Type I vs. Type II Toxin-Antitoxin Systems: Mechanisms, Applications, and Therapeutic Frontiers
This article provides a comprehensive comparison of Type I and Type II toxin-antitoxin (TA) systems for researchers and drug development professionals.
Type I vs. Type II Toxin-Antitoxin Systems: Mechanisms, Applications, and Therapeutic Frontiers
Abstract
This article provides a comprehensive comparison of Type I and Type II toxin-antitoxin (TA) systems for researchers and drug development professionals. It explores the fundamental distinctions in their genetic organization and mechanisms of action, where Type I relies on RNA-RNA interactions and Type II on protein-protein complexes. The scope extends to methodologies for studying these systems, their validated and emerging roles in phage inhibition, stress response, and bacterial persistence. We also address key challenges in the field and present a direct comparative analysis of their applications in synthetic biology and as targets for novel antibacterial strategies, synthesizing current research to guide future therapeutic development.
Defining the Core: Genetic Architecture and Mechanistic Action of Type I and Type II TA Systems
Toxin-antitoxin (TA) systems are compact genetic modules ubiquitous in prokaryotic genomes, playing critical roles in bacterial physiology, stress response, and plasmid maintenance. These two-gene operons consist of a stable toxin that disrupts essential cellular processes and a labile antitoxin that counteracts the toxin's effect. The fundamental classification of TA systems primarily hinges on the molecular nature and mode of action of the antitoxin component, which divides them into distinct types with significant functional implications. Type I systems utilize antisense RNA molecules as antitoxins that interact with toxin mRNA, while Type II systems employ proteinaceous antitoxins that directly bind and inhibit toxin proteins. Understanding the mechanistic distinctions between these systems is paramount for researchers exploring bacterial persistence, antimicrobial development, and evolutionary biology. This guide provides a structured comparison of these systems, consolidating experimental data and methodologies to serve as a resource for scientific investigation.
Table 1: Core Characteristics of Type I and Type II TA Systems
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Antisense RNA | Protein |
| Toxin Regulation | Post-transcriptional; mRNA binding inhibits translation or promotes degradation | Post-translational; direct protein-protein binding neutralizes toxin |
| Primary Function | Membrane damage, though some nucleases exist [1] | Diverse: RNase activity, DNA gyrase inhibition, etc. [2] [1] |
| Genetic Organization | Toxin and antisense RNA genes typically encoded in opposite directions or on overlapping strands | Usually co-transcribed as a bicistronic operon [1] |
| Representative Families | Hok/Sok, TisB/IstR, Ldr/Rdl, Fst/Rna [2] [1] | MazE/MazF, RelB/RelE, VapB/VapC, CcdA/CcdB [2] [3] [4] |
Mechanistic Insights: Regulation and Function
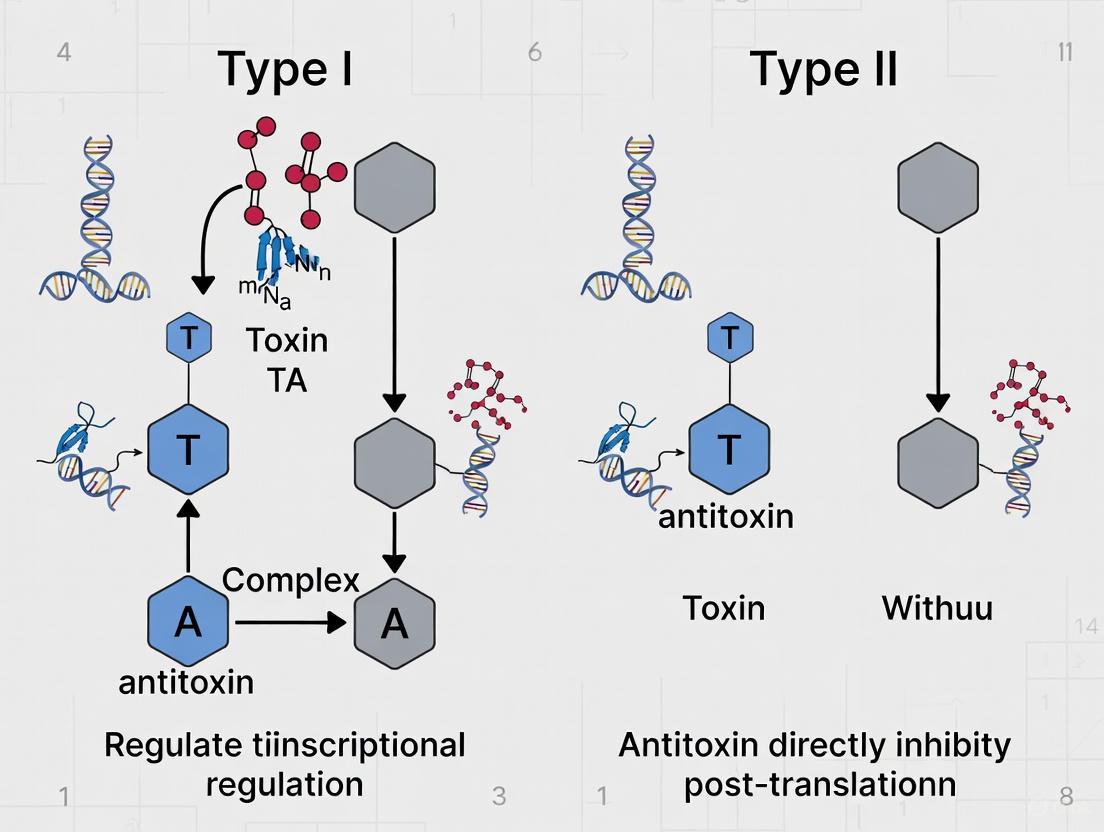
The operational divergence between Type I and Type II TA systems originates from their fundamental regulatory strategies, which are visualized in the following pathway diagram.
The diagram above illustrates the distinct regulatory pathways. In Type I systems, the antisense RNA antitoxin binds complementarily to the toxin's messenger RNA. This binding physically blocks the ribosome binding site or induces degradation of the mRNA by RNases, thereby preventing the translation of the toxin protein [1]. A well-characterized example is the Hok/Sok system, where the Sok RNA antitoxin inhibits the translation of the Hok toxin mRNA [1]. Under normal conditions, this prevents toxin synthesis. During stress, the unstable Sok RNA is rapidly degraded, freeing the Hok mRNA for translation and leading to membrane damage and potential cell death [1].
In contrast, Type II systems involve direct protein-protein interaction. The protein antitoxin binds to its cognate toxin protein, forming a stable, non-toxic complex [1]. This complex often autoregulates its own transcription by repressing the TA operon promoter. Under stress conditions, cellular proteases target and degrade the labile antitoxin protein. This releases the stable toxin, which then acts on its target to inhibit cell growth, promoting a persister state [2]. For instance, the VapC toxin in the VapBC system exhibits metal-dependent ribonuclease activity, cleaving RNA and halting protein synthesis upon activation [4].
Experimental Data and Comparative Analysis
Empirical studies and genomic surveys reveal significant differences in the distribution, mobility, and functional roles of these TA systems, summarized in the table below.
Table 2: Comparative Genomic and Functional Analysis Based on Empirical Evidence
| Analysis Dimension | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Phylogenetic Distribution | Narrow; often restricted to a single phylum or family (e.g., Ldr, TisB in Enterobacteriaceae) [1] | Broad; found across diverse bacterial phyla (e.g., MazF, RelE) [1] |
| Association with Mobile Genetic Elements | Less common; only 3 of 9 known families (Fst, Hok, XCV2162) are found on plasmids [1] | Highly common; frequently located on plasmids, genomic islands, and phages [3] [1] |
| Post-Segregational Killing (PSK) Efficacy | Variable; demonstrated for plasmid-borne systems (e.g., Hok/Sok), but chromosomal homologs may lack PSK [1] | High; a primary mechanism for plasmid stabilization; functional on both plasmids and chromosomes [5] [1] |
| Role in Bacterial Pathogenesis | Contributes to intracellular survival in specific host cell types (e.g., HokST, LdrAST in S. Typhimurium inside fibroblasts) [2] | Implicated in virulence, persistence during infection, and antibiotic tolerance (e.g., VapC2ST in S. Typhimurium) [2] [3] |
| Average Number per E. coli Genome | Information not specified in search results | Median of 23 toxin groups per strain (range 0-37) [3] |
Key Experimental Findings
- Fitness Advantage: A 2025 study on plasmid persistence demonstrated that combining an active partition system (Par) with a Type II TA system (Hok/Sok) provided a greater fitness advantage than either system alone, though the Type I Hok/Sok system was also highly effective in intracellular competitions [5].
- Functional Specialization in Pathogens: Research on Salmonella enterica revealed that distinct Type I (HokST, LdrAST, TisBST) and Type II (T4ST, VapC2ST) TA modules control bacterial fitness inside different eukaryotic host cells. Type I toxins were particularly important for survival inside fibroblasts, while VapC2ST was the dominant factor in epithelial cells [2].
- Genomic Reduction in Specialists: A large-scale genomic analysis of Escherichia coli showed that specialist, high-risk lineages (like ST131 in phylogroup B2) have undergone significant genomic reduction, which includes a diminished repertoire of TA systems, suggesting a fine-tuning of genetic content for a specific niche [3].
Essential Methodologies for TA System Research
Studying TA systems requires specific experimental protocols to validate their function, particularly their toxic nature and the neutralization by the antitoxin. The following workflow outlines a standard functional validation assay.
Detailed Experimental Protocol:
Cloning and Transformation (Step 1):
- Toxin-Only Control: The gene encoding the putative toxin is cloned into an inducible expression vector (e.g., pET, pBAD). This construct is transformed into a bacterial host (e.g., E. coli BL21). If the gene is truly toxic, obtaining clones may be difficult without the antitoxin present [6].
- TA Pair Validation: The complete TA operon, or the toxin gene alongside its cognate antitoxin (on the same or a compatible plasmid), is cloned and transformed. Successful cloning of the pair suggests the antitoxin neutralizes the toxin.
Controlled Gene Expression (Step 2):
- Transformants are grown in culture, and expression of the TA genes is induced using a chemical inducer like Isopropyl β-d-1-thiogalactopyranoside (IPTG). For Type I systems, this induces both the toxin mRNA and the antisense RNA; for Type II, it produces both the toxin and antitoxin proteins.
Phenotypic Assessment (Step 3):
- Bacterial Growth: The optical density (OD600) of cultures expressing the toxin alone versus the TA pair is monitored over time. Toxin expression alone should inhibit growth or reduce cell viability, while co-expression with the antitoxin should restore growth to near-normal levels [2] [6].
- Viability Counts: Colony Forming Unit (CFU) assays are performed by plating cultures on solid media before and after induction. A significant drop in CFU/mL upon toxin induction indicates bactericidal activity, while a static count suggests a bacteriostatic effect.
Molecular Validation (Step 4):
- Protein Analysis: Western blotting is used to confirm the expression and stability of the toxin and protein antitoxin. For Type II systems, co-immunoprecipitation can validate the physical interaction between the toxin and antitoxin proteins.
- Transcript Analysis: For Type I systems, quantitative RT-PCR (qRT-PCR) or Northern blotting confirms the expression of the antisense RNA and its impact on toxin mRNA stability or levels.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents and Materials for TA System Research
| Reagent/Material | Function and Application in TA Research |
|---|---|
| Inducible Expression Vectors (e.g., pET, pBAD) | Allows controlled, tunable expression of putative toxin genes and TA pairs for functional validation [6]. |
| Antibiotic Selection Markers | Maintains plasmid stability during culture; used in competition assays to compare fitness of different plasmid genotypes [5]. |
| Chemical Inducers (e.g., IPTG, Arabinose) | Triggers transcription from specific promoters on expression vectors to induce TA gene expression [6]. |
| Growth Assay Reagents | - Spectrophotometers for monitoring optical density (OD600).- Microplate Readers for high-throughput growth kinetics.- Agar Plates for CFU assays. |
| Molecular Biology Kits | - qRT-PCR Kits for quantifying transcript levels of toxins and antisense RNAs.- Cloning Kits for constructing TA expression plasmids.- Protein Electrophoresis & Western Blotting systems. |
| Bacterial Strains | - Cloning Strains (e.g., DH5α) for plasmid propagation.- Expression Strains (e.g., BL21) for protein production.- Clinical Isolates for studying TA systems in a pathogenic context [2] [3]. |
Toxin-antitoxin (TA) systems are small genetic elements ubiquitous in prokaryotic genomes, composed of a toxin that disrupt essential cellular processes and its cognate antitoxin that neutralizes this effect. These systems are classified into multiple types based on the nature of the antitoxin and its mechanism of action. Among these, Type I and Type II TA systems represent fundamentally distinct paradigms of genetic organization and regulation [7] [8]. While both types maintain the core toxin-antitoxin functionality, their operon structures, regulatory mechanisms, and genomic contexts differ significantly, offering valuable comparative models for studying genetic organization principles.
Type II systems, the most extensively studied, typically feature classical operon structures with both toxin and protein antitoxin genes co-transcribed on polycistronic mRNA [9] [7]. In contrast, Type I systems display more unconventional organization where protein toxins are regulated by antisense RNA antitRNAs transcribed from the opposite DNA strand [10] [8]. This structural comparison provides insights into how different regulatory strategies evolve to accomplish similar biological ends, making TA systems exceptional models for investigating the relationship between genetic architecture and function.
Fundamental Architectural Comparison: Operon Structures
The genetic organization of Type I and Type II TA systems reflects their divergent regulatory strategies, with significant implications for their functional implementation and experimental investigation.
Table 1: Core Architectural Features of Type I and Type II TA Systems
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Toxin Nature | Small protein (typically < 60 amino acids) [8] | Protein (various sizes, often enzymes) [7] |
| Antitoxin Nature | Antisense RNA [10] [8] | Protein [9] [7] |
| Genetic Organization | Genes often encoded on opposite DNA strands [8] | Classical operon structure: adjacent genes under single promoter [9] |
| Primary Regulation Level | Post-transcriptional [8] | Transcriptional & post-translational [7] |
| Antitoxin Mechanism | Binds toxin mRNA via complementarity, blocking translation or promoting degradation [8] | Protein-protein interaction directly inhibits toxin activity [7] |
| Common Genomic Locations | Plasmids, prophages, chromosomes [10] [8] | Chromosomes, plasmids, genomic islands [7] |
Type I System Organization and Regulation
Type I TA systems exhibit a distinctive genetic architecture where the toxin gene and the antisense RNA antitoxin are typically encoded on opposite strands of the DNA, producing complementary transcripts [8]. The antitoxin RNA, such as Sok in the hok/sok system, regulates toxin production through several sophisticated mechanisms including translational blockade via direct sequestration of the ribosome binding site, interaction at upstream open reading frames, or promotion of mRNA degradation by RNases [8]. This arrangement allows extremely rapid post-transcriptional responses to environmental stimuli without requiring new protein synthesis.
Type II System Organization and Regulation
Type II TA systems conform to the classical operon model first established in prokaryotic genetics [11] [12]. These systems typically feature two adjacent genes—encoding the protein antitoxin and protein toxin—transcribed together as a single polycistronic mRNA molecule under control of a shared promoter [9] [7]. The antitoxin proteins generally contain two functional domains: a DNA-binding domain that enables autoregulation by binding the operon's promoter region, and a toxin-binding domain that neutralizes the toxin through direct protein-protein interaction [7] [13]. This organization creates a tight autoregulatory loop where the TA complex itself represses its own transcription.
Functional Implications of Structural Differences
The contrasting genetic architectures of Type I and Type II TA systems directly influence their biological functions and phenotypic outcomes, with each type exhibiting specialized roles in bacterial physiology.
Table 2: Functional Specialization of Type I and Type II TA Systems
| Functional Aspect | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Mobile Genetic Element Maintenance | Stabilize prophages & plasmids [10] | Plasmid maintenance via post-segregational killing [7] |
| Stress Response | Contribute to persistence under antibiotic stress [8] | Manage oxidative, nutrient, & antibiotic stress [2] [7] |
| Virulence & Pathogenesis | Indirect modulation through persistence [8] | Direct regulation of virulence factors & survival in host cells [2] [13] |
| Biofilm Formation | Limited evidence | Significant role in biofilm development & maintenance [7] |
| Phage Inhibition | Contribute to abortive infection [8] | Defense against bacteriophage infection [7] |
| Bacterial Persistence | Important for persister cell formation [8] | Contribute to antibiotic tolerance & persistence [7] |
Functional Specialization in Pathogens
The functional specialization of TA system types is particularly evident in bacterial pathogens. In Salmonella enterica serovar Typhimurium, distinct Type I and Type II TA modules control bacterial lifestyle inside eukaryotic cells but with different specializations [2]. Type I toxins HokST, LdrAST, and TisBST, along with Type II toxins T4ST and VapC2ST, collectively promote bacterial survival inside fibroblasts. However, in epithelial cells, only the Type II toxin VapC2ST enhances bacterial fitness, demonstrating cell-type specific functional specialization [2].
In Streptococcus suis, the Type II ParDE system significantly contributes to virulence in mouse infection models, with deletion mutants showing attenuated pathogenicity [13]. The ParDE system also modulates oxidative stress response and affects susceptibility to macrophage phagocytosis, highlighting how a single Type II TA system can influence multiple aspects of bacterial pathogenesis through its regulatory functions [13].
Evolutionary Distribution Patterns
Comparative genomic analyses reveal distinct evolutionary patterns between TA system types. In Salmonella, numerous Type I TA modules show restricted distribution among serovars, with some like ibsA-sibAST found only in serovar Typhimurium [2]. This contrasts with the broader conservation of many Type II systems. Pathogenic Salmonella enterica species harbor approximately double the TA modules compared to non-pathogenic Salmonella bongori, suggesting that TA system acquisition correlates with pathogenicity [2].
In the Mycobacterium tuberculosis complex, the VapBC3 Type II TA system exhibits species-specific variations despite high overall conservation [4]. In M. bovis, a nucleotide deletion creates a truncated VapC3 toxin (109 amino acids versus 137 in M. tuberculosis) with potentially altered functional properties, as molecular docking analyses predict stronger binding affinity in the M. bovis variant [4]. This demonstrates how subtle structural variations in TA systems may contribute to host adaptation and pathogenic specialization.
Experimental Approaches and Methodologies
Standard Experimental Workflows
Research on TA systems employs specialized methodologies tailored to their unique genetic features and mechanisms of action. The following diagram illustrates a generalized experimental workflow for functional characterization of both TA system types:
Essential Research Tools and Reagents
Table 3: Research Reagent Solutions for TA System Investigation
| Reagent/Technique | Function/Application | Example Implementation |
|---|---|---|
| Inducible Expression Vectors | Controlled toxin expression to study toxicity | pRPF185-derivatives with anhydrotetracycline-inducible Ptet promoter in C. difficile [10] |
| β-galactosidase Reporter Systems | Promoter activity measurement for autoregulation studies | pTCV-Lac plasmid systems in S. suis to quantify ParDE promoter activity [13] |
| Electrophoretic Mobility Shift Assay (EMSA) | Protein-DNA binding confirmation | Confirmation of ParD antitoxin binding to ParDE promoter as dimers [13] |
| Bacterial Two-Hybrid Systems | Protein-protein interaction mapping | Study of toxin-antitoxin binding specificity and complex formation |
| Gene Deletion Techniques | Functional analysis through knockout mutants | Overlap PCR with sacB-Spc cassette for S. suis ParDE deletion [13] |
| Proteomic & Transcriptomic Analyses | Global expression profiling under stress | Identification of TA system expression inside eukaryotic host cells [2] |
Specialized Methodological Considerations
Type I TA system research requires specialized approaches to detect small, often unannotated genes encoding toxic proteins. In Clostridioides difficile, toxin genes CD0904.1, CD0956.3, and CD0977.1 were identified using tBlastn searches independent of annotation, as standard ORF prediction often misses these small coding sequences [10]. Functional validation typically involves ectopic expression with and without cognate antitoxin RNAs, demonstrating growth inhibition that is specifically neutralized by antitoxin co-expression [10].
For Type II systems, key experiments include autoregulation studies using promoter-reporter fusions and protein interaction analyses. In S. suis ParDE research, β-galactosidase assays revealed autoregulatory function, while EMSAs confirmed that the ParD antitoxin binds its promoter as dimers [13]. These approaches are crucial for establishing the classic Type II operon regulation mechanism where the TA complex represses its own transcription.
Research Applications and Therapeutic Targeting
The distinctive genetic organization of TA systems presents unique opportunities for therapeutic development and biotechnological applications. Type II systems are particularly promising targets for novel antibacterial strategies aimed at disrupting their regulatory networks or activating toxin proteins [9]. Their involvement in bacterial persistence makes them attractive for developing anti-persister therapies that could enhance the efficacy of conventional antibiotics [7].
Type I systems offer potential as biological tools for genetic engineering. In C. difficile, the toxic effects of induced Type I toxins have been leveraged to develop efficient mutagenesis tools that promote elimination of plasmid-bearing cells, significantly improving allele exchange efficiency [10]. This application demonstrates how the unique regulatory features of Type I systems can be harnessed for practical biotechnological purposes.
Comparative studies of TA system variations between closely related pathogens, such as the VapBC3 structural differences between M. tuberculosis and M. bovis, provide insights for developing species-specific therapeutic approaches [4]. Understanding how sequence variations affect TA system function may lead to targeted treatments that exploit these differences for selective antibacterial activity.
The comparative analysis of Type I and Type II TA system organization reveals how evolution has produced distinct genetic architectures to achieve core regulatory functions. Type II systems exemplify the classical operon model with protein-based regulation and transcriptional control, while Type I systems represent a more streamlined RNA-mediated regulatory strategy. These structural differences underlie functional specializations that influence bacterial stress response, pathogenesis, and evolutionary adaptation. Continuing research on both TA system types promises not only fundamental insights into genetic regulation principles but also practical applications in antimicrobial development and genetic engineering.
Toxin-antitoxin (TA) systems are genetic modules ubiquitous in bacteria and archaea, consisting of a stable toxin and a corresponding labile antitoxin. These systems are categorized into types based on the nature and neutralizing mechanism of the antitoxin. Among them, Type I and Type II TA systems represent two predominant and mechanistically distinct classes [14]. Type I toxins are typically small, hydrophobic proteins that exert their primary toxic effects by disrupting the physical and electrochemical integrity of the bacterial membrane. In contrast, Type II toxins are a diverse group of proteins that predominantly interfere with essential intracellular processes, notably nucleic acid stability and protein synthesis [15] [7]. This guide provides a detailed comparison of these distinct toxin targets, supported by experimental data and methodologies relevant to current research in microbiology and drug development.
Comparative Mechanisms of Toxin Action
The following table summarizes the core characteristics and mechanisms of Type I and Type II toxin systems.
Table 1: Core Characteristics of Type I and Type II Toxin Targets
| Feature | Type I Toxins | Type II Toxins |
|---|---|---|
| Primary Target | Bacterial cell membrane [15] | Nucleic acids & protein synthesis machinery [7] [14] |
| Nature of Toxin | Small hydrophobic proteins (often < 60 amino acids) [15] | Proteins with enzymatic activities (e.g., RNases, DNases) [7] |
| Mechanism of Action | Membrane depolarization and/or permeabilization, leading to ATP depletion and loss of viability [15]; potential pore formation [15] | Cleavage of mRNA, tRNA, or rRNA [14]; poisoning of DNA gyrase [14]; inhibition of translation [7] |
| Typical Effect | Growth arrest; cell death; "ghost" cell formation [15] | Growth arrest (bacteriostatic) [7] [14] |
| Antitoxin Type | Antisense RNA (binds toxin mRNA) [8] [14] | Protein (binds and inhibits toxin protein) [7] [14] |
| Key Example Systems | hok/sok, tisB/istR, ldr/rdl [14] |
mazEF (RNA interferase), ccdAB (DNA gyrase inhibitor), relBE (translation inhibitor) [7] [14] |
The diagram below illustrates the fundamental operational differences between Type I and Type II TA systems, from gene expression to cellular outcome.
Experimental Data and Validation
The distinct physiological impacts of these toxin types have been quantified in various experimental settings.
Table 2: Quantitative Experimental Data on Toxin Effects
| Toxin (System) | Type | Experimental Context | Key Quantitative Finding | Reference |
|---|---|---|---|---|
| HokST | I | S. Typhimurium inside eukaryotic fibroblasts | Promoted bacterial survival inside host cells [2]. | [2] |
| LdrAST | I | S. Typhimurium inside eukaryotic fibroblasts | Promoted bacterial survival inside host cells [2]. | [2] |
| TisBST | I | S. Typhimurium inside eukaryotic fibroblasts | Promoted bacterial survival inside host cells [2]. | [2] |
| VapC2ST | II | S. Typhimurium inside eukaryotic cells | Promoted bacterial survival in both fibroblasts and epithelial cells [2]. | [2] |
| Hok | I | E. coli (ectopic overexpression) | Induced membrane depolarization and killed cells within 30 minutes [15]. | [15] |
| CcdB | II | E. coli | Targets and poisons DNA gyrase, halting DNA replication [14]. | [14] |
| MazF | II | E. coli | Functions as an endoribonuclease that cleaves cellular mRNAs at specific sequences [14]. | [14] |
Detailed Experimental Protocols
To illustrate how the differential effects of these toxins are validated in a laboratory setting, here are detailed protocols for key assays.
Assessing Membrane Integrity (Type I Toxins)
Objective: To evaluate the impact of a Type I toxin on bacterial membrane potential and integrity. Principle: Membrane-depolarizing toxins dissip the transmembrane electrochemical gradient, which can be measured using fluorescent dyes like DiOC₂(3). Pore-forming activity can be assessed by monitoring the influx of small, normally impermeable molecules.
Protocol:
- Strain & Induction:
- Transform E. coli with a plasmid containing the Type I toxin gene (e.g.,
hokortisB) under a tightly regulated, inducible promoter (e.g., P~BAD~ arabinose-inducible or P~tet~ tetracycline-inducible). - Include a control strain with an empty vector or a toxin-antitoxin co-expression vector.
- Grow cultures to mid-exponential phase and induce toxin expression with the appropriate inducer.
- Transform E. coli with a plasmid containing the Type I toxin gene (e.g.,
Membrane Potential Measurement (using DiOC₂(3)):
- Harvest bacterial cells by gentle centrifugation.
- Load cells with 30 µM DiOC₂(3) in buffer and incubate for 30 minutes in the dark.
- Analyze by flow cytometry. DiOC₂(3) exhibits a concentration-dependent fluorescence shift: in cells with a strong membrane potential, it forms aggregates that emit red fluorescence; in depolarized cells, it remains as monomers that emit green fluorescence. The ratio of red to green fluorescence is a quantitative measure of membrane potential [15].
Membrane Permeabilization Assay (using SYTOX Green):
- After toxin induction, add SYTOX Green nucleic acid stain (final concentration 1 µM) to the bacterial culture.
- SYTOX Green is impermeant to intact membranes but fluoresces brightly upon binding DNA when it enters cells through membrane pores.
- Monitor fluorescence intensity over time using a plate reader or analyze by flow cytometry. An increase in fluorescence indicates a loss of membrane integrity.
Assessing Nucleic Acid Integrity (Type II Toxins)
Objective: To detect the cleavage of cellular RNA (a common target of Type II toxins) upon toxin activation. Principle: Many Type II toxins (e.g., MazF, RelE) are mRNA interferases. Their activation leads to rapid degradation of the transcriptome, which can be visualized by gel electrophoresis.
Protocol:
- Toxin Activation:
- Use a strain where the expression of a Type II toxin (e.g.,
mazF) is inducible. - Induce toxin expression in mid-exponential phase cultures. A common method to activate chromosomal TA systems is by treating cells with an antibiotic like rifampicin, which inhibits transcription and prevents synthesis of the short-lived protein antitoxin, freeing the toxin [7].
- Use a strain where the expression of a Type II toxin (e.g.,
RNA Extraction and Analysis:
- At regular time points post-induction (e.g., 0, 15, 30, 60 minutes), withdraw 1 mL of culture.
- Immediately extract total RNA using a commercial kit with DNase I treatment to remove genomic DNA contamination.
- Separate ~1 µg of total RNA on a denaturing agarose gel or a non-denaturing polyacrylamide gel.
Visualization and Interpretation:
- Stain the gel with ethidium bromide or SYBR Gold.
- Visualize under UV light. Intact RNA from uninduced cells will show clear, sharp bands for 16S and 23S rRNA. In toxin-induced cells, the general degradation of mRNA and potentially rRNA will manifest as a smear with a loss of distinct ribosomal RNA bands, indicating widespread RNA cleavage [7].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and materials used in the experimental analysis of TA systems.
Table 3: Essential Reagents for Toxin-Antitoxin Research
| Reagent/Material | Function in Research | Application Example |
|---|---|---|
| Inducible Expression Plasmids (e.g., pBAD, pET with tight promoters) | To control the timing and level of toxin gene expression, allowing for the study of toxic proteins without killing the host cell during culture maintenance [10]. | Used for ectopic overexpression of toxins Hok or MazF to study their effects [15] [14]. |
| Fluorescent Membrane Dyes (e.g., DiOC₂(3), SYTOX Green) | To quantitatively assess membrane potential and integrity in live bacterial cells via flow cytometry or fluorometry [15]. | Used in Protocol 3.1 to demonstrate Hok-induced membrane depolarization. |
| RNA Extraction & Electrophoresis Kits | To isolate high-quality, DNA-free total RNA and analyze its integrity, revealing RNA degradation activity. | Used in Protocol 3.2 to visualize the RNase activity of MazF [7]. |
| Conditional Knockout Strains (e.g., lacking specific proteases like Lon) | To study the natural activation of Type II systems, as the Lon protease is key to degrading labile protein antitoxins under stress [7]. | Used to investigate the role of RelE in stress response and persistence. |
| Antibiotics (e.g., Rifampicin) | To trigger the activation of chromosomal TA systems by halting transcription, which halts the synthesis of short-lived antitoxins [7]. | Used to activate systems like mazEF and relBE for study. |
Type I and Type II TA systems have evolved distinct and specialized strategies to modulate bacterial physiology. Type I toxins act as swift, direct disruptors of cellular energetics by compromising the membrane, a primary barrier essential for life. Type II toxins, in contrast, function as precise molecular saboteurs within the cell, halting growth by selectively disrupting the flow of genetic information and protein production. This fundamental distinction in targets—membrane integrity versus nucleic acids and protein synthesis—dictates their unique mechanisms, physiological roles, and potential applications. Understanding these differences is crucial for researchers exploring bacterial stress response, pathogenesis, and the development of novel antibacterial strategies that might exploit these native toxin systems.
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in prokaryotes, consisting of a stable toxin that disrupt vital cellular processes and a labile antitoxin that neutralizes the toxin. These systems are primarily classified based on the nature and mechanism of the antitoxin. Among these, type I and type II systems represent two fundamental paradigms for how bacteria regulate toxin activity: through translation inhibition or post-translational neutralization [16]. In type I systems, the antitoxin is an antisense RNA that binds toxin mRNA to prevent translation, while in type II systems, the antitoxin is a protein that directly binds and inhibits the toxin protein [17]. Understanding the distinct mechanisms governing these systems is crucial for fundamental bacterial physiology and developing novel antimicrobial strategies that target persistent infections.
The fundamental distinction between type I and type II TA systems lies in their regulatory logic, primarily determined by the chemical nature of the antitoxin and its mode of action.
Type I Systems: Translation Inhibition via RNA Antitoxins In type I TA systems, the antitoxin is a small non-coding RNA that acts as an antisense RNA to regulate toxin gene expression post-transcriptionally [18]. This antisense RNA binds to the messenger RNA (mRNA) of the toxin, leading to the degradation of the toxin transcript through the action of RNases and/or the direct blockade of the ribosome binding site, thereby preventing toxin translation [19] [16]. The toxin gene of type I systems typically encodes a small, hydrophobic protein that often localizes to the inner membrane and can disrupt membrane potential [20]. Well-characterized examples in Escherichia coli include tisB-istR and ldrD-rdlD [21] [20]. Once the toxin protein is synthesized, it cannot be neutralized by the RNA antitoxin, making regulation primarily pre-translational.
Type II Systems: Post-Translational Neutralization via Protein Antitoxins In type II TA systems, both the toxin and antitoxin are proteins [19]. The antitoxin gene is usually located upstream of the toxin gene, and both are organized in an operon whose expression is often auto-regulated by the toxin-antitoxin protein complex [19] [22]. The antitoxin protein directly binds to its cognate toxin protein, forming a stable complex that sterically blocks the active site of the toxin, thereby neutralizing its activity [19] [22]. Under normal growth conditions, this complex also represses the transcription of the TA operon. Under stress conditions, cellular proteases such as Lon or Clp preferentially degrade the labile antitoxin, releasing the stable toxin to act on its cellular target [19] [17]. This mechanism allows for rapid post-translational control of toxin activity.
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Small non-coding RNA (sRNA) [18] | Protein [19] |
| Core Regulatory Mechanism | Translation inhibition; mRNA degradation or ribosomal blockade [16] | Post-translational neutralization; direct protein-protein interaction [22] |
| Toxin Neutralization | Not applicable; prevention of synthesis | Direct binding and active site occlusion [22] |
| Genetic Organization | Antitoxin gene is located opposite or in cis to toxin gene [16] | Typically bicistronic operon with antitoxin upstream of toxin [19] |
| Transcriptional Autoregulation | Less common | Common; TA complex represses its own promoter [22] |
| Response to Stress | Derepression of toxin transcription and translation | Protease-mediated antitoxin degradation and toxin release [19] |
Mechanism of Action and Cellular Targets
The toxins from both type I and type II systems ultimately inhibit bacterial growth, but they employ diverse strategies to achieve this.
Type I Toxin Targets and Physiological Effects Type I toxins are typically small, hydrophobic peptides that localize to the inner membrane [20]. A primary mechanism of action is the dissipation of the proton motive force (PMF). For instance, the TisB toxin forms anion-conductive pores in the inner membrane, leading to the depolarization of both the electrical and proton gradients across the membrane [20]. This collapse of the PMF results in a dramatic reduction in intracellular ATP levels, which in turn inhibits essential processes like transcription, translation, and replication, inducing a dormant, persister state [20]. Other type I toxins, such as LdrD and HokB, have also been shown to cause membrane depolarization and ATP leakage [21].
Type II Toxin Targets and Physiological Effects Type II toxins exhibit a broader range of enzymatic activities and cellular targets, though most ultimately inhibit protein synthesis. They can be categorized into several superfamilies based on their structure and mechanism:
- RNase Toxins: Many type II toxins are sequence-specific endoribonucleases. For example, MazF cleaves free mRNA at specific sequences (e.g., ACA) and can also process 16S rRNA to create "stress ribosomes" that alter translation patterns [16]. RelE and YoeB are mRNA interferases that cleave ribosome-associated mRNAs [17] [16].
- Inhibitors of DNA Replication: Toxins like CcdB and ParE target DNA gyrase, leading to the inhibition of DNA replication and double-strand breaks [19] [17].
- Kinase Toxins: HipA phosphorylates glutamyl-tRNA synthetase, inhibiting tRNA charging and consequently translation [17]. Doc phosphorylates and inactivates the elongation factor EF-Tu, preventing tRNA delivery to the ribosome [19] [16].
- Other Mechanisms: MbcT depletes NAD+ [19], while the ζ-toxin depletes UDP-activated sugars, inhibiting cell wall synthesis [19].
Table 2: Diversity of Toxin Actions in Type I and Type II TA Systems
| System Type | Toxin Example | Primary Cellular Target | Molecular Mechanism | Physiological Outcome |
|---|---|---|---|---|
| Type I | TisB (E. coli) [20] | Inner Membrane | Forms anion channels; dissipates proton motive force | ATP depletion; growth arrest; persistence [20] |
| Type I | LdrD (E. coli) [21] | Inner Membrane | Causes membrane depolarization and ATP leakage | Growth inhibition; antibiotic tolerance [21] |
| Type II | MazF (E. coli) [21] [16] | mRNA / 16S rRNA | Ribosome-independent endoribonuclease; creates "stress ribosomes" | Global inhibition of translation; reprogramming of translation [16] |
| Type II | RelE (E. coli) [16] | mRNA | Ribosome-dependent endoribonuclease; cleaves mRNA at codon positions | Inhibition of translation during amino acid starvation [17] |
| Type II | CcdB (E. coli) [19] [17] | DNA Gyrase | Inhibits DNA gyrase, trapping it in a cleavage complex | DNA double-strand breaks; inhibition of replication |
| Type II | HipA (E. coli) [17] | Glu-tRNA Synthetase | Serine/Threonine kinase; inhibits tRNA charging | Inhibition of translation; persistence [17] |
| Type II | Doc (E. coli) [19] [16] | Elongation Factor Tu (EF-Tu) | Phosphorylation and inactivation of EF-Tu | Inhibition of translation |
Key Experimental Models and Methodologies
Research into TA systems relies on well-established genetic models and precise methodologies to dissect the complex regulation and phenotypic outcomes.
Established Model Systems
- Type I Model (LdrD): A genetically engineered E. coli model for the type I toxin LdrD involves removing the native ldrD and rdlD genes from the chromosome and reintroducing the ldrD toxin gene under the control of a tight, inducible promoter (e.g., anhydrotetracycline-aTc) [21]. This system allows controlled expression of the toxin without interference from its natural antisense RNA antitoxin, enabling the study of its physiological effects, such as growth arrest, membrane depolarization, and antibiotic tolerance [21].
- Type II Model (MazEF): A analogous model for the type II system MazEF uses controlled expression of both the MazF toxin and its protein antitoxin MazE [21]. This system demonstrates that MazF toxicity can be quenched by co-expression of MazE, highlighting the protein-protein interaction that defines type II systems. Induction of MazF alone leads to growth inhibition and increased persistence to antibiotics [21].
Critical Experimental Protocols
1. Assessing Toxin-Induced Physiological Changes:
- Membrane Depolarization Assay: The bis-(1,3-dibarbituric acid)-trimethine oxanol (DiBAC₄(3)) dye is used in flow cytometry to detect membrane potential changes. This lipophilic anionic dye enters depolarized cells, resulting in increased fluorescence. This protocol is crucial for demonstrating the action of type I toxins like TisB and LdrD [21] [20].
- Workflow: Grow bacterial cultures to mid-log phase → induce toxin expression or apply antibiotic stress → incubate → dilute cells in PBS → stain with DiBAC₄(3) → analyze fluorescence of >10,000 cells via flow cytometry [20].
- ATP Measurement: To quantify metabolic arrest, intracellular and extracellular ATP levels can be measured using luminometry-based assays (e.g., firefly luciferase). A drop in intracellular ATP is a key indicator of toxin activity, as seen with TisB and LdrD [21] [20].
2. Persistence and Tolerance Assays:
- Protocol for Post-Antibiotic Survival: Stationary-phase cultures are treated with fluoroquinolone antibiotics (e.g., ofloxacin). Crucially, toxin expression (or chemical inhibitors of transcription/translation like rifampicin/chloramphenicol) is induced only after the antibiotic treatment has concluded. Survival rates are determined by plating and counting colony-forming units (CFUs). This assay demonstrated that toxin activity during recovery is sufficient to increase persister numbers, a finding applicable to both type I (LdrD) and type II (MazF) toxins [21].
3. Genetic Interaction Studies:
- Methodology: Isogenic mutant strains (e.g., lacking recA, recB, or uvrD) are constructed and transformed with inducible toxin plasmids. The persistence of these mutants to antibiotics is compared to the wild-type strain following toxin induction. This approach identified that the persistence conferred by LdrD and MazF depends on homologous recombination (recA, recB) and nucleotide excision repair (uvrD) machinery [21].
Diagram 1: Experimental Workflow for Differentiating TA System Types. This diagram outlines the logical pathway for characterizing a TA system, from initial classification based on antitoxin nature to the distinct experimental approaches and expected outcomes for type I versus type II systems.
The Scientist's Toolkit: Essential Research Reagents
Research in TA systems utilizes a suite of specific reagents, strains, and molecular tools.
Table 3: Key Reagent Solutions for TA System Research
| Reagent / Tool | Function in Research | Specific Example(s) |
|---|---|---|
| Inducible Expression Systems | Enables controlled, high-level expression of toxic genes for functional studies. | anhydrotetracycline (aTc)-inducible promoter for LdrD [21]; Arabinose (pBAD) or IPTG-inducible systems. |
| Fluorescent Membrane Dyes | Detects changes in membrane potential, a key action of many type I toxins. | DiBAC₄(3) for flow cytometry [21] [20]. |
| ATP Measurement Kits | Quantifies metabolic activity and energy depletion upon toxin induction. | Luminometry-based kits using firefly luciferase [21]. |
| Isogenic Mutant Strains | Determines the genetic dependencies of toxin-induced phenotypes. | E. coli KEIO collection mutants (e.g., ΔrecA, ΔuvrD) [21]. |
| Protein Purification Tools | For in vitro biochemical and structural studies of type II TA complexes. | His-tag purification of complexes like DinJ-YafQ [22]; Size-exclusion chromatography (SEC). |
| Interaction Analysis Instruments | Characterizes the binding affinity and stoichiometry of type II TA protein interactions. | Isothermal Titration Calorimetry (ITC) [18]; Analytical SEC. |
| Reporter Assays | Measures transcriptional regulation and promoter activity of TA operons. | GFP fusions to promoters (e.g., plexA-gfp, pumuDC-gfp) [20]. |
Type I and type II TA systems represent two evolutionarily distinct strategies for regulating toxin activity: pre-emptively through translation inhibition or reactively through post-translational neutralization. The experimental data reveal that despite these different starting points, both systems can converge on a similar physiological outcome—bacterial growth arrest and persistence. This phenotypic convergence underscores the importance of dormancy as a survival strategy. The choice of research model and methodology is critically dependent on the system type, as delineated in this guide. A deep understanding of these regulatory mechanisms provides a foundation for targeting TA systems to combat antibiotic tolerance and persistent infections.
Prevalence and Distribution in Bacterial Chromosomes and Plasmids
Toxin-antitoxin (TA) systems are genetic modules ubiquitous in prokaryotes, composed of a stable toxin that disrupt essential cellular processes and a labile antitoxin that neutralizes the toxin [23] [14]. These systems are classified into types based on the nature and mode of action of the antitoxin. Among these, type I and type II systems represent two of the most prevalent and well-studied classes [14] [24]. Type I systems feature protein toxins regulated by antisense RNA antitoxins that inhibit toxin mRNA translation [14]. In contrast, type II systems consist of both protein toxins and protein antitoxins that form a stable complex, preventing toxin activity [23] [24]. The distribution of these systems between bacterial chromosomes and plasmids has significant implications for bacterial physiology, evolution, and pathogenicity [23] [5]. This guide provides a objective comparison of type I and type II TA systems, focusing on their prevalence, distribution, and functional characteristics, to inform research and drug development strategies.
Table 1: Fundamental characteristics of Type I and Type II TA systems.
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Toxin Nature | Protein | Protein |
| Antitoxin Nature | Antisense RNA | Protein |
| Mechanism of Inhibition | Antitoxin RNA binds toxin mRNA, blocking translation or promoting degradation [14]. | Antitoxin protein binds and neutralizes the toxin protein [23] [24]. |
| Primary Toxin Activities | Membrane depolarization, ATP synthesis inhibition [24]. | mRNA/tRNA cleavage, DNA gyrase poisoning, protein phosphorylation [23] [24]. |
| Common Localization | Found on both chromosomes and plasmids [2]. | Found extensively on both chromosomes and plasmids [23] [5]. |
Table 2: Prevalence, distribution, and functional roles of Type I and Type II TA systems.
| Aspect | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Representative Examples | hok/sok, tisB/istR, ldrD/rdlD, sprA1/sprA1as [14] [2]. |
mazEF, relBE, vapBC, ccdAB, higBA, hipBA [23] [2] [24]. |
| Role in Plasmid Maintenance | Post-segregational killing via stable toxin and unstable RNA antitoxin (e.g., hok/sok) [14] [5]. |
Post-segregational killing and plasmid stabilization (e.g., ccdAB on the F plasmid) [14] [24]. |
| Chromosomal Functions | Involved in stress response (e.g., tisB/istR in SOS response) and pathogenesis (e.g., sprA1 in S. aureus) [14] [2]. |
Linked to stress response, persistence, biofilm formation, and virulence [23] [25] [24]. |
| Distribution in Pathogens | Found in pathogens like S. aureus and S. Typhimurium, often within pathogenicity islands [14] [2]. | Highly abundant in pathogens like M. tuberculosis and ESKAPE group members [23] [24]. |
Experimental Analysis of TA System Distribution and Function
Protocol: Genomic Identification and Characterization of TA Modules
Objective: To identify and characterize the repertoire of type I and type II TA modules in a bacterial pathogen [2]. Methodology:
- Genome Sequence Analysis: Use the genome of the target pathogen (e.g., Salmonella enterica serovar Typhimurium strain SL1344) as a reference.
- Bioinformatic Screening:
- PSI-BLAST Verification: Perform iterative BLAST searches using validated toxin and antitoxin sequences as queries to identify homologs that may have been missed by standard screening [2].
- Manual Curation and Annotation: Assign names based on sequence homology to known systems. For novel modules, provide genome coordinates and designate with a systematic nomenclature (e.g., "TA-[number]-ST") [2].
- Phylogenetic Distribution Analysis: Compare the presence of identified TA modules across different strains and species (e.g., pathogenic vs. non-pathogenic Salmonella) using tools like tBLASTn against genomic databases [2].
Protocol: Assessing TA System Function in Intracellular Survival
Objective: To determine the contribution of specific type I and type II TA modules to bacterial fitness inside eukaryotic host cells [2]. Methodology:
- Proteomic and Gene Expression Analysis: Infect eukaryotic cell lines (e.g., fibroblasts, epithelial cells) with the pathogen. Analyze bacterial transcripts or proteins extracted from intracellular bacteria to identify which TA toxins are produced during infection [2].
- Mutant Construction: Generate isogenic mutant strains lacking specific type I (e.g.,
hokST,ldrAST,tisBST) or type II (e.g.,vapC2ST) toxin genes. - Intracellular Fitness Assay: Infect different eukaryotic cell lines with wild-type and mutant strains.
- Competition Assay: Co-infect cells with a 1:1 mixture of wild-type and mutant bacteria. After a set incubation period, recover intracellular bacteria and determine the competitive index (CI) by plating on selective media.
- Data Interpretation: A CI significantly less than 1 indicates the mutant has a fitness defect inside host cells, implying the deleted TA system is important for intracellular survival. Specialization of function is concluded if a toxin is essential in one cell type (e.g., fibroblasts) but not another (e.g., epithelial cells) [2].
Visualization of TA System Function and Analysis
The following diagram illustrates the fundamental mechanisms of type I and type II TA systems and their functional outcomes.
The following diagram outlines a key experimental workflow for analyzing the role of TA systems in intracellular pathogens.
Essential Research Reagents and Tools
Table 3: Key reagents, tools, and databases for TA system research.
| Reagent/Tool Solution | Type | Primary Function in Research |
|---|---|---|
| PLSDB Database [26] | Database | Provides access to a large, curated collection of plasmid sequences, aiding in the identification of plasmid-borne TA systems. |
| TADB (Toxin-Antitoxin Database) [2] | Database | A specialized web resource for the identification and analysis of type I and type II TA modules in bacterial genomes. |
| RASTA-Bacteria [2] | Software Tool | A automated tool for identifying type II TA systems in bacterial genomes. |
| pCON Model Plasmids [5] | Molecular Tool | Engineered plasmid backbones used in competition assays to study the fitness advantages conferred by TA and partition systems. |
| Specialized Eukaryotic Cell Lines (e.g., Fibroblasts, Epithelial) [2] | Biological Model | Used in intracellular infection assays to test the role of specific TA systems in pathogen survival and fitness within different host cell environments. |
| Lon Protease [24] | Enzyme | A key cellular protease responsible for degrading labile type II antitoxins under stress conditions, leading to toxin activation. |
From Bench to Biotech: Research Tools and Applications in Synthetic Biology and Medicine
Experimental Approaches for Characterizing TA System Components
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in prokaryotes, typically composed of a stable toxin that disrupt essential cellular processes and an unstable antitoxin that neutralizes the toxin under normal growth conditions [27]. These systems are classified into eight types (I-VIII) based on the nature and mechanism of the antitoxin [19] [28]. Type I systems feature protein toxins regulated by antisense RNA antitoxins that inhibit toxin mRNA translation, while Type II systems consist of both protein toxins and protein antitoxins that form complexes to inhibit toxin activity [19] [28]. The fundamental distinction between these types necessitates different experimental methodologies for their characterization, presenting unique challenges and requirements for researchers investigating their structure, function, and regulation.
This guide provides a comprehensive comparison of experimental approaches for characterizing Type I versus Type II TA system components, synthesizing current methodologies from recent scientific literature. We objectively evaluate the performance of various techniques through available experimental data and provide detailed protocols for key experiments, enabling researchers to select appropriate strategies for their specific investigations into these fascinating genetic elements.
Fundamental Differences Between Type I and Type II TA Systems
The core distinction between Type I and Type II TA systems lies in the molecular nature of their antitoxin components and their mechanisms of action, which directly dictate the experimental approaches required for their characterization.
Type I TA systems employ antisense RNA antitoxins that bind complementarily to toxin mRNA, preventing translation through occlusion of ribosome binding sites or inducing degradation of the toxin transcript [29]. These toxins are typically small, hydrophobic proteins often under 60 amino acids, with propensity for membrane association [29]. Experimental identification is challenging due to their small size and hydrophobicity, often requiring specialized computational predictions and RNA-focused molecular biology techniques.
Type II TA systems utilize protein antitoxins that directly bind to and inhibit their cognate protein toxins [19]. These systems generally feature slightly larger toxins (approximately 100 amino acids) and are often chromosomal or plasmid-encoded [19]. The protein-protein interaction nature of Type II systems makes them amenable to a wider array of standard biochemical and structural biology approaches, though specific adaptations are required to study their unique regulatory mechanisms.
Table 1: Fundamental Characteristics of Type I vs. Type II TA Systems
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin nature | Antisense RNA | Protein |
| Toxin characteristics | Small hydrophobic proteins (<60 aa) | Larger proteins (~100 aa) |
| Mechanism of inhibition | Translation inhibition via mRNA binding | Direct protein-protein interaction |
| Genomic organization | Often tandem repeats in intergenic regions | Typically operonic structure |
| Primary detection methods | RNA sequencing, computational prediction | BLAST searches, protein interaction assays |
| Stability considerations | RNA antitoxin degradation | Protein antitoxin proteolysis |
| Experimental challenges | Small protein detection, RNA secondary structure | Toxin activation control, complex purification |
Computational Identification and Bioinformatic Analysis
Specialized Approaches for Type I Systems
Computational identification of Type I TA systems presents unique challenges due to the small size of the toxin proteins and the difficulty in predicting non-coding RNA antitoxins. Successful methodologies employ specialized search parameters based on characteristic genomic features:
Tandem Repeat Detection: Identification of short open reading frames (ORFs) encoding hydrophobic proteins repeated in tandem, as many Type I loci are duplicated in intergenic regions [29]. Implementation requires in-house Perl scripts or similar pipelines to scan intergenic regions extended by 30 nucleotides into adjacent coding regions to account for potential annotation errors.
Hydrophobicity Analysis: Detection of potential transmembrane regions using two complementary approaches: TMHMM prediction for proteins >50 amino acids, and identification of at least one 15-amino-acid stretch with minimum 10 hydrophobic residues (I, V, L, F, C, M, A) for smaller proteins [29].
RNA Folding Energy Profiling: Utilization of local free-energy minima of RNA folding to detect positions of sRNA genes, leveraging the complex secondary structures characteristic of Type I antitoxin RNAs [29].
Standardized Detection of Type II Systems
Type II TA systems are more amenable to standard bioinformatic detection due to their proteinaceous nature and operonic organization:
Homology-Based Searches: Exhaustive PSI-BLAST and TBLASTN searches using known Type II toxins as queries with optimized parameters (matrix = PAM70; word size = 2; gap cost = existence 9, extension 2; no low complexity filtering) [29] [3].
Operon Structure Analysis: Identification of bicistronic operons typically featuring an upstream antitoxin gene overlapping by a few bases with the downstream toxin gene, though exceptions exist (e.g., HigA/HigB, MqsR/YgiT) [19].
Domain-Based Prediction: Recognition of conserved structural features including antitoxins with DNA-binding domains and toxin domains classified into superfamilies (ParE/RelE, MazF, HicA, VapC, HipA, FicT/Doc, AtaT/TacT, Zeta, MbcT) [19].
Table 2: Computational Detection Performance Comparison
| Method | Type I Application | Type II Application | Sensitivity | Limitations |
|---|---|---|---|---|
| Tandem repeat detection | High utility | Limited utility | Moderate (Type I) | High false positives in repetitive genomes |
| Transmembrane prediction | Essential | Not primary | High for Type I toxins | Poor performance on very short proteins |
| RNA energy profiling | Critical for antitoxin | Not applicable | Variable | Dependent on algorithm parameters |
| Homology searches | Challenging | Highly effective | High for Type II | Limited to conserved families |
| Operon structure analysis | Limited utility | Highly effective | High for Type II | Misses non-canonical organizations |
| Domain recognition | Limited utility | Highly effective | High for Type II | Requires characterized domains |
Experimental Validation and Functional Characterization
Toxin Antitoxin Interaction Mapping
Understanding the molecular interactions between toxin and antitoxin components requires different experimental approaches for Type I versus Type II systems, as illustrated in the following experimental workflow:
Protocol: Type I Toxin-Antitoxin Interaction Validation
Objective: Confirm functional interaction between Type I toxin mRNA and antisense RNA antitoxin.
Materials:
- Bacterial strains with chromosomal or plasmid-borne Type I TA loci
- GFP reporter vector system
- Primers for toxin gene amplification
- Northern blot reagents or RNA-seq capabilities
- Membrane potential-sensitive dyes (e.g., DiOC₂(3))
Methodology:
- Reporter Construct Design: Clone the toxin gene including its 5' UTR into a GFP reporter vector downstream of an inducible promoter.
- Antitoxin Co-expression: Introduce the antitoxin RNA gene either in trans on a compatible plasmid or monitor endogenous expression.
- Flow Cytometry Analysis: Measure GFP fluorescence in cells with and without antitoxin expression under induced conditions.
- RNA Interaction Validation: Perform Northern blot analysis to detect toxin mRNA and antitoxin RNA, and their interaction complex.
- Toxin Localization: Fractionate cells expressing toxin with and without antitoxin and detect toxin in membrane vs. cytoplasmic fractions.
- Membrane Function Assessment: Measure membrane potential using fluorescent dyes in toxin-overexpressing cells with and without antitoxin.
Expected Results: Successful antitoxin function should show >70% reduction in GFP fluorescence, decreased toxin mRNA levels, altered cellular localization of toxin, and maintained membrane potential compared to toxin-only controls [29] [2].
Protocol: Type II TA Complex Characterization
Objective: Purify and characterize the protein-protein complex between Type II toxin and antitoxin.
Materials:
- Cloning vectors with compatible tags (His-tag, GST-tag)
- E. coli expression strains (e.g., BL21(DE3))
- Affinity chromatography resins (Ni-NTA, glutathione agarose)
- Gel filtration columns
- Proteases (Lon, ClpAP) for degradation assays
Methodology:
- Co-expression Construct Design: Clone toxin and antitoxin genes into compatible expression vectors with different affinity tags.
- Complex Purification: Co-express both components and purify using tandem affinity chromatography.
- Stoichiometry Determination: Analyze purified complex by analytical gel filtration and multi-angle light scattering.
- DNA Binding Assessment: Perform electrophoretic mobility shift assays (EMSA) with predicted operator sequences.
- Stability Profiling: Incubate complex with relevant proteases (Lon, ClpXP) and analyze degradation kinetics by Western blot.
- Functional Assays: Test toxin activity (RNase, gyrase inhibition, etc.) with and without antitoxin using appropriate substrates.
Expected Results: Stable complex formation with defined stoichiometry (typically 2:2 or 2:4 toxin:antitoxin), specific DNA binding to operator sequences, selective antitoxin degradation by proteases, and complete inhibition of toxin activity in complexed form [19] [4].
Structural Analysis Techniques
Structural characterization provides critical insights into the molecular mechanisms of TA systems, with different technical requirements for Type I versus Type II systems.
Type I System Structural Challenges
Type I systems present particular difficulties for structural biology due to the membrane-associated nature of toxins and the RNA-based regulation. Successful approaches include:
- RNA Structure Probing: Use of selective 2'-hydroxyl acylation analyzed by primer extension (SHAPE) to determine secondary structure of antitoxin RNAs.
- Membrane Protein Crystallography: Extraction of toxin proteins with mild detergents for crystallization trials, though success rates remain low.
- Cryo-Electron Tomography: Direct visualization of toxin-induced membrane disruptions in cellular contexts.
Type II System Structural Advances
Type II systems have been more amenable to high-resolution structural analysis, with numerous complexes solved by X-ray crystallography and cryo-EM:
- Complex Crystallization: Co-crystallization of toxin-antitoxin complexes has revealed diverse interaction modes, from direct active site occlusion to allosteric inhibition [19].
- Molecular Dynamics: Simulation of antitoxin degradation and toxin release processes using structures of complexes and isolated toxins.
- Mutation Analysis: Structure-function studies through site-directed mutagenesis of key residues in both toxins and antitoxins.
Table 3: Structural Biology Method Performance for TA Systems
| Technique | Type I Applicability | Type II Applicability | Resolution | Key Insights |
|---|---|---|---|---|
| X-ray crystallography | Limited (membrane proteins) | High (soluble complexes) | Atomic (1.5-3.0Å) | Precise interaction interfaces |
| Cryo-EM | Moderate (cellular context) | High (complexes) | Near-atomic (3-4Å) | Flexible regions, larger assemblies |
| NMR spectroscopy | Limited (size constraints) | Moderate (domains) | Atomic | Dynamics, solution behavior |
| SHAPE RNA analysis | High (antitoxin RNAs) | Not applicable | Secondary structure | RNA folding, interaction sites |
| Cross-linking MS | Moderate (in vivo applications) | High (complex mapping) | Low resolution | Interaction networks, proximities |
| SAXS | Moderate (RNA structures) | High (solution structures) | Low resolution | Overall shape, conformational changes |
Functional Assessment in Biological Contexts
Protocol: Intracellular Persistence Assay
Objective: Evaluate the contribution of TA systems to bacterial persistence in infection models.
Materials:
- S. Typhimurium or E. coli strains with TA deletions/complementations
- Eukaryotic cell lines (epithelial cells, fibroblasts, macrophages)
- Gentamicin protection assay reagents
- RNA-seq or proteomic capabilities
Methodology:
- Bacterial Strain Construction: Create deletion mutants of specific TA modules (e.g., ΔldrAST, ΔtisBST for Type I; ΔvapC2ST for Type II) in relevant pathogens.
- Eukaryotic Cell Infection: Infect different cell types (epithelial cells, fibroblasts) at optimized MOI.
- Intracellular Survival Quantification: Perform gentamicin protection assays at multiple time points (2, 24, 48 hours).
- Transcriptomic/Proteomic Analysis: Isolve RNA or proteins from intracellular bacteria and analyze TA component expression.
- Complementation Tests: Reintroduce TA modules on plasmids to verify phenotype restoration.
Expected Results: Type I toxins (HokST, LdrAST, TisBST) and Type II toxins (T4ST, VapC2ST) promote bacterial survival inside fibroblasts, while only selective Type II toxins (VapC2ST) enhance fitness in epithelial cells, demonstrating host cell-specific functional specialization [2].
Research Reagent Solutions for TA System Characterization
Table 4: Essential Research Reagents for TA System Investigation
| Reagent Category | Specific Examples | Application | Type Preference |
|---|---|---|---|
| Cloning Systems | pET Duet, pCDF Duet vectors | Co-expression of toxin-antitoxin pairs | Type II |
| Affinity Tags | His-tag, GST-tag, MBP-tag | Protein purification and detection | Type II |
| RNA Detection Tools | Northern blot reagents, RNA-seq kits | Antitoxin RNA quantification | Type I |
| Membrane Assays | DiOC₂(3), NPN uptake | Toxin-induced membrane damage | Type I |
| Protein Interaction | Co-IP kits, Y2H systems, SPR chips | Toxin-antitoxin binding studies | Type II |
| Antibiotics | Gentamicin, ampicillin | Persistence and protection assays | Both |
| Protease Systems | Lon, ClpXP proteases | Antitoxin degradation studies | Type II |
| Structural Biology | Crystallization screens, cryo-EM grids | High-resolution structure determination | Type II |
The experimental characterization of TA system components requires carefully tailored approaches based on system type. Type I TA systems demand specialized computational prediction of short hydrophobic ORFs, RNA-focused methodologies for antitoxin detection, and membrane-associated functional assays. In contrast, Type II systems are more amenable to standard molecular biology techniques, including homology-based identification, protein interaction studies, and structural biology approaches.
The choice of experimental strategy should be guided by the specific research questions, available resources, and technical expertise. For comprehensive understanding, integration of multiple complementary approaches is often necessary to overcome the limitations of individual techniques. This comparative guide provides a framework for selecting appropriate methodologies to advance our understanding of these important genetic regulatory systems in prokaryotic biology.
Exploiting TA Systems for Plasmid Maintenance and Biotechnological Design
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in bacteria and archaea, composed of a stable toxin and a corresponding unstable antitoxin. These systems are strategically located on mobile genetic elements, including plasmids, as well as on bacterial chromosomes. Originally discovered as addiction modules that promote plasmid stability through post-segregational killing (PSK) of plasmid-free daughter cells, TA systems are now exploited as powerful biotechnological tools for plasmid maintenance in recombinant DNA technology and synthetic biology [30] [7] [14]. Their functional classification is based on the nature and mechanism of the antitoxin; this guide focuses on comparing the mechanisms and applications of Type I and Type II systems, the two most prominent classes utilized in biotechnology. Type I systems employ antisense RNA antitoxins that inhibit toxin mRNA translation, while Type II systems utilize protein antitoxins that directly bind and neutralize protein toxins [8] [7] [14]. Understanding their distinct characteristics is essential for selecting the appropriate system for specific biotechnological or research applications.
Comparative Analysis of Type I vs. Type II TA Systems
The fundamental distinction between Type I and Type II TA systems lies in the molecular nature of the antitoxin and its mechanism of toxin neutralization. The following table provides a structured, point-by-point comparison of their core characteristics.
Table 1: Essential Characteristics of Type I and Type II TA Systems
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Non-coding RNA (antisense RNA) [8] [14] | Protein [7] [14] |
| Toxin Neutralization Mechanism | Base-pairing with toxin mRNA; inhibits translation and/or promotes degradation [8] [14] | Protein-protein interaction; forms a complex that physically sequesters the toxin [7] [14] |
| Primary Toxic Effect | Membrane damage, loss of membrane potential, and/or cell filamentation [31] [8] | Inhibition of vital processes (e.g., translation, DNA replication) via enzymatic activity (e.g., RNase, gyrase inhibition) [7] [14] [32] |
| Common Toxin Examples | Hok, Fst, TisB, LdrD [31] [8] [14] | CcdB, MazF, VapC, RelE, HicA [31] [7] [14] |
| Typical Application | Plasmid maintenance, gene regulation studies [8] [14] | Plasmid maintenance (dominant use), counterselection in cloning, synthetic biology circuits [33] [30] [34] |
| Key Advantage | Simple genetic architecture; tight translational control [8] | Well-characterized protein components; versatility in design and engineering [33] [35] |
| Key Disadvantage | Hydrophobic toxin nature can make analysis difficult; less commonly used in commercial kits [8] [14] | Potential for cross-talk between homologous systems from different genetic elements [30] |
Mechanism of Action for Plasmid Maintenance
Both system types enforce plasmid maintenance via Post-Segregational Killing (PSK). The antitoxin is labile (degraded by nucleases for Type I RNA antitoxins or proteases for Type II protein antitoxins), while the toxin is highly stable [30] [7] [14].
In a plasmid-containing cell, continuous synthesis of the toxin and antitoxin allows the antitoxin to neutralize the toxin. After cell division, a daughter cell that fails to inherit the plasmid cannot synthesize new antitoxin or toxin molecules. The pre-existing, unstable antitoxin is rapidly degraded, allowing the stable toxin to persist and exert its lethal effect, killing the plasmid-free cell [7] [14]. The following diagram illustrates this general mechanism and highlights the key mechanistic differences between Type I and Type II systems.
Figure 1: General mechanism of Post-Segregational Killing (PSK) by TA systems. The diagram contrasts the specific regulatory pathways of Type I (RNA antitoxin) and Type II (protein antitoxin) systems in plasmid-free daughter cells, leading to cell death.
Quantitative Data on TA System Performance
The efficacy of TA systems in stabilizing plasmids and maintaining foreign genes can be quantified. Performance is often measured by plasmid retention rates over multiple generations without selection and the expression level of genes carried on the stabilized plasmid. The following table summarizes experimental data from key studies.
Table 2: Quantitative Performance of Selected TA Systems in Experimental Applications
| TA System | Type | Host Organism | Key Experimental Finding | Reported Efficacy/Quantitative Data |
|---|---|---|---|---|
| Hok/Sok [8] [14] | I | E. coli | Plasmid stabilization via PSK. | Significant reduction in plasmid loss; one of the first and most studied PSK systems. |
| CcdB/A [33] [7] [14] | II | E. coli | Positive-selection vector; kills unmodified cloning strains. | Near 100% efficiency in selecting for recombinant plasmids in commercial kits (e.g., TOPO, Gateway). |
| MazF/EF [33] [32] | II | E. coli, Plants | Used in plant biotechnology as a molecular switch; induced necrosis upon viral protease activation. | Effective containment of potyvirus (Plum Pox Virus) spread in infected plants. |
| SezAT [7] | II | Streptococcus suis | Stabilizes the SsPI-1 pathogenicity island. | Demonstrated significantly improved stability of the genetic element. |
| SgiTA [30] [7] | II | Salmonella spp. | Maintains the SGI1 resistance island. | Prevents loss of the genomic island, especially in the presence of competing IncA/C plasmids. |
Experimental Protocols for Validating TA System Function
To exploit TA systems effectively, standardized protocols are needed to confirm their functionality, particularly the toxicity of the toxin and the neutralizing capacity of the antitoxin. The following is a core methodology applicable to both Type I and Type II systems, with type-specific considerations.
Core Protocol: Toxicity and Antitoxin Neutralization Assay
This protocol outlines the key steps for validating a TA system in vivo using inducible expression vectors [10] [7].
1. Principle: The toxin gene is cloned into an inducible expression vector. Its expression should inhibit bacterial growth or cause cell death. The antitoxin gene is then provided in trans (on the same or a compatible vector); co-expression should rescue cell growth by neutralizing the toxin [10].
2. Reagents and Materials:
- Strains: Recombinant cloning strain (e.g., E. coli DH5α, BL21).
- Vectors: Plasmid with inducible promoter (e.g., pBAD, pTet, pTac).
- Growth Media: LB broth and agar plates with appropriate antibiotics.
- Inducer: Specific to the promoter (e.g., Anhydrotetracycline (ATc) for pTet, L-Arabinose for pBAD).
- Equipment: Spectrophotometer, incubator, colony counter.
3. Procedure:
A. Toxin Cloning and Induction:
- Clone the toxin gene open reading frame (ORF) into the multiple cloning site of the inducible vector to create plasmid pT.
- Transform pT into the expression host strain.
- Grow transformed cells to mid-log phase and split the culture.
- Induce toxin expression in one culture with the appropriate inducer; the other remains an uninduced control.
- Monitor optical density (OD600) over 4-8 hours. Toxicity is confirmed by a cessation of growth or a decrease in OD in the induced culture compared to the control [10].
4. Type-Specific Modifications:
- For Type I Systems: The antitoxin is an RNA. The
pAconstruct must contain the antitoxin gene under its native promoter to ensure correct synthesis of the antisense RNA. Verification often includes Northern blotting to detect the antitoxin RNA [8] [14]. - For Type II Systems: Both components are proteins. The
pAconstruct can place the antitoxin ORF under an inducible or constitutive promoter. Neutralization can be further verified by co-purification assays to demonstrate a physical protein-protein interaction [7].
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and materials required for the experimental validation and application of TA systems, based on the protocols and studies cited.
Table 3: Essential Research Reagents for TA System Work
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Inducible Expression Vectors | To control the expression of toxin and antitoxin genes for functional testing. | pBAD (arabinose-inducible), pTet (tetracycline-inducible) [10] |
| Conditional Suicide Vectors | Plasmids designed for positive selection of recombinant clones via TA system counterselection. | Vectors containing the ccdB gene (e.g., pSC-B, pDONR, pTOPO) [33] [14] |
| Chemically Competent Cells | Specialized E. coli strains for transformation, often engineered for specific TA applications. | DB3.1/E. coli gyrA mutants (resistant to CcdB toxin); standard cloning strains (killed by CcdB without insert) [14] |
| Specific Inducers | To precisely trigger the expression of genes under inducible promoters. | Anhydrotetracycline (ATc), L-Arabinose, Isopropyl β-d-1-thiogalactopyranoside (IPTG) [10] |
| Toxin-Specific Resistant Strains | Essential for propagating plasmids carrying active toxin genes. | E. coli gyrase strain (for CcdB); strains with mutated toxin targets for other systems. |
Type I and Type II TA systems offer powerful, genetically encoded solutions for enforcing plasmid stability and enabling advanced biotechnological applications. While Type II systems (particularly CcdB/A) currently dominate commercial molecular cloning kits due to their well-characterized protein components and high efficiency, Type I systems represent a versatile tool with significant potential for gene regulation and synthetic biology. The choice between them hinges on the specific requirements of the experiment. Type II systems are generally preferred for robust counterselection and plasmid addiction, whereas Type I systems offer finer, RNA-level translational control. As research continues to elucidate the intricate biology and expand the synthetic biology toolkit, the strategic exploitation of both TA system types will undoubtedly lead to more sophisticated and reliable genetic engineering technologies.
TA Systems as Targets for Novel Antibacterial Drug Development
The escalating global antibiotic resistance crisis necessitates the identification of novel targets for antimicrobial agents [36] [37]. Bacterial toxin-antitoxin (TA) systems, small genetic modules composed of a stable toxin and a labile antitoxin, have emerged as promising candidates for next-generation antibacterial strategies [37]. These systems are ubiquitous in prokaryotes and are classified into types based on the nature and mode of action of the antitoxin. Among these, type I and type II TA systems represent the most prevalent and well-studied classes, each with distinct mechanisms and therapeutic potential [1]. Type I systems feature protein toxins regulated by antisense RNA antitoxins, while type II systems consist of protein toxins neutralized by protein antitoxins [1] [19]. The exploitation of these systems aims to artificially activate the latent "molecular bombs" within bacterial cells, leading to targeted bacterial cell death or growth arrest [36] [38]. This review comprehensively compares the therapeutic potential of type I versus type II TA systems, evaluating their distribution, mechanisms, and tractability for drug development, supported by experimental data and protocols.
Comparative Analysis of Type I and Type II TA Systems
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Toxin Nature | Protein (typically small, hydrophobic) [1] [39] | Protein (diverse enzymatic activities) [19] [37] |
| Antitoxin Nature | Antisense RNA [1] [19] | Protein [36] [19] |
| Primary Regulation | Post-transcriptional, via mRNA binding and degradation [1] | Transcriptional repression & post-translational complex formation [19] [37] |
| Common Toxin Targets | Cell membrane integrity [1] [39] | mRNA translation, DNA replication, cell wall synthesis [19] [37] |
| Distribution | Often narrow, phyla-specific [1] [2] | Broad, across many phyla and mobile elements [1] [37] |
| Representative Toxins | Hok, Fst, ShoB, TisB, Ldr [1] [39] | MazF, RelE, VapC, CcdB, HipA [19] [37] |
Table 2: Therapeutic Potential for Antibacterial Drug Development
| Aspect | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Therapeutic Strategy | Exogenous delivery of toxin-derived peptides [39] | Small molecules that disrupt TA complexes or induce antitoxin degradation [38] [37] |
| Spectrum of Action | Potentially narrow, due to specific targeting and distribution [1] [28] | Could be broad or narrow, depending on the targeted system's conservation [36] [37] |
| Stage of Development | Early-stage (peptide design and in vitro validation) [39] | Early-stage (target validation and screening) [38] [37] |
| Key Challenges | Peptide solubility, delivery, and stability [39] | Specificity, potential for persistence induction, and complex regulation [36] [38] |
| Notable In Vitro Results | ShoB-derived peptides show MICs of 8–32 µM against a bacterial panel [39] | Artificial activation of YoeB and MazF toxins leads to cell lethality [38] |
Mechanisms of Action and Intracellular Regulation
The fundamental distinction between type I and type II TA systems lies in their regulatory mechanisms, which directly inform different therapeutic strategies.
Type I System Mechanisms
In type I systems, the toxin protein is encoded by an mRNA that is constitutively expressed but typically not translated due to the simultaneous expression of a complementary antisense RNA antitoxin. This antitoxin RNA binds to the toxin mRNA, preventing its translation and/or promoting its degradation [1]. Under conditions of stress or plasmid loss, the unstable antisense RNA degrades rapidly, allowing the stable toxin mRNA to be translated into a protein that often integrates into the cytoplasmic membrane, disrupting membrane potential and integrity [1] [39]. For example, the ShoB toxin in E. coli is a small hydrophobic peptide that exerts its bactericidal effect through membrane disruption [39].
Type II System Mechanisms
In type II systems, both the toxin and antitoxin are proteins encoded by a single operon. The antitoxin protein binds directly to the toxin protein, forming an inert complex that neutralizes the toxin's activity [19]. This complex also acts as a transcriptional repressor for its own operon. The antitoxin is inherently unstable and is selectively degraded by host proteases (e.g., Lon, Clp) under stress conditions. This degradation liberates the more stable toxin, which then acts on its specific cellular target [38] [19]. Type II toxins exhibit a diverse array of activities, including:
- mRNA cleavage (e.g., MazF, RelE) [19]
- DNA gyrase inhibition (e.g., CcdB, ParE) [19]
- Phosphorylation of translation factors (e.g., HipA, Doc) [19]
- NAD+ hydrolysis (e.g., MbcT) [19]
The following diagram illustrates the core regulatory workflows of both system types, highlighting key intervention points for therapeutic agents.
Diagram 1: Regulatory workflows and therapeutic intervention points in Type I and Type II TA systems.
Experimental Approaches and Research Protocols
Protocol 1: Engineering Type I Toxin-Derived Antimicrobial Peptides
A pioneering approach for harnessing type I TA systems involves the rational design of synthetic antimicrobial peptides (AMPs) based on native toxin sequences, as demonstrated with the ShoB toxin from E. coli [39]. The native ShoB peptide is highly hydrophobic and insoluble, necessitating modification for exogenous delivery.
- Step 1: Solubility Engineering. The native peptide sequence is modified by inserting multiple lysine (K) residues into the hydrophobic core and replacing cysteine with glycine to prevent dimerization. This results in a soluble analog (e.g., Peptide 1a) with reduced amphipathicity but retained α-helical structure, as confirmed by tools like AlphaFold [39].
- Step 2: Truncation Analysis. To identify the minimal active sequence, the toxin gene is truncated from either the N- or C-terminus within an expression plasmid (e.g., pET28b). Toxicity is assessed by inducing expression and measuring bacterial survival. For ShoB, the N-terminal part was more tolerant to truncation than the C-terminal core [39].
- Step 3: In Vitro Efficacy Testing. Synthetic peptides corresponding to the truncated analogs are chemically synthesized. The Minimal Inhibitory Concentration (MIC) and Minimal Bactericidal Concentration (MBC) are determined against a panel of bacterial pathogens using standard broth microdilution assays [39].
- Step 4: Resistance Propensity Assessment. The frequency of resistance development is evaluated by plating high-density bacterial cultures onto agar containing a concentration of the peptide at 4x MIC and counting the resulting colonies after incubation [39].
Protocol 2: Artificially Activating Type II TA Systems
The primary strategy for type II systems is to identify small molecules that trigger toxin activation within the bacterial cell [38] [37].
- Step 1: Target Selection and Validation. Prevalent and functional type II TA systems in clinical isolates of the target pathogen are identified via PCR and transcriptomic analyses (e.g., RT-PCR). Systems like mazEF and axe-txe are widespread in pathogens like Vancomycin-Resistant Enterococci (VRE) [38].
- Step 2: High-Throughput Screening (HTS). A library of small molecules is screened using a reporter strain where the promoter of a TA-controlled gene is fused to a detectable marker (e.g., GFP). Alternatively, a bacterial growth inhibition assay can be used to identify compounds that induce bacteriostasis [38] [37].
- Step 3: Mechanistic Validation. Hit compounds are tested for their specific mode of action:
- TA Complex Disruption: Using techniques like Surface Plasmon Resonance (SPR) or Isothermal Titration Calorimetry (ITC) to assess if the compound interferes with toxin-antitoxin binding [37].
- Antitoxin Degradation: Immunoblotting to monitor antitoxin protein levels in treated cells to determine if the compound accelerates proteolytic degradation [38].
- Transcription Repression: Electrophoretic Mobility Shift Assays (EMSAs) to test if the compound prevents the TA complex from binding its operator DNA [38].
- Step 4: Phenotypic Confirmation. The ability of the compound to induce known toxin phenotypes (e.g., mRNA cleavage for RNase toxins) is confirmed. Specificity is checked using mutant strains lacking the target TA system [38].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for TA System Research and Development
| Reagent / Solution | Function in Research | Example Application |
|---|---|---|
| Lon Protease Activators | Indirectly activates type II toxins by accelerating antitoxin degradation [38]. | Validating type II TA activation as a bactericidal strategy [38]. |
| Profile HMMs (Hidden Markov Models) | Bioinformatics tool for identifying remote TA homologs in genomic data [1]. | Discovering novel TA families and assessing their distribution across species and plasmids [1]. |
| Specialized Databases (TADB, TASmania) | Curated repositories of known and predicted TA systems [37]. | Identifying all putative TA modules in a pathogen's genome for target prioritization [2] [37]. |
| Inducible Expression Plasmids (e.g., pET28b) | Allows controlled overexpression of toxin genes for functional characterization [39]. | Assessing toxicity of wild-type vs. truncated toxin variants in vivo [39]. |
| Synthetic Antimicrobial Peptides | Exogenously applied derivatives of type I toxins [39]. | Testing direct bactericidal activity and spectrum of action of engineered peptides [39]. |
Research Implications and Future Directions
The strategic comparison of type I and type II TA systems reveals distinct and complementary paths for antibacterial development. Type II systems offer the advantage of a vast, diverse, and well-cataloged set of potential protein targets against which small molecule libraries can be screened [36] [37]. The major challenge lies in achieving sufficient specificity to avoid off-target effects and in managing potential consequences like persister cell formation, which is linked to some type II systems [36] [38]. In contrast, type I systems provide a novel source for engineering peptide antibiotics. The recent success in designing soluble, potent derivatives of the ShoB toxin demonstrates the feasibility of this approach, which may offer a lower propensity for resistance [39]. A crucial future direction will be to explore the interconnected regulatory networks between different TA systems within a single bacterial cell, as cross-regulation could impact the efficacy and consequences of targeting a single TA module [40]. Furthermore, the clinical translation of both strategies will require extensive in vivo validation to address challenges such as peptide stability and delivery for type I-based therapeutics, and the potential for unintended persistence or biofilm formation with type II-targeting compounds [36] [39] [37]. Ultimately, both types of TA systems represent promising components of the broader arsenal needed to overcome antibiotic-resistant infections.
Harnessing Phage Inhibition for Antimicrobial Strategies
Toxin-antitoxin (TA) systems are genetic modules ubiquitous in bacterial genomes, composed of a stable toxin that disrupt essential cellular processes and a labile antitoxin that counteracts the toxin's effect [41]. These systems function as sophisticated stress-response circuits and have been implicated in bacterial survival strategies, including the formation of dormant persister cells and, more recently, defense against bacteriophage (phage) predation [42] [10]. The latter function is of paramount importance in the context of harnessing phage inhibition for antimicrobial strategies. When a bacterium is infected by a phage, the resulting cellular stress can trigger the release of the TA system's toxin, leading to the premature death of the infected cell and thereby aborting the phage replication cycle to protect the bacterial population at large [41].
Among the diverse classifications of TA systems, Types I and II represent two distinct and prevalent mechanistic paradigms for regulating bacterial toxicity. A comparative analysis of their structures and functions is essential for leveraging their phage-inhibitory potential. Type I systems rely on an antisense RNA antitoxin that binds the toxin mRNA, preventing its translation and/or promoting its degradation [10] [41]. In contrast, Type II systems feature a proteinaceous antitoxin that directly binds and neutralizes the protein toxin [42] [41]. This fundamental distinction dictates their operational logic, regulatory dynamics, and, consequently, their suitability for different antimicrobial applications. This guide provides a comparative overview of Type I and Type II TA system research, detailing experimental protocols and key reagents to equip scientists with the tools for advanced antimicrobial development.
Comparative Mechanisms of Type I and Type II TA Systems
The following table summarizes the core characteristics that differentiate Type I and Type II TA systems, which are critical for understanding their unique roles in bacterial physiology and phage defense.
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Antisense RNA [10] [41] | Protein [42] [41] |
| Toxin Regulation | Post-transcriptional; mRNA translation inhibition and/or degradation [10] [41] | Post-translational; toxin protein sequestration and inactivation [42] [41] |
| Primary Functional Context | Stabilization of mobile genetic elements (e.g., prophages) [10] | Stress response, biofilm formation, phage defense, and persistence [42] |
| Common Toxin Targets | Cell membrane integrity; potential disruption of energy metabolism [10] | Protein synthesis (e.g., ribosomes), DNA replication (e.g., gyrase) [42] [41] |
| Regulatory Complexity | Controlled by small RNA interactions and often associated with riboswitches [10] | Often autoregulated at transcriptional level by the toxin-antitoxin complex [41] |
| Potential for Antimicrobial Exploitation | High (toxins are small, membrane-targeting peptides) [10] | High (toxins target essential cellular machinery) [42] [41] |
The mechanistic relationship between these components and the phage infection cycle is visually summarized in the following pathway diagram.
Phage Inhibition via TA Systems
Experimental Protocols for Analyzing TA System Function in Phage Defense
Protocol 1: Validating Functional TA Pairs via Inducible Expression
This foundational protocol is used to confirm the toxicity of a putative toxin gene and the neutralizing capability of its cognate antitoxin [10].
- Step 1: Cloning. Clone the toxin gene alone (pT) and the toxin-antitoxin pair together (pTA) into separate expression vectors under the control of an inducible promoter (e.g., anhydrotetracycline (ATc)-inducible Ptet).
- Step 2: Transformation. Introduce the constructed plasmids (pT and pTA) and an empty vector control (p) into the host bacterial strain (e.g., Clostridioides difficile 630Δerm).
- Step 3: Growth Assay on Solid Media. Plate the transformed strains on culture media (e.g., BHI plates) with and without the inducer (ATc). Incubate and observe growth after 24-48 hours.
- Step 4: Data Interpretation. Functional toxins will cause complete growth inhibition on plates containing the inducer for strains carrying pT. This growth defect is reverted in strains carrying pTA, demonstrating the antitoxin's neutralizing function. Growth should be normal in all strains without the inducer [10].
Protocol 2: Assessing TA System-Mediated Phage Inhibition
This protocol evaluates the role of a TA system in conferring resistance to phage infection.
- Step 1: Bacterial Strain Preparation. Create a set of isogenic bacterial strains: a wild-type strain, a mutant strain with the TA locus deleted (ΔTA), and a complemented mutant strain where the TA system is reintroduced on a plasmid.
- Step 2: Phage Challenge in Liquid Culture. Grow the bacterial strains to mid-exponential phase. Infect the cultures with a defined quantity of phage, expressed as a Multiplicity of Infection (MOI). An uninfected control for each strain is essential.
- Step 3: Monitoring Bacterial Growth. Monitor bacterial growth for 15-30 hours by measuring the optical density at 600 nm (OD600). The "Suppression Index" (percentage of growth inhibition caused by the phage) can be calculated from this data [43].
- Step 4: Phage Re-challenge & Resistance Index. After the initial phage exposure, harvest the cultures that show regrowth (indicating potential resistance). Re-challenge these bacterial populations with the same phage. The "Resistance Index" (percentage of growth upon re-challenge) quantifies the level of constitutive phage resistance that has developed [43].
- Step 5: Data Interpretation. A higher Suppression Index and a lower Resistance Index in the wild-type and complemented strains, compared to the ΔTA mutant, indicate that the TA system contributes to phage defense and limits the emergence of phage-resistant clones.
The Scientist's Toolkit: Essential Research Reagents
The table below catalogues key reagents and their applications for conducting research on TA systems and phage biology.
| Reagent / Solution | Function / Application |
|---|---|
| ATc-Inducible Expression System (e.g., pRPF185-derivatives) | Allows for controlled, titratable overexpression of toxin genes and TA pairs to study their effects and interactions [10]. |
| Defined Phage Stocks (e.g., DMS3vir for P. aeruginosa) | Essential for standardized phage challenge experiments to assess the phage-inhibitory role of TA systems [43]. |
| Custom Gene Deletion Mutants (e.g., ΔTA strains) | Isogenic bacterial strains lacking specific TA loci are critical for pinpointing the function of a TA system via comparative phenotyping [43]. |
| Phage-Complementarity Group (CG) Panels | Pre-defined groups of phages that use the same bacterial receptor; used to map bacterial resistance mechanisms and identify receptors targeted by TA systems [43]. |
| RNA-Sequencing & Transcriptomic Kits | For genome-wide mapping of transcription start sites (TSS) and identifying transcripts for putative antitoxin RNAs and toxin genes [10]. |
The comparative analysis of Type I and Type II TA systems reveals distinct advantages and considerations for antimicrobial strategy development. Type I systems, with their RNA-based regulation and propensity for membrane-targeting toxins, offer a rapid response mechanism that can be leveraged to trigger bacterial cell death directly [10]. Type II systems, with their broad range of essential cellular targets and complex protein-based regulation, provide a versatile and potent means to induce bacteriostasis and abort phage replication [42] [41].
The choice between focusing on Type I or Type II systems for novel antimicrobials depends on the desired outcome. Strategies aiming to directly kill bacterial populations may find Type I toxins particularly useful. In contrast, approaches designed to suppress bacterial growth and potentiate the effects of conventional antibiotics—or to robustly block phage replication in industrial settings—may benefit more from the manipulation of Type II systems. Ultimately, the experimental frameworks and reagents outlined in this guide provide a foundation for the targeted exploitation of these bacterial defense modules, turning the pathogens' own weapons into powerful tools for overcoming antimicrobial resistance.
Engineering TA Systems for Synthetic Gene Circuits and Controllable Cell Death
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in prokaryotes, consisting of a stable toxin that disrupt essential cellular processes and a labile antitoxin that neutralizes the toxin. These systems are classically known for their role in plasmid maintenance and stress response, but have recently emerged as pivotal components in synthetic biology for programming predictable cellular behaviors. For engineers designing synthetic gene circuits, the distinct regulatory mechanisms and performance characteristics of different TA types offer a versatile toolkit. Type I systems employ antisense RNA antitoxins that bind toxin mRNA to inhibit translation, while Type II systems utilize protein antitoxins that directly bind and inhibit protein toxins [1] [7]. This fundamental difference in regulation translates to unique advantages for specific applications, from biocontainment to metabolic control. This guide provides a structured comparison of Type I and Type II TA systems, focusing on their engineering parameters for synthetic circuit design, supported by experimental data and protocols relevant to researchers developing advanced genetic tools and therapeutic interventions.
Systematic Comparison of Type I and Type II TA Systems
Core Mechanisms and Genetic Architecture
The functional divergence between Type I and Type II TA systems stems from their fundamentally distinct architectures and mechanisms of action.
Type I Systems: In these systems, the toxin is a small protein, and the antitoxin is an antisense RNA that binds complementarily to the toxin's mRNA. This binding either occludes the ribosome binding site, preventing translation, or triggers degradation of the mRNA duplex by cellular RNases [1] [10]. The toxins are typically small, hydrophobic proteins that integrate into cellular membranes and cause membrane damage, though exceptions with nuclease activity exist [1]. Gene expression is often tightly controlled, with the toxin and antitoxin RNA transcribed from convergent promoters.
Type II Systems: Here, both the toxin and antitoxin are proteins. The antitoxin protein binds directly to the toxin protein, forming a stable complex that neutralizes the toxin's activity [7]. The toxin-antitoxin complex also typically acts as an autoregulatory repressor of its own operon. Toxins from Type II systems exhibit diverse functions, including targeting DNA replication, translation through RNase activity, and cell wall synthesis [7]. The antitoxin is usually metabolically unstable and degraded by host proteases like Lon, allowing for rapid toxin activation under stress conditions.
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Toxin Nature | Protein (usually small, hydrophobic) | Protein (diverse enzymatic functions) |
| Antitoxin Nature | Antisense RNA | Protein |
| Neutralization Mechanism | RNA-RNA interaction inhibiting translation | Protein-Protein interaction inhibiting activity |
| Autoregulation | Less common; often regulated by host factors | Common; TA complex represses its own promoter |
| Typical Toxin Targets | Membrane integrity [1] | Translation (RNases), DNA replication, cell wall synthesis [7] |
Distribution, Mobility, and Fitness in Genetic Elements
The evolutionary distribution and mobility of TA systems provide insights into their horizontal transfer potential and suitability for different host chassis in synthetic biology.
Bioinformatic surveys reveal that Type II TA families demonstrate broader phylogenetic distribution across bacterial phyla and are more frequently associated with mobile genetic elements (MGEs) like plasmids and phages compared to Type I systems. For instance, families like Doc, MazF/PemK, RelE/ParE, and VapC are found in all analyzed bacterial phyla [1]. In contrast, most Type I toxin families (e.g., Ldr, ShoB, TxpA, TisB) have a narrow, phyla-specific distribution and are often located exclusively on chromosomes [1].
This distribution correlates with functional advantages in plasmid persistence. Recent intracellular competition experiments demonstrate that plasmids equipped with both a partitioning system and a TA system exhibit superior fitness. Notably, a combination system featuring the Type I hok-sok module showed the highest fitness, outperforming plasmids with either system alone [5]. This suggests that while Type II systems are more widespread, certain Type I systems are highly effective for specific engineering goals like plasmid stabilization.
Table 2: Distribution and Mobility of TA Systems
| Parameter | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Phylogenetic Range | Typically narrow (often 1 phylum) [1] | Typically broad (often multiple phyla) [1] |
| Association with Plasmids | Less common (except Fst, Hok, XCV2162) [1] | Very common [1] [7] |
| Association with Prophages/Genomic Islands | Demonstrated (e.g., in C. difficile) [10] | Common [7] |
| Role in Plasmid Fitness | High fitness when combined with partitioning systems [5] | Classical "addiction" systems for plasmid maintenance [7] |
Phenotypic Outcomes and Applications in Biotechnology
Both system types induce potent growth arrest or cell death upon toxin activation, but they can elicit different physiological responses useful for synthetic biology.
Induced Phenotypes: Ectopic expression of Type I toxins (e.g., in C. difficile) leads to rapid growth inhibition and a drop in cell viability, often accompanied by morphological changes like cell elongation [10]. Similarly, Type II toxin expression (e.g., CcdB) inhibits growth by targeting essential processes like DNA gyrase [44]. Both types are implicated in biofilm formation and bacterial persistence, contributing to antibiotic tolerance [27] [44]. For example, the Type II system ccdAB in E. coli was shown to increase biofilm formation and foster persistent cells in models of recurrent urinary tract infection [44].
Synthetic Biology Applications: The compact nature of Type I systems (a short protein and a non-coding RNA) makes them easier to deliver and integrate into complex circuits. Their mechanism is well-suited for post-segregational killing in biocontainment strategies. Type II systems, with their protein-based feedback regulation, are ideal for constructing complex logic gates and fine-tuned stress response circuits [45]. The protein toxins also present targets for developing new antibacterial strategies that artificially activate toxicity [27].
Experimental Protocols for Functional Analysis
Protocol 1: Functional Validation of a TA System via Inducible Expression
This foundational protocol is used to confirm the toxicity of a putative toxin and the neutralizing capability of its cognate antitoxin.
- Cloning: Clone the toxin gene (for Type I or Type II) into an inducible expression plasmid (e.g., pRPF185 with a Ptet promoter for C. difficile [10] or pET28a with an IPTG-inducible system for E. coli [44]). For the antitoxin control, clone the toxin-antitoxin pair together into the same vector, ensuring the antitoxin (RNA or protein) is expressed from its native promoter or a constitutive one.
- Transformation: Introduce the constructed plasmid (pT for toxin-only, pTA for toxin-antitoxin) and an empty vector control into the appropriate host strain (e.g., E. coli BL21(DE3), C. difficile 630Δerm).
- Growth Assay (Solid Media): Plate the transformed strains on solid media with and without the inducer (e.g., anhydrotetracycline (ATc) or IPTG). Incubate and observe growth. A functional toxin will cause growth inhibition only in the pT strain in the presence of the inducer. This inhibition should be rescued in the pTA strain [10].
- Quantitative Growth Assay (Liquid Media): Inoculate liquid cultures of the strains and grow to exponential phase. Split the cultures and add the inducer to half. Monitor growth kinetics (OD600) and colony-forming units (CFUs) over time. Toxin expression should cause an immediate growth arrest and a drop in CFUs in the pT culture only [10].
- Morphological Analysis: Analyze induced liquid cultures by light microscopy. Some toxins, like certain Type I toxins in C. difficile, cause characteristic morphological changes such as significant cell elongation [10].
Protocol 2: Assessing the Role of a TA System in Biofilm Formation
This protocol assesses the impact of a TA system on community-level behavior, relevant for studies on persistence and antimicrobial tolerance.
- Strain Preparation: Create a strain with the toxin gene under inducible control and a control strain with an empty vector or an inactivated toxin.
- Biofilm Cultivation: Grow the strains in appropriate media in static conditions, typically in polystyrene plates or tubes, with and without a sub-lethal concentration of inducer to trigger low-level toxin expression.
- Biofilm Quantification (Crystal Violet Staining)
- After incubation (e.g., 24-48 hours), carefully remove the planktonic cells.
- Stain the adhered biofilm with a crystal violet solution (0.1% w/v) for 10-20 minutes.
- Wash away the excess stain.
- Dissolve the bound crystal violet in a solution of 30% acetic acid in ethanol.
- Transfer the solution to a new plate and measure the absorbance at 595 nm. Higher absorbance indicates more biofilm biomass [44].
- Data Analysis: Compare the biofilm formation of the toxin-induced strain versus the controls. An increase in biofilm biomass upon toxin induction suggests the TA system plays a role in biofilm development.
Visualization of TA System Logic and Experimental Workflows
Regulatory Logic of Type I and Type II TA Systems
Diagram Title: Regulatory Logic of Type I vs. Type II TA Systems
Functional Validation Workflow for TA Systems
Diagram Title: Experimental Workflow for TA System Validation
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for TA System Research and Development
| Reagent / Tool | Function / Application | Examples / Notes |
|---|---|---|
| Inducible Expression Vectors | Controlled expression of toxin or TA pairs for functional validation. | pET28a (IPTG-inducible) [44], pRPF185 (ATc-inducible) [10]. |
| Model Bacterial Strains | Hosts for cloning, toxin testing, and phenotype analysis. | E. coli BL21(DE3) [44], C. difficile 630Δerm [10]. |
| CRISPR-dCas9 Systems | Synthetic transcription platforms for building complex genetic circuits. | dCas9-VPR for strong gene activation in mammalian cells [46]. |
| Profile Hidden Markov Models (HMMs) | Bioinformatics identification of remote TA homologs in genomic data. | Used for discovering novel TA families and assessing distribution [1]. |
| Antibiotics & Selection Markers | Maintenance of plasmids and selection of successful transformants. | Kanamycin (nptII gene), Chloramphenicol (cat gene) [5] [44]. |
The choice between Type I and Type II TA systems for synthetic gene circuits hinges on the specific requirements of the application. Type I systems, with their simple RNA-based regulation and potent membrane-targeting toxins, are excellent for applications demanding compact design and rapid, irreversible action, such as biocontainment. Type II systems, offering tunable protein-based regulation, diverse enzymatic targets, and sophisticated feedback loops, provide a richer toolbox for constructing complex dynamic circuits and studying persistence. Performance data from functional assays and distribution studies provide a quantitative foundation for this selection. As synthetic biology advances, the engineering of hybrid systems that leverage the strengths of both types, perhaps combining the swift response of a Type I toxin with the programmable regulation of a Type II antitoxin, will undoubtedly unlock new frontiers in programmable cell biology and therapeutic development.
Navigating Complexities: Debated Functions and Research Challenges
Resolving Controversies in Chromosomal TA System Functions
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in bacterial chromosomes and mobile genetic elements, composed of a stable toxin and a labile antitoxin that neutralizes it. The primary controversy surrounding chromosomal TA systems stems from their evolutionary origin and primary biological function. Initially discovered as addiction modules on plasmids that ensure their stable inheritance through post-segregational killing (PSK), their presence in bacterial chromosomes in multiple copies has sparked intense debate about whether they serve genuine cellular functions or represent genomic parasites that persist due to their own selfish inheritance mechanisms [47]. While type II TA systems, where both components are proteins, have been more extensively studied, type I systems, featuring an RNA antitoxin that regulates a protein toxin, present distinct functional and evolutionary patterns that contribute uniquely to this ongoing scientific discussion [8].
This guide objectively compares type I and type II TA systems by examining their mechanisms, genomic distribution, and functional evidence, providing researchers with clarified distinctions and methodological approaches to advance the field. The resolution of these controversies requires careful consideration of their disparate characteristics, which we explore through structured comparison and experimental validation.
Comparative Analysis: Type I vs. Type II TA Systems
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Antisense RNA [8] | Protein [47] |
| Regulatory Mechanism | RNA-RNA interaction inhibiting toxin mRNA translation or stability [8] | Protein-protein interaction inhibiting toxin activity; transcriptional auto-repression [47] |
| Toxin Proteins | Typically small, hydrophobic proteins (often <60 amino acids) that damage membranes [1] [8] | Diverse enzymatic activities (RNases, DNases, gyrase inhibitors) [47] |
| Primary Initial Function | Plasmid stabilization via post-segregational killing [8] [10] | Plasmid stabilization via post-segregational killing [47] |
| Chromosomal Distribution | Often narrow, phyla-specific distribution [1] | Broad distribution across multiple phyla [1] |
| Association with Mobile Elements | Less frequently associated; some families completely chromosome-specific [1] | Frequently associated with plasmids, phages, and other mobile genetic elements [1] [47] |
| Evidence for Chromosomal Functions | Prophage stabilization [10], stress response [8] | Persister cell formation, phage defense, stress response [8] [47] |
Table 2: Genomic Distribution and Mobility Patterns
| Distribution Pattern | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Phylogenetic Range | Most families restricted to single phylum (e.g., Ldr, ShoB, Txp, TisB in Proteobacteria) [1] | Most families found across multiple phyla (e.g., Doc, MazF, RelE, VapC in all bacterial phyla) [1] |
| Representation on Mobile Elements | Lower prevalence; only 3 of 9 known families (Fst, Hok, XCV2162) found on mobile elements [1] | High prevalence; most families found on both chromosomes and mobile elements [1] |
| Presence in M. tuberculosis | Not prevalent | 76 type II TA systems in M. tuberculosis H37Rv [47] |
| Example of Narrow vs. Broad Distribution | Ldr, TisB, TxpA: Found in <5% of species within their phylum and no mobile elements [1] | MazF/PemK, RelE/ParE: Found in >70% of species within some phyla and on mobile elements [1] |
Experimental Approaches for Functional Characterization
Validating TA System Functionality
Inducible Expression and Toxin Neutralization Assay This fundamental protocol establishes whether a putative TA pair is functional by demonstrating that toxin expression inhibits growth and that co-expression of the antitoxin reverses this effect.
- Cloning and Transformation: Clone toxin genes alone (pT) and toxin-antitoxin pairs (pTA) into inducible expression vectors. For C. difficile, pRPF185-derivatives with anhydrotetracycline (ATc)-inducible Ptet promoter have been successfully used [10].
- Toxin Induction and Growth Monitoring: Plate transformed bacteria on media with and without inducer (e.g., ATc). Incubate under appropriate conditions.
- Functional Validation: Compare growth of strains carrying pT versus pTA constructs. Functional toxins completely inhibit growth with inducer, while functional antitoxins restore growth in pTA strains [10].
- Additional Phenotypic Characterization: Examine cell morphology by light microscopy. For type I toxins like CD0977.1, approximately 10% of cells exhibit increased length (≥10.5 μm) following toxin induction [10].
Assessing Prophage Stabilization
Prophage Loss Rate Quantification This protocol tests the hypothesis that TA systems stabilize mobile genetic elements in bacterial chromosomes, using prophages as a model system.
- Strain Construction: Create mutant strains with deleted TA loci within prophage regions using allele exchange methods [10].
- Growth and Propagation: Grow TA-deletion and wild-type strains for multiple generations without selective pressure.
- Prophage Loss Quantification: Monitor prophage retention using PCR targeting prophage-specific genes or by scoring phage particle production in culture supernatants [10].
- Statistical Analysis: Compare prophage loss rates between wild-type and TA-deletion strains. Significantly higher loss rates in deletion mutants support a stabilization function. In C. difficile, type I TA systems within phiCD630-1 prophage significantly contribute to its stability and heritability [10].
Experimental Workflow for TA System Validation
Research Reagent Solutions for TA System Investigation
Table 3: Essential Research Reagents and Their Applications
| Reagent / Method | Specific Examples | Function in TA Research |
|---|---|---|
| Inducible Expression Systems | pRPF185-derivatives with Ptet promoter [10] | Controlled toxin expression to demonstrate toxicity and antitoxin neutralization |
| Bioinformatic Tools | Profile hidden Markov models (HMMs), tBlastn [1] [10] | Identification of remote TA homologs, especially small type I toxins |
| Molecular Docking Software | HADDOCK [4] | Predicting TA protein interactions and functional impacts of mutations |
| Allele Exchange Methods | Type I toxin-based counter-selection [10] | Creating TA deletion mutants to study chromosomal functions |
| RNA-Seq Transcriptomics | RNA sequencing, TSS mapping [10] | Identifying antisense RNA antitoxins and expression patterns |
| Structural Prediction Tools | AlphaFold [4] | Modeling 3D structures of toxins and antitoxins for functional analysis |
Mechanisms and Pathways: Visualizing TA System Functions
Regulatory Mechanisms of Type I and Type II TA Systems
Discussion: Integrating Evidence to Resolve Controversies
The comparison between type I and type II TA systems reveals fundamental differences that inform the controversy surrounding their chromosomal functions. Type I systems demonstrate phylum-specific distribution and lower mobility, with many families (Ldr, ShoB, Txp, TisB) found in less than 5% of species within their phylum and completely absent from mobile elements [1]. This restricted distribution suggests these systems may be more integrated with host chromosome biology. In contrast, type II systems show broad phylogenetic distribution and strong association with mobile genetic elements, supporting their characterization as selfish genetic elements [1] [47].
Experimental evidence increasingly supports functional roles for both TA system types in chromosomal contexts. Type I systems contribute to prophage stabilization in C. difficile, with deletion studies demonstrating increased prophage loss rates when TA systems are disrupted [10]. Both type I and type II systems have demonstrated roles in stress response and persister cell formation, though evidence varies by specific system and bacterial species [8] [47]. The dual nature of TA systems—capable of both selfish inheritance and functional integration—may explain the persistent controversies in the field, with different systems following different evolutionary paths.
The resolution of controversies in chromosomal TA system functions requires acknowledging that these systems are not monolithic. Type I and type II TA systems exhibit distinct evolutionary patterns and potential cellular functions. Future research should focus on:
- Standardized experimental approaches across multiple bacterial species
- Comprehensive characterization of both type I and type II systems in the same genetic background
- Investigation of TA system networks rather than individual modules
- Exploration of therapeutic applications targeting TA systems in bacterial pathogens [4]
The continuing investigation of these fascinating genetic elements will undoubtedly yield new insights into bacterial physiology and potential applications for combating antimicrobial resistance.
Differentiating Between Bacteriostatic and Bactericidal Outcomes
Within the broader context of toxin-antitoxin (TA) system research, understanding the distinction between bacteriostatic and bactericidal outcomes is fundamental for both basic bacterial physiology and applied drug development. Bacteriostatic agents inhibit bacterial growth and replication without causing death, while bactericidal agents directly kill bacterial cells [48]. This distinction becomes particularly significant when studying TA systems, as the activation of different toxin types can lead to either outcome, influencing bacterial survival strategies, persistence, and response to environmental stresses. The careful characterization of these outcomes provides critical insights for exploring TA systems as novel antibacterial targets and understanding their role in bacterial pathogenesis and adaptation.
Comparative Analysis of Bacteriostatic and Bactericidal Mechanisms
Defining Characteristics and Outcomes
Table 1: Fundamental Characteristics of Bacteriostatic and Bactericidal Outcomes
| Feature | Bacteriostatic | Bactericidal |
|---|---|---|
| Primary Effect on Bacteria | Reversible growth inhibition and cellular arrest [48] | Irreversible killing and cell death [48] |
| Typical MBC/MIC Ratio | >4 [48] | ≤4 [48] |
| Dynamical Response at sub-MIC | Immediate, dose-dependent reduction in growth rate [48] | Initial growth unaffected, followed by an abrupt growth rate reduction after a dose-dependent time delay [48] |
| Population Recovery | Possible upon antibiotic removal | Not possible |
| Dependence on Host Immunity | High (requires functional immune system for clearance) | Lower (direct killing independent of immune function) |
| Common Antibiotic Examples | Chloramphenicol, Tetracycline, Erythromycin [48] | Ampicillin, Ciprofloxacin, Gentamicin [48] |
| Analogous TA System Action | Growth arrest leading to persistence [49] [50] | Post-segregational killing leading to cell death [14] |
Temporal Dynamics of Bacterial Response
The fundamental difference between bacteriostatic and bactericidal mechanisms is vividly revealed in their temporal dynamics at sub-inhibitory concentrations (sub-MIC). High-resolution growth curve analyses of Escherichia coli exposed to various antibiotics demonstrate two distinct phenotypic responses [48].
Bacteriostatic antibiotics, such as chloramphenicol and tetracycline, cause an immediate, dose-dependent reduction in the initial bacterial growth rate. The bacterial growth curves under these agents are log-linear but with slopes that deviate from the untreated control from the very early time points. Despite the slowed growth, the population typically continues to proliferate until it reaches the carrying capacity of the environment, indicative of a halted but viable state [48].
In contrast, bactericidal antibiotics, including ampicillin and ciprofloxacin, exhibit a more complex "grow-fast-then-crash" dynamic. The initial growth rate of bacteria treated with these cidals remains similar to untreated cells, even at concentrations close to the Minimum Inhibitory Concentration (MIC). However, after a dose-dependent time period (typically 1.5–3 hours), an abrupt slowdown in growth rate occurs. This delayed response suggests a mechanism of damage accumulation eventually reaching a lethal threshold, rather than an immediate metabolic inhibition [48].
Molecular Mechanisms and Cellular Targets
The differential outcomes of bacteriostatic and bactericidal actions stem from their distinct molecular targets and mechanisms, a concept that extends to the function of TA systems.
Bacteriostatic Mechanisms typically involve targeting processes essential for growth but not immediately lethal. Protein synthesis inhibitors like tetracyclines and chloramphenicol bind to ribosomal subunits, preventing translation and thereby halting proliferation without directly destroying the cell [48]. This is analogous to the action of many TA system toxins, such as the MazF toxin in type II TA systems, which is an endoribonuclease that cleaves cellular mRNAs, tRNAs, or rRNAs at specific sequences, leading to a bacteriostatic state that can facilitate persistence under stress [49] [14].
Bactericidal Mechanisms often cause irreversible, lethal damage to critical cellular structures. β-Lactam antibiotics (e.g., ampicillin) inhibit penicillin-binding proteins, disrupting cell wall synthesis and leading to osmotic lysis. Fluoroquinolones (e.g., ciprofloxacin) target DNA gyrase, causing double-stranded DNA breaks [48]. Similarly, the CcdB toxin of the type II F-plasmid TA system acts as a gyrase poison, leading to accumulation of DNA breaks and cell death, exemplifying a bactericidal outcome from a TA system [49] [14].
Experimental Protocols for Differentiating Outcomes
Standard MIC/MBC Determination Protocol
The classic method for differentiating bacteriostatic and bactericidal activity relies on determining the Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC).
Protocol:
- MIC Determination: Prepare a series of antibiotic dilutions in a liquid growth medium (e.g., Mueller-Hinton Broth) in a 96-well plate. Inoculate each well with a standardized bacterial suspension (~5 × 10^5 CFU/mL of E. coli). Incubate for 16-20 hours at 37°C. The MIC is defined as the lowest antibiotic concentration that prevents visible growth [48].
- MBC Determination: From wells showing no visible growth in the MIC assay, subculture a small volume (e.g., 10 μL) onto antibiotic-free solid agar plates. Spread the aliquot and incubate the plates for 18-24 hours. The MBC is the lowest antibiotic concentration that results in at least a 99.9% reduction (a 3-log kill) in the original bacterial inoculum [48].
- Classification: The MBC/MIC ratio is then calculated. An antibiotic is generally classified as bactericidal if the ratio is ≤ 4, and bacteriostatic if the ratio is >4 [48].
High-Resolution Growth Curve Analysis
This protocol provides dynamic, temporal data that reveals the distinct growth patterns of bacteria under bacteriostatic vs. bactericidal stress, as described in Section 2.2.
Protocol:
- Setup: Inoculate a robotic culture system (e.g., a Bioscreen C or similar) containing liquid growth medium with a diluted overnight culture of E. coli (e.g., MG1655 or BW25113) to a low optical density (OD).
- Treatment: Add a range of sub-MIC concentrations of the test antibiotic (bacteriostatic or bactericidal) to the growing cultures. Include an untreated control.
- Monitoring: Incubate the system with continuous shaking and measure the optical density (OD) at high temporal resolution (e.g., every 7-10 minutes) for a period of 10 hours or more.
- Data Analysis: Plot OD versus time to generate growth curves. Analyze the initial growth rates and the time at which any growth rate change occurs.
- Bacteriostatic signature: Immediate, dose-dependent reduction in the initial growth rate slope.
- Bactericidal signature: Initial growth rate similar to the untreated control, followed by an abrupt reduction in growth rate after a dose-dependent time delay [48].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for TA System and Antibiotic Mode-of-Action Research
| Reagent / Material | Function in Research | Specific Application Example |
|---|---|---|
| Anhydrotetracycline (ATc)-inducible Vectors | Controlled, ectopic gene expression allowing for toxin induction without endogenous regulation. | Used to overexpress type I toxin genes (e.g., CD0904.1, CD0956.3) in C. difficile to confirm toxin functionality and observe growth arrest phenotypes [10]. |
| High-Throughput Robotic Culturing Systems | Automated, high-temporal-resolution monitoring of bacterial growth dynamics under various conditions. | Essential for capturing the distinct growth trajectories of E. coli in response to sub-MIC bactericidal vs. bacteriostatic antibiotics [48]. |
| SLING / TADB / TASmania Bioinformatic Tools | In silico prediction and annotation of putative TA system loci in bacterial genomes. | SLING was used to predict 169 toxin and 290 antitoxin groups across 950 E. coli genomes, revealing sequence-type-specific patterns [3]. TADB and TASmania are databases and pipelines for genome-wide identification of TA systems [37]. |
| RNase III | Ribonuclease that cleaves double-stranded RNA. | Key enzyme in the mechanism of some type I TA systems where the antitoxin RNA and toxin mRNA duplex is cleaved, leading to RNA degradation (e.g., txpA-ratA in B. subtilis) [49]. |
| Cellular Proteases (e.g., Lon protease) | ATP-dependent proteases responsible for degrading unstable proteins. | Critical for initiating the TA system response; under stress, these proteases degrade the labile antitoxin protein (in type II systems), freeing the toxin to act on its target [3] [49]. |
The distinction between bacteriostatic and bactericidal outcomes is not merely a taxonomic classification but reflects profound differences in the dynamic interaction between an antibacterial agent and its bacterial target. The "immediate slowdown" induced by bacteriostatic agents contrasts sharply with the "grow-fast-then-crash" response to bactericidal agents, suggesting divergent underlying mechanisms of metabolic inhibition versus cumulative damage [48]. These concepts are directly mirrored in the world of toxin-antitoxin systems, where toxins can induce either a bacteriostatic, persistent state or a bactericidal, lethal outcome, depending on their specific cellular target and mode of action. A deep understanding of these differential outcomes, supported by robust experimental protocols like temporal growth analysis and MIC/MBC determination, is indispensable for researchers aiming to exploit these systems for novel antibacterial strategies, such as the artificial activation of TA toxins to combat multidrug-resistant pathogens [37].
Technical Hurdles in Studying Highly Toxic Proteins and Labile Antitoxins
Toxin-antitoxin (TA) systems are ubiquitous genetic modules in bacteria and archaea, comprising a stable toxin that inhibits essential cellular processes and a labile antitoxin that neutralizes the toxin's activity. These systems are categorized into types based on the nature and mode of action of the antitoxin. Type I systems feature protein toxins regulated by antisense RNA antitoxins that bind toxin mRNA to prevent translation. In contrast, Type II systems consist of both protein toxins and protein antitoxins that form stable complexes, with antitoxins also serving as transcriptional repressors [51] [52]. The distinct molecular characteristics of these systems present unique technical challenges for researchers, requiring specialized methodological approaches for their study. This guide objectively compares the experimental hurdles and solutions for investigating these two predominant TA system classes, providing a structured framework to aid researchers in selecting appropriate methodologies for their specific study objectives.
Fundamental Distinctions Between Type I and Type II TA Systems
The core differences between Type I and Type II TA systems extend beyond their genetic organization to their fundamental biochemical properties, which directly influence the research challenges they present. The table below summarizes the key distinguishing characteristics.
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Toxin Nature | Small hydrophobic proteins (~60 amino acids) [29] | Proteins of varying sizes and folds [7] |
| Antitoxin Nature | Antisense RNA [29] [51] | Protein [7] [51] |
| Neutralization Mechanism | mRNA-RNA interaction blocking translation [29] [52] | Protein-protein complex formation [7] [52] |
| Primary Toxin Targets | Cell membrane integrity [29] [51] | Diverse: translation, DNA replication, cell wall synthesis [7] [51] |
| Genetic Organization | Overlapping or convergent genes [51] | Typically bicistronic operon [52] |
| Regulation | Antitoxin RNA transcription [29] | Transcriptional autorepression by TA complex [53] [52] |
Technical Hurdles and Methodological Comparisons
Challenge 1: Detecting and Characterizing Highly Toxic Proteins
The inherent toxicity of toxin proteins presents a primary challenge for their cloning, expression, and purification. Unregulated expression can kill the host cells, making genetic manipulation difficult.
Table 2: Methodological Approaches for Studying Toxic Proteins
| Methodology | Application in Type I Systems | Application in Type II Systems |
|---|---|---|
| Controlled Expression | Inducible promoters essential [29] | Tightly regulated promoters required (e.g., PBAD) [2] |
| Genetic Manipulation | Toxin genes often unannotated due to small size; specialized bioinformatics needed [29] [2] | Standard molecular techniques applicable, but co-expression with antitoxin necessary [52] |
| Toxin Identification | Bioinformatics searches for short hydrophobic ORFs with transmembrane regions [29] | Homology-based searches (BLAST) with functional domain identification [7] [2] |
| Protein Production | Extreme difficulty due to membrane association and toxicity [29] | Co-purification with antitoxin partner feasible [52] |
| Functional Validation | Toxicity assays upon induced overexpression; membrane depolarization tests [29] | Target-specific assays (e.g., ribonuclease activity gels) [7] |
Challenge 2: Handling Unstable and Degradation-Prone Antitoxins
The labile nature of antitoxins is a core feature of TA system regulation but complicates their experimental characterization.
Type I Antitoxin (RNA) Challenges: The antisense RNA antitoxins in Type I systems are inherently unstable and require specialized techniques for detection. Northern blotting with specific probes is essential to detect these small RNAs and quantify their half-lives. Researchers must also account for the rapid turnover of these regulatory RNAs and their tight coupling to toxin mRNA expression [29]. The overlapping and convergent genetic organization of Type I systems adds complexity to transcriptional analyses [51].
Type II Antitoxin (Protein) Challenges: Protein antitoxins in Type II systems are structurally disordered or partially folded, making them highly susceptible to proteolytic degradation by host proteases like Lon and ClpXP [52]. This necessitates:
- Protease-Deficient Strains: Using bacterial strains lacking key proteases (e.g., Δlon) for antitoxin purification [52].
- Rapid Purification Protocols: Fast protein liquid chromatography (FPLC) at low temperatures with protease inhibitor cocktails.
- Co-expression with Toxin: Co-purifying the antitoxin with its cognate toxin partner, which often stabilizes the antitoxin within a complex [52].
- In vitro Stability Assays: Incubating purified antitoxins with bacterial proteases and monitoring degradation kinetics via Western blotting or mass spectrometry.
Challenge 3: Computational Prediction and Identification
Bioinformatic discovery of TA systems presents distinct challenges for each type, requiring specialized computational pipelines.
Table 3: Computational Approaches for TA System Identification
| Approach | Type I Systems | Type II Systems |
|---|---|---|
| Primary Strategy | Search for short, hydrophobic ORFs in intergenic regions [29] | Homology-based searches (BLAST) using known toxin/antitoxin domains [29] [2] |
| Key Features | Tandem repeats; transmembrane domains; C-terminal clusters of charged residues [29] | Operon structure; DNA-binding domains in antitoxins; toxin functional motifs [52] |
| Sequence Analysis | TBLASTN against genome sequences; manual inspection for characteristic features [29] | PSI-BLAST iterations; cluster analysis of hits [29] |
| Antitoxin Detection | Prediction of antisense RNAs; energy minimization for RNA folding [29] | Identification of palindromic sequences for operator sites [52] |
| Genomic Context | Analysis for tandem duplication and mobile genetic elements [29] | Association with pathogenicity islands, phages, and plasmids [7] [53] |
Challenge 4: Analyzing Molecular Interactions and Complex Formation
Characterizing the specific interactions between toxins and antitoxins is crucial for understanding TA system function and regulation.
Type I Interaction Analysis:
- RNA-RNA Hybridization Studies: Electrophoretic mobility shift assays (EMSAs) and Northern blot analyses to study antisense RNA-mRNA binding kinetics.
- Secondary Structure Prediction: Computational tools (e.g., mfold) to model interaction interfaces between toxin mRNA and antisense RNA [29].
- Mutational Analysis: Introducing mutations in the toxin mRNA leader sequence and assessing antitoxin binding efficiency and toxicity suppression.
Type II Interaction Analysis:
- Protein Complex Characterization: Surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and analytical ultracentrifugation to determine binding affinities and stoichiometries of toxin-antitoxin complexes.
- Crystallography and Cryo-EM: Structural determination of TA complexes to identify critical interaction interfaces and active site blocking mechanisms [52].
- Cross-linking Studies: Chemical cross-linking coupled with mass spectrometry to map interaction domains.
- DNA-Binding Assays: EMSAs to study antitoxin and TA complex binding to operator sequences and transcriptional autorepression [52].
Diagram 1: Experimental workflows for Type I versus Type II TA system analysis
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful investigation of TA systems requires specific reagents and tools to overcome the technical challenges associated with protein toxicity and antitoxin lability.
Table 4: Essential Research Reagents for TA System Studies
| Reagent/Solution | Function/Application | Examples/Specifications |
|---|---|---|
| Tightly-Regulated Expression Vectors | Controlled toxin expression; prevents host cell death | pBAD (arabinose-inducible); pTet (tetracycline-inducible); T7/lac systems [2] |
| Protease-Deficient Strains | Enhances stability of labile protein antitoxins | E. coli Δlon strains; ΔclpXP strains [52] |
| Protease Inhibitor Cocktails | Prevents antitoxin degradation during purification | Commercial mixes targeting Lon, Clp proteases; PMSF; leupeptin |
| Cross-linking Reagents | Stabilizes transient protein interactions for analysis | Formaldehyde; glutaraldehyde; DSS (disuccinimidyl suberate) |
| RNA Stabilization Solutions | Preserves labile antisense RNA antitoxins | RNAlater; TRIzol; DEPC-treated water and equipment |
| Antibodies for Detection | Detects low-abundance toxins and antitoxins | Custom antibodies against toxin/antitoxin proteins; His-tag antibodies for purified proteins |
| Bioinformatics Tools | Identifies putative TA systems in genomes | TADB; RASTA; custom BLAST parameters; RNA folding software [29] [27] |
The methodological landscape for studying toxin-antitoxin systems is characterized by distinct technical hurdles that require specialized approaches for Type I versus Type II systems. Researchers face the dual challenge of managing highly toxic proteins while working with unstable antitoxins that demand rapid processing and specific stabilization strategies. Type I systems necessitate expertise in RNA biology and membrane protein biochemistry, while Type II systems require sophisticated protein interaction analyses and structural biology capabilities. Successful experimental outcomes depend on selecting appropriate expression systems, stabilization methods, and detection techniques tailored to the specific TA system type. As research in this field advances, methodological innovations will continue to emerge, providing new tools to overcome these persistent technical challenges and further our understanding of TA system biology and their potential applications in biotechnology and antimicrobial development.
Evaluating the Role of TA Systems in Persister Cell Formation and Biofilm Development
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in bacterial genomes, typically consisting of a stable toxin that disrupts essential cellular processes and a labile antitoxin that neutralizes the toxin. These systems were initially discovered as plasmid maintenance systems but are now recognized as crucial regulators of bacterial stress responses [7]. The diversity of TA systems is reflected in their classification into multiple types (I-VIII) based on the nature and mechanism of the antitoxin [7]. Among these, type I and type II systems represent distinct evolutionary solutions to bacterial stress management, with type I utilizing RNA antitoxins and type II employing protein antitoxins [2].
This review provides a comprehensive comparison of type I and type II TA systems, focusing on their specialized roles in two critical bacterial survival strategies: biofilm development and persister cell formation. We examine experimental evidence from key pathogens, synthesize quantitative data on their functional contributions, and detail methodologies for investigating these systems. Understanding the distinct mechanisms through which these TA systems operate provides crucial insights for developing novel therapeutic strategies against chronic and persistent bacterial infections.
Comparative Analysis of Type I and Type II TA Systems
Fundamental Structural and Mechanistic Differences
Table 1: Core Characteristics of Type I and Type II TA Systems
| Feature | Type I Systems | Type II Systems |
|---|---|---|
| Toxin nature | Protein | Protein |
| Antitoxin nature | Antisense RNA (types I & III) | Protein |
| Antitoxin mechanism | Binds toxin mRNA (type I) or toxin protein (type III) to block activity | Protein-protein interaction forming TA complex |
| Genetic organization | Typically encoded in operon | Typically encoded in operon |
| Regulatory mechanism | Antitoxin RNA regulates toxin translation or activity | Antitoxin degrades faster than toxin; also acts as transcriptional repressor |
| Primary cellular targets | Membrane integrity, essential cellular processes | Translation (via mRNA cleavage), DNA replication |
| Example toxins | Hok, LdrA, TisB, IbS | MqsR, MazF, RelE, YoeB, VapC |
Type I and type II TA systems employ fundamentally different strategies for toxin regulation. In type I systems, antisense RNA antitoxins either inhibit translation of the toxin mRNA (type I) or directly bind and neutralize the toxin protein (type III) [7]. In contrast, type II systems feature protein antitoxins that form tight complexes with their cognate toxins, blocking their activity through direct protein-protein interactions [54] [7]. Under normal conditions, the antitoxin in type II systems often functions as a transcriptional repressor for the TA operon, while during stress, cellular proteases such as Lon selectively degrade the labile antitoxin, freeing the toxin to act on its cellular targets [55].
The mechanistic differences extend to their activation pathways and cellular targets. Type I toxins frequently target membrane integrity and electrochemical gradients, while type II toxins typically interfere with essential processes like translation through mRNA cleavage [54] [2]. For instance, well-characterized type II toxins including MqsR, MazF, and RelE function as sequence-specific RNases that cleave mRNA at specific recognition sites, leading to rapid inhibition of protein synthesis [54].
Distinct Roles in Biofilm Development
Table 2: Documented Roles of TA Systems in Biofilm Formation
| TA System | Type | Organism | Effect on Biofilm | Proposed Mechanism |
|---|---|---|---|---|
| MqsR/MqsA | Type II | E. coli | Reduces biofilm formation [54] | Regulates motility and autoinducer-2 quorum sensing [54] |
| MazF/MazE | Type II | E. coli | Deletion reduces biofilm [54] | Possible cell lysis provision of extracellular DNA [54] |
| YafQ/DinJ | Type II | E. coli | Deletion reduces biofilm [54] | Possible cell lysis provision of extracellular DNA [54] |
| YpjF/YfjZ | Type II | E. coli | Deletion increases biofilm [54] | Unknown (cryptic prophage origin) |
| Five TA deletions (Δ5) | Type II | E. coli | Decreases biofilm at 8h, increases at 24h [54] | Alters expression of fimbrial genes via YjgK [54] |
| Multiple systems | Type II | K. pneumoniae | Expression upregulated in strong biofilms [55] | Potential stress response and dormancy induction |
Biofilms represent a protected mode of bacterial growth that confers significant resistance to antimicrobial agents and host immune responses. The first TA system linked to biofilm formation was the type II MqsR/MqsA pair in E. coli, identified through transcriptome studies of biofilm cells [54]. Subsequent research has revealed complex, sometimes strain-specific patterns of biofilm regulation by TA systems.
The relationship between TA systems and biofilm development is demonstrated by studies where deletion of five major type II TA systems (MazF/MazE, RelE/RelB, YoeB/YefM, YafQ/DinJ, and ChpB) in E. coli resulted in reduced biofilm formation after 8 hours but increased biofilm after 24 hours [54]. This biphasic effect was linked to altered expression of fimbrial genes through the transcriptional regulator YjgK, highlighting how TA systems can influence biofilm architecture through regulation of adhesion factors [54].
In Klebsiella pneumoniae, clinical isolates that formed strong biofilms showed significantly higher expression of type II TA system genes (relBE, hipBA, vapBC, mazEF) compared to weak biofilm formers [55]. This correlation suggests that TA system activation may be a hallmark of robust biofilm development in pathogens, potentially through induction of dormant subpopulations or provision of extracellular DNA through controlled cell lysis.
Figure 1: Type II TA System Activation Pathway. Under stress conditions, cellular proteases are activated and degrade the labile antitoxin, freeing the toxin to target essential cellular processes. This activity promotes biofilm development and persister cell formation through growth arrest and other regulatory functions.
Contributions to Persister Cell Formation
Persister cells represent a dormant subpopulation that exhibits remarkable tolerance to antibiotic treatment without genetic modification. These cells are particularly abundant in biofilms and are believed to be responsible for chronic and recurrent infections [54] [55]. Both type I and type II TA systems have been implicated in persister formation, though through potentially distinct mechanisms.
The prevailing model suggests that TA systems induce a state of dormancy by targeting essential cellular processes [54] [55]. For type II systems, toxins such as MazF and RelE function as mRNA interferases that cleave cellular transcripts, leading to rapid inhibition of translation and metabolic arrest [54]. This stasis allows bacteria to survive antibiotic exposure, as most antibiotics target actively growing cells.
Evidence from Salmonella Typhimurium indicates specialization among TA systems, with different toxins promoting intracellular survival in specific host cell types [2]. The toxins HokST, LdrAST, and TisBST (type I), along with T4ST and VapC2ST (type II), were found to promote bacterial survival inside fibroblasts, while only VapC2ST provided a fitness advantage in epithelial cells [2]. This suggests that intracellular pathogens may employ distinct TA modules to adapt to varied host microenvironments.
In K. pneumoniae, analysis of persister cells isolated from biofilms revealed significantly increased expression of type II TA systems including relBE2 and vapBC, while relBE1 expression decreased compared to non-persister cells [55]. This differential expression pattern indicates that specific TA systems may be specialized for persister formation, rather than all TA systems contributing equally to this phenotype.
Experimental Approaches and Methodologies
Key Research Reagent Solutions
Table 3: Essential Research Reagents for TA System Investigation
| Reagent/Condition | Function in Research | Application Examples |
|---|---|---|
| LB broth with 1% glucose | Promotes biofilm formation in microtiter assays | K. pneumoniae biofilm assays [55] |
| Crystal violet staining | Quantifies biofilm biomass | Biofilm quantification at 570nm [55] |
| Lon protease | Key activator of type II TA systems | Degrades antitoxins during stress [55] |
| Specific antibiotics | Selective pressure for persister formation | Ciprofloxacin, ceftazidime, amikacin persister isolation [55] |
| qRT-PCR systems | Quantifies TA system gene expression | Analysis of relBE, hipBA, vapBC, mazEF expression [55] |
| Strain Δ5 (E. coli) | Multiple TA deletion strain for functional studies | Investigation of biofilm phenotypes [54] |
Protocol for Evaluating Biofilm Formation and TA System Expression
The following methodology, adapted from studies on K. pneumoniae, provides a standardized approach for investigating the relationship between TA systems and biofilm development [55]:
Biofilm Cultivation: Inoculate 180μL of Luria-Bertani (LB) broth supplemented with 1% glucose in polystyrene microtiter plates with 20μL of overnight bacterial culture. Incubate for 24-48 hours at appropriate growth temperature.
Biofilm Quantification: Remove planktonic cells and wash gently with phosphate-buffered saline. Fix biofilms with methanol and stain with 0.1% crystal violet for 15 minutes. After washing, solubilize bound dye with acetic acid and measure optical density at 570nm.
Biofilm Classification: Calculate the cutoff value (ODc) as three standard deviations above the mean OD of negative control wells. Classify isolates as: non-biofilm formers (OD ≤ ODc), weak (ODc < OD ≤ 2×ODc), moderate (2×ODc < OD ≤ 4×ODc), or strong biofilm formers (4×ODc < OD).
Gene Expression Analysis: Isclude RNA from biofilm and planktonic cells using appropriate extraction kits. Synthesize cDNA and perform quantitative real-time PCR using primers specific for target TA systems (e.g., relBE, hipBA, vapBC, mazEF). Calculate relative expression using the 2^(-ΔΔCT) method with normalization to housekeeping genes.
Persister Cell Isolation: Treat established biofilms with lethal concentrations of antibiotics (e.g., 10×MIC) for 24 hours. Wash and homogenize biofilms, then plate on antibiotic-free media to enumerate surviving persister cells.
Protocol for Intracellular Persistence Assays
For investigating TA system function during host cell infection, as demonstrated in Salmonella studies [2]:
Strain Construction: Generate deletion mutants of specific TA modules using targeted gene replacement. Complement with plasmid-based expression systems for functional rescue experiments.
Eukaryotic Cell Infection: Culture appropriate host cells (e.g., fibroblasts, epithelial cells) and infect with bacterial strains at optimized multiplicity of infection. Allow invasion to proceed for specific duration.
Intracellular Survival Assessment: At designated time points post-infection, wash eukaryotic cells and treat with gentamicin to kill extracellular bacteria. Lyse host cells and plate serial dilutions to quantify intracellular bacteria.
Proteomic Analysis: Recover bacteria from infected host cells and perform proteomic profiling to identify TA toxins produced during intracellular lifestyle.
Statistical Comparison: Compare intracellular survival rates between wild-type and TA deletion mutants to identify TA systems contributing to persistence in specific host cell types.
Figure 2: Experimental Workflow for Analyzing TA Systems in Biofilms and Persister Cells. This methodology enables correlation of TA system gene expression with biofilm formation capacity and persister cell development.
Discussion and Research Implications
The comparative analysis of type I and type II TA systems reveals both specialized functions and overlapping contributions to bacterial persistence mechanisms. While both systems can promote biofilm development and persister cell formation, they achieve these outcomes through distinct molecular mechanisms reflecting their fundamental structural differences.
Type II TA systems appear to play more diverse regulatory roles in biofilm formation, influencing processes ranging from quorum sensing to fimbrial expression [54]. The MqsR/MqsA system represents a paradigm for type II TA functionality, connecting toxin activity with broader regulatory networks through its influence on motility and autoinducer-2 quorum sensing [54]. The demonstrated ability of type II antitoxins to function as transcriptional repressors for their own operons and other genes provides a direct mechanism for influencing biofilm-related functions.
The emerging evidence that TA systems help control bacterial lifestyle inside eukaryotic cells highlights their potential importance in virulence and chronic infections [2]. The specialization of different TA modules for specific host cell types suggests that intracellular pathogens have evolved to deploy these systems strategically to overcome varied host environments. This functional specialization may explain the abundance of TA systems in pathogens like Mycobacterium tuberculosis and Salmonella enterica compared to their non-pathogenic relatives [2].
From a therapeutic perspective, TA systems represent promising targets for combating persistent infections. Strategies that inhibit TA system function could potentially sensitize biofilms to conventional antibiotics or prevent persister cell formation. However, the redundancy of these systems—where deletion of individual TA modules often produces minimal phenotypes due to functional compensation by other systems—presents a significant challenge [54] [7]. The development of broad-spectrum TA inhibitors that target multiple systems simultaneously may be necessary for effective therapeutic intervention.
Future research should focus on elucidating the precise molecular mechanisms through which different TA systems sense specific stress signals and the downstream pathways they regulate. Comparative studies across multiple bacterial species will help distinguish conserved core functions from species-specific adaptations. Additionally, more comprehensive investigations of type I systems are needed to balance the current research emphasis on type II modules. Understanding these sophisticated bacterial regulatory systems will not only advance fundamental knowledge of bacterial physiology but may also yield novel approaches for combating recalcitrant bacterial infections.
Challenges in Establishing Direct Links to Stress Response and Pathogenesis
Toxin-antitoxin (TA) systems are genetic modules ubiquitous in bacteria and archaea, comprising a stable toxin that disrupt essential cellular processes and a labile antitoxin that neutralizes the toxin. These systems are classified into types based on the antitoxin's nature and mode of action, with type I and type II being the most prevalent. While type I systems feature an RNA antitoxin that base-pairs with the toxin's mRNA to prevent translation, type II systems involve a protein antitoxin that forms a complex with the toxin protein. TA systems were initially identified for their role in plasmid maintenance but are now recognized for their potential functions in bacterial stress response, biofilm formation, persistence, and pathogenesis. However, establishing direct causal links between specific TA systems and these phenotypes, particularly stress response and virulence, presents significant challenges for researchers. This review compares the methodologies and evidence for type I versus type II TA systems, highlighting the technical and interpretive hurdles in this dynamic field.
Fundamental Distinctions Between Type I and Type II TA Systems
The core architectural and functional differences between type I and type II TA systems dictate the distinct experimental approaches required to study them. The table below summarizes their key characteristics.
Table 1: Core Characteristics of Type I and Type II TA Systems
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Non-coding antisense RNA [15] | Protein [56] |
| Toxin Regulation | Translation inhibition via mRNA binding or degradation [15] | Protein sequestration in a neutral complex [56] |
| Common Toxin Targets | Cell membrane integrity (causing depolarization) [15] | Translation (endoribonucleases), DNA replication, cell wall biosynthesis [42] [56] |
| Primary Study Challenges | Detecting small, hydrophobic toxin peptides; characterizing RNA-antitoxin interactions [15] [10] | Disentangling direct toxin effects from indirect regulatory consequences; functional redundancy [42] [57] |
These foundational differences mean that the "toolkit" for investigating one type is often not directly transferable to the other, complicating direct comparative studies.
Methodological Approaches and Experimental Evidence
Establishing Links to Stress Response
A primary function attributed to TA systems is helping bacteria cope with environmental stress, such as nutrient starvation, antibiotic exposure, or pH shifts.
Table 2: Experimental Evidence Linking TA Systems to Stress Response
| System Type | Experimental Evidence | Implied Stress Link | Key Methodological Challenge |
|---|---|---|---|
| Type I | Ectopic overexpression of toxins like Hok and TisB in E. coli causes rapid membrane depolarization and growth arrest [15]. | General stress-induced dormancy. | Demonstrating native expression levels are sufficient to cause physiological effects under realistic stress conditions [15]. |
| Type II | Transcriptional upregulation of hipBA in A. citrulli under pH and chloramphenicol stress [58]. | Antibiotic and pH stress. | Differentiating between a system's activation during stress versus its functional role in adapting to that stress [57] [58]. |
| Type II | Activation model involving polyP/Lon-mediated antitoxin degradation during stringent response [57]. | Nutrient starvation. | The "Active Regulation Model" based on polyP has been contested and partially retracted, highlighting the controversy in the field [57]. |
The following diagram illustrates a generalized experimental workflow for probing the role of a TA system in the stress response, applicable to both types with modifications at the mechanistic analysis stage.
Establishing Links to Pathogenesis
Linking TA systems to bacterial virulence and pathogenesis is a complex, multi-step process. Evidence often comes from phenotypic studies on biofilm formation and persistence, followed by validation in infection models.
Table 3: Experimental Evidence Linking TA Systems to Pathogenesis
| Pathogenesis Role | Type I Evidence | Type II Evidence |
|---|---|---|
| Biofilm Formation | Indirect evidence via prophage stabilization [10]. | Direct implication; multiple systems (e.g., HigBA, RelBE) influence biofilm production [42] [56]. |
| Persistence | Linked to membrane-associated toxins like TisB, which induce a dormant state [15]. | Highly controversial; early links in E. coli were artifacts, though systems like HipBA are associated with antibiotic tolerance [56] [57] [58]. |
| Animal/Plant Model Data | Limited direct evidence. | Transcriptional induction of vapBC and hipBA in A. citrulli during plant infection [58]. |
| Virulence Gene Regulation | Less studied. | Proposed as global metabolic managers, indirectly affecting virulence pathways [56]. |
The diagram below maps the logical pathway from a TA system's molecular action to a potential pathogenesis phenotype, showing where evidence is often circumstantial.
The Scientist's Toolkit: Essential Research Reagents and Methods
Successfully investigating TA system function requires a combination of genetic, molecular, and biochemical tools. The table below lists key reagents and their applications.
Table 4: Essential Research Reagents and Methodologies for TA System Research
| Reagent / Method | Function/Principle | Application Example |
|---|---|---|
| ATc-Inducible Plasmids | Allows controlled, ectopic overexpression of toxin genes to study their effects [10]. | Demonstrating toxicity of type I toxins CD0904.1, CD0956.3 in C. difficile [10]. |
| Markerless Gene Deletion Systems | Creation of clean knockout mutants to study the loss-of-function phenotype. | Assessing prophage stability upon deletion of type I TA systems [10]. |
| qRT-PCR | Quantifies changes in TA system transcription in response to stressors. | Measuring hipA and hipB induction in A. citrulli under pH/antibiotic stress [58]. |
| Co-transcription Assay (RT-PCR) | Determines if two genes are part of the same operon. | Confirming hipA and hipB are co-transcribed in A. citrulli [58]. |
| Antitoxin-Specific Antibodies | Detects unstable antitoxin proteins and monitors their degradation. | Crucial for testing the "Active Regulation Model" in type II systems [57]. |
| Bacterial Two-Hybrid System | Tests protein-protein interactions in vivo (e.g., toxin-antitoxin binding). | Characterizing complex formation in novel type II systems [59]. |
| Persistence Assays | Measures the fraction of bacteria surviving lethal antibiotic treatment. | Linking TA systems like HipBA to multidrug tolerance [58]. |
The journey to establish direct functional links for TA systems in stress response and pathogenesis is fraught with methodological and interpretive challenges. For type I systems, the primary hurdle is technical: reliably detecting and characterizing the expression and action of small, hydrophobic toxin peptides under native conditions. For type II systems, the challenge is more conceptual: disentangling the dense web of regulatory cross-talk and functional redundancy to assign a definitive role to a specific module. The controversy surrounding the HipBA system and persistence exemplifies this. Overcoming these challenges requires a rigorous, multi-faceted approach. Future research must move beyond correlative studies and ectopic overexpression, leveraging integrated methods that quantify expression, protein stability, and phenotypic outcomes under physiologically relevant conditions to fully elucidate the roles of these fascinating genetic elements in bacterial life and death.
Head-to-Head Analysis: Validating Functional Efficacy and Selecting Systems
Side-by-Side Comparison of Mechanism-of-Action Efficiency
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in bacterial and archaeal genomes, consisting of a stable toxin and a corresponding labile antitoxin that neutralizes it [60] [7]. These systems are classified into multiple types based on the nature and mode of action of the antitoxin. Type I systems feature protein toxins neutralized by antisense RNA antitoxins that prevent toxin translation [15]. In Type II systems, both toxin and antitoxin are proteins, with the antitoxin directly binding and inhibiting the toxin [7] [19]. Initially discovered as "plasmid addiction" systems that stabilize mobile genetic elements through post-segregational killing, chromosomal TA systems are now recognized for their roles in stress response, phage inhibition, persistence, and biofilm formation [60] [7] [61]. This guide provides a systematic, data-driven comparison of the mechanism-of-action efficiency between type I and type II TA systems for research and therapeutic development applications.
Comparative Mechanisms of Action
Core Architectural Principles
The fundamental distinction between type I and type II TA systems lies in their genetic organization and the molecular nature of their antitoxin components, which directly influences their regulatory dynamics and functional efficiency.
Mechanism-of-Action Efficiency Analysis
Efficiency in TA systems encompasses the speed of toxin activation, molecular specificity, and functional impact on bacterial physiology. The architectural differences between type I and type II systems create distinct efficiency profiles suited to different biological contexts.
Table 1: Comprehensive Efficiency Comparison of Type I vs. Type II TA Systems
| Efficiency Parameter | Type I Systems | Type II Systems | Experimental Evidence |
|---|---|---|---|
| Toxin Activation Time | Faster (minutes) due to direct RNA-based regulation | Slower (requires protein degradation) | Type I: Rapid membrane depolarization observed within 30 minutes of HokB induction [15]. Type II: Dependent on Lon protease degradation kinetics [7]. |
| Toxin Half-Life | Highly variable; toxins typically stable | Toxins generally highly stable; antitoxins labile | Type I: Hok toxin remains active long after induction [15]. Type II: CcdB toxin stable; CcdA antitoxin degraded by Lon protease [60]. |
| Molecular Target Specificity | Single target: bacterial membrane integrity | Diverse targets: mRNA, DNA gyrase, ribosomes, cell wall | Type I: TisB, HokB form membrane pores [15]. Type II: CcdB targets DNA gyrase; RelE cleaves mRNA; HipA phosphoryates Glu-tRNA synthetase [19]. |
| Regulatory Complexity | Simple RNA-RNA interactions | Complex protein-protein and protein-DNA interactions | Type I: Antisense RNA pairing with toxin mRNA [15]. Type II: Conditional cooperativity with TA complex acting as autoregulator [62]. |
| Cross-Regulation Capacity | Limited evidence of cross-talk | Extensive network interactions documented | Type II: MqsR/MqsA regulates GhoT/GhoS system; multiple type II systems form interconnected networks [40]. |
| Phage Inhibition Efficiency | Moderate | High, with specialized mechanisms | Type II: Some CRISPR-Cas elements derived from type II systems; direct abortion of phage replication [61]. |
The efficiency differential stems from fundamental mechanistic differences. Type I systems employ a straightforward regulatory approach where antisense RNA directly base-pairs with toxin mRNA, preventing ribosome binding and translation [15]. This creates a rapid-response system ideally suited for immediate membrane disruption when activated. Most type I toxins are small hydrophobic peptides with α-helical transmembrane domains that oligomerize to form pores, leading to rapid membrane depolarization and ATP depletion [15].
In contrast, type II systems operate through more complex protein-level interactions. The antitoxin protein typically contains two domains: one for DNA binding and another for toxin binding [19]. This dual functionality enables sophisticated regulatory behaviors, including conditional cooperativity where the toxin acts as a corepressor at high concentrations but derepresses at lower concentrations [62]. This complexity allows type II systems to integrate multiple environmental signals and coordinate broader physiological responses, albeit with slower activation kinetics compared to type I systems.
Experimental Analysis Protocols
Standardized Efficiency Assessment Workflow
Rigorous experimental validation is essential for comparing TA system efficiency. The following protocols represent established methodologies cited in current literature.
Key Experimental Methodologies
Toxin Activation Kinetics Protocol
Objective: Quantify the temporal dynamics of toxin activation and cellular response. Procedure:
- Clone toxin gene under inducible promoter (PBAD, Ptet) in appropriate vector [63]
- Grow bacterial culture to mid-exponential phase (OD600 ≈ 0.4-0.6)
- Induce toxin expression with optimal inducer concentration (e.g., 0.2% L-arabinose for PBAD)
- Collect samples at 0, 15, 30, 60, and 120 minutes post-induction
- Assess viability via colony forming units (CFUs) on selective media
- For type I toxins: measure membrane potential using DiOC₂(3) fluorescence [15]
- For type II toxins: monitor translation inhibition via ³⁵S-methionine incorporation [19]
Data Interpretation: Type I toxins typically show faster response (membrane depolarization within 30 minutes), while type II toxins may exhibit delayed but more sustained effects due to transcriptional feedback loops [15] [62].
Target Engagement and Specificity Profiling
Objective: Verify molecular targets and measure binding efficiency. Procedure:
- Express and purify recombinant toxin and antitoxin proteins [63]
- For type II RNase toxins: incubate toxin with total cellular RNA, separate fragments via urea-PAGE
- For type I membrane toxins: synthesize fluorescently-labeled peptides, incorporate into liposomes
- Perform electrophoretic mobility shift assays (EMSA) for protein-RNA (type I) or protein-protein (type II) interactions
- Conduct surface plasmon resonance (SPR) to determine binding kinetics (KD, kon, koff)
- For type II DNA gyrase inhibitors: assess supercoiling activity in vitro [60]
Validation: Target specificity can be confirmed through mutation of critical residues (e.g., catalytic sites in RNase toxins, hydrophobic domains in membrane toxins) [15] [19].
Research Reagent Solutions
Table 2: Essential Research Tools for TA System Investigation
| Reagent Category | Specific Examples | Research Application | Efficiency Considerations |
|---|---|---|---|
| Expression Vectors | pBAD33, pET28a, pRPF185-derivatives with Ptet [63] [10] | Controlled toxin induction | Tunable expression critical for avoiding artifacts from non-physiological levels |
| Detection Antibodies | Anti-His tag, Anti-FLAG, custom anti-toxin antibodies | Protein quantification via Western blot | Essential for monitoring toxin/antitoxin stability and complex formation |
| Protease Inhibitors | Lon protease inhibitors (e.g., levofoxacin analogs) [60] | Studying antitoxin degradation | Type II system analysis requires controlled proteolysis for physiological relevance |
| RNA Analysis Tools | Northern blot reagents, RT-qPCR kits, RNA-seq | Antitoxin RNA quantification, toxin mRNA processing | Critical for type I systems where RNA stability determines activity |
| Membrane Potential Probes | DiOC₂(3), JC-1, TMRM [15] | Assessing type I toxin activity | Rapid detection of membrane perturbation essential for kinetic studies |
| In Vitro Translation Systems | PURExpress, S30 extracts [19] | Testing type II RNase activity | Controlled systems enable precise measurement of inhibition efficiency |
| Bacterial Strains | Δlon mutants, protease-deficient variants [60] | Studying antitoxin stability | Modified hosts reveal protease dependence of type II regulation |
The comparative analysis reveals that type I and type II TA systems employ fundamentally distinct efficiency strategies suited to different physiological contexts. Type I systems demonstrate superior speed in toxin activation and membrane targeting, making them ideal for rapid response to environmental insults or phage infection. Their simple RNA-based regulation enables immediate implementation of growth arrest programs. Conversely, type II systems exhibit greater regulatory sophistication, with the capacity to integrate multiple signals through protein-level interactions and coordinate complex adaptive responses including persistence, biofilm formation, and comprehensive anti-phage defenses.
The choice between these systems in biotechnological applications depends critically on the desired temporal dynamics and specificity requirements. Type I toxins offer advantages when rapid, decisive growth inhibition is needed, while type II systems provide more tunable, multifaceted response capabilities. Future therapeutic development may leverage chimeric systems that combine the rapid activation of type I with the target diversity of type II, creating next-generation antibacterial strategies with enhanced efficiency and specificity. The expanding toolkit of research reagents continues to enable deeper mechanistic understanding, promising novel applications in antibiotic development, biofilm control, and synthetic biology.
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in prokaryotic genomes and plasmids, composed of a stable toxin that disrupt essential cellular processes and a labile antitoxin that neutralizes the toxin [23] [14]. These systems are classified into types based on the nature and mode of action of the antitoxin. In type I systems, the antitoxin is a small antisense RNA that binds the toxin's mRNA to inhibit translation, while in type II systems, the antitoxin is a protein that directly binds and inhibits the toxin protein [64] [23] [14]. The critical differentiating factor between these systems lies in their stability and degradation kinetics - specifically, the short half-life of the antitoxin component relative to the stable toxin creates a sensitive switch mechanism that allows rapid activation under stress conditions [64] [14]. This comparative analysis examines the kinetic parameters governing antitoxin half-life and toxin activation in type I versus type II TA systems, providing researchers with structured experimental data and methodologies for investigating these fundamental biological switches.
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Characteristic | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Type | Small non-coding RNA | Protein |
| Toxin Type | Small hydrophobic protein (usually <60 amino acids) | Protein (varied sizes and functions) |
| Mechanism of Neutralization | Antisense RNA binds toxin mRNA inhibiting translation | Antitoxin protein binds toxin protein inhibiting activity |
| Primary Regulatory Level | Post-transcriptional | Transcriptional & Post-translational |
| Genetic Organization | Often encoded on opposite DNA strands | Typically operon with antitoxin upstream of toxin |
| Common Toxin Targets | Membrane integrity, nucleoid condensation | mRNA translation (ribonucleases), DNA gyrase, cell wall synthesis |
Comparative Degradation Kinetics and Activation Mechanisms
The activation of TA systems fundamentally relies on the differential stability between the toxin and antitoxin components. When antitoxin degradation outpaces synthesis, the released toxin can exert its inhibitory effect on cell growth [64] [14] [65]. In type I systems, the RNA antitoxin is inherently unstable and degrades rapidly, allowing the more stable toxin mRNA to be translated when antitoxin transcription is reduced [64] [8]. For example, the Sok RNA antitoxin in the hok/sok system is highly labile, enabling rapid Hok toxin activation under stress conditions [14]. In type II systems, protein antitoxins are degraded by host proteases such as Lon and ClpXP, whose activity increases during stress [66] [65]. For instance, under amino acid starvation or oxidative stress, Lon protease activity increases, accelerating antitoxin degradation and toxin release [66].
The following diagram illustrates the fundamental regulatory differences in antitoxin degradation and toxin activation between type I and type II TA systems:
Table 2: Experimentally Measured Degradation Parameters in TA Systems
| TA System | System Type | Organism | Antitoxin Half-Life | Degradation Mechanism | Activation Signal |
|---|---|---|---|---|---|
| hok/sok | Type I | Escherichia coli | Sok RNA: <30 minutes [64] | RNase degradation | Plasmid loss, stress |
| tisB/istR | Type I | Escherichia coli | IstR-1 RNA: Short (precise data not available) [64] | RNase degradation | SOS response [14] |
| ccdAB | Type II | F-plasmid | CcdA: ~60 minutes [66] | Lon protease | Nutritional stress |
| relBE | Type II | Escherichia coli | RelB: ~60 minutes [66] | Lon protease | Amino acid starvation |
| mazEF | Type II | Escherichia coli | MazE: ~30-60 minutes [66] | ClpXP protease | Nutritional stress |
Mathematical modeling of TA system dynamics reveals how antitoxin degradation rates influence population heterogeneity. Models incorporating conditional cooperativity - where the toxin acts as a co-repressor at low concentrations but de-represses at high concentrations - demonstrate that increases in antitoxin degradation rates can trigger transitions to persistent states [65]. These models show that when the toxin translation rate exceeds twice the antitoxin translation rate, toxins accumulate in cells, leading to growth arrest [65]. The stability difference creates a bistable switch where stochastic fluctuations in antitoxin levels can push cells into a persistent state even in the absence of external stress [66] [65].
Experimental Methodologies for Kinetic Analysis
Transcriptional and Translational Fusion Assays
For type I systems, researchers commonly employ translational fusions of the toxin gene to reporter genes (e.g., gfp, lacZ) to monitor toxin expression under different conditions [67]. The antitoxin effect is measured by co-expressing the antisense RNA and quantifying changes in reporter activity. For example, in studying the hok/sok system, northern blotting is used to quantify Sok RNA antitoxin levels over time after inhibition of transcription, revealing its short half-life of less than 30 minutes [64]. For type II systems, transcriptional fusions to the TA operon promoter are used to monitor autoregulation, while protein stability is tracked using western blotting with specific antibodies or protein degradation tags [2] [67].
Proteomic and RNA Stability Measurements
Advanced proteomic approaches, particularly two-dimensional difference gel electrophoresis (2D DIGE), enable comprehensive monitoring of protein expression changes in TA system mutants [67]. In Staphylococcus aureus studies of the SprG1/SprF1 type I system, researchers extracted cellular proteins during logarithmic and stationary growth phases, labeled them with fluorescent dyes, and separated them by isoelectric focusing and SDS-PAGE [67]. Differentially expressed protein spots were identified through mass spectrometry, revealing how toxin expression alters the proteome. For RNA antitoxins in type I systems, northern blotting remains the gold standard for half-life determination, while RT-qPCR can track mRNA toxin levels [64] [8].
Genetic and Molecular Interaction Studies
Mutant complementation assays are fundamental for establishing TA system function. In Salmonella Typhimurium studies, researchers characterized 27 TA modules (5 type I and 19 type II) by creating deletion mutants and complementing them with plasmid-borne copies under inducible promoters [2]. This approach confirmed that toxins HokST, LdrAST, and TisBST (type I) and T4ST and VapC2ST (type II) promoted bacterial survival inside eukaryotic cells [2]. Molecular docking simulations provide structural insights, as demonstrated in Mycobacterium tuberculosis VapBC3 studies where HADDOCK software revealed binding affinities and interaction stability between toxins and antitoxins [4].
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for TA System Investigation
| Reagent/Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| Cloning Vectors | pCN35 series [67], pET expression systems | Genetic manipulation | TA gene expression in mutant backgrounds |
| Promoter Systems | Native TA promoters, inducible systems (Ptet, Para) | Controlled expression | Toxin induction studies |
| Protease Inhibitors | Lon protease inhibitors, ClpXP inhibitors | Protein stability assays | Antitoxin degradation studies |
| RNA Stabilization | RNAprotect, RNase inhibitors | RNA half-life measurements | Antitoxin RNA quantification |
| Protein Degradation Tags | SsrA degradation tag, Lon recognition tags | Protein turnover studies | Engineered antitoxin half-life modulation |
| Antibodies | Anti-Flag, custom toxin/antitoxin antibodies [67] | Protein detection | Western blot quantification |
| Reporter Genes | gfp, lacZ, luciferase | Promoter activity assays | Transcriptional regulation studies |
| Bioinformatics Tools | TADB, RASTA-Bacteria, HADDOCK [2] [4] | In silico analysis | TA identification and interaction prediction |
Functional Consequences in Pathogenesis
The kinetic differences between type I and type II TA systems translate to distinct functional specializations during host-pathogen interactions. Salmonella Typhimurium exemplifies this specialization, where type I toxins (HokST, LdrAST, TisBST) and type II toxins (T4ST, VapC2ST) promote bacterial survival inside fibroblasts, while only VapC2ST enhances fitness in epithelial cells [2]. This suggests that intracellular pathogens may exploit the different activation kinetics of TA types to optimize survival in varied host niches. The more rapid activation potential of type I systems (due to RNA antitoxin lability) might provide faster responses to sudden environmental changes, while type II systems offer more nuanced regulation through protein-protein interactions and transcriptional feedback [2] [64].
In Staphylococcus aureus, the SprG1/SprF1 type I system demonstrates how toxin expression can modulate host interactions by triggering the release of cytoplasmic proteins without cell lysis, potentially amplifying infection spread by modulating host inflammatory responses [67]. In Mycobacterium tuberculosis, sequence variations in VapBC3 type II systems between species affect toxin-antitoxin interaction stability, suggesting kinetic adaptations for specific host environments [4]. Molecular docking reveals that VapBC3 in M. bovis exhibits stronger binding affinity (HADDOCK score: 20.4 ± 5.4) than in M. tuberculosis (73.9 ± 11.0), indicating species-specific stabilization [4].
The experimental workflow for investigating TA system kinetics and functional roles typically follows a structured pathway from genetic identification to physiological characterization, as illustrated below:
The comparative analysis of stability and degradation kinetics in type I versus type II TA systems reveals fundamental differences in their regulatory architectures and activation timelines. Type I systems, with their RNA-based regulation and rapid antitoxin turnover, represent fast-response modules ideally suited for immediate reaction to sudden stress conditions. In contrast, type II systems, with their protein-based antitoxins and sophisticated transcriptional feedback mechanisms, provide more tunable, sustained responses to prolonged stresses. Both systems converge on the same functional outcome - transient growth arrest through toxin-mediated disruption of essential cellular processes - yet achieve this through distinct kinetic principles that may be specialized for different environmental challenges and intracellular niches. Understanding these differential kinetics provides not only fundamental biological insights but also potential therapeutic avenues for targeting persistent bacterial infections by manipulating TA system activation thresholds.
Comparative Efficacy in Phage Defense and Abortive Infection
The perpetual evolutionary arms race between bacteria and their viral predators, bacteriophages, has driven the development of sophisticated bacterial immune mechanisms [68]. Among these defenses, toxin-antitoxin (TA) systems have emerged as critical players in managing phage infection through the induction of metabolic stasis or programmed cell death [69]. These genetic modules, composed of a stable toxin and its cognate unstable antitoxin, are classified into multiple types based on the nature and mode of action of the antitoxin [44]. While TA systems were initially discovered as plasmid maintenance elements, their function has expanded to include fundamental roles in phage defense, particularly through abortive infection mechanisms that halt viral propagation [69].
The study of TA systems in antimicrobial research has gained significant momentum due to their involvement in bacterial persistence, biofilm formation, and stress response—attributes that contribute to antibiotic tolerance and recurrent infections [42] [44]. As the threat of multidrug-resistant pathogens intensifies, understanding the molecular intricacies of Type I and Type II TA systems becomes paramount for developing novel therapeutic strategies. This review systematically compares the efficacy, mechanisms, and research applications of Type I versus Type II TA systems in phage defense contexts, providing researchers with experimental insights and methodological frameworks for advancing this critical field.
Fundamental Distinctions Between Type I and Type II TA Systems
TA systems represent ubiquitous genetic elements in prokaryotic genomes that regulate bacterial physiology through coordinated toxin-antitoxin interactions [44]. Type I and Type II systems employ fundamentally different molecular strategies for toxin regulation, which in turn dictate their functional applications in phage defense and cellular homeostasis.
Type I systems are characterized by an antisense RNA antitoxin that interacts with toxin mRNA to prevent translation, often promoting degradation of the toxin transcript [10]. The toxin components are typically small hydrophobic proteins that can disrupt membrane integrity [10]. For instance, in Clostridioides difficile, five Type I TA systems located within prophage regions encode peptides of 34-47 amino acids featuring hydrophobic N-terminal regions and positively charged tails—structural characteristics that facilitate membrane association and potential disruption [10]. These toxins are silenced under normal conditions through antisense RNA binding, but environmental stressors can disrupt this equilibrium, leading to toxin expression and growth inhibition.
Type II systems employ a proteinaceous antitoxin that directly binds and neutralizes its cognate protein toxin [44] [69]. The defining feature of Type II systems is the formation of a stable toxin-antitoxin complex that regulates transcriptional repression through autoregulatory feedback loops [42]. The ccdAB system represents the prototypical Type II TA module, where the CcdB toxin targets DNA gyrase to inhibit replication, while CcdA antitoxin neutralizes this activity and regulates operon transcription [44]. Unlike Type I systems where regulation occurs at the RNA level, Type II systems primarily utilize protein-protein interactions for toxin control, with antitoxins typically degraded by host proteases like Lon, enabling rapid toxin activation under stress conditions [44].
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Antisense RNA [10] | Protein [44] |
| Toxin Nature | Protein (often membrane-acting) [10] | Protein (various cellular targets) [44] |
| Regulatory Mechanism | Post-transcriptional repression [10] | Transcriptional repression & protein complex formation [44] |
| Common Toxin Targets | Membrane integrity [10] | DNA gyrase, mRNA, cytoskeleton [44] |
| Genetic Location | Chromosomes, prophages, plasmids [10] | Chromosomes, plasmids [44] |
| Phage Defense Role | Abortive infection, prophage maintenance [10] | Abortive infection, persistence induction [69] |
The molecular distinctions between these systems directly impact their functional applications in phage defense. Type I systems typically exert their toxic effects through membrane disruption, leading to rapid cell death or stasis, while Type II systems often target essential intracellular processes like replication or translation [10] [44]. These fundamental differences inform their relative efficacy in abortive infection scenarios and their suitability for various research and therapeutic applications.
Molecular Mechanisms in Phage Defense
Abortive Infection as a Common Defense Strategy
Abortive infection represents an altruistic defense strategy in which phage-infected bacteria induce self-destruction or metabolic arrest to prevent viral replication and protect the clonal population [70] [71]. First described in the 1950s, this phenomenon has emerged as a widespread antiphage mechanism employed by diverse bacterial immune systems [71]. TA systems function as ideal mediators of abortive infection, as they can rapidly trigger growth arrest or cell death upon detecting phage-induced stress signals [69].
The AbiE system exemplifies how TA modules execute abortive infection. This widespread bicistronic operon functions through a non-interacting Type IV bacteriostatic TA mechanism, wherein the AbiEii toxin exhibits nucleotidyltransferase activity that specifically binds GTP, disrupting essential metabolic processes upon activation [69]. The system is negatively autoregulated by the AbiEi antitoxin, which contains an N-terminal winged-helix-turn-helix domain for transcriptional repression and a C-terminal domain responsible for toxin neutralization [69]. During phage infection, disruption of this delicate balance activates the toxin component, halting bacterial metabolism before phage replication completes, thereby protecting neighboring cells from infection.
Comparative Defense Mechanisms
Type I and Type II TA systems employ distinct molecular pathways to achieve abortive infection, with significant implications for their efficacy and applications:
Type I Systems typically function through membrane-targeting toxins whose expression is regulated by antisense RNAs. In C. difficile, these toxins induce growth arrest and cell elongation when overexpressed, morphological changes consistent with membrane disruption [10]. The location of Type I genes within prophage regions suggests specialized roles in maintaining lysogeny and preventing superinfection, as their toxin expression can destabilize excised prophages through post-segregational killing [10].
Type II Systems employ diverse intracellular targets depending on the specific toxin. The ccdAB system targets DNA gyrase, locking it in a cleavage complex that disrupts replication—a mechanism analogous to quinolone antibiotics [44]. Other Type II toxins, such as those in the AbiE system, function as nucleotidyltransferases that bind essential nucleotides like GTP, depleting cellular energy resources and halting metabolism [69]. MazF toxins exhibit ribonuclease activity that cleaves cellular mRNAs, preventing phage protein synthesis [42].
Table 2: Molecular Mechanisms in Phage Defense
| System Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Primary Defense Strategy | Abortive infection via membrane disruption [10] | Abortive infection via intracellular target inhibition [69] |
| Toxin Activation Trigger | Antisense RNA degradation/dilution [10] | Antitoxin degradation (e.g., by Lon protease) [44] |
| Cellular Outcome | Growth arrest, cell lysis [10] | Bacteriostasis, persistence [69] |
| Phage Replication Block | Early stage (membrane integrity) [10] | Multiple stages (replication, translation) [44] [69] |
| System Regulation | Antisense RNA transcription [10] | Transcriptional autoregulation, protein degradation [44] |
The following diagram illustrates the comparative molecular pathways of Type I and Type II TA systems in abortive infection:
Diagram 1: Comparative molecular pathways of Type I and Type II TA systems in abortive infection. Type I systems function through antisense RNA regulation of membrane-acting toxins, while Type II systems employ protein antitoxins that regulate diverse intracellular targets.
Experimental Assessment and Efficacy Metrics
Methodologies for Evaluating TA System Efficacy
Research on TA system efficacy employs standardized experimental approaches to quantify phage defense capacity, with particular emphasis on efficiency of plaquing (EOP) and abortive infection characterization:
Efficiency of Plaquing (EOP) Assays represent the gold standard for evaluating phage defense efficacy. This methodology involves comparing plaque-forming units on test strains expressing TA systems versus control strains [72]. As demonstrated in functional screens of E. coli defense systems, effective TA modules can reduce EOP by several orders of magnitude, with some systems like PD-λ-5 providing broad protection against diverse phages [72]. EOP assessments are typically complemented by plaque morphology analysis, as some systems may not completely prevent plaque formation but instead produce smaller, turbid plaques indicative of partial protection [72].
Growth and Survival Assays under phage challenge provide quantitative measures of protection. Researchers monitor bacterial growth kinetics and colony-forming units (CFUs) following phage exposure at varying multiplicities of infection (MOI) [10] [72]. Type I toxin overexpression in C. difficile induces immediate growth arrest and reduced CFUs, with approximately 10% of cells exhibiting significant elongation—a morphological indicator of toxin activity [10]. For distinguishing abortive infection from direct immunity, growth comparisons at high versus low MOI are essential, as Abi systems typically provide protection only when bacteria outnumber phages [72].
Molecular Characterization Techniques include RT-PCR for quantifying toxin and antitoxin expression, promoter activity assays using transcriptional fusions, and protein-protein interaction studies through co-purification or yeast two-hybrid systems [69]. For Type I systems, Northern blotting and RNA sequencing precisely map antisense RNA transcription and stability [10], while for Type II systems, electrophoretic mobility shift assays (EMSAs) characterize antitoxin-mediated transcriptional repression [69].
Quantitative Efficacy Comparison
Rigorous experimental analyses reveal distinct efficacy profiles for Type I and Type II TA systems:
Table 3: Experimental Efficacy Metrics of TA Systems in Phage Defense
| Efficacy Parameter | Type I TA Systems | Type II TA Systems |
|---|---|---|
| EOP Reduction Range | Up to 10³-fold (model-dependent) [10] | Up to 10⁶-fold (system-dependent) [72] |
| Protection Spectrum | Narrow (phage-specific) [10] | Narrow to broad [72] |
| Activation Kinetics | Rapid (post-transcriptional) [10] | Moderate (requires antitoxin degradation) [44] |
| Morphological Changes | Cell elongation (10% of population) [10] | Variable (system-dependent) [44] |
| Plaque Morphology | Not fully characterized | Smaller, turbid plaques [72] |
| Abortive Infection Efficiency | High within prophage context [10] | High (validated across genera) [69] |
The ccdAB Type II system demonstrates how efficacy extends beyond direct phage defense to include clinical relevance. In recurrent urinary tract infections caused by uropathogenic E. coli, ccdAB expression increases biofilm formation and persistent cell development—virulence-associated phenotypes that complicate treatment [44]. This dual functionality in both phage defense and pathogenicity underscores the multifunctional nature of TA systems and their significance in bacterial adaptation.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Advancing TA system research requires specialized reagents and methodologies tailored to the unique characteristics of these genetic elements. The following toolkit summarizes critical resources referenced in foundational studies:
Table 4: Essential Research Reagents and Methodologies for TA System Investigation
| Reagent/Methodology | Function/Application | Representative Use Case |
|---|---|---|
| pRPF185-derived Vectors | ATc-inducible expression in C. difficile [10] | Functional validation of Type I toxins [10] |
| pBAD30 Expression System | Arabinose-inducible toxin expression [69] | Controlled toxicity assays in E. coli [69] |
| pET28a Vector | IPTG-inducible protein expression [44] | Recombinant toxin-antitoxin production [44] |
| Fosmid Genomic Libraries | Functional metagenomic screening [72] | Identification of novel defense systems [72] |
| EOP Assay Protocol | Quantitative phage resistance measurement [72] | Defense efficacy quantification [72] |
| RT-PCR and 5'/3' RACE | Transcriptional start site mapping [69] | Operon structure determination [69] |
| CFU Monitoring | Bacterial survival quantification [10] | Toxin-induced bactericidal effect measurement [10] |
The experimental workflow for TA system characterization typically begins with genomic identification through sequence homology searches or functional screens, followed by cloning into appropriate expression vectors [72] [69]. Controlled toxin induction assays establish baseline toxicity, while complementary antitoxin co-expression confirms neutralization capacity [10]. Phage challenge experiments then quantify defense efficacy through EOP determinations and growth monitoring under various MOI conditions [72]. The following diagram illustrates a standardized experimental workflow for comparative TA system analysis:
Diagram 2: Experimental workflow for comparative analysis of TA systems in phage defense, from initial identification through molecular characterization and efficacy assessment.
Research Applications and Future Directions
The comparative analysis of Type I and Type II TA systems reveals distinctive advantages that inform their research applications. Type I systems offer simplified genetic organization and rapid post-transcriptional regulation, making them ideal models for studying RNA-based regulation and membrane-targeting toxins [10]. Their location within prophage regions provides insights into virus-host coevolution and lysogenic stability mechanisms. Conversely, Type II systems present more complex protein-protein interactions and diverse intracellular targets, offering platforms for investigating enzymatic inhibition and persistence mechanisms [44] [69]. Their well-characterized DNA gyrase and mRNA targets have facilitated structural studies and inhibitor design.
Beyond fundamental research, TA systems hold significant translational potential. Their role in bacterial persistence and biofilm formation positions them as targets for anti-persister therapies [44]. The ccdAB system in UPEC enhances biofilm formation and persistence development—key factors in recurrent urinary tract infections—suggesting that TA system inhibition could complement conventional antibiotics [44]. Similarly, engineering TA systems with modified specificity could create novel phage-based therapeutics that selectively target pathogenic bacteria while sparing commensal species.
Future research directions should address critical knowledge gaps in TA system biology. The precise activation mechanisms during phage infection require elucidation, as current understanding of stress signaling remains incomplete [70]. Comparative studies across diverse bacterial species would reveal evolutionary patterns and functional conservation, while structural analyses of toxin-antitoxin complexes could inform targeted manipulation [42] [69]. Additionally, the investigation of TA system combinations—both within and across types—may reveal synergistic effects that enhance phage protection, mirroring the natural accumulation of defense systems in genomic "defense islands" [68] [72].
As phage therapy advances against multidrug-resistant pathogens, understanding bacterial countermeasures like TA systems becomes increasingly crucial [73]. The ongoing molecular arms race between phage anti-defense systems and bacterial immunity underscores the dynamic nature of this evolutionary conflict [73]. By comprehensively characterizing Type I and Type II TA systems, researchers can not only advance fundamental knowledge of host-pathogen interactions but also develop novel therapeutic strategies that leverage these natural defense mechanisms for combating antibiotic-resistant infections.
Validation of Physiological Roles in Model Pathogens and Hybrid Strains
Toxin-antitoxin (TA) systems are small genetic modules ubiquitous in prokaryotes, comprising a stable toxin that disrupt essential cellular processes and a labile antitoxin that neutralizes the toxin's activity [23] [7]. These systems are historically classified into types based on the nature and mechanism of the antitoxin; in type I systems, an RNA antitoxin inhibits the translation of the toxin mRNA, whereas in type II systems, a protein antitoxin binds and directly inhibits the toxin protein [8] [7]. Initially discovered as plasmid maintenance systems, TA systems are now recognized for their potential roles in bacterial physiology, including stress response, pathogenicity, biofilm formation, and persistence [23] [7] [14]. Their abundance in the chromosomes of major bacterial pathogens has positioned them as compelling targets for research aimed at developing novel antibacterial strategies [23]. This guide provides a comparative analysis of the experimental approaches and models used to validate the physiological functions of type I and type II TA systems, with a specific focus on model pathogens and emerging hybrid strains.
Functional Comparison of TA System Types
Table 1: Fundamental Characteristics of Type I and Type II TA Systems
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Non-coding RNA (antisense) [8] [14] | Protein [7] [14] |
| Toxin Nature | Small, hydrophobic membrane protein [8] [14] | Protein (e.g., RNase, gyrase inhibitor) [7] [14] |
| Mechanism of Inhibition | Antitoxin RNA binds toxin mRNA, blocking translation or promoting degradation [8] [14] | Antitoxin protein binds toxin protein, directly inhibiting its activity [7] [14] |
| Common Genetic Organization | Overlapping or convergent genes [8] | Bicistronic operon (typically antitoxin gene precedes toxin) [7] [74] |
| Primary Validated Role | Stabilization of mobile genetic elements (plasmids, prophages) [10] [14] | Stabilization of mobile genetic elements; stress management [7] [75] |
Table 2: Experimentally Validated Physiological Roles in Pathogens
| Pathogen / Strain Model | TA System (Type) | Validated Physiological Role | Experimental Evidence |
|---|---|---|---|
| Salmonella Typhimurium | HokST, LdrAST, TisBST (I); T4ST, VapC2ST (II) [2] | Promotes bacterial survival inside eukaryotic host cells (fibroblasts) [2] | Intracellular fitness assays in infected host cells [2] |
| Clostridioides difficile 630 | CD0977.1-RCd11, CD0904.1-RCd13, CD0956.3-RCd14 (I) [10] | Contributes to prophage (phiCD630-1) stability and heritability [10] | Measurement of prophage loss rates in TA deletion mutants [10] |
| Hybrid E. coli BA1250 (aEPEC/ExPEC) | CcdAB, MazEF, PasTI, YhaV-PrlF, YoeB-YefM (II) [75] | Enhances fitness under osmotic and acid stress [75] | Growth dynamics and qPCR analysis under stress conditions [75] |
| Escherichia coli K-12 | RelBE (II) [75] | Reduced translation during nutritional stress [75] | Translation rate assays and transcript analysis during starvation [75] |
| Multiple Pathogens | HicAB (II) [74] | Biofilm formation, phage inhibition, virulence [74] | Biofilm quantification, phage infection assays, animal models [74] |
Experimental Approaches for Functional Validation
Molecular Protocols for TA System Characterization
1. Toxicity and Neutralization Assay (Ectopic Expression): This foundational protocol validates the core function of a putative TA pair [2] [10].
- Procedure: The gene encoding the putative toxin is cloned into an inducible expression plasmid (e.g., pRPF185 with a Ptet promoter). The putative antitoxin gene (for type II) or its promoter region (for type I RNA antitoxin) is cloned in tandem or on a compatible plasmid. The constructs are transformed into the host bacterium (e.g., C. difficile 630Δerm or E. coli). Growth curves and colony-forming units (CFUs) are monitored with and without induction (e.g., using anhydrotetracycline, ATc) [10].
- Expected Outcome: Induction of the toxin alone causes growth arrest or a drop in CFUs, which is reversed when the antitoxin is co-expressed, confirming a functional TA pair [10].
2. Intracellular Fitness Assay: This method tests the role of TA systems during infection of eukaryotic host cells [2].
- Procedure: Isogenic mutant strains of the pathogen (e.g., S. Typhimurium) lacking a specific TA system are created. Mutant and wild-type bacteria are used to infect cultured eukaryotic cells, such as epithelial cells or fibroblasts. At various post-infection time points, cells are lysed, and intracellular bacteria are enumerated by plating and counting CFUs.
- Expected Outcome: A statistically significant reduction in the recovery of the TA mutant compared to the wild-type strain indicates that the TA system contributes to intracellular survival or replication [2].
3. Gene Expression Profiling Under Stress: This approach investigates TA system regulation in response to environmental cues [75].
- Procedure: A bacterial strain (e.g., hybrid E. coli BA1250) is grown under various stress conditions (e.g., acid, osmotic, oxidative stress, nutritional deprivation). Samples are collected during logarithmic and stationary growth phases. Total RNA is extracted, and the relative expression levels of toxin and antitoxin genes are quantified using reverse transcription quantitative PCR (qPCR), with expression normalized to a housekeeping gene.
- Expected Outcome: Significant upregulation or downregulation of TA genes under specific stress conditions suggests their involvement in the corresponding stress response pathway [75].
Visualizing Regulatory Pathways and Workflows
Type I TA System Regulation
Figure 1: Type I TA system regulation relies on RNA-based inhibition of toxin synthesis.
Type II TA System Regulation
Figure 2: Type II TA system regulation involves protein-protein interaction and complex feedback.
Experimental Workflow for Functional Validation
Figure 3: A multi-stage experimental workflow for validating TA system functions.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for TA System Research
| Research Reagent | Function in Experimentation | Application Example |
|---|---|---|
| Inducible Plasmid Vectors | Enable controlled, ectopic expression of toxin and antitoxin genes for toxicity and neutralization assays [10]. | pRPF185-derivatives with Ptet promoter in C. difficile [10]. |
| Conditional Knockout Mutants | Allow for the study of phenotypic consequences of TA system loss under defined conditions [2]. | Studying intracellular fitness of S. Typhimurium TA mutants in host cells [2]. |
| qPCR Assays | Quantify the expression levels of TA genes in response to various environmental stresses [75]. | Profiling pasTI and mazEF expression in hybrid E. coli under acid/osmotic stress [75]. |
| Structured Biological Models | Provide a framework for interpreting gene expression dynamics and predicting system behavior [62]. | ODE models describing the negative feedback loop in type II TA systems [62]. |
The comparative analysis of validation strategies reveals that while type I and type II TA systems operate through fundamentally distinct molecular mechanisms, their investigation relies on a shared set of robust experimental paradigms. The use of model pathogens like Salmonella and C. difficile has been instrumental in linking specific TA modules to concrete physiological roles such as intracellular survival and prophage maintenance. The emerging study of hybrid pathogenic strains, such as the aEPEC/ExPEC E. coli BA1250, highlights a new frontier where TA systems may be critical for fitness across diverse host niches [75]. Future research will benefit from the continued development of specialized genetic tools and the integration of computational models with high-throughput functional data to fully unravel the complex regulatory networks governed by TA systems and assess their potential as targets for novel antimicrobials.
Selecting Optimal TA Systems for Specific Biotechnological or Therapeutic Goals
Toxin-Antitoxin (TA) systems are simple genetic modules, ubiquitous in prokaryotes, that consist of a stable toxin and its corresponding labile antitoxin. These systems are broadly categorized into eight types (I-VIII) based on the chemical nature of the antitoxin (protein or RNA) and its mechanism of toxin neutralization [7] [27]. While their biological roles in native contexts—such as stress response, phage defense, and persistence formation—are extensively studied [7] [25], their potential as programmable genetic switches has also catalyzed their adoption in biotechnology and therapeutic development. The selection between different TA types, particularly the well-characterized Type I and Type II systems, is paramount for optimizing the efficacy of applications ranging from synthetic biology circuits to antibacterial strategies.
This guide provides an objective comparison of Type I and Type II TA systems, focusing on their operational mechanisms, experimental performance data, and suitability for specific applications. It is structured to assist researchers and drug development professionals in making informed decisions by synthesizing current research findings and providing detailed, actionable methodological information.
Fundamental Comparative Analysis: Type I vs. Type II TA Systems
The core distinction between Type I and Type II TA systems lies in the nature and mode of action of the antitoxin. This fundamental difference dictates their genetic organization, regulatory dynamics, and, consequently, their suitability for various applications.
Table 1: Core Characteristics of Type I and Type II TA Systems
| Feature | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Antitoxin Nature | Antisense RNA [7] [10] | Protein [7] [25] |
| Mechanism of Toxin Neutralization | Antitoxin RNA binds toxin mRNA, inhibiting translation and/or promoting degradation [7] [10] | Antitoxin protein binds directly to toxin protein, inhibiting its activity [7] [25] |
| Typical Genetic Organization | Toxin and antitoxin genes are often overlapping or adjacent [10] | Typically organized as a bicistronic operon with antitoxin gene preceding the toxin gene [76] [7] |
| Primary Regulatory Level | Post-transcriptional [10] | Transcriptional and post-translational (e.g., protease degradation of antitoxin) [7] [25] |
| Stability of Components | Stable toxin protein; labile antisense RNA [10] | Stable toxin protein; labile antitoxin protein [7] [25] |
The following pathway diagram visualizes the fundamental functional differences and logical relationships between Type I and Type II TA systems.
Figure 1: Functional Pathways of Type I and Type II TA Systems. The diagram illustrates how different stress signals activate Type I and Type II systems through distinct mechanisms, culminating in growth arrest or cell death.
Comparative Performance in Key Applications
Maintenance of Mobile Genetic Elements
Both TA system types function as "addiction modules" to ensure the stable inheritance of plasmids, prophages, and other genomic islands, but they achieve this through different mechanistic paths.
Type II Systems: This is the classic, best-characterized function for Type II TAs. The model involves post-segregational killing (PSK) or growth inhibition. The labile antitoxin degrades faster than the stable toxin in daughter cells that lose the genetic element, leading to toxin activation and cell death [7]. Systems like
ccdABandpemIKare well-documented examples [7].Type I Systems: Evidence confirms Type I TAs also stabilize mobile elements. In Clostridioides difficile, multiple Type I TA systems (e.g.,
CD0977.1-RCd11,CD0904.1-RCd13) located within prophages were shown to contribute directly to prophage stability and heritability. Their deletion increased the rate of prophage loss, confirming a functional role in maintenance [10].
Table 2: Application-Based Performance Comparison
| Application | Type I TA Systems | Type II TA Systems |
|---|---|---|
| Maintenance of Genetic Elements | Effective for prophage and plasmid stability [10] | The gold-standard; highly effective for plasmid and genomic island maintenance [7] |
| Antiviral Tools in Plants | Limited reported use | Effective (e.g., MazF-induced death phenotype counteracted by MazE) [76] |
| Bacterial Persistence & Biofilm Formation | Implicated in stress response [10] | Strongly associated with persistence, biofilm formation, and stress response (e.g., VapBC) [7] [27] |
| Antibacterial Drug Target Potential | Toxin peptides are difficult to target; strategy relies on antisense RNA targeting | High potential; strategies include activating toxins via antitoxin inhibition [27] |
| Synthetic Biology & Biotechnology | Useful for counter-selection in genome editing [10] | Engineered as molecular switches (e.g., protease-activated) [76] |
Biotechnological Tool Development
Engineered TA systems, particularly Type II, are being developed into programmable genetic switches for sophisticated biotechnological applications.
Programmable Cell Death in Plants: The MazEF and YefM-YoeB Type II systems from bacteria have been successfully deployed in plants. Engineered versions were modified to be activated by a specific viral protease (NIapro from potyviruses). When the virus infected the plant, the protease cleaved and activated the toxin, inducing localized cell death and demonstrating potential as an antiviral tool [76].
Counter-Selection in Genome Editing: The toxicity of Type I toxins has been harnessed for genetic engineering. In C. difficile, the inducible expression of a Type I toxin gene (
CD0977.1) was used as a highly efficient counter-selection marker in allele-exchange procedures, facilitating the selection of cells that had lost the plasmid vector [10].
Experimental Protocols for Key Assays
To evaluate and utilize TA systems, robust experimental protocols are essential. Below are detailed methodologies for key functional assays.
Protocol: Testing TA System Functionality via Conditional Expression
This protocol, adapted from studies in C. difficile and Nicotiana benthamiana, is used to validate the function and cognate pairing of a TA system [10] [76].
Cloning and Genetic Construction:
- Clone the toxin gene alone (pT) and the toxin-antitoxin pair together (pTA) into inducible expression vectors (e.g., anhydrotetracycline (ATc)-inducible
Ptetpromoter). - For toxin genes cloned in bacteria, introduce an intron into the middle of the sequence to prevent accidental toxicity in the bacterial host [76].
- Ensure the antitoxin is expressed from its native promoter in the pTA construct to validate natural regulation.
- Clone the toxin gene alone (pT) and the toxin-antitoxin pair together (pTA) into inducible expression vectors (e.g., anhydrotetracycline (ATc)-inducible
Transformation and Growth Conditions:
- Transform the pT and pTA constructs into the host organism of interest (e.g., via agrobacterium infiltration for plants or standard transformation for bacteria).
- Grow transformed cells to mid-exponential phase.
Induction and Phenotypic Monitoring:
- Split the culture and add an inducer (e.g., ATc) to one half, leaving the other as an uninduced control.
- Monitor growth over time by measuring optical density (OD) and enumerating colony-forming units (CFUs).
- Expected Outcome: Expression of the toxin alone (pT + inducer) should lead to significant growth inhibition or a drop in CFUs. Co-expression of the toxin and antitoxin (pTA + inducer) should rescue growth, confirming the pair is functional and cognate [10].
Protocol: Assessing TA System-Induced Immune Responses
This protocol is based on work with the YefM-YoeB system in plants and can be adapted to other hosts to determine if toxicity triggers a defensive immune response [76].
Transient Expression:
- Express the toxin, antitoxin, or toxin-antitoxin pair in the host tissue (e.g., via agroinfiltration in plant leaves). Include empty vector controls.
Tissue Sampling and Protein Extraction:
- Before the onset of full necrosis (e.g., 5 days post-infiltration), collect tissue samples from the infiltration sites.
- Homogenize the tissue and extract total protein using a suitable buffer.
Immunoblot Analysis:
- Separate proteins by SDS-PAGE and transfer to a membrane.
- Probe the membrane with antibodies against established immune markers (e.g., Pathogenesis-Related protein 2, PR2, for salicylic acid-mediated immunity in plants).
- Use a loading control (e.g., Ponceau red staining of Rubisco) for normalization.
- Expected Outcome: A significant increase in PR2 levels in samples expressing the TA pair, compared to the toxin or antitoxin alone, indicates the induction of an effector-triggered immunity (ETI)-like response [76].
The Scientist's Toolkit: Essential Research Reagents
Successful research and development with TA systems rely on a suite of key reagents and solutions.
Table 3: Essential Research Reagents for TA System Investigation
| Reagent / Solution | Function / Application | Specific Examples / Notes |
|---|---|---|
| Inducible Expression Vectors | Controlled expression of toxin/antitoxin for functional testing without constitutive toxicity. | pRPF185 (ATc-inducible) for C. difficile [10]; pGWB402Ω for plants [76] |
| RNA Silencing Suppressor | Maximizes transient protein expression in plant systems by suppressing host gene silencing. | Co-express P19 protein [76] |
| Antibodies for Immune Markers | Detects host immune response activation via immunoblot. | Anti-PR2 antibody for salicylic acid pathway analysis [76] |
| Protease Activators | Engineered activation of toxin components in synthetic biology circuits. | Nuclear Inclusion A endopeptidase (NIapro) from potyviruses [76] |
| Strain-Specific Transformation Kits | Essential for delivering genetic constructs into model or target organisms. | Agrobacterium tumefaciens strains for plant infiltration; specialized electroporation kits for difficult bacteria. |
The choice between Type I and Type II TA systems is application-dependent. Type II systems, with their protein-based regulation and well-established role as genetic switches, currently offer greater versatility for engineered systems in biotechnology, such as programmable cell death and synthetic circuits. In contrast, Type I systems present unique opportunities for fundamental research on RNA-based regulation and have proven utility in specific contexts like genetic element maintenance and counter-selection.
Future directions will likely focus on refining the orthogonality and tunability of these systems, minimizing cross-talk in complex synthetic networks, and exploiting their structural nuances for developing novel anti-infectives that target bacterial persistence. The experimental frameworks and comparative data provided in this guide serve as a foundation for researchers to select and deploy the optimal TA system for their specific biotechnological or therapeutic goal.
Conclusion
The comparative analysis of Type I and Type II TA systems reveals a clear functional dichotomy: Type I systems primarily utilize RNA-level regulation for fast, direct control, while Type II systems employ complex protein-protein interactions for nuanced, multi-layered regulation. Both systems play crucial, though sometimes debated, roles in bacterial physiology, including phage defense, mobile genetic element stabilization, and stress adaptation. Their distinct mechanisms offer complementary toolkits for synthetic biology, from plasmid maintenance to the engineering of sophisticated genetic circuits. For therapeutic development, Type II systems, with their proteinaceous toxins and well-defined targets, present promising avenues for novel antibiotics. Future research should focus on elucidating the precise regulatory networks governing these systems in pathogens, validating their roles in infection models, and leveraging their mechanisms to design next-generation antimicrobials that overcome resistance. The integration of TA systems, particularly their proven anti-phage function, with other bacterial defense systems like CRISPR-Cas also represents a fertile ground for discovery and innovation.
References
- 1. Distribution of anti-sense regulated, type I toxin-antitoxin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5367252/]
- 2. Distinct type I and type II toxin-antitoxin modules control ... [https://www.nature.com/articles/srep09374]
- 3. Unraveling the evolutionary dynamics of toxin-antitoxin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10790755/]
- 4. Comparative in silico analysis of vapBC3 toxin-antitoxin ... [https://www.nature.com/articles/s41598-025-24448-z]
- 5. The combination of active partitioning and toxin-antitoxin ... [https://www.nature.com/articles/s41467-025-62473-8]
- 6. Discovery of Functional Toxin/Antitoxin Systems in Bacteria ... [https://www.sciencedirect.com/science/article/pii/S1097276513001299]
- 7. Biological Functions of Type II Toxin-Antitoxin Systems in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8230891/]
- 8. Type I toxin-antitoxin systems in bacteria - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11636113/]
- 9. Type II Toxin-Antitoxin Systems in Escherichia coli [https://pubmed.ncbi.nlm.nih.gov/40027916/]
- 10. Type I toxin-antitoxin systems contribute to the ... [https://www.nature.com/articles/s42003-020-01448-5]
- 11. Operon [https://en.wikipedia.org/wiki/Operon]
- 12. Operons and Prokaryotic Gene Regulation [http://www.nature.com/scitable/topicpage/operons-and-prokaryotic-gene-regulation-992]
- 13. Functional analysis of the type II toxin-antitoxin system ParDE ... [https://bmcvetres.biomedcentral.com/articles/10.1186/s12917-024-04069-w]
- 14. Toxin-antitoxin system [https://en.wikipedia.org/wiki/Toxin-antitoxin_system]
- 15. Bacterial Type I Toxins: Folding and Membrane Interactions [https://pmc.ncbi.nlm.nih.gov/articles/PMC8309996/]
- 16. Post-transcriptional regulation of gene expression in ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2014.00006/full]
- 17. Comprehensive comparative-genomic analysis of Type 2 toxin ... [https://biologydirect.biomedcentral.com/articles/10.1186/1745-6150-4-19]
- 18. Role of Antitoxin RNA Pseudoknot in Regulating Toxin ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283625004760]
- 19. Type II Toxin-Antitoxin Systems in Escherichia coli - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11869752/]
- 20. Escherichia coli type I toxin TisB exclusively controls proton ... [https://www.nature.com/articles/s41598-025-96136-x]
- 21. Toxin Induction or Inhibition of Transcription or Translation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8406316/]
- 22. Mechanisms of Toxin Inhibition and Transcriptional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4110269/]
- 23. Toxin-Antitoxin Systems in Clinical Pathogens - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4963858/]
- 24. Structure, Biology, and Therapeutic Application of Toxin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5086665/]
- 25. Toxin–antitoxins and sigma factors may optimize the fitness of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12440627/]
- 26. The PLSDB 2025 update: enhanced annotations and ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11701622/]
- 27. Characterization of toxin-antitoxin systems from public ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9424990/]
- 28. Characterization of toxin-antitoxin systems from public ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.951774/full]
- 29. Abundance of type I toxin–antitoxin systems in bacteria [https://pmc.ncbi.nlm.nih.gov/articles/PMC2887945/]
- 30. Toxin–Antitoxin Genes in Plasmids and Chromosomes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5543033/]
- 31. Distinct type I and type II toxin-antitoxin modules control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4366850/]
- 32. The Variety in the Common Theme of Translation Inhibition ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.00262/full]
- 33. Use of Bacterial Toxin-Antitoxin Systems as ... [https://pubmed.ncbi.nlm.nih.gov/39408779/]
- 34. Applications of toxin-antitoxin systems in synthetic biology [https://www.sciencedirect.com/science/article/pii/S2667370323000012]
- 35. Applications of toxin-antitoxin systems in synthetic biology [https://pubmed.ncbi.nlm.nih.gov/39629251/]
- 36. Targeting Type II Toxin-Antitoxin Systems as Antibacterial ... [https://pubmed.ncbi.nlm.nih.gov/32899634/]
- 37. Targeting Type II Toxin–Antitoxin Systems as Antibacterial ... [https://www.mdpi.com/2072-6651/12/9/568]
- 38. Artificial activation of toxin-antitoxin systems as an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3952271/]
- 39. Rational design of synthetic antimicrobial peptides based ... [https://www.nature.com/articles/s41598-025-98330-3]
- 40. Cross-Regulations between Bacterial Toxin–Antitoxin ... [https://www.sciencedirect.com/science/article/abs/pii/S0966842X20301438]
- 41. Preventing toxicity in toxin-antitoxin systems: An overview ... [https://www.sciencedirect.com/science/article/abs/pii/S0300908423001748]
- 42. Type II toxin-antitoxin systems as stress-responsive ... [https://pubmed.ncbi.nlm.nih.gov/40970936/]
- 43. A blueprint for broadly effective bacteriophage-antibiotic ... [https://www.nature.com/articles/s41467-024-53994-9]
- 44. The biological function of the type II toxin-antitoxin system ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1379625/full]
- 45. Therapeutic applications of synthetic gene/genetic circuits - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11330796/]
- 46. A synthetic transcription platform for programmable gene ... [https://www.nature.com/articles/s41467-022-33287-9]
- 47. Type II Toxin-Antitoxin Systems: Evolution and Revolutions [https://pmc.ncbi.nlm.nih.gov/articles/PMC7167474/]
- 48. Principles of bacteriostatic and bactericidal antibiotics at ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607625/]
- 49. Toxin-antitoxin systems in bacterial pathogenesis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10123168/]
- 50. Bacterial toxin-antitoxin modules: classification, functions, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8610362/]
- 51. Toxin–antitoxin systems and their role in disseminating and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5812544/]
- 52. Keeping the Wolves at Bay: Antitoxins of Prokaryotic Type II ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2016.00009/full]
- 53. Evaluating the Potential for Cross-Interactions of Antitoxins in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7354431/]
- 54. Toxin-Antitoxin Systems Influence Biofilm and Persister ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3165247/]
- 55. Biofilm establishment, biofilm persister cell formation, and ... [https://www.sciencedirect.com/science/article/abs/pii/S2452014420302600]
- 56. Emerging Roles of Toxin-Antitoxin Modules in Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6273597/]
- 57. Regulation of Type II Toxin-Antitoxin Systems [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.00895/full]
- 58. Evidence for a Functional HipBA Toxin–Antitoxin System in ... [https://www.mdpi.com/1422-0067/26/7/3366]
- 59. Novel type II toxin-antitoxin systems with VapD-like proteins [https://pmc.ncbi.nlm.nih.gov/articles/PMC11980593/]
- 60. Recent Advancements in Toxin and Antitoxin Systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3021852/]
- 61. A Primary Physiological Role of Toxin/Antitoxin Systems Is ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.01895/full]
- 62. A minimal model for gene expression dynamics of bacterial ... [https://www.nature.com/articles/s41598-021-98570-z]
- 63. Regulatory crosstalk between type I and type II toxin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4829291/]
- 64. Type I Toxin-Antitoxin Systems: Regulating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11633621/]
- 65. A General Model for Toxin-Antitoxin Module Dynamics Can ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003190]
- 66. Molecular mechanisms of multiple toxin–antitoxin systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3703989/]
- 67. Impacts of the Type I Toxin–Antitoxin System, SprG1/SprF1, ... [https://www.mdpi.com/2073-4425/12/5/770]
- 68. Overview of Phage Defense Systems in Bacteria and Their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11676413/]
- 69. A widespread bacteriophage abortive infection system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3985639/]
- 70. Abortive infection antiphage defense systems: separating ... [https://pubmed.ncbi.nlm.nih.gov/37268559/]
- 71. Abortive Infection [https://defensefinder.mdmlab.fr/wiki/general-concepts/abortive-infection]
- 72. A functional selection reveals previously undetected anti ... [https://www.nature.com/articles/s41564-022-01219-4]
- 73. The growing repertoire of phage anti-defence systems [https://www.sciencedirect.com/science/article/pii/S0966842X24001367]
- 74. The HicAB System: Characteristics and Biological Roles of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11594946/]
- 75. Chromosomal Type II Toxin–Antitoxin Systems May ... [https://www.mdpi.com/2072-6651/16/11/469]
- 76. Use of Bacterial Toxin–Antitoxin Systems as ... [https://www.mdpi.com/1422-0067/25/19/10449]