Precision Biofilm Dispersal: Harnessing CRISPRa to Activate Innate Anti-Biofilm Pathways
This article explores the transformative potential of CRISPR activation (CRISPRa) as a precision tool for combating biofilm-associated infections, a major contributor to antimicrobial resistance.
Precision Biofilm Dispersal: Harnessing CRISPRa to Activate Innate Anti-Biofilm Pathways
Abstract
This article explores the transformative potential of CRISPR activation (CRISPRa) as a precision tool for combating biofilm-associated infections, a major contributor to antimicrobial resistance. Tailored for researchers, scientists, and drug development professionals, it provides a comprehensive analysis of how CRISPRa can be programmed to upregulate key biofilm dispersal genes, such as those involved in cyclic-di-GMP signaling and quorum sensing. We cover the foundational science of biofilm dispersal mechanisms, detail the methodology for designing and delivering CRISPRa systems, address critical troubleshooting and optimization challenges, and present validation strategies that compare CRISPRa's efficacy and specificity against conventional anti-biofilm approaches. The synthesis of these insights outlines a promising pathway for developing next-generation, sequence-specific antimicrobial therapies that leverage a bacterium's own genetic machinery for biofilm eradication.
The Genetic Blueprint of Biofilm Dispersal: Identifying CRISPRa Targets
Bacterial biofilms are structured microbial communities that represent the predominant mode of bacterial life in natural, industrial, and clinical environments. These complex architectures are characterized by microbial cells embedded within a self-produced matrix of hydrated extracellular polymeric substances (EPS) that form their immediate environment [1]. The biofilm matrix is not merely a structural scaffold but a functional component that defines the biofilm lifestyle, providing mechanical stability, mediating adhesion to surfaces, and forming a cohesive, three-dimensional polymer network that interconnects and transiently immobilizes biofilm cells [1].
The architectural development of biofilms follows a defined developmental sequence beginning with initial attachment to surfaces, followed by formation of microcolonies, maturation into complex three-dimensional structures, and eventual dispersal of cells to colonize new niches [2] [3]. This lifecycle is universal across diverse bacterial species, though the specific molecular mechanisms regulating each stage can vary. The mature biofilm architecture exhibits remarkable heterogeneity, creating gradients of nutrients, oxygen, pH, and metabolic activity that support diverse physiological states within the same community [1]. This spatial organization is crucial for the functional properties of biofilms, including their enhanced tolerance to antimicrobial agents and environmental stresses.
Composition and Organization of the Biofilm Matrix
The extracellular matrix constitutes a key architectural component of biofilms, typically consisting of 97% water with the remaining composition including polysaccharides, proteins, nucleic acids, and lipids [3] [4]. This complex mixture forms a versatile external digestive system that retains extracellular enzymes close to the cells, enabling efficient metabolism of dissolved, colloidal, and solid biopolymers [1]. The matrix components work synergistically to provide protection against diverse environmental threats including desiccation, biocides, antibiotics, heavy metals, ultraviolet radiation, host immune defenses, and protozoan grazing [1].
Table 1: Major Components of the Biofilm Extracellular Matrix
| Matrix Component | Primary Functions | Representative Examples |
|---|---|---|
| Exopolysaccharides (EPS) | Structural integrity, adhesion, water retention, nutrient trapping | Cellulose, alginate, Pel, Psl |
| Proteins | Structural support, enzymatic activity, adhesion | Amyloid fibers, lectins, extracellular enzymes |
| Extracellular DNA (eDNA) | Structural integrity, horizontal gene transfer, nutrient source | Genomic DNA from lysed cells |
| Lipids and Surfactants | Hydrophobicity modulation, antimicrobial activity | Rhamnolipids, surfactins |
| Water | Solvent for nutrients/waste, medium for molecular diffusion | Hydration medium (up to 97% of matrix) |
Architectural Development Across Bacterial Species
Despite molecular differences between species, recent research has revealed that biofilm microcolony architecture is determined by conserved mechanical cell-cell interactions governed by just two key control parameters: cellular aspect ratio and cell density [5]. This remarkable conservation means that architectural phenotypes across diverse species including Vibrio cholerae, Escherichia coli, Salmonella enterica, and Pseudomonas aeruginosa can be predicted and explained through these fundamental biophysical parameters [5]. The early stages of biofilm development demonstrate that mechanical interactions between cells, rather than species-specific molecular pathways, primarily dictate the emergent three-dimensional architecture of nascent biofilms.
The following diagram illustrates the key stages of biofilm development and the architectural transitions throughout the lifecycle:
The Dispersal Lifecycle Stage: Mechanisms and Regulation
Dispersal represents the final stage in the biofilm lifecycle, serving as a critical mechanism for bacterial propagation and colonization of new niches. This highly regulated process involves the detachment of individual cells or cell clusters from the biofilm structure, transitioning them from a sessile to a motile lifestyle [2]. Biofilm dispersal can occur passively through external forces such as fluid shear or abrasion, or actively through internally regulated mechanisms triggered by environmental cues and signaling pathways [3].
The molecular regulation of dispersal involves complex networks that integrate environmental signals with intracellular signaling systems. Key regulatory mechanisms include quorum sensing systems that monitor population density, nucleotide-based secondary messengers such as cyclic di-GMP that modulate the transition between sessile and motile states, and two-component systems that sense environmental changes [6]. The intracellular concentration of cyclic di-GMP serves as a central regulator, where high levels promote biofilm formation through increased matrix production and reduced motility, while decreased concentrations trigger dispersal through downregulation of matrix components and induction of motility apparatus [6].
Environmental Triggers and Signaling Pathways
Dispersal is typically initiated in response to specific environmental conditions including nutrient availability, oxygen tension, osmotic stress, and changes in temperature or pH [2]. These external signals are integrated through complex regulatory networks that ultimately activate enzymatic effectors responsible for matrix degradation and cellular detachment. Key enzymatic activities involved in dispersal include polysaccharide depolymerases that degrade the EPS matrix, proteases that target matrix proteins and cellular adhesins, and nucleases that cleave extracellular DNA components critical for structural integrity [3].
Table 2: Regulatory Systems Controlling Biofilm Dispersal
| Regulatory System | Signaling Molecules | Mechanism of Action | Effect on Dispersal |
|---|---|---|---|
| c-di-GMP Signaling | Cyclic di-GMP | High levels promote biofilm formation; degradation triggers dispersal | Primary regulator: PDE activation reduces c-di-GMP, inducing dispersal |
| Quorum Sensing | Acyl-homoserine lactones (AHLs), autoinducer peptides | Cell-density dependent gene regulation | Can induce or inhibit dispersal based on specific system and conditions |
| Two-Component Systems | Sensor kinase/response regulator pairs | Environmental signal transduction | Activate expression of matrix-degrading enzymes and motility genes |
| Carbon Catabolite Repression | Metabolic intermediates | Nutrient availability sensing | Nutrient limitation triggers dispersal to seek new nutrient sources |
| Stringent Response | (p)ppGpp | Stress response regulation | Stress conditions induce dispersal as survival mechanism |
The following diagram illustrates the core regulatory pathways that control the transition from biofilm maturation to active dispersal:
CRISPR Activation for Precision Induction of Biofilm Dispersal
The emerging field of CRISPR-based technologies offers revolutionary approaches for precision control of biofilm dispersal through targeted genetic manipulation. While CRISPR-Cas systems naturally function as adaptive immune systems in prokaryotes, their engineered derivatives—particularly nuclease-deficient Cas9 (dCas9) variants—enable precise transcriptional regulation without permanent DNA modifications [7]. When fused to transcriptional activation domains, dCas9 forms the core of CRISPR activation (CRISPRa) systems that can specifically upregulate endogenous bacterial genes controlling dispersal mechanisms [7] [8].
CRISPRa represents a powerful synthetic biology tool for biofilm control because it allows targeted induction of natural dispersal pathways without introducing exogenous genes or causing bacterial cell death that could select for resistant mutants. This approach can be designed to activate master regulators of dispersal, including genes encoding phosphodiesterases that degrade the biofilm-promoting second messenger cyclic di-GMP, matrix-degrading enzymes, surfactants that reduce intercellular adhesion, and motility apparatus components [7]. The programmability of CRISPRa systems enables development of sequence-specific antimicrobials that can be tailored to target specific pathogens or customized for different industrial applications where biofilm control is required [3] [7].
CRISPRa System Design and Implementation
A functional CRISPRa system for biofilm dispersal requires two core components: a catalytically dead Cas9 (dCas9) protein fused to transcriptional activation domains, and guide RNAs (gRNAs) specifically targeting the promoter regions of dispersal genes [8]. The dCas9 component retains its DNA-binding capability but lacks nuclease activity, serving as a programmable DNA-targeting platform. When targeted to promoter regions by gene-specific gRNAs, the dCas9-activator fusion recruits RNA polymerase and activates transcription of the target gene [7] [6].
For effective induction of biofilm dispersal, gRNAs can be designed to target key regulatory nodes in the dispersal network, including genes encoding phosphodiesterases that reduce intracellular c-di-GMP levels, enzymes that degrade matrix components, biosynthetic genes for surfactant production, and regulators of bacterial motility [7]. The system can be delivered to bacterial populations via plasmid vectors, phage particles, or nanoparticle carriers designed to penetrate the biofilm matrix [9].
The following diagram illustrates the molecular mechanism of CRISPRa for targeted activation of biofilm dispersal genes:
Experimental Approaches and Research Methodologies
CRISPRa Delivery Systems for Biofilm Penetration
Effective delivery of CRISPRa components through the protective biofilm matrix represents a significant technical challenge that has spurred the development of innovative delivery platforms. Nanoparticle-based carriers have emerged as particularly promising vehicles due to their ability to penetrate the dense EPS matrix and protect genetic payloads from degradation [9]. Recent advances have demonstrated that lipid-based nanoparticles can achieve over 90% reduction in Pseudomonas aeruginosa biofilm biomass when delivering CRISPR-Cas9 components, while gold nanoparticle carriers have shown a 3.5-fold increase in gene-editing efficiency compared to non-carrier systems [9].
Advanced delivery platforms for CRISPRa systems include:
- Engineered phagemids: Bacteriophage-derived vectors that exploit natural phage infection mechanisms while carrying programmable CRISPRa payloads [7]
- Conjugative plasmids: Self-transmissible plasmids that enable cell-to-cell transfer of CRISPRa machinery throughout the biofilm community [8]
- Functionalized nanoparticles: Lipid, polymeric, or metallic nanoparticles engineered with surface modifications that enhance biofilm penetration and cellular uptake [9]
- Biofilm-responsive nanocarriers: Smart delivery systems that release CRISPRa payloads in response to biofilm-specific environmental cues such as reduced oxygen tension or altered pH [7]
Quantitative Assessment of Dispersal Efficacy
Rigorous quantification of dispersal outcomes requires multi-faceted analytical approaches that measure both architectural changes and transcriptional responses. Standardized methods include confocal laser scanning microscopy (CLSM) with appropriate fluorescent staining to visualize 3D biofilm architecture, crystal violet staining for biomass quantification, and flow cytometry for analysis of dispersed cells [6] [10]. These phenotypic measurements should be correlated with molecular analyses including RNA sequencing to verify transcriptional activation of target genes and qRT-PCR to quantify expression of specific dispersal markers [6].
Table 3: Key Methodologies for Analyzing Biofilm Dispersal
| Methodology | Key Parameters Measured | Technical Considerations |
|---|---|---|
| Confocal Laser Scanning Microscopy | 3D architecture, biomass, thickness, biovolume, surface coverage | Requires fluorescent reporters or staining (SYTO9, dextran conjugates) |
| Crystal Violet Staining | Total adhered biomass | High-throughput but does not distinguish live/dead cells |
| qRT-PCR | Expression of dispersal genes (e.g., phosphodiesterases, matrix enzymes) | Requires proper normalization to housekeeping genes |
| RNA Sequencing | Global transcriptional changes during dispersal | Identifies unintended effects on non-target pathways |
| Flow Cytometry | Number and viability of dispersed cells | Can be coupled with sorting for downstream analysis |
| c-di-GMP Quantification | Intracellular c-di-GMP concentrations | Correlates with dispersal activation (HPLC-MS/MS or reporter systems) |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for CRISPRa-Mediated Biofilm Dispersal Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| CRISPRa Plasmids | dCas9-activator fusions (dCas9-ω, dCas9-SoxS), gRNA expression vectors | Programmable transcriptional activation of endogenous dispersal genes |
| Delivery Vehicles | Lipid nanoparticles (LNPs), gold nanoparticles, engineered phages, conjugative plasmids | Enable penetration through biofilm matrix and intracellular delivery of CRISPRa components |
| Biofilm Stains | SYTO9/propidium iodide (Live/Dead), Congo red, calcofluor white, Alexa Fluor-dextran conjugates | Visualization of biofilm architecture, viability assessment, and EPS component staining |
| Bacterial Strains | P. aeruginosa PAO1, E. coli K-12, S. enterica Typhimurium, V. cholerae O1, relevant isogenic mutants | Model organisms for biofilm studies with well-characterized dispersal mechanisms |
| Dispersal Assay Kits | Crystal violet kits, dispersin B activity assays, c-di-GMP quantification kits | Standardized measurement of dispersal efficacy and mechanism validation |
| Gene Expression Tools | qPCR primers for dispersal genes, RNA extraction kits for biofilms, transcriptional reporter strains | Quantification of transcriptional activation following CRISPRa treatment |
Protocol: CRISPRa-Mediated Dispersal Gene Activation in Pseudomonas fluorescens Biofilms
The following detailed protocol adapts established CRISPRi methodologies in P. fluorescens [6] for CRISPRa applications targeting biofilm dispersal genes:
Strain and Plasmid Preparation:
- Transform P. fluorescens with two compatible plasmids: (1) pCRISPRa-dCas9 expressing a dCas9-activator fusion (e.g., dCas9-ω) under PtetA promoter control, and (2) pGRNA-disP expressing guide RNAs targeting the promoter region of dispersal genes (e.g., phosphodiesterases).
- Include appropriate antibiotic selection throughout (e.g., erythromycin 150 μg/mL for P. fluorescens).
Biofilm Establishment:
- Grow overnight cultures of CRISPRa-engineered strains in LB-Miller medium with appropriate antibiotics.
- Dilute cultures 1:100 in fresh medium and inoculate into flow cell chambers or 96-well plates.
- Allow biofilms to develop for 24-48 hours under continuous flow conditions (for flow cells) or static conditions (for microtiter plates).
CRISPRa Induction:
- Induce dCas9-activator expression with anhydrotetracycline (aTc) at optimized concentrations (typically 50-200 ng/mL).
- Maintain induction for 4-24 hours depending on experimental requirements.
Dispersal Quantification:
- Assess dispersal efficacy via crystal violet staining for total biomass.
- Analyze 3D biofilm architecture using CLSM with SYTO9 staining (bacterial cells) and Alexa Fluor 647-dextran (EPS matrix) [10].
- Collect dispersed cells from effluent and enumerate via plate counting or flow cytometry.
- Verify target gene activation via qRT-PCR using primers specific for induced dispersal genes.
Control Experiments:
- Include non-targeting gRNA controls to assess off-target effects.
- Utilize non-induced controls (no aTc) to measure basal dispersal rates.
- Employ strains lacking dCas9 component to control for gRNA-independent effects.
This protocol can be adapted for high-throughput screening of multiple dispersal gene targets by utilizing arrayed gRNA libraries in multiwell plate formats, enabling systematic identification of the most effective dispersal pathway activators for specific applications.
Biofilm dispersal is a critical, programmed stage of the bacterial life cycle, enabling the transition from sessile, structured communities to free-swimming planktonic cells for colonization of new niches. This process is precisely regulated by complex molecular networks. Understanding these regulators is paramount for developing novel anti-biofilm strategies, particularly within the emerging field of CRISPR activation (CRISPRa) designed to artificially induce dispersal. This technical guide details the core triumvirate of regulators—cyclic-di-GMP, quorum sensing, and nucleases—that control dispersal, providing a foundational framework for researchers and drug development professionals aiming to leverage CRISPRa for biofilm control.
Cyclic-di-GMP: The Central Biofilm Lifestyle Regulator
Molecular Mechanisms and Signaling Pathways
Cyclic-di-GMP (c-di-GMP) functions as a ubiquitous bacterial second messenger, governing the switch between motile and sessile lifestyles. Its intracellular concentration dictates phenotypic output: high levels promote biofilm formation by upregulating exopolysaccharide production and repressing motility, while low levels induce dispersal and a return to motility [11] [12].
The cellular level of c-di-GMP is dynamically controlled by the antagonistic actions of two enzymes:
- Diguanylate cyclases (DGCs): Synthesize c-di-GMP from two GTP molecules. DGCs are characterized by a conserved GGDEF domain [11].
- Phosphodiesterases (PDEs): Degrade c-di-GMP. PDEs feature EAL or HD-GYP domains, with the initial product being pGpG, which is subsequently hydrolyzed to GMP by the oligoribonuclease Orn [11].
In Pseudomonas aeruginosa, a model organism for biofilm studies, surface contact triggers specific signaling systems that modulate c-di-GMP production. The following diagram illustrates two key surface-sensing pathways:
The intracellular response to elevated c-di-GMP involves specific effectors. For instance, in P. aeruginosa, high c-di-GMP directly binds to receptors like PelD, a protein essential for the synthesis of the Pel exopolysaccharide, thereby stabilizing the biofilm matrix [11] [13]. Concurrently, it represses the expression of flagellar genes, inhibiting motility [11].
Table 1: Key Enzymes and Effectors in c-di-GMP Signaling of P. aeruginosa
| Protein/Enzyme | Domain | Function | Phenotypic Outcome |
|---|---|---|---|
| WspR | GGDEF | Diguanylate Cyclase | ↑ c-di-GMP, ↑ Pel/Psl polysaccharide production [11] |
| SadC | GGDEF | Diguanylate Cyclase | ↑ c-di-GMP in response to PilY1 signaling [11] |
| BifA | EAL | Phosphodiesterase | ↓ c-di-GMP, ↑ motility, ↓ exopolysaccharide [6] |
| DipA | EAL | Phosphodiesterase | ↓ c-di-GMP, involved in biofilm dispersion [6] |
| PelD | N/A | c-di-GMP Receptor | Binds c-di-GMP; activates Pel polysaccharide production [13] |
Quantitative Dynamics in Biofilm Development
c-di-GMP levels are not uniformly elevated throughout biofilm development but exhibit spatiotemporal heterogeneity. Measurements indicate that planktonic cells harbor less than 30 pmol/mg c-di-GMP, whereas biofilms can contain close to 100 pmol/mg c-di-GMP [11]. The increase occurs in a hierarchical, non-uniform manner, corresponding to distinct developmental stages. A crucial drop in c-di-GMP concentration, mediated by the activation of specific PDEs like DipA and BifA, is a prerequisite for the dispersal stage [6].
Quorum Sensing: Population-Dependent Dispersal Signaling
Regulatory Networks Linking QS to Dispersal
Quorum sensing (QS) is a cell-cell communication mechanism where bacteria synchronize gene expression in response to population density. Contrary to the intuitive belief that QS initiates biofilm formation, compelling evidence shows that QS activation in mature biofilms primarily triggers dispersal [14] [13]. This dispersal is mediated through the coordinated regulation of matrix components and surfactant production.
Table 2: Quorum Sensing-Regulated Factors in P. aeruginosa Biofilm Dispersal
| QS-Regulated Factor | Regulatory Mechanism | Function in Dispersal |
|---|---|---|
| Rhamnolipids | Directly activated by RhlR [13]. | Surfactants that disrupt biofilm integrity; overproduction accelerates detachment [13]. |
| Pel Exopolysaccharide | Repressed via LasI/LasR activation of TpbA phosphatase, which inhibits Pel synthesis and lowers c-di-GMP [13]. | Reduction of a key structural matrix component weakens the biofilm [13]. |
| Extracellular DNA (eDNA) | QS-induced bacterial lysis releases eDNA [13]. | While initially structural, lysis provides a dispersal mechanism for a subpopulation [13]. |
The relationship between different QS systems and their collective outcome on biofilm dispersal is complex. The following diagram maps the core QS circuitry in P. aeruginosa and its pro-dispersal effects:
Experimental Evidence and Methodologies
The role of QS in dispersal has been established through key experiments. A foundational study demonstrated that a lasI mutant formed flat, undifferentiated biofilms that were resistant to dispersion by sodium dodecyl sulfate, unlike the wild-type structured biofilms [13]. Methodologies to probe QS function now include:
- Mutant Studies: Constructing and comparing biofilm phenotypes of wild-type versus lasI, rhlI, or pqsR mutants.
- Chemical Inhibition: Using small molecule inhibitors of QS receptors (e.g., LasR antagonists) and assessing biofilm biomass and stability.
- Transcriptional Profiling: Employing RNA-seq on biofilms at various stages to map the expression of QS-regulated genes, such as rhlA and pelA.
- CRISPR Interference (CRISPRi): Utilizing dCas9 to repress QS gene expression and quantify dispersal dynamics. This approach has been successfully adapted for Pseudomonas species [6].
Nucleases: Reclaiming the Extracellular DNA Matrix
Extracellular DNA as a Dynamic Matrix Component
Extracellular DNA (eDNA) is a critical structural polymer in the biofilm matrix of many bacterial species, including Bacillus subtilis and Pseudomonas aeruginosa [15]. It contributes to biofilm integrity, adhesion, and cation sequestration, which can indirectly increase antimicrobial resistance [15]. Historically viewed as a static scaffold, recent research reveals eDNA is a dynamic metabolic reservoir.
Nuclease-Mediated Matrix Degradation and Reclamation
The controlled degradation of eDNA by secreted nucleases is a key mechanism for biofilm dispersal. In B. subtilis, a pulse of nuclease activity late in biofilm development leads to the global degradation of the eDNA matrix [15]. The key nucleases identified include:
- YhcR: A secreted Ca²⁺-dependent nuclease responsible for the primary degradation of eDNA in pellicle biofilms [15].
- NucA and NucB: Cooperate with YhcR to reclaim eDNA in colony biofilms [15].
This process allows the community to recycle the phosphate and nucleotide content of eDNA, particularly as nutrients become limited later in development [15]. The experimental workflow for identifying and characterizing these nucleases is detailed below:
The functional validation of these nucleases is critical. Mutants lacking yhcR, nucA, and nucB show impaired eDNA degradation and altered biofilm architecture, confirming their role in matrix reclamation and dispersal [15].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Investigating Biofilm Dispersal Mechanisms
| Reagent / Tool | Function/Description | Example Application |
|---|---|---|
| CRISPRi/dCas9 System | Two-plasmid system for targeted gene repression (dCas9 + gRNA) [6]. | Silencing expression of specific DGCs, PDEs, or QS regulators in diverse bacterial strains [6]. |
| Fluorescent DNA Dyes (e.g., TOTO-1) | High-affinity nucleic acid stains for imaging and quantification [15]. | Real-time visualization and measurement of eDNA dynamics within living biofilms [15]. |
| c-di-GMP Biosensors | Fluorescent or FRET-based reporters for intracellular c-di-GMP levels. | Quantifying spatiotemporal changes in c-di-GMP during biofilm development and dispersal. |
| QS Signal Molecules (e.g., 3OC12-HSL, C4-HSL) | Purified autoinducers for exogenous supplementation. | Complementing mutant studies; probing timing and threshold of QS-activated dispersal [13]. |
| DNase I (commercial) | Recombinant nuclease for exogenous matrix disruption. | Positive control for nuclease-mediated dispersal; testing biofilm susceptibility [15]. |
| Specific Nuclease Mutants (e.g., ΔyhcR) | Strains with inactivated nuclease genes [15]. | Functional validation of nuclease role in eDNA reclamation and dispersal [15]. |
| Anti-biofilm Surfactants (e.g., Rhamnolipids) | Purified biosurfactants that disrupt matrix integrity [13]. | Inducing chemical dispersion; studying synergistic effects with genetic tools. |
The molecular regulators c-di-GMP, quorum sensing, and nucleases form an integrated network controlling biofilm dispersal. Lowering c-di-GMP is a primary trigger; QS acts as a density-dependent timer that coordinates the simultaneous repression of matrix components and production of dispersal agents; and nucleases execute the physical dismantling of the eDNA scaffold.
This intricate understanding provides a robust roadmap for CRISPRa research. By designing guide RNAs to target the promoters of key dispersing genes—such as PDEs to lower c-di-GMP, QS master regulators like vfr or hapR, or secreted nucleases like yhcR—researchers can develop precision interventions to force biofilm dispersal. The quantitative data, experimental protocols, and reagent toolkit detailed herein are designed to equip scientists with the foundational knowledge to pioneer these novel, programmable anti-biofilm therapeutics.
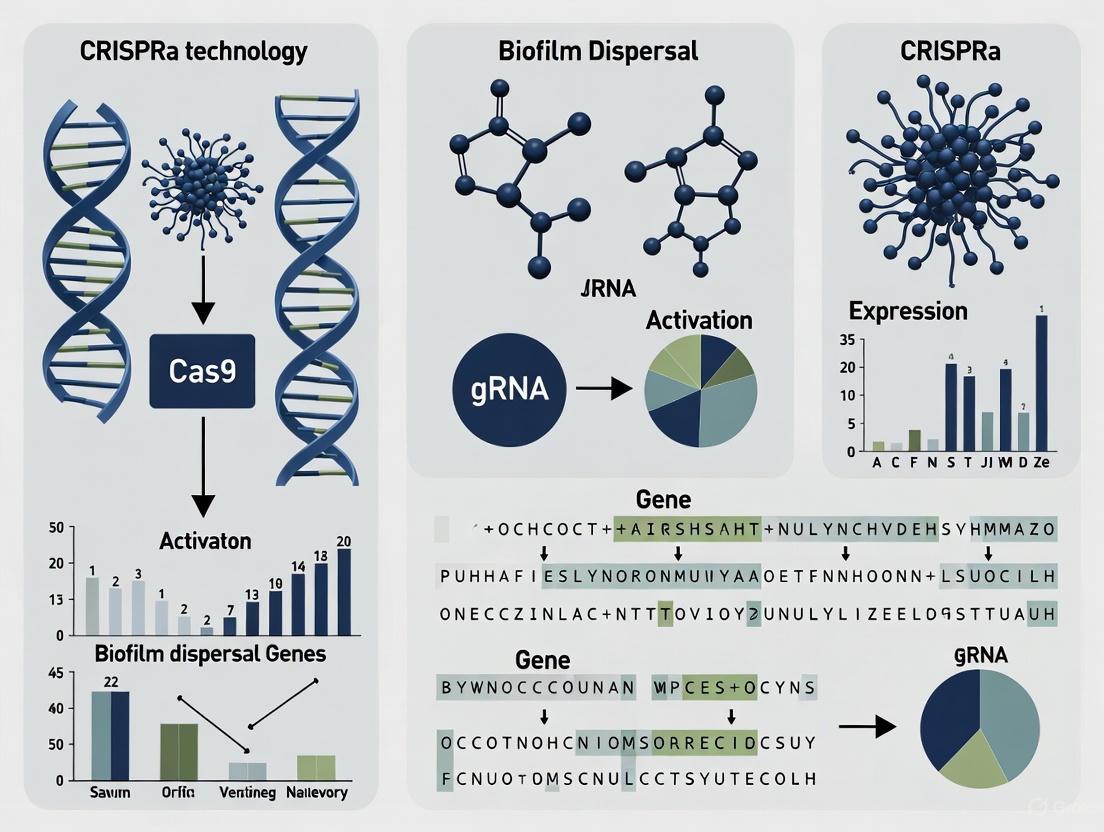
Clustered Regularly Interspaced Short Palindromic Repeats activation (CRISPRa) represents a revolutionary precision tool in genetic engineering, derived from the bacterial adaptive immune system but repurposed for programmable transcriptional control. This technical guide explores the fundamental mechanisms of CRISPRa, wherein a catalytically deactivated Cas9 (dCas9) protein is fused to transcriptional activator domains and directed by guide RNA to specific genomic loci, enabling targeted gene upregulation without DNA cleavage. Framed within the context of biofilm dispersal gene research, we examine how CRISPRa overcomes limitations of traditional genetic manipulation by allowing reversible, dose-controlled activation of endogenous genes. This review details the molecular architecture of CRISPRa systems, quantitative performance metrics across diverse applications, and comprehensive experimental protocols for implementation in biofilm research, providing researchers with the foundational knowledge and practical methodologies required to leverage this technology for combating persistent biofilm-associated infections.
The CRISPR-Cas system originated as an adaptive immune mechanism in bacteria and archaea, protecting against viral infections by storing fragments of foreign DNA and using them to recognize and cleave subsequent invasions [16] [17]. This natural system was repurposed for genome editing, with the type II CRISPR-Cas9 system emerging as a particularly powerful tool due to its simplicity and programmability [17]. The CRISPR-Cas9 system comprises a Cas9 nuclease guided by a dual RNA complex (crRNA and tracrRNA), often simplified as a single guide RNA (sgRNA), which recognizes target DNA sequences adjacent to a protospacer-adjacent motif (PAM) [17].
The evolution from nuclease-based editing to transcriptional regulation occurred with the development of catalytically dead Cas9 (dCas9), generated through point mutations in the RuvC and HNH nuclease domains [18]. This dCas9 retains DNA-binding capability but lacks cleavage activity, serving as a programmable DNA-binding platform [19]. When fused to transcriptional effector domains, dCas9 can be directed to specific genomic regions to modulate gene expression, giving rise to CRISPR interference (CRISPRi) for gene repression and CRISPR activation (CRISPRa) for gene upregulation [20] [18].
Table 1: Evolution of CRISPR Systems from Immunity to Transcription Control
| System | Key Components | Mechanism | Outcome |
|---|---|---|---|
| Native CRISPR | Cas nuclease, crRNA, tracrRNA | DNA cleavage of foreign invaders | Bacterial adaptive immunity |
| CRISPR-Cas9 | Active Cas9, sgRNA | DNA double-strand breaks | Genome editing via indels |
| CRISPRi | dCas9, repressor domains (e.g., KRAB) | Steric hindrance of transcription | Gene knockdown |
| CRISPRa | dCas9, activator domains (e.g., VPR) | Recruitment of transcriptional machinery | Gene activation |
In biofilm research, CRISPRa offers unique advantages for probing complex genetic networks controlling dispersal mechanisms. Unlike traditional gene knockouts that permanently disrupt function, CRISPRa enables reversible, titratable control over gene expression, making it ideal for studying essential genes and subtle phenotypic changes [6] [18]. This precision is particularly valuable for investigating biofilm dispersal, where timing and expression levels critically determine the transition from sessile to planktonic states.
Molecular Mechanisms and System Architectures
Core CRISPRa Components
The fundamental CRISPRa system requires two essential components: (1) a deactivated Cas9 (dCas9) protein fused to transcriptional activator domains, and (2) a guide RNA (gRNA) targeting specific promoter regions [19]. The dCas9 serves as a programmable DNA-binding scaffold, while the activator domains recruit and stimulate the cellular transcription machinery.
Figure 1: CRISPRa Mechanism for Gene Activation. The dCas9-VPR fusion protein complexed with guide RNA binds target promoter regions, recruiting transcriptional machinery to activate gene expression.
Several activator architectures have been developed with varying complexities and efficiencies:
Direct Fusion Systems (e.g., dCas9-VPR): dCas9 directly fused to a tripartite activator VP64-p65-Rta (VPR), creating a single protein component that provides strong, synergistic activation [20] [19]. This system offers simplicity with only two components (dCas9-VPR and sgRNA) but has a large gene size that can challenge viral packaging.
Protein Scaffold Systems (e.g., SunTag): dCas9 recruits multiple copies of activator domains through peptide arrays. This system leverages avidity effects for potent activation but requires multiple components [19].
RNA Scaffold Systems (e.g., SAM): Modified sgRNAs containing MS2 RNA aptamers recruit additional activator proteins (MS2-P65-HSF1) to the target site [20] [19]. While potentially more potent for some targets, this system requires three components and specialized extended sgRNAs.
Comparative studies in primary cells have demonstrated that the VPR system outperforms SAM in multiple contexts, with one study showing 97% activation of CXCR4 using VPR compared to 52% with SAM at equivalent doses [20]. The VPR system also produces more homogeneous activation across cell populations, making it particularly suitable for quantitative biofilm studies [20].
Guide RNA Design Principles
Effective gRNA design is critical for CRISPRa efficiency. Unlike CRISPR cleavage that targets coding sequences, CRISPRa requires gRNAs binding to promoter regions [19]. Optimal gRNAs typically target sites -50 to -400 base pairs upstream of the transcriptional start site (TSS) [19]. Using multiple gRNAs per gene (quadruple-guide RNAs or qgRNAs) significantly enhances activation efficacy through synergistic effects [21]. One study demonstrated that qgRNA vectors "massively increased target gene activation" compared to individual sgRNAs [21].
Table 2: Quantitative Performance of CRISPRa Systems
| System Architecture | Activation Efficiency | Key Advantages | Limitations |
|---|---|---|---|
| dCas9-VPR | 7.9-fold higher than plasmid delivery; >97% cells activated [20] | Single-component system; homogenous activation | Large size may reduce viral titer |
| dCas9-SAM | 52% cells activated at high doses [20] | Potent for some targets | Complex 3-component system |
| qgRNA-VPR | Massive increase vs. single sgRNAs [21] | Synergistic activation; robust performance | More complex cloning |
| RNA-based Delivery | 99.5% cells activated [20] | Rapid kinetics; minimal toxicity | Transient expression |
CRISPRa in Biofilm Dispersal Research: Applications and Workflows
Targeting Biofilm Dispersal Mechanisms
Biofilms are structured microbial communities embedded in an extracellular polymeric substance (EPS) that confer up to 1000-fold greater tolerance to antibiotics compared to planktonic cells [16]. The transition from biofilm to planktonic states is regulated by complex genetic networks involving signaling molecules, matrix-degrading enzymes, and regulatory proteins.
CRISPRa enables precise interrogation of these dispersal mechanisms by allowing controlled upregulation of key genes, including:
c-di-GMP signaling pathways: This ubiquitous bacterial second messenger regulates the transition between motile and sessile lifestyles [6]. High intracellular c-di-GMP promotes biofilm formation, while low levels facilitate dispersal.
Quorum sensing systems: Cell-cell communication mechanisms that coordinate biofilm development and dispersal in response to population density.
Matrix-degrading enzymes: Enzymes that breakdown EPS components, facilitating biofilm dissolution and bacterial release.
Two-component systems: Regulatory systems like GacA/S that sense environmental stimuli and trigger genetic programs controlling biofilm lifecycles [6].
Figure 2: CRISPRa Applications in Biofilm Dispersal Research. Targeted activation of dispersal genes disrupts biofilm integrity through multiple pathways, potentially enhancing anti-biofilm therapies.
Experimental Implementation Workflow
Implementing CRISPRa for biofilm studies requires a systematic approach from target selection to phenotypic validation:
Figure 3: Experimental Workflow for CRISPRa in Biofilm Research. A systematic approach from target identification to functional validation ensures reliable results.
Target Identification and gRNA Design
Identify target genes involved in biofilm dispersal pathways through literature review, transcriptomic studies of dispersal conditions, or genetic screens. For each candidate gene, design multiple gRNAs targeting regions -50 to -400 bp upstream of the TSS using specialized algorithms (e.g., CRISPRa design tools from Broad Institute) [19]. Prioritize qgRNA designs when possible, as quadruple guides demonstrate significantly higher efficacy [21].
Delivery Methods for Biofilm Systems
Effective delivery remains a critical challenge in CRISPRa implementation, particularly for biofilm systems where the extracellular matrix impedes penetration. Available strategies include:
Plasmid-based delivery: Conventional but inefficient for many bacterial species and biofilm contexts.
RNA-based delivery: Electroporation of in vitro transcribed (IVT) dCas9-VPR mRNA with synthetic sgRNAs shows high efficiency (99.5% activation) and rapid kinetics in primary cells [20]. Activation typically begins within 3 hours, peaks by 7 hours, and lasts 48 hours before declining.
Nanoparticle-mediated delivery: Inorganic and organic nanoparticles serve as effective carriers, enhancing stability and cellular uptake. Recent advances demonstrate "liposomal Cas9 formulations reduced P. aeruginosa biofilm biomass by over 90% in vitro, while gold nanoparticle carriers enhance editing efficiency up to 3.5-fold compared to non-carrier systems" [16]. These platforms can also co-deliver antibiotics or antimicrobial peptides for synergistic effects.
Bacterial conjugation and phage delivery: Engineered delivery systems tailored for microbial communities.
Validation and Phenotypic Assays
Rigorous validation is essential following CRISPRa delivery:
Transcriptional validation: Quantify mRNA levels using RT-qPCR to confirm target gene upregulation.
Protein validation: Assess protein levels via Western blot, flow cytometry (for surface proteins), or immunofluorescence.
Biofilm phenotypic assays:
- Biomass quantification: Crystal violet staining or optical density measurements
- Viability assessment: Colony-forming unit (CFU) counts under antibiotic treatment
- Structural analysis: Confocal laser scanning microscopy (CLSM) with EPS staining
- Dispersal monitoring: Time-lapse imaging of biofilm dissolution
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for CRISPRa Biofilm Research
| Reagent Category | Specific Examples | Function | Implementation Notes |
|---|---|---|---|
| CRISPRa Activators | dCas9-VPR, dCas9-SAM, dCas9-SunTag | Transcriptional activation | dCas9-VPR offers simplicity; validated in diverse systems [20] [19] |
| Guide RNA Systems | Synthetic sgRNAs, qgRNA plasmids | Target specificity | Quadruple-guide RNAs show enhanced efficacy [21] |
| Delivery Tools | Electroporation systems, lipid nanoparticles, gold nanoparticles | Component delivery | Nanoparticles enhance biofilm penetration [16] |
| Validation Assays | RT-qPCR reagents, flow cytometry antibodies, confocal microscopy | Efficacy assessment | Multi-level validation essential |
| Biofilm Assays | Crystal violet, microtiter plates, CLSM with EPS stains | Phenotypic characterization | High-content imaging reveals structural changes |
Future Perspectives and Concluding Remarks
CRISPRa technology represents a paradigm shift in our ability to precisely manipulate transcriptional programs controlling biofilm lifecycles. The integration of CRISPRa with emerging technologies—including nanoparticle delivery systems, multiplexed screening approaches, and real-time imaging—will further enhance its application in biofilm research [16] [22]. Future developments should focus on improving delivery efficiency in complex biofilm environments, expanding the CRISPRa toolbox with novel effectors and orthogonal systems, and implementing temporal control systems to map the dynamics of dispersal pathways.
For researchers investigating biofilm dispersal mechanisms, CRISPRa offers unprecedented precision to activate endogenous genes at native genomic locations, preserving natural regulatory contexts while enabling systematic gain-of-function studies. As the technology continues to evolve, CRISPRa promises to illuminate the complex genetic networks controlling the biofilm-planktonic transition, potentially revealing novel therapeutic targets for combating persistent biofilm-associated infections.
{#title Advantages of CRISPRa Over Gene Editing for Transient Phenotypic Manipulation}
{#context} This technical guide examines the strategic advantage of CRISPR activation (CRISPRa) over permanent gene editing for the transient manipulation of phenotypes, specifically within the context of pioneering research on activating biofilm dispersal genes. For researchers and drug development professionals, this non-permanent approach offers a powerful method for probing gene function and developing therapeutic interventions without the risks associated with irreversible genomic alterations.
{#introduction} In the pursuit of novel antibacterial strategies, biofilm-associated infections represent a formidable challenge due to their inherent tolerance to antibiotics [16]. While traditional CRISPR-Cas9 gene editing introduces permanent double-strand breaks to disrupt target genes [4], this approach is less suitable for controlling essential processes or achieving reversible phenotypic changes. CRISPRa, which utilizes a catalytically inactive Cas9 (dCas9) fused to transcriptional activation domains, enables precise, programmable, and transient upregulation of endogenous genes [7] [23]. This is particularly advantageous for biofilm research, where finely-tuned induction of dispersal genes can dismantle biofilms without driving resistance through lethal pressure. This guide details the mechanistic advantages, experimental protocols, and essential toolkits for implementing CRISPRa in biofilm dispersal studies.
{#section-1}
Mechanistic Advantages of CRISPRa for Transient Manipulation
CRISPRa operates through a fundamentally different mechanism than editing, focusing on transcriptional control rather than DNA cleavage. This distinction confers several critical benefits for functional genomics and therapeutic development.
Key Operational Differences
The table below summarizes the core distinctions between CRISPRa and permanent gene editing.
| Feature | CRISPRa (dCas9-Activator) | Permanent Gene Editing (Cas9 Nuclease) |
|---|---|---|
| Cas9 Form | Catalytically inactive (dCas9) [23] | Nuclease-active (Cas9) [24] |
| Primary Mechanism | Binds promoter/enhancer; recruits activation domains to upregulate transcription [7] [23] | Creates double-strand DNA breaks, leading to gene knockout via indel mutations [4] |
| Genomic Outcome | Reversible, transient gene expression change [7] | Permanent, irreversible genomic alteration [4] |
| Phenotypic Outcome | Tunable and conditional phenotype [23] | Binary (on/off) phenotype |
| Application in Biofilms | Ideal for probing essential genes, inducing transient dispersal, and studying dynamic processes [7] | Suited for validating non-essential biofilm genes through knockout [4] |
Strategic Benefits for Biofilm Dispersal Research
- Precision and Reversibility: CRISPRa allows for the precise activation of key biofilm dispersal regulators, such as genes encoding diguanylate cyclases (PDEs) that lower intracellular c-di-GMP levels [25]. This reversibility is crucial for mimicking natural biological processes and studying the dynamics of biofilm dissolution without creating permanent, potentially compensatory mutations [7].
- Targeting Essential Genes: Many genes controlling central metabolism or cellular integrity are essential for viability. Knocking them out is lethal, precluding functional study. CRISPRa can transiently modulate the expression of such genes to study their role in biofilm formation or dispersal without causing cell death [23].
- Reduced Risk of Drive Resistance: Permanent gene editing imposes strong selective pressure for escape mutants. In contrast, the transient and non-lethal nature of CRISPRa-mediated dispersal—for instance, by activating genes that promote the transition from a sessile to a motile lifestyle—poses a lower risk of driving resistance [7].
- Multiplexed Activation: CRISPRa systems can be designed with multiple guide RNAs (gRNAs) to simultaneously activate several genes in a synergistic pathway, such as targeting both a PDE and a quorum-sensing repressor to efficiently trigger biofilm dismantling [7].
{#section-2}
Experimental Workflow for CRISPRa in Biofilm Dispersal
Implementing a CRISPRa experiment to activate biofilm dispersal genes requires a structured workflow, from target identification to phenotypic validation. The following protocol and diagram outline this process.
Detailed Experimental Protocol
Target Identification and gRNA Design:
- Objective: Select target genes and design specific gRNAs.
- Methodology: Based on literature and omics data (e.g., transcriptomics of dispersed cells), identify key dispersal gene promoters (e.g., genes for PDEs like bifA or dipA in Pseudomonas [25]). Design 2-3 gRNAs targeting regions -50 to -500 bp upstream of the transcription start site for optimal activation [23].
- Validation: Use computational tools (e.g., CFDspecificity score) to predict and minimize off-target effects.
CRISPRa System Assembly:
- Objective: Construct the plasmid system expressing dCas9 and the transcriptional activator.
- Methodology: Clone the selected gRNA sequences into a suitable expression plasmid. Co-transform this with a second plasmid expressing the dCas9-activator fusion (e.g., dCas9-Sox or dCas9-RNAP) [23]. Use a inducible promoter (e.g., Ptet) for dCas9 expression to control timing.
Delivery into Bacterial Model System:
- Objective: Introduce the CRISPRa system into the target biofilm-forming bacterium.
- Methodology: For tractable models like E. coli or P. fluorescens, use electroporation or chemical transformation [25]. For less tractable clinical isolates, employ conjugation or nanoparticle-assisted delivery [16] [7].
Induction of Gene Activation and Biofilm Assay:
- Objective: Induce the CRISPRa system and quantify biofilm dispersal.
- Methodology:
- Grow biofilms of the transformed strain in a standardized system (e.g., microtiter plate, flow cell) for 24-48 hours.
- Add inducer (e.g., aTc) to activate dCas9-expression and trigger target gene transcription.
- Incubate for a further 4-24 hours to allow for phenotypic manifestation.
Validation and Phenotyping:
- Objective: Confirm gene activation and measure dispersal.
- Methodology:
- Molecular Validation: Use RT-qPCR to quantify the increase in target gene mRNA levels relative to non-induced or non-targeting gRNA controls.
- Phenotypic Validation:
- Biomass Quantification: Use crystal violet staining to measure total remaining biofilm biomass [25].
- Architectural Analysis: Use confocal laser scanning microscopy (CLSM) to visualize the 3D structure of the biofilm, noting changes in thickness and biovolume [25].
- Dispersal Quantification: Collect and plate the effluent from flow cell systems to count dispersed colony-forming units (CFUs).
{#diagram-1}
{#caption-1} CRISPRa experimental workflow for biofilm dispersal.
{#section-3}
Signaling Pathways in Biofilm Dispersal for CRISPRa Targeting
Understanding the genetic pathways controlling biofilm formation is essential for selecting effective CRISPRa targets. A primary regulatory mechanism involves the intracellular second messenger cyclic di-GMP (c-di-GMP).
The c-di-GMP Regulatory Network
In bacteria, high levels of c-di-GMP promote biofilm formation by repressing motility and stimulating the production of extracellular polymeric substances (EPS) [25]. Conversely, low levels of c-di-GMP induce biofilm dispersal and a transition to a motile, planktonic lifestyle. This switch is controlled by the antagonistic activity of two enzyme classes:
- Diguanylate Cyclases (DGCs): Synthesize c-di-GMP, promoting biofilm formation.
- Phosphodiesterases (PDEs): Degrade c-di-GMP, promoting dispersal [25].
CRISPRa can be strategically deployed to overexpress specific PDE genes (e.g., bifA, dipA), thereby artificially lowering c-di-GMP levels and triggering dispersal. This pathway and the intervention point for CRISPRa are illustrated below.
{#diagram-2}
{#caption-2} CRISPRa targets PDEs to lower c-di-GMP and induce dispersal.
{#section-4}
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of a CRISPRa experiment relies on a suite of specialized reagents and tools. The following table details the key components required for such a study.
| Research Reagent / Solution | Function & Application in CRISPRa Experiments |
|---|---|
| dCas9-Activator Plasmid | Expresses the catalytically dead Cas9 fused to transcriptional activation domains (e.g., Sox, RNAP). This is the core effector protein of the system [23]. |
| Guide RNA (gRNA) Expression Plasmid | A plasmid containing the sequence for the gRNA that targets the dCas9-activator complex to the promoter region of the specific biofilm dispersal gene (e.g., a PDE gene) [25] [23]. |
| Inducible Promoter System (e.g., Ptet) | Allows for precise temporal control over the expression of the dCas9-activator, enabling researchers to induce gene activation after the biofilm has matured [25]. |
| Nanoparticle/Delivery Vehicle | For strains resistant to standard transformation, lipid or metallic nanoparticles can be used to deliver CRISPRa components, enhancing uptake and protecting genetic material [16] [7]. |
| Confocal Laser Scanning Microscope (CLSM) | Used for high-resolution imaging of biofilms, allowing for the quantitative analysis of changes in biofilm architecture (e.g., biovolume, thickness) following CRISPRa induction [25]. |
| RT-qPCR Reagents | Essential for validating the success of the CRISPRa experiment by quantifying the upregulation of mRNA transcripts from the target dispersal gene relative to control conditions [25]. |
{#conclusion} CRISPRa represents a paradigm shift in our approach to probing and manipulating bacterial phenotypes, offering a level of precision and reversibility that is unattainable with traditional gene editing. Its application for activating biofilm dispersal genes demonstrates a powerful strategy to combat biofilm-associated infections without the selective pressure of lethal interventions. While challenges in delivery efficiency and species-specific portability remain, the continued refinement of CRISPRa systems, particularly when integrated with advanced delivery platforms like nanoparticles, promises to unlock new frontiers in antimicrobial research and therapeutic development. For scientists pursuing non-permanent phenotypic manipulation, CRISPRa is an indispensable tool in the molecular arsenal.
Engineering CRISPRa Systems for Programmable Biofilm Control
Design Principles for sgRNAs Targeting Dispersal Gene Promoters
The crisis of antibiotic-resistant bacterial infections represents one of the most urgent threats to global health, with biofilms playing a pivotal role in bacterial persistence and treatment failure [16]. Bacterial biofilms are structured communities embedded in a protective extracellular polymeric substance (EPS) that can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [16]. Within the biofilm life cycle, the dispersal phase offers a critical intervention point for novel therapeutic strategies. The emerging application of CRISPR activation (CRISPRa) technology enables precise transcriptional programming to trigger this dispersal process, potentially resensitizing persistent infections to conventional antimicrobials [26] [27]. At the heart of this approach lies the strategic design of single guide RNAs (sgRNAs) that direct CRISPRa systems to specifically activate dispersal gene promoters. This technical guide provides a comprehensive framework for designing highly efficient sgRNAs within the context of biofilm dispersal research, offering principles, protocols, and practical considerations for researchers and drug development professionals.
Biological Foundation of Biofilm Dispersal
Biofilm Architecture and Therapeutic Challenges
Biofilms are highly organized structures characterized by microcolonies interspersed with water channels that facilitate nutrient distribution and waste removal [16]. The extracellular matrix, composed primarily of polysaccharides, proteins, and extracellular DNA, forms a protective barrier that limits antibiotic penetration and maintains biofilm integrity [16]. This heterogeneous structure creates microenvironments with varying levels of nutrient availability, pH, oxygen, and waste products, contributing to microbial survival under challenging conditions [16].
The resistance mechanisms employed by bacteria within biofilms present significant therapeutic challenges. Beyond the physical barrier provided by the EPS matrix, biofilms harbor subpopulations of metabolically dormant persister cells that exhibit exceptional tolerance to antimicrobial agents [16]. This phenotypic resistance, combined with the potential for horizontal gene transfer of resistance determinants within the biofilm community, necessitates innovative approaches that target the fundamental regulatory pathways controlling biofilm maintenance and dispersal.
Molecular Regulators of Biofilm Dispersal
The transition from biofilm to planktonic lifestyle is governed by sophisticated regulatory networks that respond to environmental cues and cellular signaling molecules. Key among these are:
Cyclic di-GMP (c-di-GMP) signaling: This near-universal intracellular bacterial messenger serves as a central regulator of the transition between motile and sessile lifestyles [6]. High intracellular levels of c-di-GMP promote biofilm formation through the production of matrix components, while decreased c-di-GMP concentrations trigger dispersal. The enzymes responsible for c-di-GMP homeostasis—diguanylate cyclases (DGCs) that synthesize c-di-GMP and phosphodiesterases (PDEs) that degrade it—represent promising targets for dispersal activation [6].
Quorum Sensing (QS) pathways: Cell-to-cell communication systems regulate collective behaviors in bacteria, including biofilm development and dispersal. Targeting QS components via CRISPRa offers the potential to override native signaling and induce programmed dispersal [28].
Two-component systems (TCS): These signal transduction systems, such as the GacA/S system in Pseudomonas species, sense environmental stimuli and coordinate genetic programs that control biofilm dynamics [6].
Understanding these regulatory networks provides the biological rationale for target selection when designing sgRNAs for dispersal gene activation.
CRISPRa Systems for Gene Activation
Core CRISPRa Architecture
CRISPR activation systems employ a catalytically dead Cas9 (dCas9) protein that retains DNA-binding capability but lacks nuclease activity [26]. This dCas9 serves as a programmable platform for recruiting transcriptional activation domains to specific genomic loci. The fundamental components include:
dCas9: Engineered through point mutations in the RuvC and HNH nuclease domains to create a DNA-binding protein that does not cleave target sequences [26].
Transcriptional activators: Multipartite activation domains fused to dCas9, such as the tripartite VPR (VP64-p65-Rta) complex, which significantly enhances transcriptional activation compared to single domains [26] [27].
Guide RNA (gRNA): A single chimeric RNA molecule that combines the functions of crRNA and tracrRNA, directing dCas9 to specific DNA sequences through complementary base pairing [29].
The CRISPRa system functions as a targeted recruitment platform, bringing potent transcriptional activation machinery to specific promoter regions of genes encoding dispersal factors, thereby increasing their expression and promoting biofilm dissolution.
Optimization Strategies for Enhanced Activation
Several protein engineering approaches have been developed to enhance the efficacy of CRISPRa systems:
Multipartite activation domains: The fusion of dCas9 with multiple transcription activator domains creates a synergistic effect on gene expression. For example, the VPR system combines VP64, p65, and Rta domains, demonstrating significantly higher activation efficiency compared to single-domain constructs [26].
sgRNA scaffold engineering: Modifying the sgRNA structure to incorporate RNA aptamers (e.g., MS2, PP7) enables the recruitment of additional activation domains, creating a multivalent recruitment platform that further enhances transcriptional output [26].
Multiple sgRNA targeting: Employing several sgRNAs targeting different positions within the same promoter region can produce synergistic effects, potentially due to enhanced recruitment of transcriptional machinery or alterations in local chromatin accessibility [26].
These optimized CRISPRa systems have demonstrated robust gene activation across diverse bacterial species, making them particularly suitable for targeting biofilm dispersal genes in pathogenic organisms [27].
sgRNA Design Principles for Dispersal Gene Activation
Target Selection and Positioning
The precise positioning of sgRNA binding sites relative to the transcriptional start site (TSS) represents the most critical parameter for effective CRISPRa-mediated gene activation. Empirical studies in bacterial systems have revealed stringent requirements for functional target sites:
Table 1: Optimal sgRNA Positioning for Bacterial CRISPRa
| Parameter | Optimal Range | Key Considerations |
|---|---|---|
| Distance from TSS | 60-100 bases upstream | Narrow windows of effectiveness centered ~80 bases upstream [30] |
| Functional windows | 2-4 base periods | Activity shows 10-11 base periodicity corresponding to DNA helical turns [30] |
| Strand targeting | Non-template (NT) strand | gRNAs targeting the non-template strand generally show higher efficacy [6] |
| PAM orientation | NGG sequence for SpCas9 | Must be present adjacent to target site; dictates available targeting space [30] |
The periodicity of effective sites, corresponding to one helical turn of DNA (~10-11 bases), suggests that transcriptional activation is highly dependent on the rotational positioning of the CRISPRa complex relative to the promoter machinery [30]. This stringent positioning requirement necessitates careful mapping of TSS and systematic screening of multiple target sites within the optimal window.
Sequence-Specific Design Parameters
Beyond positional requirements, several sequence-specific factors significantly influence sgRNA efficacy:
Protospacer Adjacent Motif (PAM) Requirements: The canonical NGG PAM sequence for Streptococcus pyogenes Cas9 must be present immediately adjacent to the target site. This requirement can limit targetable positions within the optimal activation window, necessitating the use of Cas9 orthologs with alternative PAM specificities when suitable NGG sites are unavailable [30].
GC Content: Maintaining a GC content of 40-60% in the sgRNA spacer sequence ensures balanced binding stability and specificity. Guides with excessively high GC content may exhibit non-specific binding, while those with low GC content may demonstrate reduced target affinity [31].
Specificity Considerations: sgRNAs must be designed to minimize off-target activation by ensuring minimal sequence similarity to non-target genomic regions. Computational tools that perform genome-wide specificity analysis are essential for identifying guides with maximal on-target activity and minimal off-target potential [31] [29].
Secondary Structure Stability: The sgRNA itself should avoid internal hairpins or stable secondary structures that could impede Cas9 binding or complex formation. Computational prediction of sgRNA secondary structure can identify problematic designs that should be avoided [31].
These sequence-based parameters collectively determine the binding efficiency and specificity of sgRNAs, directly influencing the success of dispersal gene activation.
Target Accessibility and Chromatin Environment
While bacterial genomes lack the complex chromatin organization of eukaryotic systems, target site accessibility remains an important consideration for CRISPRa efficiency. Bacterial DNA exists in a compacted nucleoid structure where transcriptionally active regions typically demonstrate greater accessibility to DNA-binding proteins [31]. For optimal results, target sites should be located within regions of intrinsic DNA accessibility, avoiding highly constrained areas that might limit dCas9 binding.
The following diagram illustrates the key sgRNA design parameters and their relationship to transcriptional activation efficiency:
Figure 1: Key factors influencing sgRNA design for CRISPRa efficiency
Experimental Workflow for sgRNA Validation
In Silico Design and Screening
A systematic computational approach to sgRNA design significantly improves the success rate of subsequent experimental validation:
Target Gene Identification: Select dispersal genes based on their established roles in biofilm regulation, such as PDEs, surfactant producers, or matrix-degrading enzymes [6].
Promoter Mapping: Precisely identify transcriptional start sites through experimental data or computational prediction to establish the reference point for sgRNA positioning [30].
Candidate sgRNA Selection: Identify all potential sgRNA target sites within the region 40-120 bases upstream of the TSS that contain appropriate PAM sequences [30].
Specificity Analysis: Perform genome-wide alignment to eliminate guides with significant off-target potential, particularly in genetically diverse clinical isolates [29].
Secondary Structure Prediction: Evaluate both sgRNA and target DNA structures to avoid designs with stable internal structures that might impair binding [31].
This computational pipeline should generate a prioritized list of candidate sgRNAs for experimental validation, with 3-5 top candidates per target gene to account for unpredictable performance variations.
Functional Validation in Model Systems
Following computational design, sgRNAs require rigorous experimental validation to confirm their efficacy:
Table 2: sgRNA Validation Experimental Protocol
| Step | Methodology | Key Parameters | Quality Controls |
|---|---|---|---|
| Delivery System | Plasmid-based dCas9-VPR with sgRNA expression cassette [27] | Constitutive vs. inducible dCas9 expression | Transformation efficiency; plasmid retention |
| Activation Assessment | qRT-PCR measuring target gene expression [6] | Time-course measurements; multiple biological replicates | Normalization to housekeeping genes; include non-targeting sgRNA controls |
| Phenotypic Confirmation | Biofilm biomass assays (crystal violet); dispersal kinetics [6] | High-throughput screening compatible; confocal microscopy | Correlation between expression and phenotype |
| Specificity Verification | RNA-seq transcriptome analysis [27] | Genome-wide expression profiling | Identification of off-target activation events |
The experimental workflow for sgRNA validation proceeds through sequential stages of testing, beginning with transcriptional activation and culminating in functional phenotypic assessment:
Figure 2: Experimental workflow for sgRNA validation
Advanced Delivery Strategies for Therapeutic Applications
The translational application of CRISPRa-based biofilm dispersal requires efficient delivery of sgRNA and CRISPRa components to bacterial populations within biofilms. Nanoparticle-based delivery systems offer promising solutions to the challenges of in vivo implementation:
Liposomal Formulations: These have demonstrated remarkable efficacy in laboratory studies, with reports indicating over 90% reduction in Pseudomonas aeruginosa biofilm biomass when delivering CRISPR-Cas9 components [16].
Gold Nanoparticles: CRISPR-gold nanoparticle hybrids have shown a 3.5-fold increase in gene-editing efficiency compared to non-carrier systems while promoting synergistic action with antibiotics [16].
Bioresponsive Carriers: Smart delivery systems that respond to specific environmental cues (e.g., pH, enzymes) within the biofilm microenvironment can enable targeted release of CRISPRa components, enhancing specificity and reducing off-target effects [26].
These advanced delivery platforms can be engineered to co-deliver CRISPRa components with conventional antibiotics, creating a multifaceted approach that attacks bacterial populations through both genetic disruption and traditional antimicrobial mechanisms [16].
Research Reagent Solutions
The successful implementation of sgRNA design principles requires a comprehensive toolkit of specialized reagents and resources:
Table 3: Essential Research Reagents for sgRNA Development
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| CRISPRa Plasmids | dCas9-VPR expression vectors [27] | Provides transcriptional activation machinery; single-plasmid systems available for fungal pathogens [27] |
| sgRNA Cloning Systems | Gibson assembly-compatible vectors with SNR52 promoters [27] | Enables rapid, efficient sgRNA library construction |
| Biofilm Assay Kits | Crystal violet staining; confocal microscopy with EPS-binding dyes | Quantifies biofilm biomass and spatial architecture |
| Delivery Vehicles | Liposomal Cas9 formulations; gold nanoparticle carriers [16] | Enhances penetration through biofilm matrices; improves editing efficiency |
| Validation Tools | qRT-PCR reagents; RNA-seq library preparation kits | Confirms target gene overexpression and identifies off-target effects |
This reagent toolkit supports the complete workflow from sgRNA design and delivery to functional validation, enabling researchers to systematically develop and optimize sgRNAs for dispersal gene activation.
The strategic design of sgRNAs for targeting dispersal gene promoters represents a cutting-edge approach in the battle against biofilm-mediated antimicrobial resistance. The principles outlined in this technical guide emphasize the critical importance of precise target positioning within narrow windows upstream of transcriptional start sites, with consideration of sequence-specific parameters that govern binding efficiency and specificity. When implemented through a rigorous workflow of computational design and experimental validation, these design principles enable the development of highly effective sgRNAs that can activate endogenous dispersal mechanisms in bacterial biofilms. As CRISPRa technologies continue to evolve and delivery systems become increasingly sophisticated, the programmable activation of biofilm dispersal genes offers a promising pathway toward novel therapeutic interventions that could potentially resensitize persistent infections to conventional antibiotics. The ongoing optimization of these approaches will undoubtedly play a significant role in addressing the global challenge of antimicrobial resistance.
Selecting Activation Domains and dCas9 Variants for Robust Transcriptional Upregulation
The rise of multidrug-resistant bacterial infections represents one of the most pressing global health challenges, with biofilm-associated infections being particularly problematic due to their inherent tolerance to conventional antimicrobial therapies [16]. Bacterial biofilms are structured communities embedded in a self-produced extracellular polymeric substance (EPS) that can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [16]. Within the complex architecture of biofilms, bacteria exist in heterogeneous metabolic states and are protected by physical barriers that limit antibiotic penetration, making conventional treatments largely ineffective [3].
CRISPR activation (CRISPRa) systems have emerged as transformative tools for precision manipulation of bacterial gene expression networks, offering unprecedented opportunities for investigating and triggering biofilm dispersal mechanisms. These systems utilize a catalytically dead Cas9 (dCas9) fused to transcriptional activation domains, guided by sequence-specific single guide RNAs (sgRNAs) to target promoter regions of interest [32]. Unlike traditional genetic knockout approaches, CRISPRa enables reversible, tunable upregulation of endogenous genes without permanent genomic alterations, making it particularly valuable for studying essential processes and multiplexed gene regulation [7]. In the context of biofilm research, CRISPRa provides a powerful means to precisely activate master regulators of biofilm dispersal, potentially reversing the chronic infection state and resensitizing bacterial populations to antimicrobial treatments.
This technical guide examines the selection of optimal activation domains and dCas9 variants specifically for upregulating biofilm dispersal genes, providing researchers with practical frameworks for implementing these systems in antimicrobial discovery and development.
Core Components of CRISPRa Systems
dCas9 Variants and Their Properties
The foundation of any CRISPRa system is the catalytically dead Cas9 (dCas9), engineered through point mutations (D10A and H840A in Streptococcus pyogenes Cas9) that abolish nuclease activity while retaining DNA-binding capability [7]. This modified protein serves as a programmable platform for recruiting transcriptional activators to specific genomic loci. Different dCas9 variants offer distinct advantages for bacterial gene activation:
Standard dCas9: The most widely used variant provides reliable binding with minimal off-target effects. Its main limitation in bacterial systems is the requirement for a protospacer adjacent motif (PAM) sequence (NGG for SpCas9), which can constrain targetable sites in bacterial genomes [7].
dCas9 Variants with Expanded PAM Recognition: Engineered variants such as xCas9 and SpCas9-NG recognize broader PAM sequences, significantly expanding the targeting range for bacterial gene activation [7]. These are particularly valuable for targeting biofilm dispersal genes with limited suitable PAM sites in their promoter regions.
dCas9 Orthologs: Alternative Cas proteins from different bacterial species, such as dCas12a (from Francisella novicida), offer different PAM requirements and can be valuable when SpCas9 targeting is constrained [28].
Table 1: Comparison of dCas9 Variants for Bacterial Gene Activation
| dCas9 Variant | PAM Requirement | Targeting Range | Size (aa) | Best Use Cases |
|---|---|---|---|---|
| dCas9 (Sp) | NGG | Limited | 1368 | Standard applications with abundant NGG sites |
| dCas9-NG | NG | Expanded | ~1368 | Targets with constrained NGG availability |
| xCas9 | NG, GAA, GAT | Broad | ~1368 | Maximum targeting flexibility |
| dCas12a | TTTN | Complementary | ~1300 | Targeting AT-rich promoter regions |
Transcriptional Activation Domains
The efficacy of CRISPRa systems depends critically on the choice of activation domain fused to dCas9. Different domains vary in their potency, size, and suitability for bacterial systems:
Single Activation Domains: VP64, consisting of four copies of the Herpes Simplex Viral Protein 16 (VP16) minimal activation domain, represents the foundational CRISPRa architecture. While it provides moderate activation (typically 2-10 fold) and minimal metabolic burden, its efficacy may be insufficient for strongly upregulating biofilm dispersal genes that require substantial expression changes to trigger dissociation [32].
Multi-Domain Activation Systems: For enhanced transcriptional activation, engineered multi-domain systems significantly outperform single-domain approaches:
dCas9-VPR: This fusion combines VP64, p65 (a subunit of NF-κB), and Rta (from Epstein-Barr virus) in a single polypeptide, typically achieving 10-50 fold activation across diverse bacterial targets [33]. The synergistic action of these domains recruits multiple components of the transcriptional machinery simultaneously.
dCas9-SAM (Synergistic Activation Mediator): This system employs MS2 coat protein-modified sgRNAs that recruit additional activation domains (VP64 and p65-HSF1), creating a multi-component activation complex [32]. While more complex to implement, SAM typically achieves 20-100 fold activation, making it valuable for recalcitrant targets.
dCas9-p300: This fusion incorporates the catalytic core of the human p300 histone acetyltransferase, which modifies chromatin structure to create a more accessible environment for transcription [7]. While primarily used in eukaryotic systems, it shows promise for bacterial targets where chromatin structure influences transcription.
Table 2: Performance Comparison of Activation Domains in Bacterial Systems
| Activation System | Typical Fold Activation | System Complexity | Metabolic Burden | Optimal Application |
|---|---|---|---|---|
| dCas9-VP64 | 2-10x | Low | Low | Preliminary testing, essential genes |
| dCas9-VPR | 10-50x | Medium | Medium | Strong activation needs |
| dCas9-SAM | 20-100x | High | High | Recalcitrant targets, multiplexing |
| SunTag | 50-200x | High | Medium-High | Maximum activation potency |
CRISPRa System Design for Biofilm Dispersal Genes
Targeting Key Biofilm Dispersal Pathways
Effective CRISPRa-mediated biofilm disruption requires strategic targeting of master regulators within the dispersal circuitry. Several key pathways present promising targets for transcriptional activation:
Quorum Sensing Systems: In Pseudomonas aeruginosa, activation of the LasI/LasR and RhlI/RhlR quorum sensing circuits can promote biofilm dispersion through multiple mechanisms, including upregulation of rhamnolipid biosynthesis (rhlAB operon) and repression of pel exopolysaccharide production [34]. sgRNAs should target promoter regions of lasI, lasR, rhlI, and rhlR to enhance the native dispersion signaling that occurs at high cell densities.
cyclic di-GMP Signaling Network: This ubiquitous bacterial second messenger inversely regulates the transition between biofilm formation and dispersal, with low c-di-GMP levels promoting the planktonic state [6]. CRISPRa can target phosphodiesterases (PDEs) such as dipA, bifA, and rocR which degrade c-di-GMP, as well as activate regulators of PDE expression. The resultant reduction in cellular c-di-GMP concentration induces biofilm dissociation.
Matrix-Degrading Enzymes: Direct activation of genes encoding enzymes that degrade biofilm matrix components can physically dismantle the biofilm structure [35]. Targets include dispersin B (targeting polysaccharide intercellular adhesion), alginate lyase (algL), and proteases that cleave matrix proteins.
Cellular Stress Responses: Activation of toxin-antitoxin systems and other stress response pathways can induce the transition from biofilm to planktonic growth as a survival strategy [3].
Diagram 1: CRISPRa Targets for Biofilm Dispersal
gRNA Design and Delivery Considerations
Effective gRNA design is critical for CRISPRa efficacy. For transcriptional activation, gRNAs should target the non-template strand within the promoter region, typically between -50 and -500 base pairs upstream of the transcription start site (TSS) [36]. Multiple gRNAs targeting different positions within the same promoter often yield synergistic effects, with the most effective positions varying between targets. Computational tools such as CHOPCHOP and CRISPOR can assist in gRNA selection, but empirical testing of 3-5 gRNAs per target is recommended to identify optimal performers [36].
Delivery of CRISPRa components into biofilm-embedded bacteria presents unique challenges. The extracellular polymeric substance (EPS) matrix significantly impedes diffusion of genetic material, requiring specialized delivery strategies:
Conjugative Plasmids: Engineered conjugation systems can efficiently transfer CRISPRa constructs into diverse bacterial species within biofilms [7].
Phagemid Vectors: Bacteriophage-based delivery systems exploit natural phage infection mechanisms to penetrate biofilms and deliver genetic payloads [7].
Nanoparticle Carriers: Inorganic nanoparticles (e.g., gold, silica) and lipid-based nanocarriers can protect CRISPRa components from degradation and enhance biofilm penetration [16]. Recent studies demonstrate that liposomal Cas9 formulations can reduce Pseudomonas aeruginosa biofilm biomass by over 90% in vitro, while gold nanoparticle carriers enhance editing efficiency up to 3.5-fold compared to non-carrier systems [16].
Electroporation: For in vitro applications, optimized electroporation protocols can achieve efficient delivery, particularly when combined with matrix-disrupting pretreatments [32].
Experimental Framework for CRISPRa-Mediated Biofilm Dispersal
Protocol: CRISPRa System Assembly and Validation
Materials Required:
- dCas9-activator fusion plasmids (e.g., dCas9-VPR, dCas9-SAM)
- sgRNA expression backbone with appropriate bacterial resistance marker
- Target bacterial strains with known biofilm formation capability
- Conjugation or transformation reagents appropriate for target strains
- Biofilm culture vessels (e.g., flow cells, 96-well plates)
- qPCR reagents for transcriptional analysis
- Confocal microscopy equipment for biofilm visualization
Step 1: gRNA Design and Cloning
- Identify promoter regions of target biofilm dispersal genes using genome databases and literature curation.
- Design 3-5 sgRNAs per target using computational tools, focusing on the -50 to -500 region upstream of TSS.
- Synthesize oligonucleotides with appropriate overhangs for cloning into sgRNA expression vectors.
- Clone sgRNA sequences into expression vectors using Golden Gate assembly or traditional restriction-ligation methods.
- Transform cloning products into E. coli DH5α, screen colonies by PCR, and verify sequences by Sanger sequencing.
Step 2: Delivery System Assembly
- Select appropriate delivery vector based on target bacteria (conjugative plasmid for recalcitrant species, standard plasmid for tractable strains).
- Assemble complete CRISPRa system containing dCas9-activator and sgRNA expression cassettes.
- For conjugative delivery, transform assembled system into donor E. coli strain (e.g., E. coli WM3064).
Step 3: Bacterial Transformation/Conjugation
- For plasmid transformation: Prepare electrocompetent target bacteria and transform with purified CRISPRa plasmid using optimized voltage conditions.
- For conjugation: Mix donor E. coli carrying CRISPRa system with recipient bacteria at 1:2 ratio on filter membranes placed on solid media. Incubate 12-24 hours, then resuspend and plate on selective media containing appropriate antibiotics and DAP as needed.
- Validate successful transfer by colony PCR and antibiotic resistance profiling.
Step 4: Transcriptional Activation Validation
- Inoculate positive clones in liquid media and grow to mid-log phase.
- Harvest samples for RNA extraction at appropriate time points.
- Perform RT-qPCR to quantify expression changes of target genes using housekeeping genes for normalization.
- Confirm activation efficacy (minimum 5-fold increase recommended before proceeding to biofilm assays).
Protocol: Biofilm Dispersal Efficacy Assessment
Step 1: Biofilm Establishment
- Inoculate CRISPRa-containing bacteria and appropriate controls in biofilm-permissive media.
- For static biofilms: Transfer cultures to 96-well plates or cover slips and incubate for 24-72 hours to establish mature biofilms.
- For flow cell biofilms: Set up flow cell systems with continuous media perfusion to develop structurally complex biofilms over 3-5 days.
Step 2: CRISPRa Induction
- For inducible systems: Add inducer (aTc, IPTG, or arabinose depending on promoter system) at optimized concentration.
- For constitutive systems: Ensure continuous expression throughout biofilm development.
- Include appropriate controls (non-targeting sgRNA, dCas9-only).
Step 3: Biofilm Quantification and Visualization
- Quantify biofilm biomass using crystal violet staining: Fix biofilms with methanol, stain with 0.1% crystal violet, solubilize with acetic acid, and measure absorbance at 595nm.
- Assess metabolic activity using resazurin reduction assays: Incubate biofilms with resazurin solution, measure fluorescence at 560/590nm.
- Visualize biofilm architecture by confocal laser scanning microscopy (CLSM): Stain with SYTO9/propidium iodide for viability assessment, or with specific matrix component labels.
- Quantify dispersed cells: Collect effluent from biofilm systems, plate serial dilutions, and enumerate CFUs.
Step 4: Functional Assessment
- Evaluate antibiotic resensitization: Treat induced and control biofilms with relevant antibiotics at clinically achievable concentrations, assess viability reduction.
- Analyze matrix composition changes: Extract EPS from biofilms, quantify polysaccharide, protein, and eDNA content.
- Assess gene expression patterns in dispersed versus biofilm cells using RNA-seq or targeted qPCR.
Diagram 2: Experimental Workflow for CRISPRa Biofilm Studies
Research Reagent Solutions
Table 3: Essential Research Reagents for CRISPRa Biofilm Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| dCas9-Activator Plasmids | dCas9-VPR, dCas9-SAM, dCas9-TV | Transcriptional activation core | Choose based on required activation strength and bacterial compatibility |
| sgRNA Backbones | pTarget, pCRISPR, pACBSI-H | gRNA expression | Ensure compatibility with dCas9 system and host range |
| Delivery Vectors | Conjugative plasmids, Phagemids, Nanoparticles | Component delivery into biofilms | Selection critical for efficient biofilm penetration |
| Biofilm Assessment Tools | Crystal violet, Resazurin, SYTO9/PI dyes | Biofilm quantification and visualization | Multi-method approach recommended |
| Selective Antibiotics | Kanamycin, Chloramphenicol, Spectinomycin | Selection of transformants | Choose based on target bacterium resistance profile |
| Inducer Compounds | aTc, IPTG, Arabinose | Regulation of dCas9 expression | Titrate for optimal induction with minimal toxicity |
CRISPRa technology represents a paradigm shift in our approach to combating biofilm-associated infections, moving from broad-spectrum antimicrobials to precision genetic interventions. The strategic selection of activation domains and dCas9 variants, coupled with sophisticated delivery systems, enables researchers to precisely manipulate the complex genetic networks controlling biofilm dispersal. As demonstrated in recent studies, this approach can achieve remarkable efficacy, with liposomal Cas9 formulations reducing Pseudomonas aeruginosa biofilm biomass by over 90% in vitro [16].
The future of CRISPRa for biofilm control lies in advancing delivery mechanisms to overcome the physical and physiological barriers of mature biofilms, developing sophisticated multiplexing strategies to simultaneously target multiple dispersal pathways, and creating diagnostic-integrated systems that activate dispersal response only when pathogen detection thresholds are reached. As regulatory frameworks evolve to address the unique considerations of CRISPR-based antimicrobials, these precision tools hold immense potential for addressing the global crisis of biofilm-associated antimicrobial resistance.
Biofilm-associated infections represent a formidable challenge in healthcare due to their inherent resistance to conventional antibiotics, with biofilm-embedded bacteria exhibiting up to 1000-fold greater tolerance than their planktonic counterparts. The extracellular polymeric substance (EPS) matrix acts as a formidable barrier, severely limiting the penetration and efficacy of antimicrobial agents. This whitepaper examines two advanced delivery platforms—nanoparticles and bacteriophages—for their potential to penetrate biofilms and deliver precision therapeutics, with a specific focus on their application for CRISPRa (CRISPR activation) systems designed to activate biofilm dispersal genes. We evaluate mechanisms of action, summarize quantitative performance data, provide detailed experimental methodologies, and outline essential research tools for developing next-generation anti-biofilm strategies.
The Biofilm Penetration Challenge
Bacterial biofilms are structured communities of microorganisms encapsulated within a self-produced EPS matrix composed of exopolysaccharides, proteins, lipids, and extracellular DNA (eDNA) [16] [37]. This matrix forms a protective barrier that significantly contributes to antibiotic resistance through multiple mechanisms: (1) physical restriction of antibiotic diffusion; (2) creation of chemical microenvironments with altered pH and oxygen gradients that reduce antimicrobial activity; (3) presence of metabolically dormant persister cells; and (4) enhanced horizontal gene transfer of resistance determinants [38] [39].
This protective matrix presents a critical delivery challenge for any anti-biofilm therapeutic, including CRISPRa systems. The anionic, hydrophobic, and enzymatically active nature of the biofilm further complicates delivery, as it can trap, degrade, or inactivate conventional therapeutics before they reach their bacterial targets [40]. Overcoming these barriers requires delivery platforms with specific physicochemical properties that enable deep penetration and targeted release within the biofilm architecture.
Nanoparticle-Based Delivery Platforms
Mechanisms of Nanoparticle Penetration
Nanoparticles (NPs) leverage their unique physicochemical properties—including small size (typically 1-100 nm), high surface-area-to-volume ratio, and tunable surface chemistry—to overcome biofilm penetration barriers [41] [42]. Key penetration mechanisms include:
- Size-dependent passive diffusion: NPs smaller than the biofilm mesh pore size (typically 10-100 nm) can passively diffuse through the EPS matrix [38].
- Electrostatic interactions: Cationic NPs exploit electrostatic interactions with the anionic components of the EPS (e.g., DNA, polysaccharides) to enhance adhesion and retention [40].
- Intrinsic anti-biofilm activity: Certain metal NPs, including silver (Ag), zinc oxide (ZnO), and copper oxide (CuO), generate reactive oxygen species (ROS) that degrade EPS components and disrupt biofilm integrity [41] [42].
- Enzyme-functionalized delivery: NPs conjugated with EPS-degrading enzymes (e.g., DNase, dispersin B) actively break down the biofilm matrix, enhancing penetration [37].
Table 1: Performance Metrics of Selected Nanoparticles Against Biofilms
| Nanoparticle Type | Target Biofilm | Key Metrics | Delivery Mechanism |
|---|---|---|---|
| Liposomal Cas9 [16] | Pseudomonas aeruginosa | >90% reduction in biofilm biomass | Fusion with bacterial membranes; controlled release |
| Gold NP-CRISPR [16] | P. aeruginosa | 3.5x increase in editing efficiency | Enhanced cellular uptake; photothermal release |
| Cationic Liposomes [40] | Staphylococcus aureus | 4.2x improved antibiotic delivery | Electrostatic binding to anionic EPS |
| Silver Nanoparticles [42] | Mixed-species oral biofilm | 75% reduction in viable cells | ROS generation; membrane disruption |
Experimental Protocol: Evaluating NP Penetration and Efficacy
Objective: To quantify the penetration depth and anti-biofilm efficacy of nanoparticle formulations in a P. aeruginosa biofilm model.
Materials:
- Fluorescently labeled NPs: e.g., Cy5-labeled liposomes or quantum dot-tagged gold NPs.
- Biofilm growth chamber: e.g., flow cell or 96-well plate system.
- Confocal Laser Scanning Microscopy (CLSM) system with Z-stack imaging capability.
- Viability stains: SYTO 9 and propidium iodide (Live/Dead BacLight kit).
- Image analysis software: e.g., ImageJ, COMSTAT.
Procedure:
- Biofilm Cultivation: Grow P. aeruginosa PAO1 biofilms for 72-96 hours in a flow cell system with minimal media or in a static 96-well plate.
- NP Treatment: Introduce fluorescent NPs at a predetermined sub-MIC concentration to the mature biofilm. Include untreated controls.
- Penetration Analysis (CLSM):
- After 2-4 hours of incubation, acquire Z-stack images at multiple random locations.
- Use the image analysis software to create fluorescence intensity profiles across the biofilm depth (from substratum to top).
- Calculate the penetration coefficient as the ratio of fluorescence intensity at the substratum to the intensity at the biofilm surface.
- Efficacy Assessment (Live/Dead Staining):
- After 24 hours of NP exposure, stain biofilm with Live/Dead stain and image via CLSM.
- Quantify the biovolume and the ratio of dead (red) to live (green) cells.
- Gene Expression Analysis (if delivering CRISPRa):
- Harvest biofilm cells post-treatment and extract RNA.
- Perform RT-qPCR to quantify expression levels of target dispersal genes (e.g., bdlA, dipA).
Diagram 1: Workflow for evaluating nanoparticle penetration and efficacy in biofilms.
Bacteriophage-Based Delivery Platforms
Mechanisms of Bacteriophage Penetration
Bacteriophages (phages) are viruses that specifically infect and replicate within bacteria. They have evolved natural mechanisms to overcome biofilm barriers, making them promising vectors for delivering CRISPRa systems [37] [39].
- EPS-degrading enzymes: Many phages produce depolymerases that specifically hydrolyze key EPS components (e.g., polysaccharides, eDNA), creating physical passages through the matrix [37].
- Self-amplification: Upon infecting a host cell, phages undergo lytic replication, producing hundreds of progeny virions that can infect adjacent cells, creating a propagating treatment front within the biofilm [39].
- Receptor-specific targeting: Phages exhibit high specificity for bacterial surface receptors, enabling targeted delivery of CRISPRa machinery to specific species within polymicrobial biofilms without disrupting the broader microbiome [7].
A significant limitation is the narrow host range of natural phages. To overcome this, engineered phage cocktails or phage strains with broadened host ranges are being developed for more comprehensive biofilm coverage [39].
Table 2: Bacteriophage Strategies for Biofilm Control
| Phage Strategy | Mechanism of Action | Advantages | Limitations |
|---|---|---|---|
| Lytic Phage Cocktails | Bacterial lysis; biofilm structural disruption | Self-replicating; broad coverage with cocktails | Narrow host range per phage; resistance development |
| Engineered Phages with CRISPR-Cas | Delivery of genetic payloads (e.g., CRISPRa) | Precision targeting of genes; programmable | Complex construction; potential immune response |
| Phage-Derived Depolymerases | Enzymatic degradation of EPS matrix | Enhanced penetration of co-administered agents | Does not directly kill bacteria |
| Phage-Antibiotic Synergy (PAS) | Combined mechanical disruption and chemical killing | Reduced antibiotic resistance; synergistic effect | Optimization of timing/dosing required |
Experimental Protocol: Engineering Phages for CRISPRa Delivery
Objective: To engineer a lysogenic phage for the delivery of a CRISPRa system targeting the bdlA biofilm dispersal gene in P. aeruginosa.
Materials:
- Bacterial strains: P. aeruginosa PAO1 (host), susceptible phage (e.g., Pf4).
- Molecular cloning reagents: CRISPRa plasmid (dCas9-activator, guide RNA), homologous recombination system.
- Cell culture equipment: Shaking incubator, electroporator.
- Plaque assay materials: Soft agar, agar plates.
- Validation tools: PCR, Western blot, CLSM.
Procedure:
- CRISPRa Cassette Construction:
- Clone a guide RNA sequence specific to the promoter region of the bdlA gene into a plasmid containing the dCas9-activator (e.g., dCas9-VPR) system.
- Flank this cassette with homology arms matching a non-essential region of the phage genome.
- Phage Engineering:
- Introduce the CRISPRa construct into the host P. aeruginosa via electroporation.
- Infect the transformed bacteria with the wild-type Pf4 phage to allow for homologous recombination between the plasmid and the phage genome.
- Harvest the progeny phage lysate.
- Screening and Purification:
- Perform plaque assays. Pick individual plaques and screen for the integrated CRISPRa cassette using PCR.
- Amplify a positive, engineered phage clone to create a high-titer stock.
- Functional Validation:
- In vitro: Infect mature P. aeruginosa biofilms with the engineered phage. Use RT-qPCR to confirm bdlA upregulation and CLSM to quantify biofilm dissolution over 24-48 hours.
- In vivo (optional): Utilize a relevant animal model (e.g., mouse catheter infection model) to assess biofilm eradication and host response.
Diagram 2: Process for engineering a bacteriophage to deliver a CRISPRa system.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Developing Biofilm Penetration Platforms
| Reagent / Material | Function | Example Use Case |
|---|---|---|
| Cationic Liposomes (e.g., DOTAP) | Formulate NP carriers with enhanced biofilm adhesion via electrostatic interaction. | Co-encapsulate dCas9-VPR and sgRNA for CRISPRa delivery [16] [40]. |
| Gold Nanoparticles (AuNPs) | Serve as a biocompatible, photothermally-activatable delivery platform. | Conjugate with CRISPR ribonucleoproteins (RNPs); release payload via NIR laser [16]. |
| Engineered Lysogenic Phage (e.g., Pf4) | Act as a natural vector for precise gene delivery into target bacteria. | Genomic integration of CRISPRa machinery for sustained dispersal gene activation [37] [39]. |
| DNase I & Dispersin B | Degrade extracellular DNA (eDNA) and polysaccharides in EPS. | Pre-treatment to weaken biofilm matrix, enhancing NP/phage penetration [37]. |
| SYTO 9 / Propidium Iodide | Differentiate between live and dead bacterial cells via fluorescence. | Quantify bactericidal efficacy of treatments using CLSM and image analysis [38]. |
| dCas9-VPR Transcriptional Activator | Enable CRISPRa (targeted gene upregulation without DNA cleavage). | Activate expression of biofilm dispersal genes (e.g., bdlA, dipA) [7] [43]. |
Nanoparticle and bacteriophage delivery platforms offer distinct and complementary pathways to overcome the fundamental challenge of biofilm penetration. Nanoparticles provide a tunable, multi-functional platform for controlled therapeutic release, while bacteriophages offer biological precision and self-amplification capabilities. The integration of these platforms with emerging CRISPRa technologies represents a frontier in anti-biofilm research, enabling the precise activation of endogenous dispersal pathways within bacterial communities. Future work must focus on optimizing the safety, specificity, and in vivo efficacy of these combined platforms to translate them from powerful research tools into viable clinical therapeutics.
The ESKAPE pathogens—Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species—represent a critical group of multidrug-resistant bacteria responsible for the majority of hospital-acquired infections globally [44]. Among these, P. aeruginosa stands out for its remarkable ability to form biofilms and intrinsic resistance to multiple antibiotic classes, making it a prime target for novel therapeutic approaches [45] [44]. The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) systems, originally identified as an adaptive immune system in prokaryotes, have emerged as powerful tools with dual applications: as precision antimicrobials and for functional genetic studies of bacterial pathogenesis [46] [47]. While the broader thesis context focuses on CRISPR activation (CRISPRa) for activating biofilm dispersal genes, this review comprehensively examines the current in vitro applications of various CRISPR-Cas technologies, including CRISPR interference (CRISPRi) and nuclease-active systems, against P. aeruginosa and other ESKAPE pathogens.
CRISPR-Cas Mechanisms and Antimicrobial Strategies
CRISPR-Cas System Fundamentals
CRISPR-Cas systems function through three fundamental stages: adaptation, crRNA maturation, and interference [46] [47]. During adaptation, Cas proteins incorporate short sequences from invading genetic elements (spacers) into the CRISPR array. During maturation, these spacers are transcribed and processed into short CRISPR RNAs (crRNAs). Finally, during interference, Cas protein-RNA complexes recognize and cleave complementary foreign nucleic acids, providing sequence-specific immunity [46]. The system's specificity is determined by RNA-DNA interactions, making it highly adaptable for targeting specific bacterial genes [47].
Class II CRISPR systems (types II, V, and VI) utilize single effector proteins (Cas9, Cas12, and Cas13, respectively) and are most commonly employed in genetic engineering applications [46]. Critical to this targeting is the protospacer adjacent motif (PAM), a short sequence adjacent to the target DNA that is essential for recognition and cleavage [46] [47].
Antimicrobial Mechanisms of Action
CRISPR-Cas systems can be deployed against bacterial pathogens through several strategic approaches:
- Bacterial Elimination: Targeting essential or species-specific genes to induce lethal double-strand breaks, selectively eliminating pathogens while preserving the commensal microbiota [46] [47].
- Resensitization: Disrupting antibiotic resistance genes (e.g., β-lactamases, efflux pumps) to restore antibiotic efficacy without killing the bacteria [46] [48] [47].
- Virulence Attenuation: Targeting virulence factors or biofilm-associated genes to reduce pathogenicity [49] [6].
These approaches utilize different Cas protein variants, including nuclease-active Cas9, catalytically dead Cas9 (dCas9) for gene repression in CRISPRi, and base editors for point mutations [46] [47].
The following diagram illustrates the core mechanisms of different CRISPR-Cas systems used in antibacterial applications:
Case Study: CRISPRi-Mediated Gene Suppression in Pseudomonas aeruginosa
A 2022 study demonstrated the application of CRISPR interference (CRISPRi) to investigate an essential gene, PA0715, in P. aeruginosa PAO1 [49]. This gene, encoding a protein with a reverse transcriptase domain, was identified as a potential global metabolic regulator essential for bacterial survival and virulence [49].
Detailed Experimental Methodology:
CRISPRi System Construction:
- The shuttle vector pHERD20T containing an arabinose-inducible promoter was used to construct the CRISPRi system.
- The dcas9 gene was amplified using primers with restriction sites and cloned into pHERD20T to create pHERD20T-dCas9.
- sgRNA expression cassettes were designed with a constitutive promoter J23119, target-specific spacer sequences, and a terminator sequence.
- The final recombinant plasmid (pHERD20T-dCas9-PA0715 sgRNA) was transformed into P. aeruginosa PAO1 via electrotransformation [49].
Bacterial Strain and Culture Conditions:
- P. aeruginosa PAO1 wild-type and transformed strains were cultured in LB medium at 37°C under aerobic conditions.
- Gene suppression was induced with 0.02% arabinose, while control cultures received 1% glucose to repress dCas9 expression [49].
Phenotypic Assays:
- Growth curves were monitored to assess bacterial viability.
- Motility and chemotaxis were evaluated through swarming assays.
- Biofilm formation was quantified using crystal violet staining.
- Pyocyanin production was measured spectrophotometrically.
- Antibiotic susceptibility testing was performed using standard methods.
- Bacterial morphology was examined using electron microscopy [49].
In Vivo Pathogenicity Assessment:
- Galleria mellonella larvae infection model was used to evaluate bacterial pathogenicity post-gene suppression [49].
Transcriptomic Analysis:
- RNA sequencing was performed to identify differentially expressed genes following PA0715 suppression.
- Pathway enrichment analysis revealed affected metabolic and regulatory networks [49].
Key Findings and Quantitative Results
The study demonstrated that PA0715 downregulation significantly impaired multiple virulence-associated phenotypes in P. aeruginosa [49]. The table below summarizes the quantitative findings:
Table 1: Phenotypic Changes in P. aeruginosa PAO1 Following PA0715 Suppression via CRISPRi
| Phenotypic Parameter | Effect of PA0715 Suppression | Quantitative Measurement |
|---|---|---|
| Growth Rate | Significant reduction | Decreased growth curve trajectory |
| Motility and Chemotaxis | Impaired | Notable reduction in swarming diameter |
| Biofilm Formation | Reduced | Decreased crystal violet staining |
| Pyocyanin Production | Diminished | Reduced spectrophotometric absorbance |
| Antibiotic Resistance | Altered | Increased sensitivity to tested antibiotics |
| Pathogenicity in G. mellonella | Reduced | Improved larval survival rates |
Transcriptomic profiling identified 1,757 genes whose expression was altered following PA0715 suppression, including genes involved in amino acid metabolism, carbohydrate metabolism, and oxidative phosphorylation [49]. This comprehensive analysis established PA0715 as a global regulator with essential functions in P. aeruginosa physiology and pathogenicity.
Comparative Analysis Across ESKAPE Pathogens
Research has demonstrated the application of CRISPR-Cas systems across various ESKAPE pathogens, with different Cas proteins and delivery systems employed based on the target bacterium and research goals. The following table summarizes key applications and outcomes:
Table 2: CRISPR-Cas Applications Across ESKAPE Pathogens
| Pathogen | CRISPR System | Target Gene/Element | Key Findings | Delivery Method |
|---|---|---|---|---|
| P. aeruginosa | CRISPRi (dCas9) | PA0715 (reverse transcriptase) | Impaired growth, motility, biofilm formation, antibiotic resistance, and pathogenicity [49] | Electrotransformation of plasmid vector |
| P. aeruginosa | CRISPR-Cas9 | Chromosomal genes | Reduction of viable bacterial counts by 100,000-fold; cell filamentation [46] [47] | Expression vectors coding for specific crRNAs |
| K. pneumoniae | Native CRISPR-Cas3 | Resistance plasmids | ~100% elimination of resistance plasmids in vivo, reversing drug resistance [48] | Natural system activation |
| E. coli | CRISPR-Cas9 | mcr-1 and tet(X4) resistance genes | Resensitization to colistin and tigecycline; reduction of resistant bacteria to <1% [48] | Conjugative plasmids |
| Enterococcus faecalis | CRISPR-Cas9 | Self-genes | Impaired bacterial growth when targeting chromosomal genes [46] [47] | Modified native CRISPR machinery |
| Multiple ESKAPE pathogens | Cas12a/Cas13 | Pathogen detection | Development of DETECTR platform for specific pathogen identification [45] | In vitro diagnostic systems |
Delivery Systems for CRISPR-Cas Components
Nanoparticle-Mediated Delivery
The efficient delivery of CRISPR-Cas components into bacterial cells remains a significant challenge. Nanoparticles have emerged as promising vehicles due to their ability to protect genetic material and enhance cellular uptake:
- Lipid-Based Nanoparticles: Liposomal Cas9 formulations have demonstrated >90% reduction in P. aeruginosa biofilm biomass in vitro [16].
- Metallic Nanoparticles: Gold nanoparticle-CRISPR conjugates showed 3.5-fold increased editing efficiency compared to non-carrier systems [16].
- Polymeric Nanoparticles: Can be engineered with surface modifications to enhance biofilm penetration and targeted delivery [16].
These nanocarriers can facilitate co-delivery of CRISPR components with antibiotics or antimicrobial peptides, creating synergistic antibacterial effects [16].
Bacteriophage-Mediated Delivery
Engineered bacteriophages represent another promising delivery platform for CRISPR-Cas systems:
- Lytic Phages: Provide targeted delivery through natural bacterial infection mechanisms, resulting in bacterial lysis and release of CRISPR components [48].
- Temperate Phages: Can integrate CRISPR constructs into the bacterial genome for sustained expression [48].
- Phagemids: Hybrid vectors combining phage and plasmid elements for efficient delivery without integration [48].
The following diagram illustrates the workflow for a typical CRISPR-Cas experiment against biofilms, from design to assessment:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for CRISPR-Based Bacterial Studies
| Reagent/Material | Function | Examples/Specifications |
|---|---|---|
| CRISPR Plasmid Systems | Delivery of Cas proteins and guide RNAs | pHERD20T (shuttle vector), dCas9 plasmids, sgRNA expression constructs [49] |
| Inducible Promoter Systems | Controlled expression of CRISPR components | Arabinose-inducible PBAD promoter, anhydrotetracycline-inducible PtetA systems [49] [6] |
| Electrotransformation Equipment | Introduction of CRISPR constructs into bacterial cells | Bio-Rad electroporators with 2.5 kV, 5 ms parameters for Pseudomonas [49] |
| Lipid-Based Nanoparticles | Enhanced delivery of CRISPR components | Liposomal Cas9 formulations for biofilm penetration [16] |
| Metallic Nanoparticles | Improved editing efficiency and stability | Gold nanoparticle-CRISPR conjugates [16] |
| Phenotypic Assay Kits | Assessment of biofilm and virulence phenotypes | Crystal violet for biofilm biomass, specific substrates for pyocyanin quantification [49] |
| Transcriptomic Analysis Tools | Global gene expression profiling | RNA sequencing platforms, pathway analysis software [49] |
| Confocal Microscopy | High-resolution biofilm architecture imaging | CLSM for 3D biofilm structure analysis [6] |
The in vitro applications of CRISPR-Cas technologies against P. aeruginosa and other ESKAPE pathogens demonstrate remarkable potential both as investigative tools for understanding bacterial pathogenesis and as novel antimicrobial strategies. The case study on PA0715 suppression in P. aeruginosa illustrates how CRISPRi enables precise functional genomics studies of essential genes that would be inaccessible through traditional knockout approaches [49]. The ability to systematically target resistance genes, virulence factors, and biofilm-associated genes provides researchers with powerful capabilities to dissect molecular mechanisms of pathogenicity.
While significant challenges remain in delivery efficiency, specificity, and translation to clinical applications, the ongoing development of nanoparticle and bacteriophage-based delivery systems continues to advance the field [16] [48]. Future research directions should focus on optimizing these delivery platforms, exploring combinatorial approaches with conventional antibiotics, and developing more sophisticated control systems for temporal and spatial precision in gene editing. As these technologies mature, they hold substantial promise for addressing the critical threat of antimicrobial resistance, particularly for challenging pathogens like P. aeruginosa where biofilm formation and intrinsic resistance mechanisms limit conventional treatment options.
Navigating Delivery Hurdles and Ensuring Specificity in Complex Biofilms
Bacterial biofilms represent a predominant mode of microbial life, characterized by surface-associated communities embedded in a self-produced matrix of extracellular polymeric substances (EPS) [50]. This EPS matrix constitutes over 90% of the biofilm's dry mass, creating a formidable barrier that significantly limits antibiotic penetration and confers enhanced tolerance to antimicrobial treatments [50] [51]. The matrix components interact synergistically to form a complex, three-dimensional architecture that provides structural stability, facilitates nutrient retention, and protects embedded bacterial cells from host immune responses and therapeutic agents [50] [52]. Within the context of innovative antibacterial strategies, Clustered Regularly Interspaced Short Palindromic Repeats activation (CRISPRa) technology emerges as a promising approach for precision manipulation of biofilm dispersal mechanisms. This technical guide comprehensively outlines the composition-based resistance mechanisms of the EPS matrix and details advanced strategies, including CRISPRa-based interventions, designed to overcome this protective barrier for enhanced therapeutic efficacy.
Structural and Functional Architecture of the EPS Matrix
The EPS matrix is not merely a physical barrier but a dynamically functional ecosystem that confers resilience to bacterial communities. Its composition varies significantly between bacterial species and environmental conditions, but typically includes four primary components:
Exopolysaccharides: These heteropolymers form the structural scaffold of the biofilm, creating a hydrated polymer network that confers mechanical stability [51]. Their polyionic nature enables interaction with other matrix components and can sequester antimicrobial agents through charge-based interactions [51].
Extracellular DNA (eDNA): Derived from genomic DNA through controlled cell lysis, eDNA forms grid-like structures that provide structural integrity and facilitate horizontal gene transfer, including the dissemination of antibiotic resistance genes [50] [51]. In Pseudomonas aeruginosa biofilms, eDNA release is regulated by quorum sensing and iron regulation systems [50].
Proteins: The matrix contains both structural proteins that reinforce the biofilm architecture and enzymes that participate in matrix remodeling through degradation of EPS components [51]. These enzymes enable dynamic reorganization of the matrix in response to environmental cues.
Lipids and Biosurfactants: These components contribute to surface attachment, water retention, and intercellular communication within the biofilm microenvironment [51].
The EPS matrix has been conceptually categorized into three functional classes: architectural EPS (structural and signaling roles), protective EPS (defense against host immune responses and antimicrobials), and aggregative EPS (mediation of adhesion and biofilm formation) [50]. This classification provides a framework for developing targeted disruption strategies based on functional specificity.
Table 1: Primary Components of the EPS Matrix and Their Functional Roles
| Matrix Component | Primary Functions | Representative Examples |
|---|---|---|
| Exopolysaccharides | Structural scaffolding, water retention, charge-based antimicrobial sequestration | Alginate, cellulose, Pel, Psl |
| Extracellular DNA (eDNA) | Structural integrity, genetic information exchange, cation chelation | Genomic DNA fragments |
| Proteins | Structural reinforcement, enzymatic matrix remodeling, nutrient acquisition | Amyloids, proteases, dispersin B |
| Lipids & Biosurfactants | Hydrophobicity modulation, surface attachment, cellular communication | Rhamnolipids, surfactin |
Mechanisms of Antimicrobial Failure in Biofilms
The multifunctional nature of the EPS matrix contributes to antibiotic failure through several interconnected mechanisms that remain challenging to overcome in clinical settings:
Physical Barrier and Limited Diffusion
The dense, anionic polymer network of the EPS matrix significantly retards antibiotic diffusion, creating concentration gradients that result in sublethal antibiotic exposure in deeper biofilm regions [51] [52]. This limited penetration is exacerbated by binding interactions between antimicrobial agents and specific matrix components, effectively reducing the bioavailable concentration reaching bacterial cells [50].
Chemical Interactions and Inactivation
Antibiotics can undergo direct chemical inactivation through interactions with EPS components, including enzymatic degradation, chelation by eDNA, or complex formation with exopolysaccharides [51]. The matrix also creates heterogeneous microenvironments with varying pH and oxygen gradients that can negatively impact antibiotic activity [51].
Physiological Heterogeneity
Biofilms encompass metabolically diverse bacterial subpopulations, including dormant persister cells with significantly reduced metabolic activity in deeper biofilm regions [51]. These metabolically inactive cells exhibit enhanced tolerance to conventional antibiotics that primarily target active cellular processes [50] [51].
Strategic Approaches for Enhanced EPS Penetration
Matrix-Targeting Enzymatic Disruption
Enzymatic degradation of specific EPS components represents a precision approach to matrix disruption:
DNases: Degrade eDNA networks, particularly effective against P. aeruginosa biofilms where eDNA is a primary structural component [51]. Clinical applications require optimized delivery to overcome stability issues within the biofilm environment.
Dispersin B: Glycosidase hydrolase that degrades poly-N-acetylglucosamine (PNAG) polysaccharide, a key matrix component in staphylococcal and Escherichia coli biofilms [51].
Proteases: Target structural protein components and can disrupt protein-polysaccharide interactions critical for matrix integrity [51].
Table 2: Matrix-Degrading Enzymes and Their Specific Targets
| Enzyme Class | Specific Substrate | Target Biofilms | Efficacy Notes |
|---|---|---|---|
| DNase I | Extracellular DNA | P. aeruginosa, S. aureus | Reduces structural integrity, enhances antibiotic penetration |
| Dispersin B | Poly-N-acetylglucosamine (PNAG) | Staphylococci, E. coli | Detaches biofilms from surfaces, synergizes with antibiotics |
| Proteases | Matrix proteins | Multiple species | Disrupts protein-mediated cross-linking |
| Glycoside Hydrolases | Specific exopolysaccharides | Species-specific | High specificity but limited by polysaccharide diversity |
Nanoparticle-Based Penetration Platforms
Engineered nanoparticles (NPs) provide versatile platforms for enhanced biofilm penetration through multiple mechanisms:
Size-Dependent Passive Diffusion: Metal oxide nanoparticles (TiO₂, ZnO, CuO, Fe₃O₄) with precise size control can exploit matrix porosity for enhanced penetration compared to conventional antibiotics [50].
Functionalized Surface Carriers: Nanoparticles can be engineered as drug carriers with surface modifications that minimize matrix interactions, facilitating antibiotic transport to deeper biofilm regions [50] [16]. Liposomal and gold nanoparticle formulations have demonstrated particularly efficient penetration capabilities [16].
Intrinsic Antimicrobial Activity: Certain metal nanoparticles exhibit direct antimicrobial and anti-biofilm properties through generation of reactive oxygen species or metal ion release [50].
CRISPRa for Targeted Activation of Biofilm Dispersal Genes
CRISPR activation (CRISPRa) technology utilizes catalytically dead Cas9 (dCas9) fused to transcriptional activators to precisely upregulate endogenous biofilm dispersal mechanisms. This approach represents a paradigm shift from external matrix disruption to programmed biofilm auto-dispersal:
Mechanism of Action: The dCas9-activator complex is guided to specific promoter regions of dispersal genes by engineered sgRNAs, leading to targeted transcriptional enhancement without permanent genetic alterations [7] [6].
Key Targets for Activation:
- Matrix Degradation Enzymes: Activation of genes encoding endogenous proteases, glycosidases, and DNases that naturally facilitate biofilm dispersal [7].
- Quorum Sensing Systems: Precise upregulation of dispersal pathways controlled by bacterial communication systems [53] [7].
- c-di-GMP Metabolism: Targeted modulation of cyclic di-GMP signaling networks that control the transition between sessile and motile bacterial lifestyles [6].
Synergistic Combination Strategies
No single approach has demonstrated complete efficacy against mature biofilms, leading to increased investigation of combination therapies:
Enzyme-Nanoparticle Conjugates: Matrix-degrading enzymes conjugated to nanoparticles for enhanced stability and targeted delivery [51] [52].
CRISPRa-Antibiotic Combinations: CRISPRa-mediated sensitization followed by conventional antibiotic treatment [16] [7].
Quorum Sensing Inhibition with Matrix Disruption: Simultaneous interference with bacterial communication and physical matrix degradation [50] [53].
Experimental Models and Assessment Methodologies
Advanced Biofilm Quantification Platforms
Comprehensive evaluation of EPS penetration strategies requires multimodal assessment:
BiofilmQ Image Cytometry: A comprehensive software platform for automated 3D quantification of biofilm architecture and internal properties, enabling spatial resolution of penetration efficacy [54]. The system can analyze hundreds of parameters including biofilm volume, surface area, roughness coefficient, and fluorescence distribution of labeled antimicrobials within the EPS matrix [54].
Confocal Laser Scanning Microscopy (CLSM) with Fluorescent Probes: Enables real-time visualization of penetration kinetics using fluorescence-labeled antibiotics or nanoparticles in conjunction with matrix staining [55] [54].
Crystal Violet Assay: High-throughput quantification of total biofilm biomass, suitable for initial screening of anti-biofilm agents [53] [55].
Colony Forming Unit (CFU) Enumeration: Determination of viable bacterial counts following treatment, providing direct assessment of bactericidal efficacy [55].
Table 3: Standardized Experimental Protocols for Penetration Assessment
| Method | Key Measured Parameters | Protocol Considerations | Applications |
|---|---|---|---|
| BiofilmQ Analysis | 3D architecture, biovolume, fluorescence distribution, surface roughness | Requires fluorescence labeling, specialized software | High-resolution spatial mapping of penetration |
| CLSM Imaging | Penetration depth, distribution homogeneity, matrix-colocalization | Optimal dye selection critical for signal specificity | Real-time penetration kinetics |
| Microtiter Plate Assays | Total biomass, metabolic activity | High-throughput capability, limited spatial information | Initial screening of anti-biofilm agents |
| Quartz Crystal Microbalance | Real-time biomass accumulation | Sensitive to environmental vibrations | Monitoring early-stage biofilm formation |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for EPS Penetration Studies
| Reagent/Category | Specific Examples | Experimental Function |
|---|---|---|
| Matrix-Degrading Enzymes | DNase I, Dispersin B, Proteases | Selective EPS component degradation for mechanistic studies |
| Engineered Nanoparticles | Gold NPs, Liposomal carriers, Metal oxide NPs | Penetration enhancement and drug delivery evaluation |
| CRISPRa System Components | dCas9-activator plasmids, sgRNA constructs | Targeted genetic activation for dispersal mechanism studies |
| Fluorescent Reporters | SYTO dyes, FITC-conjugated dextrans, mNeonGreen | Visualization and quantification of penetration and gene expression |
| Biofilm Staining Reagents | Crystal violet, Congo red, Calcofluor white | Total biomass assessment and matrix polysaccharide staining |
Technical Protocols: Key Methodological Approaches
CRISPRi/a Implementation for Biofilm Gene Regulation
The following protocol adapts established CRISPR interference/activation methodologies for biofilm dispersal studies [53] [6]:
Stage 1: System Design and Construction
- Select target genes involved in biofilm regulation (e.g., luxS for quorum sensing, alg44 for alginate synthesis, c-di-GMP metabolic genes) [53] [6].
- Design sgRNAs complementary to target promoter regions with appropriate PAM (Protospacer Adjacent Motif) sequences (typically 5'-NGG-3' for S. pyogenes Cas9) [53].
- Clone sgRNA expression cassettes into appropriate vectors under constitutive promoters.
- For CRISPRa, utilize dCas9 fused to transcriptional activation domains (e.g., VP64-p65-Rta) [7].
Stage 2: Delivery and Expression Optimization
- Transform bacterial strains with dCas9 and sgRNA plasmids using electroporation or conjugation [53] [6].
- Induce dCas9 expression with anhydrotetracycline (aTc; typically 2μM) during early exponential phase for controlled timing [53] [6].
- Validate target gene modulation using qRT-PCR and assess knockdown/activation efficiency [53].
Stage 3: Biofilm Phenotypic Assessment
- Cultivate biofilms under conditions relevant to the specific bacterial species and research questions [55].
- Quantify biofilm biomass and architecture using crystal violet staining and confocal microscopy pre- and post-CRISPRa induction [53] [55].
- Evaluate dispersal effects through CFU enumeration and visualization of detached cells [55].
Nanoparticle Penetration Assessment Protocol
Synthesis and Functionalization:
- Prepare metal oxide nanoparticles (e.g., ZnO, TiO₂) using controlled precipitation methods to achieve optimal size (typically 20-100nm) for enhanced matrix penetration [50].
- Conjugate fluorescent tags (e.g., FITC, Cy5) for visualization and tracking.
- For targeted delivery, functionalize nanoparticle surfaces with matrix-binding ligands or enzymes [50] [16].
Penetration Kinetics Analysis:
- Establish mature biofilms (typically 48-72 hours) in flow cell systems or on relevant substrates [55] [54].
- Apply fluorescently labeled nanoparticles at clinically relevant concentrations.
- Monitor penetration depth and distribution patterns over time (0-24 hours) using confocal microscopy and quantify with BiofilmQ software [54].
- Correlate penetration efficacy with antibacterial activity through simultaneous CFU enumeration.
The persistent challenge of biofilm-mediated antimicrobial resistance necessitates innovative strategies that target the fundamental barrier properties of the EPS matrix. While enzymatic disruption and nanoparticle-mediated penetration provide immediate avenues for enhanced drug delivery, emerging technologies like CRISPRa represent a paradigm shift toward precision manipulation of fundamental biofilm processes. The integration of these approaches—combining physical matrix disruption with biological dispersal activation—holds particular promise for overcoming the multifactorial resistance mechanisms of biofilms. Future directions will likely focus on personalized therapeutic approaches informed by species-specific matrix composition analysis, development of smart delivery systems responsive to biofilm microenvironments, and integration of artificial intelligence for predictive modeling of optimal combination therapies. As characterization methodologies continue to advance, particularly in the realm of single-cell analysis within biofilm architectures, our understanding of EPS heterogeneity will enable increasingly sophisticated penetration strategies to overcome this formidable barrier to effective antimicrobial therapy.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system has revolutionized genetic research and therapeutic development by enabling precise genome modifications. This RNA-guided gene-editing tool functions through a complex of the Cas9 nuclease and a single guide RNA (sgRNA), which directs Cas9 to create double-strand breaks at specific genomic locations adjacent to a Protospacer Adjacent Motif (PAM) [56] [57]. However, a significant challenge impeding its clinical translation is the occurrence of off-target effects—unintended genetic alterations at sites other than the intended target [56] [58]. These off-target mutations primarily happen when the Cas9/sgRNA complex tolerates mismatches, bulges, or imperfect base pairing between the sgRNA and genomic DNA, potentially leading to adverse functional consequences [56].
In the context of CRISPRa (CRISPR activation) research for activating biofilm dispersal genes, controlling off-target effects is particularly critical. Unintended activation of unrelated genes could disrupt complex regulatory networks, such as quorum sensing and cyclic di-GMP signaling, which are essential for controlled biofilm dispersal [16] [6]. This guide provides a comprehensive technical framework for minimizing these risks through sophisticated gRNA design and computational prediction strategies, specifically tailored for research and drug development professionals working on biofilm-targeted therapies.
Mechanisms and Implications of Off-Target Effects
Molecular Mechanisms
Off-target effects in CRISPR/Cas9 systems arise through two primary mechanisms: sgRNA-dependent and sgRNA-independent editing [56]. sgRNA-dependent off-target activity occurs when the Cas9/sgRNA complex binds and cleaves genomic sites with significant sequence similarity to the intended target. The Cas9 enzyme can tolerate up to three mismatches between the sgRNA and genomic DNA, particularly if these mismatches are located distal to the PAM sequence [56]. Furthermore, the specific positions and types of nucleotide mismatches influence off-target potential, with some combinations exhibiting minimal impact on cleavage efficiency [56].
sgRNA-independent off-target effects present a more challenging detection problem, as they result from transient, non-specific interactions between Cas9 and DNA, or from cellular perturbations induced by the editing process itself [56]. Recent studies have also revealed that chromatin accessibility, epigenetic modifications, and nuclear organization significantly influence off-target susceptibility, creating genomic regions that are inherently more vulnerable to spurious editing events [56].
Implications for Biofilm Research
In CRISPRa applications for biofilm dispersal, off-target effects pose unique risks that can compromise experimental validity and therapeutic safety. Biofilm formation and dispersal are governed by sophisticated regulatory networks, including two-component systems (e.g., GacA/S), quorum sensing pathways, and cyclic di-GMP (c-di-GMP) signaling cascades [16] [6]. Off-target activation could inadvertently modulate these pathways, leading to:
- Unpredictable phenotypic outcomes: Altered expression of diguanylate cyclases (DGCs) or phosphodiesterases (PDEs) that regulate intracellular c-di-GMP levels, potentially reinforcing biofilm stability instead of promoting dispersal [6].
- Persister cell formation: Unintended manipulation of stress response regulators could enhance bacterial tolerance to subsequent antibiotic treatments [16].
- Horizontal gene transfer: Off-target effects on competence genes could facilitate the spread of antibiotic resistance determinants within microbial communities [16].
The table below summarizes key biofilm-related genes and their vulnerability to off-target effects:
Table 1: Biofilm-Related Genes Vulnerable to Off-Target Effects
| Gene Category | Representative Genes | Function in Biofilm Regulation | Consequences of Off-Target Modulation |
|---|---|---|---|
| Two-Component Systems | GacA, GacS | Environmental sensing and response coordination | Disrupted coordination of motility-to-sessile transition [6] |
| c-di-GMP Metabolic Enzymes | GcbA, BifA, DipA | Synthesis and degradation of c-di-GMP | Altered EPS production and biofilm architecture [6] |
| Matrix Biosynthesis | Alg44, Cellulose synthases | Production of extracellular polymeric substances | Structural integrity compromise or excessive matrix production [16] [6] |
| Quorum Sensing | LasI, LasR, RhlI, RhlR | Cell-to-cell communication and collective behavior | Premature or delayed biofilm maturation [16] |
Computational Tools for gRNA Design and Off-Target Prediction
In Silico Prediction Platforms
Bioinformatics tools are indispensable for predicting potential off-target sites during gRNA design. These platforms employ various algorithms to scan entire genomes for sequences with homology to the designed sgRNA, prioritizing sites based on their potential for off-target cleavage [56] [57]. The prediction software can be broadly categorized into alignment-based and scoring-based models, each with distinct methodological approaches and output formats.
Table 2: Major Computational Tools for gRNA Design and Off-Target Prediction
| Tool Name | Algorithm Type | Key Features | Advantages | Limitations |
|---|---|---|---|---|
| Cas-OFFinder [56] | Alignment-based | Adjustable sgRNA length, PAM types, mismatch/bulge tolerance | Wide application tolerance; comprehensive search parameters | Biased toward sgRNA-dependent off-target effects [56] |
| FlashFry [56] | Alignment-based | High-throughput analysis; provides GC content and on/off-target scores | Rapid processing of large sgRNA sets | Requires experimental validation [56] |
| CCTop [56] | Scoring-based | Predicts off-target sites based on mismatch distances to PAM | User-friendly interface with ranked predictions | Limited consideration of epigenetic factors [56] |
| DeepCRISPR [56] [57] | Scoring-based | Incorporates both sequence and epigenetic features using machine learning | Enhanced prediction accuracy through deep learning | Complex implementation for non-bioinformaticians [56] [57] |
| CRISPOR [57] [59] | Hybrid | Integrates multiple scoring algorithms; visualizes genomic context | Comprehensive functionality with intuitive visualization | May overwhelm users with extensive data output [59] |
| CHOPCHOP [57] [59] | Scoring-based | User-friendly web interface; optimized for various species | Accessibility for researchers without computational expertise | Limited advanced customization options [57] [59] |
Selection Criteria for Bioinformatics Tools
When selecting appropriate computational tools for CRISPRa biofilm research, consider the following criteria:
- Specificity scoring: Tools should provide comprehensive off-target propensity scores, such as the MIT specificity score or Cutting Frequency Determination (CFD) score, which quantify the likelihood of off-target cleavage [56].
- Genome coverage: Ensure the tool supports the relevant bacterial genomes, as many platforms are optimized for human or model organism applications [57] [59].
- Epigenetic consideration: Advanced tools like DeepCRISPR that incorporate chromatin accessibility data provide more accurate predictions for eukaryotic systems, though this may be less relevant for bacterial targets [56].
- Usability: Researchers should balance computational sophistication with practical usability, selecting tools that integrate smoothly into existing experimental workflows [57].
For comprehensive coverage, employ multiple complementary tools with different algorithmic approaches to maximize the detection of potential off-target sites [56] [57].
Experimental Protocols for Off-Target Assessment
Cell-Free Validation Methods
Following computational prediction, experimental validation is essential to identify bona fide off-target sites. Cell-free methods utilize purified genomic DNA or chromatin to detect Cas9 cleavage events in controlled environments, offering high sensitivity without confounding cellular processes [56].
Digenome-seq Protocol:
- Genomic DNA Extraction: Purify high-molecular-weight genomic DNA from target cells or bacterial strains of interest.
- In Vitro Cleavage: Incubate purified DNA (1-2 μg) with preassembled Cas9/sgRNA ribonucleoprotein (RNP) complex in appropriate reaction buffer for 4-6 hours at 37°C.
- DNA Processing: Purify the cleaved DNA and subject it to whole-genome sequencing (WGS) at high coverage (minimum 50-100×).
- Bioinformatic Analysis: Map sequencing reads to the reference genome and identify cleavage sites by detecting sequence breaks with 5' overhangs characteristic of Cas9 cleavage [56].
CIRCLE-seq Protocol:
- DNA Circularization: Shear genomic DNA to appropriate fragment sizes (500-1000 bp) and circularize using DNA ligase.
- Cas9 Treatment: Incubate circularized DNA with Cas9/sgRNA RNP complex, then linearize successfully cleaved fragments.
- Library Preparation and Sequencing: Prepare sequencing libraries from linearized fragments and perform high-throughput sequencing.
- Data Analysis: Identify off-target sites through bioinformatic detection of enriched cleavage sites compared to negative controls [56].
Table 3: Comparison of Major Off-Target Detection Methods
| Method | Sensitivity | Advantages | Limitations | Suitable for Biofilm Research |
|---|---|---|---|---|
| Digenome-seq [56] | High | Highly sensitive; comprehensive genome coverage | Expensive; requires high sequencing coverage; uses purified DNA without chromatin context | Moderate (bacterial systems have less chromatin organization) |
| CIRCLE-seq [56] | Very High | Ultra-sensitive detection; low background noise | Does not account for cellular repair mechanisms | High for initial screening |
| GUIDE-seq [56] | High | Highly sensitive; low false positive rate; works in living cells | Limited by transfection efficiency in some bacterial strains | Moderate (challenging in non-transformable isolates) |
| SITE-seq [56] | Moderate | Minimal read depth; no reference genome required | Lower sensitivity compared to other methods | Suitable for rapid screening |
| DISCOVER-seq [56] | High | Utilizes DNA repair proteins as natural markers; works in vivo | May have false positives; requires specific antibodies | Lower for bacterial systems |
Cell Culture-Based Validation
For CRISPRa applications in biofilm models, cell culture-based methods provide the most relevant assessment of off-target effects in physiological conditions.
GUIDE-seq (Genome-wide Unbiased Identification of DSBs Enabled by Sequencing) Protocol:
- Transfection: Deliver sgRNA and Cas9 along with double-stranded oligodeoxynucleotides (dsODNs) into bacterial or eukaryotic cells using appropriate transformation methods.
- Integration: Allow dsODNs to integrate into double-strand break sites during DNA repair processes (24-72 hours post-transfection).
- Genomic DNA Extraction: Harvest genomic DNA and fragment by sonication or enzymatic digestion.
- Library Preparation and Sequencing: Enrich for dsODN-integrated fragments through PCR and prepare sequencing libraries.
- Data Analysis: Identify off-target sites by mapping integration events across the genome [56].
Flow Cytometry-Based Reporter Assay for Bacterial Systems:
- Reporter Construction: Clone a fluorescent protein gene (e.g., mNeonGreen) under a constitutive promoter into the target bacterial chromosome [6].
- CRISPRa Delivery: Introduce dCas9-activator and sgRNA targeting the promoter region of the reporter gene.
- Fluorescence Measurement: Monitor fluorescence intensity over time using flow cytometry following induction of dCas9 expression.
- Data Interpretation: Compare fluorescence in experimental versus control groups to quantify off-target activation of similar promoter sequences [6].
Diagram Title: Comprehensive Workflow for Minimizing Off-Target Effects in Biofilm Research
Research Reagent Solutions for Off-Target Assessment
Table 4: Essential Research Reagents for Off-Target Analysis in CRISPRa Biofilm Studies
| Reagent/Category | Specific Examples | Function/Application | Considerations for Biofilm Research |
|---|---|---|---|
| CRISPR Delivery Systems | Plasmid vectors (pCRISPRi), Ribonucleoprotein (RNP) complexes | Introduction of CRISPR components into target cells | RNP complexes reduce off-target effects through rapid degradation [6] |
| Detection Reagents | GUIDE-seq dsODNs, Biotinylated adaptors (BLESS), Anti-MRE11 antibodies (DISCOVER-seq) | Marking double-strand breaks for subsequent identification | Bacterial cell wall modifications may be needed for efficient delivery [56] |
| Sequencing Platforms | Illumina platforms for WGS, Nanopore sequencing for structural variants | Comprehensive identification of off-target edits | Cost considerations for large-scale biofilm mutant screening [56] |
| Bioinformatics Pipelines | CasOFFinder package, CRISPResso2, MAGeCK | Analysis of sequencing data to identify and quantify off-target events | Customization needed for non-model bacterial genomes [56] [57] |
| Validation Reagents | qPCR primers for potential off-target sites, Antibodies for unintended protein expression | Confirmation of identified off-target events | Essential for verifying computational predictions in biofilm models [56] [6] |
Integrated Strategy for Biofilm Research Applications
Minimizing off-target effects in CRISPRa-mediated activation of biofilm dispersal genes requires an integrated, multi-layered approach:
Computational gRNA Design: Begin with careful gRNA selection using complementary bioinformatics tools (e.g., CRISPOR and CHOPCHOP) that optimize for both on-target efficiency and off-target minimization [57] [59]. Prioritize gRNAs with high specificity scores and minimal potential off-target sites in critical biofilm regulatory genes.
CRISPR System Selection: Consider using high-fidelity Cas9 variants (e.g., eSpCas9, SpCas9-HF1) that reduce off-target cleavage while maintaining on-target activity [56]. For biofilm research, CRISPRi (interference) systems with dCas9 can be particularly valuable for controlled gene modulation with reduced DNA damage concerns [6].
Comprehensive Off-Target Screening: Implement a tiered detection approach, starting with sensitive cell-free methods (CIRCLE-seq) followed by cell-based validation (GUIDE-seq) in relevant biofilm models [56].
Functional Validation in Biofilm Models: Ultimately, test optimized gRNAs in physiologically relevant biofilm systems, using confocal microscopy and quantitative assays to verify specific activation of dispersal genes without disruption of overall biofilm architecture [16] [6].
Diagram Title: Multi-Layered Off-Target Mitigation Strategy
This integrated framework provides researchers with a systematic approach to minimize off-target effects while developing CRISPRa therapies for biofilm control, balancing computational predictions with empirical validation in biologically relevant systems.
Optimizing Activation Kinetics and Duration for Effective Dispersal
The emergence of bacterial biofilms as a primary cause of persistent infections represents a significant challenge in clinical medicine, as biofilms can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [16]. Within the context of a broader thesis on CRISPRa (CRISPR activation) for activating biofilm dispersal genes, this technical guide addresses the critical need for precise temporal control over gene expression to achieve effective biofilm disruption. CRISPRa technology, which employs a catalytically inactive Cas9 (dCas9) fused to transcriptional activators to upregulate endogenous gene expression without altering DNA sequences, presents a transformative approach for targeting the complex genetic networks that regulate biofilm dispersal [7] [43].
The efficacy of CRISPRa-mediated biofilm dispersal is not merely a function of target gene selection but is profoundly dependent on the kinetics and duration of gene activation. Achieving therapeutically relevant outcomes requires sophisticated optimization of delivery systems, activator efficiency, and timing parameters to mirror the natural pulsatile expression patterns of dispersal genes [34]. This guide synthesizes current advancements and methodologies for optimizing these critical parameters, providing researchers and drug development professionals with a comprehensive framework for developing effective CRISPRa-based anti-biofilm strategies. We present structured quantitative data, detailed experimental protocols, and standardized visualization to facilitate the translation of this promising technology from bench to bedside.
Theoretical Foundations: Biofilm Dispersal Mechanisms and CRISPRa
Genetic Regulation of Native Biofilm Dispersal
Biofilm dispersal is a genetically programmed process critical for bacterial survival and colonization of new niches. It is tightly regulated by environmental cues and intricate signaling pathways, the understanding of which provides the foundational logic for CRISPRa intervention.
Quorum Sensing (QS) and Surfactant Production: In Pseudomonas aeruginosa, activation of the LasI/LasR and RhlI/RhlR quorum sensing systems at high cell density promotes biofilm dispersion by upregulating the synthesis of surfactant molecules like rhamnolipids. These surfactants disrupt the structural integrity of the extracellular matrix, facilitating bacterial release [34]. Exogenous addition of purified rhamnolipids to mature biofilms has been demonstrated to induce detachment, confirming their direct functional role [34].
c-di-GMP Signaling: The second messenger cyclic di-GMP serves as a central regulator of the biofilm life cycle. High intracellular c-di-GMP levels promote biofilm formation, while low levels trigger dispersal. In Vibrio cholerae, the master QS regulator HapR, activated at high cell density, controls genes encoding enzymes that degrade c-di-GMP (EAL and HD-GYP domain proteins), leading to a reduction in cellular c-di-GMP and subsequent biofilm dispersion [34].
Matrix Degradation Enzymes: The same QS system in V. cholerae activates transcription of the hap gene, encoding haemagglutinin protease (HA/P), which directly degrades protein components within the biofilm matrix and detaches cells [34].
Table 1: Key Native Genetic Pathways for CRISPRa-Targeted Biofilm Dispersal
| Target Pathway | Key Regulator/Gene | Organism | Mechanism of Action |
|---|---|---|---|
| Quorum Sensing | LasI/LasR, RhlI/RhlR | P. aeruginosa | Upregulates synthesis of matrix-disrupting rhamnolipids [34]. |
| c-di-GMP Modulation | HapR → EAL/HD-GYP proteins | V. cholerae | Reduces intracellular c-di-GMP, promoting dispersal [34]. |
| Matrix Degradation | HapR → HA protease | V. cholerae | Enzymatically degrades proteinaceous matrix components [34]. |
| Biosurfactant Production | bslA gene | B. subtilis | Produces hydrophobins that alter matrix surface properties [60]. |
CRISPRa Systems for Transcriptional Activation
Unlike nuclease-active CRISPR systems, CRISPRa uses a deactivated Cas9 (dCas9) that binds DNA but does not cut it. This dCas9 is fused to transcriptional activator domains, enabling programmable recruitment of the cellular transcription machinery to specific gene promoters.
dCas9-VPR: This is a potent synthetic activator where dCas9 is fused to a tripartite activator domain comprising VP64, p65, and Rta. The VPR system has demonstrated strong efficacy in upregulating endogenous genes, including those in bivalent chromatin states, in human cells [61]. Its effectiveness, however, can be cell-type dependent [62].
dCas9-p300: This system utilizes the catalytic core of the human p300 histone acetyltransferase, which modifies chromatin by adding acetyl groups to histone H3 lysine 27 (H3K27ac). This mark is associated with active enhancers and promoters, opening the chromatin landscape to facilitate gene transcription. Its performance can be variable, as it was found to be ineffective in stem cells in one study [61].
The selection between VPR and p300 systems is a critical first step in experimental design. VPR generally provides stronger, more direct transcriptional activation, while p300 can alter the epigenetic state for a more sustained effect, though its success is highly dependent on the pre-existing chromatin context of the target cell [61].
Optimizing CRISPRa Kinetics and Duration
The successful application of CRISPRa for biofilm dispersal hinges on the precise control of activation kinetics and duration. This involves a multi-parameter optimization of the delivery vehicle, the CRISPRa machinery, and the timing of its action.
Delivery System Optimization
Efficient delivery of CRISPRa components into bacterial cells within a biofilm is a primary bottleneck. Nanoparticles (NPs) have emerged as a superior solution, protecting genetic material and enhancing penetration through the dense extracellular polymeric substance (EPS) [16].
Table 2: Nanoparticle Delivery Systems for CRISPRa in Biofilms
| Nanoparticle Type | Key Characteristics | Reported Efficacy | Considerations |
|---|---|---|---|
| Lipid-based NPs | Biocompatible, can fuse with bacterial membranes. | >90% reduction in P. aeruginosa biofilm biomass in vitro [16]. | High encapsulation efficiency, suitable for clinical translation. |
| Gold NPs (AuNPs) | Conjugatable surface, tunable size, photothermal properties. | 3.5-fold increase in gene-editing efficiency vs. non-carrier systems [16]. | Potential for co-delivery with antibiotics or thermal activation. |
| Polymeric NPs | Controlled release kinetics, high stability. | Effective for sustained payload release (specific quantitative data not provided in search results). | Polymer biodegradability and byproduct toxicity must be assessed. |
The kinetics of activation are directly influenced by the nanoparticle's release profile. A rapid release may lead to a sharp, short-lived peak of activator, while a sustained release can prolong the duration of gene expression. Furthermore, functionalizing NP surfaces with ligands that recognize biofilm-specific markers can enhance target specificity and local concentration [7] [16].
Activator Efficiency and Timing
The choice of activator and the timing of its deployment are equally critical for mimicking natural dispersal pulses.
Activator Potency: The dCas9-VPR system has been shown to successfully activate developmentally repressed genes and those in bivalent chromatin, making it a robust choice for inducing strong, rapid transcription. In contrast, dCas9-p300's effectiveness is more constrained by the epigenetic landscape [61]. The activation level is often anti-correlated with the basal expression level of the target gene, meaning that silent or weakly expressed dispersal genes are particularly responsive to CRISPRa [61].
Temporal Control: The natural process of biofilm dispersal is often a coordinated event triggered by nutrient depletion or waste accumulation. CRISPRa systems can be designed to respond to similar cues. While not explicitly detailed in the search results, inducible promoter systems (e.g., chemically or thermally inducible) controlling the expression of the gRNA or the dCas9-activator fusion can be engineered to trigger dispersal activation at a predefined time, aligning the intervention with the most vulnerable phase of the biofilm lifecycle [7].
The duration of activation must be carefully calibrated. Prolonged, non-physiological overexpression of dispersal genes like those encoding proteases or surfactants could lead to unintended side effects, such as excessive inflammation from a sudden, massive release of bacterial components and biofilm debris into the surrounding environment. Therefore, a pulsatile activation strategy that closely mimics natural dispersal may be safer and more effective than continuous activation.
Experimental Protocols for Kinetic Analysis
To optimize and validate CRISPRa-mediated dispersal, researchers require robust, quantitative protocols for analyzing biofilm composition and dispersal kinetics.
Time-Resolved Biofilm Composition Analysis via ssNMR
Solid-state Nuclear Magnetic Resonance (ssNMR) provides a non-destructive, quantitative method to track changes in the abundance and dynamics of key biofilm components over time.
Detailed Methodology [60]:
- Sample Preparation: Grow Bacillus subtilis (or target organism) in a defined medium with 13C-labeled glycerol as the sole carbon source to achieve isotopic labeling of the biofilm matrix. Harvest biofilm samples in duplicate at critical time points (e.g., days 1-5).
- ssNMR Data Collection: Pack approximately 30 mg of intact biofilm into a 3.2-mm magic-angle spinning (MAS) rotor. Conduct experiments on a high-field spectrometer (e.g., 800 MHz).
- Use Direct Polarization (DP) with a long recycle delay (15 s) for quantitative analysis of the total carbon content (total biomass).
- Use Cross Polarization (CP) to selectively detect signals from rigid, solid-like components (e.g., core polysaccharides).
- Use DP with a short recycle delay (2 s) to selectively detect highly mobile, liquid-like components (e.g., surfactants, soluble proteins).
- Data Analysis: Integrate characteristic signals in the 1D 13C spectra for key molecular classes: proteins (carbonyl carbons, ~175 ppm), carbohydrates (anomeric carbons, ~90-110 ppm), and aliphatics (biosurfactants, ~0-50 ppm). Normalize integrals to sample weight and number of scans to calculate temporal trends in biomass density and the proportion of mobile fractions for each component.
This protocol revealed, for instance, that in a dispersing B. subtilis biofilm, a steep decline in proteins precedes that of exopolysaccharides, and a sharp rise in aliphatic carbon signals (biosurfactants) occurs on day 4 [60].
Multiplexed Single-Cell CRISPRa Screening
This protocol assesses the efficiency and specificity of thousands of gRNAs in a single experiment, identifying the best performers for activating target dispersal genes.
Detailed Methodology [62]:
- gRNA Library Design: Construct a library of hundreds of gRNAs targeting promoters and candidate enhancers of key biofilm dispersal genes (e.g., bslA, hapR, rhlAB operon). Include non-targeting control (NTC) gRNAs.
- Cell Line Engineering: Generate a stable cell line expressing the dCas9-activator (e.g., dCas9-VPR). For bacterial studies, this involves creating a heterologous expression system.
- High-Multiplicity Transfection: Transfect the gRNA library along with a transposase (e.g., piggyBac) into the activator cell line at a high library-to-transposase ratio (e.g., 20:1) to ensure most cells receive multiple gRNAs.
- Single-Cell RNA Sequencing (scRNA-seq): After a suitable incubation period (e.g., 9 days), harvest cells and partition them for scRNA-seq using a platform like 10x Genomics. This simultaneously captures the transcriptome and the gRNA barcodes from each cell.
- Computational Analysis: For each gRNA, computationally partition cells into those containing the gRNA and those without (control). Perform differential expression testing on all genes within 1 Mb of the gRNA target site. Identify "hit gRNAs" that specifically upregulate the intended target gene using an empirical false discovery rate (FDR) threshold (e.g., FDR < 0.1).
This high-throughput approach can identify gRNAs whose activity is restricted to specific conditions or cell states, which is crucial for achieving precise temporal control [62].
Pathway and Workflow Visualization
The following diagrams, generated using Graphviz DOT language and the specified color palette, illustrate the core logical relationships and experimental workflows described in this guide.
Diagram Title: Genetic Pathways for Biofilm Dispersal
Diagram Title: CRISPRa Dispersal Experiment Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPRa-Mediated Biofilm Dispersal Research
| Reagent / Material | Function / Purpose | Example & Notes |
|---|---|---|
| dCas9 Activator Plasmids | Core component for transcriptional upregulation. | dCas9-VPR: Provides strong, direct activation. dCas9-p300: Modifies chromatin state for access [61] [62]. |
| gRNA Library | Targets the CRISPRa machinery to specific genetic loci. | Custom library targeting promoters/enhancers of bslA, hapR, rhlAB, etc. Validated by multiplex screening [62]. |
| Nanoparticle Carriers | Enables efficient delivery and penetration into biofilms. | Lipid NPs: For high encapsulation and biocompatibility. Gold NPs: For high editing efficiency and conjugate stability [16]. |
| Inducible Expression System | Provides temporal control over CRISPRa component expression. | Chemical Inducers (aTc, Ara): Allow precise timing of activation onset to study kinetics [7]. |
| 13C-Labeled Substrates | Allows for quantitative, time-resolved compositional analysis via ssNMR. | 13C-Glycerol: Incorporated into EPS and biomass, enabling tracking of molecular dynamics during dispersal [60]. |
| ssNMR Instrumentation | For non-destructive, quantitative analysis of intact biofilm composition and dynamics. | High-field NMR with MAS: Resolves rigid and mobile phases within the biofilm matrix over time [60]. |
Addressing Strain-Specific Variability and Multi-Species Biofilm Challenges
Biofilms, which are structured microbial communities embedded in a self-produced extracellular polymeric substance (EPS), represent a formidable challenge in both clinical and industrial settings due to their inherent resistance to antimicrobial agents and conventional treatment strategies [16] [4]. This resistance is magnified in multi-species biofilms, where synergistic interactions between different species create a more robust and resilient structure, and is further complicated by strain-specific variability, where genetic differences between strains of the same species lead to divergent biofilm phenotypes and functional outcomes [25] [10]. Traditional broad-spectrum antimicrobials and non-targeted physical disruption methods often fail to address this complexity and can inadvertently promote further resistance.
Within this context, CRISPR Activation (CRISPRa) emerges as a transformative precision tool for biofilm control. By leveraging a catalytically inactive Cas9 (dCas9) fused to transcriptional activator domains, CRISPRa enables targeted upregulation of specific genes without permanent genomic alterations [7] [63]. This technical guide explores the application of CRISPRa for the precise activation of biofilm dispersal genes, a promising strategy within a broader research thesis aimed at developing intelligent, programmable, and species-specific biofilm mitigation strategies. This approach is particularly suited to addressing the dual challenges of strain-specific variability and multi-species complexity by offering a customizable and targeted intervention method.
Technical Foundation: CRISPRa as a Precision Tool
Core Mechanism of CRISPRa
CRISPRa systems are engineered for programmable transcriptional activation by repurposing the core targeting machinery of CRISPR-Cas systems while eliminating its nuclease activity. The fundamental components include:
- dCas9 (deactivated Cas9): A catalytically inactive form of the Cas9 protein that retains its ability to bind DNA based on guide RNA specificity but does not cleave the target DNA [7] [25].
- Transcriptional Activator Domains: Effector domains (e.g., VP64, p65AD) fused to dCas9 that recruit the host cell's transcriptional machinery to the target promoter region [63].
- Guide RNA (gRNA): A short RNA sequence that directs the dCas9-activator complex to a specific genomic locus upstream of the target gene's coding sequence [63] [25].
The system's precision stems from the complementary base pairing between the gRNA and the target DNA sequence, allowing researchers to design gRNAs that specifically target promoter regions of genes involved in biofilm dispersal, such as those encoding dispersin B, DNases, or regulators of quorum sensing (QS) pathways [7]. A significant advantage of CRISPRa is its reversibility; unlike permanent gene knockouts, its effects are transient, allowing for controlled and temporary phenotypic manipulation, which is crucial for studying essential genes and for potential therapeutic applications where permanent genetic changes are undesirable [7].
Advantages for Strain-Specific and Multi-Species Applications
CRISPRa's programmability directly addresses the core challenges of biofilm heterogeneity. The table below summarizes its key advantages for this application space.
Table 1: Advantages of CRISPRa for Complex Biofilm Challenges
| Challenge | CRISPRa Advantage | Technical Rationale |
|---|---|---|
| Strain-Specific Variability | Precision Targeting | gRNAs can be designed to target unique genomic sequences present in a specific strain but absent in closely related strains, enabling precise manipulation without affecting the broader microbial community [64]. |
| Multi-Species Complexity | Multiplexed Gene Activation | Multiple gRNAs can be co-delivered to simultaneously upregulate different dispersal genes across multiple species within a consortium, enabling coordinated disruption of the entire biofilm [7]. |
| Functional Redundancy | Pathway-Specific Override | Can activate master regulator genes or entire operons that control conserved dispersal pathways (e.g., c-di-GMP phosphodiesterases), overcoming redundant genetic controls that often exist in complex biofilms [7] [25]. |
| Phenotypic Heterogeneity | Temporal Control | Gene activation can be induced at specific time points (e.g., via inducible promoters), allowing researchers to study and trigger dispersal in dormant or persister cell subpopulations within a mature biofilm [63]. |
Quantitative Data and Experimental Evidence
Key Performance Metrics from Foundational Studies
Emerging research demonstrates the efficacy of CRISPR-based systems in modulating biofilm phenotypes. The following table consolidates quantitative findings from seminal studies, which provide a performance baseline for developing CRISPRa applications.
Table 2: Quantitative Efficacy of CRISPR-Based Systems in Biofilm Manipulation
| Study System / Organism | Intervention Type | Key Quantitative Outcome | Reference |
|---|---|---|---|
| Pseudomonas aeruginosa | Liposomal CRISPR-Cas9 | >90% reduction in biofilm biomass in vitro | [16] [65] |
| Generic Bacterial Systems | Gold Nanoparticle-CRISPR Delivery | 3.5-fold increase in gene-editing efficiency compared to non-carrier systems | [16] [65] |
| Acinetobacter baumannii ATCC19606 | Cas3 Gene Knockout (Type I-Fa system) | Significant reduction in biofilm formation and thickness; 50% survival rate in Galleria mellonella model vs. 0% for wild-type | [10] |
| Nakaseomyces glabrata | Novel CRISPRa System | Successful overexpression of genes linked to biofilm growth and stress tolerance, confirming system functionality in a fungal pathogen | [63] |
| E. coli in Mouse Gut | Phage-delivered CRISPR-Cas9 (M13) | Successful strain-specific depletion and targeted genomic deletions in vivo | [64] |
Target Genes for CRISPRa in Biofilm Dispersal
The selection of target genes is critical for a successful CRISPRa-mediated dispersal strategy. Promising candidate genes are those involved in key regulatory pathways that promote the transition from a sessile biofilm to a motile, planktonic state.
Table 3: Promising Target Gene Pathways for CRISPRa-Mediated Biofilm Dispersal
| Target Pathway | Example Genes / Operons | Proposed Mechanism of Action for Dispersal |
|---|---|---|
| Cyclic di-GMP (c-di-GMP) Metabolism | Phosphodiesterases (PDEs) containing EAL or HD-GYP domains (e.g., bifA, dipA) | Degradation of the intracellular secondary messenger c-di-GMP, a universal suppressor of motility and promoter of biofilm formation [25]. |
| Quorum Sensing (QS) Systems | Genes for autoinducer degradation, QS inhibitor proteins, or regulators of dispersal signals. | Interference with cell-cell communication that coordinates biofilm behavior, potentially triggering the dispersal phase [16] [35]. |
| Matrix-Degrading Enzymes | Dispersin B (dspB), DNases (nuc), proteases, alginate lyases. | Direct enzymatic degradation of the EPS matrix components (polysaccharides, eDNA, proteins), physically dismantling the biofilm structure [35]. |
| Stress Response Regulators | RpoS, phage-shock proteins, toxin-antitoxin systems. | Activation of stress pathways that can induce a dispersal response as a survival mechanism [63] [4]. |
Experimental Framework: A Detailed Protocol
This section provides a detailed, actionable protocol for designing and executing a CRISPRa experiment aimed at activating biofilm dispersal genes in a bacterial system, incorporating strategies to address strain-specificity.
Stage 1: In Silico Design and Vector Assembly
Step 1: Identification and Validation of Target Genes
- Utilize RNA-sequencing (RNA-seq) or proteomic data from planktonic vs. biofilm states of your target strain to identify endogenously upregulated dispersal genes.
- Select 3-5 candidate genes for screening, prioritizing master regulators (e.g., PDEs) or direct matrix-degrading enzymes.
- For strain-specific targeting, perform a comparative genomic analysis to identify unique promoter sequences in the target strain. Design gRNAs with at least 2-3 mismatches to the same region in non-target strains to ensure specificity [64].
Step 2: gRNA Design and Cloning
- Design 3-5 gRNAs for each target gene, targeting regions 50-150 base pairs upstream of the transcription start site.
- Clone the gRNA sequences into a plasmid containing a constitutive promoter (e.g., J23119 from E. coli) and the gRNA scaffold.
- For multiplexing, clone multiple gRNA expression cassettes into a single delivery vector or use a tandem gRNA array [7].
Step 3: CRISPRa System Assembly
- Assemble the dCas9-activator plasmid. A common configuration is a single plasmid expressing: a) dCas9 fused to a tripartite activator (e.g., VPR: VP64-p65-Rta) and b) the gRNA array.
- Use a medium-copy plasmid (e.g., p15A origin) with a selectable marker (e.g., Kanamycin resistance) and an inducible promoter (e.g., pTet) for dCas9 expression to prevent toxicity during cloning and allow temporal control [63].
Stage 2: Delivery and Validation
Step 4: Delivery into Target Strain
- Electroporation/Conjugation: Standard methods for plasmid delivery into competent cells. Optimize protocol for the specific strain.
- Nanoparticle-Mediated Delivery: For hard-to-transfect strains, use cationic lipid nanoparticles (LNPs) or gold nanoparticles (AuNPs) to complex with the CRISPRa plasmid and facilitate uptake. This method has been shown to enhance delivery efficiency in biofilm settings [16] [65].
- Phage-Assisted Delivery: For ultimate strain-specificity, engineer a bacteriophage (e.g., M13) to package and deliver the CRISPRa construct. This method naturally infects only specific hosts [64].
Step 5: Validation of Gene Activation
- qRT-PCR: 24-48 hours post-induction, measure mRNA levels of the target genes relative to a housekeeping gene and a non-targeting gRNA control. A successful activation should show a >5-fold increase in mRNA expression [63].
- Reporter Assays: If available, use a fluorescent reporter gene under the control of the target gene's promoter to quantify activation via flow cytometry or fluorescence microscopy.
Stage 3: Phenotypic and Community-Level Assessment
Step 6: Biofilm Dispersal Assays
- Static Biofilm Assay (Crystal Violet): Grow biofilms in 96-well plates for 48-72 hours. Induce CRISPRa and incubate for an additional 24 hours. Stain with crystal violet and measure OD590. Compare biomass reduction to controls.
- Confocal Laser Scanning Microscopy (CLSM): Grow biofilms on glass-bottom dishes. Induce CRISPRa and image at 0h, 6h, and 24h post-induction. Use stains like SYTO9 (cells, green) and dextran-conjugate (EPS, red) to visualize 3D architecture, thickness, and biovolume reduction [10].
- Dispersed Cell Quantification: Monitor the increase in optical density (OD600) or colony-forming units (CFUs) in the planktonic phase of the culture after induction, indicating active cell detachment.
Step 7: Assessment in Multi-Species Context
- Create a defined multi-species biofilm consortium including the engineered target strain and 2-3 other relevant species.
- After inducing the CRISPRa system in the target strain, use species-specific FISH (Fluorescence In Situ Hybridization) probes or strain-specific plating to quantify the relative abundance of each species within the biofilm before and after dispersal induction. This assesses the cascade effect on the community [7].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for CRISPRa Biofilm Research
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| dCas9-Activator Plasmids | Core machinery for targeted gene activation. | Addgene Kit # 1000000056 (dCas9-VPR). Ensure compatibility with your host strain's codon usage. |
| Cationic Lipid Nanoparticles (LNPs) | Enhancing plasmid delivery into biofilm-embedded cells. | Formulations like Lipofectamine 3000 or custom-made LNPs can be used for in vitro assays [16]. |
| Gold Nanoparticles (AuNPs) | Alternative nanocarrier for co-delivery of CRISPR components and antibiotics. | Can be functionalized with CRISPR plasmids and tobramycin for synergistic effects against P. aeruginosa biofilms [16] [65]. |
| Conjugative or Phagemid Vectors | For efficient and specific delivery into challenging strains. | Engineered M13 phage particles for targeted delivery to E. coli [64]. |
| Inducible Promoter Systems | For temporal control of dCas9 expression. | Anhydrotetracycline (aTc)-inducible pTet system allows precise timing of activation [25]. |
| Strain-Specific gRNA Databases | For ensuring targeting specificity and avoiding off-target effects. | Requires curated genome sequences for the target and non-target strains. Tools like CRISPOR can assist in design. |
Visualizing the Experimental Workflow and Strategy
The following diagram illustrates the core workflow and strategic logic for applying CRISPRa to biofilm dispersal, from design to phenotypic analysis.
Diagram 1: CRISPRa Experimental Workflow
The integration of CRISPRa technology for precision activation of biofilm dispersal genes represents a paradigm shift in our approach to managing complex microbial communities. By moving beyond broad-spectrum eradication to targeted, programmable intervention, this strategy offers a powerful framework to directly address the long-standing challenges of strain-specific variability and multi-species synergy in biofilms. The experimental framework and toolkit provided here equip researchers with a foundational protocol to pioneer this promising field. Success will depend on the meticulous design of strain-specific gRNAs, the development of robust delivery systems capable of penetrating diverse biofilm matrices, and the careful validation of phenotypic effects within ecologically relevant multi-species consortia. As this research advances, CRISPRa holds the potential to unlock a new class of "smart" antimicrobials that precisely manipulate microbial behavior rather than simply causing lethal toxicity, paving the way for more sustainable and effective strategies in the fight against biofilm-associated infections.
Benchmarking CRISPRa Efficacy Against Established Anti-Biofilm Strategies
In the evolving field of biofilm research, the precise quantification of dispersal is paramount for developing novel therapeutic strategies, particularly those leveraging CRISPR activation (CRISPRa) to trigger biofilm dissolution. Biofilm dispersal is a programmed process wherein cells actively leave the biofilm matrix, transitioning back to a planktonic state. For research focused on using CRISPRa to overexpress native biofilm dispersal genes, accurately measuring the ensuing breakdown is critical for evaluating therapeutic efficacy. This guide details the core quantitative metrics and methodologies for assessing biofilm dispersal, providing a technical foundation for scientists developing targeted anti-biofilm interventions. Effective measurement requires a multi-faceted approach, concurrently evaluating biomass reduction, dispersal cell viability, and matrix composition to gain a complete picture of the dispersal event [66].
Core Quantitative Metrics for Biofilm Dispersal
The assessment of biofilm dispersal rests on three pillars: measuring the remaining biofilm structure, quantifying the cells that have dispersed, and analyzing the changes in the extracellular matrix. The table below summarizes the key metrics and their significance.
Table 1: Core Metrics for Quantifying Biofilm Dispersal
| Metric Category | Specific Metric | Quantitative Measure | Technical Significance |
|---|---|---|---|
| Biomass Reduction | Total Biomass | % reduction in Crystal Violet (CV) staining (A595) [67] [66] | Quantifies overall destruction of the biofilm structure. |
| Biofilm Topography | Changes in biofilm thickness (µm) and biovolume (µm³) via CLSM [6] | Provides 3D architectural data on biofilm disintegration. | |
| Dispersal Cell Viability | Viable Dispersed Cells | Log reduction in colony-forming units (CFU/mL) of planktonic phase [67] | Measures the number and cultivability of shed cells. |
| Cell Death in Biofilm | % dead cells within residual biofilm via Live/Dead staining [67] | Assesses viability of cells that remain after a dispersal trigger. | |
| Matrix Composition | Extracellular DNA (eDNA) | Fluorescence intensity or PCR-based quantification [16] [68] | Probes the breakdown of the eDNA scaffold, a key dispersal step. |
| Polysaccharide Content | Fluorescence intensity using specific lectins or conjugates (e.g., Wheat Germ Agglutinin) [66] | Tracks degradation of the polysaccharide matrix. |
Experimental Protocols for Dispersal Quantification
Concurrent Viability and Biomass Assay
This sequential protocol allows for the dual assessment of metabolic activity and total biomass from the same biofilm sample, providing correlated data from a single experiment [66].
- Biofilm Cultivation: Grow biofilms in a standard 96-well microtiter plate under conditions optimal for the model organism (e.g., 37°C for 18-24 hours) [67] [66].
- Treatment & Dispersal Induction: Expose mature biofilms to the experimental condition, such as a CRISPRa system designed to activate a dispersal gene (e.g., a phosphodiesterase to lower c-di-GMP).
- Viability Staining:
- Aspirate the planktonic phase (which contains dispersed cells) and serially dilute it for CFU enumeration to quantify viable dispersed cells [67].
- Add a resazurin solution (e.g., 0.015% w/v in PBS) to the wells containing the remaining biofilm.
- Incubate for 30-120 minutes at ambient temperature, protected from light.
- Measure fluorescence (Ex560/Em590). The fluorescence intensity correlates with the metabolic activity of the remaining biofilm cells [66].
- Biomass Staining:
- After viability measurement, carefully remove the resazurin solution.
- Wash the wells gently with PBS to remove non-adherent cells.
- Fix the biofilm with 99% methanol for 15 minutes, then air-dry.
- Stain with a 0.1% Crystal Violet solution for 20 minutes.
- Wash thoroughly with water to remove excess dye.
- Elute the bound dye with 33% acetic acid.
- Measure the absorbance at 595 nm, which corresponds to the total remaining biofilm biomass [66].
Advanced Microscopy for Architecture and Viability
Confocal Laser Scanning Microscopy (CLSM) provides high-resolution, three-dimensional data on biofilm structure and cell viability after a dispersal event [6] [67].
- Biofilm Preparation: Grow biofilms on suitable surfaces for microscopy, such as glass-bottom dishes or coverslips.
- Live/Dead Staining: Treat the biofilm with a fluorescent viability stain, such as SYTO 9 (labels all cells green) and propidium iodide (labels dead/damaged cells red), according to manufacturer protocols.
- Image Acquisition: Use a CLSM to capture Z-stack images at multiple random locations within each sample.
- Image Analysis: Utilize biofilm analysis software (e.g., COMSTAT, ImageJ plugins) to quantify:
- Biovolume (µm³/µm²): The total volume of the biofilm per unit area.
- Average Thickness (µm): The mean depth of the biofilm.
- Viability Ratio: Calculate the ratio of dead (red) to live (green) biovolume within the remaining biofilm structure [67].
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues critical reagents and their functions for conducting dispersal quantification assays.
Table 2: Key Research Reagent Solutions for Dispersal Quantification
| Reagent / Kit | Function in Dispersal Assay | Technical Application Notes |
|---|---|---|
| Crystal Violet | Total biomass staining | Stains cells and extracellular matrix; elution with acetic acid allows for spectrophotometric quantification (A595) [67] [66]. |
| Resazurin (AlamarBlue) | Metabolic viability assay | Measures cellular metabolic activity via reduction of resazurin (blue, non-fluorescent) to resorufin (pink, fluorescent); can be used sequentially before CV staining [66]. |
| Live/Dead BacLight Kit | Dual-fluorescence viability staining | Differentiates live (SYTO 9, green) from dead/damaged (Propidium Iodide, red) cells; essential for CLSM analysis of treated biofilms [67]. |
| Wheat Germ Agglutinin (WGA)-Fluorophore | Extracellular matrix polysaccharide staining | Binds to N-acetylglucosamine residues in biofilm matrix polysaccharides (e.g., PIA/PNAG in staphylococci); enables fluorescence-based matrix quantification [66]. |
| dCas9 Transcriptional Activator | CRISPRa-based gene activation | Core component for inducing dispersal; the catalytically "dead" Cas9 (dCas9) is fused to transcriptional activators (e.g., VP64) and targeted to promoter regions of key dispersal genes [6]. |
Integrating Metrics into a CRISPRa Research Workflow
The quantification metrics described are not isolated endpoints but are integral to a cohesive research strategy for validating CRISPRa-based biofilm disruption. The following diagram illustrates the logical workflow from hypothesis to data integration.
CRISPRa Dispersal Quantification Workflow
This systematic approach, combining targeted genetic activation with multi-parameter quantitative analysis, enables robust validation of novel anti-biofilm strategies. The dispersal process itself and its key regulatory pathways, such as the central role of c-di-GMP, can be visualized to clarify the therapeutic target.
Core Biofilm Dispersal Pathway
The escalating crisis of biofilm-associated antibiotic resistance necessitates a paradigm shift from conventional antimicrobials to precision genetic therapies. This whitepaper provides a comparative analysis of three distinct therapeutic approaches: conventional antibiotics, CRISPR-Kill (a bactericidal CRISPR-Cas strategy), and CRISPR activation (CRISPRa). While conventional antibiotics offer broad-spectrum activity, their efficacy is severely limited against biofilm-protected bacteria. CRISPR-Kill enables targeted bacterial elimination but remains a destructive approach. In contrast, CRISPRa represents a transformative, non-destructive strategy that leverages the bacterial innate regulatory machinery by upregulating endogenous biofilm dispersal genes. This technical guide examines the mechanisms, efficacy, and applications of each modality, with particular focus on experimental protocols for implementing CRISPRa for biofilm dispersal within the context of advanced antibacterial drug development.
Bacterial biofilms are structured communities of microorganisms embedded in a self-produced extracellular polymeric substance (EPS) that confers inherent resistance to antimicrobial treatments [16]. The biofilm matrix creates a formidable physical and physiological barrier, reducing antibiotic penetration by up to 1000-fold compared to planktonic cells and fostering heterogeneous bacterial subpopulations with recalcitrant "persister" cells [16] [4]. This protective environment enhances horizontal gene transfer, accelerating the dissemination of antibiotic resistance genes and complicating treatment outcomes, particularly in chronic and device-associated infections [16] [65].
The limitations of conventional antibiotics have spurred the development of CRISPR-based antimicrobials, which can be categorized into two primary modalities: nuclease-active systems (including CRISPR-Kill) and nuclease-inactive systems (such as CRISPR interference and activation). While CRISPR-Kill utilizes Cas nucleases to introduce lethal double-strand breaks in bacterial genomic DNA, resulting in targeted cell death, CRISPR activation (CRISPRa) employs a catalytically inactive Cas9 (dCas9) fused to transcriptional activators to precisely upregulate endogenous bacterial genes without altering DNA sequences [7] [43]. For biofilm mitigation, CRISPRa can be programmed to activate master regulators of the biofilm dispersal cascade, offering a novel approach that leverages the bacterium's own genetic programming to dismantle biofilms reversibly.
Conventional Antibiotics
Mechanism of Action: Conventional antibiotics typically target essential bacterial processes through non-specific pharmacological interactions. Key classes include β-lactams (cell wall synthesis inhibitors), fluoroquinolones (DNA replication inhibitors), aminoglycosides (protein synthesis inhibitors), and sulfonamides (metabolic pathway antagonists). Their primary strengths—broad-spectrum activity and established clinical use—are also their fundamental limitations in biofilm contexts, as their targets are often inaccessible or metabolically inactive within biofilm microenvironments [16] [4].
Limitations in Biofilm Context: Biofilms demonstrate multi-faceted resistance mechanisms that render conventional antibiotics largely ineffective. The EPS matrix acts as a diffusion barrier, physically limiting antibiotic penetration while simultaneously creating chemical gradients that foster heterogeneous bacterial subpopulations [4]. Nutrient limitation and reduced metabolic activity in deeper biofilm layers diminish the efficacy of bactericidal antibiotics that require active cell division. Additionally, biofilms facilitate adaptive evolution through enhanced horizontal gene transfer of resistance determinants such as β-lactamase genes and efflux pump regulators [16].
CRISPR-Kill (Bactericidal CRISPR-Cas)
Mechanism of Action: CRISPR-Kill systems utilize Cas9 or Cas12 nucleases programmed with guide RNAs (gRNAs) to target essential bacterial genes or antibiotic resistance determinants for cleavage, resulting in lethal double-strand DNA breaks [7] [4]. This approach can be directed against chromosomal genes essential for viability (e.g., DNA gyrase, RNA polymerase) or plasmid-borne resistance genes (e.g., blaNDM-1, mecA), specifically eliminating targeted pathogens while sparing commensal flora.
Delivery Considerations: Effective implementation requires sophisticated delivery platforms, with nanoparticle systems demonstrating particular promise. Liposomal Cas9 formulations have achieved >90% reduction of Pseudomonas aeruginosa biofilm biomass in vitro, while gold nanoparticle carriers enhance gene-editing efficiency by 3.5-fold compared to non-carrier systems [16] [65].
CRISPR Activation (CRISPRa) for Biofilm Dispersal
Mechanism of Action: CRISPRa represents a fundamentally different approach that manipulates bacterial gene expression without causing DNA damage. The system comprises dCas9 fused to transcriptional activator domains (e.g., VP64, SoxS) programmed with sgRNAs to target promoter regions of biofilm dispersal genes [7] [43]. Unlike destructive methods, CRISPRa harnesses endogenous bacterial regulation by upregulating master controllers of the sessile-to-motile transition, including diguanylate cyclase phosphodiesterases that modulate cyclic di-GMP (c-di-GMP) signaling, quorum sensing systems that coordinate dispersal, and enzymes that degrade the EPS matrix [6].
Theoretical Advantages for Biofilm Control: By activating dispersal programs, CRISPRa induces biofilm breakdown through natural physiological processes, potentially reducing inflammatory responses associated with bacterial lysis. This approach offers reversible, tunable control and can be multiplexed to target multiple regulatory pathways simultaneously, creating a synergistic dispersal effect while minimizing resistance development [7] [43].
Table 1: Comparative Analysis of Anti-Biofilm Technologies
| Parameter | Conventional Antibiotics | CRISPR-Kill | CRISPRa for Biofilm Dispersal |
|---|---|---|---|
| Primary Mechanism | Pharmacological inhibition of essential cellular processes | Programmed cleavage of essential genes or resistance determinants | Transcriptional activation of endogenous dispersal genes |
| Specificity | Broad-spectrum, affects commensals | Sequence-specific targeting | Sequence-specific pathway modulation |
| Biofilm Penetration | Limited by EPS barrier; ~1000x reduced efficacy | Enhanced with nanoparticle carriers; 3.5x efficiency with AuNPs | Enhanced with nanoparticle carriers; tissue-dependent |
| Resistance Development | High (horizontal gene transfer enhanced in biofilms) | Moderate (requires functional delivery and target access) | Low (targets master regulators; non-lethal) |
| Bacterial Survival | Reduced but selects for resistant mutants | Eliminates targeted populations | Disperses biofilm without necessarily killing bacteria |
| Key Limitations | Limited efficacy against persisters, ecological collateral damage | Delivery challenges, potential off-target effects, host immune response to components | Complex delivery, transient effect, requires understanding of regulatory networks |
| Therapeutic Outcome | Bacteriostatic/bactericidal | Bactericidal | Biofilm dispersal without killing (potential bacteriostatic) |
| Representative Efficacy | Often <10% reduction in established biofilms | >90% biofilm biomass reduction (P. aeruginosa) | Operon-mediated potent biofilm inhibition (PFLU1114 in P. fluorescens) |
Experimental Framework for CRISPRa-Mediated Biofilm Dispersal
Target Identification and gRNA Design
Bioinformatics Pipeline:
- Identify Dispersal Gene Candidates: Focus on master regulators of biofilm dispersion, including:
- Phosphodiesterases that degrade c-di-GMP (e.g., bifA, dipA, rimA)
- Quorum sensing systems controlling dispersal (e.g., lasI, rhlI)
- Matrix-degrading enzymes (e.g., pelA, pslG, dispersin B)
- Motility regulators (e.g., flgM, fliA)
Promoter Mapping: Identify transcriptional start sites and -10/-35 promoter regions upstream of target genes using bacterial genome databases (NCBI, UniProt).
gRNA Design Criteria: Design 3-5 sgRNAs targeting regions from -400 to +50 bp relative to the transcriptional start site. Avoid sequences with off-target matches using BLAST analysis against the host genome [6] [43].
CRISPRa System Assembly
Vector Construction:
- Backbone Selection: Utilize a dual-plasmid system with compatible origins of replication and selection markers (e.g., p15A and ColE1 origins; kanamycin and chloramphenicol resistance).
- dCas9-Activator Module: Clone dCas9 (D10A, H840A mutations) fused to transcriptional activators (e.g., VP64, SoxS) under inducible control (Ptet) in plasmid 1.
- sgRNA Expression Module: Clone sgRNA expression cassettes with constitutive promoters (Pj23100) in plasmid 2 [6].
Delivery Platform Integration:
- Nanoparticle Formulation: Encapsulate CRISPRa constructs in cationic liposomes or gold nanoparticles functionalized with biofilm-penetrating peptides (e.g., DNase I, dispersin B) to enhance EPS penetration [16] [65].
Protocol: Assessing CRISPRa Efficacy in Biofilm Models
Materials and Reagents:
- Bacterial Strains: Pseudomonas aeruginosa PAO1 or relevant clinical isolates
- Culture Conditions: Mueller Hinton broth; flow cell systems for biofilm growth
- Induction: Anhydrotetracycline (aTc; 100 ng/mL) for dCas9-activator induction
- Analysis Tools: Confocal laser scanning microscopy (CLSM), crystal violet staining, RT-qPCR
Methodology:
- Biofilm Establishment:
- Grow biofilms for 48-72 hours in flow cells or 96-well plates
- For in vitro models, use CDC biofilm reactors with relevant surface materials
CRISPRa Delivery:
- Apply nanoparticle-encapsulated CRISPRa constructs at mid-exponential phase (OD600 ≈ 0.5)
- Include controls: empty nanoparticles, non-targeting sgRNA, dCas9-only
Induction and Monitoring:
- Induce with aTc at 24 hours post-biofilm maturation
- Monitor dispersal in real-time using time-lapse microscopy
Endpoint Analyses:
- Biomass Quantification: Crystal violet staining at 570 nm
- Viability Assessment: Colony-forming unit (CFU) counts from dispersed cells
- Structural Analysis: CLSM with LIVE/DEAD staining (SYTO9/propidium iodide)
- Molecular Validation: RT-qPCR for target gene expression; RNA-seq for transcriptomic profiling
- c-di-GMP Quantification: LC-MS/MS to measure intracellular c-di-GMP levels [6]
Diagram 1: CRISPRa Experimental Workflow for Biofilm Dispersal Studies
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for CRISPRa Biofilm Studies
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| dCas9-Activator Plasmids | dCas9-VP64, dCas9-SoxS, dCas9-TV | Transcriptional activation machinery; TV system (dCas9-6×TAL-2×VP64) shows enhanced activation in diverse bacterial systems |
| sgRNA Cloning Vectors | pCDF or pMB1 origin vectors with constitutive promoters | sgRNA expression; design 3-5 sgRNAs per target for screening; include non-targeting controls |
| Induction Systems | Anhydrotetracycline (aTc)-inducible Ptet, Arabinose-inducible PBAD | Tight regulation of dCas9-activator expression; aTc typically used at 50-200 ng/mL |
| Nanoparticle Delivery Systems | Cationic liposomes, Gold nanoparticles (AuNPs), Chitosan nanoparticles | Enhance delivery efficiency and biofilm penetration; AuNPs show 3.5x efficiency improvement |
| Biofilm Growth Systems | CDC biofilm reactors, Flow cells, Calgary biofilm devices | Standardized biofilm growth; flow cells enable real-time microscopy monitoring |
| Analysis Reagents | SYTO9/propidium iodide, Crystal violet, c-di-GMP ELISA kits | Biofilm viability, biomass, and signaling molecule quantification |
| Model Bacterial Strains | P. fluorescens SBW25, P. aeruginosa PAO1, E. coli MG1655 | Well-characterized biofilm formers with established genetic tools |
| Molecular Analysis Kits | RNA extraction kits with DNase treatment, RT-qPCR master mixes | Validate target gene upregulation; ensure high-quality RNA from biofilm samples |
Signaling Pathways for CRISPRa-Mediated Biofilm Dispersal
The efficacy of CRISPRa for biofilm control depends on precise targeting of key regulatory pathways that govern the transition from sessile to motile lifestyles. The most promising targets center on cyclic di-GMP (c-di-GMP) signaling, the master regulator of bacterial biofilm formation.
Diagram 2: CRISPRa Targets in Biofilm Dispersal Signaling Pathways
Discussion and Future Perspectives
The comparative analysis reveals distinctive advantages and limitations for each anti-biofilm strategy. Conventional antibiotics, despite their established clinical use, demonstrate fundamental limitations against biofilms due to penetration barriers and heterogeneous bacterial metabolism. CRISPR-Kill offers pathogen-specific elimination but shares delivery challenges with other macromolecular therapies and may face resistance through modification of PAM sequences or CRISPR evasion mechanisms.
CRISPRa represents a paradigm shift by manipulating host regulatory networks rather than introducing foreign lethal elements. The technology's ability to precisely upregulate endogenous dispersal genes—such as phosphodiesterases that degrade c-di-GMP, quorum sensing systems that coordinate dispersal, and enzymes that degrade the EPS matrix—enables non-destructive biofilm control that could mitigate resistance development [7] [6]. However, significant challenges remain in delivery efficiency, especially through complex biofilm architectures, and in achieving sufficient transcriptional activation to trigger robust dispersal responses.
Future development should focus on several key areas:
- Advanced Delivery Platforms: Engineering nanoparticles with improved biofilm penetration and bacterial uptake, potentially incorporating EPS-degrading enzymes or quorum sensing inhibitors to create synergistic effects.
- Multiplexed Targeting: Developing systems that simultaneously activate multiple dispersal pathways to enhance efficacy and prevent resistance.
- Regulatory Considerations: Addressing safety concerns related to genetically modified organisms and environmental release, potentially through self-limiting circuits that prevent horizontal gene transfer.
- Integration with Diagnostics: Combining CRISPRa therapeutics with CRISPR-based diagnostics for targeted, personalized anti-biofilm treatments.
The integration of CRISPRa with functional genomics, synthetic biology, and materials science holds exceptional promise for developing next-generation anti-biofilm strategies that could fundamentally transform our approach to persistent bacterial infections.
Within the broader thesis context of CRISPRa for activating biofilm dispersal genes, this comparative analysis demonstrates that CRISPRa offers a unique, non-destructive approach that fundamentally differs from both conventional antibiotics and CRISPR-Kill technologies. While conventional antibiotics face inherent limitations against biofilms and CRISPR-Kill employs targeted bactericidal activity, CRISPRa manipulates the bacterial innate regulatory network to programmatically induce biofilm dispersal. This strategy leverages the bacterium's own genetic programming to dismantle protective structures, potentially reducing selective pressure for resistance development. As delivery technologies advance and our understanding of biofilm regulatory networks deepens, CRISPRa-based approaches represent a promising frontier in the development of precision antimicrobials capable of addressing the persistent challenge of biofilm-associated infections.
The escalating crisis of antimicrobial resistance represents one of the most pressing challenges in modern medicine, with biofilm-associated infections playing a pivotal role in this global health threat. Biofilms—structured microbial communities encased in a self-produced extracellular polymeric substance (EPS)—demonstrate remarkable resilience to conventional antibiotics, exhibiting up to 1000-fold greater tolerance compared to their planktonic counterparts [16]. This resilience stems from a multifaceted defense system that includes physical diffusion barriers, metabolic heterogeneity, persister cell populations, and enhanced horizontal gene transfer [16] [69]. Within the context of advancing novel antimicrobial strategies, CRISPR activation (CRISPRa) systems have emerged as powerful synthetic biology tools for precise transcriptional control in bacterial pathogens. Unlike CRISPR-Cas9 which introduces double-strand breaks in DNA, CRISPRa utilizes catalytically deactivated Cas proteins fused to transcriptional activation domains to upregulate targeted endogenous genes [70].
This technical guide proposes a innovative therapeutic framework that synergistically integrates CRISPRa with antimicrobial adjuvants to combat biofilm-associated infections. The core thesis posits that targeted activation of biofilm dispersal genes via CRISPRa can fundamentally alter the biofilm phenotype, while concurrently administered adjuvants can potentiate conventional antibiotics against the newly vulnerable bacterial population. This approach represents a paradigm shift from traditional bactericidal strategies to precisely targeted phenotypic manipulation of bacterial communities. The following sections will provide a comprehensive technical foundation for developing this combinatorial approach, including mechanistic insights, experimental methodologies, and specialized research tools.
Technical Foundations: Core Concepts and Definitions
CRISPRa Systems for Bacterial Gene Activation
CRISPRa tools represent a sophisticated adaptation of the native CRISPR bacterial immune system for programmable transcriptional control. In bacteria, these systems typically employ a dCas protein (deactivated Cas9 lacking endonuclease activity) fused to transcriptional activator domains, which is guided to specific promoter regions by a programmable guide RNA (gRNA) [70]. The systematic mapping of bacterial CRISPRa systems has revealed important operational parameters, including that optimal target sites for different activators can vary by up to 200 bases in the region upstream of the transcription start site (TSS) [70]. This positional sensitivity underscores the importance of precise gRNA design for effective transcriptional activation.
Unlike eukaryotic systems where multiple activators can be combined for synergistic effects, bacterial CRISPRa systems demonstrate fundamentally different behavior, with many activator combinations producing antagonistic rather than synergistic effects [70]. This mechanistic distinction has profound implications for designing effective bacterial gene activation strategies and suggests that single-effector systems may be more reliable than complex multi-activator approaches for therapeutic applications.
Table 1: Key Components of Bacterial CRISPRa Systems
| Component | Structure/Type | Function in CRISPRa |
|---|---|---|
| dCas Protein | Catalytically deactivated Cas9 or other Cas variants | DNA-binding scaffold that targets specific genomic loci without cleaving DNA |
| Activator Domain | Omega protein, SoxS, etc. | Recruits bacterial RNA polymerase to initiate transcription |
| Guide RNA (gRNA) | ~20 nucleotide scaffold | Determines targeting specificity through complementary base pairing |
| Promoter Target | -200 to +50 bp relative to TSS | Genomic region where activation complex assembles |
Antimicrobial Adjuvants: Mechanisms and Classifications
Antimicrobial adjuvants comprise a diverse category of compounds that enhance the efficacy of conventional antibiotics through non-bactericidal mechanisms. These substances function by disrupting the structural integrity of biofilms, inhibiting resistance mechanisms, or increasing antibiotic penetration, thereby resensitizing resistant pathogens to treatment [71]. Adjuvants can be broadly categorized based on their primary mechanisms of action:
- Matrix-Targeting Agents: Enzymes such as Dispersin B (glycoside hydrolase) and DNase I degrade essential structural components of the biofilm EPS, compromising structural integrity and facilitating antibiotic penetration [35].
- Quorum Sensing Inhibitors: Natural compounds like curcumin and berberine, or synthetic analogs such as acyl homoserine lactone (AHL) analogs, disrupt bacterial communication pathways that coordinate biofilm development and maintenance [35].
- Penetration Enhancers: Surfactants including Tween 80 and Triton X-100 reduce interfacial tension, improving antibiotic diffusion through the dense biofilm matrix [71].
- Cellular Resensitizers: Compounds that target persister cells and revert them to a metabolically active, antibiotic-sensitive state [69].
The conceptual framework of this combinatorial approach can be visualized through the following workflow:
The Biofilm Challenge: Resistance Mechanisms and Therapeutic Implications
Structural and Physiological Basis of Biofilm-Mediated Resistance
The remarkable antibiotic tolerance of biofilm-associated bacteria stems from a complex interplay of physical barriers, physiological adaptations, and genetic plasticity. The biofilm lifecycle progresses through distinct phases—initial attachment, irreversible attachment, microcolony formation, maturation, and dispersion—each characterized by specific molecular processes and structural developments [69]. The mature biofilm architecture displays remarkable heterogeneity, with water channels facilitating nutrient distribution and waste removal interspersed with densely packed microbial microcolonies [16].
The extracellular polymeric substance (EPS) matrix represents a primary defense mechanism, creating a physical barrier that restricts antibiotic penetration through molecular sieving, binding, or enzymatic degradation [69]. This matrix comprises an agglomeration of various biopolymers, including polysaccharides, proteins, extracellular DNA (eDNA), and lipids, whose specific composition varies significantly between bacterial species and environmental conditions [69]. Beyond this physical barrier, biofilms employ sophisticated physiological resistance mechanisms:
- Metabolic Heterogeneity: Nutrient and oxygen gradients within the biofilm create distinct microniches, leading to subpopulations with varying metabolic states, including dormant persister cells that exhibit exceptional tolerance to antibiotics [16] [35].
- Enhanced Horizontal Gene Transfer: The dense, structured environment of biofilms dramatically accelerates the exchange of mobile genetic elements carrying antibiotic resistance genes, facilitating the rapid evolution and dissemination of resistance mechanisms [16] [69].
- Stress Response Activation: The biofilm lifestyle induces distinct patterns of gene expression compared to planktonic growth, including upregulation of efflux pumps and other stress response systems that enhance antibiotic tolerance [3] [69].
Table 2: Key Biofilm Resistance Mechanisms and Potential Intervention Points
| Resistance Mechanism | Functional Basis | Potential Intervention Strategy |
|---|---|---|
| EPS Matrix Barrier | Physical obstruction and binding of antimicrobial molecules | Matrix-degrading enzymes (Dispersin B, DNase I) |
| Metabolic Heterogeneity | Gradients create dormant persister cell subpopulations | Activation of dispersal genes to synchronize metabolism |
| Enhanced Horizontal Gene Transfer | Close cell-cell proximity facilitates plasmid exchange | CRISPR-based targeting of resistance gene transfer |
| Altered Microenvironment | Reduced pH, oxygen limitation affects antibiotic activity | Modulation of biofilm physiology via quorum sensing interference |
| Upregulated Efflux Pumps | Increased expression of multidrug efflux systems | Efflux pump inhibitors combined with antibiotic treatment |
Biofilm Dispersal Mechanisms as Therapeutic Targets
The natural dispersal phase of the biofilm lifecycle represents a critical vulnerability that can be exploited therapeutically. Biofilm dispersal occurs through both active and passive mechanisms, including seeding dispersal (active release of single cells or small clusters), erosion (continuous release of single cells), and sloughing (detachment of large biofilm fragments) [69]. These processes are regulated by complex molecular signaling networks that respond to environmental cues and intracellular second messengers.
Of particular therapeutic relevance are the enzymatic systems that degrade structural components of the biofilm matrix, including glycoside hydrolases that target expolysaccharides and DNases that degrade extracellular DNA [69]. The regulatory pathways controlling these processes, including cyclic di-GMP signaling, quorum sensing systems, and specific transcriptional regulators, represent promising targets for CRISPRa-mediated activation [69]. Natural dispersal triggers, such as nutrient limitation, oxygen availability, and nitric oxide signaling, provide additional insights into potential gene targets for therapeutic intervention [69].
Integrated Methodology: Experimental Framework for Combinatorial Therapy
CRISPRa System Design and Delivery Optimization
The development of an effective CRISPRa-based anti-biofilm therapeutic requires careful consideration of multiple design parameters and delivery strategies. The following experimental protocol outlines a systematic approach for constructing and validating CRISPRa systems targeting biofilm dispersal genes:
Phase 1: Identification and Validation of Dispersal Gene Targets
- Bioinformatic Analysis: Utilize genomic databases to identify promoter regions of known biofilm dispersal genes (e.g., glycoside hydrolases, DNases, surfactant proteins) in target pathogens such as Pseudomonas aeruginosa and Staphylococcus aureus.
- Conserved Motif Mapping: Identify conserved sequence motifs within promoter regions that may represent binding sites for natural transcriptional activators.
- gRNA Library Design: Design a comprehensive library of gRNAs targeting sites from -200 to +50 base pairs relative to the transcription start site, accounting for the positional sensitivity observed in bacterial CRISPRa systems [70].
- In Silico Specificity Screening: Perform computational off-target analysis to minimize unintended activation of non-target genes.
Phase 2: CRISPRa Construct Assembly
- Vector Selection: Utilize broad-host-range plasmids with tunable expression systems for enhanced compatibility across bacterial species.
- Activator Selection: Test multiple activator domains (e.g., Omega, SoxS) as fusions with dCas9, evaluating both single activators and combinations while noting potential antagonistic effects [70].
- Modular Cloning: Implement Golden Gate or Gibson assembly strategies for efficient combinatorial testing of different gRNA-activator configurations.
Phase 3: Delivery System Development
- Nanoparticle Formulation: Encapsulate CRISPRa constructs in lipid nanoparticles (LNPs) optimized for bacterial uptake, drawing on formulations that have demonstrated 3.5-fold increased editing efficiency in previous CRISPR antimicrobial studies [16].
- Surface Functionalization: Modify nanoparticle surfaces with pathogen-specific targeting ligands (e.g., antibodies, lectins) to enhance specificity.
- Stability Testing: Evaluate the stability of formulated nanoparticles under physiological conditions relevant to the intended application (e.g., cystic fibrosis sputum, wound exudate).
The relationship between gRNA positioning and activation efficiency follows a specific pattern:
Adjuvant Screening and Synergy Assessment
Concurrent with CRISPRa development, a systematic approach to adjuvant screening and synergy testing is essential for identifying optimal combination therapies:
Protocol 1: In Vitro Biofilm Disruption Assay
- Biofilm Cultivation: Establish standardized biofilm cultures using appropriate substrates (e.g., peg lids, flow cells) and growth conditions relevant to the target infection environment.
- Treatment Conditions:
- Group 1: CRISPRa alone
- Group 2: Adjuvant alone
- Group 3: CRISPRa + Adjuvant
- Group 4: Conventional antibiotic alone
- Group 5: CRISPRa + Adjuvant + Antibiotic
- Group 6: Untreated control
- Quantitative Assessment: Employ multiple metrics including crystal violet staining (total biomass), colony forming unit enumeration (viable cells), and confocal microscopy with live/dead staining (spatial distribution of viability).
Protocol 2: Transcriptional Response Profiling
- RNA Sequencing: Perform time-course transcriptomic analysis to evaluate CRISPRa-mediated activation of target genes and potential global transcriptional changes.
- Quorum Sensing Monitoring: Utilize reporter strains or direct measurement of signaling molecules to assess adjuvant effects on bacterial communication systems.
Protocol 3: Penetration Efficiency Measurement
- Fluorescent Tracer Studies: Employ fluorescently tagged antibiotics or dextran molecules to quantitatively assess adjuvant-mediated enhancement of biofilm penetration using fluorescence recovery after photobleaching (FRAP) techniques.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 3: Key Research Reagents for CRISPRa-Adjuvant Studies
| Reagent Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| CRISPRa Plasmids | dCas9-Omega fusion constructs, gRNA expression vectors | Programmable transcriptional activation | Ensure compatibility with target bacterial species |
| Nanoparticle Delivery Systems | Cationic lipid nanoparticles, polymer-based nanoparticles, gold nanoparticles | In vivo delivery of CRISPR components | Optimize for bacterial uptake efficiency; gold NPs shown to enhance editing efficiency 3.5-fold [16] |
| Matrix-Targeting Enzymes | Dispersin B, DNase I, proteases | EPS disruption and penetration enhancement | Determine optimal timing relative to CRISPRa administration |
| Quorum Sensing Inhibitors | AHL analogs, curcumin, cinnamaldehyde | Disruption of bacterial communication | Monitor potential effects on CRISPRa component expression |
| Bacterial Reporter Strains | GFP/lux reporters under control of dispersal gene promoters | Real-time monitoring of CRISPRa efficacy | Validate correlation between reporter signal and endogenous gene expression |
| Biofilm Assessment Tools | Crystal violet, LIVE/DEAD BacLight, Calgary biofilm device | Quantification of biofilm biomass and viability | Standardize growth conditions for reproducible results |
Technical Considerations and Implementation Challenges
The translational pathway for CRISPRa-adjuvant combination therapy presents several significant technical challenges that require careful consideration:
Delivery Optimization and Specificity
Efficient delivery of CRISPRa components to bacterial cells within biofilms remains a substantial barrier. Lipid nanoparticles (LNPs) have demonstrated promise for in vivo delivery, as evidenced by their successful use in clinical trials for hereditary transthyretin amyloidosis, where they produced sustained protein reduction exceeding 90% [72]. However, optimizing these systems for bacterial targeting rather than human hepatocytes requires substantial reformulation. Specificity concerns extend beyond off-target transcriptional activation to include potential effects on the host microbiome, necessitating careful gRNA design and potentially incorporation of additional targeting mechanisms.
Resistance Mitigation and Evolutionary Considerations
While the non-bactericidal nature of both CRISPRa and many adjuvants may reduce selective pressure for conventional resistance mechanisms, evolutionary adaptation remains a concern. Potential resistance pathways include mutations in gRNA target sequences, modification of CRISPRa component import, or compensatory mutations in dispersal regulatory networks. Combination approaches that target multiple dispersal pathways simultaneously may help mitigate this risk by raising the evolutionary barrier to resistance.
Formulation Stability and Pharmacokinetics
The development of co-formulation strategies that maintain the stability and functionality of both CRISPRa components and adjuvant compounds presents significant pharmaceutical challenges. The differing physicochemical properties of nucleic acids, proteins, and small molecule adjuvants may necessitate separate delivery systems with coordinated administration schedules. Pharmacokinetic considerations include achieving sufficient residence time at the infection site and synchronizing the peak activity of both therapeutic components.
The integration of CRISPRa-based gene activation with antimicrobial adjuvants represents a promising frontier in the battle against biofilm-mediated infections. This approach leverages the precision of synthetic biology to manipulate bacterial behavior rather than simply attempting to eradicate pathogens, potentially reducing selective pressure for resistance development while resensitizing persistent infections to conventional antibiotics. The experimental framework outlined in this technical guide provides a roadmap for researchers to systematically develop and optimize this innovative therapeutic strategy.
Future advancements in this field will likely focus on refining delivery platforms for enhanced bacterial specificity, expanding the repertoire of validated dispersal gene targets across clinically relevant pathogens, and developing sophisticated co-formulation strategies for optimal temporal control of combination therapies. As CRISPRa systems continue to evolve with improved activation efficiency and reduced off-target effects, and as new classes of antimicrobial adjuvants are discovered and characterized, this synergistic approach holds significant potential to address the growing threat of multidrug-resistant biofilm infections. The successful translation of this technology will require continued interdisciplinary collaboration between molecular biologists, pharmaceutical scientists, and clinical researchers to overcome the remaining technical challenges and realize the full therapeutic potential of this innovative approach.
Assessing Pathogen Re-Sensitization to Antibiotics Post-Dispersal
Abstract The escalating crisis of antimicrobial resistance (AMR) represents one of the most urgent threats to global health, with biofilm-associated infections being particularly challenging to treat [16]. Bacterial biofilms, structured communities encased in a self-produced extracellular polymeric substance (EPS), can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [16]. A critical, yet underexploited, vulnerability in the biofilm life cycle is the dispersal phase, where cells transition back to a free-living, planktonic state. This whitepaper provides a technical guide for a novel therapeutic strategy: using CRISPR activation (CRISPRa) to precisely trigger biofilm dispersal genes, forcing pathogens into a transiently vulnerable, antibiotic-sensitive state post-dispersal. We detail the mechanisms, experimental protocols for validating re-sensitization, and the integration of this approach with conventional antibiotics, providing a framework for researchers and drug development professionals to develop next-generation anti-infective therapies.
Biofilms are the predominant mode of growth for bacteria in both environmental and clinical settings, playing a key role in persistent infections related to medical implants, cystic fibrosis, and chronic wounds [16] [3]. The resilience of biofilms stems from multiple synergistic mechanisms: the physical barrier of the EPS matrix that limits antibiotic penetration, heterogeneous metabolic activity leading to dormant "persister" cells, and activation of general stress responses [3].
The biofilm life cycle culminates in a programmed dispersal phase, where bacteria actively escape the biofilm matrix to colonize new niches. During this transition, dispersed cells undergo significant transcriptional reprogramming. While they are in a planktonic form, they often exhibit a temporary window of increased susceptibility to antimicrobials before they establish new colonies and re-express high-level resistance mechanisms [3]. Traditional therapeutic strategies have focused on biofilm prevention or eradication. An alternative, precision medicine approach is to therapeutically induce this dispersal event, not as an endpoint, but to create a temporary therapeutic window where the newly dispersed, re-sensitized pathogens can be efficiently eradicated with conventional antibiotics.
CRISPRa technology offers an unprecedented tool to execute this strategy. By using a catalytically "dead" Cas9 (dCas9) fused to transcriptional activation domains (e.g., VP64, VPR), researchers can target and upregulate specific endogenous bacterial genes without altering the underlying DNA sequence [73]. This allows for the precise activation of key genetic regulators that control the biofilm dispersal pathway, providing a targeted method to force a dispersion event and assess the subsequent re-sensitization of the bacterial population.
Core Mechanisms: Targeting Dispersal Genes with CRISPRa
The efficacy of a CRISPRa-based dispersal strategy hinges on the precise targeting of master regulators within the biofilm genetic network. The system's core component is a dCas9 protein fused to a transcriptional activator, programmed by guide RNAs (gRNAs) to bind specific promoter or enhancer regions of target genes [73].
Table 1: Key Genetic Targets for CRISPRa-Mediated Biofilm Dispersal and Re-sensitization
| Target Gene / Pathway | Function | Proposed gRNA Target Site | Expected Outcome of Activation |
|---|---|---|---|
bdlA |
Regulatory protein controlling biofilm dispersal; often inhibited by cyclic-di-GMP [3]. | Promoter region | Triggers a cascade leading to matrix degradation and cell detachment. |
| Operons encoding DNases | Degrades extracellular DNA (eDNA), a critical structural component of the EPS matrix [3]. | Upstream of transcription start site (TSS) | Disrupts biofilm structural integrity, facilitating dissolution. |
| Operons encoding dispersin B | Glycoside hydrolase that hydrolyzes the polysaccharide poly-N-acetylglucosamine (PNAG) in the matrix [3]. | Upstream of TSS | Degrades the polysaccharide backbone of the biofilm. |
Quorum Sensing Systems (e.g., lasR) |
Master regulators of group behaviors; some circuits activate dispersal genes at high cell density [16]. | Promoter of regulatory genes | Overstimulates the dispersal module of the quorum sensing network. |
The following diagram illustrates the conceptual pathway of how CRISPRa targeting leads to biofilm dispersal and subsequent antibiotic sensitization.
Figure 1: Conceptual Pathway for CRISPRa-Induced Dispersal and Eradication. The CRISPRa system, delivered via LNPs, activates target dispersal genes within the tolerant biofilm, forcing a transition to a vulnerable planktonic state that can be eradicated with conventional antibiotics.
Experimental Protocols for Assessing Re-Sensitization
A robust experimental pipeline is required to quantify the success of CRISPRa-induced dispersal and the subsequent level of antibiotic re-sensitization. The workflow below integrates established molecular biology techniques with functional assays.
Figure 2: Workflow for Evaluating CRISPRa-Induced Re-sensitization. A sequential protocol from biofilm establishment to final antibiotic efficacy assessment.
Protocol: Multiplexed CRISPRa Screening for Dispersal Gene Identification
Objective: To identify the most potent gRNAs for activating biofilm dispersal and re-sensitization in a target pathogen (e.g., Pseudomonas aeruginosa).
Materials:
- dCas9 Activator Strain: A bacterial strain (e.g., E. coli or the target pathogen) engineered to stably express a dCas9-VPR or dCas9-VP64 fusion protein [73].
- gRNA Library: A pooled lentiviral or plasmid library encoding ~500 gRNAs targeting promoter regions of known and putative dispersal genes, quorum sensing regulators, and matrix-degrading enzyme genes. Include non-targeting control (NTC) gRNAs.
- Culture Conditions: Standard media and biofilm-promoting conditions (e.g., minimal media with flow-cell or peg-lid systems like the Calgary Biofilm Device).
Method:
- Biofilm Formation & Transduction: Grow the dCas9 activator strain in a 96-well plate or flow cell for 24-48 hours to form mature biofilms.
- CRISPRa Delivery: Transduce the biofilm with the pooled gRNA library at a high multiplicity of infection (MOI) to ensure most cells receive multiple gRNAs. For resistant pathogens, consider lipid nanoparticle (LNP) delivery, which has been shown to achieve high delivery efficiency in complex biological environments [16] [72].
- Induction & Selection: Induce dCas9 and gRNA expression (e.g., with IPTG) for 24 hours.
- Harvest Dispersed Cells: Gently collect the planktonic cells released into the supernatant and the remaining biofilm cells into separate fractions.
- Genomic DNA Extraction & Sequencing: Islect gDNA from both fractions and amplify the gRNA region via PCR for next-generation sequencing.
- Hit Identification: gRNAs that are significantly enriched in the dispersed cell fraction compared to the biofilm fraction (calculated using tools like MAGeCK or DESeq2) represent hits that successfully triggered dispersal [62].
Protocol: Quantifying Antibiotic Re-Sensitization Post-Dispersal
Objective: To measure the change in antibiotic susceptibility of CRISPRa-dispersed cells compared to untreated biofilms and planktonic cells.
Materials:
- Hit gRNA Strains: Bacterial strains containing the top-performing gRNA hits identified in Section 3.1.
- Antibiotics: A panel of relevant antibiotics (e.g., fluoroquinolones, aminoglycosides, β-lactams).
- Equipment: Microtiter plate reader, colony counter, scanning electron microscopy (SEM) or confocal laser scanning microscopy (CLSM).
Method:
- Generate Experimental Groups:
- Group A (CRISPRa-Dispersed): Biofilms treated with the hit gRNA + dCas9 activator.
- Group B (Untreated Biofilm): Biofilms treated with a non-targeting control gRNA.
- Group C (Log-phase Planktonic): Conventional planktonic cultures.
- Collect Cells: After induction, harvest dispersed cells from Group A. For Groups B and C, harvest and standardize cell density.
- Antibiotic Exposure: Expose all groups to a range of antibiotic concentrations (e.g., in a 96-well plate) for a defined period.
- Viability Assessment: Perform serial dilutions and plate for colony-forming unit (CFU) counts to determine the number of surviving cells.
- Data Analysis:
- Calculate the Minimum Inhibitory Concentration (MIC) for each group.
- Perform Time-Kill Assays to determine the rate of bactericidal action.
- The success of re-sensitization is indicated by a significant reduction in MIC and an accelerated killing rate in Group A compared to Group B, ideally approaching the susceptibility profile of Group C.
Table 2: Example Data Output from a Re-Sensitization Assay Against Ciprofloxacin
| Bacterial Population | Baseline MIC (µg/mL) | MIC Post-CRISPRa (µg/mL) | Log Reduction in CFU after 4h |
|---|---|---|---|
| Untreated Biofilm | 128 | Not Applicable | 0.5 |
| Log-Phase Planktonic | 4 | Not Applicable | 4.0 |
| CRISPRa-Dispersed (gRNA-1) | 128 | 8 | 3.5 |
| CRISPRa-Dispersed (gRNA-2) | 128 | 16 | 2.8 |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CRISPRa Biofilm Dispersal Research
| Reagent / Tool | Function | Example & Notes |
|---|---|---|
| dCas9 Activator | Catalytic core for targeted gene activation. | dCas9-VPR: A high-potency fusion (VP64-p65-Rta) for strong transcriptional upregulation [62]. |
| gRNA Expression Vector | Delivers the targeting guide RNA. | Lentiviral vectors for stable integration or high-copy plasmids for transient expression. piggyBac transposon systems offer stable genomic integration without viral components [62]. |
| Delivery Vehicle | Introduces CRISPRa components into bacterial cells. | Lipid Nanoparticles (LNPs): Ideal for in vivo delivery and hard-to-transfect biofilms; show high efficacy in clinical settings [72]. Conjugative plasmids can also be engineered for efficient delivery [74]. |
| Biofilm Assay Kits | Quantifies biofilm biomass. | Crystal Violet Staining: A standard, colorimetric method. CLSM with live/dead stains (e.g., SYTO9/PI) provides 3D visualization of biofilm architecture and viability [16]. |
| CRISPRa Screening Library | Genome-scale set of gRNAs for discovery. | Custom libraries targeting promoter regions of ~100-500 genes involved in biofilm regulation, dispersal, and antibiotic resistance [62]. |
| Antibiotic Susceptibility Testing | Measures re-sensitization efficacy. | Broth Microdilution for MIC determination according to CLSI guidelines. Time-Kill Assays for dynamic, kinetic data on bactericidal activity. |
Discussion and Future Perspectives
The integration of CRISPRa-induced biofilm dispersal with conventional antibiotics represents a paradigm shift in combating chronic infections. By moving beyond the traditional goal of biofilm eradication to a more nuanced strategy of "force dispersal, then kill," this approach leverages a key vulnerability in the biofilm life cycle. The quantitative data generated from the protocols outlined herein—such as reductions in MIC and accelerated kill rates—provide a compelling proof-of-concept for drug development.
Future work must prioritize overcoming the significant challenge of in vivo delivery. Lipid nanoparticles (LNPs), which have proven successful in delivering CRISPR therapies to human patients, are a leading candidate [72]. Furthermore, the potential for resistance evolution to the CRISPRa system itself must be considered; targeting essential genes for bacterial survival in the planktonic state or employing multiplexed gRNAs against several dispersal pathways simultaneously could mitigate this risk [74].
In conclusion, the strategic activation of biofilm dispersal genes via CRISPRa creates a temporary but critical window of opportunity to resensitize resilient pathogens to our existing arsenal of antibiotics. This targeted, mechanistic approach holds immense promise for restoring the efficacy of standard therapies and tackling some of the most tenacious infections faced in modern medicine.
Conclusion
CRISPRa represents a paradigm shift in anti-biofilm strategy, moving from broad-spectrum eradication to precise, genetic-circuit-based intervention. By synthetically activating endogenous dispersal pathways, this approach offers a powerful method to dismantle biofilms and re-sensitize embedded bacteria to conventional treatments. The key takeaways underscore the necessity of sophisticated delivery systems to navigate the biofilm matrix and the importance of comprehensive validation against complex, multi-species communities. Future directions must focus on advancing in vivo delivery platforms, integrating AI for predictive gRNA design, and navigating the regulatory pathway for these novel precision antimicrobials. Success in this field holds the potential to significantly impact the treatment of chronic infections and redefine our arsenal against antimicrobial resistance.
References
- 1. The biofilm matrix | Nature Reviews Microbiology [https://www.nature.com/articles/nrmicro2415?error=cookies_not_supported]
- 2. Microbial biofilm : formation, architecture , antibiotic resistance, and... [https://pubmed.ncbi.nlm.nih.gov/34558029/]
- 3. Beyond antibiotics: CRISPR/Cas9 triumph over biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11257871/]
- 4. Frontiers | Beyond antibiotics: CRISPR/Cas9 triumph over... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2024.1408569/full]
- 5. Shared biophysical mechanisms determine early biofilm ... architecture [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3001846]
- 6. CRISPR interference to interrogate genes that control ... [https://www.nature.com/articles/s41598-019-52400-5]
- 7. CRISPR–Cas systems as emerging tools for precision ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925021416]
- 8. A CRISPRi Gene Regulation System for Bifidobacteria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12592239/]
- 9. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://bmcmedicine.biomedcentral.com/articles/10.1186/s12916-025-04323-4]
- 10. Cas3 of type I-Fa CRISPR-Cas system upregulates ... [https://www.nature.com/articles/s42003-025-08124-6]
- 11. Controlling biofilm development through cyclic di-GMP signaling [https://pmc.ncbi.nlm.nih.gov/articles/PMC9891824/]
- 12. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling [https://www.sciencedirect.com/science/article/pii/S0021925820408981]
- 13. Biofilm dispersion and quorum sensing [https://www.sciencedirect.com/science/article/pii/S1369527414000290]
- 14. Biofilm dispersion and quorum sensing [https://pubmed.ncbi.nlm.nih.gov/24657330/]
- 15. Secreted nucleases reclaim extracellular DNA during ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11458576/]
- 16. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12366227/]
- 17. CRISPR activation: identifying and using novel genes for ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1596600/full]
- 18. CRISPRa and CRISPRi [https://www.synthego.com/guide/crispr-methods/crispri-crispra/]
- 19. CRISPRa: A Simpler Way to CRISPR Gene Activation [https://bitesizebio.com/44248/crispr-based-activation-crispra-of-genes-a-how-to-guide/]
- 20. Targeted regulation of transcription in primary cells using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8559706/]
- 21. Arrayed CRISPR libraries for the genome-wide activation ... [https://www.nature.com/articles/s41551-024-01278-4]
- 22. CRISPR activation screens: navigating technologies and ... [https://www.sciencedirect.com/science/article/pii/S0167779924000362]
- 23. CRISPR Tools To Control Gene Expression in Bacteria [https://pmc.ncbi.nlm.nih.gov/articles/PMC7117552/]
- 24. CRISPR-CAS9 Against Antibiotic Resistance-A Review [https://pubmed.ncbi.nlm.nih.gov/38702575/]
- 25. CRISPR interference to interrogate genes that control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6828691/]
- 26. Spatiotemporal control of CRISPR/Cas9 gene editing [https://www.nature.com/articles/s41392-021-00645-w]
- 27. Functional genetic characterization of stress tolerance and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10900893/]
- 28. Precision targeting of food biofilm-forming genes by microbial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9396135/]
- 29. Comprehensive approaches to design efficient gRNA for ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1579165/full]
- 30. Effective CRISPRa-mediated control of gene expression in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7113249/]
- 31. sgRNA Design Principles for Optimal CRISPR Efficiency [https://synapse.patsnap.com/article/sgrna-design-principles-for-optimal-crispr-efficiency]
- 32. Establishment of a CRISPR/dCas9 Activation Library for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12427770/]
- 33. A Neuron-Optimized CRISPR/dCas9 Activation System for ... [https://www.eneuro.org/content/6/1/ENEURO.0495-18.2019]
- 34. Biofilm dispersion and quorum sensing [https://www.sciencedirect.com/science/article/abs/pii/S1369527414000290]
- 35. Biofilms and multidrug resistance: an emerging crisis ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1625356/full]
- 36. A Neuron-Optimized CRISPR/dCas9 Activation System for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6412672/]
- 37. Bacteriophage-mediated approaches for biofilm control [https://pmc.ncbi.nlm.nih.gov/articles/PMC11491440/]
- 38. Nanotechnology-Based Drug Delivery Systems to Control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10674810/]
- 39. Bacteriophage-Mediated Control of Biofilm: A Promising ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.825828/full]
- 40. Nanomaterial-enabled anti-biofilm strategies - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d4nr04774e]
- 41. Nanoparticles targeting biofilms: A new era in combating ... [https://www.sciencedirect.com/science/article/pii/S2590097825000370]
- 42. Targeting bacterial biofilm-related genes with nanoparticle ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1387114/full]
- 43. CRISPR Activation: Identifying and Using Novel Genes for ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1596600/abstract]
- 44. Revisiting ESKAPE Pathogens: virulence, resistance, and ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2023.1159798/full]
- 45. Application of CRISPR-Cas system in the diagnosis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10470840/]
- 46. CRISPR-Cas, a Revolution in the Treatment and Study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8300728/]
- 47. CRISPR-Cas, a Revolution in the Treatment and Study of ... [https://www.mdpi.com/2079-6382/10/7/756]
- 48. CRISPR-Cas mediated targeting of resistance genes for ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813025097375]
- 49. CRISPRi-Mediated Gene Suppression Reveals Putative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9792118/]
- 50. Therapeutic Strategies against Biofilm Infections - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9866932/]
- 51. Frontiers | Innovative Strategies Toward the Disassembly of the EPS ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.00952/full]
- 52. Contemporary strategies and approaches for ... [https://www.sciencedirect.com/science/article/pii/S2095177923002794]
- 53. CRISPR Interference (CRISPRi) Inhibition of luxS Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5445563/]
- 54. Quantitative image analysis of microbial communities with ... [https://www.nature.com/articles/s41564-020-00817-4]
- 55. Quantitative and Qualitative Assessment Methods for Biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6133255/]
- 56. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 57. CRISPR-Cas9 and Its Bioinformatics Tools: A Systematic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109910/]
- 58. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 59. Unlocking the potential of CRISPR tools and databases for ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1563711/full]
- 60. Time-resolved compositional and dynamics analysis of biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11779841/]
- 61. Massively parallel characterization of CRISPR activator ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10114495/]
- 62. Multiplex, single-cell CRISPRa screening for cell type ... [https://www.nature.com/articles/s41467-024-52490-4]
- 63. Functional genetic characterization of stress tolerance and ... [https://pubmed.ncbi.nlm.nih.gov/38265239/]
- 64. Phage-delivered CRISPR-Cas9 for strain-specific ... [https://www.sciencedirect.com/science/article/pii/S2211124721014030]
- 65. Innovative approaches to combat antibiotic resistance [https://pubmed.ncbi.nlm.nih.gov/40830872/]
- 66. Combining biofilm matrix measurements with biomass and ... [https://www.nature.com/articles/ja201249]
- 67. Cell death and biomass reduction in biofilms of multidrug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7644070/]
- 68. 🧪 Monthly Insights into Biofilm Research – May 2025 [https://www.linkedin.com/pulse/monthly-insights-biofilm-research-may-2025-pawel-tulinski-phd-lvvtf]
- 69. of Mechanisms resistance in antimicrobial biofilms [https://www.nature.com/articles/s44259-024-00046-3?error=cookies_not_supported&code=b6660f1c-06ae-4a86-904a-0ff6968e4bd9]
- 70. Systematic Mapping of Bacterial CRISPRa Systems for ... [https://pubmed.ncbi.nlm.nih.gov/40693287/]
- 71. Recent Strategies to Combat Biofilms Using Antimicrobial ... [https://www.mdpi.com/2076-0817/11/3/292]
- 72. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 73. Genome-Scale CRISPR-Mediated Control of Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4253859/]
- 74. Effectiveness of CRISPR-Cas in sensitizing bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606438/]