Validating Biofilm Reduction: CRISPR Gene Editing vs. Chemical Anti-Biofilm Agents
Biofilm-associated infections pose a major therapeutic challenge due to their high tolerance to conventional antibiotics, driving the need for innovative disruption strategies.
Validating Biofilm Reduction: CRISPR Gene Editing vs. Chemical Anti-Biofilm Agents
Abstract
Biofilm-associated infections pose a major therapeutic challenge due to their high tolerance to conventional antibiotics, driving the need for innovative disruption strategies. This article provides a comprehensive methodological framework for researchers and drug development professionals to quantitatively validate the efficacy of two promising approaches: precision genetic targeting via CRISPR-Cas9 and broad-spectrum chemical treatment. We explore foundational biofilm biology and resistance mechanisms, detail established protocols for applying CRISPR and chemical agents, address optimization challenges, and present a rigorous comparative analysis of validation metrics. By synthesizing current research and emerging trends, this review aims to establish standardized benchmarks for evaluating anti-biofilm technologies and guide the development of next-generation therapeutics.
The Biofilm Challenge: Understanding Structure, Resistance, and Therapeutic Targets
Bacterial biofilms are structured communities of microbial cells enclosed within a self-produced extracellular polymeric substance (EPS) matrix, which adheres to living or non-living surfaces [1]. This matrix forms a protective fortress, often described as the "house of biofilm cells," which determines the immediate conditions of life for microorganisms by affecting porosity, density, water content, charge, and mechanical stability [2]. The biofilm architecture provides significant survival advantages, including remarkable tolerance to antimicrobial treatments, with biofilms exhibiting up to 1000-fold greater resistance to antibiotics compared to their free-floating (planktonic) counterparts [3]. This resilience poses a critical challenge in clinical and industrial settings, particularly in treating chronic infections and combating biofilm contamination in food processing facilities [1] [4].
Understanding the intricate relationship between EPS composition, biofilm ultrastructure, and antimicrobial resistance is fundamental to developing effective countermeasures. This guide objectively compares two innovative research strategies for disrupting this architecture: precision genetic editing using CRISPR/Cas systems and conventional chemical treatments. By examining their mechanisms, efficacy, and experimental validation, we provide researchers and drug development professionals with a structured analysis of these divergent approaches to biofilm control.
Deconstructing the EPS Matrix and Biofilm Architecture
Composition of the EPS Matrix
The EPS matrix is a complex, dynamic amalgamation of biopolymers that constitutes 75-90% of the biofilm's total mass, with microbial cells making up only 10-25% [1]. Contrary to early understanding, the matrix is far more than just polysaccharides.
Table 1: Key Components of the Biofilm EPS Matrix and Their Functions
| Matrix Component | Primary Functions | Examples and Microbial Sources |
|---|---|---|
| Polysaccharides | Structural scaffold, cell-cell adhesion, water retention, protection from immune response and desiccation [2] [1] | Pel, Psl, and alginate in Pseudomonas aeruginosa; cellulose in E. coli and Agrobacteria [2] [1] |
| Extracellular Proteins | Matrix stabilization, surface colonization, structural integrity, biofilm dispersal [2] [1] | Curli fibrils in E. coli; amyloid adhesins; proteases and glycosyl hydrolases for dispersal [2] |
| Extracellular DNA (e-DNA) | Structural integrity, intercellular connector, cation chelation, gene pool for horizontal transfer [2] [3] | Genomic DNA released via controlled cell lysis; forms grid-like structures in P. aeruginosa [2] |
| Lipids and Surfactants | Interface interactions, modulation of biofilm surface tension, structure dispersal [2] | Modulins in Staphylococcal biofilms [1] |
| Water | Medium for nutrient transport, enzymatic activity, maintaining hydration [1] | Up to 97% of biofilm volume [1] |
The composition is highly variable across species and environmental conditions. For instance, environmental biofilms often contain surprisingly low levels of alginate and charged polysaccharides, with proteins and e-DNA playing a more substantial structural role [2].
Ultrastructural Organization of Biofilms
The biofilm architecture is not random but a highly organized, three-dimensional structure. Its formation follows a multi-stage developmental process:
- Attachment: Planktonic cells reversibly, then irreversibly, adhere to surfaces. The Pil-Chp surface-sensing system and increased levels of the secondary messenger c-di-GMP promote this transition by restricting flagellar motility and increasing matrix production [1].
- Microcolony Formation: Attached cells proliferate and aggregate, forming clusters. Type IV pili-mediated motility is crucial for cell-cell interactions [1].
- Maturation: The community develops into a complex, heterogeneous structure characterized by "mushroom" or "tower" shapes. These structures are separated by interstitial voids and water channels that facilitate nutrient distribution and waste removal [3] [1].
- Dispersion: Cells actively or passively detach from the biofilm to colonize new surfaces, triggered by factors such as nutrient scarcity, oxygen deficiency, or enzyme-mediated matrix degradation [1].
Advanced imaging techniques like Confocal Laser Scanning Microscopy (CLSM) have revealed that the cellular arrangement within this structure is genetically determined and has profound physiological implications. For example, Pseudomonas aeruginosa cells form striations packed lengthwise across the biofilm, an arrangement that influences nutrient distribution and antibiotic tolerance. Mutants defective in type IV pilus production form "bundled" biofilms, while those with defects in global regulators or O-antigen biosynthesis exhibit "disordered" or "clustered" phenotypes, each with distinct metabolic and susceptibility profiles [5].
Comparative Analysis: CRISPR/Cas9 Gene Editing vs. Chemical Treatments
The following section provides a point-by-point comparison of two strategic approaches for biofilm disruption, based on current research data.
Table 2: Performance Comparison of CRISPR/Cas9 and Chemical Treatments for Biofilm Control
| Evaluation Parameter | CRISPR/Cas9 Gene Editing | Conventional Chemical Treatments |
|---|---|---|
| Primary Mechanism of Action | Precision targeting and disruption of specific genetic elements (e.g., resistance genes, QS systems, EPS synthesis genes) [3] [4] | Non-specific biochemical disruption of cell membranes, proteins, or matrix components [1] |
| Efficacy Against Biofilm Biomass | Liposomal Cas9 formulations reduced P. aeruginosa biofilm biomass by >90% in vitro [3]. | Variable efficacy; often requires high concentrations and fails to eradicate persistent cells [6] [4]. |
| Efficacy Against Planktonic Cells | High efficiency when successfully delivered; can be programmed to target specific pathogens [4]. | Generally high efficacy against planktonic cells, but can select for tolerant strains [3]. |
| Target Specificity | Very high; gRNA can be designed for species- or strain-specific targeting, sparing beneficial flora [4]. | Low; broad-spectrum action disrupts both pathogens and beneficial microbes [4]. |
| Penetration of EPS Matrix | Enhanced by nanoparticle carriers (e.g., gold NPs increased editing efficiency 3.5-fold) [3]. | Often limited; matrix components like alginate can bind tobramycin, eDNA can impede vancomycin [6]. |
| Impact on Antibiotic Resistance | Directly disrupts resistance genes (e.g., bla, mecA), resensitizing bacteria to antibiotics [3]. | Can accelerate resistance through selective pressure and enrichment of persister cells [3] [4]. |
| Potential for Resistance Development | Low; targets essential genetic sequences, though delivery failure can mimic resistance [4]. | High; repeated sub-lethal exposure selects for intrinsically resistant mutants [3]. |
| Key Challenges | Efficient delivery across EPS, stability of machinery, off-target effects, regulatory hurdles [3] [4]. | Inability to penetrate matrix, disruption of microbial ecology, toxicity, environmental concerns [1] [4]. |
Analysis of Comparative Data
The data reveals a fundamental divergence in strategy. Chemical treatments act as "bulldozers," applying broad-spectrum force that often fails to penetrate the biofilm core and can select for harder-to-treat residues [6] [4]. In contrast, CRISPR/Cas9 systems function as "precision scalpels," designed to inactivate the very genetic blueprints that govern biofilm resilience and antibiotic resistance [3]. The synergy of CRISPR with nanoparticle technology is particularly promising, as it directly addresses the critical challenge of EPS penetration, leveraging the intrinsic properties of nanomaterials to deliver the genetic machinery deep into the biofilm architecture [3].
Experimental Protocols for Validating Biofilm Reduction
Protocol 1: CRISPR/Cas9 Delivery via Lipid Nanoparticles (LNPs)
This protocol is adapted from studies demonstrating over 90% reduction of P. aeruginosa biofilm biomass [3].
- 1. gRNA Design and Complex Formation:
- Design gRNAs to target essential biofilm-related genes (e.g., pelA, pslD for polysaccharide synthesis in P. aeruginosa; lasI or rhlI for quorum sensing).
- Formulate ribonucleoprotein (RNP) complexes by pre-incubating purified Cas9 nuclease with the synthesized gRNA.
- 2. Nanoparticle Encapsulation:
- Encapsulate the RNP complexes into lipid nanoparticles (LNPs) using a microfluidic mixer.
- Parameters: Aqueous phase (RNP in buffer) to lipid phase (ionizable lipid, phospholipid, cholesterol, PEG-lipid) flow rate ratio of 3:1, total flow rate of 12 mL/min.
- Purify the formed LNPs via dialysis or tangential flow filtration.
- 3. Biofilm Treatment and Incubation:
- Grow 48-hour mature biofilms of target bacteria in 96-well plates or on relevant surfaces (e.g., silicone, plastic).
- Apply LNP-CRISPR formulations at a predetermined optimal concentration (e.g., 100 µg/mL total lipid) to the biofilm and incubate for 24-48 hours.
- 4. Efficacy Assessment:
- Biomass Quantification: Use crystal violet (CV) staining to measure total biofilm biomass.
- Viability Assessment: Use resazurin viability staining or perform colony-forming unit (CFU) counts after biofilm disruption.
- Structural Analysis: Use Confocal Laser Scanning Microscopy (CLSM) to visualize changes in biofilm architecture and thickness.
Protocol 2: Evaluation of Conventional Chemical Biocides
This standard protocol highlights the assessment of biofilm susceptibility, a key factor in the systematic review showing a weak correlation between biofilm biomass and antibiotic tolerance [6].
- 1. Biofilm Cultivation:
- Grow biofilms in microtiter plates or on coupons (e.g., stainless steel) for a standardized duration (e.g., 24-48 hours) in appropriate media.
- 2. Biocide Exposure:
- Prepare serial dilutions of the test biocide (e.g., sodium hypochlorite, quaternary ammonium compounds, antibiotics like tobramycin or ciprofloxacin) in fresh medium.
- Expose mature biofilms to the biocide solutions for a set contact time (e.g., 1-2 hours). Include untreated controls.
- 3. Post-Treatment Analysis:
- Metabolic Activity: Measure using the resazurin reduction assay. The signal is proportional to the number of metabolically active cells.
- Culturable Cell Count: Gently wash the biofilm to remove the biocide, disrupt the biofilm by sonication/vortexing, and plate the suspension for CFU enumeration.
- Crystal Violet (CV) Staining: Fix the biofilm with methanol, stain with CV, elute the dye, and measure absorbance to determine the remaining total biomass (cells and matrix).
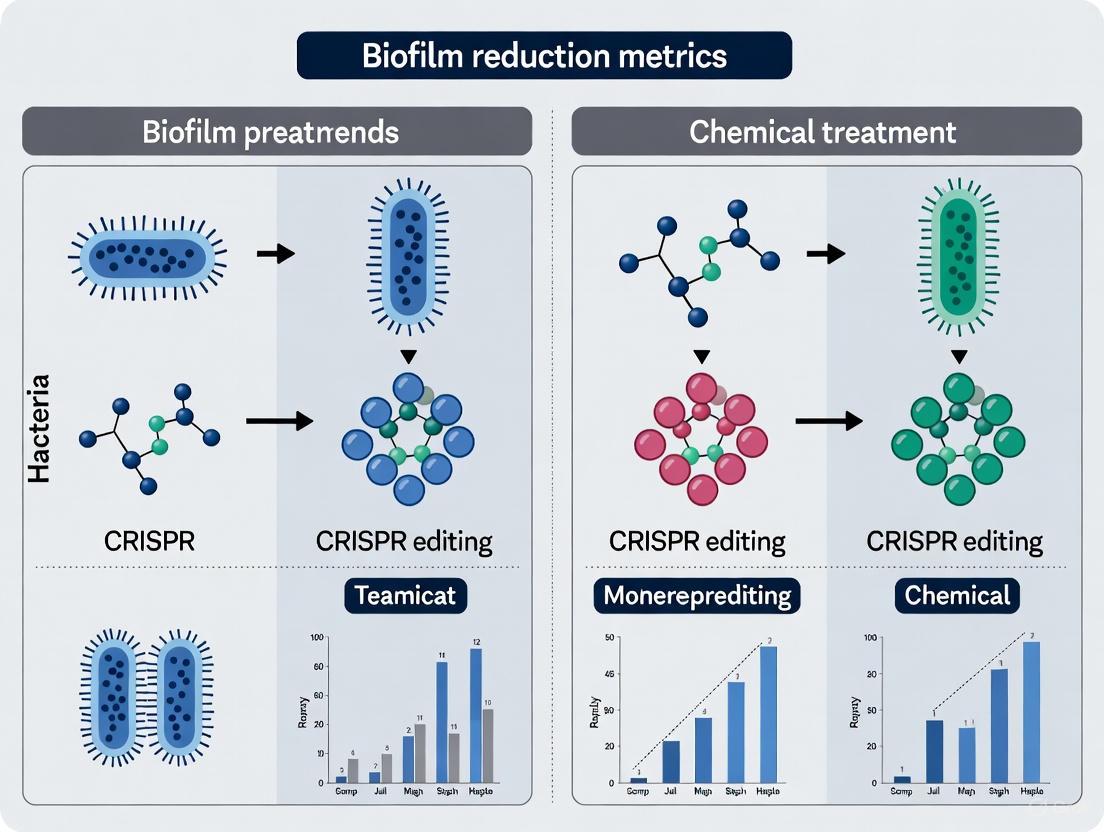
The workflow for these experimental approaches, from preparation to analysis, is summarized in the following diagram:
Experimental Workflow for Biofilm Reduction Strategies
Signaling Pathways and Molecular Mechanisms
The resilience of biofilms is governed by complex genetic networks and signaling systems. The diagrams below illustrate the key pathways targeted by the two intervention strategies.
Biofilm Regulation & CRISPR Interference
The following diagram maps the critical genetic pathways involved in biofilm formation and maturation in a model organism like P. aeruginosa, and illustrates the points of intervention for CRISPR/Cas9.
CRISPR Targets in Biofilm Genetic Pathways
Chemical Biocide Action Mechanisms
Chemical treatments exert their effects through non-specific, broad-scale mechanisms, as visualized below.
Chemical Biocide Mechanisms and Barriers
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents and Materials for Advanced Biofilm Research
| Category | Item | Function in Research | Application Example |
|---|---|---|---|
| Molecular Biology | Cas9 Nuclease | Creates double-strand breaks in target DNA sequences for gene knockout [3]. | Disruption of pelA or pslD genes in P. aeruginosa to impair EPS production [3]. |
| Guide RNA (gRNA) | Confers targeting specificity by complementary base pairing to the genomic locus of interest [3]. | Targeting quorum-sensing genes (lasI, rhlI) to inhibit biofilm maturation [4]. | |
| dCas9 (nuclease-dead) | Serves as a programmable platform for gene regulation without cutting DNA (CRISPRi/a) [4]. | Transcriptional repression of efflux pump genes to resensitize biofilms to antibiotics [4]. | |
| Nanoparticle Carriers | Lipid Nanoparticles (LNPs) | Encapsulate and protect CRISPR components, enhancing delivery and cellular uptake [3]. | Delivery of RNP complexes into P. aeruginosa biofilms, achieving >90% biomass reduction [3]. |
| Gold Nanoparticles (AuNPs) | Act as a non-viral carrier for CRISPR machinery; easily functionalized and biocompatible [3]. | Enhancing editing efficiency up to 3.5-fold compared to non-carrier systems [3]. | |
| Biofilm Assays | Crystal Violet (CV) | Stains total biofilm biomass (cells and matrix) quantitatively via absorbance measurement [6]. | Standardized metric for comparing biofilm formation across strains or treatment conditions [6]. |
| Resazurin | Viability stain; measures metabolic activity of biofilm cells fluorometrically or colorimetrically [6]. | Distinguishing between metabolic inhibition and physical biomass removal in efficacy tests [6]. | |
| Extracellular DNA (eDNA) | Critical structural component; can be targeted for disruption or used as a matrix marker [2]. | Adding DNase I to treatment regimens to degrade the eDNA scaffold and sensitize biofilms [2]. | |
| Advanced Imaging | Confocal Laser Scanning Microscopy (CLSM) | Enables 3D, non-invasive visualization of live biofilm architecture and spatial organization [5]. | Analyzing structural phenotypes (e.g., striated vs. disordered) in mutant or treated biofilms [5]. |
The "architecture of resilience" in biofilms, defined by its complex EPS composition and sophisticated ultrastructure, presents a formidable barrier to conventional antimicrobials. This comparison guide underscores a paradigm shift in R&D strategies from non-specific chemical corrosion to genetic-level deconstruction. While chemical treatments remain a practical tool, their efficacy is fundamentally limited by the very matrix they aim to destroy. The emerging CRISPR-based platforms, particularly when enhanced by nanomaterial delivery systems, offer a transformative, precision-oriented alternative capable of targeting the genetic foundations of biofilm resilience. For researchers and drug developers, the future path involves optimizing these precision tools to navigate the robust architecture of biofilms, ultimately validating new metrics for biofilm reduction that are as targeted and adaptive as the biofilms themselves.
Biofilms are structured communities of microorganisms embedded within a self-produced extracellular polymeric substance (EPS) matrix, acting as a powerful biological barrier that significantly enhances antimicrobial tolerance [7]. This protective matrix, composed of polysaccharides, proteins, and extracellular DNA (eDNA), creates a formidable obstacle to effective medical treatment, contributing to persistent infections and the global antimicrobial resistance (AMR) crisis [3] [7]. The resistance mechanisms employed by biofilm-associated bacteria are multifaceted, operating through both physical barrier-mediated protection and the formation of dormant, highly tolerant persister cell phenotypes [8] [9]. Bacterial persisters represent a metabolically dormant or slow-growing subpopulation that exhibits extreme tolerance to conventional antibiotics, which primarily target active cellular processes [8] [9]. These dormant cells can resuscitate after treatment cessation, serving as reservoirs for recurrent infections and complicating therapeutic outcomes [8]. Understanding these dual resistance mechanisms—from physical barrier function to phenotypic dormancy—is crucial for developing next-generation strategies to combat biofilm-associated infections. This guide objectively compares two innovative approaches for validating biofilm reduction metrics: precision CRISPR-based genetic editing and advanced chemical treatment strategies.
Comparative Analysis of Biofilm Reduction Strategies
Table 1: Performance Comparison of CRISPR Editing vs. Chemical Treatments for Biofilm Control
| Parameter | CRISPR-Based Gene Editing | Advanced Chemical Treatments |
|---|---|---|
| Primary Mechanism | Precision disruption of antibiotic resistance genes, quorum sensing pathways, and biofilm-regulating factors [3] | Physical/chemical disruption of biofilm matrix; direct targeting of persister cell membranes/metabolism [8] [9] [10] |
| Efficacy vs. Biofilm Biomass | Liposomal Cas9 formulations reduced P. aeruginosa biofilm by >90% in vitro [3] | Caffeine-functionalized AuNPs (Caff-AuNPs) disrupt mature biofilms and eradicate embedded dormant cells [8] |
| Efficacy vs. Persister Cells | Targets genetic basis of persistence; can be designed to reactivate dormant cells for eradication [3] [11] | Direct elimination via membrane disruption (e.g., Caff-AuNPs, AuNC@CPP) or reactivation strategies (e.g., PS+(triEG-alt-octyl)) [8] [9] |
| Specificity | High (sequence-specific gRNA targeting) [3] [11] | Variable (from broad-spectrum membrane disruption to targeted enzyme delivery) [8] [10] |
| Delivery Challenges | Significant (requires efficient delivery vectors; nanoparticles can enhance this) [3] | Moderate (nanocarriers can improve penetration and targeted release) [8] [10] |
| Synergy with Antibiotics | Enables antibiotic re-sensitization by disrupting resistance genes; nanoparticle platforms allow co-delivery [3] | High; many nanoagents designed for co-delivery, enhancing antibiotic penetration and efficacy (e.g., >40-fold reduction in required antibiotic dose) [8] [10] |
| Key Technical Hurdles | Off-target effects, delivery optimization, resistance evolution to CRISPR system [3] | Potential host cytotoxicity, stability of nanoformulations, scalable manufacturing [8] [9] |
Table 2: Quantitative Efficacy Data for Emerging Anti-Biofilm Strategies
| Therapeutic Agent / Platform | Target Organism | Experimental Model | Key Efficacy Metric | Reported Outcome |
|---|---|---|---|---|
| Liposomal Cas9 Formulations [3] | Pseudomonas aeruginosa | In vitro biofilm | Reduction in biofilm biomass | >90% reduction |
| CRISPR-Gold Nanoparticle Hybrids [3] | Model bacterial systems | In vitro delivery | Gene-editing efficiency | 3.5-fold increase vs. non-carrier systems |
| Ultrasound-Activated Nanoparticles [10] | MRSA, E. coli | In vitro biofilm & persisters | Reduction in antibiotic concentration required | >40-fold vs. biofilm; 25-fold vs. persisters |
| Caffeine-functionalized AuNPs (Caff-AuNPs) [8] | Gram-positive & Gram-negative bacteria | In vitro planktonic & biofilm-associated persisters | Bactericidal activity | Effective eradication of embedded dormant cells |
| ATP-functionalized Gold Nanoclusters (AuNC@ATP) [8] | Model bacterial systems | In vitro planktonic persisters | Reduction in persister cell population | 7-log reduction at 2.2 μM |
| Cationic Polymer PS+(triEG-alt-octyl) on PDA NPs [8] | Model bacterial systems | In vitro biofilm-associated persisters | Anti-biofilm activity | Potent clearance of persistent biofilms |
Experimental Protocols for Key Methodologies
Protocol 1: Assessing CRISPR-Cas9 Anti-Biofilm Efficacy with Nanoparticle Delivery
This protocol outlines the methodology for evaluating lipid nanoparticle (LNP)-encapsulated CRISPR-Cas9 systems targeting biofilm formation genes in P. aeruginosa [3].
Materials Required:
- Bacterial Strain: P. aeruginosa PAO1 (or other relevant biofilm-forming strain).
- CRISPR Components: LNP-encapsulated Cas9 nuclease and sgRNA targeting a biofilm-related gene (e.g., pelA for polysaccharide production or a quorum-sensing gene like lasR).
- Culture Media: Tryptic Soy Broth (TSB) or Mueller Hinton Broth (MHB).
- Biofilm Growth Substrate: 96-well polystyrene plates.
- Staining Reagent: 0.1% Crystal Violet (CV) solution.
- Destaining Solution: 30% Acetic acid.
- Detection Instrument: Microplate reader for measuring optical density at 595 nm (OD₅₉₅).
Procedure:
- Biofilm Formation: Inoculate 200 μL of diluted overnight bacterial culture (1:100 in fresh TSB) into wells of a 96-well plate. Incubate statically for 24 hours at 37°C to allow biofilm formation.
- CRISPR Treatment: Carefully aspirate planktonic cells and medium. Add LNP-encapsulated CRISPR-Cas9 constructs (e.g., at concentrations ranging from 0.1 to 10 μg/mL) in fresh medium to the pre-formed biofilms. Incubate for an additional 24 hours.
- Biofilm Quantification (CV Staining):
- Aspirate the medium and gently wash wells twice with phosphate-buffered saline (PBS) to remove non-adherent cells.
- Air-dry the plates for 45 minutes.
- Stain biofilms with 0.1% CV (150 μL per well) for 15 minutes.
- Rinse plates thoroughly under running tap water until the runoff is clear.
- Destain with 30% acetic acid (200 μL per well) for 15 minutes with gentle shaking.
- Transfer 100 μL of the destained solution to a new clean plate and measure the OD₅₉₅.
- Data Analysis: Compare the mean OD₅₉₅ of treated wells to untreated control wells. Express biofilm formation as a percentage of the control. A >90% reduction is indicative of high efficacy [3].
Protocol 2: Evaluating Metabolically Activated Nanotherapeutics Against Bacterial Persisters
This protocol details the use of light-activated, polymer-loaded nanoparticles to reactivate and kill metabolically dormant persister cells [8].
Materials Required:
- Bacterial Persisters: Prepare a persister-rich population by treating a stationary-phase culture with a high concentration of a bactericidal antibiotic (e.g., 100x MIC of ciprofloxacin for 4 hours), followed by centrifugation and washing to remove the antibiotic [9].
- Nanotherapeutic: PS+(triEG-alt-octyl) polymer loaded onto polydopamine nanoparticles (PDA NPs).
- Culture Media: Fresh cation-adjusted Mueller Hinton Broth (CA-MHB).
- Light Source: Near-Infrared (NIR) laser (e.g., 808 nm wavelength).
- Viability Stain: Resazurin dye solution or reagents for colony-forming unit (CFU) plating.
- Equipment: Microplate reader, incubator, centrifuge.
Procedure:
- Persister Cell Preparation and Treatment:
- Incubate the persister-rich cell suspension (≈10⁸ CFU/mL in PBS) with PS+(triEG-alt-octyl)PDA NPs (e.g., 50 μg/mL) for 1 hour in the dark in a multi-well plate.
- Photothermal Activation:
- Expose the plate to NIR laser light (e.g., 808 nm, 1.5 W/cm²) for 10 minutes to trigger the photothermal release of the polymer from the PDA NPs.
- Include controls without laser exposure and without nanoparticles.
- Assessment of Metabolic Activity and Viability:
- Resazurin Assay: Add resazurin solution to wells after photothermal treatment and incubate for 2-4 hours. Measure fluorescence (Ex/Em: 560/590 nm). An increase in fluorescence indicates the reactivation of bacterial metabolism.
- CFU Enumeration (Gold Standard): Serially dilute the treated and control suspensions in PBS and spot-plate onto nutrient agar plates. Incubate for 24-48 hours at 37°C and count the colonies to determine the log reduction in viable persister cells.
- Data Analysis: Calculate the percentage of metabolically reactivated cells from the resazurin assay. The log reduction in CFU/mL relative to the untreated persister control quantifies the killing efficacy. Effective treatments can achieve a >3-log reduction in viable persisters [8].
Mechanism Visualization: Signaling Pathways and Workflows
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Anti-Biofilm and Anti-Persister Studies
| Reagent / Material | Function & Application | Example Use Case |
|---|---|---|
| Lipid Nanoparticles (LNPs) | Delivery vector for encapsulating and protecting CRISPR-Cas9 components (Cas9-gRNA ribonucleoprotein or plasmid DNA), enhancing cellular uptake [3]. | Delivery of Cas9-sgRNA targeting P. aeruginosa quorum-sensing genes [3]. |
| Gold Nanoparticles (AuNPs) | Versatile nanoplatform for functionalization; can be conjugated with guide RNA, antibiotics, or bioactive molecules (e.g., caffeine). Enhances editing efficiency and facilitates combination therapy [3] [8]. | Caffeine-functionalized AuNPs (Caff-AuNPs) for direct disruption of biofilms and persisters [8]. |
| Polydopamine Nanoparticles (PDA NPs) | Bioinspired nanocarrier with excellent adhesion and photothermal properties. Allows for polymer/drug loading and light-triggered release, enhancing biofilm penetration [8]. | Delivery of PS+(triEG-alt-octyl) polymer for the "wake and kill" of dormant persisters upon NIR irradiation [8]. |
| Crystal Violet (CV) | A basic dye used for the quantitative staining of total biofilm biomass (cells and matrix). A standard, low-cost method for initial biofilm screening [6] [12]. | Quantification of biofilm formation in 96-well plates after treatment with anti-biofilm agents [12]. |
| Resazurin Viability Stain | A metabolic indicator (blue, non-fluorescent) that is reduced to resorufin (pink, fluorescent) by metabolically active cells. Used to quantify the number of viable cells within a biofilm [6]. | Monitoring the metabolic reactivation of persister cells after treatment with nanoagents [8] [6]. |
| Reactive Oxygen Species (ROS) Generating Systems | Formulations (e.g., MPDA/FeOOH-GOx@CaP) that produce hydroxyl radicals or other ROS to cause oxidative damage to cellular components, effectively killing dormant cells independent of metabolism [8] [9]. | Eradication of S. aureus and S. epidermidis persisters in prosthetic joint infection models [8]. |
| Cell-Penetrating Peptides (CPPs) | Short peptides (e.g., sequence YGRKKRRQRRR) that facilitate the translocation of cargo (e.g., nanoclusters, drugs) across bacterial membranes [8]. | AuNC@CPP nanoclusters for disrupting the proton motive force and enhancing ofloxacin efficacy against P. aeruginosa persisters [8]. |
The relentless challenge of biofilm-associated infections demands a sophisticated understanding of multifaceted resistance mechanisms, from the physical barrier of the EPS matrix to the phenotypic tolerance of persister cells. This comparison guide demonstrates that both CRISPR-based gene editing and advanced chemical nanoagents offer powerful, yet distinct, pathways for validating biofilm reduction metrics. CRISPR technology provides unparalleled precision in disrupting the genetic foundations of biofilm formation and antibiotic resistance, potentially offering a long-term solution. In parallel, chemical strategies, particularly those leveraging functionalized nanomaterials, excel at physically dismantling biofilms and directly targeting the recalcitrant persister cell subpopulation through innovative "wake and kill" or direct elimination tactics. The choice between these strategies—or their potential synergistic combination—depends on the specific research or therapeutic goals, the causative pathogen, and the clinical context. The experimental frameworks and toolkit provided here offer researchers a foundation for rigorously evaluating these next-generation anti-biofilm therapies, ultimately contributing to the global fight against antimicrobial resistance.
The global health challenge of antimicrobial resistance is profoundly exacerbated by bacterial biofilms, which are structured communities of bacteria encased in a self-produced extracellular polymeric substance (EPS) matrix [3] [7]. Biofilm-associated bacteria can be up to 1000 times more tolerant to antibiotics than their planktonic counterparts, leading to persistent infections that are notoriously difficult to eradicate [3] [13]. This resilience stems from multiple factors, including reduced metabolic activity, physical barrier properties of the EPS, and the presence of specialized "persister" cells [7]. Confronting this challenge requires a paradigm shift from traditional broad-spectrum antimicrobials toward precision strategies that selectively disrupt the fundamental regulatory networks controlling biofilm development and maintenance.
Three key regulatory systems have emerged as promising therapeutic targets for controlling biofilm-associated infections: Quorum Sensing (QS), Two-Component Systems (TCS), and cyclic di-GMP (c-di-GMP) signaling. These systems function as the master controllers of bacterial lifestyle switching, coordinating the transition from free-living planktonic cells to surface-attached biofilm communities in response to environmental cues [7] [13] [14]. This review provides a comparative analysis of two distinct approaches for targeting these systems: the genetic precision of CRISPR-based interventions versus the pharmacological approach of small molecule inhibitors, contextualized within the framework of validating biofilm reduction metrics for research and therapeutic development.
Target Systems: Mechanisms and Therapeutic Potential
Cyclic di-GMP (c-di-GMP) Signaling
Cyclic di-GMP functions as a ubiquitous bacterial second messenger that centrally regulates the transition between motile and sessile lifestyles [13] [14]. High intracellular c-di-GMP levels promote biofilm formation through multiple mechanisms: repression of flagellar motility, enhanced production of biofilm matrix components (including exopolysaccharides, proteins, and extracellular DNA), and increased antibiotic tolerance [15] [13] [16]. The molecular machinery of c-di-GMP signaling consists of diguanylate cyclases (DGCs, containing GGDEF domains) that synthesize c-di-GMP from two GTP molecules, and phosphodiesterases (PDEs, containing EAL or HD-GYP domains) that degrade the molecule [13] [14]. Pseudomonas aeruginosa alone encodes over 40 proteins with GGDEF and/or EAL domains, creating a complex, redundant regulatory network that responds to diverse environmental inputs [13].
The therapeutic potential of targeting c-di-GMP is substantial. In P. aeruginosa, the diguanylate cyclase SiaD has been identified as essential for auto-aggregation under in vivo-like conditions, such as those mimicking cystic fibrosis sputum [15]. Inhibition of SiaD by the natural compound echinacoside reduced c-di-GMP levels, decreased aggregate sizes, and potentiated tobramycin efficacy against pre-established aggregates in >80% of clinical strains tested [15]. Similarly, in Escherichia coli, genetic modulation of DgcQ expression demonstrated that c-di-GMP levels directly influence biofilm maturation capacity on biomaterial surfaces, with high c-di-GMP strains forming robust mature biofilms while low c-di-GMP strains struggled to progress beyond initial attachment [16].
Quorum Sensing (QS) Systems
Quorum Sensing enables bacterial populations to coordinate gene expression in a cell-density-dependent manner through the production, detection, and response to diffusible signaling molecules called autoinducers [7]. This intercellular communication system regulates diverse social behaviors including bioluminescence, virulence factor production, and biofilm development [7] [14]. The QS circuitry in P. aeruginosa represents one of the most extensively characterized systems, comprising Las, Rhl, and PQS hierarchies that function in a coordinated cascade to control the expression of hundreds of genes, including those encoding exopolysaccharides (Pel, Psl), biosurfactants, and secondary metabolites [7].
QS inhibition presents a compelling anti-biofilm strategy by disabling bacterial coordination without directly inducing lethal pressure. Interventions targeting QS can employ: (1) signal analogs that competitively inhibit receptor binding, (2) enzymes that degrade signaling molecules, or (3) antibodies that neutralize autoinducers [7]. The attractiveness of QS inhibition lies in its potential to attenuate virulence and biofilm formation while minimizing selective pressure for conventional resistance development.
Two-Component Systems (TCS)
Two-Component Systems represent the primary signaling mechanism by which bacteria sense and respond to environmental stimuli. A typical TCS consists of a membrane-associated histidine kinase that autophosphorylates upon detecting specific signals, and a cognate response regulator that, when phosphorylated, modulates transcription of target genes [13]. The Wsp system in P. aeruginosa exemplifies a TCS that regulates biofilm formation through c-di-GMP production. This chemosensory-like system responds to surface contact through membrane perturbation, leading to phosphorylation of the response regulator WspR, which subsequently activates its diguanylate cyclase activity to produce c-di-GMP [13]. This increased c-di-GMP pool induces production of the biofilm matrix polysaccharides Pel and Psl, cementing attachment and initiating microcolony formation [13].
Other relevant TCS include the Pil-Chp system, which senses mechanical force on type IV pili during surface attachment and activates c-di-GMP production through the diguanylate cyclase SadC [13], and the FimS-AlgR system that regulates virulence and biofilm formation in conjunction with cAMP-Vfr signaling [13]. The central positioning of TCS in transducing environmental signals into transcriptional responses makes them attractive targets for disrupting the early stages of biofilm formation.
Interventional Approaches: CRISPR vs. Chemical Targeting
CRISPR-Based Precision Targeting
The CRISPR-Cas system has evolved from a bacterial adaptive immune mechanism into a powerful programmable tool for precision genetic manipulation [3] [4] [17]. CRISPR-based antimicrobial strategies employ engineered Cas nucleases guided by synthetic RNAs to selectively target and disrupt genes essential for biofilm formation, virulence, or antibiotic resistance [3] [4]. The technology offers unprecedented sequence specificity, enabling targeted elimination of pathogens while preserving commensal microbiota—a significant advantage over broad-spectrum antibiotics [4] [18].
Multiple CRISPR platforms have been developed for biofilm control. Nuclease-active Cas9 can introduce lethal double-strand breaks in chromosomal genes encoding essential biofilm regulators [3] [4]. Alternatively, catalytically dead Cas9 (dCas9) fused to repressive or activating domains enables programmable gene silencing (CRISPRi) or activation (CRISPRa) without permanent genetic alterations [4]. More recently, RNA-targeting Cas13 effectors have been employed to degrade messenger RNAs of critical virulence genes [4]. The specificity of these systems is determined by guide RNA sequences that can be designed to target individual genes or conserved regions across multiple bacterial species.
Delivery remains a primary challenge for CRISPR-based antimicrobial applications. Nanoparticle carriers have emerged as promising vectors for protecting CRISPR components from degradation and facilitating entry into bacterial cells [3]. Lipid-based nanoparticles encapsulating Cas9 ribonucleoproteins have achieved >90% reduction in P. aeruginosa biofilm biomass in vitro [3], while gold nanoparticle conjugates have demonstrated a 3.5-fold increase in gene-editing efficiency compared to non-carrier delivery systems [3]. These hybrid platforms can also facilitate co-delivery of antibiotics or antimicrobial peptides, creating synergistic antibacterial effects [3].
Small Molecule Inhibitors
Small molecule inhibitors represent a more conventional pharmacological approach to targeting biofilm regulatory systems. These compounds typically function by binding to key enzymatic domains or receptor sites, disrupting signal transduction or synthesis. Echinacoside, a natural compound identified through virtual screening against the SiaD active site, exemplifies this approach [15]. Treatment with echinacoside reduced intracellular c-di-GMP levels, decreased aggregate sizes, and potentiated tobramycin activity against P. aeruginosa aggregates in synthetic cystic fibrosis sputum medium [15]. This synergism was demonstrated both in vitro and in vivo, with enhanced efficacy observed in 3-D alveolar epithelial cell models and murine lung infection models [15].
Small molecules targeting other regulatory systems include quorum sensing inhibitors that mimic or interfere with autoinducer signals, and two-component system inhibitors that disrupt phosphotransfer between histidine kinases and response regulators [7]. The primary advantages of small molecule approaches include well-established formulation methods, predictable pharmacokinetic profiles, and the potential for oral bioavailability. However, they may face challenges with target specificity and the development of resistance through mutation of binding sites.
Comparative Efficacy Analysis
Table 1: Comparative Analysis of CRISPR vs. Chemical Targeting Approaches
| Parameter | CRISPR-Based Approaches | Small Molecule Inhibitors |
|---|---|---|
| Mechanism of Action | Programmable DNA/RNA cleavage or gene expression modulation [3] [4] | Binding to enzymatic active sites or receptor domains [15] |
| Specificity | High sequence specificity; can distinguish between bacterial species [4] [18] | Moderate to low specificity; potential off-target effects [15] |
| Efficacy Metrics | >90% reduction in biofilm biomass (liposomal Cas9) [3]; 3.5-fold increased editing efficiency (gold nanoparticles) [3] | Reduced aggregate size; 80% of strains showed enhanced tobramycin susceptibility [15] |
| Delivery Challenges | Requires specialized nanocarriers (lipid, polymeric, or metallic nanoparticles) [3] | Conventional formulation approaches; potential penetration barriers in EPS [15] |
| Resistance Potential | Lower potential due to targeting of essential genes; escape mutants possible [4] | Moderate to high potential through mutation of binding sites [7] |
| Therapeutic Scope | Pathogen-specific elimination; microbiome preservation [4] [18] | Broad-spectrum or narrow-spectrum depending on compound [15] |
Experimental Methodologies for Biofilm Evaluation
CRISPR Workflow for Biofilm Gene Targeting
The implementation of CRISPR-based biofilm targeting follows a systematic workflow encompassing target selection, construct design, delivery optimization, and efficacy assessment [3] [4]:
Target Identification: Selection of essential biofilm regulator genes (e.g., c-di-GMP metabolism enzymes, QS regulators, TCS components) through genomic analysis and prior validation [12] [4].
gRNA Design: Computational design of guide RNA sequences with optimal specificity and minimal off-target potential. For CRISPRi/a applications, gRNAs are targeted to promoter regions to modulate transcription [4].
Delivery Vector Assembly: Construction of CRISPR-Cas cassettes in appropriate expression vectors. For nanoparticle delivery, Cas9-gRNA ribonucleoprotein complexes are preassembled and encapsulated [3].
Nanoparticle Formulation: Preparation of lipid-based, polymeric, or metallic nanoparticles loaded with CRISPR components. Characterization of size, surface charge, and encapsulation efficiency [3].
Biofilm Treatment: Application of CRISPR-nanoparticle formulations to pre-established biofilms grown in relevant models (e.g., flow cells, microtiter plates, or synthetic infection media) [3] [15].
Efficacy Assessment: Quantification of biofilm reduction using crystal violet staining (total biomass), confocal microscopy with live/dead staining (viability and structure), and colony forming unit enumeration (bacterial viability) [3] [12].
Chemical Inhibitor Evaluation Protocols
The evaluation of small molecule inhibitors targeting regulatory systems follows established pharmacological testing paradigms [15] [16]:
Compound Screening: Initial identification of candidate molecules through virtual screening against target protein structures (e.g., SiaD active site) or high-throughput phenotypic assays [15].
Dose-Response Analysis: Determination of effective concentrations (EC50) for biofilm inhibition alone and in combination with conventional antibiotics using microdilution methods in 96-well plates [15].
c-di-GMP Quantification: Measurement of intracellular c-di-GMP levels in treated versus untreated bacteria using liquid chromatography-mass spectrometry or ELISA-based methods [15].
Biofilm Architecture Analysis: Confocal laser scanning microscopy of treated biofilms using fluorescent stains (SYTO9 for cells, dextran conjugates for EPS) to visualize structural changes [12] [15].
Transcriptional Profiling: RNA sequencing or RT-qPCR analysis of genes involved in biofilm regulation to confirm mechanism of action at the molecular level [12] [16].
In Vivo Validation: Assessment of compound efficacy in relevant animal models (e.g., murine lung infection models for respiratory pathogens) [15].
Research Reagent Solutions
Table 2: Essential Research Reagents for Biofilm Regulatory Studies
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| Genetic Tools | pCas/pTargetF CRISPR-Cas9 system [16]; dCas9 repression/activation vectors [4] | Targeted gene knockout, CRISPRi/a gene regulation |
| Nanoparticle Systems | Liposomal Cas9 formulations [3]; Gold nanoparticle carriers [3] | Enhanced delivery of CRISPR components or conventional antibiotics |
| Biofilm Assay Kits | Crystal violet staining kits [12] [16]; XTT metabolic assay kits [16] | Quantification of total biofilm biomass; assessment of metabolic activity |
| Microscopy Reagents | SYTO9/green fluorescent nucleic acid stain [12]; Alexa Fluor-conjugated dextran [12] | Confocal microscopy visualization of bacterial cells and EPS matrix |
| Molecular Biology Assays | c-di-GMP ELISA kits [15]; RT-qPCR primers for biofilm genes [12] [16] | Quantification of second messenger levels; gene expression analysis |
| Specialized Growth Media | Synthetic cystic fibrosis medium (SCFM2) [15]; Lysogeny broth (LB) with supplements [16] | In vivo-like conditions for biofilm growth; routine culture with selection |
The precision targeting of key regulatory systems represents a promising frontier in combating biofilm-associated infections. CRISPR-based technologies offer unparalleled specificity for disrupting virulence and biofilm genes, while small molecule inhibitors provide familiar pharmacological properties against enzymatic targets. The comparative analysis presented herein reveals complementary strengths: CRISPR excels in pathogen-specific targeting and resistance gene elimination, whereas small molecules offer broader activity spectrum and established formulation pathways.
The most effective future strategies will likely integrate both approaches, potentially employing CRISPR to sensitize biofilms to conventional antibiotics or small molecule inhibitors. As research advances, overcoming delivery barriers for CRISPR components and optimizing the pharmacokinetic properties of regulatory inhibitors will be critical for translational success. The validated experimental frameworks and reagent systems detailed in this review provide a foundation for systematic evaluation of these emerging anti-biofilm strategies, contributing to the development of next-generation therapeutics against persistent bacterial infections.
ESKAPE pathogens represent a group of bacterial species with formidable capabilities for evading antimicrobial treatments: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species. These pathogens are responsible for the majority of nosocomial infections worldwide and pose a critical threat due to their ability to "escape" biocidal action through multiple resistance mechanisms [19]. Among these mechanisms, biofilm formation stands as a principal contributor to therapeutic failure. Biofilms are structured microbial communities encased in a self-produced extracellular polymeric substance (EPS) matrix that can exhibit 10–1000-fold greater antibiotic resistance than their planktonic counterparts [3]. The convergence of intrinsic antimicrobial resistance (AMR) and biofilm-mediated protection creates persistent, difficult-to-treat infections in clinical settings, particularly involving medical devices and compromised tissues [7]. This review systematically compares two emerging anti-biofilm strategies—CRISPR-based genetic editing and chemical anti-biofilm agents—by evaluating their efficacy metrics, mechanisms of action, and potential for clinical translation against priority ESKAPE pathogens.
Clinical Priority Assessment of ESKAPE Pathogens
The therapeutic challenge posed by ESKAPE pathogens is not uniform across species. Recent clinical surveillance data reveal distinct patterns of resistance and biofilm-forming capabilities that inform target prioritization for anti-biofilm strategies.
Table 1: Comparative Clinical Resistance and Biofilm Formation in ESKAPE Pathogens
| Pathogen | Multi-Drug Resistance (MDR) Rate | Key Resistance Markers | Biofilm Formation Capability | Strong Biofilm Producers |
|---|---|---|---|---|
| E. faecium | 90% | vanB (vancomycin), ampicillin (86.7%) | Moderate | Not specified |
| S. aureus | 10% | mecA (MRSA, 46.7%) | Moderate | Not specified |
| K. pneumoniae | High | Carbapenem (45.7%), colistin (42.9%) | High | Significant proportion |
| A. baumannii | High | Carbapenem (74.3%) | High | Significant proportion |
| P. aeruginosa | Relatively lower | Carbapenemase (variable) | Moderate (but highly structured) | Not specified |
| Enterobacter spp. | Not specified | Carbapenem (increasing) | Not specified | Not specified |
Source: PMC Analysis of 165 Clinical Isolates [19]
Among Gram-positive ESKAPE pathogens, E. faecium demonstrates alarmingly high multi-drug resistance rates (90%) compared to S. aureus (10%), with vancomycin resistance primarily mediated by the vanB gene and high-level ampicillin resistance [19]. For Gram-negative members, A. baumannii and K. pneumoniae exhibit elevated resistance to carbapenems (74.29% and 45.71%, respectively) and cephalosporins, while P. aeruginosa demonstrates relatively lower resistance profiles [19]. Of particular concern is the high rate of colistin resistance in K. pneumoniae (42.86%), impacting a last-resort antibiotic [19].
Biofilm formation prevalence is substantial across ESKAPE pathogens, with 88.5% of clinical isolates forming biofilms and 15.8% characterized as strong biofilm producers [19]. The data indicate that K. pneumoniae and A. baumannii exhibit higher biofilm-forming capabilities compared to P. aeruginosa. A significant correlation exists between biofilm formation and resistance to carbapenems, cephalosporins, and piperacillin/tazobactam (p < 0.05), suggesting biofilms play a crucial role in disseminating resistance to these antibiotic classes [19].
Comparative Efficacy of Biofilm Eradication Strategies
CRISPR-Cas9 Gene Editing Approaches
The CRISPR-Cas9 system enables precision targeting of genetic determinants underlying biofilm stability and antibiotic resistance. This technology utilizes a Cas9 nuclease and guide RNA (gRNA) complex to introduce double-strand breaks in specific DNA sequences, allowing for disruption of biofilm-related genes [3].
Table 2: CRISPR-Cas9 Anti-Biofilm Performance Against ESKAPE Pathogens
| Target Pathogen | CRISPR Delivery System | Target Genes/Functions | Biofilm Reduction Efficacy | Key Limitations |
|---|---|---|---|---|
| P. aeruginosa | Liposomal nanoparticles | quorum sensing, biofilm matrix genes | >90% biomass reduction in vitro [3] | Delivery efficiency, stability in biofilm environment |
| Multiple Gram-negative | Gold nanoparticle hybrids | antibiotic resistance genes (e.g., bla, mecA, ndm-1) | 3.5× increase in editing efficiency vs. non-carrier systems [3] | Off-target effects, resistance evolution |
| K. pneumoniae, A. baumannii | Polymer-based nanoparticles | efflux pumps, persistence pathways | Enhanced antibiotic resensitization | Limited in vivo validation |
| ESKAPE pathogens | Phage-derived vectors | virulence factors, polysaccharide synthesis | Species-specific targeting possible | Host range restrictions, immune recognition |
The integration of CRISPR-Cas9 with nanoparticle delivery platforms has significantly enhanced therapeutic potential. Liposomal Cas9 formulations have demonstrated remarkable efficacy, reducing P. aeruginosa biofilm biomass by over 90% in vitro [3]. Similarly, gold nanoparticle-CRISPR hybrids achieved a 3.5-fold increase in gene-editing efficiency compared to non-carrier systems while promoting synergistic action with conventional antibiotics [3]. These hybrid systems facilitate co-delivery of antibiotics or antimicrobial peptides, creating multifaceted approaches that attack bacterial communities through both genetic disruption and traditional antimicrobial mechanisms [3].
Chemical Anti-Biofilm Agents
Chemical approaches encompass repurposed drugs, quorum sensing inhibitors, and biofilm matrix-disrupting compounds that target the structural and regulatory integrity of biofilms.
Table 3: Chemical Anti-Biofil-m Agent Efficacy Against ESKAPE Pathogens
| Agent Category | Specific Agents | Primary Mechanism of Action | Key Efficacy Findings | Synergistic Combinations |
|---|---|---|---|---|
| Drug Repurposing | Niclosamide, Mitomycin C | Membrane disruption, QS inhibition, biofilm suppression | Antibacterial activity against resistant P. aeruginosa [20] | Multiple conventional antibiotics |
| Quorum Sensing Inhibitors | AITC, hamamelitannin analogs | Block autoinducer signaling, virulence suppression | Reduced virulence without bactericidal pressure | Potentiate vancomycin against MRSA |
| Nanoparticle-based | Metallic (Ag, Zn), lipid NPs | EPS penetration, oxidative stress, drug delivery | Enhanced biofilm penetration and retention | Antibiotic co-loading |
| Enzyme-based | DNase I, dispersin B | eDNA degradation, matrix hydrolysis | Biofilm dispersion and improved antibiotic penetration | Glycopeptides, aminoglycosides |
Drug repurposing strategies have identified compounds like niclosamide and mitomycin C that exhibit antibacterial activity through mechanisms including membrane permeability disruption, quorum sensing inhibition, and biofilm suppression [20]. Many repurposed agents demonstrate synergistic effects when combined with conventional antibiotics, potentially lowering required antibiotic concentrations and reducing selective pressure for resistance [20]. Quorum sensing inhibitors represent another promising chemical approach by targeting the cell-to-cell communication systems that coordinate biofilm development and virulence factor production without exerting direct bactericidal pressure [21].
Comparative Performance Metrics
When evaluating both strategic approaches, key differentials emerge in their potential for clinical application:
CRISPR Advantages: (1) High precision targeting of resistance genes without affecting commensals; (2) Potential reversal of existing resistance mechanisms; (3) Programmable platform adaptable to evolving threats; (4) Synergy with low-dose antibiotics [3].
Chemical Advantages: (1) Broader spectrum activity; (2) Established pharmacological and safety data for repurposed drugs; (3) Reduced development timeline and cost; (4) Simplified formulation and storage requirements [20].
Shared Challenges: (1) Limited in vivo validation data; (2) Biofilm penetration barriers; (3) Potential for resistance development even to novel mechanisms; (4) Optimization of delivery systems for target site accumulation [20] [3].
Experimental Methodologies for Anti-Biofilm Evaluation
Standardized Biofilm Cultivation Models
Reliable assessment of anti-biofilm strategies requires standardized models that recapitulate key aspects of clinical biofilms:
- Microtiter Plate Assay: High-throughput screening for initial anti-biofilm efficacy [19]
- Calgary Biofilm Device: Generates reproducible biofilm populations for susceptibility testing [7]
- Flow Cell Systems: Mimics hydrodynamic conditions of medical devices and chronic infections [7]
- MBEC (Minimum Biofilm Eradication Concentration): Standardized measurement of biofilm eradication thresholds [7]
- Polymicrobial Biofilm Models: Incorporates multi-species interactions relevant to clinical settings [22]
CRISPR-Biofilm Editing Workflow
Diagram 1: CRISPR-Biofilm Editing Experimental Workflow. The methodology begins with target identification and proceeds through construct design, delivery optimization, and comprehensive efficacy assessment.
Chemical Anti-Biofilm Screening Protocol
Standardized screening for chemical anti-biofilm agents employs a tiered approach:
- Initial Biofilm Inhibition Screening: Sub-MIC concentrations in microtiter models [20]
- Biofilm Eradication Assessment: MBEC determination against pre-formed biofilms [7]
- Mechanistic Studies: Transcriptomic analysis of quorum sensing and biofilm genes [21]
- Synergy Testing: Checkerboard assays with conventional antibiotics [20]
- Resistance Development Studies: Serial passage experiments to monitor resistance emergence [23]
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for ESKAPE Biofilm Research
| Reagent Category | Specific Products | Research Application | Technical Considerations |
|---|---|---|---|
| Biofilm Staining | Crystal violet, SYTO-9/propidium iodide (Live/Dead) | Biofilm biomass quantification, viability assessment | Crystal violet measures total biomass; fluorescence staining distinguishes viability |
| CRISPR Components | Cas9 nuclease, guide RNA constructs, nanoparticle carriers | Genetic targeting of resistance and biofilm genes | Guide RNA design critical for specificity; delivery efficiency varies by bacterial species |
| Quorum Sensing Inhibitors | Furano nes, AITC, hamamelitannin analogs | Virulence attenuation without bactericidal pressure | Sub-inhibitory concentrations to avoid resistance selection |
| Matrix Degrading Enzymes | DNase I, dispersin B, alginate lyase | EPS disruption for enhanced antibiotic penetration | Enzyme stability and activity in biofilm microenvironment |
| Microphysiological Systems | Flow cells, biofilm chips | Biofilm modeling under shear stress | Better mimics in vivo conditions than static models |
| Antibiotic Libraries | CLSI guideline antibiotics, recent clinical candidates | Resistance profiling and combination screening | Include recent antibiotics (cefiderocol, eravacycline) for comprehensive assessment |
The escalating threat of biofilm-associated infections by ESKAPE pathogens demands innovative approaches that address both microbial persistence and resistance dissemination. CRISPR-based genetic editing offers unprecedented precision in disrupting resistance determinants and biofilm regulatory networks, while chemical strategies provide broader-spectrum activity with potentially faster clinical translation. The integration of nanoparticle delivery systems significantly enhances both approaches by improving biofilm penetration and target engagement.
Future anti-biofilm development should prioritize: (1) Combination strategies that leverage the strengths of both genetic and chemical approaches; (2) Advanced delivery platforms that overcome biofilm penetration barriers; (3) Diagnostic tools that identify biofilm-associated infections early; (4) Standardized models that better recapitulate clinical biofilm heterogeneity; (5) Stewardship protocols that prevent rapid resistance emergence to novel therapies.
The comprehensive validation of biofilm reduction metrics across these platforms will be essential for translating promising in vitro results into clinical applications that address the persistent challenge of ESKAPE-associated biofilm infections.
Anti-Biofilm Arsenal: Protocols for CRISPR Editing and Chemical Treatment Application
Core Principles of gRNA Design for Bacterial Genes
Designing effective guide RNAs (gRNAs) for targeting bacterial antibiotic resistance and adhesion genes requires careful consideration of multiple biological and computational parameters. The fundamental components of the CRISPR-Cas9 system include the Cas9 nuclease and a synthetic single guide RNA (sgRNA) that directs Cas9 to specific genomic sequences through complementary base pairing [24] [25]. The gRNA recognition site typically spans approximately 20 nucleotides, with the seed sequence at the 3' end playing a critical role in target recognition specificity [24].
Two primary considerations dominate gRNA design: ensuring on-target activity (successful binding and cleavage at the intended genomic location) and minimizing off-target effects (unintended binding to partially homologous sequences) [24] [26]. Mismatches between the gRNA and target DNA, particularly in the PAM-proximal seed region, significantly reduce cleavage efficiency, though mismatches in PAM-distal positions are more tolerated [24] [27].
For biofilm-related applications, gRNAs can be designed to target essential genes involved in bacterial adhesion, extracellular polymeric substance (EPS) production, quorum sensing, and antibiotic resistance mechanisms [3] [4]. Successful disruption of these genes can resensitize bacteria to conventional antibiotics and impair biofilm formation [3].
gRNA Design Methodology and Experimental Protocols
Target Selection and Computational Design
The gRNA design process begins with identifying specific sequences within target genes that are essential for function. For antibiotic resistance genes, target conserved domains critical for antibiotic degradation or efflux; for adhesion genes, focus on regions encoding key structural motifs [3] [4]. The target must be immediately adjacent to a Protospacer Adjacent Motif (PAM) sequence (5'-NGG-3' for standard SpCas9) [27].
Computational tools are essential for predicting gRNA efficacy and specificity. Modern algorithms incorporate machine learning and neural networks trained on large datasets of gRNA performance [24]. These tools evaluate multiple parameters including GC content (optimal 40-60%), position-specific nucleotide preferences, absence of self-complementarity (which could form secondary structures), and minimal similarity to off-target sites across the genome [24] [26].
For biofilm applications, researchers have successfully designed gRNAs targeting:
- Quorum sensing genes (e.g., luxS, lasI) to disrupt cell-cell communication [4]
- Antibiotic resistance genes (e.g., bla, mecA, ndm-1) to resensitize bacteria [3]
- Adhesion genes (e.g., fimH, esp) to inhibit surface attachment [4]
- EPS production genes to compromise biofilm matrix integrity [3] [4]
Experimental Validation Protocol
Materials Required:
- Designed gRNA sequences (synthesized as crRNA:tracrRNA duplex or sgRNA)
- Cas9 nuclease (as protein, mRNA, or encoded in delivery vector)
- Appropriate delivery system (electroporation, nanoparticles, phages)
- Bacterial strains with target resistance/adhesion genes
- Antibiotics for selection pressure
- Biofilm assessment tools (crystal violet, confocal microscopy)
Procedure:
- gRNA Preparation: Synthesize designed gRNAs through in vitro transcription or commercial synthesis [26].
- CRISPR-Cas9 Delivery: Introduce Cas9 and gRNA components simultaneously using:
- Editing Validation: Isolve transformants and confirm gene editing through:
- PCR amplification of target region
- Sanger sequencing to detect indels
- Restriction fragment length polymorphism analysis if mutation creates/disrupts site
- Phenotypic Assessment: Evaluate knockout efficacy by:
Quantitative Comparison: CRISPR Editing vs. Chemical Treatments
The tables below summarize comparative performance data between CRISPR-mediated biofilm disruption and conventional chemical treatments, compiled from recent studies.
Table 1: Efficacy Metrics for Biofilm Control Strategies
| Treatment Approach | Target Specificity | Biofilm Reduction (%) | Resistance Development | Treatment Duration | Key Advantages |
|---|---|---|---|---|---|
| CRISPR-Cas9 (with nanoparticle delivery) | High (gene-specific) | 85-95% [3] | Minimal (targets DNA) | 24-48 hours | Precision targeting, resensitizes to antibiotics |
| CRISPRi (dCas9 repression) | High (gene-specific) | 70-85% [4] | None (reversible) | 12-24 hours | Tunable expression, no DNA damage |
| Chlorine-based disinfectants | Non-specific | 60-75% [4] | High (frequent) | Minutes-hours | Rapid action, low cost |
| Quaternary Ammonium Compounds | Non-specific | 50-70% [4] | Moderate | Minutes-hours | Broad spectrum, surface compatibility |
Table 2: Quantitative Performance in Biofilm-Associated Resistance Gene Targeting
| Parameter | CRISPR-Cas9 Knockout | CRISPRi Knockdown | Chemical Disinfectants |
|---|---|---|---|
| Editing Efficiency | 65-90% [25] | 70-95% (repression) [4] | Not applicable |
| Off-target Effects | 1-15% (optimized gRNAs) [24] | <5% [4] | 100% (affects all microbes) |
| Bacterial Resensitization | 3-5 log reduction in MIC [3] | 2-4 log reduction in MIC [4] | Variable, often transient |
| Biofilm Penetration | Enhanced with nanoparticles (3.5× improvement) [3] | Moderate | Good, but matrix-limited |
| Treatment Persistence | Permanent (knockout) | Temporary (during treatment) | Hours to days |
Research Reagent Solutions for CRISPR Biofilm Studies
Table 3: Essential Research Tools for gRNA Design and Validation
| Reagent/Category | Specific Examples | Function & Application | Performance Notes |
|---|---|---|---|
| gRNA Design Tools | Synthego CRISPR Design Tool, Benchling CRISPR Tool [26] | Computational gRNA selection with on/off-target scoring | Reduces design time from hours to minutes; incorporates Doench rules for efficiency prediction [26] |
| Cas9 Variants | SpCas9, eSpCas9(1.1), SpCas9-HF1 [27] | DNA cleavage with varying specificity profiles | High-fidelity variants reduce off-target effects while maintaining on-target activity [27] |
| Delivery Systems | Gold nanoparticles, Liposomal carriers [3] | Enhanced cellular uptake and biofilm penetration | Liposomal Cas9 formulations reduce P. aeruginosa biofilm by >90%; gold nanoparticles increase editing efficiency 3.5× [3] |
| Validation Reagents | T7 Endonuclease I, Surveyor Assay [25] | Detection of indel mutations at target sites | Measures editing efficiency without sequencing; confirmatory tool |
| Biofilm Assessment | Crystal violet, Confocal microscopy with fluorescent tags [3] [4] | Quantification of biofilm biomass and architecture | Enables 3D reconstruction of biofilm disruption following CRISPR treatment |
CRISPR-Cas9 technology represents a paradigm shift in biofilm control strategies, moving from non-specific chemical eradication to precision genetic targeting. The design of gRNAs for resistance and adhesion genes requires balancing multiple parameters, with the optimal approach depending on the specific experimental goals. For permanent elimination of resistance genes, CRISPR knockout approaches with high-fidelity Cas9 variants provide durable solutions, while CRISPRi offers reversible modulation for functional studies.
The integration of computational design tools with advanced delivery systems, particularly nanoparticle platforms, has significantly enhanced the efficiency and specificity of CRISPR-based biofilm interventions. When directly compared to conventional chemical treatments, CRISPR approaches demonstrate superior specificity, reduced resistance development, and the unique ability to resensitize biofilm-associated bacteria to conventional antibiotics.
As the field advances, the combination of machine learning for gRNA design with improved delivery platforms promises to further enhance the precision and efficacy of CRISPR technologies for biofilm control in both clinical and industrial settings.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system has revolutionized molecular biology by providing an unprecedented ability to edit genomes with high precision. This technology holds immense promise for treating genetic disorders, combating antibiotic-resistant infections, and advancing fundamental biological research [28]. However, the therapeutic potential of CRISPR is severely limited by a critical challenge: efficient delivery of its components into target cells [29] [28]. The CRISPR machinery—typically consisting of Cas nuclease proteins and guide RNA (gRNA)—cannot effectively enter cells independently due to its large size, negative charge, and susceptibility to degradation [29] [30].
This delivery challenge is particularly acute in the context of combating biofilm-mediated infections, where the goal is to disrupt genetic pathways controlling antibiotic resistance, quorum sensing, or biofilm formation itself [3]. Biofilms, which are structured communities of microorganisms embedded in a protective extracellular matrix, can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [3]. While CRISPR offers the potential to precisely target resistance genes within these structures, the biofilm matrix itself presents an additional barrier that delivery systems must overcome [3] [31].
Nanoparticle-based delivery systems have emerged as promising solutions to these challenges, offering advantages over both viral vectors and physical delivery methods. This guide provides a comprehensive comparison of nanoparticle carriers for CRISPR components, with particular emphasis on their application in biofilm research and therapeutic development.
Nanoparticle Platforms for CRISPR Delivery: A Comparative Analysis
Types of Nanoparticle Carriers
Various nanoparticle platforms have been developed to address the distinct requirements of CRISPR delivery, each with unique structural characteristics, loading capacities, and mechanisms of cellular interaction. The table below compares the primary nanoparticle systems used for CRISPR delivery.
Table 1: Comparison of Nanoparticle Platforms for CRISPR Delivery
| Nanoparticle Type | Key Components | CRISPR Cargo Format | Advantages | Limitations |
|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) | Ionizable lipids, phospholipids, cholesterol, PEG-lipids [30] | mRNA, RNP [30] | - Proven clinical success (COVID-19 vaccines)- Scalable production- Low immunogenicity | - Tendency to accumulate in liver- Endosomal entrapment issues [29] |
| Lipid Nanoparticle Spherical Nucleic Acids (LNP-SNAs) | LNP core with surface DNA shell [29] [32] | Cas9/gRNA RNP + DNA repair template [29] | - Enhanced cellular uptake (3× improvement)- Reduced cytotoxicity- Improved endosomal escape | - Complex synthesis- Emerging technology, limited long-term data |
| Gold Nanoparticles | Gold core with surface functionalization [3] | RNP complexes [3] | - Excellent biocompatibility- Tunable surface chemistry- Enhanced editing efficiency (3.5× improvement) [3] | - Potential long-term accumulation concerns- Limited loading capacity |
| Polymeric Nanoparticles | Cationic polymers (PEI, chitosan) [28] | DNA, RNP [28] | - High cargo capacity- Tunable degradation profiles- Cost-effective production | - Potential cytotoxicity (especially with PEI)- Heterogeneous size distribution |
| Extracellular Vesicles | Natural lipid bilayers from cells [30] | RNP, mRNA [30] | - Innate biological origin- Natural tissue targeting- Low immunogenicity | - Complex isolation and standardization- Limited production scalability |
Quantitative Performance Comparison
Recent studies have provided direct comparative data on the performance of different nanoparticle systems for CRISPR delivery, particularly in challenging applications such as biofilm disruption and genetic modification.
Table 2: Quantitative Performance Metrics of Nanoparticle CRISPR Delivery Systems
| Delivery System | Editing Efficiency (Indels) | HDR Efficiency | Biofilm Reduction | Cellular Uptake | Reference Model |
|---|---|---|---|---|---|
| Standard LNPs | 8-15% [29] | 8±4% [29] | Not reported | Baseline | Various cell lines |
| LNP-SNAs | 25-40% (2-3× improvement) [29] [32] | 21±7% (2.5× improvement) [29] | Not reported | 3× higher [29] | Various cell lines |
| CRISPR-Gold NPs | Not specified (3.5× efficiency increase) [3] | Not reported | >90% P. aeruginosa biofilm reduction [3] | Significantly enhanced | Biofilm models |
| Liposomal Cas9 | Not specified | Not reported | >90% reduction [3] | Efficient biofilm penetration | In vitro biofilm |
Experimental Protocols for Evaluating Nanoparticle-CRISPR Systems
Synthesis of LNP-SNAs for Enhanced CRISPR Delivery
The following protocol outlines the methodology for creating and testing the advanced LNP-SNA platform, which has demonstrated significant improvements in CRISPR delivery efficiency.
Table 3: Key Reagents for LNP-SNA Synthesis and Testing
| Reagent/Category | Specific Examples | Function in Experiment |
|---|---|---|
| Lipid Components | Ionizable lipids, phospholipids, cholesterol, PEG-lipids [29] | Form the nanoparticle core structure and stabilize the assembly |
| Nucleic Acids | DNA shells for SNA architecture, guide RNA, DNA repair templates [29] | Create protective surface layer and provide CRISPR functionality |
| CRISPR Components | Cas9 mRNA or protein, sgRNA, HDR templates [29] [32] | Active gene-editing machinery |
| Cell Culture | Human bone marrow stem cells, skin cells, white blood cells [29] | In vitro models for testing delivery efficiency |
| Analytical Tools | Flow cytometry, DNA sequencing, cytotoxicity assays [29] | Quantify editing efficiency and cellular health |
Procedure:
- LNP Core Formation: Prepare the lipid nanoparticle core using microfluidic mixing of ionizable lipids, phospholipids, cholesterol, and PEG-lipids in ethanol with CRISPR cargo (Cas9 ribonucleoprotein and single-guide RNA) in aqueous buffer [29].
- SNA Shell Assembly: Conjugate a dense shell of DNA strands to the LNP surface through thiol-gold chemistry or lipid-DNA conjugates. The DNA shell typically consists of short oligonucleotides (15-30 base pairs) at high density [29] [32].
- Purification and Characterization: Purify the resulting LNP-SNAs using tangential flow filtration or dialysis. Characterize the particles for size (approximately 50 nm), surface charge, and CRISPR cargo loading efficiency [29].
- Cellular Testing: Incubate LNP-SNAs with target cells (e.g., human bone marrow stem cells) at varying concentrations. Assess cellular uptake using fluorescently labeled particles [29].
- Efficacy Assessment: Measure gene-editing efficiency 48-72 hours post-delivery using next-generation sequencing of the target locus to quantify insertion-deletion mutations (indels) [29] [32].
- HDR Evaluation: When including donor DNA templates, analyze homology-directed repair efficiency using specialized reporters or sequencing methods [29].
Testing Nanoparticle-CRISPR Systems Against Bacterial Biofilms
This protocol specifically addresses the application of nanoparticle-CRISPR systems for biofilm disruption, relevant to the thesis context of validating biofilm reduction metrics.
Procedure:
- Biofilm Cultivation: Grow bacterial biofilms (e.g., Pseudomonas aeruginosa) in flow cells or 96-well plates for 24-72 hours to allow mature biofilm development with characteristic extracellular polymeric substance matrix [3].
- Nanoparticle Formulation: Encapsulate CRISPR-Cas9 components targeting antibiotic resistance genes (e.g., bla, mecA) or biofilm regulation genes (e.g., quorum sensing pathways) in selected nanoparticles (e.g., gold nanoparticles or liposomal formulations) [3].
- Treatment Application: Apply nanoparticle formulations to pre-established biofilms at defined concentrations (e.g., 100 μg/mL-1 mg/mL). Include appropriate controls (untreated, empty nanoparticles, free CRISPR components) [3].
- Biofilm Assessment: After 24-48 hours treatment, quantify biofilm biomass using crystal violet staining or confocal microscopy with fluorescent dyes. Compare reduction percentages across treatment groups [3].
- Viability and Resistance Testing: Assess bacterial viability through colony-forming unit counts and determine antibiotic susceptibility changes using minimum inhibitory concentration testing [3].
- Gene Editing Confirmation: Sequence target genomic loci to confirm precise editing of resistance genes and correlate with phenotypic changes [3].
Technical Pathways and Workflows
The following diagrams illustrate key technical pathways and experimental workflows for nanoparticle-based CRISPR delivery, particularly in the context of biofilm disruption.
LNP-SNA Architecture and Cellular Upathway
Diagram 1: LNP-SNA Cellular Internalization Pathway
CRISPR-Nanoparticle Strategy Against Biofilms
Diagram 2: Biofilm Disruption via CRISPR-Nanoparticles
Comparative Analysis: CRISPR Editing vs. Chemical Treatments for Biofilm Reduction
When evaluating biofilm reduction strategies, CRISPR-based approaches offer distinct advantages and limitations compared to conventional chemical treatments. The metrics for validation differ significantly between these modalities.
Table 4: Biofilm Reduction: CRISPR Editing vs. Chemical Treatments
| Parameter | CRISPR-Based Approaches | Conventional Chemical Treatments |
|---|---|---|
| Mechanism of Action | Precision targeting of specific genes controlling biofilm formation, resistance, or quorum sensing [3] [33] | Broad-spectrum disruption of cellular processes or physical biofilm integrity |
| Specificity | High - can target specific genetic pathways without affecting commensal bacteria [3] | Low - affects both pathogenic and beneficial microorganisms |
| Durability of Effect | Potentially permanent through heritable genetic changes [3] | Transient - requires repeated applications |
| Resistance Development | Lower potential - targets fundamental genetic elements [3] | Higher potential - selective pressure favors resistant mutants |
| Validation Metrics | - Genetic sequencing of target loci- Reduction in resistance gene transfer- Specific pathway disruption [3] [33] | - Biomass reduction- Viability counts- Metabolic activity assays |
| Delivery Challenges | Significant - requires intracellular delivery of large molecular complexes [3] [28] | Moderate - small molecules diffuse more readily through biofilm matrix |
Nanoparticle delivery systems represent a transformative approach to overcoming the critical barrier to CRISPR translation, particularly for challenging applications like biofilm eradication. The comparative data presented in this guide demonstrates that while standard lipid nanoparticles provide a foundation for CRISPR delivery, advanced systems like LNP-SNAs and CRISPR-gold nanoparticles offer significant improvements in editing efficiency and biofilm penetration.
The choice of nanoparticle platform must be guided by the specific application requirements. For biofilm disruption, the evidence indicates that gold nanoparticles and liposomal formulations currently provide the most compelling efficacy data, with demonstrated biofilm reduction exceeding 90% in model systems. However, newer technologies like LNP-SNAs show remarkable potential for genetic modification efficiency, which may translate to improved biofilm targeting as the platform develops.
When validating biofilm reduction metrics, researchers should employ a dual approach: standard quantitative assessments of biomass and viability coupled with genetic confirmation of target modification. This comprehensive validation strategy ensures that observed phenotypic changes directly result from precise genetic interventions rather than generalized toxicity. As nanoparticle delivery systems continue to evolve, their integration with CRISPR technology promises to unlock new possibilities for combating biofilm-associated infections and other challenging therapeutic targets.
Biofilms are structured communities of microorganisms embedded in a self-produced extracellular polymeric substance (EPS) matrix, which confers significant resistance to conventional antibiotics and is implicated in approximately 65-80% of all human microbial infections. [34] [35] This protective matrix creates a formidable barrier that limits antimicrobial penetration and shelters microbial cells with heterogeneous metabolic states, including dormant persister cells that exhibit extreme tolerance to treatment. [34] The escalating crisis of antimicrobial resistance has prompted intensive research into alternative strategies that target critical biofilm formation and maintenance pathways rather than directly killing bacteria, thereby potentially reducing selective pressure for resistance development. [36] [35]
Among the most promising approaches are three distinct classes of chemical agents: quorum sensing inhibitors (QSIs), which disrupt bacterial cell-to-cell communication systems; matrix-degrading enzymes, which directly dismantle the structural components of the EPS; and antimicrobial peptides (AMPs), which employ multiple mechanisms including membrane disruption and immunomodulation. [36] [37] [38] These strategies represent a paradigm shift from traditional bactericidal approaches toward anti-virulence and biofilm-disruption therapies. As research progresses, understanding the comparative efficacy, mechanisms, and appropriate applications of these agents becomes crucial for developing effective clinical interventions, especially when contrasted with emerging genetic approaches like CRISPR/Cas9 systems that target specific resistance genes. [3] This review systematically compares these three chemical agent classes through the lens of experimental validation metrics, providing researchers with structured data to inform therapeutic development.
Quorum Sensing Inhibitors (QSIs)
Mechanism of Action
Quorum sensing (QS) is a bacterial cell-to-cell communication system that utilizes small diffusible signaling molecules called autoinducers (AIs) to coordinate population-wide behaviors such as virulence factor production, biofilm formation, and antibiotic tolerance. [36] [35] As bacterial population density increases, AI concentrations rise until reaching a critical threshold that triggers changes in gene expression through receptor binding and transcriptional activation. [35] QSIs disrupt this communication through several targeted mechanisms: competitive inhibition of AI receptors, degradation of signaling molecules, interference with AI synthesis, and blockade of signal uptake systems. [36]
In Gram-negative bacteria like E. coli and Salmonella, the AI-2 signaling pathway represents a particularly important target. This system involves the LuxS-mediated synthesis of AI-2 from S-ribosyl homocysteine, its transport via the Lsr transporter complex, and intracellular phosphorylation by LsrK, leading to derepression of QS-controlled genes when AI-2-P binds the transcriptional regulator LsrR. [36] QSIs targeting this system can bind competitively to LsrB (the periplasmic AI-2 binding protein) or inhibit LuxS enzyme activity, thereby preventing signal detection and downstream virulence gene expression. [36] Similarly, in Gram-positive bacteria, QSIs typically target autoinducing peptides (AIPs) and their corresponding two-component signal transduction systems. [35]
Figure 1: Quorum Sensing Pathway and QSI Inhibition Mechanisms. QSIs disrupt bacterial communication at multiple points: inhibiting autoinducer synthesis, degrading signaling molecules, or blocking receptor binding.
Experimental Data and Efficacy
Table 1: Efficacy Metrics of Selected Quorum Sensing Inhibitors
| QSI Compound | Target Bacteria | Target Pathway | Biofilm Reduction | Key Experimental Findings |
|---|---|---|---|---|
| Halogenated furanones | E. coli, Salmonella | AI-2 signaling | 60-75% | Competitive inhibition of LsrR; downregulation of virulence genes; enhanced antibiotic susceptibility [36] |
| AHL analogs | P. aeruginosa | Las/Rhl system | 70-80% | Inhibition of virulence factor production (pyocyanin, elastase); reduced biofilm maturation [35] |
| Natural QSIs (plant-derived) | Multiple Gram-negative | Multiple QS systems | 50-65% | Synergistic effects with antibiotics; reduced EPS production without growth inhibition [39] |
| Synthetic lactonases | A. baumannii | AHL signaling | 60-70% | Degradation of AHL molecules; disruption of biofilm architecture [35] |
Experimental protocols for evaluating QSI efficacy typically involve standardized biofilm assays combined with molecular techniques. A representative methodology includes growing biofilms in 96-well plates or on relevant surfaces for 24-72 hours, treating with serially diluted QSIs, and quantifying biofilm biomass using crystal violet staining or fluorescent dyes. [36] Simultaneously, virulence factor production (e.g., pyocyanin, proteases) is measured spectrophotometrically, and QS gene expression is quantified via RT-qPCR targeting genes like lasI, lasR, rhlI, rhlR in P. aeruginosa or luxS, lsrR in E. coli and Salmonella. [36] [35] Confocal laser scanning microscopy (CLSM) with fluorescent reporter strains provides visual confirmation of biofilm architectural changes, while combination studies with sub-MIC antibiotics assess synergistic potential. [36]
Advantages and Limitations
QSIs offer significant advantages as anti-biofilm agents. Their anti-virulence approach exerts less selective pressure for resistance development compared to bactericidal agents, potentially prolonging therapeutic utility. [36] [35] Many QSIs demonstrate broad-spectrum activity against multiple bacterial species, and their ability to synergize with conventional antibiotics can resuscitate the efficacy of otherwise ineffective drugs. [36] However, QSIs face translational challenges including potential bacterial compensation through redundant signaling pathways, variable efficacy in different infection environments, and complicated pharmacokinetic profiles that may limit target site accumulation. [35] Additionally, some QSIs may exhibit host cytotoxicity at higher concentrations, necessitating careful dosing optimization.
Matrix-Degrading Enzymes
Mechanism of Action
Matrix-degrading enzymes directly target the structural integrity of the biofilm EPS, which consists primarily of exopolysaccharides, proteins, extracellular DNA (eDNA), and lipids. [37] [34] These enzymes function by catalyzing the hydrolysis of specific bonds within these macromolecules, leading to disintegration of the biofilm architecture and release of embedded bacterial cells into the planktonic state where they become more susceptible to antimicrobial agents and host immune responses. [37] The major enzyme classes include glycoside hydrolases that target exopolysaccharides, proteases that degrade protein components, and deoxyribonucleases (DNases) that cleave eDNA. [34]
The specificity of enzyme action depends on the EPS composition, which varies significantly between bacterial species and growth conditions. [37] For instance, glycoside hydrolases such as dispersin B specifically hydrolyze poly-N-acetylglucosamine (dPNAG), a key matrix component in Staphylococcus aureus, Escherichia coli, and other pathogens. [34] Proteases like proteinase K target amyloid-like proteins such as curli fibers in E. coli and Salmonella biofilms, while DNases I degrade the eDNA that provides structural stability and binding sites for cations in many biofilm types. [37] [34] The combinatorial use of multiple enzymes targeting different EPS components often produces synergistic effects due to the interconnected nature of the biofilm matrix. [37]
Experimental Data and Efficacy
Table 2: Efficacy Metrics of Selected Matrix-Degrading Enzymes
| Enzyme Class | Specific Examples | Target EPS Component | Target Bacteria | Biofilm Removal Efficacy | Key Experimental Findings |
|---|---|---|---|---|---|
| Glycoside hydrolases | Dispersin B | dPNAG | S. aureus, E. coli | 60-90% | Dose-dependent degradation; enhanced antibiotic penetration; reduction in biofilm biomass [34] |
| Proteases | Proteinase K | Amyloid proteins (curli) | E. coli, Salmonella | 50-80% | Disruption of proteinaceous matrix; effective against mature biofilms [37] [34] |
| Deoxyribonucleases | DNase I | Extracellular DNA | Multiple species | 40-70% | Reduced biofilm integrity; synergistic action with other enzymes; prevention of initial adhesion [34] |
| Alginate lyase | Alginate lyase | Alginate | P. aeruginosa | 70-85% | Specific degradation of alginate matrix; improved antibiotic efficacy in cystic fibrosis models [34] |
Standard experimental protocols for evaluating enzymatic biofilm disruption involve growing biofilms on relevant surfaces (e.g., polystyrene, medical device materials), treating with enzyme solutions at varying concentrations and exposure times, and quantifying remaining biofilm using crystal violet staining, ATP assays, or colony-forming unit (CFU) counts after disruption. [37] [34] Microscopy techniques including scanning electron microscopy (SEM) and CLSM provide visual assessment of structural changes, while enzyme synergy studies typically employ checkerboard assays with multiple enzymes and antibiotics. [37] For translational research, enzyme immobilization on catheter surfaces or nanoparticle-based delivery systems are tested for preventive efficacy against biofilm formation. [37]
Advantages and Limitations
Matrix-degrading enzymes offer several advantages as anti-biofilm agents: their high specificity minimizes damage to host tissues, they function extracellularly without requiring bacterial uptake, and resistance development is relatively uncommon. [34] Enzymes can target both growing and pre-existing biofilms, making them suitable for treatment of established infections, and their environmentally friendly profile facilitates application in food processing and industrial settings. [37] However, limitations include potential sensitivity to environmental conditions (pH, temperature, inhibitors), limited penetration into thick biofilms when applied externally, and the compositional variability of EPS between different bacterial species and strains which necessitates careful enzyme selection. [37] [34] Additionally, production costs and stability issues may present challenges for large-scale clinical application.
Antimicrobial Peptides (AMPs)
Mechanism of Action
Antimicrobial peptides are short, typically cationic peptides that exhibit broad-spectrum activity against bacteria, fungi, and viruses through multiple mechanisms of action. [38] Unlike conventional antibiotics, AMPs frequently target the bacterial membrane through electrostatic interactions with negatively charged phospholipids, leading to membrane disruption and permeability. [38] Beyond this direct membranolytic activity, many AMPs demonstrate significant anti-biofilm properties through additional mechanisms including inhibition of quorum sensing systems, suppression of genes responsible for biofilm formation, degradation of polysaccharide matrix components, and interruption of the stringent response alarmone system (p)ppGpp that promotes biofilm formation under nutrient stress. [38]
Structurally, AMPs are categorized into four groups: α-helical peptides (e.g., LL-37), β-sheet peptides (e.g., defensins), extended peptides rich in specific amino acids, and cationic loop peptides. [38] Their amphipathic nature enables interaction with both hydrophobic and hydrophilic structures, facilitating insertion into bacterial membranes. The anti-biofilm efficacy of AMPs stems from this multi-target approach, which simultaneously disrupts membrane integrity, interferes with signaling pathways, and degrades structural matrix components, making development of resistance more difficult compared to single-target agents. [38] Additionally, some AMPs demonstrate immunomodulatory functions by recruiting immune cells to infection sites, further enhancing biofilm clearance.
Experimental Data and Efficacy
Table 3: Efficacy Metrics of Selected Antimicrobial Peptides
| Antimicrobial Peptide | Class | Target Bacteria | Biofilm Reduction | Key Mechanisms | Additional Findings |
|---|---|---|---|---|---|
| LL-37 | α-helical CATHELICIDIN | P. aeruginosa, S. aureus | 65-80% | Membrane disruption, QS inhibition, eDNA binding | Suppresses virulence genes; synergistic with antibiotics; immunomodulatory [38] |
| Human β-defensin-3 | β-sheet DEFENSIN | Multiple Gram-positive | 50-70% | Membrane permeabilization, inhibition of adhesion | Effective against pre-formed biofilms; reduces initial attachment [38] |
| DJK-5 | Synthetic | Multiple Gram-negative | 70-90% | (p)ppGpp alarmone inhibition | Targets stringent response; reverses antibiotic tolerance in persister cells [38] |
| Plectasin | Fungal defensin | Gram-positive | 60-75% | Cell wall synthesis inhibition, membrane disruption | Potent against drug-resistant strains; maintains activity in biofilm environments [38] |
Methodologies for assessing AMP anti-biofilm activity include minimum biofilm inhibitory concentration (MBIC) and minimum biofilm eradication concentration (MBEC) assays using peg lids or 96-well plates, with resazurin staining or CFU enumeration for viability assessment. [38] Specific molecular techniques include RT-qPCR to measure changes in expression of biofilm-related genes (e.g., icaADBC in staphylococci, psl and pel in pseudomonads), and reporter strain assays to quantify QS inhibition. [38] Microscopy approaches including CLSM with live/dead staining and atomic force microscopy provide visualization of membrane damage and biofilm structural changes, while surface plasmon resonance can characterize AMP binding to bacterial membrane components. [38]
Advantages and Limitations
AMPs offer several advantages as anti-biofilm agents: their multiple mechanisms of action make resistance development less likely compared to conventional antibiotics, they frequently demonstrate broad-spectrum activity, and many exhibit both anti-biofilm and immunomodulatory properties. [38] Additionally, AMPs can target metabolically inactive persister cells within biofilms that are refractory to most antibiotics, and their rapid bactericidal action can prevent biofilm regeneration. [38] However, significant challenges include potential cytotoxicity toward host cells at higher concentrations, susceptibility to proteolytic degradation in physiological environments, poor pharmacokinetic profiles with rapid clearance, and manufacturing costs for complex peptide structures. [38] These limitations have spurred development of engineered synthetic analogs with improved stability and reduced toxicity.
Comparative Analysis and Research Applications
Cross-Strategy Comparison
Table 4: Comparative Analysis of Anti-Biofilm Chemical Agents
| Parameter | QS Inhibitors | Matrix-Degrading Enzymes | Antimicrobial Peptides |
|---|---|---|---|
| Primary mechanism | Interference with bacterial signaling | Enzymatic degradation of EPS matrix | Membrane disruption & multiple intracellular targets |
| Resistance potential | Low to moderate | Low | Low |
| Spectrum of activity | Species-specific | Substrate-specific (varies by EPS) | Broad-spectrum |
| Efficacy against mature biofilms | Moderate | Moderate to high | Moderate to high |
| Synergy with antibiotics | Strong | Moderate to strong | Strong |
| Toxicity concerns | Low to moderate | Low | Moderate to high (cytotoxicity) |
| Stability in vivo | Variable | Variable (protease sensitivity) | Low (protease sensitivity) |
| Optimal application context | Prophylaxis, early infection | Surface treatment, medical devices | Acute infections, topical applications |
| Development status | Preclinical/early clinical | Some clinical applications | Preclinical/early clinical |
When compared to emerging genetic approaches like CRISPR/Cas9 systems, which offer precise targeting of antibiotic resistance genes but face significant delivery challenges, these chemical agents provide more immediate therapeutic potential with simpler administration. [3] CRISPR/Cas9 systems excel at permanently disrupting resistance mechanisms but require sophisticated nanoparticle delivery platforms and face regulatory hurdles. [3] In contrast, QSIs, matrix-degrading enzymes, and AMPs offer more conventional pharmacological profiles while still representing innovative approaches to biofilm control.
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key Research Reagents for Anti-Biofilm Studies
| Reagent/Category | Specific Examples | Primary Research Application | Key Considerations |
|---|---|---|---|
| QS Reporter Strains | P. aeruginosa LasB-GFP, E. coli LsrR-GFP | QSI screening and mechanism validation | Enable real-time monitoring of QS activity; require standardization of growth conditions [36] |
| Biofilm Quantification Assays | Crystal violet, resazurin, SYTO stains | Biomass and viability measurement | Crystal violet measures total biomass; viability stains differentiate live/dead cells; complementary approaches recommended [40] |
| EPS Composition Analysis | FITC-conjugated lectins, Congo red, FRET substrates | Matrix characterization and enzyme targeting | Differentiate polysaccharide types; assess enzyme accessibility to substrates [37] [34] |
| Synergy Screening Platforms | Checkerboard microdilution, time-kill assays | Combination therapy development | Identify additive/synergistic interactions with antibiotics; complex experimental design [36] [38] |
| Advanced Imaging Tools | CLSM, SEM, atomic force microscopy | Structural and architectural analysis | Provide 3D visualization of biofilm disruption; require specialized equipment and expertise [40] |
Figure 2: Experimental Workflow for Anti-Biofilm Agent Validation. Standardized methodology for evaluating the efficacy of QSIs, matrix-degrading enzymes, and AMPs incorporates multiple complementary assessment techniques.
Research Implications and Future Directions
The comparative analysis of these anti-biofilm strategies reveals distinct yet complementary strengths, suggesting that optimal clinical approaches will likely involve combination therapies rather than monotherapies. QSIs show particular promise for prophylactic applications and early infection intervention, while matrix-degrading enzymes offer advantages for surface treatment and medical device coatings. [36] [37] AMPs provide potent broad-spectrum activity suitable for acute infections but require further engineering to address stability and toxicity concerns. [38] Future research directions include developing sophisticated delivery systems such as enzyme-functionalized nanoparticles, engineering AMP analogs with improved pharmacological properties, and exploring triple-combination approaches that simultaneously target signaling, matrix integrity, and bacterial viability. [3] [34]
The validation of biofilm reduction metrics requires special consideration of the distinct mechanisms of action for each agent class. While QSI efficacy is best measured through virulence gene expression and signal molecule quantification in addition to traditional biomass assays, enzyme effectiveness requires analysis of specific EPS component degradation, and AMP activity necessitates membrane integrity assessment and persister cell elimination metrics. [36] [37] [38] As research progresses, standardized methodologies for cross-agent comparison will be essential for translating laboratory findings into clinical applications that effectively address the persistent challenge of biofilm-associated infections.
In the pursuit of effective strategies against biofilm-associated infections, the selection of appropriate in vitro models is paramount for generating reliable, translatable data. Biofilms, structured microbial communities embedded in a self-produced extracellular polymeric substance (EPS), exhibit resistance mechanisms up to 1,000 times greater than their planktonic counterparts [41]. This resilience poses a significant challenge in medical and industrial settings, driving the need for research models that accurately simulate biofilm architecture and functional heterogeneity. The validation of novel therapeutic approaches, particularly precision tools like CRISPR-Cas systems versus broad-spectrum chemical treatments, depends fundamentally on the biofilm model employed [3] [42]. Models range from simple, high-throughput static systems to complex dynamic environments that mimic host conditions, each with distinct advantages and limitations. This guide provides an objective comparison of standardized in vitro biofilm models, detailing their experimental protocols, data output, and relevance for evaluating cutting-edge antibiofilm strategies, to aid researchers in selecting the optimal system for their specific research questions in drug development.
Biofilm Model Classifications and Core Characteristics
In vitro biofilm models are broadly categorized into static and dynamic systems, a classification reflecting fundamental differences in hydrodynamic conditions and nutrient supply that profoundly influence biofilm development and therapeutic testing outcomes [43].
Static systems, such as the microtiter plate assay, are characterized by the absence of fluid flow during biofilm growth. These systems are batch cultures where biofilms develop in a non-replenished nutrient medium, leading to progressive nutrient depletion and waste accumulation. The microtiter plate is the most ubiquitous static model, prized for its simplicity, cost-effectiveness, and high-throughput capabilities, making it ideal for initial screening of antibiofilm compounds or assessing the biofilm-forming capacity of numerous bacterial strains [43] [44]. However, a significant limitation is that the biofilms generated often cannot develop into mature, complex structures typical of natural infections, due to the lack of continuous nutrient supply and shear forces [43].
Dynamic systems, in contrast, involve a continuous flow of fresh medium over the developing biofilm. This category includes models like flow cells, drip-flow reactors, and constant depth film fermenters [45]. The continuous flow mimics in vivo conditions more closely by providing a constant supply of nutrients, removing waste products, and subjecting the biofilm to shear stress. This environment promotes the formation of mature biofilms with complex three-dimensional architectures, including microcolonies and water channels, which are critical for studying biofilm physiology and resistance in situ [41] [46]. While dynamic systems are more complex and resource-intensive, they provide superior biological relevance for evaluating treatments against established, mature biofilms.
Table 1: Comparison of Static vs. Dynamic Biofilm Model Systems
| Feature | Static Models (e.g., Microtiter Plate) | Dynamic Models (e.g., Flow Cell) |
|---|---|---|
| Hydrodynamics | No fluid flow; stagnant conditions | Continuous laminar or complex flow; shear stress present |
| Nutrient Supply | Batch culture; depletes over time | Continuous fresh medium; non-depleting |
| Throughput | High (e.g., 96- or 384-well plates) | Low to Medium (limited by setup and imaging) |
| Cost & Complexity | Low cost; technically simple | Higher cost; technically complex |
| Biofilm Architecture | Often less complex, monolayer or thin layers | Develops complex 3D structures (e.g., towers, streamers) |
| Common Applications | Initial screening, biofilm formation assays, genetic studies | Studying mature biofilms, antimicrobial penetration, spatial competition |
| Data Reproducibility | Good inter-laboratory reproducibility for CV and resazurin [44] | Can vary with flow rate and chamber geometry [46] |
Comparative Analysis of Standardized Biofilm Models
Beyond the broad static/dynamic classification, several models have been standardized for specific research applications. The key to selecting a model lies in aligning its strengths with the research objective, whether for high-throughput compound screening or the nuanced study of biofilm ecology.
The Microtiter Plate Model: The Workhorse for Screening
The microtiter plate assay is the most widely used static model. Its protocol involves incubating a bacterial inoculum in the wells of a polystyrene plate, allowing cells to adhere and form a biofilm. After incubation, non-adherent cells are removed by washing, and the adherent biofilm is quantified, typically using crystal violet (CV) staining for total biomass or resazurin for metabolic activity [43] [44]. Its primary advantage is its unparalleled utility in high-throughput screening (HTS). For instance, it has been effectively used to screen the antimicrobial effect of essential oils against S. aureus biofilms and to evaluate the efficacy of peracetic acid on E. coli biofilm elimination [43].
However, this model has critiques. The CV stain does not differentiate between live cells, dead cells, and the EPS matrix, which can lead to overestimation of viable biomass [47]. Furthermore, a key limitation is its inability to support the development of highly complex, mature biofilm structures found in vivo [43]. An interlaboratory study demonstrated that while microtiter plate methods exhibit good repeatability and reproducibility, the choice of assessment method (CV, resazurin, or plate counts) significantly impacts results, with plate counts showing the best responsiveness in antimicrobial efficacy tests [44].
Flow Cell Models: Mimicking Natural Environments
Flow cell models are dynamic systems where biofilms grow on a surface, such as a glass coverslip, under a continuous, controlled flow of medium [46]. This setup allows for real-time, non-destructive observation of biofilm development using techniques like confocal laser scanning microscopy (CLSM). The constant nutrient supply and shear force enable biofilms to progress through all developmental stages, forming intricate 3D structures like mushroom-shaped towers and sieve-like streamers that are highly relevant to natural and clinical settings [46].
The core strength of flow cells is their high biological relevance, making them ideal for studying biofilm architecture, spatial organization, and the dynamics of interspecies interactions. For example, research using porous environments with complex flow has shown how matrix-producing P. aeruginosa can create sheltered niches that allow non-producing mutants to coexist, a finding impossible to observe in static systems [46]. This model is indispensable for validating the efficacy of treatments, including CRISPR-Cas systems, against structurally mature biofilms. The main trade-offs are the low throughput, higher technical skill required, and greater resource consumption.
Advanced and Specialized Model Systems
For more niche applications, other models offer unique capabilities:
- Sorbarod Perfusion System: This model uses cellulose filters ("Sorbarods") perfused with medium, creating a gradient of nutrients and oxygen. It is particularly effective for growing dense, tissue-like biofilms and has been used to study microcosm biofilms from oral samples and the efficacy of antimicrobials like nisin [45].
- Drip-Flow Reactor (DFR): The DFR simulates low-shear environments, such as those found in chronic wounds. Biofilms grow on a surface positioned at an angle, with medium dripping over them. This model is applicable for studying biofilm growth under conditions similar to mucosal surfaces and has been used to evaluate the effect of chlorhexidine on S. mutans biofilms [45].
- Constant Depth Film Fermenter (CDFF): The CDFF maintains biofilms at a constant depth by using a rotating turntable and scraper blade. This is useful for studying biofilms over extended periods without the loss of structure due to overgrowth, and has been used in both defined-species and microcosm biofilm studies [45].
Table 2: Overview of Standardized In Vitro Biofilm Models
| Model Name | System Type | Key Feature | Best Use Case | Limitations |
|---|---|---|---|---|
| Microtiter Plate | Static | High-throughput, cost-effective | Initial compound screening, genetic studies | Limited biofilm complexity, endpoint analysis only |
| Flow Cell | Dynamic | Real-time imaging, complex 3D architecture | Studying biofilm development & structure, spatial competition | Low throughput, technically demanding |
| Calgary Biofilm Device | Static | Generates multiple, identical biofilms | Generating biofilm-specific MIC (MBIC) | Not suitable for structural analysis |
| Drip-Flow Reactor (DFR) | Dynamic | Low-shear, air-liquid interface | Modeling wound & mucosal surface biofilms | Lower throughput than microtiter plates |
| Sorbarod Perfusion | Dynamic | High-density, gradient formation | Growing dense microcosm biofilms, antibiotic penetration | Destructive sampling, less common |
| Constant Depth Film Fermenter (CDFF) | Dynamic | Controlled, uniform biofilm depth | Long-term biofilm studies, age-related studies | Complex setup and operation |
Experimental Protocols for Key Models
Microtiter Plate (96-well) Biofilm Assay with Crystal Violet
This is a foundational protocol for biomass quantification [43] [44].
- Inoculum Preparation: Grow bacteria overnight in an appropriate broth (e.g., Tryptic Soy Broth - TSB). Dilute the culture to a standardized optical density (e.g., ~5.5 Log10 CFU/mL) in fresh, often nutrient-enhanced, medium (e.g., TSB + 1% glucose).
- Biofilm Growth: Dispense 200 µL of the diluted inoculum into each well of a sterile, polystyrene, flat-bottom 96-well plate. Include negative control wells with sterile medium only. Incubate the plate statically for a defined period (e.g., 24-48 hours) at the optimal temperature for the strain (e.g., 37°C for pathogens).
- Washing: Carefully remove the planktonic culture from the wells by inverting and flicking the plate. Gently wash the adhered biofilms twice with 200-300 µL of phosphate-buffered saline (PBS, pH 7.2-7.4) to remove non-adherent cells.
- Fixation and Staining: Air-dry the plate. Add 200 µL of a crystal violet solution (e.g., 0.1% w/v) to each well and incubate at room temperature for 10-20 minutes.
- Destaining and Quantification: Carefully rinse the plate under running tap water to remove excess stain. Allow the plate to dry. Add 200 µL of a destaining solution (e.g., 30% acetic acid or 70-96% ethanol) to each well to solubilize the CV bound to the biofilm. Transfer the solubilized CV to a new plate or measure the optical density directly at 595 nm using a microplate reader.
Flow Cell Biofilm Assay for Microscopy
This protocol enables real-time observation of 3D biofilm development [46].
- Assembly and Sterilization: Assemble the flow cell system, typically consisting of a glass coverslip as the substratum, a flow channel, and inlet/outlet ports. Sterilize the entire system, often by autoclaving or flushing with sterile ethanol and water.
- Inoculation: Dilute an overnight bacterial culture to the desired density in a minimal medium or a diluted nutrient broth. Stop the flow and introduce the bacterial inoculum into the flow cell channel via the injection port, ensuring the entire channel is filled. Allow the cells to attach by incubing without flow for 1-2 hours.
- Biofilm Growth: Initiate a continuous, low-rate flow (e.g., 0.1 - 0.5 mm/s) of sterile growth medium using a peristaltic pump or syringe pump. Maintain the system at a constant temperature for the duration of the experiment (e.g., 3-5 days).
- Staining (if required): For viability assessment or specific component staining, introduce fluorescent dyes (e.g., SYTO-9 for live cells, propidium iodide for dead cells, Concanavalin A for polysaccharides) into the flow channel and incubate in the dark before resuming flow to remove excess dye.
- Image Acquisition and Analysis: Visualize the biofilm using confocal laser scanning microscopy (CLSM). Acquire Z-stacks at various positions to reconstruct the 3D architecture. Analyze images using software like ImageJ (with plugins like ComStat/ComStat2) or IMARIS to quantify parameters such as biomass, thickness, and surface coverage.
Model Selection for CRISPR vs. Chemical Treatment Research
The choice between these models is critical when the research goal is to compare the mechanistic action and efficacy of precision CRISPR-based therapies against conventional chemical treatments.
Microtiter Plates for High-Throughput CRISPR Screening: The static microtiter plate is ideal for the initial functional screening of CRISPR guide RNAs (gRNAs) or the efficacy of CRISPR-Cas delivery systems [47]. Researchers can engineer bacteria to constitutively express fluorescent or bioluminescent proteins, allowing for independent quantification of each species in a dual-species biofilm when targeted by species-specific CRISPR systems [47]. This model allows for rapid testing of hundreds of gRNA targets against genes essential for biofilm formation (e.g., for adhesion, quorum sensing, or matrix production) before moving to more complex models.
Flow Cells for Evaluating Penetration and Spatial Effects: The dynamic flow cell is essential for validating whether CRISPR-Cas systems, often delivered via nanoparticles or engineered phages, can penetrate the complex EPS matrix of a mature biofilm and reach their target cells effectively [3] [46]. While chemical disinfectants may act on the biofilm periphery, a key advantage of CRISPR is its potential to target specific genes in cells residing in the protected depths of the biofilm. Flow cells allow researchers to visualize this penetration and assess the spatial distribution of killing, which is crucial for demonstrating a superior mechanism of action over broad-spectrum chemicals.
Diagram 1: A decision workflow for selecting a biofilm model based on the mechanism of action (MOA) question being investigated, particularly relevant for CRISPR vs. chemical treatment research.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful execution of biofilm experiments requires specific reagents and materials. The following table details essential solutions for the protocols described in this guide.
Table 3: Essential Research Reagents for Biofilm Studies
| Reagent / Material | Function/Description | Example Application in Protocols |
|---|---|---|
| Crystal Violet (0.1-1% w/v) | A cationic dye that binds negatively charged cell surfaces and EPS, quantifying total adhered biomass. | Staining and quantifying biofilms in microtiter plate assays [43] [44]. |
| Resazurin Sodium Salt | A cell-permeant blue dye reduced to pink, fluorescent resorufin by metabolically active cells; measures viability. | Assessing metabolic activity of biofilm cells after treatment in microtiter plates [47] [44]. |
| Phosphate Buffered Saline (PBS) | An isotonic, non-toxic buffer used for washing and diluting cells without causing osmotic shock. | Washing away non-adherent planktonic cells in microtiter plate and flow cell protocols [43] [44]. |
| SYTO 9 / Propidium Iodide | Fluorescent nucleic acid stains for live/dead differentiation (green/red fluorescence). | Determining cell viability within 3D biofilm structures in flow cells via CLSM [47] [41]. |
| Polystyrene Microtiter Plate | A standard, treated plastic surface for cell attachment and growth in a high-throughput format. | The substrate for biofilm growth in the static microtiter plate model [43] [44]. |
| Flow Cell Chamber with Glass Coverslip | Provides a controlled flow environment and an optically clear surface for high-resolution microscopy. | The core component for growing and visualizing biofilms under dynamic conditions [46]. |
| Tryptic Soy Broth (TSB) with Glucose | A rich, general-purpose growth medium; adding 1% glucose enhances biofilm formation in many species. | Standard growth and biofilm formation medium for pathogens like S. aureus and E. coli [43] [44]. |
The journey from microtiter plates to flow cell biofilms represents a trade-off between throughput and biological relevance. For researchers validating biofilm reduction metrics for CRISPR versus chemical treatments, a synergistic approach is most powerful. The microtiter plate offers an unmatched platform for the initial high-throughput screening of CRISPR guides and antimicrobial compounds. Subsequently, the flow cell model becomes indispensable for confirming that observed efficacy translates to complex, mature biofilms and for visualizing the superior penetration and targeted killing promised by precision therapies. By understanding the capabilities and limitations of each standardized model, scientists can design robust experimental pipelines that generate reliable, translatable data, ultimately accelerating the development of next-generation antibiofilm strategies.
Overcoming Technical Hurdles: Optimization Strategies for Maximum Efficacy
The therapeutic potential of CRISPR gene editing is immense, yet its efficacy is fundamentally constrained by two interdependent challenges: the design of highly efficient guide RNAs (gRNAs) and the development of effective delivery systems. While CRISPR machinery can theoretically correct genetic defects underlying countless diseases, its practical application faces significant biological barriers. gRNAs must demonstrate high on-target activity while minimizing off-target effects, and delivery vehicles must successfully transport fragile CRISPR components to target cells without triggering immune responses or suffering degradation before reaching their destination. The convergence of artificial intelligence (AI)-optimized gRNA design with advanced nanoparticle engineering represents a transformative approach to overcoming these limitations, particularly in complex applications such as biofilm disruption where precision and efficiency are paramount.
gRNA Design Optimization with Artificial Intelligence
AI-Driven Design Frameworks
Traditional gRNA design relied on empirical rules and modest machine learning models, but these approaches often struggled to capture the complex determinants of gRNA activity and off-target effects. Recent advances in artificial intelligence, particularly deep learning, have revolutionized gRNA design by learning predictive features from large-scale CRISPR datasets. These models can ingest not only the gRNA and target DNA sequences but also additional contextual information such as chromatin accessibility, DNA methylation status, and epigenetic markers, yielding more accurate forecasts of on-target cleavage efficiency [48].
Table 1: AI Models for gRNA Design Optimization
| Model/Approach | Key Features | Performance Advantages |
|---|---|---|
| CRISPRon | Integrates gRNA sequence features with epigenomic information (e.g., chromatin accessibility) | More accurate efficiency rankings of candidate guides compared to sequence-only predictors [48] |
| CRISPR-Net | Combines convolutional neural networks (CNNs) and bidirectional gated recurrent units (GRUs) | Analyzes guides with up to four mismatches or indels relative to targets for cleavage activity prediction [48] |
| Multitask Models | Jointly learns both on-target efficacy and off-target cleavage predictions | Internalizes trade-offs in sequence features that enhance one versus the other; reveals subtle sequence motifs modulating Cas9 specificity [48] |
| Croton | Variant-aware deep learning pipeline predicting indel spectrum | Accounts for local sequence and nearby genetic variants; enables personalization of gRNA design for patient-derived cells with SNPs [48] |
Explainable AI and Safety Considerations
While AI has significantly boosted predictive performance, the "black box" nature of complex models presents challenges for clinical translation where understanding failure modes is critical for safety. Explainable AI (XAI) techniques are now being integrated to illuminate the logic behind model predictions, highlighting which nucleotide positions in the guide or target contribute most to activity or specificity. These insights not only build user confidence but can also reveal biologically meaningful patterns, such as sequence motifs that affect Cas9 binding or cleavage [48]. For therapeutic applications, comprehensive off-target evaluation remains essential, as CRISPR edits can sometimes lead to large unintended mutations or vary across genetic backgrounds.
Nanoparticle Engineering for Enhanced CRISPR Delivery
Advanced Nanoparticle Platforms
Efficient delivery of CRISPR components remains a fundamental challenge, with lipid nanoparticles (LNPs) emerging as a promising non-viral vector option. Recent innovations have significantly enhanced LNP performance through structural engineering. Northwestern University researchers have developed lipid nanoparticle spherical nucleic acids (LNP-SNAs), which carry the full CRISPR toolkit—Cas9 enzymes, guide RNA, and DNA repair templates—wrapped in a dense, protective shell of DNA [29].
Table 2: Nanoparticle Delivery Systems for CRISPR
| Delivery System | Key Characteristics | Experimental Performance |
|---|---|---|
| LNP-SNAs | DNA coating shields cargo, facilitates cell entry via receptor interactions, enables organ/tissue targeting | 3x more effective cell entry than standard LNPs; 3x boost in gene-editing efficiency; >60% improvement in precise DNA repairs; far less toxicity [29] |
| Liposomal Cas9 Formulations | Lipid-based nanoparticles encapsulating CRISPR components | >90% reduction in Pseudomonas aeruginosa biofilm biomass in vitro [3] |
| CRISPR-Gold Nanoparticle Hybrids | Gold nanoparticles as carriers for CRISPR components | 3.5x enhancement in editing efficiency compared to non-carrier systems; promotes synergistic action with antibiotics [3] |
| Standard LNPs | Traditional lipid nanoparticles without DNA coating | Natural liver affinity; used in successful in vivo trials for liver-focused diseases; enables redosing potential due to low immunogenicity [49] |
Mechanisms of Enhanced Delivery
The architectural advantages of advanced nanoparticle systems like LNP-SNAs are multifactorial. The spherical nucleic acid architecture is recognized by almost all cell types, promoting active cellular uptake and rapid internalization. The DNA shell not only provides protection but can also be engineered with sequences that target specific cell surface receptors, making delivery more selective. This structural approach contrasts with conventional LNPs, which frequently become trapped in endosomes where they cannot release their cargo. The modular nature of these platforms allows adaptation for various therapeutic applications, with seven SNA-based therapies already in human clinical trials for other applications [29].
Experimental Protocols for Validation
gRNA Efficiency Verification
Validating gRNA editing efficiency is a critical step before proceeding with downstream applications. Several methods are available, each with distinct advantages and limitations:
Next-Generation Sequencing (NGS): Considered the gold standard, targeted NGS provides extremely sensitive detection of editing outcomes through high-throughput deep sequencing of the region of interest. This method offers comprehensive data on indel patterns but requires significant time, resources, and bioinformatics expertise [50] [51].
Inference of CRISPR Edits (ICE): This user-friendly online tool uses Sanger sequencing data to determine relative abundance and levels of indels. ICE analysis provides editing efficiency scores (ICE scores) corresponding to indel frequency and detailed information on different indel types and distributions. Validation studies show high correlation with NGS results (R² = 0.96) [51].
Tracking of Indels by Decomposition (TIDE): An older decomposition method for analyzing Sanger sequencing data from edited samples. TIDE provides estimation of relative abundance of insertions or deletions but has limitations in detecting longer or more complex indels without manual parameter adjustments [51].
T7 Endonuclease 1 (T7E1) Assay: A non-sequencing based approach that detects mismatched DNA heteroduplexes through enzymatic cleavage. This method is rapid and inexpensive but provides only qualitative assessment of editing without nucleotide-level detail [51].
Biofilm Reduction Assessment
For research focused on biofilm disruption, specific methodologies are required to quantify intervention efficacy:
Biomass Quantification: Following treatment with CRISPR-nanoparticle formulations, biofilm biomass can be measured using crystal violet staining or similar approaches. Liposomal Cas9 formulations have demonstrated over 90% reduction in Pseudomonas aeruginosa biofilm biomass in vitro [3].
Resistance Gene Targeting: gRNAs can be designed to disrupt antibiotic resistance genes (e.g., bla, mecA, ndm-1) or quorum-sensing pathways (e.g., luxS, fimH in E. coli), resensitizing bacteria to conventional antibiotics [3] [52]. The success of these interventions is measured through subsequent antibiotic susceptibility testing and biofilm viability assays.
Synergistic Treatment Evaluation: Combined approaches utilizing CRISPR to disrupt resistance mechanisms alongside traditional antibiotics demonstrate enhanced efficacy. Nanoparticles can be engineered to co-deliver CRISPR components and antimicrobial agents, creating a multifaceted therapeutic strategy [3].
Comparative Performance Data
Quantitative Efficacy Metrics
The integration of optimized gRNA designs with advanced nanoparticle delivery systems produces measurable improvements in editing efficiency and functional outcomes:
Table 3: Comparative Performance of CRISPR Enhancement Strategies
| Application Context | Intervention Strategy | Efficiency Metrics | Reference |
|---|---|---|---|
| Biofilm Disruption | Liposomal Cas9 targeting resistance genes | >90% reduction in P. aeruginosa biofilm biomass in vitro | [3] |
| Gene Editing Efficiency | CRISPR-gold nanoparticle hybrids | 3.5x enhancement in editing efficiency vs. non-carrier systems | [3] |
| Cellular Delivery | LNP-SNAs vs. standard LNPs | 3x more effective cell entry; 3x boost in editing efficiency | [29] |
| Precision Editing | LNP-SNAs with repair templates | >60% improvement in precise DNA repair success rates | [29] |
| Therapeutic Protein Reduction | LNP-delivered CRISPR for hATTR (TTR protein) | ~90% reduction in disease-related protein levels sustained over 2 years | [49] |
| Therapeutic Protein Reduction | LNP-delivered CRISPR for HAE (kallikrein) | 86% reduction in target protein; 8 of 11 patients attack-free at 16 weeks | [49] |
Advantages Over Traditional Methods
Compared to traditional gene editing approaches like Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs), CRISPR-based editing offers significant advantages in simplicity, cost efficiency, and scalability. While ZFNs and TALENs can achieve high specificity through protein-based targeting, they require extensive protein engineering for each new target, making them time-consuming and expensive to develop. CRISPR systems, by contrast, simply require modification of the gRNA sequence to redirect targeting, enabling rapid prototyping and high-throughput applications [53]. The integration of AI-optimized gRNA design with nanoparticle delivery further amplifies these advantages, potentially overcoming CRISPR's historical limitations in off-target effects while maintaining its core benefits of accessibility and versatility.
Research Toolkit: Essential Reagents and Materials
Table 4: Essential Research Reagents for CRISPR-Nanoparticle Experiments
| Reagent/Material | Function | Example Applications |
|---|---|---|
| TrueGuide Synthetic gRNAs | Pre-designed, validated gRNAs for specific targets | Positive controls (e.g., targeting human HPRT, AAVS1 loci); experimental gRNAs [50] |
| GeneArt Genomic Cleavage Detection Kit | Rapid evaluation of indel formation efficiency | Estimation of CRISPR-Cas9-mediated cleavage efficiency in pooled cell populations [50] |
| Lipid Nanoparticle Formulations | Encapsulation and delivery of CRISPR components | In vitro and in vivo delivery of ribonucleoprotein complexes [29] [3] |
| Gold Nanoparticle Carriers | Enhanced delivery with intrinsic antibacterial properties | CRISPR component delivery to biofilm environments; synergistic action with antibiotics [3] |
| NGS Library Prep Kits | Preparation of sequencing libraries for edited regions | Comprehensive analysis of editing outcomes and off-target assessment [50] [51] |
| ICE or TIDE Analysis Software | Computational tools for editing efficiency quantification | Analysis of Sanger sequencing data to determine indel frequencies and types [51] |
| Crystal Violet Staining Assay | Biofilm biomass quantification | Measurement of biofilm reduction following CRISPR intervention [3] |
The convergence of artificial intelligence-optimized gRNA design with structurally advanced nanoparticle delivery systems represents a paradigm shift in CRISPR-based interventions. Experimental data consistently demonstrates that this integrated approach can enhance editing efficiency by several-fold while improving specificity and reducing off-target effects. For biofilm research and other complex applications, these developments enable unprecedented precision in targeting resistance mechanisms and pathogenic behaviors. As both computational design tools and delivery platforms continue to evolve, researchers are equipped with increasingly sophisticated methodologies to address fundamental challenges in genetic medicine, antimicrobial resistance, and therapeutic development. The standardized protocols and validation frameworks outlined here provide a roadmap for rigorous assessment of these emerging technologies across diverse application domains.
Overcoming the extracellular polymeric substance (EPS) barrier is a pivotal challenge in eradicating biofilm-associated infections. This guide compares the efficacy of two advanced strategies—CRISPR-loaded nanocarriers and synergistic physical-chemical disruption—by analyzing experimental data on their ability to penetrate the biofilm matrix and achieve targeted action.
Experimental Data at a Glance
The following table summarizes quantitative outcomes from key studies, highlighting the distinct performance profiles of each approach.
| Therapeutic Strategy | Key Experimental Findings | Biofilm Reduction / Disruption | Target Organism / Model |
|---|---|---|---|
| CRISPR/Nanoparticle Hybrids | Liposomal Cas9 formulations reduced biofilm biomass by >90% in vitro [3]. Gold nanoparticle carriers enhanced gene-editing efficiency by ~3.5-fold compared to non-carrier systems [3]. | High, precision-targeted | Pseudomonas aeruginosa ( in vitro ) [3] |
| Shockwave + Antibiotics | Shockwave treatment (120 pulses at 2 Hz) detached up to 97.5% of biofilm surface area. Combined with ciprofloxacin, it reduced bacterial viability by 40% and increased bacterial cell death to 67% [40]. | High, broad-spectrum disruption | Pseudomonas aeruginosa biofilm on silicone tube ( in vitro ) [40] |
| Anti-CRISPR: Cas3 Gene Deletion | Deletion of the cas3 gene in the Type I-Fa CRISPR system led to a significant reduction in biofilm formation and its structural thickness [12]. | N/A (Biofilm formation inhibited) | Acinetobacter baumannii [12] |
Detailed Experimental Protocols
A clear understanding of the methodologies is crucial for evaluating and reproducing these results.
Protocol for CRISPR/Nanoparticle Evaluation
This protocol focuses on testing the efficacy of lipid-based nanoparticles (LNPs) for delivering CRISPR-Cas9 components.
- 1. Nanoparticle Formulation: CRISPR-Cas9 ribonucleoproteins (RNPs) or plasmid DNA encoding Cas9 and guide RNA (gRNA) are encapsulated within lipid nanoparticles via microfluidics. The LNPs are engineered with fusogenic lipids to promote endosomal escape and can be surface-functionalized with targeting ligands (e.g., peptides that bind to biofilm components) [3].
- 2. Biofilm Cultivation: Mature biofilms are grown on relevant substrates (e.g., silicone, polystyrene) for 72-96 hours under dynamic flow conditions to mimic a realistic, three-dimensional structure [3] [40].
- 3. Treatment and Incubation: The formulated CRISPR-nanoparticles are applied to the pre-formed biofilms. Co-delivery with sub-inhibitory concentrations of antibiotics (e.g., tobramycin) is often used to produce a synergistic effect. The system is incubated for 6-24 hours [3].
- 4. Efficacy Assessment:
- Biomass Quantification: Biofilm biomass is measured using crystal violet (CV) staining, with absorbance read at 600 nm (OD600) [40].
- Bacterial Viability: Treated biofilms are disaggregated via sonication and vortexing. Serial dilutions are plated on agar to enumerate Colony Forming Units (CFU/ml) [40].
- Structural Analysis: Confocal Laser Scanning Microscopy (CLSM) with live/dead staining (SYTO9/PI) visualizes the 3D architecture and spatial distribution of live/dead cells within the biofilm [40] [12].
Protocol for Shockwave and Antibiotic Synergy
This protocol outlines a physical disruption method to enhance conventional antibiotic efficacy.
- 1. Biofilm Formation in Tubular Structure: P. aeruginosa biofilms are cultivated on the inner surface of silicone tubes for 3 days under dynamic conditions, with fresh nutrient media supplied periodically [40].
- 2. Shockwave Application: A modified intravascular lithotripsy (IVL) balloon catheter is inserted into the tubing. Biofilms are treated with 120 shockwave pulses at an energy of 4 kV and a frequency of 2 Hz in a saline bath at 37°C [40].
- 3. Antibiotic Exposure: Immediately following shockwave treatment, biofilms are perfused with a solution of 4 µg/mL ciprofloxacin for 6 hours [40].
- 4. Efficacy Assessment:
- Detachment Analysis: CV staining and Scanning Electron Microscopy (SEM) are used to qualify and quantify the removal of biofilm from the tube surface [40].
- Viability and Cell Death: CFU counts and CLSM with live/dead staining are performed to quantify bacterial killing and the proportion of dead (red-stained) cells [40].
Signaling Pathways and Workflows
The diagrams below illustrate the core mechanisms and experimental workflows for the two strategies.
The Scientist's Toolkit: Essential Research Reagents
The table below lists key materials and reagents required to implement the described experimental approaches.
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Lipid Nanoparticles (LNPs) | Carrier for in vivo delivery of CRISPR-Cas9 components; protects payload and facilitates cellular uptake [3] [49]. | Systemic delivery of CRISPR therapy to liver cells [49]. |
| Gold Nanoparticles | Metallic nanocarrier for CRISPR components; enhances editing efficiency and stability [3]. | In vitro delivery of Cas9/gRNA to bacterial biofilms [3]. |
| Shockwave IVL Catheter | Medical device generating high-pressure acoustic waves for physical biofilm disruption in tubular structures [40]. | Disrupting P. aeruginosa biofilms on silicone catheter surfaces [40]. |
| Crystal Violet (CV) Stain | Dye that binds to biomass; standard method for quantifying total biofilm formation and detachment [40] [12]. | Quantifying biofilm biomass after shockwave or CRISPR treatment [40]. |
| Live/Dead BacLight Kit (SYTO9/PI) | Fluorescent stains for differentiating live (green) and dead (red) bacterial cells via Confocal Laser Scanning Microscopy (CLSM) [40]. | Assessing bacterial viability and spatial distribution within 3D biofilm structures post-treatment [40]. |
| Ciprofloxacin | Fluoroquinolone antibiotic; used in combination therapies to target bacteria after biofilm disruption [40]. | Treating biofilms after shockwave-mediated EPS disruption [40]. |
| dCas9 (for CRISPRi/a) | Catalytically "dead" Cas9; used for gene knockdown (CRISPRi) or activation (CRISPRa) without DNA cleavage [4] [33]. | Reversible gene silencing in biofilm regulatory network studies [4] [33]. |
Biofilm-associated infections represent a formidable challenge in modern medicine, contributing significantly to the global antibiotic resistance crisis. These structured microbial communities encase themselves in a protective extracellular polymeric substance (EPS), exhibiting tolerance to antimicrobial agents up to 1,000-fold greater than their free-floating counterparts [3]. This resilience arises from a dual defense strategy: a physical barrier that limits antibiotic penetration and a physiological state featuring metabolic heterogeneity and persistent cells [3] [54]. Confronting this, the field is moving beyond monotherapeutic approaches towards innovative combinations that attack resistance simultaneously on multiple fronts. Two particularly promising strategies have emerged: CRISPR-chemical hybrid therapies, which use gene editing to disarm bacterial genetics, and antibiotic potentiation, which uses adjuvants to restore the efficacy of existing antibiotics. This guide provides a comparative analysis of these approaches, focusing on their efficacy, mechanisms, and practical application in biofilm reduction, to inform researchers and drug development professionals.
CRISPR-Chemical Hybrid Therapy: Precision Gene Editing Meets Advanced Delivery
CRISPR-Cas systems offer a revolutionary approach to combating biofilm-related resistance by enabling precise targeting and disruption of key genetic determinants. This strategy moves beyond traditional growth inhibition to selectively disable genes responsible for antibiotic resistance, virulence, and biofilm formation itself [55] [3].
Core Mechanisms of Action
The system functions as a prokaryotic adaptive immune system, repurposed for precise genetic manipulation. Its activity requires two components: the Cas nuclease, which creates double-strand breaks in DNA, and a guide RNA (gRNA), which directs the nuclease to a specific genomic target sequence [3]. When applied to biofilms, the technology can be directed against several critical targets:
- Antibiotic Resistance Genes (ARGs): By targeting and cleaving genes like bla (β-lactamase), mecA (methicillin resistance), or ndm-1 (carbapenem resistance), CRISPR-Cas can resensitize bacterial populations to existing antibiotics [55] [54].
- Quorum Sensing (QS) Pathways: Genes such as luxS are crucial for bacterial cell-to-cell communication that coordinates biofilm formation. Disrupting these pathways can prevent biofilm maturation and enhance susceptibility to antimicrobials [52].
- Biofilm Structural Genes: Targeting genes involved in adhesion (e.g., fimH in E. coli) and the production of extracellular polymeric substances can directly compromise biofilm integrity [52].
Quantitative Efficacy Data
The integration of nanoparticles as delivery vehicles has dramatically enhanced the performance of CRISPR-based antibacterials, as shown in the table below.
Table 1: Efficacy of Select CRISPR-Chemical Hybrid Therapies Against Biofilms
| Target System / Pathogen | Delivery Vehicle | Editing Efficiency / Biofilm Reduction | Key Outcome |
|---|---|---|---|
| P. aeruginosa biofilm [3] | Liposomal Cas9 formulations | >90% reduction in biofilm biomass (in vitro) | Significant disruption of biofilm structure and viability. |
| General ARG delivery [3] | Gold nanoparticle carriers | 3.5-fold increase in gene-editing efficiency | Enhanced delivery and efficacy compared to non-carrier systems. |
| E. coli biofilm [52] | CRISPR/Cas9-HDR (targeting luxS & fimH) | Significant reduction in biofilm formation | Knockout of quorum sensing and adhesion genes impaired biofilm development. |
| hATTR (Human, in vivo) [49] | Lipid Nanoparticles (LNP) | ~90% reduction in disease-related protein (TTR) | Proof-of-concept for efficient systemic in vivo delivery and effect. |
Antibiotic Potentiation: Rescuing Legacy Antibiotics with Synergistic Partners
Antibiotic potentiation refers to the strategy of using non-antibiotic agents, known as potentiators or adjuvants, to enhance the efficacy of existing antibiotics against resistant strains, including those in biofilms [56]. These compounds typically have minimal standalone antimicrobial activity but disrupt specific bacterial defense mechanisms.
Core Mechanisms of Action
Potentiators employ diverse tactics to overcome resistance, which can be categorized as follows:
- Membrane Permeabilization: Antimicrobial peptides (AMPs) like Esc(1-18) and Tachyplesin III can integrate into and disrupt bacterial membranes. This increases membrane permeability, facilitating the entry of co-administered antibiotics such as amikacin and piperacillin-tazobactam that would otherwise be excluded [57].
- Efflux Pump Inhibition: Potentiators can block bacterial efflux pumps (e.g., RND family pumps), which normally expel antibiotics like fluoroquinolones and tetracyclines from the cell. This allows intracellular antibiotic concentrations to reach lethal levels [54] [56].
- Enzyme Inhibition: A primary mechanism of resistance is enzymatic degradation of antibiotics, such as by β-lactamases. Potentiators like clavulanic acid inhibit these enzymes, protecting the co-administered antibiotic (e.g., amoxicillin) and restoring its activity [56].
- Biofilm Disruption: Some agents can interfere with the EPS matrix or quorum sensing, breaking down the physical and regulatory architecture of the biofilm. This allows antibiotics to penetrate deeper and target the embedded bacterial cells more effectively [57].
Quantitative Efficacy Data
The synergistic effects of AMP-antibiotic combinations have been demonstrated in numerous in vitro studies.
Table 2: Synergistic Efficacy of Select Antibiotic-Potentiator Combinations
| Potentiator | Antibiotic | Target Bacteria | Synergistic Outcome |
|---|---|---|---|
| Tachyplesin III [57] | Piperacillin-tazobactam | P. aeruginosa | Enhanced efficacy against biofilm-producing strains. |
| Colistin [57] | Tobramycin | P. aeruginosa | Potentiation observed in vitro. |
| Citropin 1.1 [57] | Clarithromycin, Doxycycline | R. equi | Synergistic activity demonstrated. |
| G10KHc [57] | Tobramycin | P. aeruginosa | Increased bacterial killing. |
| Gaegurin 6 [57] | Chlorhexidine, Xylitol | Oral streptococci | Improved antimicrobial effect. |
Direct Comparative Analysis: Mechanisms and Workflows
The following diagrams and tables provide a side-by-side comparison of the fundamental principles and experimental considerations for these two strategies.
Diagram 1: A comparative workflow of the two therapeutic strategies. The CRISPR-chemical hybrid path (blue) involves a precise genetic targeting process, while the antibiotic potentiation path (red) focuses on disabling resistance mechanisms.
Table 3: Strategic Comparison: CRISPR-Chemical Hybrids vs. Antibiotic Potentiation
| Aspect | CRISPR-Chemical Hybrid Therapy | Antibiotic Potentiation |
|---|---|---|
| Primary Mechanism | Targets and disrupts genetic determinants of resistance and biofilm formation [55] [3]. | Chemically inhibits resistance mechanisms (enzymes, efflux pumps) or disrupts biofilm matrix [57] [56]. |
| Key Advantage | High precision; potential for permanent genetic change and resensitization [3]. | Broadly applicable; can rapidly rescue existing antibiotics; simpler regulatory path for some agents [56]. |
| Key Challenge | Efficient and safe in vivo delivery; potential for off-target effects; microbial evolutionary pushback (e.g., anti-CRISPRs) [3] [58]. | Ensuring co-localization and synchronized pharmacokinetics; potential toxicity of combinatory regimens [57] [56]. |
| Ideal Use Case | Targeted eradication of resistant clones in chronic infections; "re-sensitizing" therapies [55] [54]. | Broad-spectrum rescue of first-line antibiotics; topical or lock-therapy applications [57] [56]. |
Experimental Protocols for Biofilm Reduction Studies
For researchers aiming to validate and compare these approaches, robust and standardized experimental models are essential. Below are detailed protocols for assessing efficacy in vitro.
Protocol: Assessing CRISPR-Cas9 Anti-Biofilm Efficacy with Nanoparticle Delivery
This protocol evaluates the ability of nanoparticle-delivered CRISPR systems to reduce pre-formed biofilms.
- Biofilm Formation: Grow a static biofilm of the target bacterium (e.g., P. aeruginosa or E. coli) in a 96-well plate or on a relevant substrate (e.g., catheter piece) for 24-48 hours [52].
- Treatment Preparation:
- Treatment Application: Gently wash the pre-formed biofilm to remove planktonic cells. Add the nanoparticle formulations to the biofilm and incubate for a set period (e.g., 4-24 hours).
- Biofilm Quantification:
- Biomass Assay: Use crystal violet staining to measure total biofilm biomass. Compare the absorbance of treated vs. control wells to calculate percentage reduction [52].
- Viability Assay: Perform an MTT assay or plate counts on homogenized biofilm to determine the number of viable cells.
- Imaging: Use Confocal Laser Scanning Microscopy (CLSM) to visualize the 3D architecture of the biofilm and assess structural disruption [3].
Protocol: Evaluating Potentiator-Antibiotic Synergy against Biofilms
This protocol determines the synergistic potential of an antibiotic-potenitiator combination using a checkerboard assay adapted for biofilms.
- Checkerboard Setup: In a 96-well plate, prepare a two-dimensional dilution series. Vary the concentration of the antibiotic along one axis and the potentiator (e.g., an AMP) along the other [57].
- Biofilm Inoculation and Incubation: Inoculate each well with a standardized bacterial suspension. Incubate to allow biofilm formation (e.g., 24 hours) in the presence of the drug combinations.
- Synergy Analysis:
- Endpoint Measurement: Quantify biofilm using crystal violet or cell viability using resazurin.
- FIC Index Calculation: Calculate the Fractional Inhibitory Concentration (FIC) index for each combination.
- FIC Index = (MIC of antibiotic in combination / MIC of antibiotic alone) + (MIC of potentiator in combination / MIC of potentiator alone)
- Interpretation: An FIC Index of ≤0.5 indicates synergy; >0.5 to 4.0 indicates additivity/indifference; and >4.0 indicates antagonism [57].
- Validation: Confirm synergy against pre-formed biofilms by treating established biofilms with the most promising combinations identified in the checkerboard assay and quantifying reduction as in Protocol 5.1.
The Scientist's Toolkit: Essential Research Reagents
Successfully implementing the aforementioned protocols requires a suite of specialized reagents and materials.
Table 4: Key Research Reagent Solutions for Anti-Biofilm Therapeutic Development
| Reagent / Material | Function / Application | Specific Examples |
|---|---|---|
| CRISPR-Cas Systems | Core gene-editing machinery for targeted genetic disruption. | Type II Cas9 (SpCas9) [55], smaller orthologues (e.g., S. uberis Cas9) for better delivery [59], Type V Cas12a [58]. |
| Nanoparticle Delivery Systems | Protect and deliver CRISPR components; enhance biofilm penetration. | Lipid Nanoparticles (LNPs) [49] [3], Gold Nanoparticles [3], Polymeric Nanoparticles [58]. |
| Guide RNA (gRNA) Design Tools | In silico design of specific gRNA sequences to minimize off-target effects. | Tools for predicting on-target efficiency and off-target activity (implied in [58] [59]). |
| Antimicrobial Peptides (AMPs) | Act as potentiators by disrupting membranes or biofilms. | Tachyplesin III, Colistin, Citropin 1.1, G10KHc, Gaegurin 6 [57]. |
| Enzyme Inhibitors | Potentiators that block antibiotic-degrading enzymes. | Clavulanic acid (β-lactamase inhibitor) [56], Taniborbactam (carbapenemase inhibitor) [54]. |
| Biofilm Assay Kits | Standardized tools for quantifying biofilm biomass and viability. | Crystal violet staining kits, Metabolic activity assays (MTT, resazurin) [52]. |
| In Vitro Biofilm Models | Systems to grow structured biofilms for therapeutic testing. | 96-well plate models [52], CDC biofilm reactors, Catheter and other medical device substrates [52]. |
Diagram 2: Molecular action sites of the two strategies. The CRISPR-chemical hybrid (top, blue background) acts inside the cell nucleus on bacterial DNA. In contrast, antibiotic potentiators (bottom, red background) act on extracellular and membrane-bound targets to disable resistance mechanisms.
Mitigating Resistance Evolution to Monotherapies
Bacterial biofilms, structured communities encased in an extracellular polymeric substance (EPS), represent a significant challenge in clinical and industrial settings due to their profound resistance to antimicrobial treatments. This resistance is multifaceted, arising from both physical barrier properties that limit antibiotic penetration and physiological heterogeneity within bacterial populations, including the presence of dormant persister cells [3] [7]. The EPS matrix, composed of polysaccharides, proteins, and extracellular DNA, creates a protected environment where bacteria can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts [3]. Traditional monotherapeutic approaches, whether based on conventional antibiotics or emerging biological agents, often prove insufficient against this robust defense system, frequently selecting for resistant variants and leading to treatment failure. Understanding the limitations of these monotherapies and exploring integrated combinatorial strategies is therefore essential for developing effective anti-biofilm interventions that mitigate resistance evolution.
Comparative Analysis of Anti-Biofilm Monotherapies
Conventional Chemical Antibiotics
Mechanism and Limitations: Conventional antibiotics typically target specific bacterial cellular processes such as cell wall synthesis, protein production, or DNA replication. However, in biofilm environments, their efficacy is substantially compromised. The EPS matrix acts as a diffusion barrier, physically limiting antibiotic penetration while also creating chemical microenvironments that neutralize certain antimicrobial compounds [3] [7]. Within biofilms, bacterial populations exhibit heterogeneous metabolic activity, with subpopulations of slow-growing or dormant persister cells that are inherently tolerant to antibiotics that require active cellular processes for their activity [3]. This physiological heterogeneity, combined with the potential for enhanced horizontal gene transfer within the biofilm matrix, accelerates the development and spread of genetic resistance determinants [3].
CRISPR-Based Antimicrobials
Precision Mechanism: CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas systems function as programmable, sequence-specific antimicrobials by targeting and inactivating essential genes, virulence factors, or antibiotic resistance genes within bacterial pathogens [3] [60]. Unlike broad-spectrum antibiotics, CRISPR-based systems can be designed to selectively eliminate specific bacterial strains based on their genetic signature, potentially preserving beneficial microbiota [4]. These systems utilize a guide RNA (gRNA) that directs the Cas nuclease to cleave target DNA sequences, resulting in lethal double-strand breaks or functional gene knockout [61] [60].
Implementation Challenges: Despite their precision, CRISPR antimicrobials face significant delivery challenges, particularly through the protective biofilm matrix [3]. Effective in vivo application requires efficient delivery vehicles such as engineered bacteriophages or nanoparticle carriers to transport CRISPR components to their intended bacterial targets [3] [60]. Additionally, bacteria can develop resistance to CRISPR systems through target sequence mutations or anti-CRISPR protein expression, potentially limiting long-term efficacy [60].
Physical Disruption Methods
Alternative Approaches: Physical methods such as shockwave therapy represent a non-chemical approach to biofilm disruption. Shockwaves are high-pressure acoustic waves that generate mechanical stress and cavitation effects, physically compromising the EPS matrix structure [40]. This disruption enhances permeability, allowing concomitant antibiotics to penetrate more effectively into the biofilm depths [40].
Table 1: Quantitative Comparison of Anti-Biofilm Monotherapy Performance
| Therapy Type | Reported Efficacy | Key Advantages | Major Limitations | Resistance Concerns |
|---|---|---|---|---|
| Conventional Antibiotics | Variable; often <50% reduction in biofilm viability [7] | Established clinical use, broad-spectrum activity | Poor biofilm penetration, metabolic heterogeneity | High; selects for genetic resistance via horizontal gene transfer [3] |
| CRISPR-Based Antimicrobials | Up to 3.5× increased editing efficiency with nanoparticle delivery [3] | Precision targeting of resistance genes, programmable | Delivery challenges through EPS, off-target effects | Moderate; potential for target mutation and anti-CRISPR mechanisms [60] |
| Shockwave Therapy | 97.5% biofilm detachment combined with antibiotics [40] | Physical disruption, enhances antibiotic penetration | Limited standalone efficacy, device-dependent | Low; physical mechanism less prone to biological resistance |
Experimental Validation: Methodologies and Metrics
CRISPR-Based Biofilm Intervention Protocols
CRISPR Interference (CRISPRi) for Gene Silencing: The catalytically inactive dCas9 protein can be employed for targeted gene repression without DNA cleavage. This approach is particularly valuable for studying essential genes and validating potential anti-biofilm targets [33].
Table 2: Essential Research Reagents for CRISPR Biofilm Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| CRISPR System Components | dCas9 (catalytically dead Cas9), Guide RNA (gRNA) | Target gene recognition and binding without cleavage [33] |
| Induction System | PtetA promoter, Anhydrotetracycline (aTc) | Controlled expression of CRISPR components [33] |
| Delivery Vehicles | Lipid-based nanoparticles, Gold nanoparticles, Engineered bacteriophages | Enhanced cellular uptake and protection of genetic material [3] |
| Biofilm Assessment Tools | Crystal violet staining, Confocal Laser Scanning Microscopy (CLSM) | Quantification of biofilm biomass and 3D architecture visualization [12] [40] |
| Viability Assays | SYTO9/PI staining, Colony-forming unit (CFU) counts | Differentiation between live/dead bacteria and quantitative viability assessment [40] |
Experimental Workflow:
- System Design: Select target genes (e.g., quorum sensing regulators, EPS production genes, antibiotic resistance determinants) and design complementary gRNAs [33].
- Vector Construction: Clone gRNA sequences into appropriate expression vectors alongside dCas9 under inducible promoters [33].
- Transformation: Introduce CRISPR constructs into target bacteria using electroporation or conjugation methods [60].
- Biofilm Formation: Allow biofilms to develop on relevant surfaces (e.g., silicone tubes, microtiter plates) under conditions mimicking natural environments [40].
- Gene Expression Modulation: Induce CRISPR system with appropriate inducer (e.g., aTc) during early or mature biofilm stages [33].
- Phenotypic Assessment: Quantify biofilm biomass, architecture, and bacterial viability using methods outlined in Table 2 [12] [40].
Chemical Treatment Protocols
Combined Shockwave and Antibiotic Therapy: This combinatorial approach physically disrupts biofilms before chemical treatment, enhancing antibiotic efficacy [40].
Experimental Workflow:
- Biofilm Development: Grow Pseudomonas aeruginosa biofilms on silicone tube interiors for 72 hours under dynamic flow conditions to simulate natural biofilm formation [40].
- Shockwave Application: Treat biofilms with focused shockwaves (120 pulses at 2 Hz) using an intravascular lithotripsy system to mechanically disrupt EPS structure [40].
- Antibiotic Exposure: Immediately follow shockwave treatment with ciprofloxacin (4 µg/mL for 6 hours) to target bacteria with compromised protection [40].
- Efficacy Assessment: Evaluate treatment success through multiple metrics: bacterial viability (CFU counts), biofilm biomass (crystal violet staining), and structural integrity (scanning electron microscopy) [40].
Diagram 1: Mechanism of action: CRISPR vs. chemical monotherapies. CRISPR precisely targets genetic elements while chemicals face multiple barriers.
Integrated Strategies to Overcome Monotherapy Limitations
The limitations of monotherapies have prompted research into integrated approaches that target multiple vulnerabilities simultaneously. The combination of CRISPR precision with nanoparticle delivery systems represents a particularly promising strategy. Lipid-based and gold nanoparticles can protect CRISPR components from degradation and enhance their penetration through biofilm matrices, with studies demonstrating up to 90% reduction in Pseudomonas aeruginosa biofilm biomass when using liposomal Cas9 formulations [3]. These hybrid systems facilitate synergistic effects by enabling co-delivery of CRISPR constructs with conventional antibiotics or other antimicrobial agents, simultaneously targeting genetic resistance determinants and exploiting physical biofilm disruption [3].
Diagram 2: Combinatorial strategy for overcoming biofilm resistance. Integrated approaches target multiple resistance mechanisms simultaneously.
The evolving challenge of biofilm-associated infections necessitates a paradigm shift from monotherapeutic approaches to integrated, multi-target strategies. While conventional antibiotics face penetration and resistance barriers, and CRISPR-based systems encounter delivery challenges, their strategic combination offers a promising path forward. The integration of physical disruption methods to compromise biofilm integrity, nanoparticle-mediated delivery to enhance therapeutic penetration, and CRISPR precision to target genetic resistance elements represents a comprehensive approach that minimizes the evolutionary pathways available for resistance development. Future research directions should focus on optimizing delivery platforms, developing predictive AI models for identifying optimal gene targets [4], and establishing standardized biofilm quantification metrics to enable direct comparison across therapeutic platforms. Through such integrated approaches, the scientific community can develop effective anti-biofilm strategies that mitigate resistance evolution and address the growing challenge of treatment-resistant infections.
Benchmarks of Success: Quantitative Metrics for Comparing Anti-Biofilm Efficacy
In the challenging fight against biofilm-associated infections and contamination, accurately measuring biofilm biomass and cellular viability is fundamental for validating new control strategies, from novel chemical treatments to advanced genetic tools like CRISPR editing. Biofilms are structured communities of microorganisms embedded in a self-produced extracellular polymeric substance (EPS) matrix, which confers inherent resistance to antimicrobials and disinfectants [62] [3]. This resilience makes biofilms a persistent problem in clinical, industrial, and food processing settings, responsible for up to two-thirds of hospital-acquired infections and recurring contamination in food production lines [62] [63].
To assess the efficacy of antibiofilm treatments, researchers rely on a suite of complementary metrics, each quantifying a distinct aspect of the biofilm lifecycle. Crystal violet (CV) staining measures total adhered biomass, including cells and the EPS matrix. Colony forming unit (CFU) counts determine the number of viable, cultivable bacteria. Metabolic assays, such as those using tetrazolium dyes, gauge the metabolic activity of the biofilm community [62] [64]. Understanding the strengths, limitations, and appropriate contexts for each method is crucial for designing robust experiments, particularly when comparing the disruptive action of precision tools like CRISPR-Cas9 against that of broad-spectrum chemical treatments [4] [3]. This guide provides a comparative overview of these core techniques, enabling researchers to select the optimal metrics for validating biofilm reduction.
Comparative Analysis of Core Metrics
The table below summarizes the primary purpose, key outputs, advantages, and limitations of the three main biofilm assessment methods.
Table 1: Comparison of Primary Biofilm Assessment Methods
| Method | Primary Measurement | Key Outputs | Fundamental Advantages | Inherent Limitations |
|---|---|---|---|---|
| Crystal Violet (CV) Staining [62] [65] | Total adhered biomass (cells & EPS) | Absorbance (590 nm) proportional to total biomass | Inexpensive, simple protocol, high-throughput, good for adhesion strength studies | Does not distinguish live/dead cells; can stain abiotic debris |
| CFU Counting [63] [64] | Number of viable, cultivable cells | Log10 CFU/cm² or CFU/mL | Gold standard for cultivable viability; direct, intuitive results | Labor-intensive; misses viable but non-culturable (VBNC) cells |
| Metabolic Assays (e.g., MTT) [62] [64] | Cellular metabolic activity | Absorbance (~570 nm for MTT) proportional to activity | Measures physiological activity, can detect early treatment effects | Activity not always equal to viability; signal can be matrix-inhibited |
The choice of metric directly influences the interpretation of an antibiofilm treatment's efficacy. For instance, a CRISPR-Cas9 system designed to disrupt a specific quorum-sensing gene might show a significant reduction in metabolic activity and CFU counts because it impairs bacterial communication and viability. However, it might result in a less pronounced reduction in CV staining if the physical structure of the biofilm and its EPS matrix remain initially intact [4] [3]. In contrast, a harsh chemical disinfectant like peracetic acid might rapidly degrade the EPS and kill cells, leading to strong reductions across all three metrics, though it may fail to eliminate all persister cells [63]. Using a combination of these metrics provides a more holistic and reliable validation of a treatment's effect, capturing both the physical dismantling of the biofilm and the functional elimination of its resident cells [62] [64].
Detailed Experimental Protocols
Crystal Violet Staining Protocol
The CV assay is a widely used, cost-effective method for quantifying total biofilm biomass. The following protocol is adapted for a standard 96-well microplate format [65].
- Reagent Preparation: Prepare a 0.1% (w/v) crystal violet solution in distilled water or phosphate-buffered saline (PBS). Stir thoroughly until completely dissolved. The solution can be stored in a dark container due to light sensitivity [65].
- Biofilm Staining:
- Growth and Treatment: Grow biofilms in a 96-well plate under desired conditions and apply experimental treatments (e.g., CRISPR-delivery nanoparticles or chemical disinfectants).
- Fixing: Gently remove the culture medium and any non-adherent cells. Wash the biofilms once with PBS. Add enough methanol to cover the biofilm (typically ~100 µL/well) and incubate for 15 minutes at room temperature.
- Staining: Remove the methanol and allow the plate to air dry completely. Add 0.1% crystal violet solution (100-150 µL/well) and incubate for 15-30 minutes at room temperature.
- Post-Staining Handling and Quantification:
- Washing: Carefully discard the stain and rinse the plate thoroughly under running tap water until the runoff is clear. This critical step removes unbound dye.
- Elution: Add 100-150 µL of a solubilization solvent (e.g., 95% ethanol, 1% acetic acid, or 10% sodium citrate) to each well. Incubate for 10-30 minutes with shaking to fully dissolve the crystal violet bound to the biofilm.
- Absorbance Measurement: Transfer 100 µL of the eluted dye to a new microplate if necessary. Measure the absorbance at 590 nm using a microplate reader. Higher absorbance values correlate with greater biofilm biomass [65].
CFU Counting Protocol
The CFU count is the definitive method for enumerating viable and cultivable bacteria within a biofilm and is often used as a benchmark for log-reduction calculations in disinfectant testing [63] [64].
- Biofilm Harvesting:
- Growth on Surfaces: Grow biofilms on relevant surfaces (e.g., stainless steel coupons, plastic, or rubber placed in a multi-well plate).
- Dislodging Cells: After treatment, transfer the biofilm-covered coupon to a tube containing a known volume of sterile PBS or neutralizing broth (to inactivate the treatment). Vigorously vortex the tube for 1-2 minutes. For more robust biofilms, sonication in a water bath (e.g., 5-10 minutes) or physical scraping with a sterile tip may be required to dislodge cells.
- Serial Dilution and Plating:
- Prepare Dilutions: Perform a 10-fold serial dilution of the harvested biofilm suspension in sterile PBS or dilution buffer.
- Plate Aliquots: Spread plate 50-100 µL aliquots from appropriate dilutions (e.g., 10⁰ to 10⁻⁵) onto nutrient-rich agar plates. Ensure even distribution.
- Incubation: Invert the plates and incubate at the optimal temperature for the organism (e.g., 37°C for 24-48 hours).
- Enumeration and Calculation:
- Count Colonies: Count the colonies on plates that contain between 30 and 300 colonies.
- Calculate CFU/mL or CFU/cm²: Calculate the CFU/mL of the original suspension using the dilution factor and plated volume. To determine surface density (CFU/cm²), divide the total CFU by the surface area of the coupon [64].
Metabolic Assay (MTT) Protocol
Metabolic assays measure the activity of cellular enzymes, providing an indicator of biofilm cell viability [62] [64].
- Principle: The yellow tetrazolium salt MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) is reduced to purple formazan by metabolically active cells, primarily by NAD(P)H-dependent oxidoreductase enzymes.
- Procedure:
- Biofilm Preparation: Grow and treat biofilms in a 96-well plate as described for the CV assay.
- MTT Incubation: Prepare a 0.5 mg/mL solution of MTT in PBS or culture medium without phenol red. Remove the medium from the biofilm wells, add the MTT solution (~100 µL/well), and incubate for 1-4 hours at 37°C in the dark.
- Solubilization: Carefully remove the MTT solution. Add 100-150 µL of an organic solvent like dimethyl sulfoxide (DMSO) to each well to dissolve the insoluble purple formazan crystals. Agitate the plate gently for a few minutes.
- Quantification: Measure the absorbance at a wavelength of 570 nm, with a reference wavelength of 630-650 nm to correct for background. The intensity of the color formed is proportional to the metabolic activity of the cells in the biofilm [64].
Experimental Design and Workflow
Integrating these metrics into a coherent experimental workflow is essential for robustly validating new antibiofilm strategies. The diagram below outlines a logical sequence for applying these assays.
Figure 1: A sequential workflow for comprehensive biofilm analysis. Note that each assay typically requires separate, destructively harvested samples to avoid interference.
When designing an experiment to compare CRISPR-based and chemical treatments, several factors are critical. First, biofilm cultivation conditions profoundly impact the results. Media composition, growth surface, and incubation time dramatically alter biofilm architecture, EPS composition, and, consequently, staining patterns and disinfectant tolerance [62] [63]. Second, defining success requires context. In the medical sector, a ≥5 log10 reduction in CFU is often the benchmark for effective disinfection, whereas a ≥4 log10 reduction may be acceptable in veterinary or food-industrial settings [63]. These targets should guide the selection of treatment concentrations and the interpretation of CFU data. Finally, the choice of analysis techniques should be hypothesis-driven. For instance, confocal laser scanning microscopy (CLSM) and scanning electron microscopy (SEM) can visually confirm the architectural disruption suggested by a drop in CV staining, providing powerful supplementary evidence [64].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents and Materials for Biofilm Research
| Item | Function/Application | Example Use in Protocol |
|---|---|---|
| Crystal Violet Powder [65] | Preparation of staining solution for total biomass quantification. | Make a 0.1% solution in PBS for the CV staining assay. |
| MTT Reagent [64] | Tetrazolium salt used to assess metabolic activity in biofilms. | Prepare a 0.5 mg/mL solution to incubate with biofilms. |
| 96-well Microtiter Plates [62] [65] | Standard platform for high-throughput biofilm growth and staining assays. | Used for CV, MTT, and other dye-based static biofilm assays. |
| Solubilization Solvents (e.g., Ethanol, DMSO) [65] | Dissolve bound CV or formazan crystals for absorbance measurement. | Ethanol (95%) elutes CV; DMSO solubilizes MTT formazan. |
| Neutralizing Broth [63] | Halts antimicrobial action after contact time for accurate CFU counting. | Used in suspension and surface tests to neutralize disinfectants like glutaraldehyde. |
| Test Surfaces (e.g., Stainless Steel Coupons) [63] [64] | Substrate for growing relevant biofilms, mimicking real-world conditions. | Coupons are placed in wells, incubated with bacteria, and harvested for CFU. |
The objective comparison of biomass and viability metrics reveals that no single assay can fully capture the complex response of a biofilm to a novel intervention. Crystal violet staining, CFU counts, and metabolic assays each provide a unique and valuable perspective. The most robust experimental designs, particularly those validating the precision of CRISPR-based tools against the brute force of chemical treatments, will strategically employ a combination of these methods. This multi-faceted approach enables researchers to confidently dissect the mechanism of action, distinguishing between the mere physical removal of biomass and the true functional eradication of the biofilm community, thereby driving the development of more effective antibiofilm strategies.
The structural integrity of bacterial biofilms, primarily governed by the extracellular polymeric substance (EPS) matrix, is a critical determinant in their resilience. Analyzing this structure is paramount in diverse fields, from clinical drug development to industrial biofilm control. This guide objectively compares the performance of two cornerstone imaging techniques—Confocal Laser Scanning Microscopy (CLSM) and Scanning Electron Microscopy (SEM)—in the context of quantifying EPS architecture and biofilm integrity. The evaluation is framed within a modern research paradigm focused on validating novel biofilm reduction strategies, contrasting the precision of CRISPR-based genetic interventions with conventional chemical treatments. We provide structured comparisons, detailed experimental protocols, and essential reagent information to support researchers in selecting and implementing the most appropriate analytical methods for their specific applications.
Technique Comparison: CLSM vs. SEM
Table 1: Core Performance Comparison of CLSM and SEM
| Feature | Confocal Laser Scanning Microscopy (CLSM) | Scanning Electron Microscopy (SEM) |
|---|---|---|
| Primary Function | 3D visualization of biofilm architecture; cell viability distribution [66] [67] | High-resolution imaging of biofilm surface morphology and ultrastructure [66] |
| Resolution | Single-cell level (lower than SEM) [66] | High resolution (typically 50-100 nm), detailed surface morphology [66] |
| Imaging Dimension | 3D and real-time 4D (time-dependent variation) [66] | Primarily 2D surface imaging (3D structure inference possible) [66] |
| Sample Environment | Hydrated, living biofilms under physiological-like conditions [66] | High vacuum, requiring extensive dehydration [66] |
| Viability Assessment | Yes, via live/dead fluorescent staining (e.g., SYTO9/PI) [67] | No, provides structural information only |
| EPS Matrix Integrity | Can be preserved during imaging; matrix components can be stained [68] | Risk of EPS collapse and biofilm shrinkage due to dehydration [66] |
| Key Quantitative Outputs | Biofilm thickness, biovolume, roughness, live/dead cell ratio [66] [67] | Qualitative assessment of surface coverage; quantitative data requires specialized software [66] [40] |
| Best Application | Assessing the physiological state and 3D structure of biofilms in situ, evaluating treatment efficacy. | Detailed analysis of biofilm-surface interactions, and morphological changes post-treatment. |
Experimental Protocols for Biofilm Analysis
Protocol for CLSM with Viability Staining
This protocol is designed for quantifying biofilm viability and 3D structure, particularly useful for evaluating antimicrobial treatments [67] [40].
- Biofilm Formation: Grow biofilms on relevant substrates (e.g., silicone tubes, glass coverslips, or plastic) under desired conditions and timeframes [40].
- Staining: Apply a fluorescent viability stain, such as the LIVE/DEAD BacLight Bacterial Viability Kit.
- Image Acquisition: Image the stained biofilm using a CLSM system (e.g., Leica Stellaris 5). Acquire Z-stacks to capture the entire 3D structure of the biofilm [67].
- Automated Image Analysis:
- Use open-source software like Fiji/ImageJ for analysis [67].
- Import the Z-stack and separate the green (SYTO9) and red (PI) channels.
- Apply an automated threshold to each channel to differentiate bacterial cells from the background.
- Use particle analysis to quantify the area or volume of live (green) and dead (red) signals.
- Calculate the percentage of dead bacteria as:
(Area/Volume of Red Signal) / (Area/Volume of Total Signal) * 100%[40]. This automated method reduces operator variability compared to manual counting [67].
Protocol for SEM Analysis of Biofilm Ultrastructure
This protocol details the steps for preparing and imaging biofilms to examine their surface morphology and integrity after treatments like shockwave therapy [40].
- Fixation: Wash the biofilm sample with phosphate-buffered saline (PBS) and fix with 2.5% glutaraldehyde in PBS for at least 2 hours at 4°C. This process cross-links and preserves the biofilm structure [40].
- Dehydration: Gradually dehydrate the fixed biofilm using a series of ethanol washes (e.g., 30%, 50%, 80%, and 100% ethanol), typically for 2 minutes at each concentration. This step removes water to prepare the sample for the microscope's vacuum [40].
- Drying: Critical Point Drying is the gold standard to minimize structural collapse associated with air drying. Alternatively, samples can be air-dried from 100% ethanol [66].
- Sputter-Coating: Coat the completely dried sample with a thin layer of conductive material, such as gold or platinum, to prevent charging under the electron beam [66].
- Image Acquisition and Analysis: Observe and image the biofilm under SEM at various magnifications. Qualitative assessment involves comparing structural features (e.g., presence of EPS, cell morphology, detachment) between treated and control samples. For quantitative analysis, specialized software can be used to measure parameters like surface coverage from the images [66] [40].
Diagram 1: Experimental workflow for biofilm structural analysis, comparing sample preparation and imaging paths for CLSM and SEM.
Application in Biofilm Reduction Research
Quantitative Data from Intervention Studies
Table 2: Efficacy Metrics of Biofilm Disruption Strategies
| Biofilm Reduction Strategy | Target / Mechanism | Analytical Technique | Key Quantitative Outcome | Experimental Reference |
|---|---|---|---|---|
| Shockwave + Ciprofloxacin | Physical matrix disruption + antibiotic | CLSM (Viability) | 67% dead bacteria [40] | P. aeruginosa biofilm in silicone tube |
| SEM / CV (Biomass) | 97.5% surface area removal [40] | |||
| Liposomal CRISPR-Cas9 | Targeted genetic disruption | Biomass Assay | >90% biofilm biomass reduction [3] | P. aeruginosa in vitro |
| CRISPR-Gold Nanoparticle | Enhanced gene editing delivery | Editing Efficiency | 3.5-fold increase in efficiency [3] | In vitro bacterial culture |
Technique Selection for Intervention Validation
Different biofilm disruption strategies necessitate specific analytical techniques for comprehensive validation.
For CRISPR & Genetic Interventions: CLSM is indispensable for assessing the physiological consequences of genetic edits. It can quantify cell death within the biofilm population following the targeted disruption of genes essential for EPS production (e.g., pel and psl genes in P. aeruginosa) [69] or quorum sensing [4]. SEM complements this by revealing if the genetic disruption has led to visible changes in the biofilm's physical structure, such as a less robust EPS matrix or failure to form microcolonies.
For Chemical & Physical Interventions: SEM excels at visualizing the physical damage caused by treatments like shockwaves, which can create microfractures in the EPS matrix [40], or by surfactants that disrupt lipid and protein components. CLSM is used post-treatment to determine whether the structural damage translates into effective bacterial killing, using live/dead stains to quantify the reduction in viable biomass [40].
Diagram 2: A decision pathway for selecting between CLSM and SEM based on the primary mechanism of the biofilm intervention being studied.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Biofilm Structural Analysis
| Reagent / Kit | Function / Application | Key Considerations |
|---|---|---|
| LIVE/DEAD BacLight Viability Kit | Differential staining of live (SYTO9) and dead (PI) bacteria in CLSM [67] [40]. | Propidium iodide can stain extracellular DNA, requiring separate channel analysis to avoid false positives [67]. |
| Glutaraldehyde Solution | Primary fixative for SEM samples; cross-links and preserves biofilm structure [40]. | Requires careful handling; fixation time and concentration are critical for optimal structural preservation. |
| Crystal Violet (CV) Stain | Total biofilm biomass quantification via colorimetric assay [64] [40]. | Does not distinguish between live and dead cells; stains both cells and EPS matrix [64]. |
| CRISPR-Cas9 System (with gRNA) | For precision targeting of biofilm-specific genes (e.g., for EPS production or quorum sensing) [4]. | Requires efficient delivery systems (e.g., nanoparticles) to penetrate the biofilm matrix [3]. |
| Lipid-based Nanoparticles | Carriers for delivering CRISPR-Cas9 components into bacterial cells within biofilms [3]. | Enhance cellular uptake and protect genetic material from degradation [3]. |
| Tryptic Soy Broth (TSB) / Agar (TSA) | Universal medium for cultivation of a wide range of bacteria (e.g., P. aeruginosa, E. coli) and subsequent CFU analysis [64] [40]. | Standardized growth conditions are vital for reproducible biofilm formation across experiments. |
Biofilms, which are structured communities of microorganisms protected by a self-produced extracellular matrix, pose a significant challenge in both clinical and industrial settings due to their inherent resistance to conventional antimicrobial treatments [40]. Validating the reduction of these resilient structures requires sophisticated metrics that can accurately quantify both physical disruption and biological pathway inhibition. The field of biofilm management is evolving beyond traditional chemical treatments toward advanced approaches, including targeted genetic interventions using CRISPR technology and innovative physical disruption methods [70] [71]. This guide provides an objective comparison between emerging CRISPR-based genetic approaches and established/novel chemical treatments for biofilm reduction, focusing on experimental validation through gene expression profiling and pathway disruption analysis. We present structured quantitative data, detailed methodologies, and analytical frameworks to enable researchers to rigorously evaluate the performance of these divergent strategies within a comprehensive validation paradigm.
The evaluation of biofilm treatment efficacy relies on multiple quantitative metrics that capture different aspects of biofilm disruption and cellular response. The following tables summarize key performance indicators for both CRISPR-mediated genetic approaches and chemical/physical treatment methods, based on recent experimental findings.
Table 1: Performance Metrics of Biofilm Physical and Chemical Treatments
| Treatment Method | Biofilm Reduction | Bacterial Viability Reduction | Key Measurement Techniques | Experimental Model |
|---|---|---|---|---|
| Shockwave + Ciprofloxacin [40] | 97.5% surface area detachment | 40% (CFU), 67% dead bacteria (CLSM) | CFU, CLSM, SEM, Crystal Violet | Pseudomonas aeruginosa in silicone tube |
| Antimicrobial Agents [70] | Dominant market share (35.2%) | Broad-spectrum efficacy | Standardized clinical assessment | Healthcare-associated infections |
| Nanoparticle-based Treatment [70] | Fastest-growing segment | Enhanced penetration capability | Market growth analysis | Various biofilm models |
Table 2: Performance Metrics of CRISPR-Based Genetic Approaches
| CRISPR Application | Editing Efficiency | Key Outcome | Validation Methods | Experimental Model |
|---|---|---|---|---|
| hATTR Treatment [49] | ~90% protein reduction | Sustained response (2+ years) | Functional assessments, protein quantification | Human clinical trial |
| HAE Treatment [49] | 86% kallikrein reduction | 8/11 patients attack-free | Protein quantification, clinical symptom tracking | Human clinical trial |
| LNP-SNA Delivery [72] | 3x improvement | Enhanced precision, reduced toxicity | Sequencing, functional assays | Various human cell lines |
| Prime Editing COL17A1 [73] | Up to 60% | Protein restoration, selective advantage | Sequencing, xenograft assessment | Patient keratinocytes |
Table 3: Analytical Techniques for Validating CRISPR Edits
| Analytical Method | Primary Application | Key Advantages | Limitations |
|---|---|---|---|
| RNA-sequencing [74] | Transcriptome-wide change detection | Identifies unanticipated transcriptional changes | More complex than targeted methods |
| Trinity Analysis [74] | De novo transcript assembly | Detects exon skipping, fusion events | Computational resource requirements |
| Sanger Sequencing [75] | Target site mutation confirmation | Standardized, accessible | Limited to PCR-amplified regions |
| T7 Endonuclease I Assay [75] | Mutation detection | No sequencing required | Lower resolution than sequencing |
Experimental Protocols for Method Validation
CRISPR Gene Editing and RNA-seq Validation
The comprehensive validation of CRISPR-mediated gene knockdown requires a multi-layered approach that extends beyond simple DNA sequencing to fully characterize on-target efficiency and unintended transcriptional consequences.
Protocol: CRISPR Knockdown and RNA-seq Analysis [74]
CRISPR Delivery and Cell Line Creation:
- Transfert target cells with plasmids encoding Cas9 nuclease and specific guide RNAs (gRNAs) using electroporation systems such as the NEON transfection system.
- For in vivo delivery, utilize lipid nanoparticles (LNPs), which show natural affinity for liver cells and enable systemic administration via IV infusion [49].
- Select successfully transfected cells using appropriate antibiotics (e.g., puromycin at 2μg/mL) and isolate single-cell clones through limiting dilution in 96-well plates.
DNA-Level Validation:
- Extract genomic DNA from monoclonal populations.
- Perform PCR amplification of the target region using flanking primers and analyze mutations via Sanger sequencing.
- For heterogeneous populations, clone PCR products using systems like the TOPO TA Cloning Kit and sequence multiple bacterial colonies to assess editing diversity.
RNA Sequencing and Analysis:
- Extract total RNA from CRISPR-treated and control cells using high-purity isolation kits.
- Prepare sequencing libraries and perform deep RNA-sequencing (minimum 50-100 million reads per sample) to ensure sufficient depth for transcript characterization.
- Process raw sequencing data through quality control (FastQC), alignment (STAR), and differential expression analysis (DESeq2).
- Conduct de novo transcript assembly using Trinity to identify aberrant transcriptional events not detectable through DNA-based methods alone, including:
- Inter-chromosomal fusion events
- Exon skipping
- Chromosomal truncations
- Unintentional modification of neighboring genes
Functional Validation:
- Perform quantitative RT-PCR on candidate genes to confirm RNA-seq findings using SYBR Green-based detection with primers spanning exon-exon junctions.
- Analyze protein expression changes via Western blotting to confirm functional knockdown when antibodies are available.
- For biofilm-specific applications, assess phenotypic consequences through biofilm formation assays and metabolic activity measurements.
Combined Shockwave and Antibiotic Treatment
The physical disruption of biofilms using shockwave technology represents a complementary approach to genetic interventions, particularly for device-related infections.
Protocol: Shockwave-Mediated Biofilm Disruption [40]
Biofilm Formation:
- Cultivate Pseudomonas aeruginosa in tryptic soy broth overnight at 37°C.
- Circulate bacterial culture through silicone tubes (inner diameter: 4mm) using a peristaltic pump system for 72 hours at 35°C with continuous fresh medium supplementation and aeration to establish mature biofilms.
Shockwave Treatment:
- Cut biofilm-colonized tubing into 3cm segments and immobilize in saline-filled tubes maintained at 37°C.
- Insert a Shockwave C2+ intravascular lithotripsy (IVL) balloon catheter into the tubing, ensuring contact with the inner wall.
- Apply shockwave treatment at 4kV energy, delivered at 2Hz frequency for 120 pulses (total duration: 60 seconds).
Antibiotic Application:
- Immediately following shockwave treatment, expose biofilms to ciprofloxacin (4μg/mL) for 6 hours at 37°C.
- Include appropriate controls: untreated biofilms and sham-treated biofilms (catheter insertion without shockwave activation).
Efficacy Assessment:
- Bacterial Viability: Detach bacteria via sonication and vortexing, then quantify viable cells through colony-forming unit (CFU) assays on tryptic soy agar plates after 24-hour incubation at 37°C.
- Biofilm Biomass: Assess biofilm detachment using crystal violet staining, with optical density measurement at 600nm after ethanol dissolution of stained biofilm.
- Structural Integrity: Visualize biofilm architecture changes using scanning electron microscopy (SEM) with standard glutaraldehyde fixation and ethanol dehydration protocols.
- Live/Dead Staining: Differentiate viable and non-viable bacteria using SYTO9/PI staining and confocal laser scanning microscopy (CLSM), with quantitative image analysis via ImageJ software.
Signaling Pathways and Workflows
CRISPR-Cas9 Mechanism and DNA Repair Pathways
The following diagram illustrates the core mechanisms of CRISPR-Cas9 gene editing and the subsequent cellular DNA repair pathways that determine editing outcomes, which are crucial for understanding both on-target and unintended effects in biofilm-related applications.
Biofilm Treatment Validation Workflow
This workflow outlines the integrated experimental approach for comparing CRISPR genetic interventions with chemical/physical treatments for biofilm disruption, highlighting key validation metrics at each stage.
Research Reagent Solutions Toolkit
Table 4: Essential Research Reagents and Tools for Biofilm and CRISPR Studies
| Reagent/Tool | Primary Function | Application Examples | Key Considerations |
|---|---|---|---|
| Lipid Nanoparticles (LNPs) [49] [72] | In vivo CRISPR delivery | Liver-targeted therapies (hATTR, HAE) | Natural liver affinity; enables redosing |
| LNP-SNAs [72] | Enhanced CRISPR delivery | Various cell types (3x efficiency improvement) | Reduced toxicity vs. standard LNPs |
| CRISPR-Cas9 Systems [74] [75] | Target gene knockout | Gene function studies, therapeutic knockdown | Specificity variants (HiFi Cas9) reduce off-targets |
| RNA-seq Reagents [74] | Transcriptome profiling | CRISPR off-target detection, pathway analysis | Deep sequencing required for fusion detection |
| Trinity Software [74] | De novo transcript assembly | Identification of aberrant splicing events | Computational resource intensive |
| Shockwave IVL System [40] | Physical biofilm disruption | Tubular structure decontamination | Requires combination with antibiotics |
| CLSM with LIVE/DEAD Staining [40] | Bacterial viability quantification | Treatment efficacy assessment | Distinguishes live/dead bacteria in biofilms |
| Crystal Violet Staining [40] | Biofilm biomass quantification | Anti-biofilm agent screening | Measures attachment, not viability |
Discussion: Integration of Validation Metrics
The comprehensive validation of biofilm reduction strategies requires correlating molecular-level interventions with phenotypic outcomes. CRISPR-based approaches offer precise genetic targeting but require sophisticated validation to account for unintended consequences, including large structural variations and transcriptomic changes that may not be detected by standard DNA sequencing methods [74] [76]. Physical and chemical methods provide immediate biofilm disruption but may lack the specificity of genetic interventions and typically require combination approaches for maximal efficacy, as demonstrated by the shockwave and antibiotic combined treatment [40].
Gene expression profiling serves as a critical bridge between these approaches, enabling researchers to identify pathway-level responses to both genetic perturbations and chemical treatments. The emerging field of structural nanomedicine, including LNP-SNA systems, demonstrates how delivery platform optimization can dramatically enhance the efficiency and safety of genetic interventions [72]. For comprehensive biofilm reduction validation, researchers should implement a multi-modal assessment strategy that integrates DNA-level editing confirmation, transcriptome-wide expression analysis, protein-level quantification, and functional phenotypic assays to fully characterize both intended and unintended consequences of intervention strategies.
When selecting between CRISPR genetic approaches and chemical/physical treatments for biofilm management, researchers must consider the specific application context, required precision, regulatory constraints, and the necessary depth of validation to ensure both efficacy and safety.
The escalating global health crisis of antimicrobial resistance is profoundly driven by the ability of bacterial pathogens to form biofilms. These structured microbial communities, encased in a protective extracellular matrix, exhibit dramatically enhanced resistance to conventional antibiotics, leading to persistent and often untreatable infections. [3] This challenge has catalyzed the exploration of novel therapeutic strategies, moving beyond traditional chemical treatments to more precise genetic interventions. Among the most promising developments is the integration of CRISPR-based gene editing systems with advanced nanoparticle delivery platforms. This paradigm shift represents a move from broad-spectrum antimicrobial activity to targeted, mechanism-based disruption of biofilm integrity and stability. The comparative analysis herein evaluates the efficacy, mechanisms, and limitations of emerging CRISPR-nanoparticle approaches against established chemical treatments, providing researchers and drug development professionals with a critical framework for therapeutic development. The objective validation of biofilm reduction metrics across these disparate modalities is essential for advancing the field and establishing standardized evaluation protocols for future anti-biofilm strategies.
Fundamental Mechanisms of Action
CRISPR-Nanoparticle Systems: Precision Genetic Targeting
The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas system, an adaptive immune mechanism in prokaryotes, has been repurposed as a powerful gene-editing tool. Its application in combating biofilms involves the precise targeting and disruption of genetic elements essential for biofilm formation, maintenance, and antibiotic resistance. [77] The system functions through a guide RNA (gRNA) that directs the Cas nuclease to specific DNA sequences, inducing double-strand breaks. [53] When deployed against biofilms, CRISPR-Cas can be programmed to target and disrupt critical genes, including those responsible for antibiotic resistance (e.g., blaNDM-1, blaKPC), quorum sensing pathways, and biofilm-regulating factors (e.g., ompA). [3] [12]
A significant breakthrough in the field has been the integration of CRISPR with nanoparticle (NP) delivery systems. Nanoparticles, including lipid-based, polymeric, and metallic varieties, serve as effective carriers that protect the CRISPR components from degradation and enhance their delivery into bacterial cells within the biofilm matrix. [3] This synergy is critical because the efficacy of CRISPR systems is wholly dependent on efficient delivery to the target cells. For instance, liposomal Cas9 formulations have demonstrated the ability to reduce Pseudomonas aeruginosa biofilm biomass by over 90% in vitro, while gold nanoparticle carriers have enhanced editing efficiency up to 3.5-fold compared to non-carrier systems. [3] These hybrid platforms can also facilitate the co-delivery of CRISPR components and antibiotics, producing synergistic antibacterial effects and superior biofilm disruption. [3]
Chemical Treatments: Broad-Spectrum Antimicrobial Activity
Chemical treatments encompass a wide range of agents, including conventional antibiotics, bacteriophages, enzymes, and surfactants. Their primary mode of action is often non-specific, targeting essential bacterial structures or functions without genetic precision. [7]
- Conventional Antibiotics: These agents, such as piperacillin/tazobactam and gentamicin, typically interfere with critical cellular processes like cell wall synthesis, protein production, or DNA replication. However, their efficacy is severely limited in biofilms due to reduced penetration through the extracellular polymeric substance (EPS) matrix and the presence of metabolically dormant "persister" cells. Biofilms can exhibit up to 1000-fold greater tolerance to antibiotics compared to their planktonic counterparts. [3]
- Bacteriophage Cocktails: Bacteriophages are viruses that infect and lyse specific bacteria. Their utility against biofilms lies in their ability to replicate at the infection site and produce enzymes (depolymerases) that degrade the EPS matrix. A 2025 study demonstrated that cocktails of lytic bacteriophages could achieve a 34.1% to 52.8% reduction in biofilm biomass of extensively drug-resistant K. pneumoniae and P. aeruginosa on various surfaces, including vascular catheters. [78]
- Enzymes and Disruptive Agents: Enzymes such as DNase, which degrades extracellular DNA (a key EPS component), and dispersin B, which targets polysaccharides, can physically destabilize the biofilm architecture. These are often used in combination with other antimicrobials to improve penetration. [7]
The following diagram illustrates the core mechanistic differences between these two strategic approaches.
Quantitative Efficacy Comparison
The following table summarizes key performance metrics for CRISPR-based and chemical anti-biofilm treatments, as reported in recent experimental studies.
Table 1: Comparative Efficacy of Anti-Biofilm Treatment Modalities
| Treatment Modality | Specific Agent/System | Target Pathogen | Reported Biofilm Reduction | Key Experimental Model |
|---|---|---|---|---|
| CRISPR-Nanoparticle | Liposomal Cas9 Formulation | Pseudomonas aeruginosa | >90% biomass reduction [3] | In vitro biofilm model |
| CRISPR-Nanoparticle | CRISPR-Gold Nanoparticle Hybrid | Bacterial Biofilms | 3.5x editing efficiency boost [3] | In vitro delivery efficiency |
| CRISPR Gene Editing | cas3 Gene Deletion | Acinetobacter baumannii | Significant reduction (p<0.05) [12] | Crystal violet staining & CLSM |
| CRISPR Gene Editing | smpB Gene Mutation | Acinetobacter baumannii | Significant reduction (p=0.0079) [79] | Crystal violet staining |
| Chemical Treatment | Lytic Bacteriophage Cocktail | K. pneumoniae (XDR) | 34.5% biomass reduction [78] | Catheter substrate, 24h treatment |
| Chemical Treatment | Lytic Bacteriophage Cocktail | P. aeruginosa (XDR) | 34.1-52.8% biomass reduction [78] | Catheter/Polystyrene, 24h treatment |
The data indicates a strong efficacy profile for CRISPR-based systems, particularly when combined with nanoparticles for enhanced delivery. The >90% biofilm biomass reduction achieved by liposomal Cas9 formulations represents a significantly higher level of efficacy compared to the ~34-53% reduction from advanced phage cocktails. [3] [78] Furthermore, genetic studies consistently show that targeted disruption of specific genes like cas3 and smpB in A. baumannii profoundly weakens biofilm formation, linking specific genetic targets to phenotypic outcomes. [12] [79]
Experimental Protocols and Methodologies
Protocol for CRISPR-Nanoparticle Anti-Biofilm Evaluation
A. Preparation of CRISPR-Loaded Nanoparticles
- Component Assembly: Synthesize or acquire the Cas9 nuclease and design guide RNAs (gRNAs) targeting specific biofilm-related genes (e.g., quorum sensing genes, antibiotic resistance genes like blaNDM-1, or virulence factors like ompA). [3]
- Nanoparticle Formulation: Encapsulate the CRISPR-Cas9 ribonucleoprotein (RNP) complex within lipid nanoparticles (LNPs). This involves mixing the RNP with lipid solutions in a specific ratio and using microfluidic devices to form stable, uniform nanoparticles. [3] Alternatively, conjugate the RNP to gold nanoparticles via thiol linkages. [3]
B. Biofilm Cultivation and Treatment
- Biofilm Formation: Grow biofilms of the target pathogen (e.g., P. aeruginosa or A. baumannii) in suitable media for 24-48 hours in static conditions on substrates like glass slides, polystyrene plates, or catheter segments to mimic clinical environments. [3] [78]
- Treatment Application: Apply the CRISPR-nanoparticle formulation to pre-formed biofilms. Include control groups treated with nanoparticles alone, free CRISPR, or standard antibiotics.
- Incubation: Incubate the treated biofilms for a defined period (e.g., 24 hours) to allow for bacterial uptake, gene editing, and phenotypic manifestation. [78]
C. Assessment of Biofilm Disruption and Viability
- Biomass Quantification (Crystal Violet Staining): Fix the biofilm with methanol, stain with 0.1% crystal violet for 15 minutes, destain with ethanol, and measure the optical density (OD) at 570-600 nm. Calculate percentage reduction compared to untreated controls. [12] [78] [79]
- Bacterial Viability (CFU Counting): Scrape the biofilm from the substrate, homogenize, serially dilute, and plate on agar. Count Colony Forming Units (CFU) after incubation to determine the reduction in viable cells. [78]
- Structural Analysis (Confocal Laser Scanning Microscopy - CLSM): Stain the biofilm with fluorescent dyes (e.g., SYTO9 for live cells, dextran-conjugates for EPS). Use CLSM to generate 3D reconstructions and quantify changes in biofilm thickness, biovolume, and structural integrity. [12]
- Editing Efficiency Confirmation: Extract genomic DNA from treated and control biofilms. Use sequencing (e.g., Sanger or NGS) to confirm the introduction of intended mutations at the target locus and to screen for potential off-target effects. [79]
Protocol for Chemical Anti-Biofilm Evaluation
A. Agent Preparation and Biofilm Cultivation
- Agent Preparation: Prepare working solutions of the chemical agent (e.g., bacteriophage cocktail at a titer of ≥10^7 PFU/mL, or antibiotic at a clinically relevant concentration). [78]
- Biofilm Formation: This step is identical to the CRISPR protocol, ensuring comparability. Grow biofilms on the chosen substrate for 24-48 hours. [78]
B. Treatment and Analysis
- Treatment Application: Apply the chemical agent to the pre-formed biofilms and incubate. For phage cocktails, a 24-hour incubation is typical. [78]
- Assessment: Use the same downstream analytical methods: crystal violet staining for total biomass, CFU counting for viability, and CLSM for structure. This methodological consistency is crucial for direct comparison. [78]
The experimental workflow for a head-to-head comparative study is visualized below.
The Scientist's Toolkit: Key Research Reagents
Successful execution of the described protocols requires a suite of specialized reagents and materials. The following table details essential solutions for anti-biofilm research.
Table 2: Essential Research Reagents for Anti-Biofilm Studies
| Reagent/Material | Function/Application | Specific Examples & Notes |
|---|---|---|
| CRISPR-Cas9 System | Precision gene editing within biofilm cells | Cas9 nuclease, gene-specific guide RNAs (gRNAs). Targets include cas3, smpB, ompA. [12] [79] |
| Nanoparticle Carriers | Delivery and protection of CRISPR components | Lipid Nanoparticles (LNPs), Gold Nanoparticles (AuNPs). Enhance stability and cellular uptake. [3] |
| Lytic Bacteriophages | Biological agent for degrading biofilms | Cocktails of phages specific to target pathogens (e.g., K. pneumoniae, P. aeruginosa). [78] |
| Crystal Violet | Histological dye for total biofilm biomass quantification | 0.1% solution; standard for colorimetric assessment after destaining. [12] [78] [79] |
| Fluorescent Stains | Visualization of biofilm components via CLSM | SYTO9 (labels live cells), Alexa Fluor-dextran (labels EPS). Enables 3D structural analysis. [12] |
| Biofilm Growth Substrates | Surfaces for in vitro biofilm formation | Glass slides, polystyrene plates, polyvinyl chloride (PVC) catheter segments. [78] |
Advantages and Limitations Analysis
CRISPR-Nanoparticle Systems
- High Precision and Specificity: Can target specific genes responsible for virulence, antibiotic resistance, and biofilm formation without indiscriminately harming beneficial microbiota or host cells. This approach aims to resensitize bacteria to antibiotics rather than simply killing them. [3]
- Potential for Synergistic Action: Nanoparticle platforms enable the co-delivery of CRISPR components and antibiotics, leading to enhanced biofilm disruption. The degradation of resistance genes can restore the efficacy of conventional drugs. [3]
Overcomes Delivery Challenges: Nanoparticles are engineered to penetrate the dense EPS matrix of biofilms, a significant barrier that limits the efficacy of many conventional antibiotics. [3]
Technical Complexity and Cost: The design, synthesis, and validation of gRNAs and nanoparticle formulations are technically demanding and currently more expensive than producing many conventional chemical agents. [3]
- Off-Target Effects: While specific, CRISPR systems can occasionally edit unintended genomic sites, potentially leading to unforeseen consequences. Ongoing research is focused on developing high-fidelity Cas variants to mitigate this risk. [80] [77]
- Immune Response and Safety: In vivo delivery may trigger immune reactions against the bacterial Cas9 protein or the nanoparticle carrier itself. Long-term safety and ecological impacts of releasing gene-editing constructs into the environment require thorough investigation. [53] [77]
- Regulatory Hurdles: As a novel biological therapy, the path to clinical approval and standardization is complex and not yet fully defined. [53]
Chemical Treatments
- Well-Established Protocols: The use of antibiotics and, increasingly, phage therapies is supported by decades of research, standardized protocols, and established (though evolving) regulatory frameworks. [7]
- Broad-Spectrum Activity: Many chemical antibiotics are effective against a wide range of bacterial species, which can be advantageous when the causative pathogen is unknown. [7]
Rapid Initial Killing: Can achieve a quick reduction in planktonic and surface-level bacteria.
Limited Biofilm Penetration and Efficacy: The EPS matrix effectively traps and impedes the diffusion of many antimicrobial agents, protecting the inner layers of the biofilm. This is a primary reason for the significantly lower percentage of biomass reduction seen in studies. [3] [78]
- Propagation of Resistance: The non-specific, lethal pressure exerted by broad-spectrum chemicals strongly selects for resistant mutants, a key driver of the AMR crisis. [3]
- Lack of Durability and Persister Cell Survival: Chemical treatments often fail to eradicate metabolically dormant persister cells, which can lead to biofilm regeneration once the treatment is discontinued. [7]
- Collateral Damage to Microbiome: Broad-spectrum agents can indiscriminately kill beneficial bacteria, disrupting the host's native microbiome and leading to secondary complications. [3]
The comparative analysis reveals a clear distinction in the operational paradigm and potential of these two strategies. Chemical treatments, while useful in certain contexts, face fundamental limitations against biofilms, particularly against the backdrop of rising multi-drug resistance. The future of biofilm treatment appears to be shifting towards mechanism-driven, precision medicine. The integration of CRISPR with nanotechnology represents a vanguard in this shift, offering the potential to target the genetic core of biofilm resilience. While challenges in delivery, safety, and regulation remain, the superior efficacy metrics and durable action seen in early studies provide a compelling rationale for continued investment and research. For the research community, the path forward involves standardizing biofilm reduction metrics across platforms, optimizing nanoparticle design for specific bacterial targets, and conducting rigorous in vivo validation studies to translate this promising technology from the lab to the clinic.
Conclusion
The comparative validation of CRISPR editing and chemical treatments reveals a complementary rather than competitive landscape for biofilm eradication. CRISPR-Cas9 offers unparalleled precision for targeting specific genetic determinants of biofilm formation and antibiotic resistance, with recent nanoparticle delivery systems enhancing its practicality. Chemical agents provide broader-spectrum matrix disruption and quorum sensing interference, often with more immediate applicability. The future of anti-biofilm therapy lies not in choosing one approach over the other, but in strategically combining their strengths—using CRISPR for precision genetic manipulation and chemical treatments for matrix disruption, potentially delivered via engineered nanocarriers. Future research must prioritize in vivo validation, standardized efficacy metrics, and overcoming delivery barriers to translate these promising technologies from the lab to the clinic, ultimately addressing the critical challenge of biofilm-associated antimicrobial resistance.
References
- 1. Microbial biofilm: formation, architecture, antibiotic resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8578483/]
- 2. The EPS Matrix: The “House of Biofilm Cells” - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2168682/]
- 3. integrating CRISPR/Cas9 and nanoparticles against biofilm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12366227/]
- 4. CRISPR–Cas systems as emerging tools for precision ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925021416]
- 5. Bacterial architects build the biofilm structures [https://www.nature.com/articles/s41579-024-01020-6]
- 6. Is Increased Biofilm Formation Associated with Decreased ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566419/]
- 7. Biofilms: structure, resistance mechanism, emerging control ... [https://pubs.rsc.org/en/content/articlehtml/2025/pm/d5pm00094g]
- 8. Antibacterial nanoagents: an emerging arsenal against ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12537890/]
- 9. Mini review: Persister cell control strategies [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1706115/full]
- 10. New ultrasound drug delivery system found to be highly ... [https://www.ox.ac.uk/news/2025-04-22-new-ultrasound-drug-delivery-system-found-be-highly-effective-against-bacterial]
- 11. Harnessing the Microbiome: CRISPR-Based Gene Editing ... [https://link.springer.com/article/10.1007/s12602-025-10573-8]
- 12. Cas3 of type I-Fa CRISPR-Cas system upregulates ... [https://www.nature.com/articles/s42003-025-08124-6]
- 13. Controlling biofilm development through cyclic di-GMP signaling [https://pmc.ncbi.nlm.nih.gov/articles/PMC9891824/]
- 14. Harnessing Cyclic di-GMP Signaling: A Strategic Approach ... [https://link.springer.com/article/10.1007/s00284-025-04091-7]
- 15. Echinacoside reduces intracellular c-di-GMP levels and ... [https://www.nature.com/articles/s41522-025-00673-2]
- 16. The role of cyclic di-GMP in biomaterial-associated infections ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0330229]
- 17. CRISPR-Cas Systems: Bridging Bacterial Immunity and ... [https://www.mdpi.com/2673-8007/5/4/118]
- 18. CRISPR/Cas‑mediated targeted gene editing of ... [https://www.spandidos-publications.com/10.3892/wasj.2025.349]
- 19. Comparative Analysis of Biofilm Formation and Antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382965/]
- 20. A bibliometric analysis and systematic review of drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12571837/]
- 21. Biofilm, resistance, and quorum sensing: The triple threat in ... [https://www.sciencedirect.com/science/article/pii/S2950194625003462]
- 22. Biofilm and Outer Membrane Vesicle Formation in ESKAPE ... [https://www.mdpi.com/1422-0067/26/20/9857]
- 23. ESKAPE pathogens rapidly develop resistance against ... [https://www.nature.com/articles/s41564-024-01891-8]
- 24. gRNA Design: How Its Evolution Impacted on CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10741458/]
- 25. Mechanism and Applications of CRISPR/Cas-9-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8388126/]
- 26. How To Design Guide RNA for CRISPR [https://www.synthego.com/guide/how-to-use-crispr/design-grna-crispr/]
- 27. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 28. Challenges in delivery systems for CRISPR-based genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8316527/]
- 29. CRISPR's efficiency triples with DNA-wrapped nanoparticles [https://news.northwestern.edu/stories/2025/09/crisprs-efficiency-triples-with-dna-wrapped-nanoparticles]
- 30. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 31. Nanoparticles approach to eradicate bacterial biofilm- ... [https://www.sciencedirect.com/science/article/abs/pii/S0045653521030757]
- 32. A general genome editing strategy using CRISPR lipid ... [https://pubmed.ncbi.nlm.nih.gov/40906807/]
- 33. CRISPR interference to interrogate genes that control ... [https://www.nature.com/articles/s41598-019-52400-5]
- 34. Strategy to combat biofilms: a focus on biofilm dispersal ... [https://www.nature.com/articles/s41522-023-00427-y]
- 35. Quorum Sensing Inhibitors: An Alternative Strategy to Win ... [https://www.mdpi.com/1420-3049/29/15/3466]
- 36. Mechanisms of Inhibition of Quorum Sensing as an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9143355/]
- 37. Extracellular matrix-degrading enzymes as a biofilm control ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10533455/]
- 38. Action of Antimicrobial Peptides against Bacterial Biofilms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6317029/]
- 39. Natural product-based inhibitors of quorum sensing [https://www.sciencedirect.com/science/article/pii/S2405580825001980]
- 40. Investigation of shockwave treatment for disruption ... [https://www.nature.com/articles/s41598-025-19955-y]
- 41. Current Status of In Vitro Models and Assays for ... [https://www.mdpi.com/2227-9059/7/2/34]
- 42. Beyond antibiotics: CRISPR/Cas9 triumph over biofilm- ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2024.1408569/full]
- 43. An overview of various methods for in vitro biofilm formation [https://pmc.ncbi.nlm.nih.gov/articles/PMC10533769/]
- 44. Interlaboratory study for the evaluation of three microtiter ... [https://www.nature.com/articles/s41598-021-93115-w]
- 45. In vitro model systems for exploring oral biofilms [https://pmc.ncbi.nlm.nih.gov/articles/PMC10505481/]
- 46. Flow environment and matrix structure interact to determine ... [https://elifesciences.org/articles/21855]
- 47. A High-Throughput Microtiter Plate Screening Assay to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10536743/]
- 48. Artificial Intelligence for CRISPR Guide RNA Design [https://arxiv.org/html/2508.20130v1]
- 49. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 50. Verification of CRISPR Gene Editing Efficiency [https://www.thermofisher.com/us/en/home/life-science/genome-editing/genome-editing-learning-center/genome-editing-resource-library/crispr-validated-protocols/verification-crispr-gene-editing-efficiency.html]
- 51. How to Pick the Best CRISPR Data Analysis Method for ... [https://www.synthego.com/guide/how-to-use-crispr/analysis-methods/]
- 52. Reduction of biofilm formation of Escherichia coli by ... [https://www.sciencedirect.com/science/article/pii/S1876034123001818]
- 53. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 54. Beyond the Resistome: Molecular Insights, Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561093/]
- 55. The Application of the CRISPR-Cas System in Antibiotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9356603/]
- 56. Antibiotic potentiators as a promising strategy for ... [https://www.nature.com/articles/s44259-025-00112-4]
- 57. Synergistic action of antimicrobial peptides and antibiotics [https://pmc.ncbi.nlm.nih.gov/articles/PMC11322369/]
- 58. Advancements in CRISPR/Cas systems for disease treatment [https://www.sciencedirect.com/science/article/pii/S2211383525003132]
- 59. CMN Weekly (14 March 2025) - Your ... [https://crisprmedicinenews.com/news/cmn-weekly-14-march-2025-your-weekly-crispr-medicine-news/]
- 60. Reinvigorating AMR resilience: leveraging CRISPR–Cas ... [https://tropmedhealth.biomedcentral.com/articles/10.1186/s41182-025-00728-2]
- 61. CRISPR Sequencing: Introduction, Workflow, and ... [https://www.cd-genomics.com/resource-crispr-sequencing-introduction-workflow-and-applications.html]
- 62. Critical Assessment of Methods to Quantify Biofilm Growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6022921/]
- 63. Evaluation of Biofilm Cultivation Models for Efficacy Testing ... [https://www.mdpi.com/2076-2607/11/3/761]
- 64. Biofilm formation and analysis of EPS architecture ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996925006118]
- 65. Crystal violet staining protocol [https://www.abcam.com/en-us/knowledge-center/cell-biology/crystal-violet-staining-protocol]
- 66. Microscopy Methods for Biofilm Imaging: Focus on SEM ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7828176/]
- 67. Biofilm viability checker: An open-source tool for automated ... [https://www.nature.com/articles/s41522-021-00214-7]
- 68. Detection techniques for extracellular polymeric substances in ... [https://bioresources.cnr.ncsu.edu/resources/detection-techniques-for-extracellular-polymeric-substances-in-biofilms-a-review/]
- 69. Contemporary strategies and approaches for ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11022105/]
- 70. Biofilm Treatment Market Trends, Share & Forecast, 2025- ... [https://www.coherentmarketinsights.com/industry-reports/biofilm-treatment-market]
- 71. Biofilms Treatment Market Size, Share, Trends, Report 2035 [https://www.marketresearchfuture.com/reports/biofilms-treatment-market-27062]
- 72. Scientists just made CRISPR three times more effective [https://www.sciencedaily.com/releases/2025/09/250907024543.htm]
- 73. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 74. Techniques for Validating CRISPR Changes Using RNA- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12027141/]
- 75. A simple validation and screening method for CRISPR/Cas9 ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0312722]
- 76. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 77. Advancements in CRISPR/Cas systems for disease treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC12254762/]
- 78. Efficacy of Phage Cocktails Against Biofilms Formed by ... [https://pubmed.ncbi.nlm.nih.gov/41229989/]
- 79. CRISPR/Cas9-targeted smpB mutation revealing roles in ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0329638]
- 80. Recent applications, future perspectives, and limitations of ... [https://www.sciencedirect.com/science/article/pii/S216225312500188X]