Single-Cell Techniques for Bacterial Persisters: From Mechanisms to Therapeutic Breakthroughs
Bacterial persisters, a subpopulation of dormant, antibiotic-tolerant cells, are a major cause of recurrent and chronic infections, posing a significant challenge in clinical settings.
Single-Cell Techniques for Bacterial Persisters: From Mechanisms to Therapeutic Breakthroughs
Abstract
Bacterial persisters, a subpopulation of dormant, antibiotic-tolerant cells, are a major cause of recurrent and chronic infections, posing a significant challenge in clinical settings. This article provides a comprehensive overview of how advanced single-cell technologies are revolutionizing the study of these elusive cells. We explore the foundational biology of persisters and their distinction from resistant bacteria, detail cutting-edge methodological approaches including microfluidics, fluorescent biosensors, and single-cell RNA sequencing, and discuss optimization strategies to overcome technical hurdles. By comparing and validating insights gained from these techniques, we highlight their collective power in uncovering the molecular mechanisms of persistence. This synthesis aims to equip researchers and drug development professionals with the knowledge to leverage single-cell analysis for developing novel anti-persister therapies and improving infectious disease treatment outcomes.
Understanding the Persister Phenotype: Why Single-Cell Analysis is Essential
The failure of antibiotic therapy often stems not from conventional genetic resistance, but from the remarkable ability of a subpopulation of bacterial cells to enter a transient, dormant state. Within the context of single-cell bacterial research, it is crucial to distinguish between three key survival phenotypes: antibiotic resistance, antibiotic tolerance, and the viable but non-culturable (VBNC) state. Antibiotic resistance is typically defined by heritable genetic changes that enable bacteria to grow in the presence of an antibiotic, quantified by the minimum inhibitory concentration (MIC) [1]. In contrast, antibiotic tolerance and persistence are non-heritable, phenotypic traits that allow bacteria to survive prolonged exposure to lethal antibiotic concentrations without growing [2] [3]. The VBNC state represents a condition where cells are viable and metabolically active but cannot form colonies on routine media that would normally support their growth, and are capable of returning to a culturable state under favorable conditions [2] [4]. This application note, framed within a broader thesis on single-cell techniques, provides researchers with clear definitions, quantitative distinctions, and detailed protocols essential for studying these elusive bacterial subpopulations.
Defining the Phenotypes: Resistance, Tolerance, Persistence, and the VBNC State
The following table summarizes the core characteristics that differentiate these key survival strategies.
Table 1: Key Characteristics of Bacterial Survival Phenotypes
| Feature | Antibiotic Resistance | Antibiotic Tolerance/Persistence | VBNC State |
|---|---|---|---|
| Heritability | Heritable (genetic) | Non-heritable (phenotypic) | Non-heritable (phenotypic) |
| Growth on Media | Grows in drug presence | Cannot grow during drug exposure, but culturable after | Non-culturable on routine media |
| Primary Metric | Increased MIC [1] | Increased MDK (Minimum Duration of Killing) [1] | Loss of culturability, maintained viability [2] |
| Mechanism | Target modification, efflux pumps, enzyme inactivation | Dormancy, slowed metabolism, toxin-antitoxin systems [5] [3] | Profound metabolic shutdown, morphological changes [6] [4] |
| Population | Entire population | A small subpopulation (persisters) or entire population (tolerance) [2] [3] | Can be a large fraction or entire population [2] |
| Resuscitation | Not applicable | Resumes growth upon antibiotic removal | Can resuscitate under specific environmental cues [6] [4] |
The Relationship Between Persisters and VBNC Cells
A critical concept emerging from recent research is that these dormant states are not entirely distinct but may exist on a continuum of dormancy [2]. In this model, VBNC cells are in a deeper state of dormancy compared to persister cells. While both are dormant and exhibit low metabolic activity, the defining difference lies in culturability: persister cells can resume growth on standard laboratory media once the antibiotic is removed, whereas VBNC cells require specific resuscitation conditions to regain culturability [2] [4]. Some researchers have even suggested that "persisters" and "VBNC" cells may be variants of the same phenomenon [4]. The following diagram illustrates this continuum and the environmental triggers that drive bacterial populations between these states.
Quantitative Distinctions and Key Research Metrics
For researchers, quantifying these phenotypes is paramount. The table below outlines the essential experimental parameters and their interpretations.
Table 2: Core Experimental Metrics for Differentiating Survival Phenotypes
| Experimental Metric | Definition & Measurement | Interpretation and Significance |
|---|---|---|
| Minimum Inhibitory Concentration (MIC) | The lowest antibiotic concentration that prevents visible growth [1]. | A raised MIC indicates antibiotic resistance. Tolerant/persister and VBNC populations do not typically show a change in MIC. |
| Minimum Duration for Killing (MDK) | The time required to kill 99% of the bacterial population at a high antibiotic concentration (>>MIC) [1]. | An increased MDK is the hallmark of tolerance. The biphasic killing curve, with a subpopulation surviving longer, defines persistence [2] [7]. |
| Culturability vs. Viability | Culturability is measured by colony-forming units (CFU). Viability is assessed via methods like PMA-qPCR (see Protocol 2) or fluorescence-based viability kits [8]. | A population with high viability but low or zero culturability is indicative of the VBNC state [2] [8]. |
| Resuscitation Window | The time period during which VBNC cells can revert to a culturable state upon receiving a specific stimulus (e.g., temperature upshift, nutrient addition) [6]. | A defined resuscitation window confirms the VBNC state, distinguishing it from cell death. The window can be extended by certain metabolites like lactate [6]. |
Essential Single-Cell Protocols for Persister and VBNC Research
Protocol 1: Assessing Single-Cell Persister Recovery Kinetics
This protocol, adapted from Wilmaerts et al., details the steps to isolate and study the recovery of persister cells at the single-cell level, which is crucial for understanding the heterogeneity within the persister subpopulation [7].
Key Research Reagent Solutions:
- LB Medium: Standard lysogeny broth for culturing E. coli.
- Antibiotic Stock Solution: e.g., Amikacin at 10 mg/mL in milli-Q water, filter-sterilized. The specific antibiotic and concentration must be optimized for the bacterial strain.
- 10 mM MgSO₄ Solution: Used for optical density adjustments without promoting bacterial growth.
Procedure:
- Determine MIC: Perform a broth microdilution susceptibility test to determine the Minimum Inhibitory Concentration (MIC) for the antibiotic and bacterial strain being used [7].
- Time-Kill Assay: Treat a stationary-phase culture with a high concentration of antibiotic (e.g., 10x MIC) for a duration determined to reach the "persister plateau," where the killing rate slows significantly. Sample at intervals to monitor the decline in CFU/mL [7].
- Persister Isolation: After antibiotic treatment, wash the cells by centrifugation to remove the antibiotic. Resuspend the cell pellet in fresh, pre-warmed medium.
- Single-Cell Recovery and Monitoring: Serially dilute the persister suspension to a concentration that yields individual cells. Using spectrophotometry or, ideally, single-cell techniques like time-lapse microscopy, monitor the lag time and growth resumption of individual persister cells to quantify heterogeneity in recovery kinetics [7].
Protocol 2: Detection and Quantification of VBNC Cells using PMA-qPCR
This protocol is essential for studying VBNC cells, as it differentiates them from both culturable and dead cells. It is based on methods used for Campylobacter jejuni but can be adapted for other species [8].
Key Research Reagent Solutions:
- Propidium Monoazide (PMA): A DNA-intercalating dye that selectively enters membrane-compromised dead cells. Upon photoactivation, it forms covalent bonds with DNA, inhibiting its amplification in PCR.
- Specific Primers: e.g., primers targeting the rpoB gene for C. jejuni [8].
- DNA Extraction Kit: e.g., Presto Mini gDNA Bacteria Kit.
Procedure:
- Induce VBNC State: Subject a bacterial culture to a specific stressor (e.g., 7% NaCl for osmotic stress, low temperature) until CFU counts on solid media drop to zero.
- PMA Treatment: Add an optimized concentration of PMA (e.g., 20 µM) to the sample and incubate in the dark. Then, expose the sample to a high-intensity halogen light source to photoactivate the dye.
- DNA Extraction and qPCR: Extract genomic DNA from the PMA-treated sample. Perform qPCR with species-specific primers.
- Quantification: The qPCR signal from the PMA-treated, non-culturable sample represents the VBNC population, as PMA has suppressed the signal from dead cells with compromised membranes. The number of culturable cells (CFU/mL) is subtracted from the total viable count (from PMA-qPCR) to estimate the VBNC cell count [8].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Persister and VBNC Research
| Research Reagent / Material | Function in Experimental Workflow |
|---|---|
| Propidium Monoazide (PMA) | Differentiates viable cells (with intact membranes) from dead cells (with compromised membranes) in molecular assays like qPCR [8]. |
| Viability Stains (e.g., LIVE/DEAD BacLight) | Used in conjunction with flow cytometry or microscopy to visually assess cell membrane integrity and viability at the single-cell level. |
| Specific Resuscitation Signals (e.g., Lactate) | Used to trigger the resuscitation of VBNC cells back to a culturable state. For example, lactate extends the resuscitation window for Vibrio parahaemolyticus [6]. |
| 96-well Microtiter Plates | Standard platform for high-throughput assays, including MIC determinations and growth curve analyses [7]. |
| Filters for Antibiotic Sterilization | 0.22 µm filters for preparing sterile antibiotic stock solutions to avoid contamination [7]. |
The precise distinction between antibiotic resistance, tolerance, persistence, and the VBNC state is fundamental for advancing research into chronic and recurrent bacterial infections. The definitions, quantitative metrics, and single-cell protocols detailed in this application note provide a framework for researchers to accurately identify and characterize these phenotypes. Moving forward, leveraging these tools will be critical for elucidating the molecular mechanisms driving dormancy and for developing novel therapeutic strategies that effectively target all dormant bacterial subpopulations.
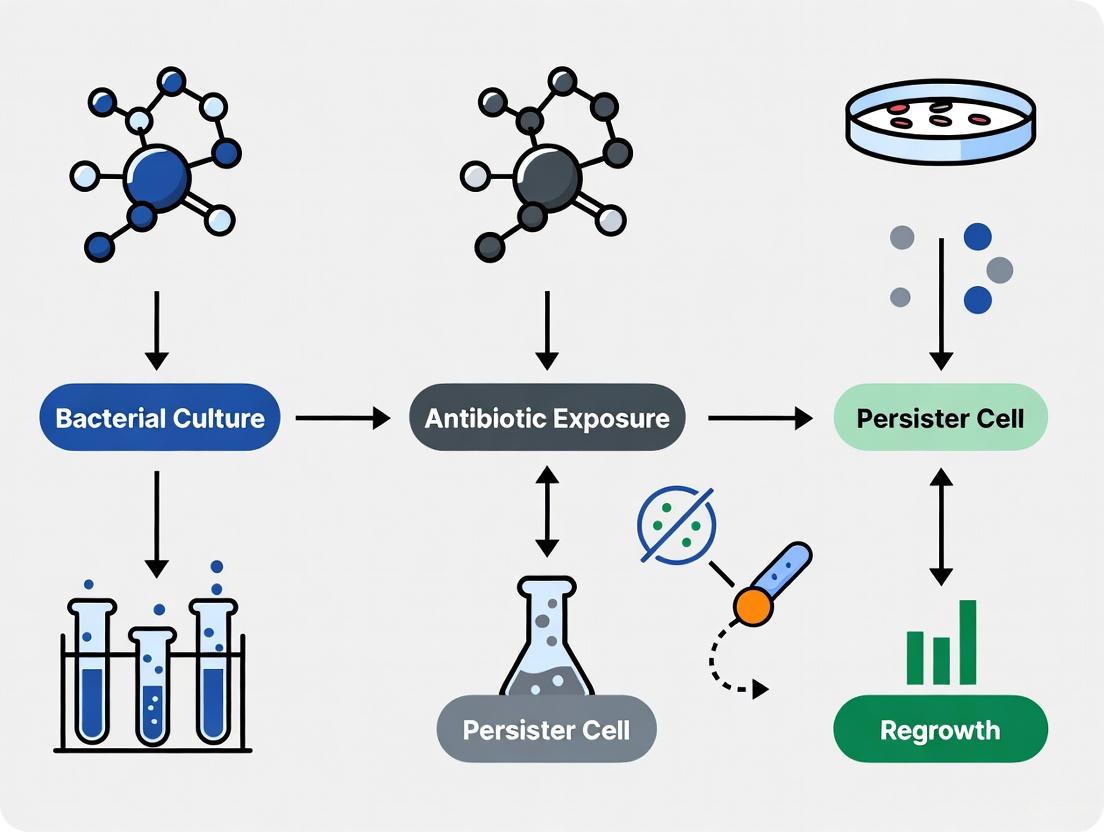
Bacterial persisters represent a non-genetic, phenotypic variant of a bacterial population that exhibits exceptional tolerance to high doses of conventional antibiotics and can regrow once antibiotic pressure is removed [9]. These cells are not antibiotic-resistant, as demonstrated by their unchanged Minimum Inhibitory Concentration (MIC) compared to normal cells, but they survive treatment by entering a transient, slow-growing or non-growing dormant state [3] [9]. This phenomenon is a significant clinical concern because persisters are implicated in the recalcitrance of chronic and recurrent infections, leading to treatment failures and prolonged therapeutic courses [10] [11]. They are considered a major culprit behind relapsing infections such as tuberculosis, recurrent urinary tract infections, cystic fibrosis-associated lung infections, and infections linked to biofilms on medical implants [3] [10]. Understanding and targeting persisters is therefore a critical frontier in the fight against persistent bacterial infections. This application note details the core concepts, quantitative profiles, and advanced single-cell methodologies essential for researching this challenging subpopulation of bacteria.
Quantitative Profiling of Bacterial Persisters
A comprehensive understanding of bacterial persistence requires a grasp of its quantitative aspects across different bacterial species, antibiotics, and growth conditions. The following tables consolidate key data from scientific surveys to provide a reference for experimental design and interpretation.
Table 1: Bacterial Pathogens Known to Form Persisters in Clinical Contexts
| Bacterial Species | Associated Disease(s) | Primary Tools for Persister Analysis |
|---|---|---|
| Mycobacterium tuberculosis | Tuberculosis | CFU, Single-Cell Raman Spectroscopy (SCRS), Transcriptomics [9] |
| Pseudomonas aeruginosa | Cystic fibrosis lung infections, chronic suppurative otitis media | Colony-Forming Unit (CFU) [9] [11] |
| Staphylococcus aureus | Osteomyelitis, endocarditis, infections of indwelling devices | CFU, Fluorescence-Activated Cell Sorting (FACS), Signature-Tagged Mutagenesis (STM) [9] |
| Escherichia coli | Recurrent urinary tract infections, sepsis | CFU, FACS, Imaging Flow Cytometry (IFC), Microfluidic Culture, Microscopy [9] |
| Borrelia burgdorferi | Lyme disease | CFU, Microscopy [9] |
| Salmonella enterica | Acute gastroenteritis, systemic infections | CFU [9] |
| Candida albicans | Oral, gastrointestinal, and vaginal infections; Septicemia | CFU [9] [11] |
Table 2: Survey of Persister Levels and Influencing Factors
| Factor Category | Observation | Quantitative Example / Note |
|---|---|---|
| Antibiotic Class | Persistence levels vary significantly by mechanism of action. Membrane-active antibiotics typically yield the fewest persisters [12]. | The median percentage of surviving cells can span five orders of magnitude, from as low as 7 × 10⁻⁴% in P. putida to 100% in E. faecium under certain conditions [12]. |
| Growth Phase | Persister fractions are generally lower in exponentially growing cells and higher in stationary-phase cultures and biofilms [12] [11]. | In E. coli, the proportion of persisters is low during the log phase and increases significantly in the stationary and death phases [11]. |
| Strain Variation | Different strains of the same species can exhibit vastly different persister fractions, and this is often antibiotic-specific [13]. | An E. coli strain may show a high persister fraction with one antibiotic but a low fraction with another, even if the two drugs have similar modes of action [13]. |
| Gram Staining | Persistence is generally observed to be more common in Gram-positive bacteria compared to Gram-negatives [12]. | This observation is based on a broad survey across multiple species and antibiotic treatments [12]. |
Experimental Protocols for Persister Research
Protocol 1: Isolation and Analysis of Persisters via Flow Cytometry and Fluorescent Protein Dilution
This protocol leverages flow cytometry to distinguish and quantify persister, Viable But Non-Culturable (VBNC), and dead cell subpopulations following beta-lactam antibiotic treatment [14].
1. Principle: Ampicillin, a beta-lactam antibiotic, primarily kills growing cells by inhibiting cell wall synthesis. Non-growing persister and VBNC cells survive this treatment intact. By pre-loading cells with a fluorescent protein and monitoring its dilution upon resumption of growth, one can track the resuscitation of persisters at the single-cell level [14].
2. Research Reagent Solutions:
| Item | Function/Brief Explanation |
|---|---|
| E. coli strain with IPTG-inducible fluorescent protein (e.g., mCherry) cassette | Enables tracking of cell division through dilution of the fluorescent protein in daughter cells. |
| Ampicillin | Beta-lactam antibiotic used to lyse growing, sensitive cells and enrich for persisters. |
| IPTG (Isopropyl β-d-1-thiogalactopyranoside) | Inducer for the expression of the fluorescent protein. |
| Luria-Bertani (LB) Broth/Agar | Standard culture medium for growing E. coli. |
| Flow Cytometer | Instrument for quantifying and characterizing fluorescent cell populations at high throughput. |
| Propidium Iodide (PI) or similar viability stain | Optional; stains dead cells with compromised membranes, providing an additional viability parameter. |
3. Procedure:
- Step 1: Fluorescent Protein Induction. Inoculate an overnight culture of the reporter strain in LB medium with the appropriate inducer (e.g., IPTG) to ensure high fluorescent protein expression in all cells [14].
- Step 2: Experimental Culture and Antibiotic Treatment. Dilute the overnight culture in fresh, pre-warmed LB medium (with IPTG) and grow to mid-exponential phase (OD₆₀₀ ~0.25). Treat the culture with a lethal dose of ampicillin (e.g., 100 µg/mL) for a sufficient time (e.g., 3 hours) to achieve a biphasic kill curve, ensuring the death of non-persister cells [14].
- Step 3: Wash and Resuscitation. Pellet the cells, wash thoroughly with fresh LB medium to remove the antibiotic and IPTG, and resuspend in fresh LB broth. This step removes the antibiotic pressure and allows persisters to resuscitate.
- Step 4: Flow Cytometry Analysis. Monitor the cells over time using a flow cytometer. Measure forward scatter (FSC, indicative of cell size) and fluorescence intensity.
- Resuscitating Persisters: Cells that begin to divide will dilute their fluorescent protein, showing a decrease in fluorescence intensity over time. Their FSC may also increase due to cell elongation.
- VBNC Cells: These intact, live cells will retain high fluorescence as they do not divide.
- Dead Cells/Debris: Will typically exhibit low FSC and fluorescence [14].
- Step 5: Data Analysis. Calculate the initial number of resuscitating persisters and their doubling time based on the decay of fluorescence and cell count data from the flow cytometer [14].
Figure 1: Flowchart of the flow cytometry-based persister resuscitation protocol.
Protocol 2: Single-Cell Dynamics of Persisters Using Microfluidics
This protocol utilizes microfluidic devices to track the pre- and post-antibiotic exposure history of individual persister cells, providing unparalleled insight into their heterogeneous behaviors [15].
1. Principle: A microfluidic device with a membrane-covered microchamber array (MCMA) traps single cells and small microcolonies, allowing for continuous, high-resolution microscopy. The device permits rapid medium exchange, enabling researchers to observe the same cells before, during, and after antibiotic challenge, directly linking a cell's prior state to its survival outcome [15].
2. Research Reagent Solutions:
| Item | Function/Brief Explanation |
|---|---|
| Microfluidic Device (e.g., MCMA) | Platform for long-term, single-cell imaging under controlled and dynamically changing conditions. |
| Wild-type or Fluorescent Reporter Bacterial Strains | For observation; reporter strains can reveal physiological states (e.g., stress response). |
| Lethal Concentrations of Antibiotics | e.g., Ampicillin (200 µg/mL) or Ciprofloxacin (1 µg/mL) for E. coli MG1655. |
| Controlled Environment Microscope | System for maintaining temperature and conducting time-lapse imaging. |
| Cell Culture Media | Rich (e.g., LB) and/or defined minimal media for growing cells in the device. |
3. Procedure:
- Step 1: Device Priming and Cell Loading. Prime the microfluidic device with the appropriate growth medium. Load a diluted bacterial culture (from either exponential or stationary phase) into the device, allowing cells to be trapped in the microchambers [15].
- Step 2: Pre-Treatment Imaging. With fresh medium flowing, image the trapped cells for several hours to establish a baseline growth history for each cell. Track parameters like division time, cell size, and morphology.
- Step 3: Antibiotic Exposure. Switch the medium flow to one containing a lethal concentration of antibiotic. Continue time-lapse imaging for the duration of the treatment (e.g., 3-8 hours). Observe the lysis of sensitive cells and the behavior of surviving cells.
- Step 4: Post-Antibiotic Recovery. Switch the flow back to fresh, drug-free medium. Continue imaging for an extended period (e.g., 24-48 hours) to monitor for regrowth (resuscitation) of any surviving persister cells.
- Step 5: Lineage Tracking and Phenotype Classification. Analyze the image data to reconstruct the lineage of every persister cell that regrew. Correlate their post-antibiotic behavior (e.g., continuous growth, growth arrest, filamentation, L-form like morphology) with their pre-antibiotic growth state (growing or non-growing) [15].
Figure 2: Workflow for analyzing persister dynamics using microfluidic single-cell analysis.
The Scientist's Toolkit: Key Reagent Solutions
The following table consolidates essential reagents and tools for advanced persister research, as featured in the protocols and literature.
Table 3: Key Research Reagent Solutions for Persister Studies
| Category | Item | Specific Function in Persister Research |
|---|---|---|
| Single-Cell Analysis Platforms | Microfluidic Devices (e.g., MCMA) | Enables long-term, high-resolution tracking of individual cell lineages before, during, and after antibiotic stress [15]. |
| Flow Cytometer | High-throughput quantification and sorting of cell subpopulations based on size, granularity, and fluorescence [9] [14]. | |
| Critical Reagents | Beta-lactam Antibiotics (e.g., Ampicillin) | Used to enrich for persisters by selectively lysing growing, cell wall-synthesizing cells [14]. |
| Fluorescent Protein Reporter Systems (e.g., mCherry, GFP) | Visualize cell growth, division, and physiological states via protein expression and dilution [14] [15]. | |
| Metabolic Inhibitors (e.g., Arsenate) | Used to manipulate cellular ATP levels and study the relationship between metabolism and persistence [14]. | |
| Detection & Viability Tools | Colony-Forming Unit (CFU) Assays | The gold standard for quantifying culturable, resuscitating persister cells [9]. |
| Viability Stains (e.g., Propidium Iodide) | Distinguishes cells with compromised membranes (dead) from those with intact membranes (live) [14]. | |
| Single-Cell Raman Spectroscopy (SCRS) | Provides a label-free biochemical fingerprint of individual cells, useful for identifying dormant states [9]. |
Phenotypic heterogeneity is a fundamental survival strategy in which genetically identical bacterial cells within a clonal population exhibit diverse physiological states. This bet-hedging strategy ensures that a subset of cells, known as persisters, can survive transient environmental stresses such as antibiotic exposure, even though the entire population remains genetically susceptible [16] [17]. These persister cells are typically characterized by a transient, low-metabolism, or dormant state that reduces the efficacy of conventional antibiotics which predominantly target active cellular processes [3] [18]. The clinical significance of persisters is profound, as they underlie the challenges in treating chronic and recurrent infections, including tuberculosis, urinary tract infections, and biofilm-associated infections [3] [17]. Understanding and investigating phenotypic heterogeneity is therefore critical for developing more effective therapeutic strategies against persistent bacterial infections.
This Application Note provides a structured framework for studying bacterial phenotypic heterogeneity and persistence, with a specific focus on single-cell analytical techniques. We summarize current methodologies, provide detailed protocols for key experiments, and outline the essential reagents and tools required for a comprehensive research workflow in this field.
Core Concepts and Classifications
Phenotypic heterogeneity manifests in distinct types of persister cells, primarily classified based on their formation mechanisms. Table 1 summarizes the defining characteristics of the three established types of bacterial persisters.
Table 1: Classification and Characteristics of Bacterial Persister Cells
| Persister Type | Formation Trigger | Metabolic/Growth State | Key Regulatory Factors |
|---|---|---|---|
| Type I (Triggered) | Environmental stress; Stationary phase [3] [18] | Pre-existing, non-growing [3] [18] | Toxin-Antitoxin (TA) systems, RpoS [3] [19] |
| Type II (Stochastic) | Spontaneous, stochastic process [3] [18] | Slow-growing throughout population lifecycle [3] [18] | (p)ppGpp alarmone, TA systems [3] [20] |
| Type III (Specialized) | Specific antibiotic-induced stress signals [18] | Not necessarily slow-growing; mechanism-specific [18] | e.g., low catalase-peroxidase (for isoniazid persistence) [18] |
The formation and survival of these persister subpopulations are governed by a complex interplay of molecular mechanisms. The stress alarmone ppGpp emerges as a central regulator, integrating various stress signals to induce a multidrug-tolerant state [20]. Its action is often mediated through Toxin-Antitoxin (TA) modules, which function as downstream effectors. In these systems, the toxin component (e.g., MqsR) can disrupt essential processes like translation by cleaving mRNA, promoting dormancy [19] [20]. The general stress response sigma factor RpoS also plays a critical role, with studies showing that suppression of the RpoS-mediated stress response can dramatically increase persistence [19]. Furthermore, other pathways including quorum sensing, drug efflux pumps, and the SOS response to DNA damage contribute to the formation of the persistent state [3] [18]. The following diagram illustrates the relationships between these key mechanisms and their convergence on the persister cell state.
Single-Cell Technologies for Phenotypic Heterogeneity Analysis
Population-averaged measurements often mask rare persister subpopulations. Single-cell technologies are therefore indispensable for dissecting phenotypic heterogeneity. The following table compares the key quantitative applications of major single-cell techniques used in persister research.
Table 2: Single-Cell Technologies for Analyzing Phenotypic Heterogeneity and Persistence
| Technology | Key Measurable Parameters (Quantitative Data) | Typical Persister Frequency Detected | Key Advantages / Applications |
|---|---|---|---|
| Flow Cytometry | Fluorescence intensity (gene expression), cell size, complexity [17] | N/A | High-throughput analysis and sorting of rare cell populations [16] [17] |
| Microfluidics | Single-cell growth rates, lag time, resuscitation dynamics, real-time gene expression [16] [17] | N/A | Long-term, temporal monitoring of individual cells under controlled conditions [16] |
| Single-Cell RNA-Seq (scRNA-seq) | Global transcriptomes of individual bacteria; 1,000+ transcripts per cell under optimal conditions [21] | N/A | Unbiased discovery of transcriptional states and subpopulations without prior genetic engineering [21] |
| Fluorescent Reporters & Biosensors | Relative mRNA/protein abundance, intracellular ATP levels, metabolite concentrations (e.g., c-di-GMP) [17] | N/A | Dynamic, live-cell imaging of metabolic activity and gene expression heterogeneity [17] |
| Raman Spectroscopy | Intracellular biochemical composition at the single-cell level [16] | N/A | Label-free analysis of metabolic state and antimicrobial response [16] |
| Microcolony-seq | Inherited transcriptomic profiles from single progenitor cells [22] | N/A | Links initial single-cell state to downstream phenotypic inheritance and fitness [22] |
Application Note: Tracking Stochastic Persistence with Microfluidics and Fluorescent Reporters
Objective: To investigate the formation and resuscitation of stochastic (Type II) persister cells at the single-cell level.
Background: Type II persisters arise spontaneously in growing cultures without an external trigger. This protocol uses a microfluidic platform to trap and monitor individual cells, coupled with a fluorescent growth reporter to distinguish dormant from active cells.
Materials:
- Bacterial Strain: Genetically tractable strain of interest (e.g., E. coli).
- Growth Medium: Appropriate liquid and solid media.
- Microfluidic System: Commercially available cell-asay chip or custom-designed PDMS device.
- Fluorescent Reporter: Plasmid or chromosomal fusion of a constitutive promoter to a stable fluorescent protein (e.g., P~const~-GFP).
- Antibiotic: Cidal antibiotic of choice (e.g., ampicillin or ofloxacin).
- Imaging System: Inverted microscope with an environmental chamber (maintained at 37°C), high-resolution camera, and time-lapse capability.
Protocol:
- Strain Preparation: Transform the bacterial strain with the P~const~-GFP reporter construct. Grow an overnight culture in the appropriate medium.
- Chip Priming and Loading: Dilute the overnight culture 1:100 in fresh medium and grow to mid-exponential phase (OD~600~ ≈ 0.3-0.5). Follow the manufacturer's instructions to prime the microfluidic chip with medium. Load the bacterial culture into the chip, allowing cells to be trapped in the growth chambers.
- Pre-Treatment Imaging: Continuously perfuse the chip with fresh, pre-warmed medium. Initiate time-lapse imaging, capturing phase-contrast and fluorescence images of the trapped cells every 15-30 minutes for 2-3 hours to establish baseline growth and fluorescence.
- Antibiotic Treatment: Switch the perfusion medium to one containing the antibiotic at the desired concentration (e.g., 5-10x MIC). Continue time-lapse imaging for the duration of the treatment (e.g., 4-8 hours).
- Post-Treatment Recovery: Switch the perfusion back to fresh, antibiotic-free medium. Continue time-lapse imaging for 12-24 hours to monitor for cell resuscitation and regrowth.
- Data Analysis:
- Growth Rate: Use phase-contrast images to calculate the elongation rate of individual cells before antibiotic treatment.
- Persistence Identification: A cell is classified as a persister if it maintains membrane integrity (does not lyse) during antibiotic exposure but ceases to elongate. Fluorescence intensity should remain stable, indicating a halt in protein synthesis.
- Resuscitation: Identify persister cells that resume elongation and division in the recovery phase. Quantify the lag time (time from antibiotic removal to first division) for each resuscitated persister.
Key Signaling Pathways and Workflows
The ppGpp-Mediated Persistence Pathway
The ppGpp pathway is a central hub regulating the bacterial persistence response. The following diagram details the key molecular players and their interactions leading to the formation of a persistent, multidrug-tolerant cell.
Workflow for Single-Cell RNA-Sequencing of Bacterial Persisters
Single-cell RNA-sequencing (scRNA-seq) enables unbiased transcriptomic profiling of individual bacteria, revealing the heterogeneity within a population. The workflow below outlines the major steps from cell preparation to data analysis, specifically tailored for the challenging task of capturing rare persister cells.
Detailed Protocol Steps:
- Culture & Antibiotic Treatment: Grow the bacterial culture to the desired phase. Treat with a cidal antibiotic at an appropriate concentration (e.g., 5-10x MIC) for a defined period to enrich for, but not exclusively isolate, persisters. Include a viability stain (e.g., propidium iodide) if sorting will be used.
- Single-Cell Isolation: Critically, standard bacterial scRNA-seq requires sorting single cells into multi-well plates containing lysis buffer [21]. Commercial droplet-based systems (e.g., 10x Genomics) are optimized for larger eukaryotic cells and may not be suitable for most bacteria. Fluorescence-Activated Cell Sorting (FACS) can be used to sort cells based on specific markers or viability stains.
- Cell Lysis & mRNA Capture: Lyse sorted cells in a buffer containing detergents. For poly(A)-independent transcriptome capture, use random hexamers instead of oligo-d(T) primers during reverse transcription to avoid bias against non-polyadenylated bacterial mRNAs [21].
- cDNA Synthesis & Library Prep: Perform reverse transcription to generate cDNA. Amplify the cDNA using a method like Smart-seq2 for high sensitivity [21]. Prepare sequencing libraries with unique barcodes for each cell to enable multiplexing.
- High-Throughput Sequencing: Sequence the libraries on an Illumina platform to a sufficient depth to detect low-abundance transcripts characteristic of dormant cells.
- Bioinformatic Analysis: Process raw sequencing data (quality control, adapter trimming). Map reads to the reference genome. Generate a gene expression matrix (genes x cells). Perform downstream analyses: clustering, differential expression, and trajectory inference to identify persister subpopulations and their transcriptional signatures.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for Studying Phenotypic Heterogeneity
| Reagent / Material | Function / Application | Specific Examples / Notes |
|---|---|---|
| Fluorescent Protein Reporters | Visualizing gene expression heterogeneity and protein localization in live cells [17] | P~const~-GFP for growth reporting; multi-reporter constructs for simultaneous tracking of multiple genes [17] |
| Metabolic Biosensors | Quantifying metabolic activity and metabolite levels at single-cell resolution [17] | "QUEEN" biosensor for intracellular ATP levels; riboswitch-based biosensors for c-di-GMP [17] |
| Viability and Staining Probes | Differentiating live/dead cells and assessing cellular components [17] | SYTOX Green for nucleic acid staining in dead cells; HADA for probing peptidoglycan synthesis [17] |
| Microfluidic Devices | Long-term, high-resolution imaging and manipulation of single cells in controlled environments [16] [17] | Commercial cell-asay chips or custom PDMS devices for antibiotic exposure and recovery studies |
| scRNA-seq Kits & Reagents | Profiling the global transcriptome of individual bacterial cells [21] | Protocols must be adapted for bacteria, typically using poly(A)-independent methods with random hexamers [21] |
| Toxin Expression Plasmids | Inducing persistence mechanisms in a controlled manner for functional studies [19] | Plasmids for inducible expression of toxins like MqsR to study their role in dormancy [19] |
The study of phenotypic heterogeneity and bacterial persistence has been revolutionized by single-cell technologies that move beyond population-level averages to reveal the behavior of rare but critically important cell states. As detailed in this application note, techniques ranging from microfluidics and advanced fluorescence microscopy to single-cell RNA-sequencing provide the necessary resolution to dissect the formation, maintenance, and resuscitation of persister cells. A comprehensive understanding of the molecular mechanisms—centered on ppGpp, TA systems, and other stress response pathways—combined with these powerful analytical tools is paving the way for novel therapeutic strategies designed to eradicate persistent infections. The protocols and resources outlined herein provide a foundational framework for researchers embarking on this challenging yet vital area of microbiological research.
Single-cell analysis has revolutionized our understanding of bacterial persistence, revealing extraordinary heterogeneity in how individual bacterial cells survive antibiotic exposure. This application note details the core biological pathways governing persister formation—toxin-antitoxin (TA) systems, the (p)ppGpp alarmone, and the stringent response—within the context of modern single-cell research. We provide experimental protocols and quantitative frameworks that leverage cutting-edge single-cell techniques to dissect these pathways, enabling researchers to move beyond bulk population studies and uncover the molecular basis of phenotypic heterogeneity in bacterial populations.
Quantitative Data Synthesis
Table 1: Key Quantitative Parameters in Persister Pathways
| Parameter | Experimental System | Measured Value | Biological Impact | Citation |
|---|---|---|---|---|
| GTP Persister Threshold | B. subtilis with fluorescent GTP reporter | Rapid growth-dormancy switch when GTP drops below critical threshold | Single-cell persister formation | [23] [24] |
| Persister Frequency (Wild-type) | B. subtilis exposed to vancomycin | ~0.1% of population | Basal persistence level in exponentially growing cells | [23] |
| Triggered Persister Frequency | B. subtilis under starvation conditions | ~50% of population (500-fold increase) | Maximum induced persistence | [23] |
| TA System Impact on Survival | L. pneumophila ΔgndRX under genotoxic stress | Shift to viable but non-culturable (VBNC) state | Non-canonical TA function promoting survival | [25] |
| Bioenergetic Stress Persistence Enhancement | E. coli with constitutive ATP hydrolysis (pF1) + ciprofloxacin | Significant increase in persister fractions | Link between energy stress and persistence | [26] |
| Alarmone Synthetase Contribution | B. subtilis Rel/Sas mutants | Rel essential for triggered persistence; Rel+SasB for spontaneous | Specialization of alarmone production pathways | [23] |
Table 2: Single-Cell Experimental Observations of Persister Dynamics
| Observation | Experimental System | Methodology | Key Finding | Citation |
|---|---|---|---|---|
| Pre-exposure Growth States | E. coli + ampicillin or ciprofloxacin | Microfluidics + single-cell tracking | Most persisters were growing before antibiotic treatment | [27] |
| Heterogeneous Survival Dynamics | E. coli at single-cell level | High-throughput time-lapse imaging | Multiple survival modes: L-form-like growth, responsive arrest, filamentation | [27] |
| Metabolic Heterogeneity in Macrophages | L. pneumophila in human macrophages | BATLI software backtracking | Early mitochondrial changes (Δψm, mROS) predict bacterial replication | [28] |
| Replication Inefficiency | L. pneumophila in hMDMs | Automated confocal microscopy + single-cell analysis | Only 17±8% of infected macrophages supported bacterial replication | [28] |
| Contact-Dependent Survival | L. pneumophila wild-type and ΔgndRX co-culture | Inter-strain co-culture assays | Wild-type cells confer enhanced survival to ΔgndRX in contact-dependent manner | [25] |
Pathway Visualization
Diagram 1: Alarmone-GTP Persistence Switch
Diagram 2: Stringent Response Network Integration
Diagram 3: Single-Cell Analysis Workflow
Experimental Protocols
Protocol 1: Single-Cell Persistence Analysis Using Microfluidics
Application: Tracking persister cell histories and heterogeneous survival dynamics at single-cell resolution [27]
Materials:
- Bacterial strain: Wild-type E. coli or other target pathogen
- Growth medium: Appropriate rich and defined media
- Antibiotics: Ampicillin (100 µg/mL), ciprofloxacin (varies by MIC)
- Microfluidic device: CellASIC ONIX or similar perfusion system
- Imaging system: Phase-contrast and fluorescence microscope with environmental control
- Analysis software: Custom tracking algorithms or commercial packages
Procedure:
- Culture preparation: Grow bacterial culture to mid-exponential phase (OD600 ≈ 0.3-0.5) in appropriate medium.
- Device loading: Dilute culture to ~10⁶ cells/mL and load into microfluidic device according to manufacturer's protocol.
- Antibiotic treatment: Initiate perfusion with medium containing lethal antibiotic concentration (typically 5-10× MIC).
- Time-lapse imaging: Acquire images every 10-30 minutes for 24-48 hours using phase-contrast and fluorescence microscopy.
- Cell tracking: Use automated tracking software to follow individual cells and lineages throughout experiment.
- Outcome classification: Categorize cells based on survival dynamics:
- Continuous growth and fission (L-form-like)
- Responsive growth arrest
- Post-exposure filamentation
- Cell death and lysis
- Backtracking analysis: Reconstruct pre-treatment history of persister cells to identify precursor states.
Applications: This protocol enables researchers to distinguish between different modes of persistence based on single-cell histories and pre-exposure growth states, revealing that most persisters derive from growing cells rather than dormant subpopulations.
Protocol 2: GTP Level Monitoring During Persister Formation
Application: Visualizing the alarmone-GTP switch in single cells using fluorescent reporters [23] [24]
Materials:
- Bacterial strain: B. subtilis expressing fluorescent GTP reporter
- Growth medium: Appropriate defined medium
- Antibiotics: Vancomycin, ciprofloxacin, kanamycin at determined MICs
- Starvation triggers: SHX (500 µM) for amino acid starvation, other nutrient limitations
- Imaging system: Spinning-disk confocal or high-throughput microscope
- Analysis software: BATLI or similar backtracking analysis tool [28]
Procedure:
- Strain validation: Confirm GTP reporter functionality and calibration using known GTP modulators.
- Experimental setup: Grow reporter strain to early exponential phase in defined medium.
- Stress application:
- Spontaneous persistence: Dilute and grow in fresh medium without additional stress
- Triggered persistence: Add SHX (500 µM) or allow growth into stationary phase
- Antibiotic-induced: Sub-MIC antibiotic exposure
- Time-lapse imaging: Monitor GTP fluorescence and cell growth at 5-15 minute intervals.
- Threshold determination: Identify critical GTP threshold value correlating with persistence commitment.
- Single-cell analysis: Track individual cells and correlate GTP dynamics with survival outcomes.
- Validation: Confirm persistence through antibiotic challenge and regrowth assays.
Applications: This protocol enables direct visualization of the critical GTP threshold that triggers persister formation, demonstrating that all three persistence pathways (spontaneous, triggered, antibiotic-induced) converge on this common alarmone-GTP switch mechanism.
Protocol 3: Host-Pathogen Single-Cell Analysis Using BATLI
Application: Predicting bacterial replication outcomes in infected macrophages through backtracking analysis [28]
Materials:
- Host cells: Human monocyte-derived macrophages (hMDMs)
- Bacterial strain: L. pneumophila expressing constitutive GFP
- Dyes: Hoechst (nuclear stain), Cell Tracker Blue (cytoplasmic stain), mitochondrial probes (TMRM for Δψm, MitoSOX for mROS)
- Equipment: High-throughput confocal microscope, 384-well microplates
- Software: BATLI (Backtracking Analysis of Time-Lapse Images)
Procedure:
- Macrophage preparation: Differentiate hMDMs in 384-well microplates for 5-7 days.
- Infection: Infect macrophages with GFP-expressing L. pneumophila at MOI 10.
- Staining: Load mitochondrial probes and live-cell tracking dyes before infection.
- Time-lapse imaging: Acquire images hourly for 18 hours post-infection.
- Single-cell segmentation: Use BATLI to identify and track individual infected macrophages.
- Parameter quantification: Measure bacterial area (GFP fluorescence), mitochondrial membrane potential (Δψm), and mitochondrial ROS production (mROS) for each cell.
- Backtracking analysis: Categorize cells based on infection outcome (bacterial replication vs. restriction) and reconstruct metabolic parameter histories.
- Predictive modeling: Train machine learning models using early timepoint data to predict later replication outcomes.
Applications: This protocol enables identification of early metabolic predictors of bacterial replication success in host cells, achieving 83% accuracy in predicting L. pneumophila replication outcomes by 5 hours post-infection based on mitochondrial parameters.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Single-Cell Persistence Research
| Reagent Category | Specific Examples | Function/Application | Key References |
|---|---|---|---|
| Genetic Reporters | Fluorescent GTP biosensors, constitutive GFP/RFP | Visualizing metabolic states and bacterial localization in single cells | [23] [28] |
| Metabolic Probes | TMRM (Δψm), MitoSOX (mROS), Cell Tracker dyes | Monitoring mitochondrial function and metabolic activity in host and bacterial cells | [28] |
| Stress Inducers | DL-serine hydroxamate (SHX), arsenate, CCCP | Inducing stringent response and bioenergetic stress in controlled manner | [23] [29] |
| Single-Cell Platforms | Microfluidic devices, 384-well microplates | Maintaining individual cell tracking and high-throughput experimentation | [28] [27] |
| Analysis Software | BATLI, custom tracking algorithms | Backtracking analysis and outcome prediction from time-lapse data | [28] |
| TA System Tools | Pan-TA deletion strains, inducible toxin expression | Dissecting specific TA system functions in persistence | [25] [3] |
Technical Notes and Applications
Interpretation of Single-Cell Data
Single-cell analysis consistently reveals that persistence is not merely a binary dormant state but encompasses a continuum of metabolic states and survival strategies. Researchers should expect significant heterogeneity even within genetically identical populations, with multiple distinct pathways leading to antibiotic survival. The alarmone-GTP switch represents a convergent mechanism that can be triggered through different upstream signaling events.
Troubleshooting Common Issues
- Low persister frequencies: Use appropriate stress conditions (starvation, sub-MIC antibiotics) to increase persister yields for single-cell analysis
- Cell tracking challenges: Optimize dye concentrations and imaging intervals to balance phototoxicity with temporal resolution
- Reporter functionality: Validate metabolic reporters under control conditions before persistence experiments
- Macrophage heterogeneity: Use primary macrophages rather than cell lines and account for donor-to-donor variability
Applications in Drug Discovery
These protocols enable identification of novel anti-persister targets, including:
- Alarmone synthetase inhibitors [30]
- Metabolic reprogramming approaches to reverse persistence [31]
- Combination therapies that target both growing and persistent subpopulations
- Host-directed therapies that modulate metabolic interactions [28]
The integration of single-cell analysis with molecular pathway dissection provides unprecedented resolution for understanding and combating bacterial persistence, offering new opportunities for therapeutic intervention against chronic and recurrent infections.
Historical Context and Evolution of Persister Research
Bacterial persisters represent a fascinating and clinically significant subpopulation of cells that survive antibiotic treatment without genetically acquired resistance. These dormant phenotypic variants are a major contributor to recurrent and chronic infections, posing a substantial challenge in clinical settings [3] [32]. This Application Note traces the evolution of persister research from its initial discovery to contemporary single-cell analytical approaches, providing researchers with both historical context and practical methodologies for investigating this complex phenomenon. The content is framed within a broader thesis on single-cell techniques, highlighting how technological advances have progressively unveiled the mechanisms underlying bacterial persistence.
Within isogenic bacterial populations, persisters constitute a small fraction (typically 0.001–1%) that transiently exhibits multidrug tolerance through metabolic dormancy or reduced growth rates rather than genetic resistance mechanisms [33] [3]. This transient, non-heritable nature has made persisters particularly challenging to study, requiring sophisticated approaches capable of capturing rare physiological states within heterogeneous populations.
Historical Timeline of Key Discoveries
Table 1: Major Milestones in Persister Research
| Year | Key Discovery | Researcher(s) | Significance |
|---|---|---|---|
| 1942 | Initial observation of bacterial survival after penicillin exposure | Gladys Hobby | First documentation of phenotypic tolerance to antibiotics [3] |
| 1944 | Naming of "persisters" and description of their characteristics | Joseph Bigger | Formal conceptualization of the persister phenotype and proposal for intermittent treatment [3] [34] |
| 1970s | Description of "antibiotic tolerance" in pneumococcal mutants | Alexandre Tomasz | Distinction between different forms of bacterial survival under antibiotic pressure [3] |
| 1983 | Identification of first high-persistence (hip) mutant in E. coli | Harris Moyed | Genetic evidence for persistence mechanisms via hipA gene discovery [3] |
| 2000 | Link between persistence and biofilm infections | Kim Lewis | Established clinical relevance to chronic infections [3] |
| 2004–Present | Single-cell technologies and molecular mechanisms | Multiple groups | Elucidation of persister formation mechanisms and heterogeneity [33] [15] [35] |
The historical journey of persister research reveals a pattern of initial discovery, prolonged neglect, and renewed interest driven by technological advances. The phenomenon was first observed in 1942 when Gladys Hobby discovered that penicillin killed approximately 99% of bacterial cells, leaving 1% survivors [3]. This finding was systematically investigated by Joseph Bigger in 1944, who coined the term "persisters" and recognized their clinical significance, even proposing an intermittent treatment strategy that foreshadowed modern therapeutic approaches [3].
Despite these astute observations, persister research remained relatively dormant for several decades, overshadowed by the excitement surrounding new antibiotic discovery and the more immediately pressing issue of genetic resistance [36]. The field experienced a renaissance beginning in the 1980s with Moyed's identification of the first high-persistence (hip) mutant in E. coli, providing crucial evidence that persistence had a genetic component and could be systematically studied [3]. The clinical relevance of persisters became unequivocally established when Kim Lewis demonstrated their connection to biofilm-associated chronic infections in 2000, explaining why these infections often relapse after antibiotic therapy [3] [32].
Evolution of Methodological Approaches
From Population-Level to Single-Cell Analysis
Early persister research relied predominantly on population-level assays such as biphasic killing curves, which revealed the presence of persisters through a characteristic pattern of rapid initial killing followed by a plateau phase representing the surviving subpopulation [3] [37]. While these approaches confirmed the existence of persistence, they offered limited insight into the underlying heterogeneity or mechanisms.
The advent of single-cell technologies has revolutionized the field by enabling researchers to investigate rare persister cells within complex populations. Key methodological transitions include:
- Microscopy and Microfluidics: Advanced imaging platforms combined with microfluidic devices allow continuous observation of individual bacterial cells before, during, and after antibiotic exposure [15]. The membrane-covered microchamber array (MCMA) device, for instance, enables researchers to monitor over one million individual cells simultaneously under controlled conditions [15].
- Flow Cytometry: This approach facilitates high-throughput analysis and sorting of persister cells based on physiological parameters such as membrane potential, metabolic activity, or reporter gene expression [33].
- Single-Cell RNA Sequencing (scRNA-seq): Cutting-edge transcriptomic techniques like PETRI-seq have enabled comprehensive profiling of persister cell states, revealing that diverse persister types converge toward a transcriptional state characterized by translational deficiency [35].
Key Single-Cell Techniques and Applications
Table 2: Single-Cell Methodologies for Persister Research
| Technique | Key Features | Applications in Persistence Research | References |
|---|---|---|---|
| Microfluidics | Long-term imaging of individual cells under controlled environments; membrane-covered microchambers for medium exchange | Tracking persister cell histories; heterogeneous survival dynamics; correlation between pre-exposure growth and persistence [15] | |
| Flow Cytometry | High-throughput multiparameter analysis; cell sorting based on physiological markers | Isolation of persister subpopulations; analysis of metabolic heterogeneity; quantification of persistence frequency [33] [38] | |
| Fluorescent Biosensors | Gene expression reporters; protein-FP fusions; FRET-based physiological sensors | Monitoring transcriptional and translational heterogeneity; quantifying metabolic activity; stress response pathways [33] | |
| Single-Cell RNA Sequencing | Comprehensive transcriptome profiling of individual cells; identification of rare cell states | Defining persister-specific transcriptional signatures; identifying key regulatory pathways [35] | |
| Raman Spectroscopy | Label-free analysis of biochemical composition; monitoring metabolic activity | Identification of physiological states associated with persistence; non-invasive tracking of dormancy depth [33] |
Contemporary Experimental Protocols
Protocol: Single-Cell Persister Dynamics Using Microfluidics
This protocol adapts methodologies from [15] for investigating persister cell histories at single-cell resolution.
Materials and Equipment
- Strain: E. coli MG1655 (or relevant bacterial species)
- Microfluidic Device: Membrane-covered microchamber array (MCMA)
- Culture Media: Appropriate broth medium (e.g., LB, M9 minimal medium)
- Antibiotics: Ampicillin (200 µg/mL), ciprofloxacin (1 µg/mL) or target antibiotic
- Imaging System: Time-lapse fluorescence microscope with environmental control
- Analysis Software: Image analysis platform (e.g., ImageJ, Matlab) for single-cell tracking
Procedure
- Device Preparation: Fabricate MCMA device with 0.8-µm deep microchambers on glass coverslip; coat with cellulose semipermeable membrane via biotin-streptavidin bonding [15].
- Cell Loading: Introduce mid-exponential phase bacterial culture (OD600 ≈ 0.3-0.5) into microchambers at appropriate dilution to achieve 1-10 cells per chamber.
- Pre-treatment Monitoring: Flow fresh medium through device and record baseline growth for 2-3 hours at appropriate temperature (e.g., 37°C for E. coli) with images captured at 5-10 minute intervals.
- Antibiotic Treatment: Switch medium to antibiotic-containing solution (e.g., 200 µg/mL ampicillin, 12.5×MIC) for defined treatment period (typically 3-8 hours).
- Post-treatment Recovery: Replace with fresh antibiotic-free medium and monitor for regrowth for 12-24 hours.
- Image Analysis: Track individual cell lineages using automated tracking software; classify survival dynamics based on morphological changes and division events.
Key Parameters and Data Analysis
- Quantification of Survival Dynamics: Categorize persister behaviors as (1) continuous growth with L-form-like morphology, (2) responsive growth arrest, or (3) post-exposure filamentation [15].
- Correlation Analysis: Link pre-exposure growth characteristics (division time, cell size) to survival probability.
- Persistence Frequency Calculation: Determine as percentage of cells surviving antibiotic treatment capable of resuming growth.
Protocol: Persister Recovery Kinetics via Spectrophotometry and Flow Cytometry
This protocol, adapted from [38], enables quantification of persister resuscitation dynamics and physiological states.
Materials
- Bacterial Strains: Target organism with appropriate genetic background
- Antibiotics: Bactericidal antibiotics relevant to study
- Culture Media: Appropriate broth medium
- Flow Cytometer: Equipped with 488-nm laser and appropriate filter sets
- Viability Stains: SYTOX Green, membrane potential-sensitive dyes (e.g., DiOC2(3))
- Microplate Reader: For high-throughput spectrophotometric measurements
Procedure
Persister Isolation:
- Grow bacterial culture to desired phase (exponential, stationary)
- Treat with lethal antibiotic concentration (5-10×MIC) for predetermined time
- Wash cells to remove antibiotic using centrifugation or filtration
- Confirm persister isolation by plating and CFU enumeration
Recovery Kinetics Monitoring:
- Resuspend persister cells in fresh pre-warmed medium
- Distribute into 96-well plates for spectrophotometric reading
- Monitor OD600 at regular intervals (every 15-30 minutes) for 12-24 hours
- Include controls (untreated cells, medium only)
Single-Cell Physiological Analysis:
- Sample recovering culture at designated timepoints (0, 2, 4, 8 hours)
- Stain with viability indicators (SYTOX Green) and metabolic dyes
- Analyze by flow cytometry with appropriate gating strategies
- Correlate physiological states with resuscitating capability
Data Interpretation
- Lag Time Determination: Calculate duration from antibiotic removal to resumption of growth
- Heterogeneity Assessment: Identify distinct subpopulations based on physiological parameters
- Correlation Analysis: Link resuscitation kinetics with pre-treatment conditions or genetic background
Visualization of Key Concepts and Workflows
Single-Cell Analysis Workflow for Persister Research
Diagram 1: Comprehensive workflow for single-cell analysis of bacterial persisters, integrating multiple technological approaches.
Molecular Mechanisms of Persister Formation
Diagram 2: Molecular pathways and cellular responses involved in persister cell formation, highlighting key regulatory mechanisms.
Research Reagent Solutions
Table 3: Essential Research Reagents for Persister Investigations
| Reagent Category | Specific Examples | Function/Application | Key References |
|---|---|---|---|
| Fluorescent Reporters | GFP, mCherry, CFP transcriptional fusions; RpoS-mCherry | Monitoring gene expression heterogeneity; stress response pathways; promoter activity in single cells [33] [15] | |
| Viability and Metabolic Probes | SYTOX Green, Hoechst 33342, DiOC2(3), ATP biosensors (QUEEN, iATPSnFR) | Distinguishing live/dead cells; DNA content analysis; membrane potential; metabolic activity monitoring [33] | |
| Biosensors | FRET-based sensors; riboswitch-based biosensors (e.g., c-di-GMP); O-propargyl-puromycin (OPP) | Quantifying intracellular metabolites; second messenger dynamics; protein synthesis rates [33] | |
| Genetic Tools | CRISPRi libraries; transposon mutant collections; fluorescent protein fusion libraries | Genome-wide functional screens; targeted gene knockdown; protein localization studies [35] | |
| Specialized Growth Media | Defined minimal media; nutrient-limited conditions; stress-inducing supplements | Controlling growth rates; inducing persistence; mimicking host environments [15] [35] |
The historical evolution of persister research demonstrates a remarkable trajectory from phenomenological observation to mechanistic understanding at single-cell resolution. Early population-level studies correctly identified the existence and clinical significance of persisters but lacked the tools to probe their underlying heterogeneity. Contemporary single-cell technologies have revealed that persistence is not a unitary phenomenon but encompasses diverse survival strategies influenced by antibiotic class, growth history, and stochastic cellular processes [15] [35].
Future research directions will likely focus on several key areas: First, the integration of multi-omics approaches at single-cell resolution will further elucidate the molecular networks controlling persister formation and resuscitation. Second, the development of more sophisticated microfluidic platforms that better mimic host environments will enhance the clinical relevance of experimental findings. Finally, the translation of basic mechanistic insights into therapeutic strategies that specifically target persister cells holds promise for addressing the persistent challenge of chronic and recurrent infections.
The methodologies and protocols outlined in this Application Note provide researchers with practical tools for investigating bacterial persistence using state-of-the-art single-cell approaches, contributing to the ongoing effort to understand and ultimately overcome this significant clinical problem.
The Single-Cell Toolbox: Techniques for Isolating, Observing, and Profiling Persisters
The relentless challenge of antibiotic persistence, a phenomenon where a small, dormant sub-population of bacteria survives antibiotic treatment, represents a significant hurdle in treating recurrent infections. Unlike genetic resistance, persistence is a transient, phenotypic state, making it difficult to detect and eradicate [39] [33]. Understanding the underlying mechanisms of bacterial persistence requires tools that can dissect cellular processes at the single-cell level, as these rare persister cells are often masked in population-averaged studies. Fluorescent biosensors and reporters have emerged as indispensable instruments in this endeavor, providing real-time, dynamic insights into the gene expression, metabolic activity, and signaling events that define the persister state. This Application Notes and Protocols document details the use of these powerful tools, framing them within the context of a broader thesis on single-cell techniques for analyzing bacterial persisters. It is designed to equip researchers, scientists, and drug development professionals with the methodologies to illuminate the hidden heterogeneity within bacterial populations and accelerate the development of anti-persister therapies.
Technical Specifications of Key Biosensors and Reporters
The following table summarizes the core characteristics of several advanced biosensors and reporters pivotal for single-cell persister research.
Table 1: Key Fluorescent Biosensors and Reporters for Single-Cell Analysis
| Biosensor/Reporter Name | Detection Target | Technology/Design | Key Performance Metrics | Primary Application in Persister Research |
|---|---|---|---|---|
| QUEEN-2m [40] | ATP concentration | Single fluorescent protein (cpEGFP) fused to bacterial ATP-binding protein; ratiometric. | ( K_d ) ~2 mM (at 25°C); Dynamic range ~3.0; Insensitive to growth rate changes. | Quantifying absolute ATP levels to assess metabolic dormancy and heterogeneity in persister cells. |
| FRET-based Pyruvate Sensor [41] | Pyruvate concentration | FRET pair (CFP/YFP) flanking pyruvate-binding domain of PdhR. | ( EC_{50} ) ~400 µM (in permeabilized cells); Responds within seconds to metabolic shifts. | Monitoring rapid fluctuations in central metabolism and glycolytic oscillations upon perturbation. |
| RGB-S Reporter [42] | Multimodal stress response (RpoS, SOS, RpoH) | Three orthogonal fluorescent proteins (RFP, GFP, BFP) under control of distinct stress promoters. | Reports on physiological stress (RFP), genotoxicity (GFP), and cytotoxicity (BFP) simultaneously. | Identifying heterogeneous stress responses in biofilms and characterizing multimodal effects of anti-persister compounds. |
| Riboswitch-based TPP Biosensor [43] | Thiamin pyrophosphate (TPP) metabolism | thiC riboswitch aptamer domain controlling GFP expression. | Detects TPP concentrations as low as 50-100 nM; ~2-fold dynamic range in regulation. | Interrogating metabolic pathway activity and nutrient status at a single-cell level. |
Application Notes & Detailed Protocols
Protocol 1: Single-Cell ATP Quantification in Bacterial Persisters using QUEEN-2m
Background: Persister cells are characterized by a marked reduction in metabolic activity. Direct measurement of intracellular ATP concentration is a critical metric for assessing this metabolic dormancy. The QUEEN-2m biosensor is ideal for this purpose due to its ratiometric nature, insensitivity to bacterial growth rate, and affinity suitable for physiological ATP ranges [40].
Materials:
- Bacterial Strain: E. coli expressing QUEEN-2m biosensor from an appropriate plasmid vector.
- Growth Medium: Suitable defined medium (e.g., M9 minimal medium) with a carbon source.
- Antibiotic: For selection of the biosensor plasmid.
- Inducer: If the biosensor is under inducible control.
- Imaging Equipment: Epifluorescence or confocal microscope equipped with:
- Excitation filters: 400/10 nm and 480/20 nm.
- Emission filter: 510/20 nm.
- 40x or higher magnification oil-immersion objective.
- Microfluidic Flow Cell or Live-Cell Imaging Chamber: For environmental control during time-lapse imaging.
- Image Analysis Software: e.g., ImageJ/FIJI with ratiometric analysis plugins.
Procedure:
- Culture Preparation: Grow the sensor strain to the desired growth phase (e.g., mid-exponential or stationary phase, where persister levels are higher [39]) in the appropriate medium with antibiotic selection.
- Sample Loading: For continuous observation, immobilize cells in a microfluidic flow chamber pre-warmed to 37°C. Alternatively, prepare an agarose pad for fixed-endpoint imaging.
- Antibiotic Challenge (Optional): To isolate and study persisters, perfuse the chamber with medium containing a high concentration of a bactericidal antibiotic (e.g., ampicillin or ciprofloxacin) for a defined period. Wash with fresh medium to remove the antibiotic before imaging survivors.
- Ratiometric Image Acquisition:
- Acquire two images for each field of view and time point: one with 400 nm excitation and one with 480 nm excitation, both using the 510 nm emission filter.
- Set acquisition parameters (exposure time, gain) to avoid pixel saturation and keep them constant throughout the experiment.
- Collect time-lapse data at regular intervals (e.g., every 5-10 minutes) to monitor dynamic changes.
- Image and Data Analysis:
- Background Subtraction: Subtract the background fluorescence from both the 400ex and 480ex image stacks.
- Ratio Calculation: Create a ratio image (R = Intensity({400ex}) / Intensity({480ex})) for each time point.
- Single-Cell Quantification: Define regions of interest (ROIs) around individual cells and extract the mean ratio value (R) for each cell over time.
- ATP Calibration: To convert ratio values to absolute ATP concentrations, perform an in vivo calibration using cells with known ATP levels (e.g., by titrating with metabolic inhibitors like KCN as described in [40]).
Troubleshooting:
- Low Signal-to-Noise Ratio: Ensure the biosensor is expressed at sufficient levels. Use a stronger promoter or increase inducer concentration.
- Cell Photobleaching: Minimize light exposure by using lower excitation intensity or longer intervals between time points.
- Inconsistent Ratios: Verify that the microscope light source and filters are stable. Use control cells expressing a non-ratiometric fluorescent protein to check for illumination artifacts.
Protocol 2: Profiling Multimodal Stress Responses in Biofilms using the RGB-S Reporter
Background: Biofilms are a major reservoir for persisters, and cells within a biofilm experience heterogeneous microenvironments, leading to varied stress responses. The RGB-S reporter allows simultaneous monitoring of three major stress regulons (RpoS, SOS, RpoH) in live cells, providing a high-content readout of a cell's physiological state [42].
Materials:
- Bacterial Strain: E. coli harboring the RGB-S reporter plasmid (PosmY::mRFP1, PsulA::GFPmut3b, PgrpE::mTagBFP2) [42].
- Growth Medium: LB or defined medium.
- Antibiotic: Kanamycin for plasmid selection.
- Stressors: Compounds of interest (e.g., glyphosate for RpoS, nalidixic acid for SOS, 2-propanol for RpoH).
- Imaging Equipment: Confocal microscope capable of sequential imaging with laser lines and filters for RFP (ex: 561 nm, em: 570-620 nm), GFP (ex: 488 nm, em: 500-550 nm), and BFP (ex: 405 nm, em: 430-470 nm).
- Flow Cytometer (Optional): For high-throughput single-cell analysis.
Procedure:
- Biofilm Cultivation: Grow the RGB-S reporter strain in a flow cell or on a coverslip in a drip-flow reactor to form a mature biofilm (e.g., 3-5 days).
- Stressor Exposure: Expose the biofilm to the stressor of interest by perfusing with medium containing the compound at the desired concentration.
- Time-Lapse Confocal Imaging:
- Image the biofilm at multiple locations and time points (e.g., 0, 1, 2, 4 hours post-exposure).
- Use sequential scanning mode to avoid cross-talk between the three fluorescent channels.
- Acquire Z-stacks to capture the three-dimensional structure of the biofilm.
- Image Analysis:
- Preprocessing: Apply a mild background subtraction to each channel.
- Segmentation: Use automated or semi-automated cell segmentation tools (e.g., in Ilastik or CellProfiler) to identify individual bacterial cells within the biofilm.
- Intensity Extraction: For each segmented cell, measure the mean fluorescence intensity in the RFP, GFP, and BFP channels.
- Data Visualization and Interpretation:
- Create scatter plots (e.g., RFP vs. GFP) to visualize subpopulations with different stress response signatures.
- Overlay fluorescence data on the biofilm structure to correlate stress response with spatial location (e.g., interior vs. periphery).
Troubleshooting:
- Spectral Cross-Talk: Optimize sequential scanning settings and verify that signal in one channel is not detected in another using control samples with single fluorophores.
- Weak BFP Signal: BFP maturation can be sensitive. Ensure the microscope detectors are optimized for the blue spectrum, and consider using a more photostable BFP variant if needed.
- Plasmid Loss: Maintain antibiotic selection throughout the experiment to ensure plasmid retention, especially in long-term biofilm cultures [42].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Fluorescent Biosensor Experiments
| Item/Category | Function/Description | Example Product/Source |
|---|---|---|
| Genetically Encoded Biosensors | Core reagents for detecting specific metabolites or signaling events in live cells. | QUEEN series (ATP) [40]; FRET-based pyruvate sensor [41]; RGB-S reporter plasmid [42]. |
| Microfluidic Cultivation Devices | Provides precise control over the cellular microenvironment, enabling long-term imaging and antibiotic perturbation. | Commercial flow cells (e.g., CellASIC ONIX2); lab-made PDMS devices. |
| Live-Cell Imaging Media & Supplements | Maintains cell viability and function during microscopy, without inducing autofluorescence. | Phenol-red free imaging media; buffering agents (e.g., HEPES). |
| High-Sensitivity Cameras | Essential for detecting low-light fluorescence signals from single cells, especially dim or small bacterial cells. | Electron-Multiplying CCD (EMCCD) or scientific CMOS (sCMOS) cameras. |
| Metafluor/ImageJ FIJI Software | Industry-standard and open-source software for ratiometric image analysis, cell segmentation, and fluorescence quantification. | Molecular Devices MetaFluor; NIH ImageJ/FIJI. |
Visualizing Experimental Workflows and Signaling Pathways
Single-Cell Analysis Workflow for Bacterial Persisters
The following diagram illustrates a generalized workflow for using fluorescent biosensors to investigate bacterial persisters, from sample preparation to data analysis.
Key Signaling Pathways in Bacterial Persistence
Bacterial persistence is regulated by an interconnected network of signaling pathways that respond to metabolic and environmental stress. The following diagram summarizes the core pathways involving toxin-antitoxin (TA) systems, alarmones, and second messengers, as revealed by biosensor studies [39] [33] [42].
Bacterial persisters are a transient, phenotypically variant subpopulation within an isogenic culture that exhibit remarkable tolerance to high doses of conventional antibiotics without acquiring heritable genetic resistance [3] [37]. These metabolically dormant or slow-growing cells are now recognized as a primary cause of chronic and recurrent infections, posing a significant challenge in clinical settings [44] [3]. Unlike resistant bacteria, persisters do not exhibit an elevated Minimum Inhibitory Concentration (MIC) and resume normal growth once antibiotic pressure is removed, leading to disease relapse [37].
The study of persister cells has been historically challenging due to their low frequency (typically 10⁻⁶ to 10⁻³) in bacterial populations and their non-genetic, transient nature [15] [45]. Traditional bulk-culture methods lack the resolution to isolate or observe these rare cells, limiting our understanding of their formation and survival mechanisms [46]. The emergence of microfluidic technologies coupled with high-resolution live-cell imaging has revolutionized this field by enabling real-time tracking of individual bacterial cells before, during, and after antibiotic exposure [15] [47] [46]. This application note details standardized protocols for using microfluidic devices to dissect the heterogeneous dynamics of bacterial persisters at single-cell resolution, providing researchers with robust methodologies to advance both fundamental knowledge and therapeutic development.
Background and Significance
The Persister Phenotype: Definitions and Clinical Relevance
Persister cells were first identified by Joseph Bigger in 1944 when he observed that penicillin failed to sterilize a Staphylococcus culture [3] [37]. These cells exhibit biphasic killing kinetics in time-kill assays, characterized by an initial rapid decline in viable cells followed by a much slower death rate of the persistent subpopulation [37]. This phenomenon is distinct from antibiotic resistance, as persisters survive antibiotic treatment through passive mechanisms primarily linked to reduced metabolic activity and growth arrest, rather than active defense mechanisms [3].
Clinically, persisters are implicated in a wide range of persistent infections including tuberculosis, recurrent urinary tract infections, Lyme disease, and biofilm-associated infections on medical implants [3]. Their presence necessitates prolonged antibiotic therapies, which in turn contributes to the development of genetic resistance [37]. Eradicating persisters remains a formidable challenge in infectious disease management, as most conventional antibiotics target actively growing cells.
The Power of Single-Cell Analysis
Population-level studies mask the significant heterogeneity in individual cell responses to antibiotic stress [46]. Microfluidic platforms overcome this limitation by enabling:
- Long-term tracking of single-cell lineages across multiple generations
- Precise control of microenvironmental conditions and antibiotic gradients
- High-throughput observation of rare persister cells among millions of susceptible cells
- Real-time monitoring of morphological and physiological changes during stress adaptation
Recent studies utilizing microfluidics have challenged the classical dogma that persisters are exclusively dormant cells generated before antibiotic exposure. Observations of over one million individual Escherichia coli cells revealed that many persisters were actively growing before treatment with ampicillin or ciprofloxacin, and exhibited diverse survival strategies including continuous growth with L-form-like morphologies, responsive growth arrest, or post-exposure filamentation [15] [45].
Experimental Protocols
Microfluidic Device Fabrication: The MCMA Platform
The Membrane-Covered Microchamber Array (MCMA) device has been successfully implemented for persister studies in E. coli and can be adapted for other bacterial species [15] [45].
Materials and Equipment
- Photomasks: High-resolution CAD files printed on polymer slides
- Silicon wafers: 4-inch diameter
- Photoresist: SU-8 2002 and SU-8 2025
- PDMS: Polydimethylsiloxane base and curing agent
- Semipermeable membrane: Cellulose membrane functionalized with biotin
- Glass coverslips: No. 1.5 thickness for high-resolution imaging
- Spin coater
- UV exposure system
- Plasma cleaner
- Soft bake and hard bake ovens
Fabrication Procedure
Wafer Preparation: Clean a 4-inch silicon wafer with 100% isopropanol and bake at 120°C for 15 minutes to dehydrate [48].
Photolithography for Microchambers:
- Apply SU-8 2002 photoresist (3 mL) to the wafer using a spin coater at 400 rpm for 30 seconds to achieve a 3-μm layer [48].
- Soft bake at 95°C for 2 minutes [48].
- Expose through the microchamber photomask with 100 mJ/cm² UV at 365 nm [48].
- Perform post-exposure bake at 95°C for 2 minutes [48].
- Develop in SU-8 developer for 15-30 seconds, rinse with isopropanol, and dry with compressed air [48].
- Hard bake at 150°C for 15 minutes [48].
Photolithography for Flow Channels:
- Apply SU-8 2025 photoresist (3 mL) at 2,500 rpm for 30 seconds to achieve a 40-μm layer [48].
- Soft bake at 95°C for 7 minutes [48].
- Align the flow channel photomask with the existing microchamber layer.
- Expose with 160 mJ/cm² UV at 365 nm [48].
- Perform post-exposure bake at 65°C for 2 minutes followed by 95°C for 6 minutes [48].
- Develop for 45-60 seconds, rinse with isopropanol, and dry [48].
- Hard bake at 150°C for 15 minutes [48].
PDMS Device Casting:
Device Assembly:
- Peel off the cured PDMS from the mold and cut to size.
- Create inlet and outlet ports using a biopsy punch.
- Treat PDMS and glass coverslip with oxygen plasma for 60 seconds.
- Bond PDMS to glass coverslip immediately after plasma treatment.
- Functionalize the device with biotinylated cellulose membrane using streptavidin-biotin bonding to create semipermeable covers for microchambers [15] [45].
Bacterial Strain Preparation and Loading
Materials and Reagents
- Bacterial strains: Wildtype E. coli MG1655 or other relevant strains
- Fluorescent reporters: GFP, mCherry, or other fluorescent proteins for lineage tracking
- Culture media: LB, M9 minimal media, or other appropriate growth media
- Antibiotics: For selection when using plasmid-based reporters
- Syringe pumps: For precise medium delivery
- Microfluidic tubing: Compatible with syringe pumps and device ports
Strain Engineering Protocol
Chromosomal Reporter Integration:
- For metabolic activity reporters, fuse GFP to endogenous genes of interest (e.g., rpoS for stress response) via homologous recombination [15] [45].
- Include degron sequences (e.g., CLN2-PEST) for protein destabilization to enable dynamic response monitoring [48].
- For lineage tracing, constitutively express a fluorescent protein with nuclear localization signal (NLS) to track cell divisions [48].
- Validate reporter functionality and strain fitness compared to wildtype.
Culture Conditions for Persister Studies:
Device Loading and Conditioning:
- Dilute bacterial culture to appropriate density (typically 10⁶-10⁷ cells/mL) in fresh medium.
- Inject bacterial suspension into the microfluidic device using a syringe pump at low flow rate (1-5 μL/min).
- Allow cells to settle in microchambers for 15-30 minutes.
- Initiate continuous medium flow at 5-10 μL/min to remove non-adhered cells and establish stable environmental conditions.
- Allow cells to adapt to the microfluidic environment for 2-3 hours before initiating experiments.
Time-Lapse Imaging of Antibiotic Treatment
Materials and Equipment
- Inverted microscope: Equipped with high-precision stage, temperature control, and CO₂ incubation if needed
- Objective lens: 60× or 100× oil-immersion objective with high numerical aperture
- Camera: High-sensitivity EMCCD or sCMOS camera
- Laser or LED light source: For fluorescence excitation
- Environmental chamber: To maintain constant temperature during imaging
- Image acquisition software: With multi-position and focus-maintenance capabilities
Imaging Protocol
Pre-treatment Baseline Imaging:
- Acquire phase contrast and fluorescence images of the entire microchamber array every 10-15 minutes for 2-3 hours to establish baseline growth dynamics and identify individual cell lineages [15].
Antibiotic Treatment:
- Prepare antibiotic solutions at desired concentrations in fresh growth medium.
- Switch medium reservoir to antibiotic-containing medium using a fluidics switch or multi-channel pump.
- Continue time-lapse imaging with 10-15 minute intervals throughout treatment duration (typically 4-8 hours).
Post-treatment Recovery Monitoring:
- Switch back to antibiotic-free medium to assess resuscitation of persister cells.
- Continue imaging for 12-24 hours to monitor regrowth of surviving cells.
Image Acquisition Parameters:
- Phase contrast: Minimal exposure to minimize phototoxicity
- Fluorescence: Optimize exposure time based on reporter brightness while minimizing photobleaching
- Multiple positions: Program stage positions to capture 10-20 microchambers per experiment
- Z-stacking: If needed for proper cell segmentation, acquire 3-5 Z-slices with 0.5-1 μm spacing
Data Analysis and Persister Identification
Software Tools
- Image analysis: Cell segmentation and tracking using specialized software (e.g., PhyloCell, ImageJ plugins, or custom MATLAB/Python scripts)
- Data visualization: R, Python, or commercial graphing software
- Statistical analysis: Appropriate tests for significance (e.g., log-rank test for survival analysis)
Analysis Workflow
Cell Segmentation and Tracking:
- Apply automated cell segmentation algorithms to identify individual cells in each frame.
- Track cells through divisions to establish lineage relationships.
- Curate tracking results manually to correct errors.
Growth Rate Calculation:
- Measure cell length or area over time for each tracked cell.
- Calculate instantaneous growth rates using derivative or exponential fit methods.
Persister Identification Criteria:
- Survival through the entire antibiotic treatment period (typically 4-8 hours)
- Ability to resume growth and division after antibiotic removal
- Normal susceptibility of progeny to the same antibiotic (confirmed by separate assays)
Classification of Persister Dynamics:
- Categorize persisters based on pre-treatment state (growing vs. non-growing)
- Classify based on survival strategy during treatment (L-form transition, filamentation, growth arrest, etc.)
Technical Specifications and Data Analysis
Key Experimental Parameters for Persister Studies
Table 1: Standardized experimental parameters for microfluidic persister studies
| Parameter | Recommended Specifications | Notes and Variations |
|---|---|---|
| Microchamber dimensions | 0.8 μm depth, 10-20 μm width [15] | Adjust based on bacterial size; M. tuberculosis may require taller chambers |
| Medium flow rate | 5-10 μL/min [48] | Balance between nutrient supply and mechanical stress |
| Antibiotic concentrations | Amp: 200 μg/mL (12.5× MIC) [15]; Cip: 1 μg/mL (32× MIC) [15] | Validate MIC for specific strains and growth conditions |
| Treatment duration | 4-8 hours [15] | Extend for slow-acting antibiotics or tolerant strains |
| Imaging interval | 10-15 minutes [15] | Shorter intervals for rapid response studies |
| Temperature control | 37°C for mammalian pathogens | Adjust for environmental isolates |
| Total experiment duration | 24-48 hours | Limited by phototoxicity and nutrient depletion |
Quantitative Analysis of Persister Dynamics
Table 2: Key metrics for quantitative analysis of persister cells
| Metric | Calculation Method | Biological Significance |
|---|---|---|
| Persister frequency | Number of persisters / total cells before treatment × 100% | Baseline persistence level of population |
| Pre-treatment growth rate | Exponential fit of cell size over 2-3 hours before treatment | Correlation between growth state and persistence |
| Time to regrowth | Duration from antibiotic removal to first division | Depth of dormancy or damage extent |
| Morphological dynamics | Quantitative shape descriptors (aspect ratio, curvature) | Identification of survival strategies (L-forms, filaments) |
| Lineage relationship | Tracking of mother-daughter pairs through treatment | Inheritance patterns of persistence |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key research reagent solutions for microfluidic persister studies
| Reagent/Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Microfluidic devices | MCMA [15], Pneumatic valve arrays [46] | Single-cell confinement and environmental control |
| Bacterial strains | E. coli MG1655 [15], M. tuberculosis [46], Clinical isolates | Model organisms and clinically relevant strains |
| Fluorescent reporters | GFP, mCherry, RpoS-mCherry [15] [45] | Metabolic activity, stress response, and lineage tracing |
| Antibiotics | Ampicillin [15], Ciprofloxacin [15], Isoniazid [46] | β-lactams, fluoroquinolones, and species-specific drugs |
| Culture media | LB, M9 minimal media [48], 7H9 for mycobacteria [46] | Defined or rich media depending on experimental goals |
| Detection biosensors | RpoS-mCherry [15], ATP-based reporters [44] | Stress response and metabolic state monitoring |
Visualization of Experimental Workflows and Biological Pathways
Microfluidic Persister Analysis Workflow
Workflow for Persister Analysis - This diagram illustrates the complete experimental pipeline from device preparation through data analysis.
Molecular Mechanisms of Persister Formation
Persister Formation Pathways - This diagram shows the key molecular pathways leading to persister formation and antibiotic tolerance.
Applications and Future Directions
The integration of microfluidics with live-cell imaging has enabled unprecedented insights into persister biology, revealing heterogeneous survival strategies that depend on both antibiotic class and cellular pre-history [15] [45]. These platforms are particularly valuable for:
Mechanistic Studies: Elucidating the molecular pathways underlying persister formation through real-time monitoring of fluorescent reporters for stress responses, metabolic activity, and protein expression [15] [48].
Therapeutic Screening: Evaluating the efficacy of novel anti-persister compounds, including nanoagents that directly target dormant cells or reactivate them for eradication [44].
Combination Therapy Optimization: Testing antibiotic sequencing and combination strategies to prevent persistence and minimize resistance evolution [3] [46].
Clinical Isolate Characterization: Profiling persistence levels of clinical isolates to inform treatment strategies and understand treatment failure mechanisms [3] [37].
Future developments in this field will likely focus on increasing throughput through parallelization, integrating multi-omics approaches with single-cell dynamics, and developing more sophisticated biosensors for metabolic state and antibiotic target engagement. These advances will further establish microfluidics as an indispensable tool in the ongoing battle against persistent bacterial infections.
Single-cell RNA sequencing (scRNA-seq) represents a transformative technological advancement that enables the quantification of gene expression at the resolution of individual cells. While extensively applied in eukaryotic systems, its implementation in bacterial research has historically been challenging due to technical barriers including the absence of polyadenylated tails on bacterial mRNA, exceptionally high ribosomal RNA (rRNA) content, and low starting RNA quantities [49] [50]. Recent methodological breakthroughs are now empowering researchers to apply scRNA-seq to bacterial systems, revealing unprecedented insights into phenotypic heterogeneity, antibiotic persistence, and biofilm dynamics [49] [33] [3]. This Application Note details the integration of scRNA-seq methodologies within bacterial persister research, providing structured experimental protocols, analytical workflows, and resource guidance to investigate transcriptional heterogeneity in these clinically relevant subpopulations.
scRNA-seq in Bacterial Persister Research: Methodological Approaches
Bacterial persisters represent a subpopulation of cells that survive antibiotic treatment despite being genetically identical to their susceptible counterparts. These cells exhibit phenotypic heterogeneity, encompassing variations in metabolic activity, growth rates, and persistence levels, which are recalcitrant to traditional bulk transcriptomic analyses [33] [3]. Single-cell technologies are critical to dissect this heterogeneity, as they can identify rare cell states, characterize distinct transcriptional programs, and elucidate the mechanisms underlying persistence [33] [50].
Two primary methodological frameworks have been developed for bacterial scRNA-seq: plate-based and microfluidics-based barcoding approaches. Plate-based systems (e.g., 96- or 384-well plates) offer accessibility but impose limitations on throughput. Microfluidics-based approaches (e.g., the 10x Genomics Chromium platform) enable high-throughput analysis of thousands of cells but require specialized instrumentation [49] [51]. A significant innovation is the development of split-pool barcoding, which labels individual cells with a unique combination of oligonucleotide barcodes through successive rounds of pooling and splitting, eliminating the need for physical cell separation or expensive equipment [49] [50].
A central challenge in bacterial scRNA-seq is the overwhelming abundance of rRNA, which can constitute >90% of sequencing reads, thereby limiting the detection of informative mRNA transcripts [49] [52]. Recent protocols have integrated efficient rRNA depletion strategies to markedly improve mRNA detection sensitivity. The table below summarizes the performance of current state-of-the-art methods.
Table 1: Performance Comparison of Bacterial scRNA-seq Methods with rRNA Depletion
| Method Name | rRNA Depletion Strategy | Reported mRNA Detection Rate | Key Advantages |
|---|---|---|---|
| RiboD-PETRI [52] | Subtractive hybridization with probes and magnetic beads | Up to 92% (S. aureus, stationary phase) | Cost-effective, equipment-free, high mRNA detection |
| BaSSSh-seq [49] | Enzyme-free subtractive hybridization | Specific efficiency not reported; "significantly reduces rRNA" | Optimized for low-metabolic-activity cells (e.g., biofilm) |
| M3-seq [52] | RNase H digestion post-DNA probe hybridization in cells | ~65% | Effective rRNA reduction |
| BacDrop [52] | RNase H digestion post-DNA probe hybridization in cells | ~61% | Effective rRNA reduction |
| MATQ-DASH [52] | Cas9-mediated targeted rRNA depletion | ~30% | CRISPR-based precision |
Detailed Experimental Protocol: BaSSSh-seq for Biofilm and Persister Analysis
The following protocol, based on the BaSSSh-seq (Bacterial scRNA-seq with split-pool barcoding, second strand synthesis, and subtractive hybridization) method, is optimized for studying transcriptional heterogeneity in complex bacterial communities like biofilms, which are a known reservoir for persister cells [49] [3].
Cell Preparation and Fixation
- Harvesting: Grow bacterial cultures (e.g., Staphylococcus aureus) to the desired growth phase (planktonic exponential/stationary or biofilm). For biofilms, gently wash to remove loosely attached cells before scraping.
- Fixation and Permeabilization: Resuspend the cell pellet in a fixative solution (e.g., 4% formaldehyde) for 15-30 minutes at room temperature to preserve RNA and cellular integrity. Quench the fixation, wash, and subsequently treat cells with a permeabilization buffer (e.g., containing lysozyme and Triton X-100) to degrade the cell wall and membrane, enabling reagent access [49] [50].
Split-Pool Barcoding and cDNA Synthesis
- First Barcoding Round (Reverse Transcription): Distribute the fixed and permeabilized cells into a 96-well plate, where each well contains a unique barcoded primer. The primer consists of a cell barcode, a unique molecular identifier (UMI), and a random hexamer sequence for unbiased mRNA capture. Perform reverse transcription inside the cells.
- Pooling and Washing: Pool all cells from the 96 wells, then split them into a new 96-well plate for the second barcoding round.
- Second Barcoding Round (Ligation): In this new plate, perform a ligation reaction to attach a second well-specific barcode to the cDNA.
- Third Barcoding Round (Ligation): Repeat the pooling, splitting, and ligation steps a final time to attach a third barcode.
- Cell Lysis and cDNA Purification: After the final barcoding round, lyse the cells to release the barcoded cDNA. Purify the full-length, barcoded cDNA using streptavidin magnetic beads, which bind to a biotin tag on the terminal barcode [49].
Second Strand Synthesis and rRNA Depletion
- Second Strand Synthesis: Replace the inefficient template-switching approach with a random-primed second strand synthesis reaction. This drastically improves cDNA yield and library quality [49].
- rRNA Depletion (Subtractive Hybridization): Hybridize the double-stranded cDNA library with a pool of biotinylated DNA probes that are reverse-complementary to the conserved sequences of all major rRNA species. Add streptavidin magnetic beads to bind the probe-rDNA complexes. The mRNA-derived cDNA, remaining in the supernatant, is collected for library construction [49] [52].
Library Construction and Sequencing
Amplify the final, rRNA-depleted cDNA library via PCR, adding Illumina-compatible adapters and sample indices. The library is then sequenced on a high-throughput platform (e.g., Illumina NovaSeq) [51].
Table 2: Research Reagent Solutions for Bacterial scRNA-seq
| Item | Function | Example/Note |
|---|---|---|
| Fixative Solution | Preserves cellular RNA content and integrity instantly. | 4% Formaldehyde. |
| Permeabilization Buffer | Disrupts cell wall/membrane for intracellular access. | Contains Lysozyme & Triton X-100; concentration requires species-specific optimization [50]. |
| Barcoded Primers | Uniquely labels all transcripts from a single cell during reverse transcription. | Contains Cell Barcode, UMI (for digital counting), and Random Hexamer [49]. |
| Streptavidin Magnetic Beads | Purifies barcoded cDNA and depletes rRNA-derived cDNA. | Used in multiple cleanup and enrichment steps [49] [52]. |
| Biotinylated rRNA Probes | Hybridizes to rRNA-derived cDNA for depletion. | Probe set must be designed to span rRNA sequences of target organism(s) [52]. |
| Microfluidic Chip (e.g., Chromium X) | Partitions single cells into nanoliter-scale reactions for barcoding. | Essential for high-throughput, droplet-based methods [51]. |
Diagram 1: Bacterial scRNA-seq Workflow (e.g., BaSSSh-seq). The process involves cell fixation, combinatorial barcoding in plates, and key enzymatic steps to generate rRNA-depleted sequencing libraries.
Data Analysis and Visualization Workflow
Following sequencing, raw data is processed to generate a gene expression matrix (cells x genes). The subsequent analytical workflow involves several critical steps to extract biological meaning from the data [53] [54].
- Quality Control (QC) and Filtering: Assess each cellular barcode based on three key metrics: (i) total counts per barcode, (ii) number of genes detected per barcode, and (iii) the fraction of mitochondrial (if applicable) or other QC gene counts. Filter out barcodes with low counts/genes (indicating poor-quality cells or empty droplets) or exceptionally high counts/genes (indicating potential doublets) [53].
- Normalization and Feature Selection: Normalize the data to account for differences in sequencing depth between cells. Identify highly variable genes (HVGs) that drive heterogeneity across the population for downstream analysis [53].
- Dimensionality Reduction: Reduce the high-dimensional data using Principal Component Analysis (PCA). Subsequently, non-linear methods like t-Distributed Stochastic Neighbor Embedding (t-SNE) or Uniform Manifold Approximation and Projection (UMAP) are used to visualize cells in 2D or 3D space, where similar cells cluster together [53] [54] [55].
- Clustering and Cluster Annotation: Apply graph-based clustering algorithms to the reduced dimensions to identify groups of transcriptionally similar cells. These clusters are then annotated based on the expression of known marker genes to define cell states (e.g., persister, actively growing, stress-responsive) [53].
- Trajectory Inference (Pseudotime Analysis): For dynamic processes like persister formation or regrowth, trajectory inference tools (e.g., Monocle, PAGA) can be used to order cells along a pseudotemporal continuum, reconstructing the sequence of transcriptional changes that occur during state transitions [49] [54].
Diagram 2: scRNA-seq Data Analysis Pipeline. Key computational steps transform raw sequencing data into insights about cellular heterogeneity and dynamics.
Application in Investigating Bacterial Persisters
The application of scRNA-seq to bacterial persister research is yielding profound insights. A landmark study using BaSSSh-seq on Staphylococcus aureus biofilms captured extensive transcriptional heterogeneity and identified distinct biofilm subpopulations that displayed differential responses to immune cell pressure, providing a new level of resolution in understanding biofilm-associated persistence [49].
Furthermore, single-cell studies have challenged the classical dogma that persisters are exclusively derived from pre-existing, non-growing cells. Observations of over a million individual E. coli cells revealed that under certain conditions (e.g., treatment with ciprofloxacin), many persisters were in fact actively growing before antibiotic exposure and exhibited diverse survival dynamics, including continuous growth with morphological changes and responsive growth arrest [15]. This highlights the power of scRNA-seq and single-cell tracking to deconstruct the complex and heterogeneous nature of bacterial persistence, moving beyond bulk population averages.
Single-cell RNA sequencing has emerged as a powerful tool to dissect the transcriptional heterogeneity underlying bacterial persistence. The ongoing refinement of wet-lab protocols—particularly in cellular barcoding and rRNA depletion—coupled with advanced computational analytical frameworks, now provides researchers with a robust methodology to identify, characterize, and understand persister cells at an unprecedented resolution. The integration of these techniques is poised to accelerate the discovery of novel therapeutic targets and strategies aimed at eradicating persistent infections.
Bacterial persisters are a subpopulation of genetically susceptible cells that exhibit a transient, non-growing, or slow-growing dormant state, enabling them to survive exposure to lethal concentrations of antibiotics [3]. This phenotype is a significant clinical challenge, underlying chronic and recurrent infections, treatment failures, and contributing to the development of antibiotic resistance [3] [33]. Unlike antibiotic resistance, which is genetic and heritable, persistence is a form of phenotypic heterogeneity where persister cells are transiently tolerant and can resuscitate once antibiotic pressure is removed [3].
The study of persisters is complicated by their rarity, often constituting only 0.001–1% of a bacterial population, and their transient physiological state [33]. Flow cytometry and its advanced counterpart, imaging flow cytometry (IFC), are powerful single-cell technologies that overcome the limitations of population-averaged techniques. They enable high-throughput quantification, identification, and morphological characterization of these rare persister cells based on light scattering, fluorescence, and imagery [56] [33]. Mass cytometry is a related high-parameter technique that uses metal-tagged antibodies for detection. This application note details how these cytometric technologies are applied in bacterial persister research, providing structured experimental data and validated protocols.
Key Characteristics of Persisters and the Role of Cytometry
Persisters exhibit distinct characteristics that cytometric techniques are uniquely positioned to probe at a single-cell level:
- Metabolic Diversity and Dormancy: Persisters can be broadly categorized. Type I persisters are non-growing cells induced by external environmental factors, while Type II persisters are slow-growing cells generated spontaneously without external triggers [3]. Their metabolic states exist on a continuum from shallow to deep persistence [3]. Flow cytometry can assess metabolic activity and membrane integrity using fluorescent dyes [38].
- Morphological Changes: Entry into a persistent state is often accompanied by morphological changes. For example, rod-shaped bacteria may transition to smaller coccoid forms during slow growth or stress [56]. Imaging Flow Cytometry allows for high-throughput, quantitative analysis of these morphological shifts—such as cell size, shape, and elongation—at the single-cell level, linking morphology to physiological phenotypes like the viable but non-culturable (VBNC) state [56].
- Phenotypic Heterogeneity: Clonal bacterial populations can contain subpopulations with varying physiological states, a bet-hedging strategy to survive environmental threats [33]. Single-cell techniques like flow cytometry are essential to resolve this heterogeneity, which is often masked by population-based methods [33].
Experimental Protocols
The following protocols outline how flow cytometry can be integrated into standard persister research workflows.
Protocol 1: Imaging Flow Cytometry for Morphological Phenotyping of Bacterial Persisters
This protocol, adapted from Frontiers in Cellular and Infection Microbiology, details the use of IFC to characterize morphological changes in bacterial persisters [56].
- Key Applications: Characterizing and quantifying changes in cell size, shape, and granularity; identifying and classifying dormant cell phenotypes (e.g., VBNC cells); monitoring resuscitation post-antibiotic treatment [56].
- Bacterial Models: Validated for Bacillus subtilis, Lactiplantibacillus plantarum, Pediococcus acidilactici, and Escherichia coli [56].
Procedure:
- Culture and Stress Induction:
- Grow bacterial cultures to the desired growth phase (e.g., exponential or stationary) in appropriate media and conditions [56].
- To induce persistence, expose cultures to a lethal dose of a bactericidal antibiotic (e.g., 100 µg/ml ampicillin for E. coli DH5α). The minimum inhibitory concentration (MIC) should be determined beforehand [56].
- Sample Preparation for IFC:
- Collect samples at predetermined time points (e.g., hourly during growth, and at 6h and 16h post-antibiotic exposure) [56].
- Dilute samples to a standardized optical density (e.g., OD600 of 0.2) in an appropriate buffer to ensure optimal event rate for the cytometer [56].
- (Optional) Stain cells with fluorescent viability (e.g., propidium iodide for membrane integrity) or metabolic dyes (e.g., carboxyfluorescein diacetate succinimidyl ester for esterase activity) to correlate morphology with physiological state [56] [57].
- Data Acquisition on Imaging Flow Cytometer:
- Use an imaging flow cytometer (e.g., an Annis or Luminex instrument).
- Set up the instrument to capture brightfield and side-scatter (SSC) images for every cell.
- If fluorescent staining is used, configure the appropriate laser excitation lines and emission filters.
- Acquire data for a minimum of 10,000-50,000 events per sample to ensure adequate statistical power for identifying rare subpopulations.
- Data Analysis:
- Use the instrument's software (e.g., FlowJo, IDEAS) for analysis.
- Gating: Create a gating strategy to select single, intact cells based on aspect ratio and intensity in brightfield and SSC images [56].
- Morphological Feature Extraction: Calculate features like cell area, diameter, length, width, and eccentricity for each cell from the brightfield images.
- Population Identification: Use scatter plots of morphological features (e.g., Area vs. Aspect Ratio) to identify and gate distinct morphological subpopulations corresponding to normal, filamentous, or coccoid cells.
- Quantification: Report the frequency and morphological statistics of each gated population.
Protocol 2: Tracking Persister Recovery via Flow Cytometry
This protocol, based on methods from STAR Protocols, describes how to monitor the physiological states of persister cells as they recover from antibiotic treatment [38].
- Key Applications: Quantifying the kinetics of persister resuscitation; analyzing physiological heterogeneity (metabolic activity, membrane integrity) during recovery; elucidating genes and mechanisms involved in persister survival [38].
Procedure:
- Persister Isolation:
- Subject a bacterial culture to a high concentration of a bactericidal antibiotic (e.g., 50x MIC) for several hours to kill the majority of the population [38].
- Remove the antibiotic by washing the cells via centrifugation (e.g., 5 minutes at 8000 rpm) and resuspend the pellet in fresh, pre-warmed media [56] [38].
- Recovery and Staining:
- Incubate the resuspended cells under optimal growth conditions to allow for resuscitation.
- At regular time intervals post-antibiotic removal, collect aliquots for analysis.
- Stain samples with a combination of fluorescent dyes, for example:
- A viability dye like propidium iodide (PI) to label cells with compromised membranes.
- A metabolic dye like carboxyfluorescein diacetate succinimidyl ester (CFDA-SE or CFSE) that is cleaved by intracellular esterases in metabolically active cells [57].
- Flow Cytometric Analysis:
- Acquire data on a standard flow cytometer equipped with lasers and filters appropriate for your chosen fluorescent dyes.
- Set up a plot of metabolic dye fluorescence (e.g., FITC channel for CFSE) versus viability dye fluorescence (e.g., PE-Cy5 channel for PI).
- Data Interpretation:
- Dead/Lysed Cells: PI-positive, low metabolic fluorescence.
- Dormant Persisters: PI-negative, low metabolic fluorescence (viable but metabolically inactive).
- Resuscitating/Active Cells: PI-negative, high metabolic fluorescence (viable and metabolically active).
- Tracking the shift of the population from dormant to active states over time provides a quantitative measure of recovery kinetics.
Data Presentation and Analysis
The high-throughput nature of cytometry generates complex, multi-parametric data. The tables below summarize key experimental parameters and data analysis strategies.
Table 1: Summary of Key Experimental Parameters from Cited Studies
| Bacterial Species | Stress Inducer | Concentration | Key Cytometric Readouts | Reference |
|---|---|---|---|---|
| Escherichia coli DH5α | Ampicillin | 100 µg/ml | Cell morphology, VBNC state identification, resuscitation monitoring | [56] |
| Staphylococcus aureus | Oxacillin, Moxifloxacin | 50x MIC | Proteome labeling via BONCAT, metabolic activity, membrane integrity | [57] |
| Bacillus subtilis | Growth phase (Stationary) | N/A | Cell size, shape characterization, phenotypic heterogeneity | [56] |
| Lactiplantibacillus plantarum | Acidic Environment | N/A | Filamentation, cell integrity, metabolic activity | [56] |
Table 2: Essential Research Reagent Solutions for Cytometric Persister Analysis
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Viability Dyes (e.g., Propidium Iodide) | Labels cells with compromised membranes; distinguishes live/dead cells. | Used in combination with metabolic dyes to identify intact but dormant persisters [57]. |
| Metabolic Dyes (e.g., CFDA-SE, CFSE) | Measures enzymatic activity as a proxy for metabolic state. | Tracking the resuscitation of persisters from a metabolically inactive state to an active one [57] [38]. |
| BONCAT Probes (e.g., Aha, Hpg) | Bio-Orthogonal Non-Canonical Amino Acid Tagging; labels newly synthesized proteins. | Identifying the proteome expressed during persistence and recovery; retrieved via click chemistry for MS analysis [57]. |
| Nucleic Acid Stains (e.g., Hoechst 33342) | Labels DNA content; can be used for cell cycle or ploidy analysis. | Correlating genome copy number with persistence to antibiotics like fluoroquinolones [33]. |
| Fluorescent Reporter Plasmids | Reports on gene expression and transcriptional heterogeneity. | Studying heterogeneity in genes related to persistence, growth, and stress responses [33]. |
| Antibiotic Analogs (e.g., OPP) | Labels active cellular processes like translation. | Measuring single-cell translation rates in persister subpopulations [33]. |
Visualization of Experimental Workflow:
The following diagram illustrates the logical workflow for a typical cytometric analysis of bacterial persisters, integrating the protocols and reagents described.
Advanced Applications: Integration with Other Technologies
Flow and mass cytometry are often integrated with other powerful single-cell technologies to provide a more comprehensive view of persister biology:
- Fluorescent Biosensors and Reporters: Genetically encoded fluorescent reporters allow researchers to monitor gene expression heterogeneity (e.g., from stress response promoters) in real-time using flow cytometry [33]. FRET-based biosensors can probe protein-protein interactions and signaling molecules in single cells [33].
- BONCAT and Proteomics: As demonstrated in S. aureus, BONCAT uses non-canonical amino acids to label proteins synthesized during persistence. These proteins can be isolated via click chemistry and identified by Liquid Chromatography-Mass Spectrometry (LC-MS), providing deep insight into the active proteome of this rare cell state [57].
- Raman Spectroscopy and Microfluidics: These complementary techniques can provide information on biochemical composition and allow for long-term, time-lapse imaging of individual persister cells, respectively [33].
Flow cytometry, imaging flow cytometry, and mass cytometry are indispensable tools in the modern microbiologist's toolkit for studying bacterial persisters. Their power lies in the ability to move beyond population averages and dissect the rare and transient phenotypic heterogeneity that defines persistence. The detailed protocols and data analysis frameworks provided in this application note empower researchers to quantitatively monitor morphological changes, physiological states, and recovery dynamics of persister cells. The integration of these cytometric techniques with advanced molecular probes and other 'omics' technologies promises to further unravel the complex mechanisms of persistence, accelerating the path toward novel therapeutic strategies to combat chronic and recurrent bacterial infections.
Raman Spectroscopy and Other Label-Free Methods for Functional Analysis
Bacterial persisters are a subpopulation of cells that exhibit transient, non-inheritable tolerance to multiple antimicrobials. These phenotypic variants survive lethal antibiotic concentrations in a non-growing or slow-growing state and can resume growth after treatment cessation, contributing to chronic infections and treatment relapse [58] [3] [37]. Research on persisters presents significant technical challenges due to their low abundance, transient phenotype, and non-genetic nature [58] [37].
Label-free analytical techniques, particularly Raman spectroscopy, have emerged as powerful tools for investigating persister cells without requiring genetic modification or external labels. These methods provide intrinsic molecular "fingerprints" that reveal biochemical composition and metabolic activity at the single-cell level [58] [59] [60]. This application note details how Raman spectroscopy and related approaches are advancing bacterial persister research within the broader context of single-cell analysis techniques.
Technical Principles of Raman Spectroscopy
Raman spectroscopy is a non-intrusive analytical technique that probes molecular vibrations to generate comprehensive biochemical profiles of individual cells. When monochromatic laser light interacts with a sample, most photons scatter elastically (Rayleigh scattering), but approximately 1 in 10 million photons scatter inelastically, resulting in energy shifts that provide specific molecular bond information [61].
The resulting Raman spectrum serves as a molecular "fingerprint" with peaks corresponding to specific cellular components including proteins, lipids, nucleic acids, and metabolites [58] [60]. This label-free approach enables non-destructive analysis of living cells under physiological conditions, making it particularly valuable for studying transient physiological states like bacterial persistence.
Technical Enhancements and Variations
Surface-Enhanced Raman Spectroscopy (SERS) utilizes plasmonic nanostructures (typically gold or silver nanoparticles) to amplify Raman signals by factors of 10⁶-10⁹, enabling detection of low-abundance metabolites and single-cell analysis [61] [62]. SERS has been successfully applied to detect quorum-sensing molecules like pyocyanin in Pseudomonas aeruginosa and purine metabolites for antibiotic susceptibility testing [61] [62].
D₂O-Ramanometry integrates Raman spectroscopy with heavy water (D₂O) labeling to quantify metabolic activity. When D₂O is incorporated into cellular biosynthesis, carbon-deuterium (C-D) bonds form, detectable as distinct Raman shifts from carbon-hydrogen (C-H) bonds. The C-D/C-H ratio provides a quantitative measure of metabolic activity at single-cell resolution [58].
Application to Bacterial Persister Research
Identification and Characterization of Persisters
Single-cell Raman spectroscopy (SCRS) enables discrimination of persisters from normal cells based on intrinsic biochemical profiles. Research on Escherichia coli persisters induced by ampicillin treatment (100 μg/mL, 32× MIC, 4 hours) revealed notable differences in Raman band intensities related to major cellular components and metabolites [58]. Multivariate analysis techniques including principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE) successfully classified E. coli and persister cells into distinct projective zones based on their spectral profiles [58].
Table 1: Key Raman Spectral Differences Between E. coli and Persister Cells
| Cell Type | Biochemical Characteristics | Classification Outcome | Metabolic Activity (D₂O uptake) |
|---|---|---|---|
| Normal E. coli | Standard Raman band intensities for cellular components | Distinct projective zone in PCA/t-SNE | Lower metabolic activity |
| Ampicillin Persisters | Altered intensity in bands for proteins, lipids, metabolites | Separate projective zone in PCA/t-SNE | Higher metabolic activity |
Metabolic Activity Assessment
Contrary to the historical view of persisters as completely dormant cells, SCRS studies reveal complex metabolic heterogeneity. D₂O-Ramanometry demonstrated that E. coli persisters exhibit higher metabolic activities than untreated cells [58]. After antibiotic removal, persister cells showed a temporal pattern of D₂O intake distinct from non-persister cells, providing insights into their resuscitation dynamics [58].
The following diagram illustrates the experimental workflow for identifying and analyzing bacterial persisters using D₂O-Ramanometry:
Antibiotic Susceptibility Testing and Resistance Detection
Raman spectroscopy enables rapid antibiotic susceptibility testing (AST) by detecting phenotypic responses to antimicrobial agents. Studies demonstrate that SCRS can identify "antibiotic effect signatures" in susceptible E. coli cells after just 2-hour exposure to antibiotics at clinical breakpoint concentrations [59]. These spectral changes, accompanied by increased intercellular heterogeneity, can be detected through principal component analysis [59].
SERS-based approaches can determine minimum inhibitory concentrations (MIC) in approximately 1 hour by monitoring antibiotic-induced changes in purine metabolite secretion patterns [62]. This method significantly reduces the time-to-result compared to conventional growth-based AST (24-48 hours). Furthermore, deep learning approaches applied to Raman spectra can distinguish methicillin-resistant and susceptible Staphylococcus aureus (MRSA/MSSA) with 89±0.1% accuracy [60].
Table 2: Raman Spectroscopy Applications in Antimicrobial Research
| Application | Methodology | Time Required | Key Outcome |
|---|---|---|---|
| Persister Identification | SCRS + Multivariate Analysis | 4h antibiotic treatment + analysis | Distinct spectral profiles for persisters |
| Metabolic Activity (D₂O-Ramanometry) | C-D band detection in D₂O-labeled cells | 2-4h incubation + measurement | Higher metabolism in persisters than normal cells |
| Antibiotic Susceptibility Testing | Spectral changes after antibiotic exposure | 2h exposure + analysis | "Antibiotic effect signature" detection |
| MIC Determination | SERS of purine metabolites | ~1h total | Accurate MIC values for various antibiotics |
| Resistance Detection (MRSA/MSSA) | Deep Learning + Raman Spectroscopy | Acquisition + algorithm processing | 89.1±0.1% classification accuracy |
Experimental Protocols
Protocol 1: Identification of Bacterial Persisters Using SCRS and D₂O-Ramanometry
Principle: This protocol utilizes single-cell Raman spectroscopy to distinguish persisters based on intrinsic biochemical profiles and measures their metabolic activity via D₂O incorporation [58].
Materials:
- Bacterial strain: Escherichia coli ATCC 25922
- Luria-Bertani (LB) broth and agar plates
- Ampicillin sodium salt (100 μg/mL working concentration)
- Deuterium oxide (D₂O, 99.9% atom % D)
- Phosphate buffered saline (PBS, pH 7.4)
- Raman spectrometer with microscope (532 nm or 785 nm laser)
- Microfluidic device or concentration chamber for single-cell analysis
Procedure:
- Culture Preparation: Inoculate E. coli from frozen stock onto LB agar plates and incubate overnight at 37°C. Pick a single colony into 5 mL liquid LB broth and culture with shaking (200 rpm) for 12 hours at 37°C.
- Persister Induction: Dilute overnight culture 1:100 into fresh LB broth and culture to mid-exponential phase (OD₆₀₀ ≈ 0.5). Add ampicillin to final concentration of 100 μg/mL (32× MIC) and incubate for 4 hours at 37°C with shaking.
- Cell Washing: Centrifuge bacterial suspension at 5,000 × g for 5 minutes and wash twice with PBS to remove antibiotic and debris.
- D₂O Labeling: Resuspend bacterial pellet in LB medium prepared with 70% D₂O and incubate for 30-60 minutes at 37°C.
- Raman Measurement:
- Deposit 2-3 μL of bacterial suspension onto aluminum-coated slides or Raman-compatible substrates.
- Acquire Raman spectra from individual cells using 532 nm or 785 nm laser excitation with 1-10 seconds integration time.
- Collect spectra from at least 50-100 individual cells per sample to account for heterogeneity.
- Data Analysis:
- Pre-process spectra: subtract background fluorescence, normalize to internal standard (e.g., CH stretching band at 2930 cm⁻¹).
- Perform multivariate analysis (PCA, t-SNE) to classify spectral profiles.
- Calculate C-D/C-H ratio from band intensities at 2040-2300 cm⁻¹ (C-D) and 2800-3100 cm⁻¹ (C-H) to quantify metabolic activity.
Expected Results: Persister cells will exhibit distinct Raman spectra compared to normal cells, with altered band intensities for proteins, lipids, and nucleic acids. D₂O incorporation rates will reveal heterogeneous metabolic activities among persister subpopulations.
Protocol 2: Ultra-Rapid Antibiotic Susceptibility Testing via SERS
Principle: This protocol leverages SERS detection of purine metabolites to determine antibiotic susceptibility profiles within 1 hour, based on antibiotic-induced changes in bacterial stringent response and purine secretion [62].
Materials:
- Clinical bacterial isolates or reference strains
- Mueller Hinton Broth (MHB)
- Antibiotic stock solutions at appropriate concentrations
- Gold nanoparticle colloid (60 nm diameter)
- Silicon wafer substrates or glass slides
- Phosphate buffered saline (PBS, pH 7.4)
- SERS spectrometer with 785 nm laser excitation
Procedure:
- Antibiotic Exposure:
- Prepare doubling dilutions of antibiotic in MHB (0, 2×, 4×, 8×, 16× MIC expected values) in 50 mL tubes.
- Add bacterial inoculum (10⁸ CFU/mL) to each antibiotic concentration and incubate for 30 minutes at 37°C.
- Sample Preparation:
- Centrifuge 1 mL of each bacterial suspension at 5,000 × g for 5 minutes.
- Wash bacterial pellets twice with deionized water to remove media components.
- Resuspend cells in 20 μL deionized water.
- SERS Substrate Preparation:
- Deposit 2 μL of gold nanoparticle colloid onto silicon wafer substrate.
- Allow to air dry for 5-10 minutes.
- SERS Measurement:
- Apply 2 μL of bacterial suspension onto prepared SERS substrate.
- Acquire SERS spectra using 785 nm laser excitation with 5-10 seconds integration time.
- Collect 20-30 spectra from different spots for each sample.
- Data Analysis:
- Normalize spectra to the total intensity or internal standard.
- Monitor intensity changes of purine metabolite peaks (650-750 cm⁻¹ region).
- Determine MIC as the lowest antibiotic concentration showing significant purine signal reduction compared to untreated control.
Expected Results: Susceptible strains will show dose-dependent reduction in purine metabolite signals with increasing antibiotic concentrations, while resistant strains will maintain consistent signal levels across concentrations.
The following diagram illustrates the SERS-based antibiotic susceptibility testing workflow:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Raman Spectroscopy of Bacterial Persisters
| Reagent/Material | Function/Application | Example Specifications |
|---|---|---|
| Deuterium Oxide (D₂O) | Metabolic activity labeling for D₂O-Ramanometry | 99.9% atom % D, sterile-filtered |
| Gold Nanoparticles | SERS substrate for enhanced signal detection | 60 nm diameter, citrate-stabilized |
| Raman-Compatible Substrates | Sample platform for SCRS measurements | Aluminum-coated slides, silicon wafers |
| Mueller Hinton Broth | Standard medium for antibiotic susceptibility testing | CLSI-compliant formulation |
| Reference Bacterial Strains | Method validation and quality control | ATCC 25922 (E. coli) and other relevant strains |
| Antibiotic Reference Standards | Persister induction and AST | USP-grade, known potency |
Integration with Other Single-Cell Techniques
Raman spectroscopy complements other single-cell approaches in bacterial persister research. Proteomic studies using Bio-Orthogonal Non-Canonical Amino Acid Tagging (BONCAT) have identified widespread translational changes in Staphylococcus aureus persisters, including alterations in purine and amino acid metabolism, stress response systems, and transporter activities [63]. High-throughput proteomics of E. coli persisters has revealed proteins important for postantibiotic recovery, such as AhpF (alkyl hydroperoxide reductase) and OmpF (outer membrane porin) [64].
These multi-modal approaches provide comprehensive insights into persister physiology, revealing that persistence mechanisms vary across bacterial genotypes and antibiotic exposures rather than following a universal pathway [63] [64].
Raman spectroscopy and related label-free methods provide powerful capabilities for investigating bacterial persisters at single-cell resolution. These approaches enable researchers to identify persister cells based on intrinsic biochemical profiles, quantify their metabolic activities, determine antibiotic susceptibility phenotypes, and elucidate mechanisms of persistence and resuscitation. The techniques outlined in this application note—particularly SCRS, D₂O-Ramanometry, and SERS-based AST—offer robust methodologies for advancing our understanding of bacterial persistence and developing more effective therapeutic strategies against chronic and recurrent infections.
Navigating Technical Challenges in Single-Cell Persister Research
Bacterial persisters are a subpopulation of cells that are genetically susceptible to antibiotics but can survive lethal antibiotic concentrations by entering a transient, dormant state [3] [37]. These cells typically constitute only 0.001–1% of a bacterial population, posing significant technical challenges for their study [33]. Their isolation and enrichment are critical steps in understanding the mechanisms underlying phenotypic heterogeneity and developing treatments for chronic, recurrent infections [3]. This application note details established and emerging methodologies for enriching and isolating these rare persister cells, framed within the context of single-cell analytical techniques.
Core Concepts and Definitions
Distinguishing Persister Phenomena
Persisters are often confused with other bacterial survival states. The table below clarifies key concepts and their relationships to persistence.
Table 1: Key Concepts in Bacterial Survival and Their Relation to Persistence
| Term | Definition | Genetic Basis | MIC Change | Population Heterogeneity |
|---|---|---|---|---|
| Antibiotic Resistance | Ability to grow in the presence of an antibiotic due to genetic mutations [37]. | Heritable genetic changes [37]. | Increased [37]. | Homogeneous (entire population is resistant). |
| Antibiotic Tolerance | Ability of a bulk population to survive transient antibiotic exposure without genetic change; characterized by slower killing [65] [37]. | Non-heritable, phenotypic [65]. | Unchanged [37]. | Homogeneous (entire population exhibits tolerance) [37]. |
| Antibiotic Persistence | Ability of a small subpopulation to survive lethal antibiotic treatment due to a dormant, non-growing state [33] [3]. | Non-heritable, phenotypic [37]. | Unchanged [37]. | Heterogeneous (a small subpopulation exhibits the trait) [33] [37]. |
| Viable But Non-Culturable (VBNC) | A deep-dormancy state where cells are alive but cannot proliferate on standard media without specific resuscitation stimuli [3] [37]. | Non-heritable, phenotypic [37]. | Unchanged (assumed). | Can be heterogeneous or homogeneous. |
Quantitative Landscape of Persistence
The prevalence of persisters varies significantly across bacterial species and in response to different antibiotics. The following table consolidates quantitative data on persister fractions from a broad survey of the literature.
Table 2: Persister Fractions Across Bacterial Species and Antibiotic Classes [12]
| Bacterial Species | Number of Antibiotics Tested | Typical Persister Fraction (Median) | Notes |
|---|---|---|---|
| Escherichia coli | 32 | Varies by antibiotic and condition | Most extensively studied model organism. |
| Staphylococcus aureus | 18 | Varies by antibiotic and condition | Includes MRSA strains. |
| Pseudomonas aeruginosa | 16 | Varies by antibiotic and condition | Common in cystic fibrosis infections. |
| Acinetobacter baumannii | Not specified | ~0.01% | Lowest among species with ≥20 data points. |
| Enterococcus faecium | Not specified | Up to 100% in some conditions | Exhibits very high levels of survival. |
| All Species (Range) | 54 | 7 × 10⁻⁴% to 100% | Highlights immense variation. |
Antibiotic Class Impact: Membrane-active antibiotics (e.g., polymyxins) generally result in the lowest persister fractions, while other classes can select for significantly higher survival rates [12].
Methodologies for Persister Enrichment and Isolation
A successful strategy for obtaining persisters involves first enriching their proportion in a population and then isolating them for downstream analysis.
Enrichment Strategies
Antibiotic Killing and Physiological Separation
The most common enrichment method is the application of a lethal dose of a bactericidal antibiotic to kill the majority of the population, leaving the non-growing persisters alive.
- Protocol: Standard Time-Kill Assay for Persister Enrichment [38]
- Culture Preparation: Grow a culture of the target bacterium to the desired growth phase (e.g., mid-exponential or stationary phase). The growth phase is a critical factor, as stationary phase cultures often contain a higher initial fraction of persisters [12].
- Antibiotic Exposure: Treat the culture with a high concentration (typically 10-100x the MIC) of a selected bactericidal antibiotic (e.g., ampicillin, ciprofloxacin).
- Incubation: Incubate the culture under optimal growth conditions for a defined period (usually 3-6 hours) to allow for complete killing of non-persister cells.
- Termination of Antibiotic Activity: Remove the antibiotic. This can be achieved by:
- Washing: Centrifuging the culture and resuspending the pellet in fresh, antibiotic-free medium.
- Enzyme Inactivation: Adding a neutralizing enzyme (e.g., penicillinase for β-lactams) [66].
- Dilution: Diluting the culture into a large volume of fresh medium.
- Validation: Plate serial dilutions of the treated culture on antibiotic-free agar to quantify the number of surviving colony-forming units (CFUs), which represent the enriched persister population.
Physiological and Genetic Induction
Persister levels can be increased by exploiting the biological mechanisms that control dormancy.
- Nutrient Starvation: Incubating cells in saline or phosphate-buffered saline (PBS) for several hours induces a state of phenotypic tolerance that can enrich for persister-like cells [65].
- Stationary Phase Enrichment: Harvesting cells from the stationary phase of growth naturally yields a higher percentage of persisters compared to exponential phase cultures [12].
- Genetic Manipulation: Utilizing mutant strains with defects in key metabolic or regulatory pathways (e.g.,
hipA7ormetG2mutants in E. coli) can increase the baseline frequency of persister formation by several orders of magnitude, simplifying enrichment [65] [3].
Isolation Techniques
Following enrichment, persisters must be physically separated from dead cells and debris for single-cell analysis.
Fluorescence-Activated Cell Sorting (FACS)
FACS is a powerful technique for isolating persisters based on physiological markers.
- Principle: Cells are stained with fluorescent dyes that report on viability, membrane integrity, or metabolic activity and are then physically separated into multi-well plates or tubes based on their fluorescence signature.
- Protocol: Staining and Sorting for Low Metabolic Activity
- Staining: Incubate the antibiotic-treated culture with a fluorescent dye such as:
- SYTOX Green: Stains DNA in cells with compromised membranes (dead cells). Persisters with intact membranes will exclude the dye [33].
- Fluorescent dyes for metabolic activity (e.g., CFDA, CTC): Identify cells with low enzymatic activity or respiration, a hallmark of persisters.
- Gating Strategy: Using a flow cytometer, establish a gate for the population of interest—e.g., SYTOX Green-negative (viable) and low metabolic fluorescence (dormant).
- Sorting: Sort the gated population directly into lysis buffer for molecular analysis or into nutrient broth for recovery studies [33].
- Staining: Incubate the antibiotic-treated culture with a fluorescent dye such as:
Microfluidics and Single-Cell Analysis
Microfluidic platforms allow for the immobilization and long-term observation of individual cells, enabling the direct identification and study of persisters without the need for prior sorting.
- Application: Single cells can be trapped in micro-wells or channels, exposed to pulses of antibiotics, and monitored for survival and regrowth, directly identifying persisters based on their phenotype [33].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for Persister Research
| Reagent/Tool | Function/Principle | Key Application |
|---|---|---|
| Fluorescent Biosensors (e.g., QUEEN-ATP, iATPSnFR) | Genetically encoded sensors that measure intracellular ATP concentrations via fluorescence ratio imaging [33]. | Quantifying metabolic dormancy at the single-cell level. |
| Membrane Integrity Probes (e.g., SYTOX Green, Propidium Iodide) | Nucleic acid stains that cannot penetrate intact membranes; selectively stain dead cells [33]. | Differentiating viable persisters from dead cells in a population. |
| Metabolic Activity Dyes (e.g., CTC, CFDA) | Fluorescent compounds that require enzymatic conversion or membrane potential for fluorescence activation [33]. | Identifying dormant, low-activity persister cells via flow cytometry. |
| Click Chemistry Probes (e.g., OPP) | Puromycin analog that incorporates into nascent peptides; via click chemistry, it can be linked to a fluorophore to report on translation rates [33]. | Detecting and isolating cells with low levels of protein synthesis. |
| Microfluidic Devices | Platforms with channels and chambers for manipulating picoliter to nanoliter fluid volumes and immobilizing single cells [33]. | Tracking the formation and resuscitation of individual persister cells over time. |
Visualizing Experimental Workflow and Molecular Mechanisms
The following diagrams outline the core experimental workflow for persister isolation and the key molecular mechanisms that can be exploited for their enrichment.
Diagram 1: Experimental Workflow for Persister Research. This diagram outlines the sequential phases of persister studies, from initial enrichment via multiple methods to final isolation and analysis using single-cell techniques.
Diagram 2: Key Molecular Pathways in Persister Formation. This diagram illustrates the core molecular mechanisms, including toxin-antitoxin systems and the stringent response, that converge to induce the dormant state responsible for antibiotic tolerance in persisters [3] [37].
Single-cell RNA sequencing (scRNA-seq) has revolutionized the study of cellular heterogeneity in eukaryotic systems. Its application to bacterial physiology, particularly in the context of persister cell research, promises to unravel the mechanisms underlying phenotypic heterogeneity and antibiotic tolerance [67] [3]. Bacterial persisters—metabolically dormant, genetically susceptible subpopulations that survive antibiotic treatment—represent a significant clinical challenge in combating chronic and recurrent infections [3] [33]. However, technical hurdles related to bacterial cell structure and RNA biology have historically limited the application of scRNA-seq in prokaryotic systems [67] [50]. This application note details the major challenges and corresponding methodological advances in bacterial scRNA-seq, with a specific focus on investigating persister cell formation and survival mechanisms.
Technical Hurdles and Methodological Advances
The adaptation of scRNA-seq for bacterial systems requires overcoming several distinct technical challenges that stem from fundamental differences between prokaryotic and eukaryotic cell biology. The table below summarizes the primary hurdles and corresponding solutions that have been developed.
Table 1: Key Technical Hurdles in Bacterial scRNA-seq and Current Solutions
| Technical Hurdle | Impact on scRNA-seq | Current Solutions |
|---|---|---|
| Robust Cell Wall Lysis [50] | Inefficient RNA release due to Gram-positive (thick peptidoglycan) or Gram-negative (outer membrane) structures. | Optimization of lysis buffers (e.g., Triton X-100, EDTA, lysozyme); mechanical disruption (vortexing, sonication) [50] [49]. |
| Low RNA Quantity & Quality [67] [50] | A single bacterial cell contains ~0.1 pg of total RNA (100-200x less than eukaryotic cells), with mRNA comprising only 4-5%. | Reverse transcription with random hexamers; polyadenylation of bacterial mRNA; whole transcriptome amplification [50]. |
| Lack of Poly-A Tails [50] [68] | Precludes use of standard oligo(dT) primers for mRNA enrichment. | Use of random hexamer primers for cDNA synthesis; template-switching mechanisms [50] [49]. |
| High Ribosomal RNA Content [50] [68] | rRNA can constitute >80% of total RNA, drastically reducing sequencing depth for informative transcripts. | CRISPR-based depletion (e.g., DASH/scDASH); RNase H-mediated digestion; subtractive hybridization [68] [49] [69]. |
| Short mRNA Half-Life [50] | Bacterial mRNA typically lasts only minutes, requiring immediate stabilization. | Rapid cell fixation and permeabilization; use of RNA stabilization reagents immediately upon collection [50]. |
Cell Wall Lysis and Single-Cell Separation
Effective lysis of individual bacterial cells is a critical first step. The rigid, complex structure of bacterial cell walls, whether Gram-positive or Gram-negative, resists standard mammalian lysis protocols [50]. Early bacterial scRNA-seq protocols often relied on physical separation methods like fluorescence-activated cell sorting (FACS), laser capture microdissection (LCM), or micromanipulation to isolate individual cells into plates [50]. More recent high-throughput methods, such as the BaSSSh-seq (bacterial scRNA-seq with split-pool barcoding, second strand synthesis, and subtractive hybridization) protocol used for Staphylococcus aureus biofilms, employ split-pool barcoding which bypasses the need for physical isolation before lysis [49]. This method involves fixing and permeabilizing cells first, then labeling their RNA with combinatorial barcodes through successive rounds of splitting and pooling [49]. Lysis is then achieved with optimized buffers, often containing lysozyme to degrade peptidoglycan, and can be supplemented by brief vortexing or sonication [50] [49].
RNA Capture and cDNA Synthesis
Capturing the sparse and unstable mRNA from a single bacterial cell is a major technical challenge. The extremely low abundance of bacterial mRNA is compounded by its lack of a 3' poly(A) tail, which is the standard anchor for cDNA synthesis in eukaryotic scRNA-seq [50] [68]. To address this, bacterial scRNA-seq methods universally use random hexamer primers for reverse transcription, enabling unbiased capture of all RNA species, including non-polyadenylated mRNA [50] [49]. Following RNA capture, a common innovation in protocols like BaSSSh-seq is the use of random primed second strand synthesis to generate double-stranded cDNA libraries. This approach has been shown to be significantly more efficient than template-switching mechanisms, which can lead to substantial transcript loss, especially for the low-abundance RNA found in bacterial biofilms [49].
Ribosomal RNA Depletion
The overwhelming abundance of ribosomal RNA (rRNA) in bacterial cells (≥80% of total RNA) poses a significant problem for sequencing efficiency, as it drastically reduces the read depth for informative mRNA transcripts [50] [68]. Several strategies have been developed to deplete rRNA, each with distinct advantages.
- CRISPR-Based Depletion (DASH/scDASH): The DASH (Depletion of Abundant Sequences by Hybridisation) method uses Cas9 nuclease complexed with a library of single-guide RNAs (sgRNAs) designed against target rRNA sequences. This complex is applied to the pooled cDNA library after amplification, cleaving the rRNA-derived cDNA fragments. The remaining, undigested cDNA is then selectively PCR-amplified [68] [69]. The scDASH variant has been adapted for single-cell total RNA-seq, achieving a 70% reduction in rRNA sequencing coverage and a 3.5-fold enrichment of informative reads [68] [70].
- Enzyme-Free Subtractive Hybridization: The BaSSSh-seq protocol employs a subtraction-based method that uses biotinylated DNA oligomers complementary to rRNA sequences. These oligomers hybridize to the target rRNA-cDNA in the library, and the complexes are then removed using streptavidin-coated magnetic beads. This method reduces reagent costs and avoids potential biases introduced by enzymatic treatments [49].
- RNase H-Mediated Depletion: This method involves hybridizing DNA oligos to rRNA sequences in the RNA or cDNA, followed by digestion with RNase H, which specifically cleaves the RNA strand in RNA-DNA hybrids. While effective, this method can be challenging to implement inside fixed cells prior to barcoding, potentially leading to cell loss [49].
Table 2: Comparison of Primary rRNA Depletion Methods for Bacterial scRNA-seq
| Method | Mechanism | Key Component(s) | Efficiency | Pros/Cons |
|---|---|---|---|---|
| scDASH [68] [69] [70] | Post-amplification CRISPR/Cas9 cleavage of rRNA cDNA. | Cas9 nuclease, rRNA-specific sgRNA library. | ~70% reduction in rRNA reads. | Pro: High specificity; post-library synthesis. Con: Requires design and synthesis of multiple sgRNAs. |
| Subtractive Hybridization [49] | Post-synthesis hybridization and magnetic bead removal. | Biotinylated rRNA probes, Streptavidin beads. | Significant reduction (specific % not stated). | Pro: Enzyme-free; cost-effective. Con: Requires optimization of hybridization conditions. |
| RNase H-Mediated [49] | Digestion of rRNA in RNA-DNA hybrids. | DNA oligos, RNase H enzyme. | ~50% reduction in rRNA reads. | Pro: Well-established mechanism. Con: Risk of off-target digestion; can be difficult to perform on fixed cells. |
Application to Bacterial Persister Research
The ability to profile transcriptomes at single-cell resolution is particularly transformative for studying bacterial persisters. These cells are rare, transient, and exist in a state of phenotypic heterogeneity within a genetically identical population, making them invisible to bulk genomic and transcriptomic analyses [3] [33] [15]. Bacterial scRNA-seq enables researchers to directly investigate the transcriptional states that lead to and define the persister phenotype.
A key application is the identification of rare transcriptional subpopulations within a larger community, such as a biofilm, that may have enhanced survival potential [49]. For instance, when S. aureus biofilms were exposed to different types of immune cells (macrophages, neutrophils, granulocytic myeloid-derived suppressor cells), scRNA-seq revealed distinct transcriptional responses within specific biofilm subpopulations, highlighting how pathogens adapt to immune pressure at a cellular level [49]. Furthermore, scRNA-seq can help characterize the metabolic and stress response pathways that are active in persister cells, moving beyond classical models that assume all persisters are entirely dormant [33]. Single-cell studies have shown that persisters can exhibit diverse survival dynamics, including continuous growth with morphological changes or responsive growth arrest, depending on the antibiotic and pre-exposure history [15].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Bacterial scRNA-seq
| Reagent / Material | Function | Example Application / Note |
|---|---|---|
| Lysozyme [50] | Enzymatic degradation of the bacterial peptidoglycan cell wall. | A key component of lysis buffers for Gram-positive bacteria. |
| Triton X-100 & EDTA [50] | Detergent and chelating agent used to disrupt cell membranes and aid lysozyme activity. | Common components of a bacterial single-cell lysis buffer. |
| Random Hexamer Primers [50] [49] | Primers for reverse transcription that bind randomly to all RNA transcripts, essential for capturing non-polyadenylated bacterial mRNA. | Used in place of oligo(dT) primers in virtually all bacterial scRNA-seq protocols. |
| Cas9 Nuclease & sgRNA Library [68] [69] | Core components of the DASH/scDASH method for targeted depletion of rRNA sequences from cDNA libraries. | sgRNAs are designed to tile across the entire length of target rRNA genes (e.g., 16S). |
| Biotinylated Oligonucleotides & Streptavidin Beads [49] | Used in subtractive hybridization methods to capture and remove rRNA sequences magnetically. | Provides an enzyme-free alternative for rRNA depletion. |
| Fixation/Permeabilization Reagents (e.g., formaldehyde, methanol) [49] | To crosslink and preserve cellular contents and create pores in the cell wall for reagent entry. | Critical for split-pool barcoding methods where barcoding occurs inside intact cells. |
| Template Switching Oligo (TSO) | Facilitates the addition of a universal PCR handle during cDNA synthesis, used in some protocols (e.g., Smart-seq2). | Note: Newer methods (e.g., BaSSSh-seq) are moving to second-strand synthesis due to higher efficiency [49]. |
The following diagram illustrates a generalized, integrated workflow for bacterial scRNA-seq that incorporates solutions to the key hurdles of lysis, RNA capture, and rRNA depletion.
Detailed Protocol: Key Steps for Bacterial scRNA-seq (Adapted from BaSSSh-seq and scDASH)
Sample Preparation and Fixation:
- Grow bacterial culture under conditions known to induce persister formation (e.g., stationary phase, specific stressor).
- Fix cells immediately by adding formaldehyde to a final concentration of 1-3% and incubating for 15-60 minutes at room temperature.
- Permeabilize cells by washing and resuspending in ice-cold 70% methanol for at least 30 minutes on ice. This is critical for allowing reagents to access the RNA in subsequent steps [49].
Split-Pool Barcoding (for plate-based methods):
- Distribute permeabilized cells into a 96-well plate where each well contains a unique barcode oligo with a random hexamer sequence.
- Perform reverse transcription to incorporate the first barcode into the cDNA.
- Pool the cells, mix thoroughly, and redistribute into a new plate for a second round of barcode ligation. Repeat this process for a third round to achieve a combinatorial barcode unique to each cell [49].
Cell Lysis and RNA Capture:
- After barcoding, lyse the cells by resuspending in a lysis buffer containing Triton X-100 (e.g., 0.2%), EDTA (e.g., 1 mM), and lysozyme (e.g., 1 mg/mL). Incubate at 37°C for 15-30 minutes with agitation [50] [49].
- Purify the barcoded RNA/cDNA molecules using streptavidin magnetic beads (if the barcodes are biotinylated).
cDNA Library Construction:
- Generate double-stranded cDNA using random primed second strand synthesis. This is more efficient for bacterial samples than template switching [49].
- Amplify the final cDNA library with PCR using primers specific to the universal handles added during barcoding.
rRNA Depletion (using scDASH):
- Design and synthesize a library of sgRNAs tiling the target rRNA sequences (e.g., 16S, 23S) [68] [69].
- Complex the sgRNAs with Cas9 nuclease to form ribonucleoproteins (RNPs).
- Incubate the pooled cDNA library with the RNP mixture. Cas9 will cleave the cDNA fragments derived from rRNA.
- Perform a final PCR amplification to enrich the non-cleaved, informative cDNA fragments for sequencing [68] [70].
The methodological breakthroughs in bacterial scRNA-seq—specifically in overcoming the hurdles of cell wall lysis, RNA capture, and rRNA depletion—have opened a new frontier in microbiology. By enabling high-resolution profiling of transcriptional heterogeneity, these techniques provide a powerful lens through which to study bacterial persister cells. The continued refinement and application of these protocols will be instrumental in deciphering the molecular pathways of antibiotic tolerance and survival, ultimately informing the development of novel therapeutic strategies to eradicate persistent infections.
The study of bacterial persisters represents a significant challenge in microbiology and therapeutic development. These rare, transient phenotypic variants exhibit remarkable tolerance to antibiotic treatments despite genetic susceptibility, contributing substantially to chronic and recurrent infections [3] [37]. A key characteristic of persisters is their metabolic dormancy and phenotypic heterogeneity, which allows them to survive environmental stresses that eliminate their genetically identical counterparts [3]. This physiological state is inherently fragile and can be easily altered by experimental perturbations, potentially leading to misleading conclusions about persister biology and mechanisms.
Maintaining physiological relevance during single-cell analysis is therefore paramount. Traditional bulk measurement techniques mask the critical heterogeneity within bacterial populations, while conventional single-cell approaches often introduce significant perturbations through sample preparation, cell sorting, and analysis procedures [33]. These perturbations can alter metabolic states, gene expression profiles, and ultimately the very persister phenotypes researchers seek to understand. This Application Note provides detailed protocols and methodologies designed to minimize experimental perturbations during single-cell analysis of bacterial persisters, ensuring the preservation of their native physiological states throughout the investigative process.
Key Challenges in Single-Cell Analysis of Bacterial Persisters
Technical Hurdles in Microbial Single-Cell RNA Sequencing
Applying single-cell RNA sequencing (scRNA-seq) to bacterial systems presents unique challenges that differ substantially from eukaryotic applications. Bacterial mRNA lacks polyadenylated tails, necessitating alternative capture methods, and the total RNA content per bacterial cell is dramatically lower—typically 10- to 100-fold less than in mammalian cells [71]. Furthermore, bacterial cell walls are structurally diverse and often require harsh enzymatic or mechanical disruption that can significantly alter transcriptional profiles and cellular physiology [71]. Perhaps most critically, ribosomal RNA (rRNA) constitutes over 90% of bacterial RNA content, overwhelming mRNA signals without effective depletion strategies [49].
Preservation of Native Physiological States
The transient nature of the persister phenotype introduces additional complexities. Persister cells exist along a continuum of "dormancy depth," with varying metabolic activities and persistence capabilities [3]. Their physiological states are exquisitely sensitive to environmental changes, meaning that standard laboratory procedures—including centrifugation, chemical fixation, and even changes in temperature or nutrient availability—can trigger transitions between persistent and non-persistent states [33] [37]. This sensitivity demands experimental approaches that minimize both physical and chemical perturbations throughout the entire analytical workflow.
Advanced Methodologies for Perturbation-Minimized Single-Cell Analysis
Bacterial Single-Cell RNA Sequencing with Minimal Perturbation
The BaSSSh-seq (Bacterial Single-cell RNA sequencing with Split-pool barcoding, Second strand synthesis, and Subtractive hybridization) methodology represents a significant advancement in microbial transcriptomics that effectively addresses key technical challenges while maintaining physiological relevance [49]. This protocol optimizes RNA capture from bacterial cells with low metabolic activity, such as those found in persister populations and biofilms.
Key Features of BaSSSh-seq:
- Split-pool barcoding: Labels individual cells without sophisticated commercial equipment, enhancing accessibility
- Random hexamer capture: Provides unbiased transcript detection without requiring prior knowledge of genomic targets
- Second strand synthesis: Replaces inefficient template switching to significantly improve transcript yield
- Enzyme-free rRNA depletion: Utilizes subtractive hybridization to reduce rRNA contamination without additional enzymatic steps on fixed cells
Table 1: Comparison of Single-Cell RNA Sequencing Approaches for Bacterial Persister Studies
| Method Feature | BaSSSh-seq | Plate-based Methods | Microfluidics-based Methods |
|---|---|---|---|
| Cell Throughput | High | Limited (96-384 wells) | High |
| Equipment Requirements | Standard lab equipment | Standard lab equipment | Specialized commercial instruments |
| rRNA Depletion Efficiency | High (subtractive hybridization) | Variable | Moderate (Cas9/RNase H) |
| RNA Capture Method | Random hexamers | Targeted probes or random hybridization | Variable |
| Applicability to Biofilms | Optimized | Limited | Limited to planktonic cells |
| Cell Loss During Processing | Minimized | Significant | Significant with additional enzymatic steps |
Experimental Protocol: BaSSSh-seq for Bacterial Persister Analysis
Sample Preparation and Cell Handling
- Gentle Harvesting: Culture bacterial persisters using established models (stationary phase cultures, antibiotic enrichment, or biofilm models). Avoid centrifugation; instead, use gentle filtration or passive settling to harvest cells.
- Minimal Perturbation Fixation: Fix cells with 1% formaldehyde for 15 minutes at room temperature with gentle agitation. Quench with 125mM glycine for 5 minutes.
- Permeabilization Optimization: Permeabilize cells using lysozyme (1mg/mL) in TE buffer for 10 minutes on ice. Monitor permeabilization efficiency microscopically with SYTOX Green nucleic acid stain.
- Cell Integrity Verification: Verify cell wall integrity and membrane potential using fluorescent probes (e.g., membrane potential-sensitive dyes) to ensure persister phenotypes are maintained.
Split-Pool Barcoding and Library Preparation
- Barcoding Reaction Setup: Resuspend fixed, permeabilized cells in reverse transcription mixture containing random hexamers with barcodes in 96-well plate format.
- Three-Round Barcoding: Perform three rounds of barcoding with pooling and mixing steps between rounds. Include complementary oligos during pooling to block unreacted barcodes and prevent non-specific ligations.
- Cell Handling Between Rounds: Filter, vortex, and briefly sonicate cells between barcoding steps to decrease doublet formation.
- Transcript Capture and Purification: Lyse cells and purify captured transcripts using streptavidin magnetic beads leveraging the biotin tag on the 5'-end of the terminal barcode oligo.
- Second Strand Synthesis: Perform random primed second strand synthesis to generate double-stranded cDNA libraries. This approach significantly improves transcript yield compared to template switching methods.
- rRNA Depletion: Implement subtractive hybridization for enzyme-free rRNA depletion to significantly reduce rRNA contamination (from >90% to manageable levels) without additional enzymatic steps.
Quality Control and Sequencing
- Library Quality Assessment: Verify library quality using Bioanalyzer or TapeStation analysis. Expect typical yields of 10-50pg/μL for persister samples.
- Sequencing Parameters: Sequence libraries on appropriate platforms (Illumina NovaSeq or NextSeq) with 75bp paired-end reads recommended for bacterial transcriptomes.
- Bioinformatic Analysis: Process data using customized pipelines that account for barcode decoding, UMI deduplication, and normalization specific to bacterial scRNA-seq data.
Integration of Complementary Single-Cell Technologies
Fluorescence-Based Approaches with Minimal Intervention
While scRNA-seq provides comprehensive transcriptional profiling, fluorescence-based methods offer complementary insights with potentially lower perturbation when properly implemented.
Fluorescent Biosensor Implementation
- Multi-Reporter Constructs: Engineer plasmids with multiple fluorescent reporters to simultaneously monitor different transcriptional activities with minimal spectral overlap [33]. For persister studies, focus reporters on key pathways: toxin-antitoxin systems, stringent response elements, and metabolic activity markers.
- Protein-FP Fusions: Utilize fluorescent protein fusions to track protein localization and abundance without antibody-based detection that requires cell fixation [33]. Implement controls to verify that fusion proteins do not alter native protein function or persistence formation.
- Small Molecule Probes: Apply minimally perturbing fluorescent probes for key cellular components:
- Nucleic acid stains (Hoechst 33342, DAPI) for DNA content analysis
- Fluorescently labeled amino acids for peptidoglycan synthesis tracking
- O-propargyl-puromycin (OPP) analogs for translation rate monitoring [33]
FRET-Based Pathway Monitoring Implement Förster Resonance Energy Transfer (FRET) pairs to monitor protein interactions and signaling pathways central to persister formation without cellular disruption [33]. Key applications include:
- Chemotaxis signaling pathway dynamics
- Toxin-antitoxin module interactions
- Stress response pathway activation
Advanced Computational Integration Methods
Reference Mapping Algorithms Leverage computational approaches that map new single-cell data to well-curated reference datasets, reducing analytical variability and enhancing detection of subtle persister-specific signatures [72]. Implementation steps include:
- Reference Atlas Construction: Build persister-specific reference atlases from carefully validated datasets using methods like Seurat, Symphony, or scArches [72].
- Query Dataset Processing: Apply reference-defined transformations to new experimental data for standardized comparative analysis.
- Differential State Identification: Utilize algorithms like MELD (Manifold Enhancement of Latent Dissimilarity) to quantify perturbation effects at single-cell resolution across the transcriptional manifold [73].
Table 2: Research Reagent Solutions for Perturbation-Minimized Single-Cell Analysis
| Reagent Category | Specific Products/Systems | Function in Persister Analysis | Perturbation Considerations |
|---|---|---|---|
| Cell Viability Probes | SYTOX Green, Propidium Iodide, FM 4-64 | Membrane integrity assessment | Concentration-dependent toxicity; minimal staining recommended |
| Metabolic Probes | Resazurin, CTC, ATP biosensors (QUEEN, iATPSnFR) | Metabolic activity monitoring at single-cell level | Some dyes may affect bacterial metabolism; validate with controls |
| rRNA Depletion Reagents | Subtractive hybridization probes, Cas9 guides | mRNA enrichment for bacterial scRNA-seq | Enzyme-free methods preferred to maintain native states |
| Fixation Reagents | Formaldehyde, Methanol, Ethanol | Cellular structure preservation | Optimize concentration and duration to minimize epitope damage |
| Permeabilization Agents | Lysozyme, Polymyxin B nonapeptide | Cell wall disruption for probe access | Species-specific optimization required to maintain viability |
| Barcoding Oligonucleotides | Split-pool barcodes, UMIs | Single-cell identification in sequencing | Validate non-interference with cellular processes |
Molecular Mechanisms of Persister Formation: Key Signaling Pathways
Understanding the molecular basis of persister formation is essential for designing physiologically relevant analytical approaches. Current research indicates that persistence arises through multiple interconnected mechanisms rather than a single pathway.
Toxin-Antitoxin (TA) Systems play a central role in persister formation through controlled metabolic modulation. Type I TA modules (TisB/istR, hokB/sokB) disrupt proton motive force and inhibit ATP synthesis, while type II systems (HipAB) phosphorylate aminoacyl-tRNA synthetases, activating the stringent response via RelA and increasing (p)ppGpp alarmone levels [37]. These coordinated actions induce metabolic quiescence characteristic of persister cells.
The Stringent Response mediated by (p)ppGpp signaling represents another critical pathway. This signaling molecule redirects cellular resources from growth to maintenance by modulating transcription and translation, promoting the dormant state associated with persistence [37]. Additional interconnected mechanisms include SOS response to DNA damage, quorum sensing for population-level coordination, and biofilm-associated signaling that provides a protective microenvironment for persister enrichment [3] [37].
Applications in Bacterial Persister Research and Therapeutic Development
The perturbation-minimized approaches outlined in this Application Note enable more accurate investigation of persister biology and enhance therapeutic development efforts targeting these recalcitrant cell populations.
Mechanism of Action Studies for Anti-Persister Compounds
Implement single-cell approaches to evaluate how candidate compounds affect persister physiology without the averaging effects of bulk measurements. Key applications include:
- Metabolic Reactivation Tracking: Monitor resuscitation kinetics at single-cell resolution using optimized ATP biosensors and metabolic probes
- Membrane Permeabilization Assessment: Quantify compound-induced membrane damage using time-lapsed fluorescence microscopy with minimally perturbing dyes
- Transcriptional Response Profiling: Apply BaSSSh-seq to characterize heterogeneous responses to anti-persister compounds across persister subpopulations
Biofilm Persister Dynamics
Biofilms represent a critical context for persister studies, as these structured communities protect and promote persister formation [49] [3]. Implement the following approaches for biofilm persister analysis:
- In Situ Spatial Mapping: Combine gentle biofilm dissociation methods with single-cell RNA sequencing to preserve native transcriptional states
- Microenvironment Analysis: Correlate persister distributions with localized metabolic gradients and signaling molecule concentrations
- Immune Interaction Profiling: Apply BaSSSh-seq to study how biofilm persisters respond to immune cell interactions, revealing differential responses to macrophages, neutrophils, and granulocytic myeloid-derived suppressor cells [49]
Maintaining physiological relevance through minimized perturbations is not merely a technical consideration but a fundamental requirement for meaningful single-cell analysis of bacterial persisters. The methodologies detailed in this Application Note—particularly the BaSSSh-seq protocol and complementary fluorescence-based approaches—provide robust frameworks for investigating persister biology while preserving native physiological states. As single-cell technologies continue to evolve, further innovations in perturbation reduction will undoubtedly enhance our understanding of these elusive bacterial subpopulations and accelerate the development of effective therapeutic strategies against persistent infections.
Optimizing Protocols for Diverse Bacterial Species and Clinical Isolates
Bacterial persisters constitute a subpopulation of cells that are genetically susceptible to antibiotics but can transiently survive lethal antibiotic treatment by entering a dormant or slow-growing state [3] [37]. These cells are increasingly recognized for their role in chronic and recurrent infections, including tuberculosis, cystic fibrosis-associated pneumonia, and recurrent urinary tract infections [33] [3]. Research into persisters is complicated by their low abundance (typically 0.001–1% of a population), non-heritable phenotype, and transient nature [33] [37]. Single-cell techniques have emerged as powerful tools to overcome these challenges, enabling the isolation and analysis of these rare cells within heterogeneous bacterial populations.
This application note provides optimized protocols and methodologies for studying persisters across diverse bacterial species and clinical isolates, with particular emphasis on single-cell approaches that can capture the phenotypic heterogeneity central to bacterial persistence.
Technical Insights into Single-Cell Persister Analysis
Core Principles and Challenges
The fundamental characteristic distinguishing persisters from resistant cells is their transient, non-genetic nature. Unlike resistant bacteria that possess stable genetic mutations, persisters exhibit tolerance only temporarily and revert to being fully antibiotic-susceptible once they resume growth [3] [37]. This phenotypic heterogeneity represents a bet-hedging strategy that enhances population survival in fluctuating environments [33]. When exposed to lethal antibiotic concentrations, bacterial populations typically exhibit biphasic killing curves, with rapid killing of normal cells followed by a slower decline as persisters survive treatment [37].
Several key challenges complicate persister research across diverse species:
- Low abundance: Persisters are rare in bacterial populations, necessitating enrichment strategies or highly sensitive detection methods [33] [37].
- Transient phenotype: The persister state is temporary, making it difficult to maintain during experimental procedures [37].
- Species-specific variability: Molecular mechanisms underlying persistence vary significantly between species, requiring optimized protocols [3].
- Clinical isolate limitations: Many clinical isolates are genetically intractable, restricting the use of tools that require genetic manipulation [33].
Research Reagent Solutions for Persister Studies
Table 1: Essential research reagents for single-cell persister studies
| Reagent Category | Specific Examples | Function/Application | Compatibility Notes |
|---|---|---|---|
| Viability Stains | SYTOX Green, Hoechst 33342, DAPI | Nucleic acid staining to assess membrane integrity and DNA content [33] | Compatible with diverse species; fixed cells |
| Metabolic Probes | O-propargyl-puromycin (OPP) | Incorporation into nascent peptides to measure translation rates [33] | Incompatible with intrinsically puromycin-resistant Gram-negative species [33] |
| Biosensors | QUEEN ATP biosensor, iATPSnFR | Quantify intracellular ATP concentrations at single-cell resolution [33] | Requires optimization for bacterial systems [33] |
| Fluorescent Reporters | Multi-reporter constructs, transcriptional FP fusions | Monitor gene expression heterogeneity and protein localization [33] | Primarily for genetically tractable strains [33] |
| Antibiotic Analogs | Fluorescently-labeled antibiotic derivatives | Visualize antibiotic binding and target engagement [33] | Species-dependent binding efficiency |
| Cell Wall Probes | Fluorescent D-amino acids (FDAAs) | Label nascent peptidoglycan to assess cell wall synthesis [33] | Broad compatibility across species |
Single-Cell Methodologies for Persister Research
Advanced Imaging and Tracking Technologies
Single-cell microscopy and tracking technologies have revolutionized our understanding of persister dynamics by enabling direct observation of individual cells before, during, and after antibiotic treatment.
Microfluidics-Based Single-Cell Tracking
Recent advances in microfluidics allow for unprecedented observation of persister cell histories. A 2025 study visualized over one million individual E. coli cells to track their responses to lethal antibiotic doses [27]. The protocol revealed that most persisters from exponentially growing populations were actively growing before antibiotic treatment, challenging the traditional view that persistence is exclusively linked to pre-existing dormancy [27].
Key findings from microfluidics approaches:
- Persisters exhibit heterogeneous survival dynamics including continuous growth with L-form-like morphologies, responsive growth arrest, or post-exposure filamentation [27].
- The relationship between pre-treatment growth status and persistence varies by antibiotic class: with ciprofloxacin, all persisters were growing before treatment, even in stationary phase cultures [27].
- Microfluidics enables continuous monitoring of single cells throughout antibiotic treatment and recovery, capturing transient phenotypic states [27].
High-Throughput Live-Cell Imaging
For intracellular pathogens, high-throughput live-cell imaging provides insights into host-pathogen interactions at single-cell resolution. A recent study on Legionella pneumophila infection in human macrophages utilized spinning-disk high-throughput confocal microscopy to track thousands of infected cells over time [28]. The methodology employed the BATLI (Backtracking Analysis of Time-Lapse Images) software tool for retrospective analysis of individual infected cells, correlating early metabolic changes with subsequent infection outcomes [28].
Workflow for high-throughput intracellular persister studies:
- Infect human monocyte-derived macrophages with GFP-expressing bacteria at MOI 10
- Stain host cells with Hoechst and Cell Tracker Blue for nucleus and cytoplasm detection
- Acquire time-lapse images at hourly intervals up to 18 hours post-infection
- Quantify bacterial replication by measuring area occupied by intracellular bacteria
- Use backtracking analysis to correlate early host cell parameters with bacterial replication outcomes [28]
This approach demonstrated that only 17±8% of L. pneumophila-infected macrophages supported bacterial replication, highlighting the importance of single-cell analysis for capturing this heterogeneity [28].
Molecular Profiling Techniques
Fluorescence-Based Biosensing
Fluorescent biosensors enable real-time monitoring of physiological parameters in live persister cells at single-cell resolution.
Table 2: Fluorescent biosensing approaches for persister studies
| Target | Biosensor Technology | Mechanism | Information Obtained |
|---|---|---|---|
| Gene Expression | Fluorescent reporter plasmids [33] | Transcriptional fusions with FPs | Heterogeneity in gene expression patterns |
| Protein Localization | Protein-FP fusions, FRET pairs [33] | FP fused to protein of interest | Protein abundance, localization, interactions |
| Metabolic Status | QUEEN ATP biosensor [33] | Ratio-metric ATP sensing | Intracellular ATP concentrations |
| Second Messengers | Riboswitch-based biosensors [33] | Aptamer-Spinach fusion with DFHBI | c-di-GMP signaling dynamics |
| Translation Rates | OPP-fluorophore conjugates [33] | Click chemistry on incorporated OPP | Single-cell protein synthesis rates |
Implementation considerations:
- Multi-reporter constructs with spectral separation enable simultaneous monitoring of multiple cellular processes [33].
- FRET-based biosensors require careful optimization of expression levels to avoid artifacts [33].
- Riboswitch-based biosensors offer advantages for studying signaling molecules in live cells without genetic manipulation [33].
RNA Detection Methods
Fluorescent in situ hybridization (FISH) techniques allow detection of specific RNA transcripts in individual bacterial cells. Recent advances include:
- par-seqFISH: Combines multiple RNA probes to analyze heterogeneous gene expression based on geographic location within microbial populations [33].
- Living FISH: Protocols for binding specific RNA molecules in living, non-fixed bacteria, enabling longitudinal studies of gene expression in persisters [33].
Optimized Protocols for Diverse Bacterial Species
Protocol 1: Rapid Isolation from Clinical Samples
For bacteremia and bloodstream infections, timely pathogen isolation is critical. This protocol enables efficient bacterial isolation from blood samples within 30 minutes, achieving over 70% efficiency even at low bacterial concentrations (1-10 bacteria/0.3 mL blood) [74].
Key advantages:
- Maintains bacterial viability with no notable change in growth lag times
- Validated for clinically relevant species including E. coli, Klebsiella pneumoniae, and Staphylococcus aureus [74]
- Compatible with downstream identification methods (molecular, spectrometry-based)
- Uses commonly available laboratory equipment without disrupting standard workflows [74]
Workflow:
- Process fresh blood samples without culture enrichment
- Apply optimized isolation technique (specific methodology not detailed in source)
- Recover bacteria with preserved viability for immediate analysis
- Proceed to identification or phenotypic characterization
This rapid isolation protocol significantly reduces diagnostic delays compared to traditional culture-based methods, which typically require several days [74].
Protocol 2: Single-Cell Persister Recovery Kinetics
This protocol quantifies post-antibiotic persister recovery kinetics and physiological states at single-cell resolution using spectrophotometry and flow cytometry [38].
Key steps:
- Persister enrichment: Use susceptibility tests and time-kill assays to obtain persister cells
- Technical setup: Configure equipment for continuous monitoring
- Data acquisition: Employ spectrophotometry for bulk measurements and flow cytometry for single-cell analysis of physiological states [38]
Applications:
- Elucidation of genes and mechanisms involved in persister survival
- Characterization of heterogeneity in resuscitation times within persister populations
- Identification of distinct physiological states during recovery from persistence
Protocol 3: Metabolic Tracing in Host-Pathogen Systems
For intracellular bacteria, this protocol tracks metabolic interactions between pathogens and host cells at single-cell resolution.
Experimental framework:
- Infect primary human macrophages (hMDMs) with GFP-expressing bacteria
- Monitor infection progression and metabolic parameters in parallel across thousands of cells
- Use automated confocal microscopy with microtiter plates for high-throughput data collection
- Apply backtracking analysis to identify early predictive markers of infection outcome [28]
Key findings enabled by this approach:
- Early changes in mitochondrial membrane potential (Δψm) and mitochondrial ROS production are associated with macrophages that subsequently support bacterial replication [28]
- Machine learning models trained on these data achieve 83% accuracy in predicting L. pneumophila replication outcomes by 5 hours post-infection [28]
- Mitochondrial functions serve as predictive biomarkers for intracellular bacterial replication success [28]
Data Visualization and Analysis
Experimental Workflow for Single-Cell Persister Studies
The following diagram illustrates a comprehensive workflow for single-cell persister analysis, integrating multiple approaches discussed in this application note:
Molecular Mechanisms of Persister Formation
The molecular pathways underlying persister formation represent potential targets for anti-persister therapeutic development. The following diagram summarizes key mechanisms:
Comparative Analysis of Single-Cell Technologies
Table 3: Comparison of single-cell techniques for persister research
| Technique | Resolution | Throughput | Key Applications | Limitations | Compatibility with Clinical Isolates |
|---|---|---|---|---|---|
| Microfluidics [27] | Single-cell dynamic tracking | High (millions of cells) | Persister history tracking, resuscitation kinetics | Specialized equipment required | Moderate (requires culturing) |
| Live-Cell Imaging [28] | Single-cell spatial and temporal | Medium (thousands of cells) | Intracellular persister dynamics, host-pathogen interactions | Photo-toxicity potential | High with primary cells |
| Flow Cytometry [33] [38] | Single-cell snapshot | Very high | Physiological state classification, cell sorting | Limited temporal information | High |
| Raman Spectroscopy [33] | Single-cell metabolic | Medium | Metabolic heterogeneity, antibiotic response | Complex data interpretation | High |
| Fluorescent Biosensors [33] | Single-cell molecular | Variable | Pathway activity, metabolic status | Genetic manipulation often required | Low for unmodified isolates |
| Mass Spectrometry [33] | Single-cell (with specialized equipment) | Low | Metabolic profiling, compound identification | Technically challenging | High |
The optimized protocols presented in this application note provide a comprehensive toolkit for investigating bacterial persisters across diverse species and clinical isolates. Single-cell technologies are particularly valuable for capturing the inherent heterogeneity of persister populations and elucidating the dynamic transitions between physiological states. Integration of these approaches—from rapid clinical isolation to high-resolution single-cell analysis—enables researchers to overcome the challenges posed by the low abundance and transient nature of persister cells. As these methodologies continue to evolve, they promise to accelerate the development of novel therapeutic strategies targeting persistent infections, ultimately addressing a critical unmet need in clinical management of bacterial diseases.
Data Analysis and Computational Tools for Interpreting Complex Single-Cell Datasets
The study of bacterial persisters—metabolically dormant, antibiotic-tolerant subpopulations within genetically homogeneous cultures—presents significant analytical challenges due to their rarity and transient nature. Single-cell RNA sequencing (scRNA-seq) has emerged as a transformative technology for dissecting this heterogeneity, moving beyond bulk measurements that mask rare cell states [3]. Recent methodological breakthroughs have enabled researchers to profile transcriptional heterogeneity in complex bacterial communities, including biofilms, at unprecedented resolution [49] [52]. These advances are particularly crucial for understanding persistent infections, as biofilms contain persister cells that underlie many chronic and recurrent bacterial diseases [3].
The application of scRNA-seq to bacterial systems requires specialized computational and experimental approaches distinct from eukaryotic single-cell analysis. Bacterial mRNA lacks polyadenylated tails, is short-lived, and is vastly outnumbered by ribosomal RNA, creating unique challenges for transcript capture and detection [49] [52]. This application note details integrated experimental and computational workflows specifically designed to overcome these hurdles, enabling comprehensive characterization of bacterial persister cells and their transcriptional programs within heterogeneous populations.
Computational Tools for Single-Cell Transcriptomics
Foundational Analysis Frameworks
The computational analysis of single-cell RNA sequencing data relies on established frameworks that form the foundation of most analytical pipelines. These tools handle essential tasks including data normalization, dimensionality reduction, clustering, and visualization.
Table 1: Foundational Computational Frameworks for scRNA-seq Analysis
| Tool | Primary Function | Language | Key Features | Applicability to Bacterial Persisters |
|---|---|---|---|---|
| Scanpy [75] | Large-scale scRNA-seq analysis | Python | Scalable to millions of cells; integrates with scVI-tools and Squidpy | Ideal for large datasets from complex biofilm communities |
| Seurat [75] [76] | Single-cell analysis and integration | R | Robust data integration across batches; supports multi-modal data | Useful for comparing persisters across conditions and timepoints |
| Cell Ranger [75] | Raw data processing | Pipeline | Processes raw FASTQ files to gene-barcode matrices; uses STAR aligner | Foundational processing for 10x Genomics platform data |
| SingleCellExperiment [75] | Data container and ecosystem | R/Bioconductor | Standardized data structure; enables method interoperability | Promotes reproducible analysis across research groups |
These foundational tools enable researchers to perform essential quality control, filter out damaged cells and doublets based on metrics like total UMI counts, numbers of detected genes, and mitochondrial content fraction, and identify distinct cell states present within heterogeneous bacterial populations [76]. The choice between platforms often depends on researcher preference, with Seurat dominating the R ecosystem and Scanpy serving as the primary Python-based framework, both offering extensive functionality for standard single-cell analytical workflows.
Advanced Analytical Tools
Beyond foundational analyses, specialized tools address more complex biological questions relevant to bacterial persistence, such as trajectory inference, RNA velocity, and spatial context.
Table 2: Advanced Analytical Tools for scRNA-seq Data
| Tool | Primary Function | Key Features | Application in Persister Research |
|---|---|---|---|
| scVI-tools [75] | Deep generative modeling | Probabilistic framework; superior batch correction; transfer learning | Modeling rare persister cell states; integrating multiple experiments |
| Monocle 3 [75] | Trajectory inference | Graph-based abstraction of lineages; UMAP compatibility | Reconstructing paths to persistence and resuscitation |
| Velocyto [75] | RNA velocity | Quantifies spliced/unspliced transcripts; predicts future states | Limited in bacteria but may inform resuscitation dynamics |
| CellBender [75] | Ambient RNA removal | Deep learning to distinguish signal from noise | Critical for droplet-based assays with low-persistence fractions |
| Harmony [75] | Batch correction | Efficient dataset integration; preserves biological variation | Integrating data across experimental replicates and conditions |
| Squidpy [75] | Spatial analysis | Spatial neighborhood graphs; ligand-receptor interactions | Contextualizing persisters within structured biofilm communities |
These advanced tools enable researchers to move beyond static snapshots of cellular heterogeneity toward dynamic models of bacterial persistence. For instance, trajectory inference with Monocle 3 can reconstruct the transcriptional paths through which normal bacterial cells enter persistent states and subsequently resuscitate, potentially identifying key regulatory decision points [75]. Similarly, the batch correction capabilities of Harmony and scVI-tools are essential for integrating data across multiple experimental conditions, time courses, and antibiotic treatments to distinguish consistent persistence signatures from technical artifacts.
Experimental Protocols for Bacterial Single-Cell Analysis
Sample Preparation and Single-Cell Barcoding
The accurate identification and transcriptional profiling of bacterial persisters requires specialized sample preparation methodologies that preserve the fragile and transient states of these rare cell types while enabling efficient mRNA capture.
Protocol: Bacterial Single-Cell Profiling with BaSSSh-seq [49]
Cell Fixation and Permeabilization
- Harvest bacterial cultures during late exponential or early stationary phase, when persister frequencies are typically highest
- Fix cells with 4% formaldehyde for 20 minutes at room temperature to preserve transcriptional states
- Permeabilize cells with lysozyme (10 mg/mL) and Triton X-100 (0.1%) for 15 minutes at 37°C to enable probe access
- Quench fixation with 1.25M glycine and wash cells twice with PBS
Split-Pool Barcoding (BaSSSh-seq)
- Utilize split-pool barcoding to label individual cells without specialized microfluidic equipment
- Perform initial reverse transcription with random hexamers to capture diverse RNA species
- Execute two sequential ligation reactions with barcode oligonucleotides
- Between each barcoding step, pool, vortex, and briefly sonicate cells to minimize doublet formation
- Block unreacted barcodes with complementary oligos during pooling to prevent non-specific ligations
cDNA Synthesis and rRNA Depletion
- Lyse barcoded cells and purify captured transcripts using streptavidin magnetic beads
- Generate double-stranded cDNA using random primed second strand synthesis (replacing inefficient template switching)
- Implement subtractive hybridization for rRNA depletion using probes targeting ribosomal sequences
- Recover mRNA-enriched cDNA for library preparation and sequencing
This protocol addresses key challenges in bacterial scRNA-seq, including the need for specialized barcoding approaches compatible with bacterial cell walls and the critical requirement for efficient ribosomal RNA depletion to enable meaningful mRNA detection from these transcript-sparse samples [49].
Ribosomal RNA Depletion for Enhanced mRNA Detection
The overwhelming abundance of ribosomal RNA in bacterial cells presents a significant challenge for single-cell transcriptomics, requiring specialized depletion strategies to achieve meaningful mRNA coverage.
Protocol: RiboD-PETRI for Enhanced mRNA Recovery [52]
Probe Design and Hybridization
- Design a comprehensive set of probe primers spanning all regions of bacterial rRNA sequences
- Ensure 3' ends are reverse complementary to rRNA-derived cDNA (r-cDNA) for specific recognition
- Engineer 5' ends to complement a biotin-labeled universal primer for subsequent capture
- Hybridize probes to cDNA library following template switching and RNase H treatment
rRNA Depletion and mRNA Enrichment
- Add pre-treated streptavidin magnetic beads to hybridized rRNA-derived cDNA
- Incubate for 30 minutes at room temperature with gentle rotation
- Separate r-cDNA-probe-bead complexes using magnetic separation
- Collect supernatant containing enriched mRNA-derived cDNA
- Proceed to library construction and sequencing
The RiboD protocol dramatically improves mRNA detection rates from approximately 5-10% to over 80-90% across various bacterial species and growth phases, enabling more comprehensive characterization of transcriptional heterogeneity within bacterial populations, including rare persister subpopulations [52]. This enhancement is particularly crucial for studying bacterial persisters, which typically exhibit low overall transcriptional activity, making efficient mRNA capture essential for meaningful analysis.
Visualization of Experimental Workflows
The following diagrams illustrate key experimental and computational workflows for single-cell analysis of bacterial persisters, created using Graphviz DOT language with the specified color palette.
Figure 1: BaSSSh-seq Experimental Workflow. This diagram outlines the key steps in the Bacterial Single-cell RNA Sequencing with split-pool barcoding method, from sample preparation through computational analysis [49].
Figure 2: Computational Analysis Pipeline. This workflow illustrates the standard processing steps for single-cell RNA sequencing data, from raw data processing through advanced analytical applications [76].
Research Reagent Solutions
The following table details essential research reagents and their specific functions in single-cell studies of bacterial persisters.
Table 3: Essential Research Reagents for Single-Cell Persister Studies
| Reagent/Category | Specific Function | Application Notes |
|---|---|---|
| Fixation Reagents (Formaldehyde) [49] | Preserves transcriptional states at time of collection | Critical for capturing transient persister phenotypes; standard 4% concentration |
| Permeabilization Agents (Lysozyme, Triton X-100) [49] | Enables probe access to intracellular RNA | Concentration optimization required for different bacterial species |
| Barcoding Oligonucleotides [49] [52] | Labels individual cells for single-cell resolution | Split-pool approach enables high-throughput without specialized equipment |
| rRNA Depletion Probes [52] | Enriches mRNA by removing abundant rRNA | Essential for bacterial scRNA-seq; RiboD method achieves >90% depletion efficiency |
| Viability Stains (SYTOX, FM4-64) [77] [78] | Distinguishes live from dead cells | Critical for flow cytometry-based persister identification and sorting |
| Fluorescent Reporters [77] [33] | Tracks cell growth and protein dilution | Enables monitoring of persister resuscitation at single-cell level |
| Metabolic Probes (ATP biosensors) [33] | Measures metabolic activity in single cells | QUEEN and iATPSnFR sensors quantify ATP in live cells |
These reagents form the foundation of robust single-cell experiments focused on bacterial persistence. The careful selection and optimization of these components is particularly important when working with rare persister cells, where efficient capture and minimal technical variation are essential for obtaining meaningful biological insights.
Integrated Data Analysis Workflow
The interpretation of complex single-cell datasets from bacterial persistence studies requires an integrated analytical approach that connects experimental data with biological insights.
Protocol: Integrated Analysis of Persister scRNA-seq Data [76]
Experimental Design Considerations
- Define specific scientific questions and appropriate controls
- Determine necessary cell numbers based on expected persister frequency (typically 0.001-1%)
- Plan for sufficient replication across biological conditions
- Include time-course designs to capture persistence and resuscitation dynamics
Data Processing and Quality Control
- Process raw sequencing data with appropriate pipelines (Cell Ranger, CeleScope)
- Perform rigorous quality control to remove damaged cells and doublets
- Filter cells based on UMI counts, detected genes, and mitochondrial content
- Normalize data to account for technical variability in sequencing depth
Cell State Identification and Characterization
- Perform dimensionality reduction (PCA, UMAP) to visualize cellular heterogeneity
- Cluster cells using graph-based or density-based approaches
- Identify marker genes distinguishing persister from non-persister subpopulations
- Annotate cell states based on transcriptional signatures
Advanced Analytical Applications
- Apply trajectory inference to model transitions into and out of persistence
- Conduct regulatory network analysis to identify key persistence regulators
- Perform cross-species or cross-condition comparisons to identify conserved mechanisms
- Integrate with complementary data types (proteomics, metabolomics) where available
This integrated workflow enables researchers to move from raw sequencing data to biological insights about bacterial persistence mechanisms, potential therapeutic targets, and dynamics of treatment failure and relapse in chronic infections.
The integration of advanced single-cell technologies with specialized computational tools has dramatically enhanced our ability to study bacterial persisters, moving from population-level observations to mechanistic understanding of these rare but clinically important cell states. The methodologies detailed in this application note—including optimized wet-lab protocols for bacterial scRNA-seq, computational workflows for data analysis, and essential research reagents—provide a foundation for investigating persistence across different bacterial species and experimental conditions. As these tools continue to evolve, particularly with improvements in rRNA depletion efficiency [52] and multi-omic integration capabilities [75], researchers will gain increasingly refined insights into the molecular mechanisms underlying bacterial persistence and its role in chronic infections. These advances will ultimately support the development of novel therapeutic strategies specifically targeting persister cells, addressing a critical unmet need in the management of persistent bacterial infections.
Synthesizing Insights: How Single-Cell Data Validates and Transforms Our Understanding
Correlating Single-Cell Observations with Population-Level Kill Curves
Within a genetically identical bacterial population, a small subset of cells, known as persisters, can survive lethal doses of antibiotics without acquiring genetic resistance. This phenomenon is a significant cause of antibiotic treatment failure and chronic infections. Traditional population-level studies, exemplified by time-kill curves, reliably quantify the extent of tolerance but cannot reveal the underlying heterogeneity. Single-cell technologies now bridge this gap, enabling researchers to directly observe and characterize the rare persister cells that are responsible for the second, non-killable phase of the biphasic kill curve. This Application Note details the methodologies for integrating these two levels of analysis, providing a comprehensive framework for understanding bacterial persistence.
Quantitative Data Correlation: Linking Population and Single-Cell Metrics
The correlation between population-level kill curves and single-cell observations is foundational. The kill curve identifies the "what" and "when"—the fraction of surviving cells over time—while single-cell analysis explains the "who" and "why"—the identity and physiology of the survivors.
Table 1: Correlating Kill Curve Phases with Single-Cell Observations
| Kill Curve Phase | Population-Level Observation | Corresponding Single-Cell Observation | Quantitative Single-Cell Data |
|---|---|---|---|
| Initial Rapid Killing | Logarithmic decline in Colony Forming Units (CFUs). | Lysis of the majority, antibiotic-sensitive cell population. | Flow cytometry shows a loss of membrane integrity in >99% of cells [77]. |
| Persister Plateau | CFU counts stabilize at a low, persistent level. | Enrichment of dormant persister and VBNC cells. | A distinct transcriptional state is identified via scRNA-seq, comprising 1.2% - 36% of wild-type populations in specific models [35]. |
| Post-Antibiotic Resuscitation | Regrowth of the population upon antibiotic removal. | Resumption of cell division in persister cells. | Persister cells initiate resuscitation within 1 hour in fresh media, with a measured doubling time of 23.3 ± 2.54 minutes, comparable to normal cells [77]. |
Table 2: Single-Cell Transcriptomic Signatures of Persister Cells
| Characteristic | Description | Example Genetic Markers |
|---|---|---|
| Translational Deficiency | A dominant signature of downregulated ribosomal and protein synthesis genes. | Ribosomal protein genes are significantly downregulated [35]. |
| Stress Response Signatures | Upregulation of genes involved in specific stress response pathways. | Upregulation of rmf (ribosome modulation factor), mdtK (drug efflux pump), yhaM (cysteine detoxification), and genes upregulated by the envelope stress activator PspF [35]. |
| Convergent State | Persisters from diverse genetic backgrounds (metG*, hipA7) converge to a similar transcriptional state. |
This state is distinct from standard growth phases (exponential, stationary, lag) [35]. |
Experimental Protocols
The following protocols provide a roadmap for generating and integrating population-level kill curves with single-cell data.
Protocol 1: Population-Level Time-Kill Assay
This foundational protocol determines the bactericidal activity of an antibiotic and identifies the "persister plateau" [7].
- Determine Minimal Inhibitory Concentration (MIC): Perform a broth microdilution assay according to EUCAST guidelines. The MIC is the lowest antibiotic concentration that completely inhibits growth [7].
- Prepare Inoculum: Grow the bacterial strain of interest (e.g., E. coli BW25113) to stationary phase in a suitable medium like Lysogeny Broth (LB). Adjust the culture to a standardized optical density (OD625) equivalent to a 0.5 McFarland standard (~1.5 x 10^8 CFU/mL) [7].
- Initiate Antibiotic Treatment: Add a lethal concentration of antibiotic (typically ≥10x MIC) to the culture. For example, use 100 µg/mL amikacin for E. coli BW25113 (MIC = 8 µg/mL) [7].
- Sample and Enumerate Viable Cells: At predetermined timepoints (e.g., 0, 2, 4, 6, 8, 24 hours), remove culture samples.
- Serially dilute the samples in a neutral buffer.
- Plate the dilutions onto antibiotic-free agar plates.
- Incubate plates and count resulting colonies to calculate CFU/mL.
- Plot Time-Kill Curve: Graph log10(CFU/mL) versus time. The plot will typically show a biphasic pattern, with the stabilization point defining the persister fraction.
This protocol leverages flow cytometry to monitor the resuscitation and physiological state of persister cells isolated from the kill curve assay [77].
- Isolate Persister Cells: Following the time-kill assay (e.g., after 3 hours of ampicillin treatment), pellet the cells by centrifugation. Wash the pellet to remove the antibiotic thoroughly [77].
- Stain for Viability and Metabolism: Resuspend the washed cell pellet in fresh media. Use a combination of fluorescent stains, such as:
- Membrane integrity dye: e.g., SYTOX Green ( stains dead cells).
- Metabolic activity dye: e.g., CTC (5-cyano-2,3-ditolyl tetrazolium chloride) or similar.
- Protein dilution marker: For pre-induced cultures, tracking dilution of a fluorescent protein like mCherry can directly indicate cell division [77].
- Acquire Single-Cell Data: Analyze the cell suspension using a flow cytometer equipped with appropriate lasers and filters. Collect data for forward scatter (FSC, indicative of cell size), side scatter (SSC, indicative of granularity), and all fluorescence channels.
- Monitor Resuscitation Kinetics: Transfer the stained cells into fresh, antibiotic-free liquid media and incubate. Take samples at regular intervals (e.g., every 30-60 minutes) for flow cytometric analysis to track the emergence of metabolically active, dividing subpopulations [77].
Protocol 3: Transcriptomic Profiling of Persisters via Single-Cell RNA-Seq
This advanced protocol characterizes the unique gene expression profile of persister cells, providing mechanistic insights [79] [35].
- Cell Fixation and Permeabilization: After antibiotic treatment (at the persister plateau), immediately fix the cells with formaldehyde and permeabilize them with lysozyme to make the RNA accessible [79].
- Combinatorial Cell Indexing (e.g., M3-seq or PETRI-seq):
- Round 1 Indexing: Distribute cells across a multi-well plate. In each well, perform in situ reverse transcription with primers containing a well-specific barcode (BC1) and a Unique Molecular Identifier (UMI) [79].
- Pool and Re-distribute: Pool all cells and then re-partition them into droplets for a second round of indexing.
- Round 2 Indexing: In droplets, ligate a second cell barcode (BC2) to the cDNA. The combination of BC1 and BC2 uniquely identifies each cell [79].
- rRNA Depletion: To maximize sequencing efficiency for informative mRNA, perform ribosomal RNA depletion. This can be achieved post-amplification using an RNase H-based method with species-specific DNA probes [79] [80].
- Library Preparation and Sequencing: Construct sequencing libraries from the rRNA-depleted, cell-barcoded cDNA and sequence on a high-throughput platform.
- Bioinformatic Analysis: Process the data to demultiplex cells based on their combinatorial barcodes, align reads to the reference genome, and generate a gene expression matrix for each cell. Use clustering algorithms to identify distinct transcriptional states, such as the persister cluster [35].
Visualizing the Experimental and Analytical Workflow
The following diagram illustrates the integrated workflow from population-level killing to single-cell analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of these integrated experiments requires a suite of specialized reagents and tools.
Table 3: Key Research Reagent Solutions
| Reagent / Tool | Function | Application Notes |
|---|---|---|
| Lethal Dose Antibiotics (e.g., Ampicillin, Amikacin, Ciprofloxacin) | To eliminate growing, non-persister cells and enrich for the tolerant persister subpopulation. | Concentration must be determined empirically; typically ≥10x MIC. Use from a prepared, filter-sterilized stock solution [7]. |
| Viability Stains (e.g., SYTOX Green, Propidium Iodide) | To distinguish cells with compromised membranes (dead) from those with intact membranes (live). | Critical for flow cytometry to gate on the intact, non-lysed population after antibiotic treatment [77] [56]. |
| Metabolic Activity Probes (e.g., CTC, CFDA-AM) | To assess the metabolic state of cells, identifying dormant versus active persisters. | Used in conjunction with viability stains in flow cytometry to provide a functional profile of persister cells [56]. |
| Fixation/Permeabilization Reagents (e.g., Formaldehyde, Lysozyme) | To preserve cellular RNA and break down the cell wall for intracellular RNA access in scRNA-seq. | Essential first step for bacterial scRNA-seq protocols like M3-seq and PETRI-seq [79]. |
| Combinatorial Indexing Kits (e.g., M3-seq, PETRI-seq) | To label the RNA from thousands of individual cells with unique barcodes in a high-throughput, pool-based manner. | Enables transcriptome-wide profiling of rare persister cells without the need for physical cell isolation [79] [35]. |
| rRNA Depletion Probes (e.g., species-specific DNA probes for RNase H treatment) | To selectively remove abundant ribosomal RNA sequences from sequencing libraries, dramatically enriching for mRNA. | Greatly increases the sensitivity and cost-effectiveness of bacterial scRNA-seq [79] [80]. |
The synergistic application of population-level kill curves and single-cell technologies represents a powerful paradigm shift in persister research. While kill curves provide the essential quantitative framework for identifying and measuring persistence, single-cell methods like flow cytometry and scRNA-seq illuminate the underlying heterogeneity, physiological states, and molecular mechanisms. The protocols and tools detailed in this Application Note provide a clear path for researchers to dissect this complex phenotype, ultimately accelerating the discovery of novel therapeutic strategies to target and eliminate persistent bacterial infections.
Bacterial persisters are a subpopulation of genetically drug-susceptible cells that enter a transient, non-growing or slow-growing state, enabling them to survive lethal concentrations of antibiotics and other environmental stresses. Upon removal of the antibiotic pressure, these cells can resuscitate and re-establish a susceptible population, contributing to chronic and relapsing infections [3]. Unlike antibiotic resistance, which involves heritable genetic mutations that elevate the Minimum Inhibitory Concentration (MIC), persistence is a non-inheritable phenotypic tolerance characterized by a biphasic killing curve in time-kill assays [81] [3]. This phenomenon is observed across all major bacterial pathogens and is implicated in biofilm-associated infections, tuberculosis, and recurrent urinary tract infections [33] [82].
The study of persisters requires a shift from population-level analyses to single-cell techniques, as these rare cells (often comprising only 0.001–1% of a population) are masked by bulk measurements [33]. This Application Note details the core molecular mechanisms of persistence in four key bacterial species—Escherichia coli, Mycobacterium tuberculosis, Pseudomonas aeruginosa, and Staphylococcus aureus—and provides standardized protocols for their single-cell analysis, serving as a practical resource for researchers and drug development professionals.
Comparative Mechanisms of Persister Formation
The formation of persister cells is governed by a complex interplay of molecular pathways that show both conserved and species-specific features. The table below provides a comparative summary of the key mechanisms.
Table 1: Core Molecular Mechanisms of Persister Formation Across Bacterial Species
| Species | Key Molecular Mechanisms & Regulators | Role of Toxin-Antitoxin (TA) Modules | Primary Stress Responses | Notable Phenotypic Features |
|---|---|---|---|---|
| E. coli | HipA toxin kinase, RelE, MazF mRNA endonucleases [83] [84] | Central role; >10 Type II TA modules (e.g., hipBA, relBE, mazEF) can induce dormancy [84]. | Stringent Response, SOS Response [84] | Dormancy induced by translation inhibition or ATP depletion [84]. |
| M. tuberculosis | Lipid metabolism shifts, DosR regulon (hypoxia), ~80 putative TA modules [82] [84] | High redundancy; ectopic expression of RelE homologs (e.g., Rv1246c) increases drug tolerance [84]. | Stringent Response, Hypoxic Response, Nutrient Starvation [82] | Deep dormancy; association with lipid bodies; morphological heterogeneity [82] [84]. |
| P. aeruginosa | RelA/SpoT (stringent response), HigBA TA system, SOS response [85] [86] | HigBA overexpression increases persisters 1000-fold; role in biofilm tolerance [85] [86]. | Stringent Response, SOS Response, Low Energy [86] | High persister levels in biofilms; filamentation upon β-lactam exposure [85]. |
| S. aureus | Rsh/RelP/RelQ for (p)ppGpp synthesis, profound transcriptomic reprogramming [81] | Role is less defined compared to Gram-negatives; not the primary driver. | Stringent Response, Cell Wall Stress, Heat Shock [81] | Intracellular persistence within host cells; multi-drug tolerance after single antibiotic exposure [81]. |
The following diagram synthesizes the core mechanistic pathways leading to persister formation and their interactions across these bacterial species.
Essential Reagents and Single-Cell Techniques
Dissecting the heterogeneous nature of persister cells necessitates tools that provide resolution at the single-cell level. The table below catalogs key reagents and their applications in persister research.
Table 2: Research Reagent Solutions for Single-Cell Persister Analysis
| Reagent Category | Specific Examples | Primary Function in Persister Research | Example Application |
|---|---|---|---|
| Viability Stains | LIVE/DEAD BacLight (SYTO9/PI), Propidium Iodide (PI) [83] [81] | Distinguish viable from membrane-compromised cells. | Assessing bacterial viability after antibiotic treatment in macrophages [81]. |
| Metabolic Probes | Redox Sensor Green (RSG) [85] | Report on cellular metabolic (reductase) activity. | Identifying metabolically active but non-growing persisters in planktonic P. aeruginosa [85]. |
| Biosensors | "QUEEN" ATP biosensor [33], O-propargyl-puromycin (OPP) [33] | Quantify intracellular ATP levels or measure translation rates via click chemistry. | Correlating dormancy with low energy levels; monitoring protein synthesis shutdown in single cells [33]. |
| Fluorescent Reporters | GFP, transcriptional/translational FP fusions [33] [81] | Report on gene expression or protein localization. | Monitoring bacterial replication and growth arrest in intracellular S. aureus using fluorescence dilution [81]. |
| Nucleic Acid Stains | Hoechst 33342, DAPI, SYTOX Green [33] | Visualize and quantify DNA content. | Linking higher genome copy number to persistence in E. coli [33]. |
| FISH Probes | par-seqFISH probes [33] | Detect specific RNA transcripts in single, live cells. | Elucidating heterogeneous gene expression based on spatial location in microcolonies [33]. |
The workflow for a typical single-cell persister analysis integrates these reagents and technologies, as shown in the following protocol diagram.
Detailed Experimental Protocols
Protocol 1: Time-Kill Curve Assay for Persister Quantification
This foundational protocol is used to confirm the presence of a persister population by demonstrating biphasic killing [85] [81] [38].
Application Notes:
- This assay is the gold standard for differentiating bactericidal activity from a persistent phenotype.
- The surviving fraction after the first killing phase consists of persisters. The concentration and class of antibiotic should be carefully selected based on the MIC for the strain [85].
Procedure:
- Inoculum Preparation: Grow the bacterial culture to the desired growth phase (e.g., mid-exponential or stationary). For P. aeruginosa, stationary phase cultures can contain ~1% persisters [83].
- Antibiotic Exposure: Add a high concentration of bactericidal antibiotic (typically 5-10x MIC) to the culture. Maintain a control culture without antibiotic.
- Sampling and Plating: At predetermined time points (e.g., 0, 2, 4, 8, 24 hours), remove aliquots from the treated and control cultures.
- Washing: Centrifuge the aliquots and wash the pellet with sterile phosphate-buffered saline (PBS) or medium to remove the antibiotic. This step is critical to prevent antibiotic carryover.
- Viable Count: Serially dilute the washed samples and plate on antibiotic-free solid medium. Incubate the plates for the appropriate time and count the colony-forming units (CFU).
- Data Analysis: Plot the log(_{10})(CFU/mL) against time. A biphasic curve, characterized by an initial rapid decline followed by a plateau, indicates the presence of persisters.
Protocol 2: Single-Cell Physiological Profiling via Flow Cytometry
This protocol uses metabolic stains to profile the physiological state of persisters at the single-cell level, complementing the CFU-based data [85].
Application Notes:
- Redox Sensor Green (RSG) measures general metabolic activity, while propidium iodide (PI) indicates membrane integrity. Persisters can exhibit a range of metabolic states, from fully dormant to moderately active [85] [3].
- This method can be combined with fluorescent reporter constructs to link physiology to the expression of specific genes.
Procedure:
- Sample Treatment: Expose a bacterial culture to a bactericidal antibiotic (e.g., at 5x MIC for 4 hours) to enrich for persisters [85].
- Staining: Concentrate the cells by centrifugation and resuspend in a buffer containing a pre-optimized concentration of RSG and PI. Incubate in the dark for 15-30 minutes.
- Flow Cytometry Analysis: Analyze the stained cells using a flow cytometer. Use untreated and killed control populations to set the gates for RSG (metabolically active) and PI (dead) signals.
- Data Interpretation: Persisters are typically identified as a subpopulation that is PI-negative (viable) but may show reduced RSG signal (low metabolic activity) compared to the bulk of the untreated population.
Protocol 3: Fluorescence Dilution for Intracellular Persister Growth Kinetics
This powerful protocol tracks the replication of individual bacteria inside host cells, confirming their non-growing state during persistence [81].
Application Notes:
- This method is ideal for studying pathogens known for intracellular survival, such as S. aureus and M. tuberculosis.
- It directly demonstrates the non-growing, dormant state of persisters and its reversibility.
Procedure:
- Reporter Strain Preparation: Use a bacterial strain (e.g., S. aureus) harboring an inducible fluorescent protein (e.g., GFP) construct.
- Fluorescence Induction: Grow the bacteria in the presence of an inducer (e.g., anhydrotetracycline) to load cells with GFP.
- Infection and Antibiotic Challenge: Infect mammalian cells (e.g., J774 macrophages) with the pre-induced bacteria. After infection, wash away extracellular bacteria and add a high concentration of a relevant antibiotic (e.g., oxacillin) to the culture medium.
- Fluorescence Monitoring: At various time points post-treatment, analyze infected cells by flow cytometry or fluorescence microscopy. In non-replicating persisters, the GFP signal remains high. In replicating bacteria, the fluorescence is diluted with each cell division.
- Phenotype Reversion: To confirm the transient nature of persistence, remove the antibiotic from a subset of cultures and monitor the fluorescence dilution, which will indicate the resumption of growth.
The comparative analysis of E. coli, M. tuberculosis, P. aeruginosa, and S. aureus reveals that persistence is a multifactorial phenotype with a common outcome—dormancy and antibiotic tolerance—but achieved through diverse molecular mechanisms. While the stringent response and TA modules are recurring themes, their relative importance varies, with E. coli and M. tuberculosis exhibiting high redundancy in TA systems, while S. aureus persistence is heavily driven by global transcriptomic reprogramming.
The path forward for developing effective anti-persister therapies lies in leveraging the single-cell technologies and protocols outlined herein. Future efforts should focus on:
- Targeting Wake-Up Pathways: Developing compounds that stimulate persister cells to resuscitate, making them vulnerable to conventional antibiotics [82].
- Energy-Consuming Systems: Targeting proton motive force or ATP levels, as metabolic dormancy is a cornerstone of persistence [33] [3].
- Novel Drug Combinations: Using insights from persister physiology to design rational combination therapies that attack multiple dormant subpopulations simultaneously [82] [3].
A deep, mechanism-based understanding of persistence across species is paramount for translating single-cell observations into novel therapeutic strategies that can ultimately overcome chronic and relapsing bacterial infections.
The study of bacterial persisters—metabolically dormant cells that survive antibiotic treatment—is fundamentally enhanced by multi-omics integration. Analyzing transcriptomics, proteomics, and metabolomics data in concert provides a comprehensive view of biological systems that single-omics analyses cannot achieve, revealing complex patterns and interactions underlying the persister phenotype [87]. This approach is particularly valuable for understanding phenotypic heterogeneity in clonal bacterial populations, where rare, transient persister cells contribute to recurrent infections and treatment failure [33] [3]. The integration of these different molecular layers enables researchers to identify key regulatory networks, biomarker signatures, and potential therapeutic targets for eradicating persistent infections.
Core Integration Strategies and Methodologies
Categories of Integration Approaches
Multi-omics integration strategies can be broadly classified into three principal approaches, each with distinct methodologies and applications for persister research [87].
Table 1: Multi-Omics Integration Approaches
| Integration Approach | Core Concept | Key Methods | Application in Persister Research |
|---|---|---|---|
| Combined Omics Integration | Independently analyzes each omics dataset in an integrated manner to create a unified biological narrative. | Pathway enrichment analysis, Interactome analysis, Compound-reaction-enzyme-gene networks. | Identifying dysregulated inflammatory, metabolic, and stress response pathways in persister cells. |
| Correlation-Based Integration | Applies statistical correlations between different omics datasets and represents relationships as networks. | Gene co-expression analysis (e.g., WGCNA), Gene-metabolite correlation networks, Similarity Network Fusion (SNF). | Discovering co-regulated gene and metabolite modules associated with antibiotic tolerance and dormancy. |
| Machine Learning Integrative Approaches | Uses computational models to learn joint representations from multiple omics datasets for prediction and classification. | Factor analysis (e.g., MOFA+), Variational Autoencoders, Bayesian models, Neural Networks. | Predicting persister cell states, classifying patient infection outcomes, and identifying biomarker panels. |
Data Integration Specifics for Transcriptomics, Proteomics, and Metabolomics
The integration of transcriptomic, proteomic, and metabolomic data requires specific techniques to link these related but distinct molecular layers [87]:
- Linking Transcriptomics and Proteomics: Correlation-based approaches and pathway/co-expression analysis are used to identify relationships between gene expression and protein abundance, which are often discordant due to post-transcriptional regulation.
- Linking Proteomics and Metabolomics: This integration is powerful for biomarker discovery and understanding disease states, as it can directly reveal alterations in metabolic pathways driven by enzyme (protein) activity.
- Linking Transcriptomics and Metabolomics: Co-expression analysis and network-based techniques can connect upstream regulatory events (transcript levels) with downstream functional outcomes (metabolite abundance).
Figure 1: Multi-Omics Integration Workflow. This diagram outlines the process from sample preparation through data generation from transcriptomics, proteomics, and metabolomics, to computational integration and final biological insight.
Application Notes: Protocol for an Integrated Multi-Omics Study of Bacterial Persisters
Experimental Workflow for Persister Multi-Omics
The following protocol describes an integrated approach to investigate the molecular basis of bacterial persistence, from persister isolation to multi-omics data generation and integration.
Table 2: Key Research Reagent Solutions for Persister Multi-Omics
| Reagent/Material | Function | Application Example |
|---|---|---|
| O-propargyl-puromycin (OPP) | A puromycin analog that incorporates into nascent peptides; can be modified via click chemistry with a fluorophore to measure single-cell translation rates. | Assessing heterogeneous protein synthesis in bacterial populations to identify dormant persisters [33]. |
| Fluorescent Biosensors (e.g., QUEEN, iATPSnFR) | Genetically encoded sensors that change fluorescence upon binding specific intracellular metabolites (e.g., ATP). | Quantifying metabolically quiescent persister cells with low ATP levels at single-cell resolution [33]. |
| Hoechst 33342 / DAPI / SYTOX Green | Nucleic acid-binding fluorescent dyes for staining DNA content. | Correlating genome copy number with persistence to antibiotics like fluoroquinolones [33]. |
| Fluorescently Labelled Amino Acids | Incorporated into newly synthesized peptidoglycan to report on cell wall biosynthesis rates. | Probing cell wall activity heterogeneity and its role in persister formation [33]. |
| Riboswitch-Based Biosensors | Engineered RNA elements that change conformation and fluorescence upon binding specific ligands (e.g., c-di-GMP). | Detecting intracellular second messenger signals regulating virulence and persistence [33]. |
Figure 2: Experimental protocol for persister multi-omics, from culture to validation.
Detailed Procedural Steps
Persister Cell Isolation:
- Grow a bacterial culture to stationary phase to enrich for Type I persisters or use a mid-log phase culture for Type II persisters [3].
- Expose the culture to a high concentration of a bactericidal antibiotic (e.g., 10x the Minimum Inhibitory Concentration (MIC) of a fluoroquinolone or aminoglycoside) for 4-6 hours. This kills the majority of the population while selecting for persisters.
- Remove the antibiotic thoroughly by repeated centrifugation and washing with fresh medium or phosphate-buffered saline (PBS) [38].
Multi-Omics Sampling and Data Generation:
- Transcriptomics: Extract total RNA from persister and control (untreated) samples. Use RNA sequencing (RNA-Seq) to identify differentially expressed genes (DEGs). For example, in a radiation stress model, 1,595 genes were upregulated and 1,242 downregulated in a high-stress group [88].
- Proteomics: Lyse cells and extract proteins. Digest proteins with trypsin and analyze peptides using Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS). Identify and quantify differentially expressed proteins (DEPs). A study on diabetic ulcers found 464 upregulated and 419 downregulated proteins [89].
- Metabolomics/Lipidomics: Quench metabolism rapidly (e.g., using cold methanol). Extract metabolites and lipids from the supernatant. Analyze using LC-MS and/or Nuclear Magnetic Resonance (NMR) spectroscopy. A multi-omics study identified 1,304 metabolites, with lipids (32.1%) and organic acids (20.2%) being predominant [89].
Bioinformatic Processing and Integration:
- Data Processing: Perform quality control on raw data. Map sequences to reference genomes and quantify features (genes, proteins, metabolites). Conduct differential analysis to identify significant changes in persisters versus controls.
- Correlation-Based Integration:
- Perform Weighted Gene Co-expression Network Analysis (WGCNA) on transcriptomics data to identify modules of co-expressed genes [87].
- Correlate module "eigengenes" (representative expression profiles) with normalized metabolomics data to link gene modules to metabolite abundance patterns [87].
- Construct a gene-metabolite interaction network using Pearson correlation coefficients. Visualize the network in Cytoscape to identify key regulatory hubs connecting transcripts and metabolites [87] [88].
- Pathway and Joint Analysis:
- Use Joint-Pathway Analysis to overlay DEGs, DEPs, and differentially abundant metabolites onto KEGG or Reactome pathways. This reveals interconnected pathways disrupted in persisters, such as amino acid biosynthesis, lipid metabolism, and energy pathways, as seen in radiation and diabetic ulcer studies [89] [88].
- Perform Gene Ontology (GO) enrichment analysis on DEGs to identify perturbed biological processes (e.g., "immunoglobulin production," "cell adhesion") [88].
Analysis of a Representative Multi-Omics Workflow
The power of this integrated approach is exemplified by a study investigating total-body irradiation in mice, which shares similarities with antibiotic stress in triggering a conserved stress response [88]. In this model:
- Transcriptomics revealed dysregulation of 2,837 genes in the high-dose group. GO enrichment highlighted disruptions in immune response and cell adhesion pathways.
- Metabolomics/Lipidomics showed significant alterations in amino acids, phospholipids (PC, PE), and carnitine levels.
- Joint-Pathway and STITCH Interaction Analysis integrated these findings, demonstrating that the stress response resulted in coordinated changes in amino acid, carbohydrate, lipid, nucleotide, and fatty acid metabolism [88]. This integration provided a systems-level understanding of the molecular interactions underlying the stress response, which would have been obscured in a single-omics analysis.
The Scientist's Toolkit: Essential Computational Tools for Integration
The computational integration of multi-omics data relies on a diverse set of software tools, chosen based on whether the data is matched (from the same cell) or unmatched (from different cells) [90].
Table 3: Computational Tools for Multi-Omics Data Integration
| Tool Name | Methodology | Data Types | Integration Capacity |
|---|---|---|---|
| MOFA+ | Factor Analysis | mRNA, DNA methylation, Chromatin accessibility | Matched |
| Seurat v4/v5 | Weighted Nearest Neighbour; Bridge Integration | mRNA, Protein, Chromatin accessibility, Spatial | Matched & Unmatched |
| TotalVI | Deep Generative Model | mRNA, Protein | Matched |
| GLUE | Graph Variational Autoencoder | Chromatin accessibility, DNA methylation, mRNA | Unmatched |
| LIGER | Integrative Non-negative Matrix Factorization | mRNA, DNA methylation | Unmatched |
| Cobolt / MultiVI | Multimodal Variational Autoencoder / Probabilistic Modelling | mRNA, Chromatin accessibility | Mosaic |
Integrating transcriptomic, proteomic, and metabolomic findings is no longer optional for a deep understanding of complex biological phenomena like bacterial persistence. The synergistic application of these layers, guided by the protocols and tools outlined, enables researchers to move beyond descriptive observations to uncover the functional mechanisms and key regulatory networks that define the persister state. This holistic view is critical for translating molecular data into novel therapeutic strategies capable of overcoming the challenge of chronic and recurrent bacterial infections.
The rise of persistent bacterial infections poses a significant challenge to global health, primarily driven by bacterial persisters—metabolically dormant cells that survive antibiotic treatment despite genetic susceptibility. These phenotypically heterogeneous subpopulations contribute to chronic and relapsing infections, with traditional bulk analysis methods failing to capture their unique characteristics. The integration of advanced single-cell technologies has revolutionized our ability to identify and validate novel molecular targets within these rare cell populations, creating a critical bridge between basic discovery and therapeutic development. This application note details cutting-edge methodologies and protocols for leveraging single-cell approaches in bacterial persister research, providing a structured framework for target validation from initial discovery to therapeutic assessment.
Single-Cell Technologies for Target Discovery in Bacterial Persisters
Bacterial Single-Cell RNA Sequencing Platforms
Recent advances in bacterial scRNA-seq have enabled unprecedented resolution in studying persister heterogeneity. These technologies are crucial for identifying novel molecular targets by revealing transcriptional variation within persister subpopulations that bulk RNA-seq would miss.
Table 1: Comparison of Bacterial Single-Cell RNA Sequencing Methods
| Method | Key Features | mRNA Detection Rate | Throughput | Applications in Persister Research |
|---|---|---|---|---|
| BaSSSh-seq | Split-pool barcoding, subtractive hybridization | Not specifically reported | High (30,000+ cells) | Biofilm transcriptional heterogeneity, immune response studies [49] |
| RiboD-PETRI | Ribosomal cDNA depletion, equipment-free | 54-92% (across species) | High (60,000 cells) | Biofilm subpopulation identification, persister markers [52] |
| MicroSPLiT | Poly A polymerase mRNA enrichment | ~7% | Moderate | Transcriptional heterogeneity in planktonic cultures [52] |
| M3-seq | RNase H rRNA depletion post-hybridization | ~65% | Moderate | Bacterial stress responses [52] |
| BacDrop | RNase H rRNA depletion in cells | ~61% | Moderate | Population heterogeneity [52] |
| MATQ-DASH | Cas9-mediated rRNA depletion | ~30% | Moderate | Transcriptional profiling [52] |
The development of BaSSSh-seq represents a significant advancement, specifically optimized for capturing transcriptional diversity in bacterial populations with low metabolic activity, such as biofilms and persisters. This method employs split-pool barcoding to label individual cells without requiring sophisticated commercial equipment, random hexamers for unbiased RNA capture, and an enzyme-free rRNA depletion method based on subtractive hybridization to significantly reduce rRNA contamination while increasing sequencing depth [49]. When applied to Staphylococcus aureus biofilms, BaSSSh-seq successfully identified transcriptionally distinct subpopulations and their differential responses to various immune cell pressures, including macrophages, neutrophils, and granulocytic myeloid-derived suppressor cells [49].
RiboD-PETRI addresses the fundamental challenge in bacterial scRNA-seq: the lack of polyadenylated tails on bacterial mRNA and the overwhelming abundance of rRNA. By integrating a ribosomal RNA-derived cDNA depletion protocol into PETRI-seq, this method achieves dramatically improved mRNA detection rates—from as low as 3.9% in standard PETRI-seq to 92% in RiboD-PETRI for stationary phase S. aureus [52]. This enhanced sensitivity enables more precise identification of rare transcriptional signatures within persister subpopulations.
Fluorescent Biosensors and Reporter Systems
Fluorescence-based techniques remain fundamental for studying phenotypic heterogeneity in bacterial persisters at the single-cell level, providing real-time monitoring of physiological states and persistence dynamics.
Table 2: Fluorescent Biosensors for Persister Research
| Biosensor Type | Target/Application | Key Features | Examples |
|---|---|---|---|
| Transcriptional Reporters | Gene expression heterogeneity | Multi-reporter constructs for simultaneous transcript monitoring | Plasmid-based fluorescent reporters [33] |
| Protein-FP Fusions | Protein abundance and localization | Genome-wide fusion libraries; FRET for protein interactions | Whole-genome fluorescent fusion libraries in E. coli [33] |
| Small Molecule Probes | Metabolic activity, cellular structures | Nucleic acid staining, metabolic incorporation | Hoechst 33342 (DNA), OPP (translation) [33] |
| ATP Biosensors | Metabolic activity, energy status | Single-excitation ratiometric measurements | QUEEN, iATPSnFR [33] |
| Riboswitch Biosensors | Second messenger signaling | Conformational changes upon ligand binding | c-di-GMP biosensors [33] |
Fluorescent reporter plasmids enable monitoring of relative transcription levels of persistence-associated genes, with recent advances including "multi-reporter" constructs that overcome traditional limitations of spectral overlap [33]. For instance, the combination of Fluorescence Dilution (FD) reporters with inducible fluorescent proteins allows direct quantification of bacterial proliferation dynamics at the single-cell level, distinguishing nongrowing persisters from their growing counterparts [91].
The development of the pSCRATCH (plasmid for Selective CRISPR Array expansion To Check Heritage) system represents an innovative approach that combines fluorescence-based detection with genomic recording. This molecular tool records the state of antibiotic persistence in the genome of Salmonella persisters by leveraging a modified CRISPR-Cas system that incorporates spacers into chromosomal CRISPR arrays specifically in nongrowing cells [91]. This system enables discrimination between treatment failure originating from persistence versus resistance in infection models, providing a powerful method for validating persistence-specific targets.
Experimental Protocols for Target Validation
Protocol: Bacterial Single-Cell RNA Sequencing with BaSSSh-seq
Principle: This protocol enables high-throughput transcriptional profiling of bacterial persisters at single-cell resolution, capturing heterogeneity within biofilm and persister populations [49].
Materials:
- Fixed and permeabilized bacterial cells from persister assays
- Split-pool barcoding oligonucleotides
- Reverse transcription reagents
- Ligation reagents
- Streptavidin magnetic beads
- Second strand synthesis reagents
- Subtractive hybridization probes for rRNA depletion
- Library preparation and sequencing reagents
Procedure:
- Cell Preparation and Fixation:
- Harvest persister cells following antibiotic treatment and resuspend in appropriate fixative
- Permeabilize cells to enable oligonucleotide entry while maintaining RNA integrity
Split-Pool Barcoding:
- Perform initial reverse transcription with random hexamers to capture RNA transcripts
- Execute first ligation reaction with barcode oligonucleotides
- Pool and mix cells, then redistribute for subsequent barcoding rounds
- Perform second and third ligation reactions with distinct barcode sets
- Between each step, filter, vortex, and briefly sonicate cells to minimize doublet formation
cDNA Processing:
- Lyse cells and purify captured transcripts using streptavidin magnetic beads
- Perform random-primed second strand synthesis to generate double-stranded cDNA
rRNA Depletion:
- Implement subtractive hybridization with specifically designed rRNA probes
- Remove rRNA-derived cDNA using magnetic separation
- Recover mRNA-derived cDNA from supernatant
Library Construction and Sequencing:
- Amplify cDNA libraries with appropriate PCR handles
- Prepare sequencing libraries following standard protocols
- Sequence on appropriate platform (e.g., Illumina)
Validation: Compare transcriptional profiles with bulk RNA-seq data (expected correlation r=0.84) and validate identified subpopulations using fluorescent reporters or functional assays [49] [52].
Protocol: Tracking Persister Progeny with pSCRATCH
Principle: This protocol uses a CRISPR-based genomic recorder to permanently mark persister cells and track their progeny, enabling differentiation between relapse due to persistence versus resistance [91].
Materials:
- pSCRATCH plasmid or appropriate derivatives
- Bacterial strain with deleted endogenous cas genes but intact CRISPR arrays
- Arabinose and IPTG inducers
- Appropriate antibiotics for selection
- PCR reagents for spacer acquisition detection
- qPCR reagents for plasmid copy number quantification
Procedure:
- Strain Construction:
- Introduce pSCRATCH plasmid into target bacterial strain
- Verify proper regulation of RepL (arabinose-inducible) and Cas1-Cas2 (IPTG-inducible) expression
Persister Marking:
- Preload bacteria with high pSCRATCH copy number by growing with arabinose inducer
- Transfer to non-inducing conditions and expose to antibiotic treatment
- During or after antibiotic exposure, induce Cas1-Cas2 expression with IPTG
- Allow spacer acquisition to occur in nongrowing persisters maintaining high plasmid copy number
Spacer Acquisition Detection:
- Isolate surviving cells after antibiotic treatment
- Perform colony PCR using primers flanking CRISPR array insertion sites
- Detect spacer acquisition by increased amplicon size
- Sequence acquired spacers to verify origin from pSCRATCH plasmid
Progeny Tracking:
- Culture spacer-positive clones without inducers
- Monitor stability of acquired spacers over multiple generations
- Use spacer presence as heritable marker for persister lineage in subsequent experiments
Validation: Confirm that spacer acquisition correlates with nongrowing状态 by parallel fluorescence dilution assays. Verify that growing cells diluted the high plasmid copy number and thus did not acquire spacers despite Cas1-Cas2 induction [91].
Protocol: Metabolic Profiling of Persisters via Aminoglycoside Potentiation
Principle: This protocol measures metabolic capabilities of persisters by exploiting metabolite-enabled aminoglycoside killing, providing insights into persister physiology for target validation [92].
Materials:
- Carbon sources (e.g., glycerol, glucose) and metabolic inhibitors
- Aminoglycoside antibiotics (e.g., kanamycin)
- Phenotype microarrays (if available)
- Flow cytometry equipment (optional)
- Standard microbiological culture materials
Procedure:
- Persister Enrichment:
- Treat exponential-phase cultures with high concentrations of bactericidal antibiotics (e.g., ampicillin)
- Collect surviving cells after appropriate treatment duration
- Wash to remove antibiotics and debris
Metabolic Potentiation Assay:
- Resuspend persister-enriched population in buffer containing test carbon sources
- Add aminoglycoside antibiotic (e.g., 25 μg/ml kanamycin)
- Incubate at appropriate temperature for designated time
- Include controls without carbon sources and without antibiotics
Viability Assessment:
- Plate serial dilutions on appropriate agar media
- Incubate and enumerate colony-forming units
- Alternatively, use flow cytometry with viability stains for rapid assessment
Data Analysis:
- Calculate persister killing efficiency for each carbon source
- Compare with negative controls to identify metabolites that stimulate persister metabolic activity
- Prioritize carbon sources that consistently potentiate aminoglycoside killing across persister types
Validation: Confirm that metabolic stimulation alone (without antibiotic) does not significantly affect persister viability. Verify that metabolic inhibitors block the potentiation effect [92].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Single-Cell Persister Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Fluorescent Reporters | pFCcGi, pSCRATCH | Monitoring bacterial growth and persistence at single-cell level | Inducer concentration optimization required [91] |
| Barcoding Oligonucleotides | Split-pool barcodes | Single-cell transcriptome labeling | Minimize doublet formation through protocol optimization [49] |
| rRNA Depletion Reagents | RiboD probes, Cas9 guides | Enhancing mRNA detection in bacterial scRNA-seq | Efficiency varies by bacterial species and growth phase [49] [52] |
| Metabolic Probes | O-propargyl-puromycin (OPP) | Measuring translation activity in single cells | Incompatible with intrinsically puromycin-resistant species [33] |
| ATP Biosensors | QUEEN, iATPSnFR | Monitoring metabolic activity and energy status | May require optimization for bacterial expression [33] |
| CRISPRi Components | dCas9, sgRNA libraries | Genome-scale functional genomics | sgRNA design critical for effective repression [93] |
Visualization of Experimental Workflows
BaSSSh-seq Workflow for Bacterial scRNA-seq
pSCRATCH System for Persister Lineage Tracking
Integrating Target Validation into Therapeutic Development
The transition from single-cell discoveries to therapeutic development requires rigorous target validation frameworks. The use of Target Product Profiles (TPPs) provides a strategic planning tool that defines essential attributes required for a clinically successful drug, including target patient population, efficacy and safety requirements, dosing regimen, and cost considerations [94]. For anti-persister therapeutics, TPPs should specifically address the need to eradicate dormant subpopulations and prevent relapse, which may differ from conventional antibiotics.
CRISPR interference (CRISPRi) pooled screening enables genome-scale functional genomics in bacteria, providing a powerful approach for target validation. This method demonstrates superior performance compared to transposon mutagenesis, particularly for short genes and non-coding RNAs [93]. Essential genes identified through CRISPRi screening represent promising targets for anti-persister therapies, especially when their inhibition demonstrates cidal rather than cytostatic effects—a critical consideration for persistent infections where host immune responses may be compromised [94].
The integration of single-cell technologies throughout the validation pipeline strengthens the connection between target identification and therapeutic development. Single-cell approaches confirm target expression and function in heterogeneous persister populations, assess target essentiality across different physiological states, and enable monitoring of target engagement and response to intervention at the cellular level. This comprehensive validation framework ensures that potential therapeutic targets address the unique challenges posed by bacterial persistence, ultimately contributing to more effective treatments for chronic and relapsing infections.
Single-cell technologies have revolutionized the study of bacterial persisters, a dormant subpopulation of cells that exhibits multidrug tolerance and contributes significantly to chronic and recurrent infections [17] [37]. Unlike antibiotic-resistant bacteria, persisters lack genetic mutations and instead enter a transient, slow-growing or metabolically inactive state that protects them from conventional antibiotics that target active cellular processes [37] [5]. This phenotypic heterogeneity within isogenic bacterial populations presents a substantial challenge for research and therapeutic development.
The emergence of sophisticated single-cell analysis platforms has enabled researchers to investigate these rare bacterial subpopulations with unprecedented resolution. These technologies allow scientists to move beyond bulk population measurements that mask critical cellular heterogeneity and instead examine the physiological states, transcriptional profiles, and metabolic activities of individual persister cells [17]. This application note provides a comprehensive benchmarking of available single-cell technologies, detailed experimental protocols for persister research, and practical guidance for selecting appropriate methodologies based on specific research objectives in the context of bacterial persistence.
Technology Landscape and Comparative Analysis
The single-cell technology landscape encompasses diverse platforms with distinct operational principles, capabilities, and limitations. Understanding these characteristics is essential for selecting the most appropriate technology for investigating bacterial persisters.
Table 1: Comparison of High-Throughput and High-Accuracy Single-Cell Technologies
| Factor | High-Throughput (Droplet/Microwell Microfluidics) | High-Accuracy (Image-Based Cell Dispensing) |
|---|---|---|
| Best For | Large-scale single-cell sequencing of homogeneous populations [95] | User-controlled single-cell omics, rare-cell studies, single-cell proteomics/metabolomics [95] |
| Throughput | Up to 40,000 cells per run [95] | 100s-1,000s of individually selected cells [95] |
| Single-Cell Accuracy | Lower; high multiplet risk (many empty droplets or multiplets) [95] | Near-zero multiplet risk; image verification of isolated cells [95] |
| Subpopulation Targeting | Requires fluorescence-activated cell sorting (FACS) prior to analysis [95] | Selection based on morphology and/or fluorescence (1-4 channels) [95] |
| Dead Volume | Significant [95] | Minimal to negligible [95] |
| Flexibility/Workflow Tunability | Limited to standardized kits and reagents [95] | Fully customizable workflows with environmental controls [95] |
| Sample Versatility | Homogenous cell sizes with standard biological properties [95] | Any cell type, any size, including mixed populations [95] |
Table 2: Overview of Single-Cell Omics Technologies for Bacterial Research
| Technology | Key Principle | Primary Application in Persister Research | Key Limitations |
|---|---|---|---|
| scRNA-seq (BaSSSh-seq) | Split-pool barcoding with subtractive hybridization for rRNA depletion [49] | Capturing transcriptional heterogeneity in biofilms and responses to immune pressure [49] | Traditionally challenging due to low bacterial mRNA abundance and lack of polyadenylated tails [49] |
| Fluorescent Biosensors | Reporter plasmids, protein-FP fusions, FRET, small molecule probes [17] | Monitoring gene expression heterogeneity, protein localization, and metabolic activities [17] | Limited to genetically tractable species; FP fusion may disrupt native protein function [17] |
| Live-Cell Imaging + BATLI | High-throughput time-lapse microscopy with backtracking analysis software [28] | Correlating early metabolic dynamics with future infection outcomes in single cells [28] | Requires specialized analysis software; complex data interpretation [28] |
| Microfluidics-Based Platforms | Droplet-based encapsulation (10X Genomics) or microwell arrays (BD Rhapsody) [95] [96] | High-throughput transcriptomic profiling of thousands of single cells [95] | High multiplet rates; limited sensitivity (gene dropout); large sample input requirements [95] |
Key Research Reagent Solutions
Essential reagents and tools form the foundation of reliable single-cell research on bacterial persisters. The following table catalogues critical solutions mentioned in the literature.
Table 3: Essential Research Reagents and Tools for Single-Cell Persister Studies
| Reagent/Tool | Function/Application | Specific Examples |
|---|---|---|
| Fluorescent Reporters & Biosensors | Marking and monitoring gene expression, protein localization, and metabolic states in live cells [17] | - Transcriptional reporter plasmids [17]- Protein-FP fusions (e.g., whole-genome fusion libraries) [17]- FRET pairs (e.g., CheY-CheZ for chemotaxis) [17]- QUEEN and iATPSnFR ATP biosensors [17] |
| Viability and Staining Probes | Differentiating cell states, visualizing structural components, and assessing viability [28] [17] | - Nucleic acid stains (DAPI, SYTOX Green, Hoechst 33342) [28] [17]- Metabolic indicator dyes (e.g., for Δψm, mROS) [28]- O-propargyl-puromycin (OPP) for translation monitoring [17] |
| Single-Cell Barcoding Platforms | Labeling transcripts from individual cells for sequencing | - 10X Genomics Chromium [95] [97]- BD Rhapsody [95]- Split-pool barcoding (BaSSSh-seq) [49] |
| Specialized Software & Algorithms | Data analysis, trajectory inference, and image analysis for single-cell data [28] [97] | - BATLI (Backtracking Analysis of Time-Lapse Images) [28]- SEURAT, Galaxy Europe Single Cell Lab [97]- t-SNE, GPLVM for dimensionality reduction [97] |
Detailed Experimental Protocols
Protocol 1: Bacterial Single-Cell RNA Sequencing (BaSSSh-seq) for Biofilm Analysis
BaSSSh-seq enables transcriptomic profiling of individual bacterial cells within biofilms, which are known reservoirs for persisters [49]. This protocol is particularly valuable for uncovering transcriptional heterogeneity and identifying persister subpopulations.
Diagram 1: Bacterial scRNA-seq Workflow (BaSSSh-seq)
Procedure:
Sample Preparation and Cell Isolation:
- Grow bacterial biofilms under conditions relevant to persistence (e.g., late stationary phase, nutrient limitation) [37].
- Gently dissociate biofilms using mild enzymatic (e.g., DNase I, dispersin B) and/or mechanical methods to obtain single-cell suspensions while preserving integrity.
- Filter the suspension through a series of membranes (e.g., 5μm, then 1μm) to remove aggregates and debris.
Cell Fixation and Permeabilization:
- Fix cells with 1-4% formaldehyde for 15-30 minutes at room temperature to preserve RNA and cellular structure.
- Quench the fixation reaction with 1.25M glycine.
- Pellet cells and wash twice with 1X PBS.
- Permeabilize cells using lysozyme (10mg/mL) in TE buffer for 15-30 minutes at 37°C to allow barcode entry [49].
Split-Pool Barcoding (Diagram 1, detailed steps):
- Round 1 - Reverse Transcription: Resuspend cells in reverse transcription mix containing random hexamers and the first set of barcoded oligonucleotides. Incubate to capture and barcode cellular RNA [49].
- Pool, Split, and Block: Pool all cells, then redistribute into new tubes. Use complementary oligos to block unreacted barcodes and prevent non-specific ligation in subsequent steps [49].
- Round 2 - Ligation: Incubate cells with the second set of barcoded oligos via ligation.
- Pool, Split, and Block: Repeat the pooling, splitting, and blocking process.
- Round 3 - Ligation: Perform the final barcoding with the third set of barcoded oligos via ligation. Each cell now carries a unique combination of three barcodes on its transcripts [49].
Cell Lysis and cDNA Purification:
- Lyse cells using a strong detergent (e.g., SDS) and proteinase K.
- Purify barcoded cDNA using streptavidin magnetic beads, which bind to the biotin tag on the terminal barcode [49].
rRNA Depletion and Library Preparation:
- Perform enzyme-free rRNA depletion via subtractive hybridization with biotinylated rRNA-specific probes, significantly reducing rRNA contamination (from >90% to more manageable levels) [49].
- Generate double-stranded cDNA using random-primed second strand synthesis, which is more efficient than template switching for bacterial samples [49].
- Amplify the cDNA library via PCR and prepare for sequencing on an Illumina platform.
Protocol 2: Predicting Infection Outcomes via Live-Cell Imaging and Metabolic Tracking
This protocol combines time-lapse microscopy with machine learning to track metabolic dynamics in single infected macrophages and predict which cells will support bacterial replication—a key question in persister research [28].
Diagram 2: Metabolic Dynamics Infection Prediction
Procedure:
Cell Culture and Infection Setup:
- Differentiate human primary macrophages (e.g., monocyte-derived macrophages) in 384-well microplates to enable high-throughput imaging [28].
- Infect macrophages at a high multiplicity of infection (MOI~10) with GFP-expressing bacteria (e.g., Legionella pneumophila). Include a T4SS-deficient mutant (e.g., ΔdotA) as a control for non-replicating bacteria [28].
- Centrifuge plates briefly to synchronize infection.
Metabolic Staining and Live-Cell Imaging:
- Simultaneously with infection, stain cells with fluorescent dyes for:
- Nuclei: Hoechst 33342
- Cytoplasm: Cell Tracker Blue
- Mitochondrial Membrane Potential (Δψm): Tetramethylrhodamine methyl ester (TMRM)
- Mitochondrial ROS (mROS): MitoSOX Red [28]
- Acquire time-lapse images hourly for up to 18 hours post-infection using a high-throughput confocal microscope, maintaining environmental control (37°C, 5% CO₂).
- Simultaneously with infection, stain cells with fluorescent dyes for:
Image Analysis with BATLI Software:
- Use custom BATLI (Backtracking Analysis of Time-Lapse Images) software to track individual infected cells across all time points [28].
- Quantify bacterial replication by measuring the area occupied by GFP-expressing bacteria at each time point. Define replication as: (bacterial area at 18hpi) - (bacterial area at 0hpi) > 15μm² [28].
- Export kinetic data for metabolic parameters (Δψm and mROS fluorescence intensity) for each tracked cell.
Machine Learning Model Training and Prediction:
- Use the backtracked data to train an explainable machine-learning classifier (e.g., random forest). Use early time-point metabolic data (e.g., 0-5 hours post-infection) as features and the final infection outcome (bacterial replication vs. restriction) as the label [28].
- Validate model performance using cross-validation, achieving ~83% accuracy in predicting which macrophages will support bacterial replication before replication begins [28].
Analysis and Data Interpretation
Effective data analysis is crucial for extracting biological insights from single-cell persister studies. The massive datasets generated require specialized bioinformatics approaches.
Data Quality Control and Preprocessing: For scRNA-seq data, perform rigorous quality control including assessment of library size, number of detected genes per cell, and mitochondrial or spike-in RNA percentages [97]. Filter out low-quality cells that may represent broken cells or empty barcodes.
Dimensionality Reduction and Clustering: Use principal component analysis (PCA) followed by advanced algorithms like t-distributed Stochastic Neighbor Embedding (t-SNE) or Gaussian Process Latent Variable Modeling (GPLVM) to visualize high-dimensional data in two or three dimensions [97]. Cluster cells based on transcriptomic profiles to identify distinct subpopulations, including potential persister states.
Trajectory Inference and Regulatory Networks: Apply trajectory inference tools (e.g., Monocle, PAGA) to reconstruct cellular dynamics and transitions, such as the entry into or exit from the persister state [49] [97]. Integrate with iModulon analyses to visualize transcriptional regulatory networks across heterogeneous subpopulations [49].
Backtracking Analysis for Temporal Dynamics: For live-cell imaging data, use backtracking approaches to correlate early cellular parameters with later outcomes, identifying predictive biomarkers of persistence [28].
Single-cell technologies provide powerful and complementary approaches for dissecting the complex biology of bacterial persisters. The optimal choice of technology depends heavily on the specific research question, with high-throughput microfluidics platforms offering scalability for large cell numbers, while high-accuracy image-based dispensing enables precise isolation and analysis of rare persister cells. The detailed protocols for BaSSSh-seq and live-cell imaging with metabolic tracking provide robust methodologies for investigating persister formation, heterogeneity, and resuscitation.
As these technologies continue to evolve, integration with artificial intelligence and machine learning will further enhance our ability to predict persister behavior and identify novel therapeutic targets. By selecting appropriate single-cell technologies and implementing optimized experimental workflows, researchers can overcome the challenges posed by bacterial persistence and develop more effective strategies for combating recalcitrant infections.
Conclusion
The application of single-cell techniques has fundamentally shifted our understanding of bacterial persistence from a monolithic concept of dormancy to a dynamic spectrum of heterogeneous physiological states. By enabling the direct observation of rare persister cells, these methods have validated key molecular mechanisms, such as the central role of (p)ppGpp and toxin-antitoxin modules, while also revealing unexpected survival strategies, including continuous growth and L-form transitions. The convergence of microfluidics, single-cell omics, and advanced biosensors provides an unprecedented, multi-dimensional view of persister biology. The future of combating persistent infections lies in leveraging these detailed insights to design novel therapeutic strategies that either prevent persister formation, force their metabolic reawakening to sensitize them to conventional antibiotics, or directly target persister-specific vulnerabilities. As these technologies continue to mature and become more accessible, they hold the promise of delivering targeted, effective treatments for some of the most recalcitrant bacterial infections, ultimately translating single-cell discoveries into clinical breakthroughs.
References
- 1. Epistasis between antibiotic tolerance, persistence, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6642377/]
- 2. Relationship between the Viable but Nonculturable State and Antibiotic Persister Cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6153661/]
- 3. Bacterial persisters: molecular mechanisms and ... [https://www.nature.com/articles/s41392-024-01866-5]
- 4. The importance of the viable but non-culturable state in human bacterial pathogens - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4040921/]
- 5. Mini review: Persister cell control strategies [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1706115/full]
- 6. Bacterial dormancy: A subpopulation of viable but non ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7837498/]
- 7. Protocol for assessing single-cell persister recovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11015153/]
- 8. Detection and Quantification of Viable but Non-culturable ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2019.02920/full]
- 9. Molecular mechanism and application of emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11568608/]
- 10. Role of persister cells in chronic infections [https://pubmed.ncbi.nlm.nih.gov/21459912/]
- 11. Bacterial Persister Cells and Development of Antibiotic ... [https://www.frontierspartnerships.org/journals/british-journal-of-biomedical-science/articles/10.3389/bjbs.2024.12958/full]
- 12. A Quantitative Survey of Bacterial Persistence in the Presence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7460088/]
- 13. Quantitative analysis of persister fractions suggests different ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/1471-2180-13-25]
- 14. Flow-cytometry analysis reveals persister resuscitation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7346475/]
- 15. Observation of persister cell histories reveals diverse ... [https://elifesciences.org/articles/79517]
- 16. - Single Technologies to Study Cell and... Phenotypic Heterogeneity [https://pubmed.ncbi.nlm.nih.gov/34835403/]
- 17. - Single Technologies to Study Cell and... Phenotypic Heterogeneity [https://www.mdpi.com/2076-2607/9/11/2277]
- 18. From Dormancy to Eradication: Strategies for Controlling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11942268/]
- 19. Bacterial persistence increases as environmental fitness ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3323757/]
- 20. Molecular Mechanisms Underlying Bacterial Persisters [https://www.sciencedirect.com/science/article/pii/S0092867414002979]
- 21. - Single RNA-sequencing reports... | Nature Microbiology cell [https://www.nature.com/articles/s41564-020-0774-1?error=cookies_not_supported&code=369ec85c-4190-4b2a-b55d-ea1d9fba5c9b]
- 22. Uncovering phenotypic inheritance from single cells with ... [https://pubmed.ncbi.nlm.nih.gov/40865524/]
- 23. A shared alarmone–GTP switch controls persister ... [https://www.nature.com/articles/s41564-025-02015-6]
- 24. A shared alarmone-GTP switch controls persister formation in ... [https://pubmed.ncbi.nlm.nih.gov/40374742/]
- 25. A bacterial toxin-antitoxin system involved in an unusual ... [https://pubmed.ncbi.nlm.nih.gov/40825874/]
- 26. Bioenergetic stress potentiates antimicrobial resistance ... [https://www.nature.com/articles/s41467-025-60302-6]
- 27. Observation of persister cell histories reveals diverse modes ... [https://pubmed.ncbi.nlm.nih.gov/40356339/]
- 28. Backtracking metabolic dynamics in single cells predicts ... [https://www.nature.com/articles/s41467-025-64225-0]
- 29. Stringent response-mediated ferroptosis-like death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12542694/]
- 30. Targeting the bacterial stringent response to combat ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1663548/full]
- 31. Metabolite-driven reprogramming of bacterial persisters [https://www.sciencedirect.com/science/article/abs/pii/S1368764625001256]
- 32. Bacterial Persister Cells and Development of Antibiotic ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11335562/]
- 33. Single-Cell Technologies to Study Phenotypic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8620850/]
- 34. Persisters—as elusive as ever - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4939303/]
- 35. Identification and genetic dissection of convergent persister ... [https://www.nature.com/articles/s41586-024-08124-2]
- 36. A Historical Perspective on Bacterial Persistence [https://pubmed.ncbi.nlm.nih.gov/26468095/]
- 37. Molecular mechanism and application of emerging ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-024-03628-3]
- 38. Protocol for assessing single-cell persister recovery ... [https://www.sciencedirect.com/science/article/pii/S2666166724001497]
- 39. Metabolic aspects of bacterial persisters - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4205924/]
- 40. Diversity in ATP concentrations in a single bacterial cell ... [https://www.nature.com/articles/srep06522]
- 41. Dynamic fluctuations in a bacterial metabolic network [https://www.nature.com/articles/s41467-023-37957-0]
- 42. A three-colour stress biosensor reveals multimodal ... [https://www.nature.com/articles/s41522-023-00424-1]
- 43. Fluorescent riboswitch-controlled biosensors for the ... [https://www.nature.com/articles/s41598-024-61980-w]
- 44. Antibacterial nanoagents: an emerging arsenal against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12537890/]
- 45. Observation of persister cell histories reveals diverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12074638/]
- 46. Microfluidic dose–response platform to track the dynamics ... [https://www.nature.com/articles/s41598-022-24175-9]
- 47. Microfluidics to Meet Antibiotic Resistance: A Growing ... [https://www.preprints.org/manuscript/202511.0470]
- 48. Microfluidics for single-cell lineage tracking over time to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7757727/]
- 49. Bacterial single-cell RNA sequencing captures biofilm ... [https://www.nature.com/articles/s41467-024-54581-8]
- 50. Bacterial single-cell transcriptomics: Recent technical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10477369/]
- 51. Single cell RNA-seq: An introductory overview and tools for ... [https://www.10xgenomics.com/blog/single-cell-rna-seq-an-introductory-overview-and-tools-for-getting-started]
- 52. An improved bacterial single-cell RNA-seq reveals biofilm ... [https://elifesciences.org/articles/97543]
- 53. Current best practices in single‐cell RNA‐seq analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6582955/]
- 54. Introduction to Single-Cell RNA Sequencing Data Analysis [https://www.rna-seqblog.com/introduction-to-single-cell-rna-sequencing-data-analysis-2/]
- 55. Structure-preserving visualization for single-cell RNA-Seq ... [https://www.nature.com/articles/s42003-023-04662-z]
- 56. The Application of Imaging Flow Cytometry for ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2021.716592/full]
- 57. Using BONCAT to dissect the proteome of S. aureus persisters [https://pmc.ncbi.nlm.nih.gov/articles/PMC12482189/]
- 58. Single-cell Raman spectroscopy identifies Escherichia coli ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9386477/]
- 59. Culture-free Antibiotic-susceptibility Determination From ... [https://www.nature.com/articles/s41598-018-22392-9]
- 60. Rapid identification of pathogenic bacteria using Raman ... [https://www.nature.com/articles/s41467-019-12898-9]
- 61. Surface-Enhanced Raman Scattering Spectroscopy for ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2018.00143/full]
- 62. Ultra-rapid, quantitative, label-free antibiotic susceptibility ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025003972]
- 63. Using BONCAT to dissect the proteome of S. aureus ... [https://pubmed.ncbi.nlm.nih.gov/40920103/]
- 64. High-Throughput Proteomics Identifies Proteins With ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2019.00378/full]
- 65. Antibiotic-persistent bacterial cells exhibiting low-level ROS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12345145/]
- 66. Quantitative analysis of persister fractions suggests different ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3682893/]
- 67. Recent advances in single-cell RNA sequencing of bacteria [https://www.sciencedirect.com/science/article/abs/pii/S138917232500026X]
- 68. Effective ribosomal RNA depletion for single-cell total ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7812930/]
- 69. CRISPR/Cas9-based depletion of 16S ribosomal RNA ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09724-4]
- 70. scDASH: a rRNA depletion method for single-cell total ... [https://www.rna-seqblog.com/scdash-a-rrna-depletion-method-for-single-cell-total-rna-seq/]
- 71. Advances and challenges in single-cell RNA-seq of ... [https://www.sciencedirect.com/science/article/abs/pii/S136952742030120X]
- 72. The future of rapid and automated single-cell data analysis using reference mapping - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11184658/]
- 73. Quantifying the effect of experimental perturbations at single-cell resolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8122059/]
- 74. A simple, low-cost, and highly efficient protocol for rapid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12380869/]
- 75. Top 10 Bioinformatics Tools for scRNA-seq in 2025 [https://neovarsity.org/blogs/top-bioinformatics-tools-for-scrna-seq]
- 76. Data analysis guidelines for single-cell RNA-seq in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9716519/]
- 77. Flow-cytometry analysis reveals persister resuscitation ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-020-01888-3]
- 78. The identification of Pseudomonas aeruginosa persisters ... [https://pubmed.ncbi.nlm.nih.gov/36287586/]
- 79. Single-cell massively-parallel multiplexed microbial ... [https://www.nature.com/articles/s41564-023-01462-3]
- 80. An Advanced Bacterial Single-cell RNA-seq Reveals ... [https://elifesciences.org/reviewed-preprints/97543v2/reviews]
- 81. Intracellular Staphylococcus aureus persisters upon ... [https://www.nature.com/articles/s41467-020-15966-7]
- 82. Targeting Persisters for Tuberculosis Control - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3346619/]
- 83. Specialized Persister Cells and the Mechanism of Multidrug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC532439/]
- 84. High Persister Mutants in Mycobacterium tuberculosis [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0155127]
- 85. Pseudomonas aeruginosa persister cell formation upon ... [https://www.nature.com/articles/s41598-022-20323-3]
- 86. Tolerance and Persistence of Pseudomonas aeruginosa in ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.02057/full]
- 87. Strategies for Comprehensive Multi-Omics Integrative Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592251/]
- 88. Metabolomics and transcriptomics based multi-omics ... [https://www.nature.com/articles/s41540-023-00305-5]
- 89. Integrated transcriptomic, proteomic, and metabolomic ... [https://pubmed.ncbi.nlm.nih.gov/40620796/]
- 90. A Guide to Multi-omics Integration Strategies [https://frontlinegenomics.com/a-guide-to-multi-omics-integration-strategies/]
- 91. Tracking the progeny of bacterial persisters using a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11494289/]
- 92. Establishment of a Method To Rapidly Assay Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3754326/]
- 93. Pooled CRISPR interference screening enables genome ... [https://www.nature.com/articles/s41467-018-04899-x]
- 94. Target Validation: Linking Target and Chemical Properties ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3182078/]
- 95. High-Throughput vs. High-Accuracy in Single-Cell Omics [https://www.cellenion.com/high-throughput-vs-high-accuracy-in-single-cell-omics/]
- 96. Current and Future Perspectives of Single-Cell Multi-omics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11974510/]
- 97. Clinical application of single‐cell RNA sequencing in disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12576031/]