensive polyphasic framework that incorporates multiple lines of evidence [76]. This integrated approach ensures that taxonomic decisions reflect both genomic relatedness and phenotypic coherence.
Genomic metrics should be interpreted alongside:
- Phenotypic characteristics: Growth requirements, metabolic capabilities, and morphological features [16]
- Chemotaxonomic data: Cell wall composition, fatty acid profiles, and other biochemical markers [76]
- Phylogenetic analysis: 16S rRNA gene sequences (for initial grouping) and core genome phylogenies [16]
Decision Framework for Species Delineation
The following workflow illustrates how genomic metrics integrate with other data types in a polyphasic taxonomic study:
Applications and Case Studies
Resolving Taxonomic Complexes
Genomic metrics have proven particularly valuable for clarifying taxonomic relationships within complex groups:
- Enterobacter cloacae complex: ANI and dDDH analyses helped resolve classification conflicts, supporting a subspecies-based classification scheme that correlated better with virulence genes and capsule typing patterns [75]
- Streptomyces species: MLSA distance values showed strong correlation with ANI and dDDH, enabling development of a refined decision framework for species delineation in this genus [76]
- Mariniflexile species: ANI values (85.42-85.86%) and dDDH values (27.0-27.8%) between a novel rhizosphere strain and marine isolates provided clear evidence for proposing a novel species [16]
Database Curation and Quality Control
The implementation of genomic metrics has revealed significant issues with strain identification in public databases. One study found that less than 30% of sequenced genomes labeled with validly published names actually belonged to the corresponding type strains [29]. This highlights the critical importance of:
- Verifying strain identities through culture collection catalogs
- Prioritizing sequencing of type strains to establish reference datasets
- Applying ANI and dDDH analyses to correct misidentified genomes in public databases
The correlation between traditional DDH values and modern genomic metrics has successfully enabled a paradigm shift in prokaryotic taxonomy. The established thresholds of >95-96% for ANI and >70% for dDDH provide robust, reproducible standards for species delineation that mirror the historical gold standard while offering substantial advantages in speed, accuracy, and data reuse.
As genomic sequencing becomes increasingly accessible, these in silico methods will continue to form the cornerstone of polyphasic taxonomic approaches, enabling researchers to build upon cumulative databases and establish a more stable, predictive classification system for prokaryotes. Proper implementation of these tools—with attention to quality control, appropriate thresholds for specific taxonomic groups, and integration with phenotypic data—will ensure continued progress in microbial systematics.
Prokaryotic taxonomy is undergoing a profound transformation, moving from phenotype-based classifications to a robust, sequence-based framework grounded in evolutionary relationships. The Genomic Species Concept has emerged as a unified framework, leveraging whole-genome data to delineate species with unprecedented resolution and objectivity. This paradigm shift addresses the limitations of single-marker gene analysis and incorporates the complex realities of prokaryotic genomics, including horizontal gene transfer and pangenome diversity. By integrating genomic data with established principles, this concept provides a stable, predictive, and scalable taxonomy essential for modern microbiology, from clinical diagnostics to bioprospecting.
The classification of prokaryotes has long been a challenging endeavor, historically reliant on observable phenotypic characteristics such as morphology, biochemical tests, and physiological attributes [19]. However, these properties often fail to reveal true evolutionary relationships, leading to artificial groupings [78]. The advent of molecular genetics provided the first tools for a more natural, phylogenetic classification. Comparative 16S rRNA gene sequencing, pioneered by Woese, revolutionized the field by revealing the three-domain structure of life and offering a universal molecular chronometer [19].
Despite its utility, the 16S rRNA gene lacks sufficient resolution for precise species-level demarcation, as organisms with highly similar 16S sequences can represent distinct genomic species [78] [19]. The contemporary solution, polyphasic taxonomy, strives for a consensus by integrating phenotypic, genotypic, and phylogenetic data [78] [4]. Yet, the rapid accumulation of whole-genome sequences is now propelling a decisive shift toward a genome-based taxonomy [79] [19]. The Genomic Species Concept formalizes this shift, defining a species as a monophyletic group of strains whose genomes are more similar to each other than to those of any other group, as measured by robust genomic metrics. This framework satisfies the need for an evolutionary-based, portable, and highly discriminatory system capable of classifying both cultivated isolates and uncultivated organisms recovered through metagenomics [19].
The Conceptual Framework: From Phenotype to Genome
The Limitations of Pre-Genomic Concepts
Traditional prokaryotic taxonomy relied heavily on pragmatic, purpose-built definitions. The Biological Species Concept, which defines species based on reproductive isolation, is largely inapplicable to asexual prokaryotes that exhibit widespread horizontal gene transfer [80] [81]. Prior to genomics, the gold standard for species delineation was DNA-DNA hybridization, with a recommended threshold of 70% similarity [78] [79]. Although useful, this method is technically cumbersome, not easily reproducible, and provides no information on evolutionary relationships [78].
The introduction of 16S rRNA gene sequence analysis (≥97% identity for species demarcation) provided a universal and portable tool [78]. It successfully established a broad phylogenetic framework but proved inadequate for distinguishing between closely related species, as it represents only a tiny fraction (∼0.05%) of the total genome [19]. This limitation often resulted in the grouping of genetically distinct organisms.
The Rise of the Genomic Species Concept
The Genomic Species Concept is founded on the principle that the complete genome sequence is the ultimate reference standard for determining phylogeny and taxonomy [79] [19]. This concept leverages several key advantages of whole-genome data:
- Comprehensive Resolution: Genome sequences provide significantly greater phylogenetic signal than any single gene, enabling the resolution of both deep evolutionary relationships and recent divergences [19].
- Objectivity and Reproducibility: Genomic metrics, such as Average Nucleotide Identity (ANI), offer highly reproducible and objective criteria for species delineation [19] [73].
- Handling of Uncultured Diversity: Metagenome-assembled genomes can be systematically incorporated into the taxonomic framework, finally allowing the "uncultured majority" to be classified [19].
This conceptual transition is summarized in the following workflow, which depicts the integration of genomic data into the modern polyphasic taxonomy approach.
Genomic Delineation: Metrics and Methodologies
A cornerstone of the Genomic Species Concept is the availability of standardized, quantitative metrics to replace older, less reproducible methods. The following table summarizes the key genomic standards used for species and subspecies delineation.
Table 1: Genomic Metrics for Species Delineation in Prokaryotes
| Metric | Description | Threshold for Same Species | Replaces/Correlates With |
|---|---|---|---|
| Average Nucleotide Identity (ANI) | The average nucleotide identity of all orthologous genes shared between two genomes [80] [73]. | ≥95% [80] [73] | ~70% DNA-DNA Hybridization [80] |
| digital DNA-DNA Hybridization (dDDH) | An in silico simulation of the laboratory DDH experiment [73]. | >70% [73] | 70% wet-lab DDH [73] |
| 16S rRNA Gene Identity | Percentage identity of the small subunit ribosomal RNA gene. | ≥97% (but not sufficient alone) [78] | N/A |
| G+C Content Difference | Difference in the guanine-cytosine content of the genomes. | <1% within a species [73] | Historical phenotypic clustering |
Among these, ANI has become the most widely accepted and robust standard due to its clear biological interpretation and computational efficiency. The comparison between a novel isolate and a type strain is a critical step in classification, as visualized below.
Laboratory Protocol: Genome Sequencing for Taxonomic Classification
This protocol details the steps from a bacterial isolate to a taxonomic designation using whole-genome sequencing.
1. DNA Extraction and Quality Control
- Reagent: Wizard Genomic DNA Purification Kit or equivalent.
- Procedure: Extract high-molecular-weight genomic DNA from a pure culture. Assess DNA purity and integrity using spectrophotometry (e.g., A260/A280 ratio ~1.8-2.0) and gel electrophoresis.
- Critical Step: Ensure DNA is free of contaminants that could inhibit sequencing.
2. Library Preparation and Sequencing
- Reagent: ThruPLEX DNA-Seq Kit or similar for Illumina compatibility.
- Procedure: Fragment DNA, repair ends, add indexing adapters, and PCR-amplify the library. Quantify the final library accurately.
- Sequencing Platform: Perform paired-end sequencing (e.g., 2x150 bp) on an Illumina Hi-Seq/Mi-Seq platform [73]. For complete genomes, consider long-read technologies (PacBio, Oxford Nanopore).
3. Genome Assembly and Annotation
- Software: Use tools like Fastp for read trimming and quality control [73]. Perform de novo assembly with SPAdes, Unicycler, etc.
- Quality Assessment: Check assembly completeness and contamination with CheckM. Annotate the genome using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) or RAST.
4. Phylogenomic and Comparative Analysis
- Average Nucleotide Identity (ANI): Calculate ANI using the OrthoANIU algorithm against the Type Strain Genome Server (TYGS) or a local database [73].
- digital DDH: Calculate dDDH using the GGDC web server [73].
- Phylogenomic Treeing: Identify a set of core, single-copy genes. Create a multiple sequence alignment and infer a maximum-likelihood tree (e.g., with IQ-TREE) to visualize monophyly [16].
Table 2: Key Reagents, Tools, and Databases for Genomic Taxonomy
| Item/Resource | Function/Description | Application in Taxonomy |
|---|---|---|
| Wizard Genomic DNA Purification Kit | Isolation of high-quality genomic DNA from bacterial cultures. | The foundational step for obtaining template DNA for genome sequencing [73]. |
| Illumina Sequencing Platform | High-throughput platform for generating short-read sequence data. | Provides the raw data for genome assembly; the most common data source for modern taxonomy [73]. |
| Fastp Software | A tool for fast and quality-controlled processing of sequencing data. | Performs adapter trimming and quality filtering to ensure clean data for assembly [73]. |
| Type Strain Genome Server (TYGS) | A web service for prokaryotic species identification and classification. | The primary platform for performing digital DDH calculations against type strains [73]. |
| OrthoANIU Algorithm | A program for calculating Average Nucleotide Identity (ANI). | Used for precise genomic species delineation against a reference database [80] [73]. |
| CheckM | A tool for assessing the quality and completeness of genome assemblies. | Evaluates metagenome-assembled genomes (MAGs) and isolate genomes prior to taxonomic analysis [19]. |
| GTDB (Genome Taxonomy Database) | A public database providing a standardized bacterial and archaeal taxonomy based on genomics. | Essential resource for phylogenomic placement and accessing curated reference genomes [73]. |
Case Study: Resolving theBacillus velezensisComplex
The power of the Genomic Species Concept is exemplified in the reclassification of the Bacillus subtilis group. Strains initially identified as different species or subspecies (e.g., B. amyloliquefaciens subsp. plantarum, B. methylotrophicus) based on phenotypic traits and limited genetic data were later found to belong to a single genomic species: Bacillus velezensis [73].
A recent study characterized nine Bacillus strains isolated from soil in Brazil. Initial identification by MALDI-TOF MS suggested they belonged to the B. subtilis group, but precise classification was unclear [73]. Whole-genome sequencing and subsequent ANI analysis revealed that the strains shared 95% to 98.04% ANI with the B. velezensis type strain NRRL B-41580, while dDDH values ranged from 89.3% to 91.8%, firmly placing them within the B. velezensis species boundary [73]. Phylogenomic analysis confirmed that the strains formed a monophyletic clade with B. velezensis NRRL B-41580 with a 100% bootstrap value, demonstrating the cohesive power of this approach to accurately group and identify strains with high biotechnological potential [73].
The Genomic Species Concept represents the culmination of a long search for a natural, evolutionary-based framework for prokaryotic taxonomy. By leveraging the comprehensive information within whole genomes, it overcomes the limitations of phenotypic and single-gene approaches, providing a unified, objective, and scalable system. As sequencing technologies continue to advance and computational tools become more sophisticated, this framework will be essential for organizing the exploding diversity of the microbial world, ultimately strengthening research and development across microbiology, ecology, and biotechnology.
Bacterial taxonomy, the science of classifying and naming microorganisms, has evolved significantly from its early reliance on morphological and phenotypic characteristics. The advent of molecular biology introduced powerful genetic tools, yet single-gene analyses, like 16S rRNA sequencing, often lack the resolution to distinguish between closely related species, leading to historical misclassifications [15] [82]. To overcome these limitations, the field has increasingly adopted a polyphasic approach, which integrates genomic, phenotypic, and phylogenetic data to create a robust and holistic taxonomic framework [15]. This methodology is essential for correcting long-standing errors and accurately delineating taxonomic boundaries. This Application Note details the protocols and presents a contemporary case study demonstrating how a polyphasic approach successfully resolved the misclassification between the genera Zhongshania and Marortus, leading to the description of a novel species [15].
## The Polyphasic Toolkit: Core Components and Protocols
A polyphasic taxonomic study synthesizes data from multiple, independent methodologies. The following table summarizes the key components and their specific roles in resolving taxonomic ambiguities.
Table 1: Core Components of a Polyphasic Taxonomic Study
| Component | Primary Function | Key Taxonomic Insight |
|---|---|---|
| 16S rRNA Gene Phylogeny [15] | Initial placement and assessment of evolutionary relationships. | Provides a first estimate of genus-level affiliation; cannot reliably delineate species. |
| Whole-Genome Sequencing [15] | Provides comprehensive genetic data for high-resolution comparisons. | Enables calculation of definitive genomic metrics like ANI and dDDH for species demarcation. |
| Phylogenomic Analysis [82] | Reconstructs evolutionary history using hundreds of core genes. | Offers a highly robust and reliable phylogenetic tree compared to single-gene trees. |
| Digital DNA-DNA Hybridization (dDDH) [15] | Estimates genome similarity between two strains. | A value ≥70% is a standard threshold for defining a bacterial species [15]. |
| Average Nucleotide Identity (ANI) [15] | Calculates the average nucleotide identity of shared genes between two genomes. | A value ≥95% is a widely accepted genomic standard for species demarcation [15]. |
| Phenotypic Characterization [15] | Assesses morphological, physiological, and biochemical traits. | Determines flagellar type, optimal growth conditions (temp, pH, salinity), and substrate utilization. |
| Chemotaxonomic Analysis [15] | Profiles specific cellular components. | Identifies major respiratory quinones (e.g., Q-8), polar lipids, and fatty acids (e.g., C16:0). |
### Detailed Experimental Protocols
#### Protocol 1: 16S rRNA Gene Sequencing and Phylogenetic Analysis
Purpose: To obtain an initial phylogenetic placement of the isolate.
- DNA Extraction: Cultivate the bacterial strain (e.g., on Marine Agar for marine bacteria) and extract genomic DNA using a commercial kit, such as the DNeasy PowerSoil Pro kit (QIAGEN) [15].
- PCR Amplification: Amplify the nearly full-length 16S rRNA gene using universal primers (e.g., 27F and 1492R). A standard PCR protocol should be used.
- Sequencing and Assembly: Purify the PCR product and perform Sanger sequencing using additional internal primers (e.g., 518F, 805R). Assemble the resulting sequence reads into a consensus sequence using software like BioEdit.
- Phylogenetic Tree Construction:
- Compare the assembled sequence against databases like EzBioCloud or GenBank to identify closely related type strains [15].
- Perform a multiple sequence alignment with tools like CLUSTAL W.
- Construct phylogenetic trees using neighbor-joining, maximum-likelihood, and maximum-parsimony algorithms in software such as MEGA X [15].
- Assess the robustness of the tree topology with bootstrap analysis (e.g., 1000 replicates) [15].
#### Protocol 2: Whole-Genome Sequencing and Phylogenomics
Purpose: To perform high-resolution genomic comparisons for accurate species and genus delineation.
- Genome Sequencing: Submit high-quality genomic DNA for whole-genome sequencing using an Illumina (short-read) and/or Oxford Nanopore (long-read) platform to produce a draft or complete genome assembly [15].
- Genome Annotation: Annotate the assembled genome using automated pipelines (e.g., RAST, Prokka) to identify coding sequences, RNAs, and other genomic features [15].
- Calculation of Genomic Relatedness Indexes:
- Average Nucleotide Identity (ANI): Use tools such as OrthoANIU or FastANI to calculate the ANI between the query genome and the genomes of related type strains [15]. The 95% threshold is a key species boundary.
- Digital DNA-DNA Hybridization (dDDH): Calculate dDDH values using the Genome-to-Genome Distance Calculator (GGDC) [15]. The 70% threshold corresponds to the traditional DNA-DNA hybridization standard for species definition.
- Phylogenomic Tree Construction: Extract a set of universal, single-copy core genes (e.g., using UBCG - Up-to-date Bacterial Core Gene) from the genomes of interest [15]. Align the concatenated sequences and construct a maximum-likelihood tree with high bootstrap support.
#### Protocol 3: Phenotypic and Chemotaxonomic Characterization
Purpose: To provide complementary, non-genetic data that reflects the organism's functional and ecological identity.
- Morphology: Use microscopy (e.g., Cryo-TEM) to determine cell shape, size, and flagellation [15].
- Physiology: Determine optimal growth conditions for temperature, pH, and salinity by cultivating the strain in a range of media [15].
- Chemotaxonomy:
- Respiratory Quinones: Analyze quinones using mass spectrometry-based methods (e.g., HPLC-MS).
- Polar Lipids: Profile polar lipids via two-dimensional thin-layer chromatography.
- Fatty Acid Methyl Esters (FAMEs): Analyze cellular fatty acids using gas chromatography [15].
The integration of data from these protocols is a critical, multi-step process. The workflow below visualizes the logical sequence and how findings from each branch inform the final taxonomic conclusion.
## Case Study: Resolving theZhongshaniaandMarortusComplex
A recent study exemplifies the power of the polyphasic approach to resolve taxonomic confusion between the genera Zhongshania and Marortus within the family Spongiibacteraceae [15]. Initially, Marortus luteolus ZX-21T was distinguished from Zhongshania species based on lineage and phenotypic features. However, subsequent phylogenomic analyses suggested significant overlap, indicating potential misclassification [15].
### Application of the Polyphasic Toolkit
Researchers re-evaluated the type strains of all recognized species under uniform conditions, employing the protocols outlined above.
Genomic Findings: The decisive genomic data is summarized in the table below.
Table 2: Genomic Relatedness Metrics for Zhongshania and Marortus Type Strains [15]
| Strain 1 | Strain 2 | dDDH Value (%) | ANI Value (%) | Interpretation |
|---|---|---|---|---|
| Zhongshania marina DSW25-10T | Marortus luteolus ZX-21T | >70% | >95% | Belong to the same species [15] |
| Zhongshania marina DSW25-10T | Zhongshania aquimaris CAU 1632T | <70% | 73.31 - 78.57% | Represent distinct species [15] |
| Zhongshania antarctica ZS5-23T | Zhongshania aliphaticivorans SM-2T | <70% | 73.31 - 78.57% | Represent distinct species [15] |
Phenotypic and Chemotaxonomic Findings:
- Morphology: Cryo-TEM revealed that both Z. marina DSW25-10T and M. luteolus ZX-21T possessed a single polar flagellum, contradicting the earlier description of M. luteolus as having numerous flagella [15].
- Physiology: The strains shared highly similar optimal growth conditions (30°C, pH 6.5, 0-2% NaCl) [15].
- Chemotaxonomy: All strains in the cluster shared major respiratory quinone (Q-8), polar lipids (PE, PG, DPG), and the major fatty acid (C16:0) [15].
The polyphasic data provided conclusive evidence: the high dDDH and ANI values (>70% and >95%, respectively) between Z. marina and M. luteolus confirmed they are a single species [15]. The supporting phenotypic and chemotaxonomic data validated this genomic finding. Consequently, the study proposed Marortus luteolus as a later heterotypic synonym of Zhongshania marina [15].
Furthermore, the same polyphasic workflow identified a novel isolate, BJYM1T, as a new species. Its genomic relatedness (ANI 73.31-78.57%; dDDH <70%) and distinct metabolic pathways (e.g., unique cobalt and ferrous iron transporters) clearly differentiated it from its closest relatives, leading to the description of Zhongshania aquatica sp. nov. [15].
## The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Polyphasic Taxonomy
| Item | Function/Application |
|---|---|
| Marine Agar (MA; Difco) [15] | Standard medium for the cultivation and isolation of marine bacteria. |
| DNeasy PowerSoil Pro Kit (QIAGEN) [15] | DNA extraction kit optimized for microbial cells, used to obtain high-quality genomic DNA for PCR and sequencing. |
| Universal 16S rRNA Primers (27F, 1492R, etc.) [15] | Oligonucleotides for PCR amplification of the 16S rRNA gene for initial phylogenetic analysis. |
| CLUSTAL W Software [15] | Tool for performing multiple sequence alignments of 16S rRNA or core gene sequences. |
| MEGA X Software [15] | Integrated tool for conducting phylogenetic analysis and building evolutionary trees. |
| FastANI/OrthoANIU Software [83] [15] | Computational tools for rapid calculation of Average Nucleotide Identity between genomes. |
| Genome-to-Genome Distance Calculator (GGDC) [15] | Online tool for calculating digital DNA-DNA hybridization (dDDH) values. |
| Cryo-Transmission Electron Microscope (Cryo-TEM) [15] | Advanced imaging for high-resolution visualization of ultrastructural features like flagella. |
The case of Zhongshania and Marortus powerfully illustrates that reliance on limited data, whether single-gene phylogenies or incomplete phenotypic profiles, is insufficient for accurate taxonomic classification. The polyphasic approach, by integrating high-resolution genomic standards like ANI and dDDH with detailed phenotypic and chemotaxonomic characterization, provides an unambiguous framework to correct historical misclassifications and establish a stable, predictive taxonomy [15] [82]. This rigorous methodology is fundamental for all microbial research, ensuring that scientific discoveries in ecology, biotechnology, and drug development are built upon a foundation of correctly identified organisms.
Application Note: A Polyphasic Framework for Strain Identification and Validation
The discovery and development of novel drugs and Live Biotherapeutic Products (LBPs) demand rigorous validation methodologies to ensure product safety, efficacy, and quality. A polyphasic approach, which integrates phenotypic, genotypic, and chemotaxonomic data, has emerged as the gold standard for the precise identification and characterization of bacterial strains. This framework is crucial for applications ranging from discovering novel antimicrobials to developing defined microbial consortia for therapeutic use. This application note details a validated, polyphasic protocol for bacterial identification, framing it within the context of modern drug and LBP development pipelines.
A Polyphasic Workflow for Strain Validation
The following workflow outlines the key stages of a comprehensive polyphasic characterization, from initial isolation to final validation and deposition. This process ensures the accurate identification and functional understanding of a bacterial strain for critical applications.
Experimental Protocols
Protocol 1: Whole Genome Sequencing and Phylogenomic Analysis
Purpose: To obtain comprehensive genomic data for precise strain identification and phylogenetic placement.
Materials:
- Purified genomic DNA (>50 ng/µL in TE buffer)
- Illumina DNA Prep kit or similar
- Illumina sequencing platform (e.g., Hi-Seq 2500)
- High-performance computing cluster
- Bioinformatics software: GTDB-tk, OrthoANI, TYGS
Method:
- DNA Extraction and Quality Control: Extract high-molecular-weight DNA using a commercial kit (e.g., Wizard Genomic DNA Purification Kit). Assess purity and concentration via spectrophotometry (A260/A280 ratio of ~1.8-2.0) and confirm integrity by agarose gel electrophoresis [84].
- Library Preparation and Sequencing: Fragment DNA and construct paired-end libraries using a ThruPLEX DNA-Seq Kit. Sequence on an Illumina Hi-Seq 2500 platform to a minimum depth of 100x coverage [84].
- Genome Assembly and Annotation: Assemble raw reads into contigs using SPAdes. Assess assembly quality using QUAST. Annotate the assembled genome with PROKKA to identify coding sequences, rRNA, and tRNA genes [16].
- Phylogenomic Calculation:
- Calculate Average Nucleotide Identity (ANI) using OrthoANI with the proposed novel strain and all closely related type strains. An ANI value of <95% supports novel species designation [16] [84].
- Perform digital DNA-DNA Hybridization (dDDH) using the Type Strain Genome Server (TYGS). A dDDH value of <70% supports novel species designation [16] [84].
- Phylogenetic Tree Construction: Generate a phylogenomic tree based on a set of core single-copy genes (e.g., 1,347 genes) using maximum-likelihood methods in RAxML. Include relevant type strains and use an appropriate outgroup. Support node robustness with 1000 bootstrap replicates [16].
Protocol 2: Phenotypic and Chemotaxonomic Profiling
Purpose: To determine phenotypic and chemotaxonomic traits that differentiate the strain from its closest relatives.
Materials:
- Marine Agar and other growth media
- API ZYM and API 20NE test strips (bioMérieux)
- GC-MS system for fatty acid analysis
Method:
- Morphological and Growth Characterization: Describe colony morphology, size, and pigmentation after growth on Marine Agar for 72 hours. Determine optimal growth temperature (e.g., 25-30°C) and pH (e.g., 7.0-8.0) and salt tolerance (e.g., 0-4% NaCl) in liquid media [16].
- Biochemical Assays: Inoculate API ZYM and API 20NE strips according to the manufacturer's instructions. Record reactions after specified incubation periods. Typical positive reactions for novel strains may include α-galactosidase, alkaline phosphatase, and N-acetyl-β-glucosaminidase [16].
- Chemotaxonomic Analysis: Harvest cells from mid-log phase cultures grown on standard media. Extract and analyze fatty acid methyl esters (FAMEs) following the Sherlock Microbial Identification System protocol. Identify major fatty acids (e.g., iso-C15:0, C15:0, iso-C17:0 3-OH) by comparing retention times to standards [16].
Critical Validation Parameters and Thresholds
The following table summarizes the key genomic thresholds and phenotypic characteristics used to validate a novel bacterial species, using Mariniflexile rhizosphaerae TRM1-10T as an exemplar [16].
Table 1: Quantitative Thresholds for Novel Species Validation
| Parameter | Recommended Cut-off for Novel Species | Exemplar Data: Mariniflexile rhizosphaerae TRM1-10T |
|---|---|---|
| Average Nucleotide Identity (ANI) | < 95% [84] | 85.86% vs. M. soesokkakense RSSK-9T [16] |
| digital DNA-DNA Hybridization (dDDH) | < 70% [84] | 27.8% vs. M. soesokkakense RSSK-9T [16] |
| 16S rRNA Gene Similarity | < 98.7-99.0% | 96.9% vs. M. soesokkakense RSSK-9T [16] |
| Difference in G+C Content | ≤ 1% [84] | Data consistent with genus [16] |
| Major Fatty Acid Differences | Presence/absence or significant ratio differences | Presence of specific fatty acids (e.g., iso-C15:0) [16] |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Polyphasic Bacterial Identification
| Reagent / Kit | Function | Application Context |
|---|---|---|
| Wizard Genomic DNA Purification Kit | High-quality DNA extraction | Prepares pure, integral DNA for WGS and PCR [84]. |
| ThruPLEX DNA-Seq Kit | Library preparation for NGS | Creates sequencing-ready libraries from low-input DNA [84]. |
| API ZYM / API 20NE Strips | Standardized biochemical profiling | Provides reproducible phenotypic data for taxonomic discrimination [16]. |
| Sherlock Microbial ID System | Fatty Acid Methyl Ester (FAME) analysis | Generates chemotaxonomic fingerprints for species-level identification [16]. |
| MARINE AGAR | Standardized growth medium | Supports the cultivation of diverse marine and halotolerant bacteria for phenotypic studies [16]. |
Application Note: Validation in Live Biotherapeutic Products (LBPs) and AI-Driven Discovery
The field of Live Biotherapeutic Products (LBPs) represents a paradigm shift in therapeutics, using live organisms to treat or prevent diseases. Concurrently, Artificial Intelligence (AI) is revolutionizing drug discovery. Both fields demand robust, novel validation frameworks to ensure safety and efficacy. For LBPs, this begins with stringent strain identification as outlined in the polyphasic framework, while AI leverages large-scale data to predict and optimize drug candidates.
Validation Workflow for Live Biotherapeutic Products
The path from a candidate bacterial strain to an approved LBP involves multiple, stringent validation checkpoints. This workflow integrates the initial polyphasic identification with specific safety and efficacy assessments required for therapeutic application.
Experimental Protocols
Protocol 3: LBP Strain Safety and Functional Validation
Purpose: To screen a candidate LBP strain for safety and functionality as per regulatory expectations.
Materials:
- Annotated genome sequence of the candidate strain
- Caco-2 cell lines (for in vitro translocation assays)
- GC-MS or LC-MS for metabolomic analysis (e.g., SCFA measurement)
Method:
- Genomic Safety Screening: Interrogate the annotated genome for the presence of antibiotic resistance genes (ARGs) using the CARD database. Crucially, distinguish between intrinsic and acquired/horizontally transferable ARGs. Screen for virulence factors using the VFDB database [85].
- In Vitro Functional Assays:
- Pathogen Inhibition: Co-culture the LBP strain with target pathogens (e.g., Clostridioides difficile) to assess growth inhibition.
- Barrier Function: Use a Caco-2 cell monolayer model to assess the strain's ability to improve epithelial barrier integrity (e.g., via Transepithelial Electrical Resistance measurement) and its potential for translocation [86].
- Immunomodulation: Treat immune cells (e.g., peripheral blood mononuclear cells) with heat-killed bacteria or cell-free supernatants of the LBP strain and measure cytokine production (e.g., IL-10, TNF-α) via ELISA.
- Metabolite Profiling: Using GC-MS, analyze the production of key metabolites like short-chain fatty acids (acetate, propionate, butyrate) in culture supernatants, which are linked to immune modulation and gut health [86].
Protocol 4: AI-Enhanced Validation in Drug Discovery
Purpose: To utilize AI tools for validating and prioritizing novel drug targets and lead compounds.
Materials:
- Publicly available databases (e.g., PubChem, ChEMBL, Protein Data Bank)
- AI/ML platforms (e.g., Atomwise, Insilico Medicine)
- High-performance computing resources
Method:
- Target Identification and Validation: Use natural language processing (NLP) models to mine biomedical literature and genomic databases to identify novel disease-associated targets. Validate targets using AI models that predict protein-ligand binding affinities [87].
- Virtual Screening and Compound Validation: Employ deep learning models, such as convolutional neural networks (CNNs) or generative adversarial networks (GANs), to screen millions of compounds in silico. This validates the potential of a compound as a hit candidate before synthesis. For instance, AI platforms can predict the binding of molecules to a target protein (e.g., the SARS-CoV-2 spike protein) within days [87].
- Predicting Experimental Outcomes: Train machine learning models on historical data from high-throughput screening assays to predict key parameters like toxicity, pharmacokinetics (PK), and pharmacodynamics (PD), thereby validating compounds for progression to in vitro and in vivo testing [87].
Validation Standards for LBPs and AI in Drug Development
Table 3: Validation Benchmarks for Advanced Therapeutics
| Field | Validation Parameter | Standard / Requirement |
|---|---|---|
| Live Biotherapeutic Products (LBPs) | Strain Identification | Whole Genome Sequencing (WGS) as the gold standard for genus, species, and strain-level identification [85]. |
| Safety Assessment | Mandatory screening for transferable antibiotic resistance genes and virulence factors [85]. | |
| Efficacy Validation | High-quality, double-blind, placebo-controlled Randomized Controlled Trials (RCTs) with pre-registration [85]. | |
| Product Quality | Viable count (CFU) specified at end of shelf-life; full process quality control under GMP [85]. | |
| AI in Drug Discovery | Data Integrity | Adherence to ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, Available) [88]. |
| Model Validation | Transparent reporting of model performance, training data, and limitations for regulatory evaluation [87] [89]. | |
| Regulatory Alignment | Use of AI should be described in submissions to regulatory bodies like the FDA, which has seen over 500 submissions with AI components [89]. |
The Scientist's Toolkit: LBP and AI Research Solutions
Table 4: Essential Tools for LBP and AI-Enhanced Drug Discovery
| Tool / Platform | Function | Application Context |
|---|---|---|
| CARD Database | Antibiotic Resistance Gene Repository | Essential for genomic safety screening of LBP candidate strains [85]. |
| AlphaFold Protein Structure Database | Protein Structure Prediction | Provides high-accuracy protein models for AI-driven molecular docking and target validation [87]. |
| AI Platforms (e.g., Atomwise) | CNN-based Virtual Screening | Accelerates hit identification by predicting molecular interactions for thousands of compounds [87]. |
| cGMP Manufacturing Facilities | Scalable, Quality-Assured Production | Ensures consistent, contaminant-free manufacturing of LBPs for clinical trials [90] [85]. |
Conclusion
The polyphasic taxonomic approach represents the most robust and pragmatic framework for bacterial classification, synthesizing phenotypic, genotypic, and phylogenetic data to build a stable and evolutionarily coherent system. The key takeaway is that no single method is sufficient; confidence in identification is achieved through consensus across multiple techniques. The future of microbial taxonomy is firmly rooted in genomics, with whole-genome sequencing and comparative genomics providing unprecedented resolution. For biomedical and clinical research, this evolving framework is crucial. It enables the precise identification of pathogens, ensures the accurate characterization of probiotics and live biotherapeutics, and unlocks the vast potential of uncultured microbial diversity through metagenomics, directly impacting drug discovery, diagnostics, and our understanding of the microbial world.
References
- 1. Comparison of Phenotypic and Genotypic Techniques for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC105261/]
- 2. Evolving strategies in microbe identification—a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11412244/]
- 3. Identifying Bacteria Through Look, Growth, Stain and Strain [https://asm.org/articles/2020/february/identifying-bacteria-through-look,-growth,-stain]
- 4. Polyphasic approach of bacterial classification [https://pmc.ncbi.nlm.nih.gov/articles/PMC3450112/]
- 5. Polyphasic Analysis [https://microbiosci.creative-biogene.com/polyphasic-analysis.html]
- 6. Advances in Bacterial Classification: From Phenotypic ... [https://www.ijpsjournal.com/article/Advances+in+Bacterial+Classification+From+Phenotypic+Traits+to+Genomic+Signatures]
- 7. The Comparison of Methods Experiment [https://www.westgard.com/lessons/basic-method-validation/lesson23.html]
- 8. New Insights into the Taxonomy of Bacteria in the Genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9148247/]
- 9. Polyphasic Approach to Microbial Identification [https://www.americanpharmaceuticalreview.com/Featured-Articles/113054-Polyphasic-Approach-to-Microbial-Identification/]
- 10. Understanding molecular identification and polyphasic ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701214001353]
- 11. A polyphasic strategy incorporating genomic data for the ... [https://pubmed.ncbi.nlm.nih.gov/24505076/]
- 12. #7: Moving targets: Recent changes in bacterial taxonomy ... [https://www.linkedin.com/pulse/moving-targets-recent-changes-bacterial-taxonomy-dr-tim]
- 13. Phylogenetic Inference - Stanford Encyclopedia of Philosophy [https://plato.stanford.edu/entries/phylogenetic-inference/]
- 14. A polyphasic taxonomy analysis reveals the presence of an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10174453/]
- 15. Polyphasic and phylogenomic reevaluation of ... [https://www.nature.com/articles/s41598-025-08302-w]
- 16. Polyphasic and comparative genomic characterization of a ... [https://www.nature.com/articles/s41598-025-18301-6]
- 17. 4 Visualization and annotation of phylogenetic trees: ggtree [https://guangchuangyu.github.io/ggtree-book/chapter-ggtree.html]
- 18. Chapter 4 Phylogenetic Tree Visualization [https://yulab-smu.top/treedata-book/chapter4.html]
- 19. Prokaryotic taxonomy and nomenclature in the age of big ... [https://www.nature.com/articles/s41396-021-00941-x]
- 20. Polyphasic taxonomy, a consensus approach to bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC239450/]
- 21. 16S and ITS rRNA Sequencing | Identify bacteria & fungi ... [https://www.illumina.com/areas-of-interest/microbiology/microbial-sequencing-methods/16s-rrna-sequencing.html]
- 22. 16S rRNA Gene Sequencing for Bacterial Identification in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2045242/]
- 23. 16S rRNA Methodology and Analysis Guide [https://cores.emory.edu/eicc/_includes/documents/sections/resources/16s_methodology_2024-01-24.html]
- 24. Introduce to 16S rRNA and 16S rRNA Sequencing [https://www.cd-genomics.com/blog/introduce-to-16s-rrna-and-16s-rrna-sequencing/]
- 25. 16S rRNA Sequencing Guide [https://blog.microbiomeinsights.com/16s-rrna-sequencing-guide]
- 26. Evaluation of 16S rRNA gene sequencing for species and ... [https://www.nature.com/articles/s41467-019-13036-1]
- 27. A Comprehensive Protocol for Bayesian Phylogenetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12021591/]
- 28. A Polyphasic Taxonomic Approach for Designation and ... [https://www.sciencedirect.com/science/article/pii/B9780128148495000095]
- 29. Shifting the genomic gold standard for the prokaryotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2776425/]
- 30. DNA DNA Hybridization - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/dna-dna-hybridization]
- 31. Digital DNA-DNA hybridization for microbial species ... [https://environmentalmicrobiome.biomedcentral.com/articles/10.4056/sigs.531120]
- 32. DNA–DNA hybridization [https://en.wikipedia.org/wiki/DNA%E2%80%93DNA_hybridization]
- 33. Genome sequence-based species delimitation with ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-60]
- 34. New insights into the relationship between the average ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1359021/full]
- 35. GGDC - Leibniz Institute DSMZ [https://ggdc.dsmz.de/]
- 36. Multilocus sequence analysis (MLSA) in prokaryotic ... [https://pubmed.ncbi.nlm.nih.gov/25959541/]
- 37. rpoB, a promising marker for analyzing the diversity of ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-019-1546-z]
- 38. A Multilocus Sequence Analysis Scheme for Phylogeny of ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2017.01321/full]
- 39. Identification of Clinical Aeromonas Species by rpoB and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4298543/]
- 40. A Novel Oligonucleotide Pair for Genotyping Members of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6481054/]
- 41. Average nucleotide identity — the backbone of modern ... [https://www.nature.com/articles/s41576-025-00911-5]
- 42. and long-read whole-genome sequencing for microbial ... [https://pubmed.ncbi.nlm.nih.gov/41222256/]
- 43. Whole-genome sequencing of 490640 UK Biobank ... [https://www.nature.com/articles/s41586-025-09272-9]
- 44. Polyphasic taxonomy and genome mining of Desertivibrio ... [https://www.sciencedirect.com/science/article/pii/S2666517425000999]
- 45. Exploration of the genetic landscape of bacterial dsDNA ... [https://pubmed.ncbi.nlm.nih.gov/39878503/]
- 46. Ultima Genomics UG 100 platform vs. Illumina NovaSeq X ... [https://www.illumina.com/science/genomics-research/articles/ultima-ug-100-vs-illumina-novaseq-x-wgs-data.html]
- 47. A Guide to De Novo Genome Assembly [https://www.cd-genomics.com/a-guide-to-de-novo-genome-assembly.html]
- 48. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 49. Duganella hordei sp. nov., Duganella caerulea sp. ... [https://link.springer.com/article/10.1007/s10482-025-02160-2]
- 50. Techniques and Methods for Fatty Acid Analysis in ... [https://www.mdpi.com/2297-8739/12/2/41]
- 51. Comprehensive profiling of conjugated fatty acid isomers ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267022002380]
- 52. Advances in Chemical and Biological Methods to Identify ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6560418/]
- 53. Characterization and selection of probiotic lactic acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10196247/]
- 54. Complete genome sequencing and probiotic characterization ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-03757-3]
- 55. Use of 16S rRNA Gene for Identification of a Broad Range of ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0117617]
- 56. Sequencing of Bacterial Genomes: Principles and Insights ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3927574/]
- 57. Robust taxonomic classification of uncharted microbial ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1817-x]
- 58. Sequencing 101: Metagenome-assembled genomes [https://www.pacb.com/blog/sequencing-101-metagenome-assembled-genomes/]
- 59. Metagenome-Assembled Genomes (MAGs): Advances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114606/]
- 60. From cultured to uncultured genome sequences [https://pmc.ncbi.nlm.nih.gov/articles/PMC4611022/]
- 61. Integration of metagenome-assembled genomes with ... [https://www.nature.com/articles/s41467-025-64950-6]
- 62. gcMeta 2025: a global repository of metagenome ... [https://pubmed.ncbi.nlm.nih.gov/41171134/]
- 63. Six factors affecting reproducibility in life science research ... [https://www.nature.com/articles/d42473-019-00004-y]
- 64. Are Your Laboratory Data Reproducible? The Critical Role of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11996692/]
- 65. Why standardisation threatens reproducibility [https://blogs.bmj.com/openscience/2018/03/27/why-standardisation-threatens-reproducibility/]
- 66. Experimental design and quantitative analysis of microbial ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1359-z]
- 67. Automated whole genome sequencing platform for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12077132/]
- 68. Strain-level bacterial typing directly from patient samples ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9950433/]
- 69. Polyphasic discrimination of Shewanella seohaensis from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12442382/]
- 70. Ten simple rules to colorize biological data visualization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7561171/]
- 71. Polyphasic Taxonomy Approach for Microbial Identification [https://live-biotherapeutic.creative-biolabs.com/polyphasic-taxonomy-approach-for-microbial-identification.htm]
- 72. Polyphasic taxonomy analyses of bacterial isolates [https://bio-protocol.org/exchange/minidetail?id=5913879&type=30]
- 73. The research on the identification, taxonomy, and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1544934/full]
- 74. Genomic Metrics Applied to Rhizobiales (Hyphomicrobiales) [https://pmc.ncbi.nlm.nih.gov/articles/PMC8026895/]
- 75. Genome sequence-based species classification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11237491/]
- 76. New Insights Into the Threshold Values of Multi-Locus ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.910277/full]
- 77. In-depth Analysis of Average Nucleotide Identity (ANI) [https://www.bocsci.com/resources/in-depth-analysis-of-average-nucleotide-identity-ani.html?srsltid=AfmBOoqARxVRq7ot3D-w3KqmA7pajlmbQmp0VJLr2juh801PAy3rSg4G]
- 78. Stepping stones towards a new prokaryotic taxonomy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1764938/]
- 79. Towards a prokaryotic genomic taxonomy [https://www.sciencedirect.com/science/article/abs/pii/S0168644504000889]
- 80. The Prokaryotic Species Concept and Challenges - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK558821/]
- 81. 2.1: Species Concepts [https://bio.libretexts.org/Courses/University_of_California_Davis/BIS_2B%3A_Introduction_to_Biology_-_Ecology_and_Evolution/02%3A_Biodiversity/2.01%3A_Species_Concepts]
- 82. An innovative approach to resolve the structure of complex ... [https://pubmed.ncbi.nlm.nih.gov/27924912/]
- 83. A quick alternative method for resolving bacterial taxonomy ... [https://genomics.peercommunityin.org/articles/rec?id=6]
- 84. The research on the identification, taxonomy, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11962042/]
- 85. Shaping the future of probiotics, live biotherapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12527994/]
- 86. Editorial: Live Biotherapeutic Products: where are we? [https://www.frontiersin.org/journals/microbiomes/articles/10.3389/frmbi.2025.1664282/full]
- 87. Transformative Role of Artificial Intelligence in Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406033/]
- 88. How to be prepared for Pharmaceutical Validation in 2025 [https://www.gillsprocess.com/blog-1/2024/12/11/how-to-be-prepared-for-pharmaceutical-validation-in-2025]
- 89. Artificial Intelligence for Drug Development [https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/artificial-intelligence-drug-development]
- 90. Live Biotherapeutic Products Whitepaper 2025 [https://novotech-cro.com/whitepapers/live-biotherapeutic-products-whitepaper-2025]